

Abstract
Immune-related adverse events, particularly severe toxicities such as myocarditis (MC), are major challenges to immune checkpoint inhibitor (ICI) utility in anti-cancer therapy1. The pathogenesis of ICI-myocarditis (ICI-MC) is poorly understood. Pdcd1−/−Ctla4+/− mice recapitulate clinicopathologic features of ICI-MC, including myocardial T cell infiltration2. Single cell RNA/T cell receptor (TCR) sequencing on the cardiac immune infiltrate of Pdcd1−/−Ctla4+/− mice identified clonal effector CD8+ T cells as the dominant cell population. Treatment with anti-CD8, but not anti-CD4, depleting antibodies rescued survival of Pdcd1−/−Ctla4+/− mice. Adoptive transfer of immune cells from mice with MC induced fatal MC in recipients which required CD8+ T cells. α -myosin, a cardiac specific protein absent from the thymus3,4, was identified as the cognate antigen source for three MHC-I restricted TCRs derived from mice with fulminant MC. Peripheral blood T cells from three patients with ICI-MC were expanded by α-myosin peptides, and these α-myosin expanded T cells shared TCR clonotypes with diseased heart and skeletal muscles, indicating that α-myosin may be a clinically important autoantigen in ICI-MC. These studies underscore the critical role for cytotoxic CD8+ T cells, are the first to identify a candidate autoantigen in ICI-MC and yield new insights into ICI toxicity pathogenesis.
Immune checkpoint inhibitors (ICIs) have drastically altered the treatment landscape and prognosis for many cancers. However, not all patients respond to treatment and many experience immune-related adverse events (irAEs), especially when ICIs are used in combination. Thus, preventing, diagnosing, and treating irAEs are urgent clinical challenges. Currently, clinically actionable biomarkers of response and toxicity are limited and the mechanistic basis of irAEs is poorly defined.
Myocarditis (MC) is an uncommon irAE, affecting <1% of ICI-treated patients, but has a mortality rate of nearly 50%1,5. Combination ICI therapy (with anti-PD-1 and anti-CTLA4) is the most well-established risk factor for ICI-myocarditis (ICI-MC)6-9. ICI-MC is pathologically characterized by predominance of T lymphocytes and macrophages in the heart and often co-occurs with myositis, with early studies suggesting common clonotypes of T lymphocytes in both tissues5. These data suggest the possibility of shared target antigens driving T lymphocyte expansion and activation, which would be critical for pathogenesis; however, experimental data have been lacking.
Generally, mice treated with ICIs do not replicate the full spectrum of irAEs seen in patients, limiting research on mechanisms of toxicity. We recently described a mouse model of ICI-MC in which C57BL6/J mice with homozygous knockout of Pdcd1 and heterozygous deletion of Ctla4 die prematurely and specifically due to MC, recapitulating clinical and pathological features of ICI-MC2. Severe inflammation is specific to the heart in these mice. By flow cytometry, the myocardial immune infiltrate is primarily composed of CD8+ T cells, similar to patients with ICI-MC. Furthermore, treatment with abatacept, a CTLA4 fusion protein, attenuates MC and increases survival in the mice, consistent with early clinical data from patients with ICI-MC treated with abatacept2,10. Here we utilize this mouse model of ICI-MC to characterize the immune infiltrates, establish CD8+ T cells as necessary for disease, and identify α-myosin as a cognate antigen for the most abundant TCRs in MC. Furthermore, we extend these findings to human disease and find that α-myosin expanded TCRs are present in inflamed cardiac and skeletal muscles in patients with ICI-MC.
Cardiac clonal CD8+ T cells are abundant
Fulminant MC affects 50% of Pdcd1−/−Ctla4+/− mice and was characterized by histologic destruction of the myocardial architecture (Fig. 1a, b)2. Incidence was higher in female mice, in line with data in patients that female sex is a risk factor for ICI-MC2,9. The myocardial immune infiltrate in affected mice was primarily composed of CD8+ T cells and did not differ by sex (Extended Data Fig. 1a)2. No significant antibody deposits or B220+ B cells were observed in the hearts of mice with MC, supporting further study of T cells at the site of disease (Extended Data Fig. 1b). We used single cell RNA and TCR sequencing to characterize sorted CD45+ infiltrating immune cells from six healthy wild type mouse hearts and four hearts from Pdcd1−/−Ctla4+/− mice affected by MC. Dimensionality reduction with uniform manifold approximation and projection (UMAP), clustering with Louvain and cluster cell type annotation assisted by SingleR showed distinct immune cell populations in MC compared to control (Fig. 1c; Extended Data Fig. 2a). The largest difference was seen in the activated T cell cluster, which made up 34% of the MC immune cells, and only 2% of the control immune cells. Markers of activation such as Ccl5, Ccl4, Tigit, Nkg7, and Gzmb were upregulated in the T cell clusters in MC compared to control T cell clusters (Extended Data Fig. 2b). Conversely, markers of naïve status such as Ccr7, Lef1 and Sell were upregulated in control T cells. Activation markers were also upregulated in other clusters, including myeloid cell subsets, in the MC samples (Extended Data Fig. 2c). Aw112010, a long noncoding RNA essential for the orchestration of mucosal immunity during infections and in colitis, was strongly upregulated in several clusters in the MC samples11,12. In contrast, B lymphocytes made up most of the immune cells in the control heart, consistent with previous studies13-15.
Figure 1. Single Cell RNA/TCR sequencing reveals abundant clonal effector CD8+ T cells in ICI-MC.

A) Phenotypic summary of mice with Pdcd1 and Ctla4 genetic loss. Pdcd1−/−Ctla4+/+ mice do not have an overt phenotype. Mice with complete loss of Ctla4 have a fatal lymphoproliferative disorder, regardless of Pdcd1 genotype. Pdcd1−/−Ctla4+/− mice develop fulminant MC. B) H&E of cardiac tissue from a healthy wild type mouse and a Pdcd1−/−Ctla4+/− mouse with MC. Scale bar represents 200μm. Representative of n=10 animals per genotype. C) Dimensionality reduction with UMAP of scRNAseq on sorted CD45+ immune cells from control wild type mouse hearts (n=6) compared to hearts (n=4) of Pdcd1−/−Ctla4+/− mice with MC (n= 2509 cells per genotype). Cell type annotations were assisted by singleR and are quantified on the right. D) UMAP is subset on cells with Cd3e expression >1.5 and presence of a TCR, and then clustered using the Louvain algorithm (n=1266 cells). The proportion of the control and MC T cells in each cluster is quantified on the right. E) Expression of key T cell identity genes Cd8a and Cd4 are shown for each T cell cluster. F) TCR density is a measure of how many of the 100 nearest neighbors share the same TCR α and β chain. TCR density is shown for each cluster and split by control or MC. G) Differential gene expression between cluster 0 T cells and all other T cell clusters (1,2,3, and 4). Higher expression in cluster 0 is indicated by positive fold change. Red indicates FDR-corrected p-value (q-value) <0.05. Black indicates not significant. h) Violin plots shown expression of key genes by clonality and sample. Clonal is defined as > 2 cells with the same TCR α and β chains. No clonal cells are seen in the control sample. Identity genes are light blue. Naïve T cells genes are dark blue. T cell activation genes are red.
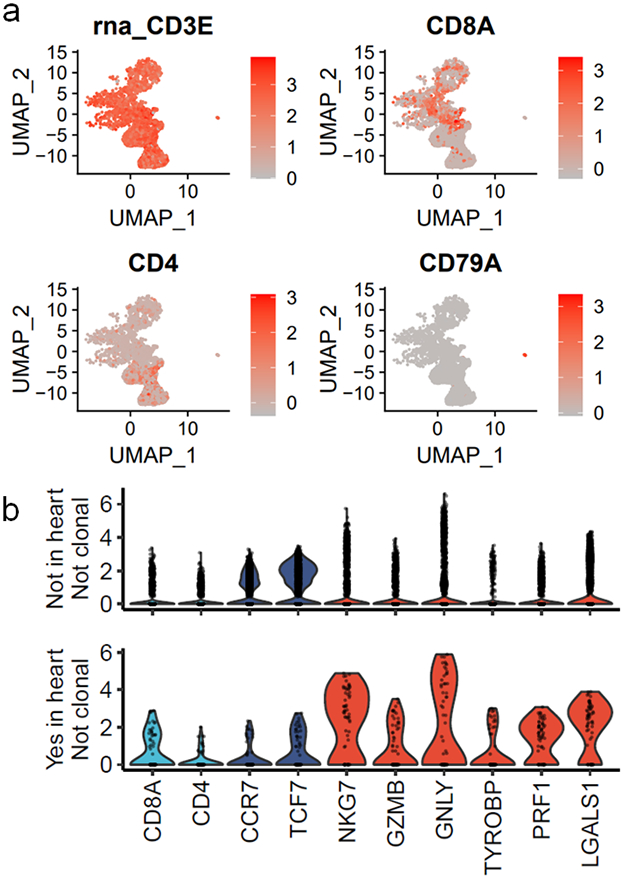
To further characterize T lymphocytes, we performed dimensionality reduction and clustering on Cd3e+ and TCR+ cells, which showed differential cluster abundance in MC compared to control (Fig. 1d). Differential gene expression analyses and plotting of key identity genes revealed distinct cluster identities (Fig. 1e; Extended Data Fig. 3a). Cluster 0 cells were activated effector CD8+ T cells expressing Gzmb, Ifng, and Nkg7. Cluster 1 cells were resting CD8+ T cells, expressing Ccr7, Sell, and Klf2. Cluster 2 cells were CD4+ T cells expressing Cd4, Ccr7, and Cd40lg. Cluster 3 cells were proliferative CD8+ T cells expressing Mki67, Cdk1 and Tk1. Cluster 4 cells were Cd24a expressing T cells and comprised a small fraction of the total T cells. CD24 is upregulated with TCR signaling and is necessary for T cell proliferative capacity16,17. Cluster 0 (effector CD8+ T cells) comprised the majority of the MC cells. Cluster 3 (proliferating CD8+ T cells) and cluster 0 were enriched in the MC sample relative to the control sample. Cluster 0 and cluster 3 also had the highest TCR density, defined as the number of neighboring cells with the same TCR α and β chain. High TCR density was limited to the MC sample (Fig. 1f). Genes upregulated in cluster 0 T cells included Ccl5, Nkg7, Ccl4, Cxcr6, Lag3, and Prf1 (Fig. 1g; Extended Data Fig. 3b). We also sought to investigate genes associated with tissue residency, as intravascular immune cells were not excluded in our tissue preparation protocol. We found substantial expression of Cd69, Itgb1, Itgal, Cxcr3, Cxcr6, and Runx3 in MC T cell clusters, and lower levels of these genes in control T cells (Extended Data Fig. 3c)18,19. These data suggest that MC T cells were CD8+, tissue-resident, 15effector and proliferating.
We next sought to assess the clonality of TCRs in the MC samples using both bulk and single cell TCR sequencing. Cardiac tissue from affected Pdcd1−/−Ctla4+/− mice had lower Shannon diversity than splenic tissue from healthy wild type mice or mice with MC, indicating a higher degree of clonal TCRs (Extended Data Fig. 3d), which did not differ by sex. No clonal (>2 cells with the same TCR clonotype) cells were identified by single cell TCR sequencing of healthy cardiac immune infiltrate. In contrast, 63% of TCR+ cells in the MC samples represented clonal TCRs (Extended Data Fig. 3e). Comparing gene expression by clonality showed that non-clonal cells from control samples expressed Cd8a, Cd4, and markers associated with naïve status, rather than activation. In contrast, clonal cells from MC samples expressed Cd8a and cytotoxicity genes such as Nkg7 and Gzmb, but not Cd4 or markers of naïve status (Fig. 1h). These data show that there was a large population of highly activated, clonally expanded CD8+ T cells in murine ICI-MC.
CD8+ T cells are necessary for myocarditis
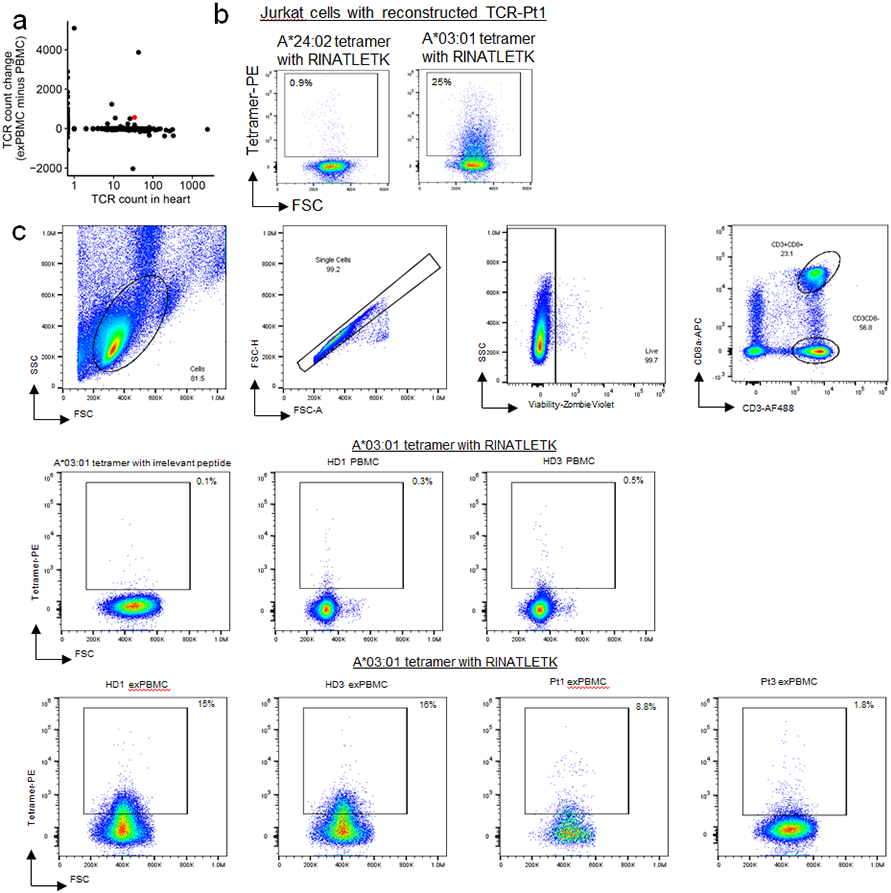
Early administration of corticosteroids, which are immunosuppressive through a variety of mechanisms, is associated with better survival in patients with ICI-MC, but severe cases can be refractory to steroids20,21. In our murine model, dexamethasone treatment did not attenuate MC or extend survival, consistent with a severe phenotype (Extended Data Fig. 4a). Using anti-CD8 and anti-CD4 depleting antibodies (starting at 21 days of age and administered three times weekly, confirmation shown in Extended Data Fig. 4b), we tested whether depletion of these cell subsets would attenuate MC and affect survival of Pdcd1−/−Ctla4+/− mice. Depletion of CD8+ cells, but not CD4+ cells, significantly rescued survival in these mice (Fig. 2a). Corroborating these data, adoptive transfer of whole splenocytes, but not splenocytes from which CD8+ cells were depleted (confirmed in Extended Data Fig. 4c), from Pdcd1−/−Ctla4+/− mice with MC to Rag1−/− recipients was sufficient to induce fatal MC (Fig. 2b-c). The single fatality in the CD8-depleted arm was likely due to a bowel obstruction with no evidence of MC histologically. Immunohistochemistry showed abundant cardiac infiltration of CD3+ and CD8+ cells, and limited CD4+ cells and F4/80+ cells, in the whole splenocyte recipients but not the CD8-depleted recipients (Fig. 2d; Extended Data Fig. 4d). We performed TCR β chain sequencing on the cardiac tissue of one donor mouse (Donor) and four whole splenocyte recipients (Rec1, Rec2, Rec3, and Rec4; Fig. 2e). High numbers (>2000) of TCR reads were seen in all sequenced hearts, indicating significant T cell infiltration, as expected from histology (Extended Data Fig. 4e). In 4/4 recipient mice, the most clonal TCR β chain occupied greater than 65% of the total cardiac TCR repertoire, indicating strong monoclonal expansion. The most clonal TCR β chain (CDR3: CASSLRRGEQYF) in the donor heart (which comprised 37% of the donor cardiac repertoire) was expanded in three of four recipients (Rec1, 3, 4). Interestingly, in one recipient mouse (Rec2), a low frequency TCR from the donor was expanded and occupied the majority of the TCR β chain repertoire (CDR3: CASSLGGTVQDTQYF). This high degree of expansion from donor to recipient cardiac tissue suggests a single TCR clonotype may drive MC in the recipient animals. Together, these results strongly indicated that CD8+ T lymphocytes are necessary for the development of MC.
Figure 2. CD8+ T cells are necessary for MC.

A) Pdcd1−/−Ctla4+/− mice were treated with anti-CD4, anti-CD8 or control antibodies. Antibody treatments were started at 21 days of age and administered three times weekly. Time is measured since birth, but no animals are censored prior to the start of the experiment at day 21. P =0.03, anti-CD8 v control, p=0.02, anti-CD8 v. anti-CD4, two-sided cox proportional hazard tests. Risk tables show size of groups. B) Whole splenocytes or splenocytes from which CD8 cells were depleted from Pdcd1−/−Ctla4+/− mice with MC were transferred to Rag1−/− recipient mice. Day 0 is the day of adoptive transfer. P=0.0017, two-sided cox proportional hazard test. Risk tables show size of groups. C) Representative H&E from CD8 depleted splenocyte recipients compared to whole splenocyte recipients. Only cardiac sections are shown. Scale bars show 50μm. Representative of n=10 animals per group. D) Representative IHC for CD8 on cardiac sections from CD8 depleted splenocyte recipients compared to whole splenocyte recipients. Scale bars show 50μm. Representative of n=10 animals per group. E) TCR β chain sequencing on cardiac tissue from a donor Pdcd1−/−Ctla4+/− mouse (Donor, in bold) and Rag1−/− whole splenocyte recipients (Rec1-4). The top ten most abundant TCRs from the donor plus the most abundant TCR from Rec2 are shown. Flow between samples indicates shared TCRs. Bolded CDR3s indicate most clonal TCRs.
Myocarditis TCRs recognize α-myosin
Next, we aimed to identify the cognate antigen for clonal murine TCRs. For antigen discovery, we tested five TCRs derived from single cell RNA/TCR sequencing and selected on the basis of their abundance. These TCRs (TCR-A and TCRs 1-4) were primarily associated with cluster 0 effector CD8+ T cells and cluster 3 proliferating T cells (Fig. 3a). We also included two TCRs which were expanded the hearts of recipient mice in adoptive transfer experiments (TCRs B and C). TCR-B was the most abundant TCR in the heart of the donor and three recipients (β CDR3: CASSLRRGEQYF). TCR-C was the most abundant TCR in the heart of recipient 2 (β CDR3: CASSLGGTVQDTQYF; Fig. 2e). CDR3 amino acid sequences, V genes, and J genes are shown in Table 1. These TCRs were reconstructed using Stitchr, cloned, and retrovirally transduced into Jurkat nuclear factor of activated T cells (NFAT)-GFP reporter cells22-24. Syngeneic bone marrow derived dendritic cells were used as antigen presenting cells (APCs).
Figure 3. α-myosin is an MHC-I restricted autoantigen in murine MC.
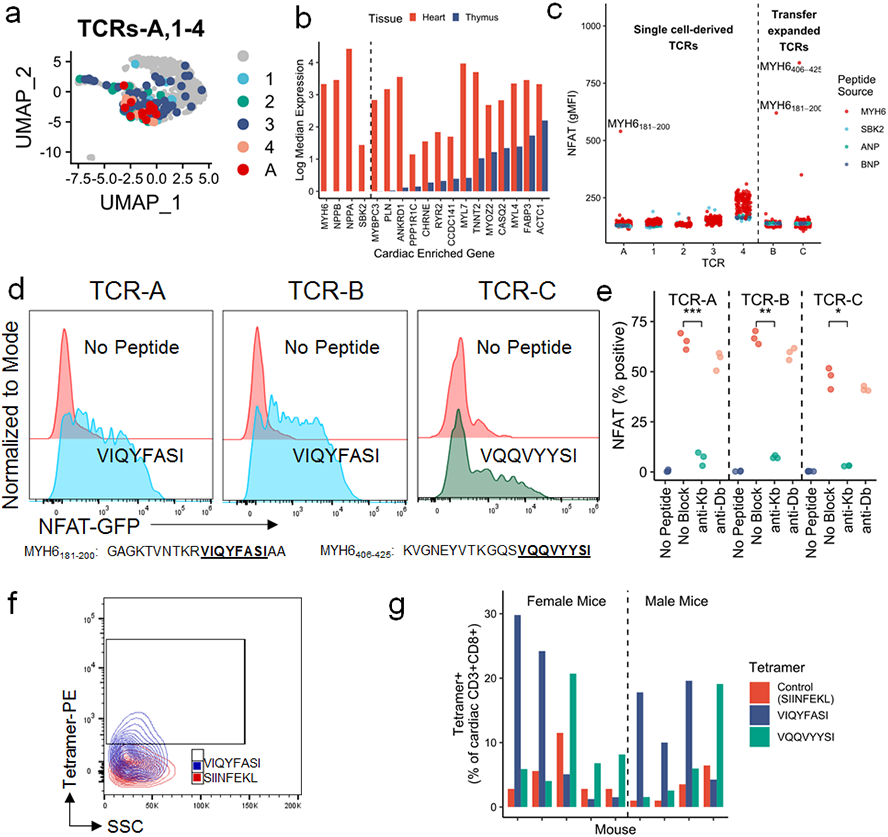
A) TCRs-A, 1-4, used for antigen discovery, shown on the same plot as Fig. 1d. Grey cells do not express TCRs A or 1-4. b) Log median expression of 18 cardiac enriched genes in the heart (red) and thymus (blue). Genes to the left of the dashed line have no detectable expression in thymic APCs. c) NFAT-GFP reporter activity, measured by flow cytometry and shown as geometric mean fluorescence intensity, of all TCR cell lines stimulated independently with 172, 10-20aa SBK2, ANP, BNP, or α-myosin peptides. TCRs to the left of the dotted line are derived from single cell sequencing data (see Fig. 3a). TCRs to the right of the dotted line were selected due to expansion in adoptive transfer experiments (see Fig. 2e). TCRs named with letters (A-C) have a cognate antigen identified whereas TCRs named with numerals (1-4) do not have an identified cognate antigen. Top α-myosin peptide hits are labeled. d) Representative (of n=3 independent replicates) flow cytometry histograms of each TCR cell line co-cultured with BMDCs and stimulated with 10μg/mL predicted cognate peptide relative to no peptide. Peptide sequences are shown in the table. e) Each TCR cell line was co-cultured with EL-4 APCs and 10μg/mL cognate peptide (VIQYFASI for TCRs A and B; VQQVYYSI for TCR C; except for no peptide controls) with or without 10μg/mL of anti-Kb or anti-Db blocking antibody. NFAT-GFP reporter activity is shown as percent of live cells. n=3 biological replicates. P=0.00035 (TCR-A), p=0.004 (TCR-B), p=0.013 (TCR-C), two-sided t-tests for no block to anti-Kb, adjusted for multiple comparisons. f) Representative flow cytometry of SIINFEKL (red) and VIQYFASI (blue) loaded H2-Kb tetramer staining on cardiac CD3+CD8+ cells. g) Quantification of control, VIQYFASI, and VQQVYYSI H2-Kb-tetramer staining in cardiac CD3+CD8+ cells in individual mice. Each group of three bars represents one mouse with MC. n=9 mice (n=5 female; n=4 male).
Table 1.
Summary of TCR CDR3, V and J genes for TCRs used in antigen discovery experiments.
| TCR ID | TCR Source |
β CDR3 | TRBV | TRBJ | α CDR3 | TRAV | TRAJ | Antigen |
|---|---|---|---|---|---|---|---|---|
| A | Murine single cell sequencing | CSAAWGGSAETLYF | TRBV1 | TRBJ2-3 | CAVSDRGSALGRLHF | TRAV7-3*04 | TRAJ18 | MYH6191-198 (VIQYFASI) |
| 1 | Murine single cell sequencing | CASSPGQGAYAEQFF | TRBV12-2 | TRBJ2-1 | CAVSSGYGSSGNKLIF | TRAV7-5 | TRAJ32 | Unknown (Not in α-myosin, SBK2, ANP, BNP) |
| 2 | Murine single cell sequencing | CASKTGYNYAEQFF | TRBV19 | TRBJ2-1 | CALNTGYQNFYF | TRAV4-4-DV10 | TRAJ49 | Unknown (Not in α-myosin, SBK2, ANP, BNP) |
| 3 | Murine single cell sequencing | CASGGLGGPSQNTLYF | TRBV12-2 | TRBJ2-4 | CALERSTGNYKYVF | TRAV13-1 | TRAJ40 | Unknown (Not in α-myosin, SBK2, ANP, BNP) |
| 4 | Murine single cell sequencing | CASSDAGYAEQFF | TRBV13-3 | TRBJ2-1 | CALGDSNYQLIW | TRAV6-6 | TRAJ33 | Unknown (Not in α-myosin, SBK2, ANP, BNP) |
| B | Adoptive transfer (Donor, Rec1,3,4) | CASSLRRGEQYF | TRBV15 | TRBJ2-7 | CALERASGSWQLIF | TRAV13-1 | TRAJ22 | MYH6191-198 (VIQYFASI) |
| C | Adoptive Transfer (Rec2) | CASSLGGTVQDTQYF | TRBV12-2 | TRBJ2-5 | CALGDRNNAGAKLTF | TRAV6D-6 | TRAJ39 | MYH6418-425 (VQQVYYSI) |
| TCR_pt1 | Pt 1 exPBMC (single cell sequencing) | CASSPYQSSGANVLTF | TRBV9 | TRBJ2-6 | CALSDRYGGATNKLIF | TRAV19 | TRAJ32 | MYH6443-451 (RINATLETK) |
We used a candidate autoantigen approach for TCR screening. Analysis of published RNA sequencing data on thymic APCs showed four cardiac enriched genes (genes where expression in the heart was significantly enriched relative to other tissues) with no detectable expression in the thymus (MYH6, NPPA, NPPB, SBK2; Fig. 3b)4. Lack of thymic expression would be predicted to enable self-reactive T cells to escape negative selection, an important mechanism of self-tolerance. Expression of these genes was also low to absent in the thymus of Pdcd1−/−Ctla4+/− mice and did not differ by sex (Extended Data Fig.5a). We used a library of 172 overlapping peptides, covering all of α-myosin (encoded by Myh6), ANP (encoded by Nppa), BNP (encoded by Nppb) and SBK2 (encoded by Sbk2; Extended Data Table 1). Three TCR cell lines, including both expanded TCRs (B and C), had NFAT activity in response to α-myosin peptides. None of the other three tested cardiac proteins activated any of the TCR cell lines (Fig. 3c). MYH6 (α-myosin) has been confirmed by other groups to not be expressed in the thymus in mice or humans and has been shown to be an MHC-II restricted autoantigen in mouse models3,25,26. Interestingly, 4/5 single cell-derived TCR cell lines did not recognize any of the tested cardiac peptides (denoted TCRs 1-4 to differentiate from TCRs A-C where cognate antigens were identified). These data suggest two important possibilities: 1) the presence of “bystander” T cells which are attracted to the site of inflammation but are not specific for disease-causing antigens or 2) the possibility that these TCRs might recognize other cardiac antigens which are important in disease pathogenesis. The bystander TCR hypothesis is well supported by prior literature in tumor immunology showing that a minority of tumor-infiltrating T cells are likely to be tumor specific27-31.
TCRs A and B activated NFAT reporters in response to the same α-myosin peptide (MYH6181-200), whereas TCR-C had NFAT activity against a distinct α-myosin peptide (MYH6406-425; Fig. 3c). From these 20 amino acid peptides, we used TepiTool to identify the most likely immunogenic epitopes32 and re-screened these epitopes against the reporter TCR lines. TCRs A and B recognized MYH6191-198 (VIQYFASI), while TCR-C recognized MYH6418-425 (VQQVYYSI; Fig. 3d). VIQYFASI and VQQVYYSI both had strong predicted binding to H2-Kb (Extended Data Table 2). The tyrosine and phenylalanine residues at position five of the peptides are known to be key binding epitopes for H2-Kb33. In line with these predictions, antibody blocking of H2-Kb, but not H2-Db, abrogated NFAT reporter activity for all three cell lines (Fig. 3e). Using an empty H2-Kb tetramer loaded with either VIQYFASI or VQQVYYSI, compared to a tetramer loaded with an irrelevant peptide (SIINFEKL), we found that 6-30% of the cardiac infiltrating CD8+ T cells were specific for one of the two α-myosin peptides in nine additional Pdcd1−/−Ctla4+/− mice with MC (Fig. 3f,g; Extended Data Fig. 5b). We did not identify any mice with MC lacking VIQYFASI or VQQVYYSI tetramer positive CD8+ T cells in their hearts, demonstrating the ubiquity of α-myosin reactive T cells in this murine model. Notably, high levels of α-myosin tetramer positive CD8+ T cells were confined to the hearts (Extended Data Fig. 5c). These data strongly suggest that α-myosin is an important MHC-I restricted autoantigen in murine immune checkpoint deficiency MC.
α-myosin TCRs are found in ICI-MC patients
We next aimed to test the relevance of α-myosin as a potential autoantigen in humans, using three healthy donors and three patients with histologically-proven fulminant ICI-MC. ICI-MC patient information is summarized in Table 2. First, we tested whether it was possible to expand α-myosin specific T cells from peripheral blood mononuclear cells (PBMCs). PBMCs were stimulated with α-myosin peptides or control cytomegalovirus, Epstein-Barr virus and influenza (CEF) peptides (in healthy donor PBMCs only) for 14 days to generate expanded PBMCs (exPBMC). TCR β chain sequencing was used to assess expansion. Shannon diversity decreased from pre-expansion PBMC to α-myosin exPBMC for healthy donors and ICI-MC patients, indicating clonal expansion of α-myosin specific T cells. Interestingly, Shannon diversity did not change from baseline to CEF peptide expansion, suggesting that α-myosin is a strong stimulus for clonal T cell expansion (Fig. 4a). For all donors, both α-myosin and CEF stimulation resulted in expansion of some individual TCR clonotypes. This expansion can be seen by comparing the frequency of each TCR β chain in the baseline PBMC compared to the exPBMC of the same patient (Extended Data Fig. 6a, b). These data suggest that both healthy donors and ICI-MC patients have peripheral α-myosin specific T cells capable of expansion in certain conditions.
Table 2.
Summary of MC patient information.
| Pt | Age | Sex | ICI history |
Primary Tumor |
Disease tissue TCR sequencing |
Brief Clinical Course |
|---|---|---|---|---|---|---|
| 1 | 75 | M | Ipilimumab + Nivolumab | Renal cell carcinoma | Cardiac biopsy; Autopsy: diaphragm, psoas | Pt 1 presented to the emergency department with chest pain 3wks post initiation of ICI and was found to have ventricular tachycardia (VT), and elevated troponin. EMB confirmed MC. Pt’s clinical course was complicated by cardiogenic shock, acute hypoxic respiratory failure and acute renal failure, despite high dose steroids. The pt and family declined further aggressive treatment with curative intent and opted for palliative extubation. |
| 2 | 64 | M | Nivolumab | Small cell lung cancer | Autopsy: RV, LV, IVS, deltoid, diaphragm | Pt2 was admitted to the hospital with recurrent VT and elevated troponin. Pt was found to have a dilated RV on echo. Prednisone treatment with initiated. EMB was complicated by RV perforation leading to acute cardiac tamponade, left atrial thrombus, and rapid clinical deterioration. Family opted for palliative extubation. |
| 3 | 78 | M | Pembrolizumab | Lung adenocarcinoma | Cardiac biopsy | Pt3 was evaluated for fatigue and myalgias and was found to have elevated troponin. The patient was admitted, started on high dose steroids and MC was confirmed by EMB. Patient recovered with steroid treatment and did not experience recurrence of MC. The patient died 5 months later in home hospice due to complications related to a hemothorax which was potentially related to underlying malignancy. |
Figure 4. α-myosin expanded TCRs are present in cardiac and skeletal muscle of patients with ICI-MC.

a) Shannon diversity of TCR beta chain repertoires. Dashed lines connect blood samples within same donor. P-values represent two-sided Wilcoxon tests. n=6 PBMC, n= 6 α-myosin exPBMC, n=3 CEF exPBMC, n=5 heart from 3 patients (multiple regions for pt2), n=4 skeletal muscle from 2 patients, n=3 rejection from 3 cardiac transplant patients, n=8 ICI-colitis from 8 patients, n=4 Crohn’s from 4 patients. b) ICI-MC patient tissues. RV= right ventricle. LV = left ventricle. IVS = interventricular septum. c) Change in TCR counts from PBMC to α-myosin exPBMC plotted by abundance of the same TCR beta chain in the autologous inflamed cardiac tissue. Minimal change is less than a 50 read count change. Not present means not found in either PBMC or exPBMC. d) Dimensionality reduction with UMAP on scRNAseq of CD3+ patient 1 exPBMC. Groups are divided by whether the TCR beta chain expressed by that cell is present in the patient’s heart and whether that TCR is clonal (expressed by >2 cells in exPBMC). e) Proportion of single cell sequenced exPBMCs that are clonal, stratified by whether that TCR is present in the heart. P<0.0001 by two-sided Fisher’s Exact test. f) Violin plots of key genes by presence or absence in heart and clonality in exPBMC. Identity genes are light blue. Naïve genes are dark blue. Activation genes are red. g) Flow cytometry histogram of TCR-Pt1 reporter cell line co-cultured with autologous LCLs and stimulated with 10μg/mL RINATLETK peptide relative to no peptide. (n=3 replicates) h) Scatter plot showing CD3+CD8+ Pt1 exPBMC stained with irrelevant peptide or RINATLETK on HLA-A:03*01 tetramer. i) Quantification of RINATLETK on HLA-A:03*01 tetramer staining across samples, compared to irrelevant peptide. n=2 healthy donors for baseline PBMC. n=6 exPBMC (1 replicate for 2 ICI-MC patients and 2 replicates for 2 healthy donors).
To assess whether α-myosin expanded TCR clones might be involved in cardiac and skeletal muscle toxicity, we compared TCR β chain repertoires in the heart and inflamed muscle to those overrepresented in α-myosin exPBMC relative to unexpanded PBMC. We performed bulk TCR β chain sequencing on formalin-fixed paraffin embedded tissues from endomyocardial biopsy (patient 1 and 3) and autopsy material (patients 1 and 2). Tissue samples from each MC patient are summarized in Fig. 4b. High numbers of total TCR reads (>1500) were obtained in all samples, consistent with high T cell infiltration (Extended Data Fig. 6c). Shannon diversity was lower in the hearts and skeletal muscles of patients with ICI-MC compared to inflamed colonic tissue of patients with ICI-colitis or Crohn’s disease, indicating the high clonality of TCR repertoires seen in ICI-MC compared to another highly T cell infiltrated immunotherapy toxicity34 (Fig. 4a). Biopsy samples of acute cellular rejection following cardiac transplantation were also included as cardiac-specific comparison TCR repertoires. These samples had low Shannon diversity, consistent with T cell-mediated anti-cardiac autoimmunity (Fig. 4a).
Plotting the degree of α-myosin expansion (count in α-myosin exPBMC minus count in pre-expansion PBMC, with expansion shown in red) against the abundance of the same TCR β chain in the autologous inflamed tissues of ICI-MC patients, shows that α-myosin expanded TCRs are present in inflamed hearts from all three patients (Fig. 4c; Extended Data Fig. 8a). Some α-myosin expanded TCRs were abundant in the inflamed heart and skeletal muscles (Fig. 4c; Extended Data Fig. 7), suggesting that α-myosin may be a relevant disease antigen for ICI-MC and myositis. We performed single cell RNA/TCR sequencing on the sorted CD3+ exPBMC from patient 1. Gene expression analysis of TCR+ cells showed expression of CD3E in all cells (including both CD8A and CD4 expressing cells), and a very small population of residual CD79A expressing B cells (Extended Data Fig. 8a). We further filtered cells based on a shared TCR β chain with the cardiac TCR repertoire (overlap with bulk β chain sequencing) which would be expected to be enriched for disease-relevant TCRs. Following dimensionality reduction with UMAP, the cells clustered distinctly by group (Fig. 4d). Of the cells with TCR clonotypes shared with the heart, a significantly higher proportion were clonal in the exPBMC relative to cells with TCRs not present in the heart (Fig. 4e) and these clonal cells had high expression of CD8A (Fig. 4f; Extended Data Fig. 8b). Clonal cells in the exPBMC are expected to be enriched for α-myosin specificity. Clonal cells with TCRs present in the heart also have high expression of markers of activation such as NKG7, GZMB, and GNLY (Fig. 4f; Extended Data Fig. 8b).
To confirm that α-myosin expansion generates clonal TCRs specific for α-myosin, we aimed to map the epitope specificity and MHC restriction of a TCR found in both the exPBMC and cardiac tissue of patient 1 (TCR-Pt1; Extended Data Fig. 9a). HLA types for ICI-MC patients and healthy donors are shown in Extended Data Table 3. We reconstructed and transduced TCR-Pt1 into Jurkat NFAT-GFP reporter cells (TCR CDR3 and gene information is shown in Table 1). Using the same approach as for murine TCR cell lines of testing against the α-myosin 20 amino acid peptide library, followed by TepiTool-guided epitope narrowing, we found that TCR-Pt1 activated the NFAT reporter specifically to MYH6443-451 (RINATLETK; Fig. 4g). This peptide has strong predicted binding to HLA-A*03:01 using TepiTools. In line with this prediction, the TCR-Pt1 cell line stained with a tetramer with MYH6443-451 loaded on HLA-A*03:01 but not a tetramer with the same peptide loaded on HLA-A*24:02 (Extended Data Fig. 9b). Pt1 carries both of these HLA-A alleles. We next tested the MYH6443-451 on A*03:01 tetramer against exPBMC of healthy donors and ICI-MC patients carrying HLA-A*03:01. We found high prevalence of MYH6443-451 specific T cells in the exPBMC of ICI-MC patient 1 and two healthy donors and low but detectable MYH6443-451 specific T cells in the exPBMC of ICI-MC patient 3 (Fig. 4h,i; Extended Data Fig. 9c). Patient 2 was not assessed because they lacked the HLA-A*03:01 allele. Thus, CD8+ cytotoxic T cells specific for α-myosin are present in the blood and diseased hearts of patients with fulminant ICI-MC, and may also be present in healthy individuals.
Tumor-specific MYH6 expression
The importance of α-myosin as an autoantigen in ICI-MC raises the possibility that tumor cells may aberrantly express MYH6 and tumor reactive T cells specific for α-myosin epitopes may develop and lead to an increased risk for ICI-MC. It has been previously published by our group that a patient with fulminant ICI-MC had aberrant melanoma-specific expression of muscle transcripts, including MYH65. Using a previously published dataset of RNA sequencing on melanoma tumors treated with ICI, we found that 37/91 tumors expressed low but detectable levels of MYH6 (Extended Data Fig. 10a)35. Despite treatment with ICI, none of these patients developed clinically significant MC or myositis. However, very few patients with MYH6 expression were treated with combination anti-CTLA4 and anti-PD-1, which is a known risk factor for ICI-MC. Given the rarity of MC and the comparatively low patient numbers examined here, these data are insufficient to determine whether tumor-specific MYH6 expression may be a risk factor for ICI-MC. HLA type may be an important modifying factor. Importantly, tumor-specific MYH6 expression is not unique to this dataset. Analysis of The Cancer Genome Atlas (TCGA) melanoma cohort shows that 250 of 363 tumors had detectable MYH6 expression (Extended Data Fig. 10b).
Discussion
Immunotherapy toxicities are important limitations to the use of ICIs. Here we present a new perspective on ICI-MC as an antigen-driven, cytotoxic T cell mediated toxicity. We used a genetically altered mouse to model a human drug toxicity and found that Pdcd1−/−Ctla4+/− mice recapitulate many of the important clinicopathologic features of ICI-MC seen in humans, including severe cardiac inflammation predominately comprised of CD8+ T cells. The mice also have significant conduction abnormalities and preserved ejection fraction2. Pharmacologic animal models have not replicated these important features. MRL mice treated with anti-PD-1 and anti-CTLA-4 for 8 weeks develop a mild MC that is only evident histologically2. Monkeys treated with anti-PD-1 and anti-CTLA-4 develop mild to moderate inflammation in all organs examined including the heart, where mild CD4+ T cell infiltration is seen36.
We show in our mouse model that MC is characterized by cytotoxic CD8+ T cells with highly clonal TCRs, and that CD8+ cells are necessary for the development of MC. Though CD4 depletion did not rescue survival, it is unknown what role CD4+ T cells play, particularly in the initiation of MC, which is more difficult to determine in a genetic model and is a limitation of our study. The CD8 dependence of our model is in contrast to the CD4 dependence of MC seen in Pdcd1−/−Lag3−/− mice, raising the possibility of distinct mechanisms of MC with different immune checkpoint deficiencies37,38. Three of the most clonal TCRs, derived from independent mice, recognized α-myosin epitopes. Strikingly, α-myosin specific TCRs expanded when transferred to recipient mice and occupied greater than 65% of the highly inflamed cardiac TCR repertoire at the time of death from MC. Lack of thymic expression suggests that α-myosin specific T cells may escape central tolerance mechanisms3. α-myosin specific T cells can be expanded from the blood of healthy donors and patients with ICI-MC. This finding, likely not unique to α-myosin, suggests that the presence of α-myosin specific T cells in the periphery is common in humans. Furthermore, we found T cells specific for the exact epitope MYH6443-451 on HLA-A*03:01 could be expanded from the blood in all four tested patients with the HLA-A*03:01 allele. It is currently unknown whether particular HLA alleles may be a risk factor for ICI-MC. Several studies have recently examined associations of HLA alleles and response or toxicity to ICI, but more work is needed, particularly for rare toxicities like MC 39-42. Additionally, the effect of the microbiome on ICI-MC risk is unknown and should be explored in future work43,44.
α-myosin expanded TCRs overlapped with TCR repertoires in the diseased hearts and skeletal muscle of three patients with ICI-MC. Although presence of shared clones is insufficient to establish causality, these data suggest that α-myosin may be an important autoantigen in ICI-MC. The presence of other high frequency TCRs in the hearts that were not enriched in the α-myosin expanded repertoires could point to other relevant antigens, particularly by the time MC has become severe.
Prior studies have shown α-myosin to be an MHC-II restricted autoantigen in mouse models, primarily using transgenic TCRs or α-myosin vaccination approaches, where α-myosin is used to initiate an immune response3,45-50. Our studies are the first to identify MHC-I restricted α-myosin epitopes in a spontaneous murine model of MC which is dependent on CD8+ T cells. Furthermore, to the best of our knowledge, our studies are among the first to identify a candidate autoantigen for an immunotherapy toxicity in humans. We have also identified novel exact TCR-peptide:MHC interactions for three murine TCRs and one human TCR. Knowledge of the most relevant disease antigens may allow for antigen-directed approaches to suppressing inflammation without sacrificing anti-tumor efficacy such as tolerogenic vaccines. Identification of α-myosin as an autoantigen may also guide identification of biomarkers to predict which patients are at higher risk for MC, such as surveillance of peripheral α-myosin reactive T cells or identification of pre-existing autoantibodies.
Methods
Mice.
Pdcd1−/−Ctla4+/− mice were maintained as previously described2. Female mice were primarily (but not exclusively) used in these studies due to their higher incidence of myocarditis. Rag1−/− mice were purchased from The Jackson Laboratory (#002216)51. For the generation of survival curves, events were defined as either death (i.e., mice found dead) or identification of mice requiring euthanasia (e.g., due to lethargy, moribund, dyspnea, weight loss). All mice were housed at Vanderbilt University Medical Center vivarium, an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)–accredited, specific pathogen-free (SPF) animal facility. All experiments were performed in accordance with Vanderbilt University Medical Center Institutional Animal Care and Use Committee (IACUC) guidelines. Mice were on 12-h light–dark cycles which coincided with daylight in Nashville, TN. The mouse housing facility was maintained at 20–25 °C and 30–70% humidity.
Preparation of cardiac dissociates for single-cell RNA/TCR sequencing.
Single-cell suspensions were obtained from murine hearts by mincing followed by enzymatic digestion with 125 U/mL DNase I (Worthington; cat no. LS002138) and 250 U/mL Collagenase 3 (Worthington; cat no. LS004182). Dissociated hearts were filtered through a 30μm filter. Red blood cells were lysed using ACK lysing buffer (KD Medical/MediaTech; cat no. NC0274127). Single-cell suspensions were either used fresh or cryopreserved in 10% DMSO 90% FBS. Prior to sorting, cells were stained with Alex Flour 488 anti-mouse CD45 (BioLegend; clone 30-F11; cat no. 103122, dilution 1:1000) for 20 minutes at 4°C. Following staining and washing with PBS, cells were resuspended in PBS with DAPI (1:20,000). Live CD45+ immune cells were sorted by fluorescence-activated cell sorting on AF488-positive DAPI-negative events. The wildtype control sample consisted of pooled, without hashing, cardiac immune infiltrates from six female animals, in order to obtain sufficient cells as the healthy heart has a low frequency of cardiac immune cells. The myocarditis sample consisted of four inflamed hearts from female Pdcd1−/−Ctla4+/− mice. Inflammation was confirmed by flow cytometry for CD45. Only mice with CD45+ cells comprising greater than ten percent of the total single cells were included. Mice ranged from three to six weeks in age. One inflamed heart was run as an individual sample on the 10X Genomics chromium platform. The additional three inflamed hearts were hashed together using Total Seq C reagents according to the manufacturer’s instructions (BioLegend: TotalSeq™-C0301 cat# 155861, TotalSeq™-C0302 cat# 155863, TotalSeq™-C0303 cat# 155865).
Single-cell RNA/TCR sequencing.
Each sample (targeting 5,000 – 15,000 cells/sample) was processed for single cell 5' RNA and TCR sequencing utilizing the 10X Chromium system. Libraries were prepared following the manufacturer's protocol. The libraries were sequenced using the NovaSeq 6000 with 150 bp paired end reads. RTA (version 2.4.11; Illumina) was used for base calling and analysis was completed using 10X Genomics Cell Ranger software. Data were analyzed in R using the filtered h5 gene matrices in the Seurat package52-54. Briefly, samples were subset to include cells with greater than 200 but less than 3000 unique transcripts to exclude likely non-cellular RNA reads and doublets. Cells with greater than 15% of reads coming from mitochondrial transcripts were also excluded as likely dying cells. For murine hearts, hash tag oligos were deconvoluted using HTODemux with positive quantile set at 0.85. Samples were downsized so that equivalent numbers of cells originating from healthy wild type or myocarditis Pdcd1−/−Ctla4+/− cardiac infiltrating immune cells were included (2509 cells per genotype of origin). Ten clusters were identified using a resolution of 0.4. UMAP was used for dimensionality reduction with 15 nearest neighbors and minimum distance of 0.5. SingleR was used to assist with cell type annotation of clusters. Clonal is defined as more than two cells with the same TCR clonotype (defined by unique combinations of CDR3 regions). T cells were subset on expression of Cd3e > 1.5 and presence of a TCR (n=1266 cells). Clustering with the Louvain algorithm at resolution of 0.3 yielded five distinct clusters. Additional subclustering yielded small clusters of <15 cells. Differential gene expression analyses were used to identify clusters. TCR density is defined as the number of the 100 nearest cell neighbors expressing the same TCR clonotype (alpha and beta chain).
T cell receptor sequencing.
TCR sequencing and clonality quantification was assessed in formalin-fixed paraffin embedded (FFPE) or snap frozen samples of murine hearts or spleens. All human samples were derived from FFPE or isolated PBMC. For FFPE, RNA was extracted from 10μm sections using the Promega Maxwell 16 FFPE RNA kits and the manufacturer’s protocol. TCRs were sequenced using the TCR Immunoverse all chain assay following the manufacturer’s protocol (Invitae/ArcherDX). Sequencing results were evaluated using the Archer Immunoverse analyzer. CDR3 sequences and frequency tables were extracted from the manufacturers’ analysis platform and imported into R for analysis using the Immunarch package (https://immunarch.com) in R.
Antibody-mediated depletion and dexamethasone treatment.
Female Pdcd1−/−Ctla4+/− mice were randomly assigned to control or dexamethasone treatment at 21 days of age. Mice were treated with 1mg/kg/day of dexamethasone. Experiment was concluded when mice reached 115 days of life. Female Pdcd1−/−Ctla4+/− mice were randomly assigned to control, anti-CD8a, or anti-CD4 injections at 21 days of age. Mice were injected intraperitoneally three times a week with 200μg of anti-CD4 (BioXCell, Cat# BE0003-1, clone GK1.5) or anti-CD8 (BioXCell, Cat# BE0061, clone 2.43) depleting antibodies or vehicle, all in a maximum volume of 100μL. Treatment lasted until 90 days of age. Peripheral blood was sampled via tail prick for assessment of depletion efficiency at week 3. In order to detect an anticipated mortality difference of 50% (for control) to 5% (for an intervention which rescues mortality) with an alpha of 0.05 and 80% power, a sample size of 14 animals per group is needed.
Adoptive transfer.
Splenocytes were isolated from Pdcd1−/−Ctla4+/− mice with myocarditis by manual dissociation, filtering, and red blood cell lysis. Myocarditis of the donor mice was confirmed by either H&E or dissociation of the heart and flow cytometry for CD45+ immune cells. A portion of each spleen underwent CD8 depletion using magnetic bead isolation (Miltenyi CD8 (TIL) MicroBeads, Mouse, Cat# 130-116-478). One million whole or CD8 depleted splenocytes were injected into each Rag1−/− recipient mouse in 100μL PBS via tail vein injection. Mice were monitored for death or signs of distress. At death or euthanasia, hearts, spleens, livers, lungs, and kidneys were stained by H&E and evaluated microscopically. In order to detect an anticipated mortality difference of 50% (for whole splenocyte transfer) to 1% (for CD8 depleted splenocyte transfer) with an alpha of 0.05 and 80% power, a sample size of 11 animals per group is needed.
Histology and pathology.
Formalin-fixed tissues were processed routinely into paraffin blocks, sectioned at 5μm, and stained with H&E by standard protocols in Vanderbilt University Medical Center's Translational Pathology Shared Resource (TPSR) core laboratory. To further characterize the mononuclear cardiac infiltrates detected by light microscopy, a panel of IHC markers was employed. IHC staining was performed in the TPSR using standard, validated protocols for chromogenic IHC. All steps besides dehydration, clearing, and coverslipping were performed on the Leica Bond-Max IHC autostainer (Leica Biosystems Inc.). Slides were deparaffinized. Antigen retrieval was performed using EDTA (CD markers) or proteinase K (F4/80) or on the Bond Max using their Epitope Retrieval 2 solution for 20 minutes (CD45R and IgG). Slides were incubated with primary antibodies as indicated below. Secondary antibody labeling was performed for all markers except CD3 and IgG by incubating in rabbit anti-rat antibody (BA-4001, Vector Laboratories, Inc.) for 15 minutes at a 1:650 dilution. Immunolabeling by rabbit antibody was visualized using the Bond polymer refine detection system (#DS9800, Leica Biosystems, Inc.). Slides were then dehydrated, cleared, and coverslipped. For primary antibodies, anti-CD3 (Abcam, Ab16669) was used at 1:250 dilution, anti-CD4 (eBioscience, 14–9766–82) was used at 1:1,000 dilution, anti-CD8 (eBioscience, 14–0808–82) was used at 1:1,000 dilution, anti-F4/80 (Novus Biologicals, NB600–404) was used at 1:900 dilution, and anti-CD45R/B220 (cat# 553086, BD Pharmingen, clone RA3-6B2) was used at 1:20,000 dilution. For IgG, slides were placed in the Biotin Blocking System (Ref# x0590, DAKO, Carpinteria, CA) for 10 minutes each. Slides were incubated in biotinylated goat anti-mouse IgG (Cat# BA-9200, Vector Laboratories, Inc., Burlingame, CA) for 15 minutes at a 1:2000 dilution. The Bond Intense R detection system (Cat#DS9263, Leica Biosystems, Buffalo Grove, IL) was used for visualization. IHC quantification is reported as number of positive cells per high power field (40x), averaged over three high power fields per slide. Slide images were obtained using Olympus cellSens Standard v1.17.
RNA preparation and qPCR analysis.
Flash frozen mouse tissue (cardiac ventricle or thymus) was homogenized using a TissueLyser II (Qiagen) for 2 minutes at 30Hz. RNA was harvested from dissociated tissue using the Maxwell 16 automated workstation (Promega) and LEV simplyRNA Tissue Kit (Promega, Cat# AS1280). RNA was then analyzed for concentration by a NanoDrop 2000 (Thermo Fisher Scientific) before cDNA synthesis using SensiFAST cDNA Synthesis Kit (Bioline, Meridian Bioscience, Inc., Catalog BIO-65054) with 1 μg of RNA per sample. cDNA and SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, Catalog 1725270) were then combined with target-specific primers on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The following primer pairs were used: Sbk2 forward 5’- CTCTGAGCCCAGAAATGCCA- 3’ reverse 5’- AATGTGTTCCAGGGCAGAGG- 3’; Nppb forward 5’- CGTTTGGGCTGTAACGCACTG- 3’ reverse 5’- TCACTTCAAAGGTGGTCCCAG- 3’; Nppa forward 5’- TCTGATGGATTTCAAGAACCTGCT- 3’ reverse 5’- ACACACCACAAGGGCTTAGG- 3’; Myh6 forward 5’- AACCAGAGTTTGAGTGACAGAATG - 3’ reverse 5’- ACTCCGTGCGGATGTCAA- 3’; β-actin forward 5’- ACGGCCAGGTCATCACTATTG- 3’ reverse 5’- AGGATTCCATACCCAAGAAGGAA- 3’. Three technical replicates were used for each reaction. Beta-actin was used as the housekeeping gene. Data were analyzed using the delta Ct method in which the number of cycles needed to amplify the gene of interest is normalized to the number of cycles needed to amplify the housekeeping gene.
Flow cytometry for murine cell populations and Jurkat reporter cells.
Samples were run on an Attune NxT Acoustic Focusing Cytometer (Life Technologies). Data were collected using the Attune NxT Software v3.2.1. Analysis was performed in FlowJo v10.6. Gating was first done on forward scatter and side scatter to exclude debris. Doublets were excluded by gating on FSC area versus FSC height. DAPI was used to exclude dead cells from analyses. Antibodies used: CD45-PerCP/Cy5.5 (BioLegend, cat# 103132, clone 30-F11, dilution 1:400), CD3-AF488 (BioLegend, cat# 100210, clone 17A2, dilution 1:200), CD4-APC (BioLegend, cat# 100412, clone GK1.5, dilution 1:100), CD8a-PE/Cy7 (BioLegend, cat# 100722, clone 53-6.7, dilution 1:400), Thy1.1 (BioLegend, cat# 202506, Clone OX-7, dilution 1:1000 for AF488, 1:500 for APC/Cy7) and TCR-beta chain-PE (BioLegend, cat# 109208, Clone H57-597, dilution 1:200).
TCR sequences and cloning.
TCR sequences were generated from CDR3 regions, V genes and J genes using Stitchr55. Paired TCR alpha and beta sequences were derived from single cell sequencing or from the adoptive transfer experiments in which pairing of chains could be inferred from bulk sequencing as single alpha and beta chains made up the majority of each repertoire. Alpha genes and beta genes were separated using a T2A sequence. Restriction digest sites were added to either end. Full TCR gene blocks were synthesized as custom orders from Genewiz. Full TCR block sequences can be found in supplemental material. TCR sequences were cloned into MSCV-IRES-Thy1.1 DEST vector. MSCV-IRES-Thy1.1 DEST was a gift from Anjana Rao (Addgene plasmid # 17442; http://n2t.net/addgene:17442 ; RRID:Addgene_17442)56. Retrovirus was made using the platA retroviral packaging cell line (Cell BioLabs RV-102). Jurkat-TCR-ko-CD8+-NFAT-GFP reporter cells were a gift from Dr. Peter Steinberger. Reporter cells were retrovirally transduced with TCRs of interest. TCR expression was confirmed via flow cytometry for Thy1.1 and TCR-beta chain. Retrovirally transduced cells were sorted on the WOLF cell sorter (NanoCellect) for Thy1.1-AF488. Cells were confirmed to be >90% Thy1.1 positive post-sort prior to use in downstream assays.
Antigen discovery.
Jurkat-NFAT-GFP cell lines with reconstructed TCRs were used for antigen screening. Syngeneic (derived from C57BL6 mice) bone marrow derived dendritic cells (BMDCs) were used as APCs. BMDCs were generated by flushing femurs and tibias from mice with PBS, filtering the cells though a 70μM filter, lysing RBCs, and plating in RMPI + 10% FBS + 1% HEPES + 20ng/mL GM-CSF (ProSpec Cat# CYT-222). BMDCs were polarized in GM-CSF containing media for 9 days (replacing the media at days 3 and 6) prior to harvesting the adherent fraction via mechanical dissociation using a cell scrapper and cryopreservation for future experiments. For antigen discovery, BMDCs were thawed into GM-CSF containing media in flat bottom plates the day before adding TCR cell lines and peptides. Cells were plated at a ratio of 1 TCR cell to 3 BMDCs. The alpha myosin, ANP, BNP, and SBK2 peptide library was generated as 20aa peptides with 5aa overlaps from GenScript. Due to insolubility in aqueous solution, two 20aa peptides were replaced by three 10aa peptides each. Sequences of all 172 peptides are shown in Extended Data Table 1. Peptides were added at a concentration of 10μg/mL and co-cultures were incubated overnight. TCR cell lines were stained with DAPI to assess viability and analyzed via flow cytometry for NFAT-GFP reporter activity. For the human TCR (TCR-Pt1), autologous LCLs were used as APCs. TCR-Pt1 was screened against 130 alpha-myosin peptides.
MHC blocking.
Jurkat TCR cell lines were co-cultured with EL-4 cells as APCs with or without 10μg/mL cognate peptide overnight. EL-4 cells were a gift from Dr. Simon Mallal. Blocking antibodies (anti-Db clone 28-14-8S or anti-Kb clone B8-24-3) were added to cells at a concentration of 10μg/mL for 1 hour prior to adding peptides. Blocking antibodies were generously provided by Dr. John Sidney. TCR cell lines were stained with Thy1.1-APC/Cy7 (BioLegend, cat# 202506, Clone OX-7, dilution 1:500) to differentiate from EL-4s and with DAPI to assess viability and analyzed via flow cytometry for NFAT-GFP reporter activity.
Patients.
All studies were conducted in accordance with the Declaration of Helsinki principles under protocols approved by the Vanderbilt University Medical Center (VUMC) Institutional Review Board. Healthy donors provided informed consent under an institutionally approved protocol (IRB# 030062). Myocarditis patients and families provided informed consent for research use of biospecimens and clinical data (IRB# 191213). ICI-colitis and Crohn’s patients provided informed consent for research use of biospecimens and clinical data (IRB# 09109). Cardiac transplant and heart failure patients provide informed consent for research use of biospecimens and clinical data (IRB #200551). Patients in RNA-sequencing cohort were previously published35. An additional melanoma gene expression dataset was generated by the TCGA Research Network: https://www.cancer.gov/tcga and accessed using cBioPortal57,58.
Generation of LCLs from PBMC.
EBV-transformed lymphoblastoid B cell lines were generated from cryopreserved donor PBMCs by infection with EBV virus stock59,60. Approximately, 1-3 × 106 PBMCs in RPMI-1640 media supplemented with non-heat inactivated 20% fetal bovine serum (FBS), 1 μg/mL cyclosporin A (CSA; Sigma-Aldrich C1832), and 2.5 μg/mL CpG (Invitrogen ODN2006) were infected with filtered EBV stock and cultured for 2-3 weeks, until clusters of cells were visible by light microscopy.
PBMC expansion.
PBMCs were isolated from EDTA collection tubes and processed using a Ficoll gradient. Antigen-specific PBMC expansion was adapted from previously described protocols61,62. Fresh or cryopreserved PBMCs were stimulated with 130 pooled alpha-myosin peptides at a final concentration of 400ng/mL of each peptide or a pool of control CMV, EBV, and flu (CEF) peptides (AnaSpec, AS-61036-003). PBMCs were cultured in CTS OpTmizer medium (CTS OpTmizer T Cell Expansion SFM with CTS supplement A1048501, substituted with 2mM L-glutamine, and 2% human serum, Sigma-Aldrich, H3667) with cytokine supplementation (25ng/mL each of rhIL-2, rhIL-7 and rhIL-15, Peprotech). For myocarditis patients 1,2, and 3, expansion cultures were also supplemented with autologous LCLs to serve as antigen presenting cells at a ratio of 1 APC per 10 PBMC. For healthy donors, expansion was done directly from fresh, not cryopreserved, blood. Peptides were only added on the first day of culture. On day 3, additional media with cytokines was added. On day 7, cells were transferred to a new culture dish with fresh media with cytokines. Cells were analyzed or cryopreserved on day 14.
Single cell sequencing of exPBMC.
Expanded PBMCs (exPBMC) from patient 1 were prepared for single cell sequencing as follows. exPBMC were incubated with Human TruStain FcX™ (Fc Receptor Blocking Solution; BioLegend cat# 422302, dilution 1:100) for 5 minutes on ice, then washed and incubated with human anti-CD3-AF488 (BioLegend, cat# 300319, clone HIT3a, dilution 1:200) for 30 minutes on ice, and then washed and resuspended to a concentration of 5x105 cells/mL. SYTOX AADvanced™ Ready Flow™ Reagent (Invitrogen, cat# R37173, dilution 2 drops per 1mL) was used following the manufacturer’s instructions to exclude dead cells. CD3+ live cells were sorted on the WOLF cell sorter (Nanocellect). Cells were sequenced and data were analyzed as described above. Data were analyzed in R using the filtered h5 gene matrices in the Seurat package52-54. Briefly, samples were subset to include cells with greater than 200 but less than 4000 unique transcripts to exclude likely non-cellular RNA reads and doublets. Cells with greater than 15% of reads coming from mitochondrial transcripts were also excluded as likely dying cells. Clonal is defined as more than two cells with the same TCR clonotype (defined by unique combinations of CDR3 regions). For exPBMC, 5,816 cells with TCR reads were analyzed. To identify TCRs overlapping with the cardiac repertoire, beta CDR3 sequences were used.
Tetramer staining and flow cytometry.
Murine tetramer staining was done using an empty H-2Kb-PE tetramer (Tetramer Shop). Empty tetramer was loaded with peptide by incubating 5μL of empty tetramer with 0.5μL of 200μM peptide solution (all peptides custom ordered from GenScript, in this case VIQYFASI, VQQVYYSI or SIINFEKL as an irrelevant peptide control which is known to bind to H-2Kb strongly) for at least 30 minutes at 4°C. Loaded tetramers were centrifuged at 3300xg for 5 minutes prior to use. Murine hearts were dissociated as above. Cells were first stained with Zombie Violet (BioLegend, cat # 423113, dilution 1:1000) for 15 minutes at room temperature in PBS with no added protein. Cells were then incubated with TruStain FcX block (BioLegend, cat # 101319, clone 93, dilution 1:100) for 5 minutes at room temperature. Cells were washed in PBS + 2%FBS. Cells were stained with 5μL of loaded tetramer in 50 μL PBS+2% FBS for 20 minutes at 37°C. Without washing 20 μL of solution containing desired surface antibodies was added and cells were incubated for 20 minutes at 4°C. Surface antibodies used were anti-CD3-APC (BioLegend, cat# 100235, clone 17A2, dilution 1:400) and anti-CD8a-FITC (Thermo Fisher, cat # MA5-16759, clone KT15, dilution 1:200). Samples were washed and then analyzed.
Human tetramer staining was done using empty tetramers easYmer HLA-A*03:01 (Eagle Biosciences, cat # 1016-01-20) or easYmer HLA-A*24:02 (Eagle Biosciences, cat # 1020-01-20). Monomers were loaded with peptide following the manufacturer’s instructions and incubated for 48 hours at 18°C. Tetramers were produced by adding 2.1 μL of Streptavidin-PE antibody (BioLegend, cat# 405203) and incubating for at least 1 hour at 4°C. Cells were first stained with Zombie Violet (BioLegend, cat # 423113, dilution 1:1000) for 15 minutes at room temperature in PBS with no added protein. Cells were then incubated with TruStain FcX block (BioLegend, cat # 422301, dilution 1:200). Cells were stained with tetramer in PBS with 2% FBS for 20 minutes at room temperature. Cells were then washed and stained with surface antibodies anti-CD3-AF488 (BioLegend, cat# 300319, clone HIT3a, dilution 1:200) and anti-CD8a-APC (BioLegend, Cat # 301014, clone RPA-T8, dilution 1:200) for 20 minutes at 4°C. Samples were washed in PBS with 2% FBS. Samples were run on an Attune NxT Acoustic Focusing Cytometer (Life Technologies). Analysis was performed in FlowJo. Gating was first done on forward scatter and side scatter to exclude debris. Doublets were excluded by gating on FSC area versus FSC height. Zombie Violet was used to exclude dead cells from analyses.
Statistical analysis.
All statistical analyses were performed in R v4.1.1. All single-cell statistical analyses were calculated in R using the Seurat package52-54. Visualization and graph generation was performed in R. Shannon diversity was calculated using the R package vegan63. The R package immunarch was used for evaluating TCR repertoires64. P-value cut-offs displayed on plots correspond to “ns” equals p>0.05, * equals 0.01<p< 0.05, ** equals 0.001<p<0.01, *** equals 0.0001<p<0.001, **** equals p<0.0001.
Extended Data
Extended Data Figure 1. Myocardial immune infiltrate does not differ by sex.

a) Quantification of immunohistochemistry (IHC) for CD8 and CD4 in male and female Pdcd1−/−Ctla4+/− mice with MC. Cells are counted as number of positive cells per high power (40x) field (HPF). Each data point represents an average of three high power fields per mouse. n=4 female mice, n=4 male mice. Box plots show the median, first and third quartiles. The whiskers extend to the maxima and minima but no further than 1.5 times the inter-quartile range. b) Representative IHC for IgG and B220 (CD45R) in hearts of mice with MC and positive control staining in spleen. Images are representative of n=8 independent Pdcd1−/−Ctla4+/− mice with MC (n=4 male; n=4 female). Scale bars represent 50μm.
Extended Data Figure 2. MC is characterized by activated immune cells and clonal T cells.
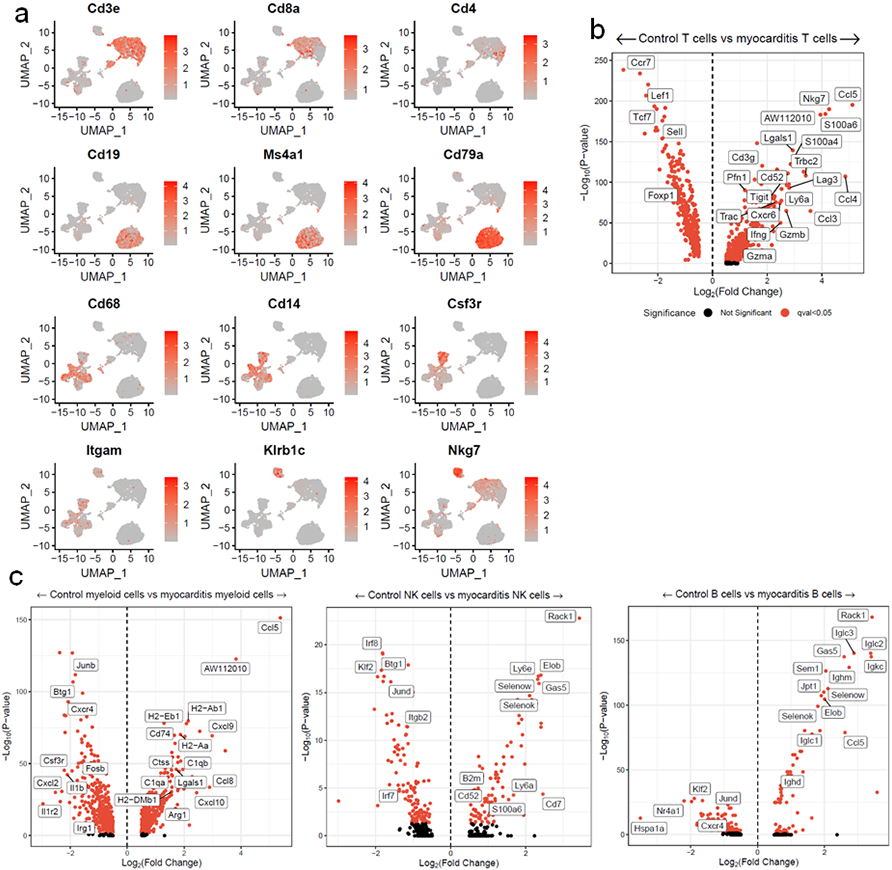
a) Gene expression of key identity genes, showing cell types of clusters. b) Differential gene expression for T, c) myeloid, B and NK cells in MC compared to control cardiac CD45+ cells. Red indicates FDR-corrected p-value (q-value) <0.05. Black indicates not significant.
Extended Data Figure 3. T cells in MC are effector or proliferating, tissue-resident and clonal.

a) Expression of key T cell genes by cluster in single cell data. b) Differential gene expression for Cluster 0 vs. Cluster 3 T cells. Red indicates FDR-corrected p-value (q-value) <0.05. Black indicates not significant. c) Violin plots show expression of key tissue residency associated genes by cluster and MC vs. control. d) Shannon diversity on bulk TCR sequencing beta chain repertoires. Color indicates sex. Shape indicates whether the tissue was derived from a control wild type mouse (open circle) or a Pdcd1−/−Ctla4+/− mouse with MC (filled circle). P=0.0002, two-sided Wilcoxon test. Box plots show the median, first and third quartiles. The whiskers extend to the maxima and minima but no further than 1.5 times the inter-quartile range. e) TCR counts in single cell data. MC sample is divided by mouse of origin. Clonal TCRs are found in all 4 sequenced hearts.
Extended Data Figure 4. Confirmation of cell type depletion.
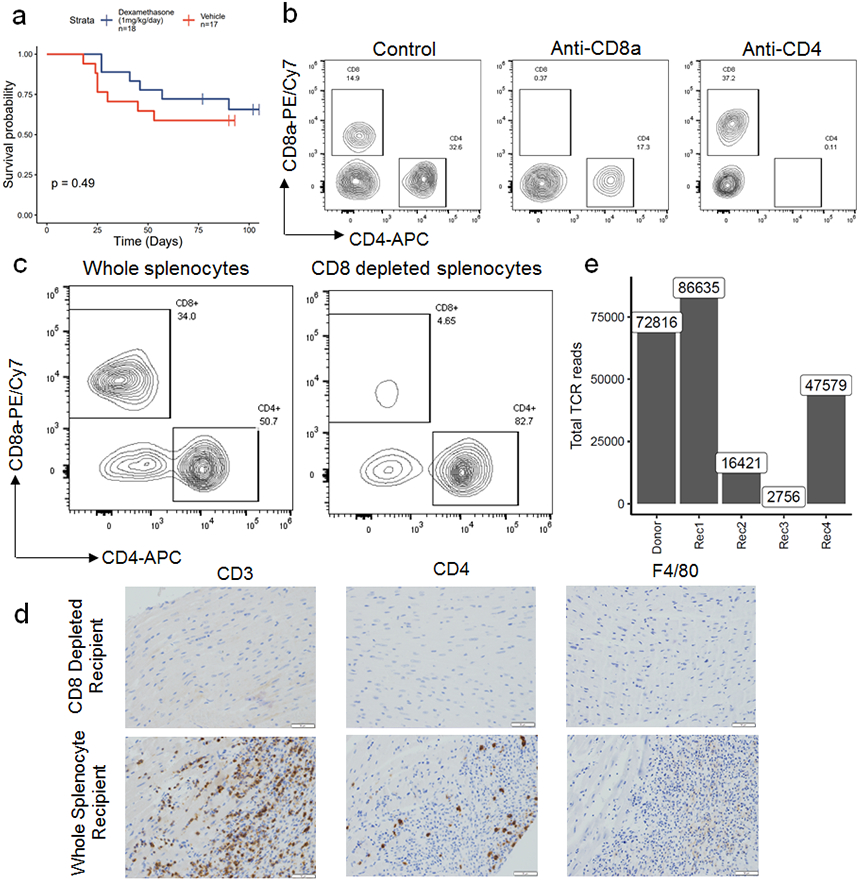
a) Female Pdcd1−/−Ctla4+/− mice were treated with dexamethasone (1mg/kg/day; n=18) or vehicle (n=17) starting at 21 days of life. Time is measured since birth. P=0.49, two-sided cox proportional hazard test. b) Representative flow cytometry gated on live CD45+ cells isolated from blood of different treatment groups, at week 3 of treatment. c) Representative flow cytometry on CD8 depleted (via magnetic beads) compared to whole splenocytes used for adoptive transfer. d) Representative immunohistochemistry on hearts of a CD8 depleted splenocyte recipient compared to a whole splenocyte recipient. Only cardiac sections are shown. Scale bars show 50μm. Representative of n=10 animals per group. e) Total TCR reads for cardiac TCR beta chain sequencing on donor and recipient hearts.
Extended Data Figure 5. Thymic expression of Myh6 and flow cytometry gating for murine α-myosin tetramers.

a) Gene expression for Myh6, Nppa, Nppb, and Sbk2 in the heart and thymus of n=3 each male and female Pdcd1−/−Ctla4+/− mice. Gene expression is normalized to beta-actin. Gene expression is plotted as 2^-(Ct gene of interest minus Ct of beta-actin). Box plots show the median, first and third quartiles. The whiskers extend to the maxima and minima but no further than 1.5 times the inter-quartile range. b) Gating strategy for H2-Kb tetramers on murine heart samples. Debris, doublets and dead cells (Zombie Violet positive) are excluded. CD3+CD8+ cells are used for tetramer analysis. Staining for Control (SIINFEKL) H2-Kb, and VQQVYYSI H2-Kb tetramers are shown. c) Quantification of spleen tetramer positive CD3+CD8+ cells, by sex of the mouse. The spleens used in this experiment correspond to the mice show in Fig. 3f, which all have α-myosin tetramer positive MC.
Extended Data Figure 6. TCR sequencing on exPBMC shows expansion of α-myosin and CEF specific TCRs.

a) Comparison of TCR beta chain abundance in α-myosin exPBMC and pre-expansion PBMC for all patients. Each plot is within the same patient only. Color represents change from PBMC to exPBMC. Minimal change is less than a 50 read count change. b) Comparison of TCR beta chain abundance in CEF exPBMC and pre-expansion PBMC for all healthy donors. Color represents change from PBMC to CEF exPBMC. Minimal change is less than a 50 read count change. c) Total TCR reads for biopsy (heart), autopsy, and PBMC samples from patients 1, 2 and 3.
Extended Data Figure 7. α-myosin expanded TCRs are found in the hearts and skeletal muscles of patients with ICI-MC.
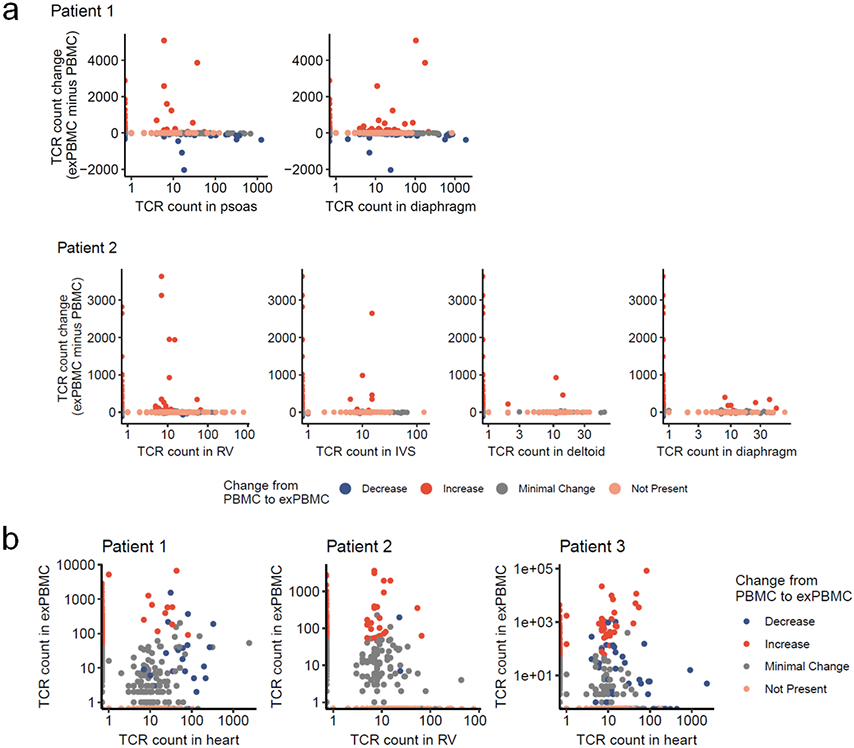
a) Change in TCR counts from PBMC to α-myosin exPBMC plotted by abundance of the same TCR beta chain in the autologous inflamed cardiac or skeletal muscle tissue of each patient. Minimal change is less than a 50 read count change. Not present means not found in either PBMC or exPBMC, but present in indicated tissue. b) Comparison of TCR beta chain abundance in α-myosin exPBMC and heart (biopsy for patient 1 and 3 or right ventricle for patient 2). Color represents change from PBMC to exPBMC. Minimal change is less than a 50 read count change. Not present means not found in either PBMC or exPBMC, but present in heart.
Extended Data Figure 8. Purity analysis for single cell sequencing on exPBMCs from patient 1.
a) Gene expression is shown on single cell sequencing of CD3 sorted exPBMCs from patient 1. b) Violin plots of key gene expression by presence or absence in cardiac TCR repertoire and clonality in exPBMC. Identity genes are shown in light blue. Genes associated with naïve T cells are shown in dark blue. Genes associated with T cell activation are shown in red.
Extended Data Figure 9. TCR from Pt 1 exPBMC recognizes α-myosin epitope.
a) TCR-Pt1, which was cloned and transduced into Jurkat NFAT-GFP reporter cells, is shown in red on the same plot show in Fig 4c. This shows the expansion of this TCR in the exPBMC and abundance in the heart. b) Representative flow cytometry scatter plots are shown for the TCR-pt1 Jurkat cell line is stained with A*24:02 tetramer with RINATLETK or A*03:01 tetramer with RINATLETK. c) Full flow cytometry gating strategy for human PBMC and exPBMC tetramer staining. Debris, doublets and dead cells (Zombie Violet positive) are excluded. CD3+CD8+ cells are used for tetramer analysis. Tetramer staining for all samples is shown.
Extended Data Figure 10. Tumor-specific MYH6 expression.

a) MYH6 transcripts per million are shown for n=91 pre-treatment RNA-sequencing melanoma samples. Bars are colored by what ICI treatment the patient received. b) MYH6 expression is shown for n=363 melanoma samples accessed from TCGA. Samples to the right of the dotted lines have detectable MYH6 expression.
Extended Data Table 1.
Amino acid sequences for α-myosin, ANP, BNP and SBK2 peptides included in peptide library.
| Name | Sequence | Protein |
|---|---|---|
| peptide_1 | MTDAQMADFGAAAQYLRKSE | Alpha-myosin |
| peptide_2 | LRKSEKERLEAQTRPFDIRT | Alpha-myosin |
| peptide_3 | FDIRTECFVPDDKEEYVKAK | Alpha-myosin |
| peptide_4 | YVKAKWSREGGKVTAETEN | Alpha-myosin |
| peptide_5 | AETENGKTVTIKEDQVMQQN | Alpha-myosin |
| peptide_6 | VMQQNPPKFDKIEDMAMLTF | Alpha-myosin |
| peptide_7 | AMLTFLHEPAVLYNLKERYA | Alpha-myosin |
| peptide_9 | FCVTVNPYKWLPVYNAEWA | Alpha-myosin |
| peptide_10 | AEWAAYRGKKRSEAPPHIF | Alpha-myosin |
| peptide_11 | PPHIFSISDNAYQYMLTDRE | Alpha-myosin |
| peptide_12 | LTDRENQSILITGESGAGKT | Alpha-myosin |
| peptide_13 | GAGKTVNTKRVIQYFASIAA | Alpha-myosin |
| peptide_14 | ASIAAIGDRSKKENPNANKG | Alpha-myosin |
| peptide_15 | NANKGTLEDQIIQANPALEA | Alpha-myosin |
| peptide_16 | PALEAFGNAKTVRNDNSSRF | Alpha-myosin |
| peptide_17 | NSSRFGKFIRIHFGATGKLA | Alpha-myosin |
| peptide_18 | TGKLASADIETYLLEKSRVI | Alpha-myosin |
| peptide_20 | IFYQILSNKKPELLDMLLVT | Alpha-myosin |
| peptide_21 | MLLVTNNPYDYAFVSQGEVS | Alpha-myosin |
| peptide_23 | LATDSAFDVLSFTAEEKAGV | Alpha-myosin |
| peptide_24 | EKAGVYKLTGAIMHYGNMKF | Alpha-myosin |
| peptide_25 | GNMKFKQKQREEQAEPDGTE | Alpha-myosin |
| peptide_26 | PDGTEDADKSAYLMGLNSAD | Alpha-myosin |
| peptide_27 | LNSADLLKGLCHPRVKVGNE | Alpha-myosin |
| peptide_28 | KVGNEYVTKGQSVQQVYYSI | Alpha-myosin |
| peptide_29 | VYYSIGALAKSVYEKMFNWM | Alpha-myosin |
| peptide_30 | MFNWMVTRINATLETKQPRQ | Alpha-myosin |
| peptide_31 | KQPRQYFIGVLDIAGFEIFD | Alpha-myosin |
| peptide_32 | FEIFDFNSFEQLCINFTNEK | Alpha-myosin |
| peptide_33 | FTNEKLQQFFNHHMFVLEQE | Alpha-myosin |
| peptide_34 | VLEQEEYKKEGIEWEFIDFG | Alpha-myosin |
| peptide_35 | FIDFGMDLQACIDLIEKPMG | Alpha-myosin |
| peptide_36 | EKPMGIMSILEEECMFPKAS | Alpha-myosin |
| peptide_37 | FPKASDMTFKAKLYDNHLGK | Alpha-myosin |
| peptide_38 | NHLGKSNNFQKPRNVKGKQE | Alpha-myosin |
| peptide_39 | KGKQEAHFSLVHYAGTVDYN | Alpha-myosin |
| peptide_40 | TVDYNIMGWLEKNKDPLNET | Alpha-myosin |
| peptide_41 | PLNETWGLYQKSSLKLMAT | Alpha-myosin |
| peptide_42 | KLMATLFSTYASADTGDSGK | Alpha-myosin |
| peptide_43 | GDSGKGKGGKKKGSSFQTVS | Alpha-myosin |
| peptide_44 | FQTVSALHRENLNKLMTNLK | Alpha-myosin |
| peptide_45 | MTNLKTTHPHFVRCIIPNER | Alpha-myosin |
| peptide_46 | IPNERKAPGVMDNPLVMHQL | Alpha-myosin |
| peptide_47 | VMHQLRCNGVLEGIRICRKG | Alpha-myosin |
| peptide_48 | ICRKGFPNRILYGDFRQRYR | Alpha-myosin |
| peptide_49 | RQRYRILNPAAIPEGQFIDS | Alpha-myosin |
| peptide_50 | QFIDSRKGAEKLLGSLDIDH | Alpha-myosin |
| peptide_51 | LDIDHNQYKFGHTKVFFKAG | Alpha-myosin |
| peptide_52 | FFKAGLLGLLEEMRDERLSR | Alpha-myosin |
| peptide_53 | ERLSRIITRIQAQARGQLMR | Alpha-myosin |
| peptide_54 | GQLMRIEFKKIVERRDALLV | Alpha-myosin |
| peptide_56 | VKNWPWMKLYFKIKPLLKSA | Alpha-myosin |
| peptide_57 | LLKSAETEKEMANMKEEFGR | Alpha-myosin |
| peptide_58 | EEFGRVKDALEKSEARRKEL | Alpha-myosin |
| peptide_59 | RRKELEEKMVSLLQEKNDLQ | Alpha-myosin |
| peptide_60 | KNDLQLQVQAEQDNLNDAEE | Alpha-myosin |
| peptide_61 | NDAEERCDQLIKNKIQLEAK | Alpha-myosin |
| peptide_62 | QLEAKVKEMTERLEDEEEMN | Alpha-myosin |
| peptide_63 | EEEMNAELTAKKRKLEDECS | Alpha-myosin |
| peptide_64 | EDECSELKKDIDDLELTLAK | Alpha-myosin |
| peptide_65 | LTLAKVEKEKHATENKVKNL | Alpha-myosin |
| peptide_66 | KVKNLTEEMAGLDEIIAKLT | Alpha-myosin |
| peptide_67 | IAKLTKEKKALQEAHQQALD | Alpha-myosin |
| peptide_68 | QQALDDLQAEEDKVNTLTKS | Alpha-myosin |
| peptide_69 | TLTKSKVKLEQQVDDLEGSL | Alpha-myosin |
| peptide_70 | LEGSLEQEKKVRMDLERAKR | Alpha-myosin |
| peptide_71 | ERAKRKLEGDLKLTQESIMD | Alpha-myosin |
| peptide_72 | ESIMDLENDKLQLEEKLKKK | Alpha-myosin |
| peptide_73 | KLKKKEFDISQQNSKIEDEQ | Alpha-myosin |
| peptide_74 | IEDEQALALQLQKKLKENQA | Alpha-myosin |
| peptide_75 | KENQARIEELEEELEAERTA | Alpha-myosin |
| peptide_76 | AERTARAKVEKLRSDLSREL | Alpha-myosin |
| peptide_77 | LSRELEEISERLEEAGGATS | Alpha-myosin |
| peptide_78 | GGATSVQIEMNKKREAEFQK | Alpha-myosin |
| peptide_79 | AEFQKMRRDLEEATLQHEAT | Alpha-myosin |
| peptide_80 | QHEATAAALRKKHADSVAEL | Alpha-myosin |
| peptide_81 | SVAELGEQIDNLQRVKQKLE | Alpha-myosin |
| peptide_82 | KQKLEKEKSEFKLELDDVTS | Alpha-myosin |
| peptide_83 | DDVTSNMEQIIKAKANLEKV | Alpha-myosin |
| peptide_84 | NLEKVSRTLEDQANEYRVKL | Alpha-myosin |
| peptide_85 | YRVKLEEAQRSLNDFTTQRA | Alpha-myosin |
| peptide_86 | TTQRAKLQTENGELARQLEE | Alpha-myosin |
| peptide_87 | RQLEEKEALISQLTRGKLSY | Alpha-myosin |
| peptide_88 | GKLSYTQQMEDLKRQLEEEG | Alpha-myosin |
| peptide_89 | LEEEGKAKNALAHALQSSRH | Alpha-myosin |
| peptide_90 | QSSRHDCDLLREQYEEEMEA | Alpha-myosin |
| peptide_91 | EEMEAKAELQRVLSKANSEV | Alpha-myosin |
| peptide_92 | ANSEVAQWRTKYETDAIQRT | Alpha-myosin |
| peptide_93 | AIQRTEELEEAKKKLAQRLQ | Alpha-myosin |
| peptide_94 | AQRLQDAEEAVEAVNAKCSS | Alpha-myosin |
| peptide_95 | AKCSSLEKTKHRLQNEIEDL | Alpha-myosin |
| peptide_96 | EIEDLMVDVERSNAAAAALD | Alpha-myosin |
| peptide_97 | AAALDKKQRNFDKILAEWKQ | Alpha-myosin |
| peptide_98 | AEWKQKYEESQSELESSQKE | Alpha-myosin |
| peptide_99 | SSQKEARSLSTELFKLKNAY | Alpha-myosin |
| peptide_100 | LKNAYEESLEHLETFKRENK | Alpha-myosin |
| peptide_101 | KRENKNLQEEISDLTEQLGE | Alpha-myosin |
| peptide_102 | EQLGEGGKNVHELEKIRKQL | Alpha-myosin |
| peptide_103 | IRKQLEVEKLELQSALEEAE | Alpha-myosin |
| peptide_104 | LEEAEASLEHEEGKILRAQL | Alpha-myosin |
| peptide_105 | LRAQLEFNQIKAEIERKLAE | Alpha-myosin |
| peptide_106 | RKLAEKDEEMEQAKRNHLRM | Alpha-myosin |
| peptide_108 | ETRSRNEALRVKKKMEGDLN | Alpha-myosin |
| peptide_109 | EGDLNEMEIQLSQANRIASE | Alpha-myosin |
| peptide_110 | RIASEAQKHLKNSQAHLKDT | Alpha-myosin |
| peptide_111 | HLKDTQLQLDDAVHANDDLK | Alpha-myosin |
| peptide_112 | NDDLKENIAIVERRNNLLQA | Alpha-myosin |
| peptide_113 | NLLQAELEELRAWEQTERS | Alpha-myosin |
| peptide_114 | QTERSRKLAEQELIETSERV | Alpha-myosin |
| peptide_115 | TSERVQLLHSQNTSLINQKK | Alpha-myosin |
| peptide_116 | INQKKKMESDLTQLQTEVEE | Alpha-myosin |
| peptide_117 | TEVEEAVQECRNAEEKAKKA | Alpha-myosin |
| peptide_118 | KAKKAITDAAMMAEELKKEQ | Alpha-myosin |
| peptide_119 | LKKEQDTSAHLERMKKNMEQ | Alpha-myosin |
| peptide_120 | KNMEQTIKDLQHRLDEAEQI | Alpha-myosin |
| peptide_121 | EAEQIALKGGKKQLQKLEAR | Alpha-myosin |
| peptide_122 | KLEARVRELENELEAEQKRN | Alpha-myosin |
| peptide_123 | EQKRNAESVKGMRKSERRIK | Alpha-myosin |
| peptide_124 | ERRIKELTYQTEEDKKNLMR | Alpha-myosin |
| peptide_125 | KNLMRLQDLVDKLQLKVKAY | Alpha-myosin |
| peptide_126 | KVKAYKRQAEEAEEQANTNL | Alpha-myosin |
| peptide_127 | ANTNLSKFRKVQHELDEAEE | Alpha-myosin |
| peptide_128 | DEAEERADIAESQVNKLRAK | Alpha-myosin |
| peptide_129 | KLRAKSRDIGAKKMHDEE | Alpha-myosin |
| 19A | KSRVIFQLKA | Alpha-myosin |
| 19B | FQLKAERNYH | Alpha-myosin |
| 19C | ERNYHIFYQI | Alpha-myosin |
| 55A | DALLVIQWNI | Alpha-myosin |
| 55B | IQWNIRAFMG | Alpha-myosin |
| 55C | RAFMGVKNWP | Alpha-myosin |
| SBK2_1 | MPGKQSEDKPMEVSTVEDGG | SBK2 |
| SBK2_2 | VEDGGDEGLGGLTVEELQQG | SBK2 |
| SBK2_3 | ELQQGQEAALALEDMMALSA | SBK2 |
| SBK2_4 | MALSAQTLVQTEVEELYEEV | SBK2 |
| SBK2_5 | LYEEVRPLGQGRFGRVLLVT | SBK2 |
| SBK2_6 | VLLVTHRQKGTPLALKQLPK | SBK2 |
| SBK2_7 | KQLPKQSTSLRGFLYEFCVG | SBK2 |
| SBK2_8 | EFCVGLSLGTH SAIVTAYG1 | SBK2 |
| SBK2_9 | TAYGIGIESANSYSFLTEPV | SBK2 |
| SBK2_10 | LTEPVLHGDLITFIQPKVGL | SBK2 |
| SBK2_11 | PKVGLPQPAAQRCAAQLASA | SBK2 |
| SBK2_12 | QLASALEHIHSHGLVYRDLK | SBK2 |
| SBK2_13 | YRDLKPENVLVCDPACQRVK | SBK2 |
| SBK2_14 | CQRVKLTDFGHTRPRGTLLR | SBK2 |
| SBK2_15 | GTLLRLTGPPIPYTAPELCA | SBK2 |
| SBK2_16 | PELCAPPPLPEGLPIQPSLD | SBK2 |
| SBK2_17 | QPSLDAWALGVLIFCLLTGY | SBK2 |
| SBK2_18 | LLTGYFPWDQPLVEVDPFFE | SBK2 |
| SBK2_19 | DPFFEDFLIWQASGQPQDRP | SBK2 |
| SBK2_20 | PQDRPQPWYSLSPAADTLLW | SBK2 |
| SBK2_21 | DTLLWGLLDPHPRKRNPVGS | SBK2 |
| SBK2_22 | NPVGSIKSYLGQPWKQREGE | SBK2 |
| SBK2_23 | QREGEAEELATELREDGWRG | SBK2 |
| SBK2_24 | DGWRGGQEAAKGEQPAC | SBK2 |
| NPPA_26 | FWLPGHIGANPVYSAVSNTD | ANP |
| NPPA_27 | VSNTDLMDFKNLLDHLEEKM | ANP |
| NPPA_28 | LEEKMPVEDEVMPPQALSEQ | ANP |
| NPPA_29 | ALSEQTEEAGAALSSLPEVP | ANP |
| NPPA_30 | LPEVPPWTGEVNPPLRDGSA | ANP |
| NPPA_31 | RDGSALGRSPWDPSDRSALL | ANP |
| NPPA_32 | RSALLKSKLRALLAGPRSLR | ANP |
| NPPA_33 | PRSLRRSSCFGGRIDRIGAQ | ANP |
| NPPA_34 | RIGAQSGLGCNSFRYRR | ANP |
| BNP_35 | MDLLKVLSQMILFLLFLYLS | BNP |
| BNP_36 | FLYLSPLGGHSYPLGSPSQS | BNP |
| BNP_37 | SPSQSPEQFKMQKLLELIRE | BNP |
| BNP_38 | ELIREKSEEMAQRQLLKDQG | BNP |
| BNP_39 | LKDQGLTKEHPKRVLRSQGS | BNP |
| BNP_40 | RSQGSTLRVQQRPQNSKVTH | BNP |
| BNP_41 | SKVTHISSCFGHKIDRIGSV | BNP |
| BNP_42 | RIGSVSRLGCNALKLL | BNP |
Extended Data Table 2.
Prediction scores for binding of α-myosin peptides to MHC-I molecules in C57BL/6 mice generated by TepiTool.
| Seq# | Peptide start | Peptide end | Peptide | Percentile rank | Allele |
|---|---|---|---|---|---|
| 1 | 190 | 198 | RVIQYFASI | 0.01 | H-2-Kb |
| 1 | 113 | 120 | MIYTYSGL | 0.01 | H-2-Kb |
| 1 | 650 | 660 | SALHRENLNKL | 0.02 | H-2-Db |
| 1 | 644 | 652 | SSFQTVSAL | 0.03 | H-2-Kb |
| 1 | 613 | 621 | SSLKLMATL | 0.04 | H-2-Db |
| 1 | 282 | 290 | RNYHIFYQI | 0.04 | H-2-Kb |
| 1 | 191 | 198 | VIQYFASI | 0.04 | H-2-Kb |
| 1 | 764 | 771 | KVFFKAGL | 0.05 | H-2-Kb |
| 1 | 824 | 832 | MGVKNWPWM | 0.06 | H-2-Db |
| 1 | 322 | 330 | ASIDDSEEL | 0.06 | H-2-Db |
| 1 | 827 | 834 | KNWPWMKL | 0.06 | H-2-Kb |
| 1 | 613 | 621 | SSLKLMATL | 0.06 | H-2-Kb |
| 1 | 1759 | 1767 | KAITDAAMM | 0.07 | H-2-Db |
| 1 | 644 | 652 | SSFQTVSAL | 0.07 | H-2-Db |
| 1 | 1242 | 1250 | KAKANLEKV | 0.07 | H-2-Db |
| 1 | 730 | 738 | AAIPEGQFI | 0.08 | H-2-Db |
| 1 | 684 | 692 | GVMDNPLVM | 0.08 | H-2-Db |
| 1 | 479 | 486 | INFTNEKL | 0.08 | H-2-Kb |
| 1 | 909 | 917 | QUKNKIQL | 0.09 | H-2-Db |
| 1 | 1893 | 1901 | TNLSKFRKV | 0.11 | H-2-Kb |
| 1 | 421 | 428 | VYYSIGAL | 0.12 | H-2-Kb |
| 1 | 356 | 363 | AIMHYGNM | 0.12 | H-2-Kb |
| 1 | 148 | 157 | SEAPPHIFSI | 0.13 | H-2-Db |
| 1 | 1890 | 1898 | QANTNLSKF | 0.13 | H-2-Db |
| 1 | 155 | 165 | FSISDNAYQYM | 0.14 | H-2-Db |
| 1 | 279 | 287 | KAERNYHIF | 0.14 | H-2-Db |
| 1 | 311 | 319 | YAFVSQGEV | 0.14 | H-2-Db |
| 1 | 487 | 495 | QQFFNHHMF | 0.16 | H-2-Db |
| 1 | 149 | 157 | EAPPHIFSI | 0.17 | H-2-Db |
| 1 | 113 | 121 | MIYTYSGLF | 0.17 | H-2-Kb |
| 1 | 243 | 251 | SRFGKFIRI | 0.18 | H-2-Kb |
| 1 | 649 | 657 | VSALHRENL | 0.18 | H-2-Kb |
| 1 | 282 | 291 | RNYHIFYQIL | 0.18 | H-2-Kb |
| 1 | 724 | 732 | YRILNPAAI | 0.19 | H-2-Db |
| 1 | 487 | 497 | QQFFNHHMFVL | 0.2 | H-2-Db |
| 1 | 335 | 342 | SAFDVLSF | 0.2 | H-2-Kb |
| 1 | 765 | 772 | VFFKAGLL | 0.2 | H-2-Kb |
| 1 | 147 | 157 | RSEAPPHIFSI | 0.21 | H-2-Db |
| 1 | 1520 | 1528 | EGGKNVHEL | 0.21 | H-2-Db |
| 1 | 666 | 674 | TTHPHFVRC | 0.21 | H-2-Kb |
| 1 | 431 | 439 | SVYEKMFNW | 0.21 | H-2-Kb |
| 1 | 650 | 657 | SALHRENL | 0.21 | H-2-Kb |
| 1 | 1587 | 1595 | QAKRNHLRM | 0.22 | H-2-Db |
| 1 | 682 | 692 | APGVMDNPLVM | 0.22 | H-2-Db |
| 1 | 1563 | 1570 | AQLEFNQI | 0.22 | H-2-Kb |
| 1 | 418 | 425 | VQQVYYSI | 0.22 | H-2-Kb |
| 1 | 1674 | 1682 | AIVERRNNL | 0.23 | H-2-Kb |
| 1 | 881 | 891 | SLLQEKNDLQL | 0.24 | H-2-Db |
| 1 | 681 | 690 | KAPGVMDNPL | 0.24 | H-2-Db |
| 1 | 260 | 268 | ASADIETYL | 0.25 | H-2-Db |
| 1 | 1791 | 1799 | TIKDLQHRL | 0.25 | H-2-Kb |
| 1 | 454 | 461 | RQYFIGVL | 0.25 | H-2-Kb |
| 1 | 1490 | 1500 | YEESLEHLETF | 0.26 | H-2-Db |
| 1 | 284 | 291 | YHIFYQIL | 0.26 | H-2-Kb |
| 1 | 764 | 772 | KVFFKAGLL | 0.26 | H-2-Kb |
| 1 | 420 | 428 | QVYYSIGAL | 0.26 | H-2-Kb |
| 1 | 834 | 841 | LYFKIKPL | 0.29 | H-2-Kb |
| 1 | 355 | 363 | GAIMHYGNM | 0.29 | H-2-Kb |
| 1 | 76 | 84 | VMQQNPPKF | 0.3 | H-2-Db |
| 1 | 487 | 494 | QQFFNHHM | 0.3 | H-2-Kb |
| 1 | 125 | 133 | VNPYKWLPV | 0.3 | H-2-Kb |
| 1 | 1291 | 1299 | RQLEEKEAL | 0.31 | H-2-Db |
| 1 | 657 | 664 | LNKLMTNL | 0.31 | H-2-Kb |
| 1 | 320 | 330 | SVASIDDSEEL | 0.32 | H-2-Db |
| 1 | 1207 | 1215 | EQIDNLQRV | 0.32 | H-2-Db |
| 1 | 834 | 842 | LYFKIKPLL | 0.32 | H-2-Kb |
| 1 | 271 | 278 | KSRVIFQL | 0.32 | H-2-Kb |
| 1 | 545 | 553 | SDMTFKAKL | 0.32 | H-2-Kb |
| 1 | 1492 | 1500 | ESLEHLETF | 0.33 | H-2-Db |
| 1 | 384 | 391 | KSAYLMG | 0.33 | H-2-Kb |
| 1 | 437 | 444 | FNWMVTRI | 0.35 | H-2-Kb |
| 1 | 1861 | 1869 | KNLMRLQDL | 0.35 | H-2-Kb |
| 1 | 1257 | 1265 | QANEYRVKL | 0.35 | H-2-Kb |
| 1 | 157 | 165 | ISDNAYQYM | 0.37 | H-2-Kb |
| 1 | 417 | 425 | SVQQVYYSI | 0.37 | H-2-Kb |
| 1 | 490 | 497 | FNHHMFVL | 0.37 | H-2-Kb |
| 1 | 833 | 841 | KLYFKIKPL | 0.39 | H-2-Kb |
| 1 | 123 | 131 | VTVNPYKWL | 0.39 | H-2-Kb |
| 1 | 441 | 448 | VTRINATL | 0.39 | H-2-Kb |
| 1 | 883 | 891 | LQEKNDLQL | 0.4 | H-2-Db |
| 1 | 1409 | 1416 | VNAKCSSL | 0.41 | H-2-Kb |
| 1 | 261 | 269 | SADIETYLL | 0.42 | H-2-Db |
| 1 | 1484 | 1494 | FKLKNAYEESL | 0.42 | H-2-Kb |
| 1 | 322 | 331 | ASIDDSEELL | 0.43 | H-2-Db |
| 1 | 1239 | 1247 | QIIKAKANL | 0.43 | H-2-Kb |
| 1 | 79 | 87 | QNPPKFDKI | 0.43 | H-2-Kb |
| 1 | 649 | 657 | VSALHRENL | 0.44 | H-2-Db |
| 1 | 607 | 615 | VGLYQKSSL | 0.44 | H-2-Kb |
| 1 | 467 | 474 | EIFDFNSF | 0.44 | H-2-Kb |
| 1 | 431 | 440 | SVYEKMFNWM | 0.44 | H-2-Kb |
| 1 | 562 | 570 | NNFQKPRNV | 0.44 | H-2-Db |
| 1 | 355 | 363 | GAIMHYGNM | 0.45 | H-2-Db |
| 1 | 288 | 298 | YQILSNKKPEL | 0.45 | H-2-Db |
| 1 | 487 | 496 | QQFFNHHMFV | 0.46 | H-2-Db |
| 1 | 93 | 101 | LTFLHEPAV | 0.46 | H-2-Kb |
| 1 | 823 | 832 | FMGVKNWPWM | 0.47 | H-2-Db |
| 1 | 1655 | 1663 | TQLQLDDAV | 0.47 | H-2-Db |
| 1 | 242 | 251 | SSRFGKFIRI | 0.47 | H-2-Kb |
| 1 | 1258 | 1265 | ANEYRVKL | 0.48 | H-2-Kb |
| 1 | 1240 | 1247 | IIKAKANL | 0.48 | H-2-Kb |
| 1 | 920 | 928 | KVKEMTERL | 0.49 | H-2-Kb |
| 1 | 1479 | 1486 | LSTELFKL | 0.5 | H-2-Kb |
| 1 | 614 | 621 | SLKLMATL | 0.5 | H-2-Kb |
| 1 | 592 | 602 | MGWLEKNKDPL | 0.51 | H-2-Db |
| 1 | 971 | 980 | HATENKVKNL | 0.51 | H-2-Db |
| 1 | 1783 | 1792 | RMKKNMEQTI | 0.51 | H-2-Db |
| 1 | 388 | 396 | LMGLNSADL | 0.52 | H-2-Db |
| 1 | 229 | 237 | EAFGNAKTV | 0.52 | H-2-Db |
| 1 | 594 | 602 | WLEKNKDPL | 0.52 | H-2-Db |
| 1 | 149 | 157 | EAPPHIFSI | 0.52 | H-2-Kb |
| 1 | 1831 | 1839 | EQKRNAESV | 0.53 | H-2-Db |
| 1 | 156 | 165 | SISDNAYQYM | 0.53 | H-2-Db |
| 1 | 389 | 397 | MGLNSADLL | 0.53 | H-2-Db |
| 1 | 227 | 237 | ALEAFGNAKTV | 0.53 | H-2-Kb |
| 1 | 164 | 174 | YMLTDRENQSI | 0.54 | H-2-Db |
| 1 | 1223 | 1230 | KSEFKLEL | 0.54 | H-2-Kb |
| 1 | 321 | 330 | VASIDDSEEL | 0.56 | H-2-Db |
| 1 | 1544 | 1553 | SALEEAEASL | 0.56 | H-2-Db |
| 1 | 311 | 321 | YAFVSQGEVSV | 0.57 | H-2-Db |
| 1 | 487 | 495 | QQFFNHHMF | 0.57 | H-2-Kb |
| 1 | 251 | 259 | IHFGATGKL | 0.57 | H-2-Kb |
| 1 | 816 | 824 | IQWNIRAFM | 0.59 | H-2-Kb |
| 1 | 488 | 496 | QFFNHHMFV | 0.59 | H-2-Kb |
| 1 | 999 | 1009 | KALQEAHQQAL | 0.6 | H-2-Db |
| 1 | 816 | 824 | IQWNIRAFM | 0.6 | H-2-Db |
| 1 | 1109 | 1117 | KLKENQARI | 0.6 | H-2-Db |
| 1 | 709 | 716 | KGFPNRIL | 0.6 | H-2-Kb |
| 1 | 1446 | 1454 | KKQRNFDKI | 0.61 | H-2-Db |
| 1 | 816 | 823 | IQWNIRAF | 0.62 | H-2-Kb |
| 1 | 283 | 291 | NYHIFYQIL | 0.62 | H-2-Kb |
| 1 | 1379 | 1388 | TDAIQRTEEL | 0.64 | H-2-Db |
| 1 | 124 | 131 | TVNPYKWL | 0.64 | H-2-Kb |
| 1 | 687 | 695 | DNPLVMHQL | 0.64 | H-2-Kb |
| 1 | 768 | 778 | KAGLLGLLEEM | 0.66 | H-2-Db |
| 1 | 289 | 299 | QILSNKKPELL | 0.66 | H-2-Db |
| 1 | 709 | 717 | KGFPNRILY | 0.67 | H-2-Db |
| 1 | 487 | 494 | QQFFNHHM | 0.68 | H-2-Db |
| 1 | 1643 | 1652 | KHLKNSQAHL | 0.68 | H-2-Db |
| 1 | 599 | 607 | KDPLNETW | 0.69 | H-2-Db |
| 1 | 486 | 494 | LQQFFNHHM | 0.69 | H-2-Kb |
| 1 | 532 | 540 | MSILEEECM | 0.7 | H-2-Db |
| 1 | 69 | 77 | VTIKEDQVM | 0.7 | H-2-Db |
| 1 | 1201 | 1209 | SVAELGEQI | 0.71 | H-2-Db |
| 1 | 1380 | 1388 | DAIQRTEEL | 0.72 | H-2-Db |
| 1 | 1488 | 1497 | NAYEESLEHL | 0.72 | H-2-Db |
| 1 | 190 | 198 | RVIQYFASI | 0.72 | H-2-Db |
| 1 | 1649 | 1657 | QAHLKDTQL | 0.72 | H-2-Db |
| 1 | 1723 | 1732 | TSLINQKKKM | 0.72 | H-2-Db |
| 1 | 1437 | 1444 | SNAAAAAL | 0.72 | H-2-Kb |
| 1 | 597 | 607 | KNKDPINETW | 0.73 | H-2-Db |
| 1 | 1328 | 1335 | KNALAHAL | 0.73 | H-2-Kb |
| 1 | 1675 | 1682 | IVERRNNL | 0.73 | H-2-Kb |
| 1 | 1757 | 1767 | AKKAITDAAMM | 0.75 | H-2-Db |
| 1 | 642 | 649 | KGSSFQTV | 0.75 | H-2-Kb |
| 1 | 479 | 489 | INFTNEKLQQF | 0.76 | H-2-Kb |
| 1 | 550 | 558 | KAKLYDNHL | 0.76 | H-2-Kb |
| 1 | 1764 | 1771 | AAMMAEEL | 0.77 | H-2-Db |
| 1 | 645 | 652 | SFQTVSAL | 0.78 | H-2-Kb |
| 1 | 244 | 251 | RFGKFIRI | 0.79 | H-2-Kb |
| 1 | 514 | 522 | FGMDLQACI | 0.8 | H-2-Db |
| 1 | 763 | 771 | TKVFFKAGL | 0.8 | H-2-Kb |
| 1 | 544 | 553 | ASDMTFKAKL | 0.8 | H-2-Kb |
| 1 | 426 | 436 | GALAKSVYEKM | 0.81 | H-2-Db |
| 1 | 1830 | 1839 | AEQKRNAESV | 0.82 | H-2-Db |
| 1 | 1562 | 1570 | RAQLEFNQI | 0.82 | H-2-Db |
| 1 | 1519 | 1528 | GEGGKNVHEL | 0.83 | H-2-Db |
| 1 | 824 | 834 | MGVKNWPWMKL | 0.84 | H-2-Db |
| 1 | 157 | 165 | ISDNAYQYM | 0.85 | H-2-Db |
| 1 | 488 | 495 | QFFNHHMF | 0.85 | H-2-Kb |
| 1 | 769 | 778 | AGILGLIEEM | 0.86 | H-2-Db |
| 1 | 282 | 290 | RNYHIFYQI | 0.87 | H-2-Db |
| 1 | 485 | 495 | KLQQFFNHHMF | 0.88 | H-2-Db |
| 1 | 1157 | 1165 | GATSVQIEM | 0.89 | H-2-Db |
| 1 | 467 | 477 | EIFDFNSFEQl | 0.89 | H-2-Kb |
| 1 | 822 | 832 | AFMGVKNWPWM | 0.9 | H-2-Db |
| 1 | 332 | 340 | ATDSAFDVL | 0.9 | H-2-Db |
| 1 | 469 | 477 | FDFNSFEQL | 0.9 | H-2-Db |
| 1 | 428 | 436 | LAKSVYEKM | 0.91 | H-2-Kb |
| 1 | 651 | 660 | ALHRENLNKl | 0.92 | H-2-Db |
| 1 | 1102 | 1110 | LALQLQKKL | 0.94 | H-2-Kb |
| 1 | 667 | 674 | THPHFVRC | 0.94 | H-2-Kb |
| 1 | 260 | 268 | ASADIETYL | 0.94 | H-2-Kb |
| 1 | 644 | 651 | SSFQTVSA | 0.95 | H-2-Kb |
| 1 | 1679 | 1687 | RNNLLQAEL | 0.95 | H-2-Kb |
| 1 | 267 | 275 | YLLEKSRVI | 0.96 | H-2-Db |
| 1 | 283 | 290 | NYHIFYQI | 0.96 | H-2-Kb |
| 1 | 468 | 477 | IFDFNSFEQL | 0.97 | H-2-Kb |
| 1 | 1551 | 1561 | ASLEHEEGKIL | 0.98 | H-2-Db |
| 1 | 1301 | 1308 | SQLTRGKL | 0.98 | H-2-Kb |
| 1 | 1102 | 1110 | LALQLQKKL | 0.99 | H-2-Db |
| 1 | 667 | 675 | THPHFVRCI | 0.99 | H-2-Kb |
Extended Data Table 3.
HLA types for healthy donors and ICI-MC patients.
| Subject Identifier | HLA-A Condensed/Putative Allele Result |
HLA-B Condensed/Putative Allele Result |
HLA-C Condensed/Putative Allele Result |
HLA-DPB1 Condensed/Putative Allele Result |
HLA-DQA1 Condensed/Putative Allele Result |
HLA-DQB1 Condensed/Putative Allele Result |
HLA-DRB1 Condensed/Putative Allele Result |
HLA-DRB3 Condensed/Putative Allele Result |
HLA-DRB4 Condensed/Putative Allele Result |
HLA-DRB5 Condensed/Putative Allele Result |
|---|---|---|---|---|---|---|---|---|---|---|
| Myocarditis Pt 1 | 03:01:01G + 24:02:01G | 07:02:01G + 51:01:01G | 01:02:01G + 07:02:01G | 02:02:01G + 04:01:01G | 01:02:01G + 05:01:01G | 03:01:01i + 06:02:01i | 11:01:01i + 15:01:01i | 02:02:01i + NP | NP + NP | 01:01:01i + NP |
| Myocarditis Pt 2 | 02:01:01G + 24:02:01G | 39:06:02G + 40:02:01G | 07:02:01G + 15:02:01G | 03:01:01G + 06:01:01G | 03:01:01G + 04:01:01G | 03:01:01i + 04:02:01i | 04:01:01i + 08:01:01i | NP + NP | 01:01:01i + NP | NP + NP |
| Myocarditis Pt 3 | 01:01:01G + 03:01:01G | 07:02:01G + 08:01:01G | 07:01:01G + 07:02:01G | 03:01:01G + 04:01:01G | 01:02:01G + 05:01:01G | 02:01:01i + 06:02:01i | 03:01:01i + 15:01:01i | 01:01:02i + NP | NP + NP | 01:01:01i + NP |
| Healthy Donor 1 | 03:01:01G + 23:01:01G | 07:02:01G + 44:03:01G | 04:01:01G + 07:02:01G | 02:01:02G + 02:01:02G | 01:02:01G + 02:01:02G | 02:02:01i + 06:02:01i | 07:01:011 + 15:01:01i | NP + NP | 01:01:01i + NP | 01:01:01i + NP |
| Healthy Donor 2 | 02:06:01G + 30:01:01G | 14:02:01G + 48:01:01G | 08:02:01G + 08:03:01G | 02:01:02G + 04:01:01G | 01:01:01G + 03:01:01G | 03:04:01i + 05:01:01i | 01:02:01i + 04:03:01i | NP + NP | 01:01:01i + NP | NP + NP |
| Healthy Donor 3 | 03:01:01G + 66:01:01G | 41:02:01G + 57:01:01G | 06:02:01G + 17:01:01G | 04:01:01G + 04:01:01G | 01:02:01G + 05:01:01G | 03:01:01i + 05:02:01i | 13:03:01i + 16:01:01i | 01:01:02i + NP | NP + NP | 02:02:01i + NP |
Supplementary Material
Supplemental Methods. Full nucleotide sequences for reconstructed TCRs.
Acknowledgements
We thank the patients and families who donated tissue to this study. We thank M. Madden and M. Korrer for assistance with sample acquisition. We thank members of the J.M.B. laboratory and B.I. Reinfeld for their constructive input. We thank S. Mallal, P. Steinberger and J. Sidney for cell lines and reagents. This work was supported by F30CA236157, T32GM007347 (MLA), R01HL141466(JJM), R01HL155990 (JJM), R01CA227481(DBJ, JMB), R01HL156021 (JMB, JJM), P30 AI110527 (SM), Susan and Luke Simons Directorship in Melanoma, Van Stephenson Melanoma Research Fund, and James C. Bradford Melanoma Fund (DBJ). W. Meijers is supported by the Mandema-Stipendium of the Junior Scientific Masterclass 2020- 10 of the University Medical Center Groningen and by the Dutch Heart Foundation (Dekker grant 03-005-2021-T005). S Zhang is supported by National Natural Science Foundation of China (82100677). JP Allison is a CPRIT Distinguished Scholar in Cancer Research. We acknowledge the Translational Pathology Shared Resource supported by NCI/NIH Cancer Center Support Grant P30CA068485 and the Shared Instrumentation Grant S10 OD023475-01A1 for the Leica Bond RX. The Vanderbilt VANTAGE Core, including A. Jones and L. Raju, provided technical assistance for this work. VANTAGE is supported in part by a CTSA Grant (5UL1 RR024975-03), the Vanderbilt Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126) and the NIH/NCRR (G20 RR030956). Figures 1a and 4b were created with BioRender.com.
Footnotes
Conflict of Interest Disclosure
M.L. Axelrod is listed as a coinventor on a provisional patent application for methods to predict therapeutic outcomes using blood-based gene expression patterns, that is owned by Vanderbilt University Medical Center, and is currently unlicensed. S.C. Wei is an employee of Spotlight Therapeutics, a consultant for BioEntre, and an inventor on a patent for a genetic mouse model of autoimmune adverse events and immune checkpoint blockade therapy (PCT/US2019/050551) pending to Board of Regents, The University of Texas System. K. Amancherla serves on the Data Safety Monitoring Board for ACI Clinical. J.C. Rathmell is a founder, scientific advisory board member, and stockholder of Sitryx Therapeutics, a scientific advisory board member and stockholder of Caribou Biosciences, a member of the scientific advisory board of Nirogy Therapeutics, has consulted for Merck, Pfizer, and Mitobridge within the past three years, and has received research support from Incyte Corp., Calithera Biosciences, and Tempest Therapeutics. P.B. Ferrell receives research support from Incyte Corporation. D.B.Johnson has served on advisory boards or as a consultant for BMS, Catalyst Biopharma, Iovance, Jansen, Mallinckrodt, Merck, Mosaic ImmunoEngineering, Novartis, Oncosec, Pfizer, and Targovax, has received research funding from BMS and Incyte, and has patents pending for use of MHC-II as a biomarker for immune checkpoint inhibitor response, and abatacept as treatment for immune-related adverse events. J.P. Allison reports personal fees from Achelois, Adaptive Biotechnologies, personal fees from Apricity Health, personal fees from BioAtla, Candel Therapeutics, personal fees from Codiak BioSciences, personal fees from Dragonfly Therapeutics, Earli, Enable Medicine, personal fees from Hummingbird, personal fees from ImaginAb, personal fees from Jounce Therapeutics, personal fees from Lava Therapeutics, personal fees from Lytix Biopharma, personal fees from Marker Therapeutics, PBM Capital, Phenomic AI, personal fees from BioNTech, and personal fees from Polaris Pharma, Time Bioventures, Trained Therapeutics, Two Bear Capital, Venn Biosciences outside the submitted work; in addition, J.P. Allison has a patent for a genetic mouse model of immune checkpoint blockade induced immune-related adverse events pending to The University of Texas MD Anderson Cancer Center; and have received royalties from intellectual property licensed to BMS and Merck. J. Moslehi has served on advisory boards for Bristol Myers Squibb, Takeda, Audentes, Deciphera, Janssen, Immuno-Core, Boston Biomedical, Amgen, Myovant, Kurome Therapeutics, Star Therapeutics, ProtinQure, Pharmacyclics, Pfizer, Mallinckrodt Pharmaceuticals, Silverback Therapeutics, Cytokinetics, and AstraZeneca. J. M. Balko receives research support from Genentech/Roche, and Incyte Corporation, and is an inventor on provisional patents regarding immunotherapy targets and biomarkers in cancer. No disclosures were reported by the other authors.
Data Availability.
Sequencing data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE213486. Source data are provided for all studies involving animals.
References
- 1.Wang DY, Salem J-EE, Cohen JV., et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018;4(12):1721–1728. http://www.embase.com/search/results?subaction=viewrecord&from=export&id=L624011231. Accessed October 15, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Meijers WC, Axelrod ML, et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor–Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2020;11(3):614–639. doi: 10.1158/2159-8290.cd-20-0856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lv H, Havari E, Pinto S, et al. Impaired thymic tolerance to α-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011;121(4):1561–1573. doi: 10.1172/JCI44583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gabrielsen ISM, Helgeland H, Akselsen H, et al. Transcriptomes of antigen presenting cells in human thymus. PLoS One. 2019;14(7):e0218858. doi: 10.1371/JOURNAL.PONE.0218858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375(18):1749–1755. doi: 10.1056/NEJMoa1609214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu J-RR, Florido R, Lipson EJ, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc Res. 2019;115(5):854. https://academic.oup.com/cardiovascres/advance-article/doi/10.1093/cvr/cvz026/5304411. Accessed March 4, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salem JE, Manouchehri A, Moey M, et al. Spectrum of cardiovascular toxicities of immune checkpoint inhibitors: A pharmacovigilance study. Lancet Oncol. 2018;19(12):1579. doi: 10.1016/S1470-2045(18)30608-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moslehi J, Lichtman AH, Sharpe AH, Galluzzi L, Kitsis RN. Immune checkpoint inhibitor–associated myocarditis: manifestations and mechanisms. J Clin Invest. 2021;131(5). doi: 10.1172/JCI145186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamami Y, Niimura T, Okada N, et al. Factors Associated With Immune Checkpoint Inhibitor–Related Myocarditis. JAMA Oncol. 2019;5(11):1635–1637. doi: 10.1001/JAMAONCOL.2019.3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salem J-E, Allenbach Y, Vozy A, et al. Abatacept for Severe Immune Checkpoint Inhibitor–Associated Myocarditis. N Engl J Med. 2019;380(24):2377–2379. doi: 10.1056/NEJMc1901677 [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Bam M, Becker W, Nagarkatti PS, Nagarkatti M. Long Noncoding RNA AW112010 Promotes the Differentiation of Inflammatory T Cells by Suppressing IL-10 Expression through Histone Demethylation. J Immunol. 2020;205(4):987–993. doi: 10.4049/jimmunol.2000330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson R, Kroehling L, Khitun A, et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature. 2018;564(7736):434–438. doi: 10.1038/s41586-018-0794-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adamo L, Rocha-Resende C, Lin CY, et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI insight. 2020;5(3). doi: 10.1172/JCI.INSIGHT.134700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bönner F, Borg N, Burghoff S, Schrader J. Resident cardiac immune cells and expression of the ectonucleotidase enzymes CD39 and CD73 after ischemic injury. PLoS One. 2012;7(4). doi: 10.1371/JOURNAL.PONE.0034730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martini E, Kunderfranco P, Peano C, et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation. 2019;140(25):2089–2107. doi: 10.1161/CIRCULATIONAHA.119.041694 [DOI] [PubMed] [Google Scholar]
- 16.Li O, Zheng P, Liu Y. CD24 expression on T cells is required for optimal T cell proliferation in lymphopenic host. J Exp Med. 2004;200(8):1083–1089. doi: 10.1084/JEM.20040779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hubbe M, Altevogt P. Heat-stable antigen/CD24 on mouse T lymphocytes: evidence for a costimulatory function. Eur J Immunol. 1994;24(3):731–737. doi: 10.1002/EJI.1830240336 [DOI] [PubMed] [Google Scholar]
- 18.Szabo PA, Miron M, Farber DL. Location, location, location: Tissue resident memory T cells in mice and humans. Sci Immunol. 2019;4(34). doi: 10.1126/SCIIMMUNOL.AAS9673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fonseca R, Burn TN, Gandolfo LC, et al. Runx3 drives a CD8+ T cell tissue residency program that is absent in CD4+ T cells. Nat Immunol 2022 238. 2022;23(8):1236–1245. doi: 10.1038/s41590-022-01273-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Zlotoff DA, Awadalla M, et al. Major adverse cardiovascular events and the timing and dose of corticosteroids in immune checkpoint inhibitor-associated myocarditis. Circulation. 2020;141(24):2031–2034. doi: 10.1161/CIRCULATIONAHA.119.044703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335(1):2. doi: 10.1016/J.MCE.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heather JM, Spindler MJ, Alonso MH, et al. Stitchr: stitching coding TCR nucleotide sequences from V/J/CDR3 information. Nucleic Acids Res. 2022;1(1256879):13–14. doi: 10.1093/nar/gkac190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosskopf S, Leitner J, Paster W, et al. A Jurkat 76 based triple parameter reporter system to evaluate TCR functions and adoptive T cell strategies. Oncotarget. 2018;9(25):17608–17619. doi: 10.18632/oncotarget.24807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jutz S, Leitner J, Schmetterer K, et al. Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: Simultaneous measurement of NF-κB, NFAT and AP-1. J Immunol Methods. 2016;430:10–20. doi: 10.1016/j.jim.2016.01.007 [DOI] [PubMed] [Google Scholar]
- 25.Gil-Cruz C, Perez-Shibayama C, de Martin A, et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science (80- ). 2019;366(6467):881–886. doi: 10.1126/science.aav3487 [DOI] [PubMed] [Google Scholar]
- 26.Massilamany C, Gangaplara A, Steffen D, Reddy J. Identification of novel mimicry epitopes for cardiac myosin heavy chain-α that induce autoimmune myocarditis in A/J mice. Cell Immunol. 2011;271(2):438–449. doi: 10.1016/J.CELLIMM.2011.08.013 [DOI] [PubMed] [Google Scholar]
- 27.Meier SL, Satpathy AT, Wells DK. Bystander T cells in cancer immunology and therapy. Nat Cancer 2022 32. 2022;3(2):143–155. doi: 10.1038/S43018-022-00335-8 [DOI] [PubMed] [Google Scholar]
- 28.Maurice NJ, McElrath MJ, Andersen-Nissen E, Frahm N, Prlic M. CXCR3 enables recruitment and site-specific bystander activation of memory CD8+ T cells. Nat Commun 2019 101. 2019;10(1):1–15. doi: 10.1038/S41467-019-12980-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simoni Y, Becht E, Fehlings M, et al. Bystander CD8 + T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–579. doi: 10.1038/S41586-018-0130-2 [DOI] [PubMed] [Google Scholar]
- 30.Maurice NJ, Taber AK, Prlic M. The Ugly Duckling Turned to Swan: A Change in Perception of Bystander-Activated Memory CD8 T Cells. J Immunol. 2021;206(3):455–462. doi: 10.4049/JIMMUNOL.2000937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheper W, Kelderman S, Fanchi LF, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. 2019;25(1):89–94. doi: 10.1038/S41591-018-0266-5 [DOI] [PubMed] [Google Scholar]
- 32.Paul S, Sidney J, Sette A, Peters B. TepiTool: A pipeline for computational prediction of T cell epitope candidates. Curr Protoc Immunol. 2016;2016:18.19.1–18.19.24. doi: 10.1002/cpim.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammenseet H-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. 1991. [DOI] [PubMed] [Google Scholar]
- 34.Luoma AM, Suo S, Williams HL, et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell. 2020;0(0):1–17. doi: 10.1016/j.cell.2020.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DB, Nixon MJ, Wang Y, et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight. 2018;3(24). http://www.ncbi.nlm.nih.gov/pubmed/30568030. Accessed December 21, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji C, Roy MD, Golas J, et al. Myocarditis in cynomolgus monkeys following treatment with immune checkpoint inhibitors. Clin Cancer Res. 2019;25(15):4735–4748. doi: 10.1158/1078-0432.CCR-18-4083/74931/AM/MYOCARDITIS-IN-CYNOMOLGUS-MONKEYS-FOLLOWING [DOI] [PubMed] [Google Scholar]
- 37.Woo SR, Turnis ME, Goldberg MV., et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi: 10.1158/0008-5472.CAN-11-1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okazaki T, Okazaki IM, Wang J, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. 2011;208(2):395. doi: 10.1084/JEM.20100466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (80- ). 2018;359(6375):582–587. doi: 10.1126/science.aao4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naranbhai V, Groha S, Braun DA, et al. HLA-A*03 and response to immune checkpoint blockade in cancer: an epidemiological biomarker study. Lancet Oncol. 2021;0(0). doi: 10.1016/S1470-2045(21)00582-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Correale P, Saladino RE, Giannarelli D, et al. HLA Expression Correlates to the Risk of Immune Checkpoint Inhibitor-Induced Pneumonitis. Cells. 2020;9(9). doi: 10.3390/CELLS9091964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hasan Ali O, Berner F, Bomze D, et al. Human leukocyte antigen variation is associated with adverse events of checkpoint inhibitors. Eur J Cancer. 2019;107:8–14. doi: 10.1016/J.EJCA.2018.11.009 [DOI] [PubMed] [Google Scholar]
- 43.McCulloch JA, Davar D, Rodrigues RR, et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat Med 2022. February 2022:1–12. doi: 10.1038/s41591-022-01698-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrews MC, Duong CPMM, Gopalakrishnan V, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. 2021;27(8):1432–1441. https://www.nature.com/articles/s41591-021-01406-6. Accessed November 2, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van der Borght K, Scott CL, Martens L, et al. Myocarditis Elicits Dendritic Cell and Monocyte Infiltration in the Heart and Self-Antigen Presentation by Conventional Type 2 Dendritic Cells. Front Immunol. 2018;9:2714. doi: 10.3389/fimmu.2018.02714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rieckmann M, Delgobo M, Gaal C, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest. August 2019. doi: 10.1172/jci123859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JH, Kim TH, Park HE, et al. Myosin-primed tolerogenic dendritic cells ameliorate experimental autoimmune myocarditis. Cardiovasc Res. 2014;101(2):203–210. doi: 10.1093/CVR/CVT246 [DOI] [PubMed] [Google Scholar]
- 48.Tajiri K, Imanaka-Yoshida K, Tsujimura Y, et al. A New Mouse Model of Chronic Myocarditis Induced by Recombinant Bacille Calmette-Guèrin Expressing a T-Cell Epitope of Cardiac Myosin Heavy Chain-α. Int J Mol Sci. 2021;22(2):1–16. doi: 10.3390/IJMS22020794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hua X, Hu G, Hu Q, et al. Single-Cell RNA Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis. Circulation. May 2020. doi: 10.1161/circulationaha.119.043545 [DOI] [PubMed] [Google Scholar]
- 50.Taylor JA, Havari E, McInerney MF, Bronson R, Wucherpfennig KW, Lipes MA. A Spontaneous Model for Autoimmune Myocarditis Using the Human MHC Molecule HLA-DQ8. J Immunol. 2004;172(4):2651–2658. doi: 10.4049/jimmunol.172.4.2651 [DOI] [PubMed] [Google Scholar]
Methods only extended references
- 51.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–877. doi: 10.1016/0092-8674(92)90030-G [DOI] [PubMed] [Google Scholar]
- 52.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. 2015;33(5):495–502. doi: 10.1038/nbt.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heather JM, Spindler MJ, Alonso MH, et al. Stitchr: stitching coding TCR nucleotide sequences from V/J/CDR3 information. bioRxiv. December 2021:2021.12.20.473544. doi: 10.1101/2021.12.20.473544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu Y, Borde M, Heissmeyer V, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375–387. doi: 10.1016/J.CELL.2006.05.042 [DOI] [PubMed] [Google Scholar]
- 57.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269). doi: 10.1126/SCISIGNAL.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oh HM, Oh JM, Choi SC, et al. An efficient method for the rapid establishment of Epstein-Barr virus immortalization of human B lymphocytes. Cell Prolif. 2003;36(4):191–197. doi: 10.1046/J.1365-2184.2003.00276.X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Granato M, Santarelli R, Farina A, et al. Epstein-Barr Virus Blocks the Autophagic Flux and Appropriates the Autophagic Machinery To Enhance Viral Replication. J Virol. 2014;88(21):12715. doi: 10.1128/JVI.02199-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wölfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc 2014 94. 2014;9(4):950–966. doi: 10.1038/nprot.2014.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eberhardt CS, Kissick HT, Patel MR, et al. Functional HPV-specific PD-1+ stem-like CD8 T cells in head and neck cancer. Nat 2021 5977875. 2021;597(7875):279–284. doi: 10.1038/s41586-021-03862-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oksanen J, Blanchet FG, Friendly M, et al. Package “vegan” Title Community Ecology Package Version 2.5-7. 2020. [Google Scholar]
- 64.Nazarov V, immunarch.bot, Rumynskiy E. immunarch: An R Package for Painless Bioinformatics Analysis of T-Cell and B-Cell Immune Repertoires. June 2020. doi: 10.5281/ZENODO.3893991 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods. Full nucleotide sequences for reconstructed TCRs.
Data Availability Statement
Sequencing data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE213486. Source data are provided for all studies involving animals.