

Abstract
Background
Loss of the transcription factor GLI-Similar 3 (GLIS3) function causes congenital hypothyroidism (CH) in both humans and mice due to decreased expression of several thyroid hormone (TH) biosynthetic genes in thyroid follicular cells. Whether and to what extent, GLIS3 regulates thyroid gene transcription in coordination with other thyroid transcriptional factors (TFs), such as PAX8, NKX2.1 and FOXE1, is poorly understood.
Methods
PAX8, NKX2.1, and FOXE1 ChIP-Seq analysis with mouse thyroid glands and rat thyrocyte PCCl3 cells was performed and compared to that of GLIS3 to analyze the co-regulation of gene transcription in thyroid follicular cells by these TFs.
Results
Analysis of the PAX8, NKX2.1, and FOXE1 cistromes identified extensive overlaps between these TF binding loci and those of GLIS3 indicating that GLIS3 shares many of the same regulatory regions with PAX8, NKX2.1, and FOXE1, particularly in genes associated with TH biosynthesis, induced by thyroid stimulating hormone (TSH), and suppressed in Glis3KO thyroid glands, including Slc5a5 (Nis), Slc26a4, Cdh16, and Adm2. ChIP-QPCR analysis showed that loss of GLIS3 did not significantly affect PAX8 or NKX2.1 binding and did not cause major alterations in H3K4me3 and H3K27me3 epigenetic signals.
Conclusions
Our study indicates that GLIS3 regulates transcription of TH biosynthetic and TSH-inducible genes in thyroid follicular cells in coordination with PAX8, NKX2.1, and FOXE1 by binding within the same regulatory hub. GLIS3 does not cause major changes in chromatin structure at these common regulatory regions. GLIS3 may induce transcriptional activation by enhancing the interaction of these regulatory regions with other enhancers and/or RNA Polymerase II (Pol II) complexes.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13578-023-00979-8.
Keywords: GLIS3, NKX2.1, PAX8, FOXE1, Gene transcription, Thyroid follicular cells, PCCl3, Thyroid hormone biosynthesis, TSH, NIS
Introduction
Thyroid hormone (TH) biosynthesis in thyroid follicular cells is regulated by thyroid stimulating hormone (TSH) released by the pituitary [1, 2]. Interaction of TSH with the TSH receptor (TSHR), a G-protein-coupled receptor, causes activation of several protein kinase pathways that subsequently lead to increased transcription of several genes critical for TH biosynthesis, including the sodium–iodide symporter (NIS; SLC5A5), dual oxidase 2 (DUOX2), thyroglobulin (TG), thyroid peroxidase (TPO), and pendrin (PDS; SLC26A4) [3, 4]. Impairments in TH biosynthesis lead to thyroid dyshormonogenesis, one type of congenital hypothyroidism (CH) [5–9].
Paired box 8 (PAX8), NK2 homeobox 1 (NKX2.1 or TTF1), forkhead box E1 (FOXE1 or TTF2), and hematopoietically expressed homeobox (HHEX) are among the transcription factors (TFs) that have been implicated in the regulation of several TH biosynthetic genes as well as embryonic thyroid gland development [3, 4, 6, 10–13]. Mutations in these genes are causally linked to defects in thyroid gland development (thyroid dysgenesis), the major cause of CH [3, 4, 7, 10, 14–16].
Recently, we identified the Krüppel-like zinc finger TF, GLI-Similar 3 (GLIS3), as an additional critical regulator of TH biosynthesis [17–19]. Loss of GLIS3 function in humans and mice causes a syndrome characterized by neonatal diabetes and CH [18–26]. In Zebrafish, glis3 was found to be critical for early thyroid development [27]. Single nucleotide polymorphisms (SNPs) in human GLIS3 have been associated with increased risk of CH and thyroid dysfunction [28–36].
GLIS3 directly regulates the transcription of several TH biosynthesis-related genes, including Slc5a5, Slc26a4, and Duoxa2 [18, 19]. Loss of GLIS3 function greatly reduces the expression of TH biosynthesis-related genes, thereby providing a causal mechanism for the development of CH in Glis3KO mice and likely in humans as well [19]. Comprehensive hypergeometric optimization of motif enrichment (HOMER) analysis of our GLIS3 ChIP-Seq data revealed that binding sites for members of the PAX, NKX, and FOX TF families regularly localize near GLIS3 binding loci. We hypothesized that GLIS3 regulates thyroid gene transcription in coordination with TFs with well-established regulatory functions in the thyroid gland, such as PAX8, NKX2.1, and FOXE1. To obtain further support for this hypothesis and to analyze the extent by which GLIS3 co-regulates transcription with these thyroid TFs, we performed PAX8, NKX2.1, and FOXE1 ChIP-Seq analyses in the mouse thyroid gland and rat thyrocyte PCCl3 cells. These analyses indicated that GLIS3 co-regulates the transcription of several genes critical for TH biosynthesis and/or induced by TSH, with these thyroid TFs by binding within the same regulatory hub in these genes. We further show that loss of GLIS3 does not cause major changes in the binding of PAX8 or NKX2.1 or the open chromatin structure at these common regulatory regions. GLIS3 may induce transcriptional activation by enhancing the interaction of regulatory regions with RNA Polymerase II (Pol II) complexes.
Methods
Mice
Glis3-EGFP mice (C57BL/6-Glis3<tm3(Glis3-EGFP)Amj>) expressing a GLIS3-EGFP fusion protein and Glis3-deficient (Glis3KO) mice (B6.Glis3<tm3(mCherry)AmJ>) were described previously [19, 37, 38]. Mice were routinely fed a NIH-31 normal diet (ND; Harlan, Madison, WI). For ChIP-Seq, 8–10 weeks-old Glis3-EGFP mice were fed a low-iodine diet (LID; TD.95125 diet, Harlan) for 2 weeks before thyroid glands were collected for analysis. All animal studies followed guidelines outlined by the NIH Guide for the Care and Use of Laboratory Animals and protocols were approved by the Institutional Animal Care and Use Committee at the NIEHS.
Cell culture and Western blot analysis
Rat thyrocyte PCCl3 cells (ATCC CRL-1468) were cultured in Coon’s/F12 as described previously [19]. PCCl3-pIND20-Glis3 cells, expressing a doxycycline (Dox)-inducible GLIS3 tagged with Flag and HA at N- and C-terminus, respectively, were generated via pIND20-Flag-Glis3-HA lentivirus infection and subsequent puromycin selection as described previously [39].
ChIP-Seq
ChIP-Seq was carried out as described previously [19, 39]. Briefly, thyroid glands from Glis3-EGFP mice fed a LID for 2 weeks, were isolated and homogenized in PBS using a Tekmar Tissumizer Homogenizer (Tekmar Company, Cincinnati, OH). Homogenate was cross-linked in 1% formaldehyde for 10 min and the reaction subsequently quenched by the addition of 125 mM glycine for 10 min. In the case of PCCl3-pIND20-Glis3-HA, cells were treated for 24 h with 100 ng/ml Dox and then collected and processed for cross-linking and quenching as described above for the thyroid gland. The cross-linked tissues or cells were washed two times with PBS, resuspended in lysis buffer A for 10 min, pelleted, and resuspended in lysis buffer B for 10 min. Samples were subsequently sheared in lysis buffer C for 40 min using an S220 focused-ultrasonicator (Covaris, Woburn, MA). After centrifugation, the cleared chromatin supernatant was incubated with a PAX8 (NBP1-32440, Novus Biologicals) or NKX2.1 (ab76013, Abcam) antibody for tissue ChIP, and HA (#3724, Cell Signaling), NKX2.1 or FOXE1 (PA02000, Biopat) antibody for PCCl3 ChIP. For ChIP-Seq for histone marks, thyroid gland from 4-week-old WT and Glis3KO mice fed ND and LID for 6 days were collected and processed as above. Then chromatin supernatant was incubated with H3K4me3 (ab8580, Abcam) and H3K27me3 (ab6002, Abcam) antibodies. After an overnight incubation at 4 °C, washed Dynabeads Protein G (ThermoFisher Scientific) were added and the mixture rotated for 3 h at 4 °C. After subsequent washes, ChIPed-DNA was eluted, reverse cross-linked, incubated with proteinase, and DNA fragments purified using a PCR purification kit (Qiagen). Libraries were synthesized using a NEXTflex Rapid DNA-Seq kit (PerkinElmer, Austin, TX). Sequencing was performed with NovaSeq 6000, NextSeq 500 or MiSeq (Illumina, San Diego, CA).
ChIP-seq data analysis
Raw sequence reads were filtered to remove any entries with a mean base quality score < 20. Adapters were removed by Cutadapt v1.12 and single-end reads mapped against the mm10 or rn6 reference assembly via Bowtie v1.2, with only uniquely mapped hits accepted [40, 41]. Duplicate mapped reads were removed via MarkDuplicates.jar (using flag REMOVE_DUPLICATES = TRUE) from the Picard tool suite v1.110. Initial peak calls for TFs from mouse thyroid samples were made by HOMER (v4.10.3) with parameters “-style factor -fdr 0.00001 -F 8”, comparing each ChIP sample against the respective input sample. Initial peak calls for TFs from PCCl3 samples were made by HOMER (v4.10.3) with parameters “-style factor -fdr 0.001 -F 4”, comparing each ChIP sample against the respective input sample. For all TFs, peaks were re-sized to 300 bp centered on the called peak midpoints prior to downstream analysis. Enriched motifs were identified by HOMER ‘findMotifsGenome’ at “-size given” and all other parameters default. For the purposes of the genomic context of peak summaries, TSS proximal is defined as the region from − 1 kb to the annotated transcriptional start site (TSS), Upstream as the region from − 5 kb to − 1 kb relative to TSS, Genebody as the region from TSS to transcription end site (TES), and Intergenic as all other genomic locations. To establish a collapsed set of regions for direct comparison of ChIP-seq signal from multiple TFs, a combined peak set was defined via BEDTools (v2.29.2) merge function with parameter “-d 50”. Each collapsed peak was then scored as positive or negative for each TF according to the probability that the overlapping fragment count would be observed at random (cutoff set at 1e−6).
Peak calls for histone marks were made by HOMER (v4.10.3) with parameters “-region -size 500 -minDist 1000 -L 0” for H3K4me3 and “-region -size 1000 -minDist 2500 -L 0” for H3K27me3. For each histone mark, the called peaks were collapsed to a single unified peak set defined as the union of peak calls from individual samples. Tukey box-and-whisker plots of histone modification ChIP-seq signal were generated with R package ggplot2 (v3.3.2), in which the box indicates the 25th through 75th percentile and the whiskers denote values up to 1.5 * inter-quartile range from the 25th or 75th percentile. Counts of mapped ChIP-seq reads (extended to the estimated fragment length of 150 bp) overlapping ± 1 kb flanks relative to annotated TSS at genes of interest were determined via BEDTools (v2.29.2) coverage function using the ‘-counts’ flag, then normalized by reads overlapping ± 1 kb flanks relative to all RefSeq TSS.
RNA-seq
4-week-old WT mice were fed an ND (n = 4) and an LID (n = 4) for 6 days. Thyroid RNA was extracted using a RNAqueous-Micro total RNA isolation kit (ThermoFisher Scientific). TruSeq Stranded mRNA kit and TruSeq RNA Library preparation kit (Illumina Inc., San Diego, CA) were used to make libraries for RNA-Seq. Sequencing reads were obtained using a NextSeq500 or a NovaSeq 6000 Sequencing System (Illumina). Differential gene expression analysis was carried out through edgeR package. Genes with a minimum of 1.5-fold expression difference and FDR of less than 0.05 were considered as differentially expressed.
Pathway analysis
Pathway analysis was performed via DAVID Bioinformatics Resources 6.8 (http://david.abcc.nbcifcrf.gog/), ToppGene (http://toppgene.cchmc.org) for KEGG, Reactome, and Biocarta pathway analyses.
ChIP Q-PCR analysis
ChIP Q-PCR was performed with thyroid glands isolated from WT and Glis3KO mice fed a ND using NKX2.1 and PAX8 antibodies. Q-PCR reactions were carried out in triplicate on three independent samples using StepOnePlus Real-time PCR system (Applied Biosystem). Gapdh and Tpo (at − 1.9 kb) were used as negative controls. Primer sequences are listed in Additional file 5: Table S1.
Data availability
The PAX8 and NKX2.1 ChIP-seq data from the thyroid glands and the GLIS3-HA, NKX2.1, and FOXE1 ChIP-seq data from PCCl3 cells generated in this study were deposited in the NCBI Gene Expression Omnibus (GEO) database under accession #GSE20777. RNA-seq data from WT mice fed an ND and an LID were deposited under accession #GSE20775. GLIS3 ChIP-Seq data from mouse thyroid glands and RNA-Seq data of thyroid glands from WT and Glis3KO mice fed an LID used in this study were from GSE103297. The PAX8 ChIP-Seq data from PCCl3 cells were obtained from GSE26938.
Statistical analysis
Data are presented as mean ± standard deviation (SD) and were analyzed by one-way ANOVA.
Results
TF binding motifs near GLIS3 binding loci
Comprehensive analysis of our GLIS3 ChIP-Seq data of the mouse thyroid gland [19] by HOMER indicated that binding motifs of the NKX, FOX, and PAX TF family are frequently localized near GLIS binding sites (GLISBS) (Fig. 1A). Since NKX2.1, FOXE1, and PAX8 have well-established roles in the regulation of gene expression in thyroid follicular cells [12, 13, 42, 43], we hypothesized that GLIS3 regulates transcription of a subset of target genes in these cells in coordination with these TFs.
Fig. 1.

Global analysis of GLIS3, NKX2.1, and PAX8 genomic binding in mouse thyroid glands. A–C HOMER analysis, heatmap, and ChIP-Seq read density of GLIS3 (A), NKX2.1 (B), and PAX8 (C) binding data. Heatmap of the 2 kb region is centered on each of the binding peaks identified. D Genomic context of the GLIS3, NKX2.1, and PAX8 peaks within the whole mouse genome (mm10)
To obtain support for this hypothesis, we performed NKX2.1 and PAX8 ChIP-Seq analyses with thyroid glands from mice fed a low iodine diet (LID), in which TSH blood levels are highly elevated [44]. As far as we know, this is the first time ChIP-Seq analyses with thyroid glands and endogenous PAX8 and NKX2.1 have been performed. These analyses identified 29,464 NKX2.1 and 41,044 PAX8 binding peaks. De novo motif analysis of the NKX2.1-enriched sequences identified, in addition to the NKX binding motif, consensus binding sites for members GLIS, PAX, and FOX families were among the top motifs (Fig. 1B). Motif analysis of PAX8 ChIP-Seq data identified a PAX binding sequence as the top binding motif together with binding motifs for NKX, GLIS, and FOX family members (Fig. 1C). NKX2.1 and PAX8 binding loci were most highly enriched within the gene body and intergenic regions, while 5–8% were localized within 1 kb upstream of TSS (TSS proximal) (Fig. 1D).
Comparison of GLIS3, PAX8 and NKX2.1 binding revealed substantial overlaps between GLIS3 binding loci and those of NKX2.1 and PAX8 (Fig. 2A, B). The majority of GLIS3 binding loci contained both NKX2.1 and PAX8 binding regions (referred to as G+N+P+). Subsets of GLIS3 binding regions overlapped with those of either NKX2.1 or PAX8 (G+N+P− or G+N−P+, respectively), while some of the GLIS3, NKX2.1 and PAX8 binding regions did not exhibit any overlap (G+N−P−, G−N+P−, and G−N−P+, respectively). These observations are consistent with the concept that the transcription of subsets of GLIS3 target genes are regulated in coordination with PAX8 and NKX2.1, and that some genes are regulated by only one or two of the three TFs.
Fig. 2.

GLIS3, NKX2.1 and PAX8 binding to the mouse thyroid gland genome partially overlap. A The number of collapsed binding regions that are positive for GLIS3, NKX2.1, and PAX8 signal, and the percent overlap between them are indicated. B Heatmap showing overlap between GLIS3, NKX2.1, and PAX8 binding. Heatmap of the 2 kb region centered on each of the binding regions with ChIP-seq signal normalized to 10 million reads. C Venn diagram showing the number of target genes with GLIS3, NKX2.1, and/or PAX8 binding regions. D Venn diagram showing the overlap of G+N+P+ genes with genes up- or down-regulated in the thyroid gland of Glis3KO-LID mice compared to that of WT-LID mice. E KEGG analysis of the 4502 G+N+P+ genes
Although enhancers can reside within the gene body and very distant from TSSs, we limited our analysis to genes with binding peaks within 5 kb up- or downstream from the TSS. This analysis identified binding of GLIS3, NKX2.1 and PAX8 near 5240, 8975 and 8832 genes, respectively (Fig. 2C). The majority (85.9%) of the GLIS3-bound genes shared binding both with NKX2.1 and PAX8 (G+N+P+), 9.9% with NKX2.1 only (G+N+P−) and 2.4% with PAX8 only (G+N−P+), while 1.8% bound GLIS3 only (G+N−P−) (Fig. 2C). A summary of GLIS3, NKX2.1, and PAX8 bound genes is presented in Additional file 6: Table S2A.
GLIS3, NKX2.1, and PAX8 binding to TH biosynthetic and TSH-induced genes
We previously reported that loss of GLIS3 function particularly suppresses the expression of a subset of genes that are required for TH biosynthesis, induced when mice are fed an LID or known to be induced by TSH [11, 19, 45, 46]. Therefore, we were interested in determining which of these differentially expressed genes were regulated by GLIS3 in coordination with PAX8 and/or NKX2.1. Among the 4502 G+N+P+ genes (Fig. 2C, D), 345 and 196 genes were, respectively, down- or up-regulated in Glis3KO mice fed an LID (Glis3KO-LID) compared to WT mice fed an LID (WT-LID) thyroid (Additional file 6: Table S2B, C). KEGG pathway analysis of the 345 down-regulated G+N+P+ genes identified TH biosynthesis as the top pathway (Fig. 2E). No pathway was found to be significantly associated with G+N+P+ up-regulated genes. Table 1 shows GLIS3, PAX8, and NKX2.1 binding to several gene clusters in relation to their repression in Glis3KO-LID thyroid and their induction in WT-LID thyroid. This comparison shows that GLIS3, PAX8, and NKX2.1 bound to many genes critical for TH biosynthesis, including Slc5a5, Slc26a4, Tpo, Tg, Iyd, and Duoxa2, and several genes known to be induced by TSH, such as Adm2, Sod3, Dio1, and Cdh16 [45–49]. The expression of several of these genes (e.g., Slc5a5, Slc26a4, Adm2, Sod3, Dio1, Cdh16) was significantly repressed in Glis3KO-LID mice compared to WT-LID and induced in the thyroid of WT-LID mice compared to WT-ND (Table 1). Other thyroid genes, including Tpo, Iyd, and Duoxa2, were induced in WT-LID thyroid, but not significantly affected by the loss of GLIS3 function, while the expression of certain GLIS3, PAX8, and NKX2.1 bound genes, including Tg, Txnrd1, and Duox2, were not significantly altered in WT-LID thyroid nor suppressed in Glis3KO-LID (Table 1).
Table 1.
GLIS3, NKX2.2, and PAX8 binding to nearby genes (within 5 kb regions from TSS) in comparison to their in/decreased expression in Glis3KO-LID or WT-LID thyroid
| Category | geneSYM | RNA-Seq FC: LID-WT vs ND-WT | P value | RNA-Seq FC: Glis3KO-LID vs WT-LID | P value | GLIS3 target | NKX2.1 target | PAX8 target |
|---|---|---|---|---|---|---|---|---|
| TH synthesis | Slc5a5 | 12.50 | 3.01E−60 | − 15.73 | 3.81E−28 | + | + | + |
| Slc26a4 | 6.45 | 1.99E−29 | − 21.32 | 1.79E−14 | + | + | + | |
| Tpo | 6.08 | 2.25E−64 | NS | + | + | + | ||
| Tshr | 2.74 | 1.36E−08 | NS | − | + | + | ||
| Iyd | 1.75 | 5.79E−09 | NS | + | + | + | ||
| Slc16a2 | − 1.68 | 2.69E−05 | − 2.49 | 2.84E−33 | + | + | − | |
| Duox1 | − 2.39 | 9.95E−04 | NS | − | + | + | ||
| Duoxa1 | − 2.84 | 7.79E−07 | NS | − | + | + | ||
| Duox2 | NS | NS | + | + | + | |||
| Tg | NS | NS | + | + | + | |||
| Duoxa2 | 1.72 | 2.47E−05 | NS | + | + | + | ||
| TSH induced | Adm2 | 155.33 | 1.17E−140 | − 271.48 | 1.03E−267 | + | + | + |
| Sod3 | 11.30 | 4.24E−90 | − 18.80 | 5.28E−113 | + | + | + | |
| Cdh13 | 9.32 | 1.10E−138 | − 10.59 | 2.90E−95 | − | − | − | |
| Dio1 | 3.26 | 4.44E−49 | − 2.32 | 7.68E−10 | + | + | + | |
| Cdh16 | 2.57 | 5.35E−15 | − 8.17 | 4.03E−68 | + | + | + | |
| Pde4d | 2.53 | 5.40E−09 | NS | − | − | − | ||
| Ano1 | 2.49 | 2.84E−06 | NS | − | − | − | ||
| Kcnq1 | 2.07 | 8.66E−07 | − 2.24 | 6.78E−13 | − | + | + | |
| Thyroid function | Txnrd1 | 1.88 | 1.74E−12 | NS | + | + | + | |
| Txnrd2 | − 1.58 | 3.07E−05 | NS | + | + | + | ||
| Gnas | − 1.61 | 1.04E−06 | NS | + | + | + | ||
| Fam20c | − 2.30 | 1.92E−05 | NS | + | + | + | ||
| Clcn5 | NS | NS | − | − | − | |||
| Dio2 | NS | NS | − | − | − | |||
| Gnaq | NS | NS | + | + | + | |||
| Gpx1 | NS | NS | + | + | + | |||
| Kcne2 | NS | NS | − | − | − | |||
| Txn1 | NS | NS | + | + | + | |||
| Txn2 | NS | NS | − | − | − | |||
| Thyroid TFs | Glis3 | 2.10 | 2.22E−07 | NS | + | + | + | |
| Hhex | NS | NS | + | + | + | |||
| Pax8 | NS | 2.03 | 2.01E−15 | + | + | + | ||
| Nkx2-1 | NS | NS | + | + | + | |||
| Foxe1 | NS | NS | + | + | + | |||
| ECM | Adamts8 | 22.02 | 3.27E−03 | − 57.44 | 2.54E−84 | − | − | − |
| Itga2 | 14.29 | 5.60E−68 | − 47.14 | 4.50E-58 | − | − | − | |
| Col4a1 | 4.43 | 1.24E−34 | − 8.55 | 1.37E−91 | − | + | + | |
| Cdh5 | 3.58 | 9.83E−30 | − 3.98 | 5.85E−47 | − | − | − | |
| Col18a1 | 3.48 | 2.55E−19 | − 8.22 | 1.05E−105 | + | + | + | |
| Col1a1 | 3.36 | 7.67E−15 | − 37.69 | 2.92E−37 | − | − | − | |
| Col4a2 | 3.36 | 1.86E−25 | − 4.80 | 1.70E−44 | − | + | + | |
| Col13a1 | 3.18 | 1.95E−46 | NS | − | + | − | ||
| Col5a2 | 3.13 | 1.42E−21 | − 12.68 | 1.55E−78 | − | − | − | |
| Col1a2 | 2.85 | 6.95E−14 | − 17.43 | 2.35E−46 | − | − | − | |
| Col3a1 | 2.76 | 2.04E−13 | − 37.92 | 1.10E−53 | − | − | − | |
| Col5a3 | 2.67 | 9.43E−11 | − 5.56 | 5.77E−53 | + | + | + | |
| Col11a2 | 2.49 | 8.43E−17 | − 2.27 | 3.60E−06 | − | − | − | |
| Col6a1 | 2.38 | 4.39E−11 | − 3.78 | 3.77E−06 | − | − | − | |
| Col16a1 | 2.17 | 1.84E−14 | − 5.91 | 1.75E−60 | − | + | + | |
| Col6a2 | 2.15 | 1.47E−07 | NS | − | + | + | ||
| Col14a1 | 2.02 | 8.30E−10 | − 10.66 | 4.56E−34 | − | − | − | |
| Col12a1 | 1.90 | 3.66E−06 | NS | + | + | + | ||
| Col15a1 | 1.44 | 4.16E−03 | − 4.68 | 1.73E−47 | + | + | + | |
| Itgb4 | NS | NS | + | + | + | |||
| Cdh4 | NS | − 2.34 | 1.47E−18 | + | + | + | ||
| Inflammation | Ccl2 | 21.36 | 4.44E−49 | − 37.84 | 7.27E−25 | − | − | − |
| Ccl7 | 12.97 | 2.13E−46 | − 85.29 | 7.65E−47 | − | − | − | |
| Ccl17 | 3.64 | 7.08E−04 | − 29.32 | 1.01E−14 | − | − | − | |
| Ccl6 | 3.05 | 2.62E−11 | − 3.23 | 2.15E−17 | − | − | − | |
| Ccl9 | 2.69 | 1.15E−10 | − 3.20 | 4.10E−15 | + | + | + | |
| Ccl8 | 2.39 | 6.85E−04 | − 4.47 | 2.19E−06 | − | − | − | |
| Ccl12 | 2.22 | 1.10E−03 | − 30.15 | 1.65E−18 | − | − | − | |
| Il6 | 5.62 | 7.32E−07 | − 14.73 | 2.75E−10 | − | − | − |
+: indicates binding of respective TF; −: indicates no binding
NS no significant change
The genome browser tracks in Fig. 3A indicate the shared locations of the binding of endogenous PAX8, NKX2.1, and GLIS3 in several genes critical for TH biosynthesis, including Slc5a5, Slc26a4, Duoxa2, Iyd, Tpo, and Tg, and Slc16a2. In several genes (e.g., Slc5a5, Slc26a4, Tpo, Tg), PAX8, NKX2.1, and GLIS3 bound within the same region of the proximal promoter. In Slc16a2 and Iyd only NKX2.1 and GLIS3 shared binding to the proximal promoter region, while NKX2.1 and PAX8 bound to Tshr, but not GLIS3. In several genes (e.g., Tpo, Tg, Slc26a4, Iyd, Slc16a2) binding of these 3 TFs overlapped in more than one region suggesting that their transcription may be controlled by multiple regulatory regions (Fig. 3A). These observations support the hypothesis that GLIS3 regulates gene transcription in coordination with several other thyroid TFs. In the case of Duoxa2, the GLIS3/PAX8/NKX2.1 binding region is in intron 1 of Duoxa2, which is within a 35 kb region on mouse chromosome 2 that also encompasses Duox2 and Duoxa1 (Fig. 3A). We cannot rule out that his enhancer region might play a role in the transcriptional regulation of all three genes.
Fig. 3.

GLIS3, NKX2.1, and PAX8 share binding loci within the regulatory regions of several TH biosynthetic genes. A Colocalization of GLIS3, NKX2.1, and/or PAX8 ChIP-seq loci in genes critical for TH biosynthesis. The NUE region in Slc5a5 (Nis) is indicated. B, C NKX2.1 and PAX8 analysis at Cdh16 (− 0.2 kb), Slc5a5 (− 2.8 kb), and Tpo (− 0.1 kb) with thyroid glands from WT and Glis3KO mice. Binding to Gapdh and Tpo (− 1.9 kb) served as negative controls
The transcriptional regulation of Slc5a5 has been extensively studied in thyroid follicular cell lines and reported to be controlled by the proximal promoter and a region, referred to as Nis upstream enhancer (NUE, located − 2.8 kb from the TSS) that has been reported to bind several TFs, including PAX8 and NKX2.1 [13, 14, 43, 50–55]. The Slc5a5 genome browser tracks show the localization of binding peaks for endogenous GLIS3, PAX8, and NKX2.1 within the NUE region, whereas no major binding was observed within the proximal promoter region (Fig. 3A). These observations are consistent with the view that NUE is a major enhancer region driving the activation Slc5a5 transcription by GLIS3 in the thyroid of mice fed an LID. As Scl5a5 is one of the genes most strongly regulated by GLIS3 (Table 1), this raised the question whether GLIS3 binding was required for the binding of PAX8 and NKX2.1 to the NUE region. To investigate this, we compared NKX2.1 and PAX8 binding to the NUE region in thyroids isolated from WT and Glis3KO mice. ChIP Q-PCR analysis demonstrated that NKX2.1 and PAX8 occupancy at the NUE region was not significantly affected by the absence of GLIS3 (Fig. 3B, C) indicating that GLIS3 is not required for PAX8 or NKX2.1 binding to the NUE regulatory region. The lack of GLIS3 also did not significantly affect PAX8 or NKX2.1 binding to the proximal promoters of Cdh16 and Tpo (region − 0.1 kb upstream from TSS). No significant binding of PAX8 or NKX2.1 was observed to Gapdh or the − 1.9 kb upstream region of Tpo, which served as negative controls (Fig. 3B, C). ChIP-Seq analysis of several epigenetic markers showed no significant differences in the level of H3K4me3 and H3K27me3 signals in GLIS3-bound genes that were differentially expressed between Glis3KO-LID and WT-LID thyroid glands (Additional file 1: Fig. S1A). Moreover, little difference in H3K4me3 and H3K27me3 signals was observed at the NUE region of Slc5a5 between WT and Glis3KO thyroid glands, although some small changes in these signals were observed at its proximal promoter (Additional file 1: Fig. S1B). Together, these observations suggest that in the absence of GLIS3 genomic regions, such as NUE, remain accessible and in an active (open) state.
Genome browser tracks in Fig. 4A show the overlap between the binding of endogenous PAX8, NKX2.1, and GLIS3 to regulatory regions of several other thyroid genes reported to be induced by TSH, induced in WT-LID, and repressed in Glis3KO-LID thyroids, including Adm2, Sod3, Dio1, and Cdh16 [45–48].
Fig. 4.

Genome browser tracks of several genes showing overlap of GLIS3, NKX2.1, and PAX8 binding loci in mouse thyroid gland. A Genes known to be induced by TSH. B Collagen and chemokine genes
GLIS3, NKX2.1, and PAX8 binding to extracellular matrix (ECM) and inflammatory genes.
Next, we examined whether any differentially expressed ECM and inflammatory genes are co-regulated by GLIS3, PAX8, and/or NKX2.1. In contrast to TH biosynthetic genes, relatively few ECM and inflammatory genes (e.g., Col18a1, Ccl9) showed binding of all 3 TFs, while a few genes (e.g., Col4a1, Col16a1) bound PAX8 and NKX2.1, but not GLIS3 (Table 1). Of course, we cannot rule out that these TFs might regulate some of these genes by binding distant enhancers. The genome browser tracks in Fig. 4B show overlaps between GLIS3, PAX8, and NKX2.1 binding regions in Col18a1, Col5a3, Col15a1, Col4a2, and Ccl9. The transcription of other differentially expressed ECM and inflammatory genes, such as Ccl7, that do not show GLIS3, PAX8, and NKX2.1 binding, are likely regulated by other TFs.
Our ChIP-Seq analysis further identified binding peaks of all three TFs within the same genomic region(s) of Glis3, Pax8, and Nkx2.1, as well as two other thyroid TF genes, FoxE1 and Hhex (Additional file 2: Fig. S2) suggesting transcriptional regulation of each other consistent with previous observations [3, 4, 56].
GLIS3 regulates a subset of TH biosynthetic genes in coordination with FOXE1
Since HOMER also identified FOX binding motifs near GLISBS (Fig. 1A), we were interested in examining co-regulation of TH biosynthetic genes by GLIS3 and FOXE1. Since several attempts to perform FOXE1 ChIP-Seq analysis in mouse thyroid glands were unsuccessful, we performed FOXE1, NKX2.1, and GLIS3-HA ChIP-Seq analysis in rat thyrocyte PCCl3 cells and compared these data with that of PAX8 in PCCl3 cells reported previously [43]. Nuclear expression of GLIS3-HA and endogenous FOXE1 in PCCl3 was confirmed by immunofluorescence staining (Additional file 3: Fig. S3). Consistent with our de novo motif analysis of the GLIS3 ChIP-Seq data from the mouse thyroid gland, analysis of the GLIS3-enriched sequences identified GLISBS as the top motif as well as consensus binding motifs for NKX, FOX, and PAX family members (Fig. 5A). Similarly, de novo motif analysis of the NKX2.1, PAX8 and FOXE1-enriched sequences identified, in addition to their own consensus binding motif, binding motifs of the three other TFs (Fig. 5B–D). The genomic contexts of the GLIS3, FOXE1, PAX8, and NKX2.1 binding peaks are shown in Fig. 5E. Comparison of GLIS3, PAX8, NKX2.1, and FOXE1 binding revealed substantial overlaps between the GLIS3 binding loci and the other three TFs (Fig. 6A, B). Most FOXE1-binding loci (74.8%) were in the proximity of GLIS3-binding loci. The different clusters of GLIS3, NKX2.1, PAX8 and/or FOXE1 bound genes (based on regions within 5 kb from the TSS) are presented in Additional file 7: Table S3. The data showed that GLIS3, NKX2.1, PAX8, and FOXE1 (G+N+P+F+) were bound near each other in 3118 genes, including several TH biosynthetic and TSH-responsive genes [e.g., Scl5a5 (NUE region), Duoxa2, Duox2, Cdh16, as well as Pax8, Nkx2.1, Hhex, and Foxe1], but not several other G+N+P+ thyroid genes (e.g., Slc16a2, Tshr, and Dio1). The genome browser tracks display the co-localization of GLIS3, NKX2.1, PAX8, and FOXE1 binding peaks in Scl5a5, Cdh16, Duoxa2, and Adm2 (Fig. 6C) and Glis3, Nkx2.1, Pax8, and Foxe1 (Additional file 4: Fig. S4) in PCCl3 cells. FOXE1 binding did not overlap with GLIS3, PAX8, and NKX2.1 binding at the proximal promoter of Adm2 but did show an overlap within a downstream intergenic region. The GLIS3, NKX2.1, and PAX8 binding patterns in PCCl3 were very similar to those as observed in mouse thyroid (Fig. 3A; Additional file 2: Fig. S2). However, binding of GLIS3, NKX2.1, and PAX8 in PCCl3 cells did not always match those observed in the mouse thyroid gland, including their binding to Tpo, Glis3, and Iyd (Table 2). This might be due to epigenomic differences between mouse thyroid follicular cells in vivo and immortalized rat thyrocyte PCCl3 cells, for which the gene expression profile is likely different from thyroid follicular cells in vivo.
Fig. 5.

Global analysis of GLIS3, NKX2.1, PAX8, and FOXE1 genomic binding in rat thyroid follicular PCCl3 cells. A–D HOMER analysis, heatmap, and ChIP-Seq read density of GLIS3 (A), PAX8 (B), FOXE1 (C), and (D) NKX2.1 binding data. E Genomic context of the GLIS3, PAX8, FOXE1, and NKX2.1 peaks within the whole rat genome (rn6)
Fig. 6.

GLIS3, NKX2.1, PAX8, and FOXE1 binding to the PCCl3 genome partially overlap. A Percent NKX2.1, PAX8, and FOXE1 binding loci overlapping with those of GLIS3. B Heatmaps of the 2 kb region centered on each of the binding regions with ChIP-seq signal normalized to 10 million reads shows overlaps between GLIS3, NKX2.1, PAX8, and FOXE1 binding. C Genome browser tracks of several thyroid genes showing overlap of GLIS3, NKX2.1, PAX8, and FOXE1 binding loci in PCCl3 cells
Table 2.
GLIS3, NKX2.2, PAX8, and FOXE1 to nearby genes (within 5 kb regions of TSS) in rat thyroid follicular PCCl3 cells
| Category | geneSYM | rGLIS3 target | rNKX2.1 target | rPAX8 target | rFOXE1 target |
|---|---|---|---|---|---|
| TH synthesis | Slc5a5 | + | + | + | + |
| Slc26a4 | − | + | + | + | |
| Tpo | − | + | − | − | |
| Tshr | + | + | + | − | |
| Iyd | − | − | − | − | |
| Slc16a2 | + | + | + | − | |
| Duox1 | − | + | − | − | |
| Duoxa1 | + | + | − | − | |
| Duox2 | + | + | + | + | |
| Tg | − | + | − | − | |
| Duoxa2 | + | + | + | + | |
| TSH induced | Adm2 | + | + | + | − |
| Sod3 | + | + | + | + | |
| Cdh13 | − | + | − | − | |
| Dio1 | + | + | + | − | |
| Cdh16 | + | + | + | + | |
| Pde4d | + | + | + | + | |
| Ano1 | + | + | + | − | |
| Kcnq1 | − | − | − | − | |
| Thyroid function | Txnrd1 | + | + | + | + |
| Txnrd2 | + | + | + | − | |
| Gnas | + | + | + | − | |
| Fam20c | + | + | + | + | |
| Clcn5 | − | − | − | − | |
| Dio2 | − | − | − | − | |
| Gnaq | + | + | + | − | |
| Gpx1 | + | + | − | + | |
| Kcne2 | − | − | − | − | |
| Txn1 | + | + | + | − | |
| Txn2 | + | + | + | − | |
| Thyroid TFs | Glis3 | − | − | − | − |
| Hhex | + | + | + | + | |
| Pax8 | + | + | + | + | |
| Nkx2-1 | + | + | + | + | |
| Foxe1 | + | + | + | + |
Discussion
Comprehensive HOMER analysis of our GLIS3 ChIP-Seq data from the mouse thyroid gland indicated that members of the PAX, NKX, and FOX families frequently bind in the proximity of GLIS3 binding loci. We proposed that GLIS3 regulates the transcription of certain thyroid genes in coordination with PAX8, NKX2.1, and FOXE1, which have well-established regulatory functions in the thyroid, including thyroid gland development and TH biosynthesis [4, 7, 10, 12, 13, 30]. And although binding of PAX8, NKX2.1, and FOXE1 to a few thyroid genes, such as Slc5a5, has been demonstrated by in silico and ChIP-PCR analyses, global ChIP-Seq analysis of the binding of endogenous PAX8, NKX2.1, FOXE1 has not been previously reported. Our GLIS3, PAX8, NKX2.1, and FOXE1 cistrome analyses with mouse thyroid gland and/or thyrocyte PCCl3 cells identified considerable overlaps in binding of these four TFs indicating that GLIS3 shares many of its promoter/enhancer regions with PAX8, NKX2.1, and/or FOXE1, including genes related to thyroid biosynthesis/function and/or induced by TSH (Figs. 2 and 6). Our data are consistent with previous in silico and ChIP-PCR analyses indicating that binding motifs for these thyroid TFs localize near each other within the regulatory region of certain TH biosynthetic genes [4, 42, 57]. Thyroid target genes are regulated by different combinations of these TFs thereby forming distinct clusters, e.g., G+N+P+F+, G+N+P+F−, etc. (Additional file 7: Table S3A). Although the expression of some G+N+P+ genes was not or only moderately changed, genes most highly induced in WT-LID thyroid (versus WT-ND) or repressed in Glis3KO-LID thyroid (versus WT-LID), including Slc5a5, Slc26a4, Adm2, Sod3, and Cdh16, all showed shared GLIS3, PAX8, NKX2.1 binding (Table 1; Fig. 3A) and in PCCl3 cells, frequently overlap with FOXE1 binding sites (Table 2; Fig. 6C). Together, these findings support our hypothesis that GLIS3 regulates these genes in coordination with PAX8, NKX2.1, and/or FOXE1 and further demonstrate that GLIS3 binding to these shared regulatory regions is essential for optimal expression of these genes and their induction by TSH. Except for a few genes (e.g., Col18a1, Col5a3, Ccl9) (Table 1; Fig. 4B), GLIS3, PAX8, and NKX2.1 did not bind near many of the differentially expressed ECM and chemokine genes suggesting that they do not play a direct role in the transcriptional regulation of many of these genes. However, we cannot rule out that they might be regulated by distant binding sites as may be the case for Ccl7, which has an NKX2.1 binding site 62 kb upstream from its TSS. Interestingly, in thyroid follicular cells colocalization of GLIS3, PAX8, and NKX2.1 binding peaks was also found in Glis3, Pax8 and Nkx2.1, as well as in Foxe1 and Hhex, suggesting that these TFs may coregulate each other’s expression. This is consistent with previous studies showing that during thyroid development these TFs are part of an integrated regulatory network in which each of them controls the expression of other members [3, 4, 56]. In mice, NKX2.1 and PAX8 are expressed at an earlier stage of embryonic thyroid development [4, 12] than GLIS3 (manuscript in preparation). This would be consistent with the hypothesis that during embryonic thyroid development Glis3 transcription is directly regulated by NKX2.1 and PAX8.
An interesting question was whether GLIS3 is required for the binding of PAX8 and NKX2.1 to common regulatory regions and affects the open/closed chromatin structure. Comparison of the binding of PAX8 and NKX2.1 to the NUE region of Slc5a5 and the proximal promoter of Cdh16 and Tpo in the thyroid gland from WT and Glis3KO mice by ChIP-PCR indicated that lack of GLIS3 has no significant effect on the binding of PAX8 and NKX2.1 to the NUE and the two other proximal promoter regions. These data suggest that GLIS3 does not greatly affect the open/closed chromatin structure at these regions. This was supported by our analysis of histone markers, H3K4me3 and H3K27me3, which reflect active and inactive transcription, showing little difference in the intensity of the signals around the NUE region and in the meta-analysis of H3K4me3 and H3K27me3 between WT-LID and Glis3KO-LID. However, we cannot rule out that GLIS3 binding alters the chromatin of certain genes. Future studies using scATAC-seq analysis might establish whether GLIS3 has any effect on chromatin structure.
GLIS3 is particularly critical for the transcriptional regulation of several genes that are highly induced in WT-LID thyroid and repressed in Glis3KO-LID thyroid, including Slc5a5, Slc26a4, Cdh16, Adm2, and Sod3 (Fig. 7). We hypothesize that GLIS3 boosts the transcriptional activation of these genes by recruiting additional transcriptional mediators at these regulatory hubs and promoting the interaction with TFs bound to other regulatory hubs through changes in 3D chromatin organization (genome loop formation) thereby enhancing the interaction with polymerase II complexes (Fig. 7) [58]. The transcriptional mediator WWTR1 (TAZ), which interacts directly with GLIS3 and enhances its transcriptional activity [59], might be one of such proteins. WWTR1, which can bind several other thyroid TFs, including the Hippo pathway TF, TEAD1, might facilitate interactions between the NUE-bound transactivation complex with that associated with the Slc5a5 proximal promoter [13, 60]. Additional studies are needed to establish the role of GLIS3 in these interactions.
Fig. 7.
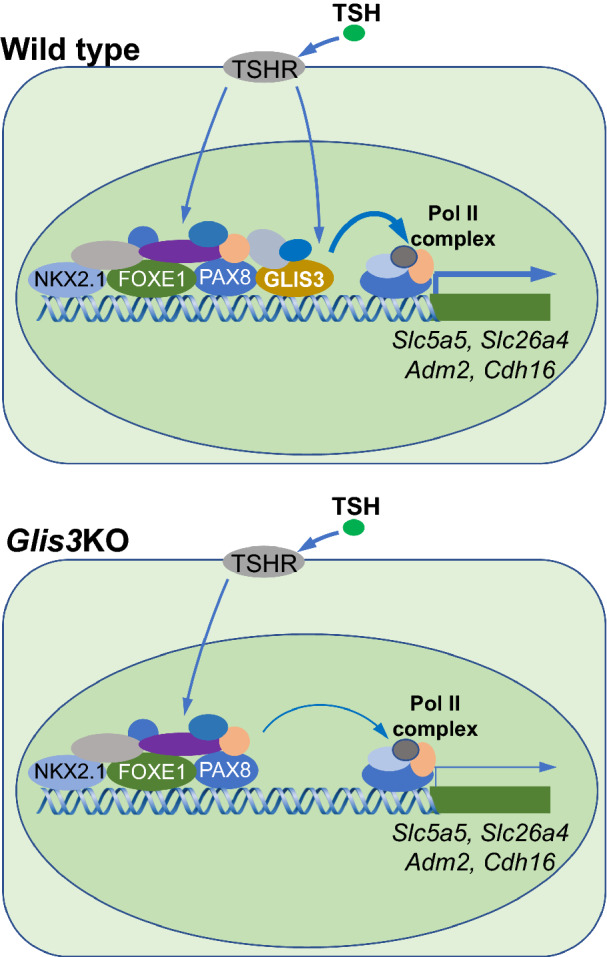
Schematic view of GLIS3 regulation in the expression of target genes with other TFs. GLIS3 does not affect PAX8 or NKX2.1 binding nor the open/closed chromatin structure at their regulatory regions of several GLIS3 target genes but may promote transcriptional activation by enhancing the interaction of regulatory regions with Pol II complexes
Conclusions
Our data supports the hypothesis that GLIS3 regulates the transcription of many thyroid genes, particularly TH biosynthesis-related and TSH-inducible genes, in coordination with several other thyroid follicular cell-associated TFs by interacting within the same distinct regulatory regions. Although GLIS3 is critical for the TSH-dependent induction of several TH biosynthetic genes, it does not affect the binding of PAX8 or NKX2.1 nor the open/closed chromatin structure. GLIS3 may induce transcriptional activation by promoting the interaction of regulatory regions with other enhancer sites and/or with RNA Polymerase II (Pol II) complexes.
Supplementary Information
Additional file 1: Figure S1. ChIP-Seq analysis of H3K4me3 and H3K27me3 in thyroid glands from WT and Glis3KO mice fed a ND or LID. A Tukey box- and whisker plots of H3K4me3 and H3K27me3 ChIP-seq signals associated with GLIS3 target genes that are either down- or up-regulated in Glis3KO-LID compared to WT-LID thyroids. B Genome browser tracks of H3K4me3 and H3K27me3 histone markers at the Slc5a5 and Tg loci in thyroid glands from WT-ND, WT-LID, Glis3KO-ND, and Glis3KO-LID mice.
Additional file 2: Figure S2. Genome browser tracks of Glis3, Nkx2.1, Pax8, Foxe1, and Hhex loci showing colocalization of GLIS3, NKX2.1, and/or PAX8 ChIP-seq signal in mouse thyroid glands.
Additional file 3: Figure S3. Nuclear expression of endogenous FOXE1 and exogenous GLIS3-HA in PCCl3 cells. Green, FOXE1 or GLIS3-HA; Blue, Dapi.
Additional file 4: Figure S4. Genome browser tracks of Glis3, Nkx2.1, Pax8, and Foxe1 loci showing colocalization of GLIS3, NKX2.1, PAX8 and/or FOXE1 ChIP-seq signal in rat thyrocyte PCCl3 cells.
Additional file 5: Table S1. List of primers used in ChIP Q-PCR.
Additional file 6: Table S2. A Lists of genes binding GLIS3, NKX2.1, and/or PAX8 as indicated (within 5 kb from TSS). ChIP-Seq analysis was performed with thyroid glands from mouse fed an LID. G+N+P+, G+N+P−, etc. are defined as described in “Results” section. B Total list of G+N+P+ genes that are down-regulated in Glis3KO-LID thyroid gland compared to WT-LID thyroid. C Total list of G+N+P+ genes that are up-regulated in Glis3KO-LID thyroid gland compared to WT-LID thyroid.
Additional file 7: Table S3. Lists of genes binding GLIS3, NKX2.1, PAX8, and/or FOXE1 as indicated (within 5 kb from TSS). ChIP-Seq analysis was performed with rat thyrocyte PCCl3 cells. G+N+P+, G+N+P−, etc. are defined as described in “Results” section. PAX8 data were obtained from [43].
Acknowledgements
We would like to acknowledge Laura Miller Degraff at NIEHS for managing the mouse colonies.
Abbreviations
- GLIS3
Gli-similar 3
- GLISBS
GLIS binding sites
- CH
Congenital hypothyroidism
- TH
Thyroid hormone
- TF
Transcriptional factors
- TSH
Thyroid stimulating hormone
- TSHR
TSH receptor
- NIS
Sodium–iodide symporter
- NUE
Nis upstream enhancer
- DUOX2
Dual oxidase 2
- TG
Thyroglobulin
- TPO
Thyroid peroxidase
- PDS
Pendrin
- PAX8
Paired box 8
- NKX2.1
NK2 homeobox 1
- FOXE1
Forkhead box E1
- HHEX
Hematopoietically expressed homeobox
- HOMER
Hypergeometric optimization of motif enrichment
- LID
Low iodine diet
- ND
Normal diet
- ChIP
Chromatin immunoprecipitation
Author contributions
HSK: design and perform experiments; analysis data; write the manuscript. SAG: data analysis; edit manuscript. RJ: analysis data and edit the manuscript. PS: data analysis; edit manuscript. AMJ: design experiments; analysis data; write the manuscript. All authors read and approved the final manuscript.
Funding
Open Access funding provided by the National Institutes of Health (NIH). AMJ research was supported by the Intramural Research Program of the NIEHS, NIH Z01-ES-101585 and PS was supported by PID2019-105303RB-100/AEI/10.13.039/501100011033 from MICIN and P2022/BMD-7379 iTironet2-CM (Comunidad de Madrid), Spain.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. Data have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession numbers GSE20777, GSE20775, GSE103297and GSE26938.
Declarations
Ethics approval and consent to participate
All studies and procedures involving mice were approved by the NIEHS Institutional Animal Care and Use Committee (IACUC) and by the National Institute of Health (NIH) guidelines.
Consent for publication
The publication of this manuscript has been approved by all authors.
Competing interests
The authors declare that they have on competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zoeller RT, Tan SW, Tyl RW. General background on the hypothalamic–pituitary–thyroid (HPT) axis. Crit Rev Toxicol. 2007;37(1–2):11–53. doi: 10.1080/10408440601123446. [DOI] [PubMed] [Google Scholar]
- 2.Mendoza A, Hollenberg AN. New insights into thyroid hormone action. Pharmacol Ther. 2017;173:135–145. doi: 10.1016/j.pharmthera.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nilsson M, Fagman H. Development of the thyroid gland. Development. 2017;144(12):2123–2140. doi: 10.1242/dev.145615. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Marquez A, Carrasco-Lopez C, Fernandez-Mendez C, Santisteban P. Unraveling the complex interplay between transcription factors and signaling molecules in thyroid differentiation and function, from embryos to adults. Front Endocrinol. 2021;12:654569. doi: 10.3389/fendo.2021.654569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravera S, Reyna-Neyra A, Ferrandino G, Amzel LM, Carrasco N. The sodium/iodide symporter (NIS): molecular physiology and preclinical and clinical applications. Annu Rev Physiol. 2017;79:261–289. doi: 10.1146/annurev-physiol-022516-034125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Citterio CE, Targovnik HM, Arvan P. The role of thyroglobulin in thyroid hormonogenesis. Nat Rev Endocrinol. 2019;15(6):323–338. doi: 10.1038/s41574-019-0184-8. [DOI] [PubMed] [Google Scholar]
- 7.Kostopoulou E, Miliordos K, Spiliotis B. Genetics of primary congenital hypothyroidism—a review. Hormones. 2021;20(2):225–236. doi: 10.1007/s42000-020-00267-x. [DOI] [PubMed] [Google Scholar]
- 8.Grasberger H, Refetoff S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr. 2011;23(4):421–428. doi: 10.1097/MOP.0b013e32834726a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran C, Schoenmakers N, Visser WE, Schoenmakers E, Agostini M, Chatterjee K. Genetic disorders of thyroid development, hormone biosynthesis and signalling. Clin Endocrinol. 2022;97(4):502–514. doi: 10.1111/cen.14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mio C, Grani G, Durante C, Damante G. Molecular defects in thyroid dysgenesis. Clin Genet. 2020;97(1):222–231. doi: 10.1111/cge.13627. [DOI] [PubMed] [Google Scholar]
- 11.Jang D, Marcus-Samuels B, Morgan SJ, Klubo-Gwiezdzinska J, Neumann S, Gershengorn MC. Thyrotropin regulation of differentiated gene transcription in adult human thyrocytes in primary culture. Mol Cell Endocrinol. 2020;518:111032. doi: 10.1016/j.mce.2020.111032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez LP, Lopez-Marquez A, Santisteban P. Thyroid transcription factors in development, differentiation and disease. Nat Rev Endocrinol. 2015;11(1):29–42. doi: 10.1038/nrendo.2014.186. [DOI] [PubMed] [Google Scholar]
- 13.Riesco-Eizaguirre G, Santisteban P, De la Vieja A. The complex regulation of NIS expression and activity in thyroid and extrathyroidal tissues. Endocr Relat Cancer. 2021;28(10):T141–T165. doi: 10.1530/ERC-21-0217. [DOI] [PubMed] [Google Scholar]
- 14.Nitsch R, Di Dato V, di Gennaro A, de Cristofaro T, Abbondante S, De Felice M, Zannini M, Di Lauro R. Comparative genomics reveals a functional thyroid-specific element in the far upstream region of the PAX8 gene. BMC Genom. 2010;11:306. doi: 10.1186/1471-2164-11-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoupa A, Kariyawasam D, Carre A, Polak M. Update of thyroid developmental genes. Endocrinol Metab Clin N Am. 2016;45(2):243–254. doi: 10.1016/j.ecl.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Persani L, Rurale G, de Filippis T, Galazzi E, Muzza M, Fugazzola L. Genetics and management of congenital hypothyroidism. Best Pract Res Clin Endocrinol Metab. 2018;32(4):387–396. doi: 10.1016/j.beem.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Jetten AM. GLIS1-3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell Mol Life Sci. 2018;75(19):3473–3494. doi: 10.1007/s00018-018-2841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scoville DW, Kang HS, Jetten AM. Transcription factor GLIS3: critical roles in thyroid hormone biosynthesis, hypothyroidism, pancreatic beta cells and diabetes. Pharmacol Ther. 2020;215:107632. doi: 10.1016/j.pharmthera.2020.107632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang HS, Kumar D, Liao G, Lichti-Kaiser K, Gerrish K, Liao XH, Refetoff S, Jothi R, Jetten AM. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J Clin Invest. 2017;127(12):4326–4337. doi: 10.1172/JCI94417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimitri P. The role of GLIS3 in thyroid disease as part of a multisystem disorder. Best Pract Res Clin Endocrinol Metab. 2017;31(2):175–182. doi: 10.1016/j.beem.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Dimitri P, Habeb AM, Garbuz F, Millward A, Wallis S, Moussa K, Akcay T, Taha D, Hogue J, Slavotinek A, et al. Expanding the clinical spectrum associated with GLIS3 mutations. J Clin Endocrinol Metab. 2015;100(10):E1362–E1369. doi: 10.1210/jc.2015-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, Charon C, Nicolino M, Boileau P, Cavener DR, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38(6):682–687. doi: 10.1038/ng1802. [DOI] [PubMed] [Google Scholar]
- 23.Habeb AM, Al-Magamsi MS, Eid IM, Ali MI, Hattersley AT, Hussain K, Ellard S. Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatr Diabetes. 2012;13(6):499–505. doi: 10.1111/j.1399-5448.2011.00828.x. [DOI] [PubMed] [Google Scholar]
- 24.London S, De Franco E, Elias-Assad G, Barhoum MN, Felszer C, Paniakov M, Weiner SA, Tenenbaum-Rakover Y. Case report: neonatal diabetes mellitus caused by a novel GLIS3 mutation in twins. Front Endocrinol. 2021;12:673755. doi: 10.3389/fendo.2021.673755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, Kiyonari H, Shiba D, Nishio S, Mochizuki T, et al. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. 2009;583(12):2108–2113. doi: 10.1016/j.febslet.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 26.Splittstoesser V, Vollbach H, Plamper M, Garbe W, De Franco E, Houghton JAL, Dueker G, Ganschow R, Gohlke B, Schreiner F. Case report: extended clinical spectrum of the neonatal diabetes with congenital hypothyroidism syndrome. Front Endocrinol. 2021;12:665336. doi: 10.3389/fendo.2021.665336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rurale G, Marelli F, Duminuco P, Persani L. Glis3 as a critical regulator of thyroid primordium specification. Thyroid. 2020;30(2):277–289. doi: 10.1089/thy.2019.0196. [DOI] [PubMed] [Google Scholar]
- 28.Fu C, Luo S, Long X, Li Y, She S, Hu X, Mo M, Wang Z, Chen Y, He C, et al. Mutation screening of the GLIS3 gene in a cohort of 592 Chinese patients with congenital hypothyroidism. Clin Chim Acta. 2018;476:38–43. doi: 10.1016/j.cca.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 29.Porcu E, Medici M, Pistis G, Volpato CB, Wilson SG, Cappola AR, Bos SD, Deelen J, den Heijer M, Freathy RM, et al. A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. PLoS Genet. 2013;9(2):e1003266. doi: 10.1371/journal.pgen.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Filippis T, Gelmini G, Paraboschi E, Vigone MC, Di Frenna M, Marelli F, Bonomi M, Cassio A, Larizza D, Moro M, et al. A frequent oligogenic involvement in congenital hypothyroidism. Hum Mol Genet. 2017;26(13):2507–2514. doi: 10.1093/hmg/ddx145. [DOI] [PubMed] [Google Scholar]
- 31.Teumer A, Chaker L, Groeneweg S, Li Y, Di Munno C, Barbieri C, Schultheiss UT, Traglia M, Ahluwalia TS, Akiyama M, et al. Genome-wide analyses identify a role for SLC17A4 and AADAT in thyroid hormone regulation. Nat Commun. 2018;9(1):4455. doi: 10.1038/s41467-018-06356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi T, Nakamura A, Nakayama K, Hishimura N, Morikawa S, Ishizu K, Tajima T. Targeted next-generation sequencing for congenital hypothyroidism with positive neonatal TSH screening. J Clin Endocrinol Metab. 2020;105(8):e2825-33. doi: 10.1210/clinem/dgaa308. [DOI] [PubMed] [Google Scholar]
- 33.Zhang RJ, Zhang JX, Du WH, Sun F, Fang Y, Zhang CX, Wang Z, Wu FY, Han B, Liu W, et al. Molecular and clinical genetics of the transcription factor GLIS3 in Chinese congenital hypothyroidism. Mol Cell Endocrinol. 2021;528:111223. doi: 10.1016/j.mce.2021.111223. [DOI] [PubMed] [Google Scholar]
- 34.Park KS. Analysis of worldwide carrier frequency and predicted genetic prevalence of autosomal recessive congenital hypothyroidism based on a general population database. Genes. 2021;12(6):863. doi: 10.3390/genes12060863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perdas E, Gadzalska K, Hrytsiuk I, Borowiec M, Fendler W, Mlynarski W. Case report: neonatal diabetes mellitus with congenital hypothyroidism as a result of biallelic heterozygous mutations in GLIS3 gene. Pediatr Diabetes. 2022;23(6):668–674. doi: 10.1111/pedi.13341. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Li X, Wang X, Han M, Zhao D, Wang F, Liu S. Mutation screening of eight genes and comparison of the clinical data in a Chinese cohort with congenital hypothyroidism. Endocrine. 2023;79(1):125–134. doi: 10.1007/s12020-022-03188-4. [DOI] [PubMed] [Google Scholar]
- 37.Kang HS, Takeda Y, Jeon K, Jetten AM. The spatiotemporal pattern of Glis3 expression indicates a regulatory function in bipotent and endocrine progenitors during early pancreatic development and in Beta, PP and ductal cells. PLoS ONE. 2016;11(6):e0157138. doi: 10.1371/journal.pone.0157138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang HS, Chen LY, Lichti-Kaiser K, Liao G, Gerrish K, Bortner CD, Yao HH, Eddy EM, Jetten AM. Transcription factor GLIS3: a new and critical regulator of postnatal stages of mouse spermatogenesis. Stem Cells. 2016;34(11):2772–2783. doi: 10.1002/stem.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeon K, Kumar D, Conway AE, Park K, Jothi R, Jetten AM. GLIS3 transcriptionally activates WNT genes to promote differentiation of human embryonic stem cells into posterior neural progenitors. Stem Cells. 2019;37(2):202–215. doi: 10.1002/stem.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–12. [Google Scholar]
- 41.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Palma T, Conti A, de Cristofaro T, Scala S, Nitsch L, Zannini M. Identification of novel Pax8 targets in FRTL-5 thyroid cells by gene silencing and expression microarray analysis. PLoS ONE. 2011;6(9):e25162. doi: 10.1371/journal.pone.0025162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz-Llorente S, Carrillo Santa de Pau E, Sastre-Perona A, Montero-Conde C, Gomez-Lopez G, Fagin JA, Valencia A, Pisano DG, Santisteban P. Genome-wide analysis of Pax8 binding provides new insights into thyroid functions. BMC Genom. 2012;13:147. doi: 10.1186/1471-2164-13-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukuda H, Yasuda N, Greer MA. Acute effects of thyroxine, triiodothyronine, and iodide on thyrotropin secretion. Endocrinology. 1975;97(4):924–931. doi: 10.1210/endo-97-4-924. [DOI] [PubMed] [Google Scholar]
- 45.Nagasaki S, Fukui M, Asano S, Ono K, Miki Y, Araki S, Isobe M, Nakashima N, Takahashi K, Sasano H, et al. Induction of adrenomedullin 2/intermedin expression by thyroid stimulating hormone in thyroid. Mol Cell Endocrinol. 2014;395(1–2):32–40. doi: 10.1016/j.mce.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 46.de Cristofaro T, Di Palma T, Fichera I, Lucci V, Parrillo L, De Felice M, Zannini M. An essential role for Pax8 in the transcriptional regulation of cadherin-16 in thyroid cells. Mol Endocrinol. 2012;26(1):67–78. doi: 10.1210/me.2011-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsumoto C, Ito M, Yamada H, Yamakawa N, Yoshida H, Date A, Watanabe M, Hidaka Y, Iwatani Y, Miyauchi A, et al. Genes that characterize T3-predominant Graves’ thyroid tissues. Eur J Endocrinol. 2013;168(2):137–144. doi: 10.1530/EJE-12-0507. [DOI] [PubMed] [Google Scholar]
- 48.Laatikainen LE, Castellone MD, Hebrant A, Hoste C, Cantisani MC, Laurila JP, Salvatore G, Salerno P, Basolo F, Nasman J, et al. Extracellular superoxide dismutase is a thyroid differentiation marker down-regulated in cancer. Endocr Relat Cancer. 2010;17(3):785–796. doi: 10.1677/ERC-10-0021. [DOI] [PubMed] [Google Scholar]
- 49.Ito M, Toyoda N, Nomura E, Takamura Y, Amino N, Iwasaka T, Takamatsu J, Miyauchi A, Nishikawa M. Type 1 and type 2 iodothyronine deiodinases in the thyroid gland of patients with 3,5,3′-triiodothyronine-predominant Graves’ disease. Eur J Endocrinol. 2011;164(1):95–100. doi: 10.1530/EJE-10-0736. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez-Mendez C, Santisteban P. A critical balance between PAX8 and the Hippo mediator TAZ determines sodium/iodide symporter expression and function. Thyroid. 2022;32(3):315–325. doi: 10.1089/thy.2021.0191. [DOI] [PubMed] [Google Scholar]
- 51.Ohno M, Zannini M, Levy O, Carrasco N, di Lauro R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol. 1999;19(3):2051–2060. doi: 10.1128/MCB.19.3.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez LP, Lopez-Marquez A, Martinez AM, Gomez-Lopez G, Santisteban P. New insights into FoxE1 functions: identification of direct FoxE1 targets in thyroid cells. PLoS ONE. 2013;8(5):e62849. doi: 10.1371/journal.pone.0062849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chun JT, Di Lauro R. Characterization of the upstream enhancer of the rat sodium/iodide symporter gene. Exp Clin Endocrinol Diabetes. 2001;109(1):23–26. doi: 10.1055/s-2001-11021. [DOI] [PubMed] [Google Scholar]
- 54.Pasca di Magliano M, Di Lauro R, Zannini M. Pax8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci USA. 2000;97(24):13144–13149. doi: 10.1073/pnas.240336397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costamagna E, Garcia B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J Biol Chem. 2004;279(5):3439–3446. doi: 10.1074/jbc.M307138200. [DOI] [PubMed] [Google Scholar]
- 56.Parlato R, Rosica A, Rodriguez-Mallon A, Affuso A, Postiglione MP, Arra C, Mansouri A, Kimura S, Di Lauro R, De Felice M. An integrated regulatory network controlling survival and migration in thyroid organogenesis. Dev Biol. 2004;276(2):464–475. doi: 10.1016/j.ydbio.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 57.Lopez-Marquez A, Fernandez-Mendez C, Recacha P, Santisteban P. Regulation of foxe1 by thyrotropin and transforming growth factor beta depends on the interplay between thyroid-specific, CREB and SMAD transcription factors. Thyroid. 2019;29(5):714–725. doi: 10.1089/thy.2018.0136. [DOI] [PubMed] [Google Scholar]
- 58.Lim B, Levine MS. Enhancer-promoter communication: hubs or loops? Curr Opin Genet Dev. 2021;67:5–9. doi: 10.1016/j.gde.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang HS, Beak JY, Kim YS, Herbert R, Jetten AM. Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol Cell Biol. 2009;29(10):2556–2569. doi: 10.1128/MCB.01620-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim CL, Choi SH, Mo JS. Role of the Hippo pathway in fibrosis and cancer. Cells. 2019;8(5):468. doi: 10.3390/cells8050468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. ChIP-Seq analysis of H3K4me3 and H3K27me3 in thyroid glands from WT and Glis3KO mice fed a ND or LID. A Tukey box- and whisker plots of H3K4me3 and H3K27me3 ChIP-seq signals associated with GLIS3 target genes that are either down- or up-regulated in Glis3KO-LID compared to WT-LID thyroids. B Genome browser tracks of H3K4me3 and H3K27me3 histone markers at the Slc5a5 and Tg loci in thyroid glands from WT-ND, WT-LID, Glis3KO-ND, and Glis3KO-LID mice.
Additional file 2: Figure S2. Genome browser tracks of Glis3, Nkx2.1, Pax8, Foxe1, and Hhex loci showing colocalization of GLIS3, NKX2.1, and/or PAX8 ChIP-seq signal in mouse thyroid glands.
Additional file 3: Figure S3. Nuclear expression of endogenous FOXE1 and exogenous GLIS3-HA in PCCl3 cells. Green, FOXE1 or GLIS3-HA; Blue, Dapi.
Additional file 4: Figure S4. Genome browser tracks of Glis3, Nkx2.1, Pax8, and Foxe1 loci showing colocalization of GLIS3, NKX2.1, PAX8 and/or FOXE1 ChIP-seq signal in rat thyrocyte PCCl3 cells.
Additional file 5: Table S1. List of primers used in ChIP Q-PCR.
Additional file 6: Table S2. A Lists of genes binding GLIS3, NKX2.1, and/or PAX8 as indicated (within 5 kb from TSS). ChIP-Seq analysis was performed with thyroid glands from mouse fed an LID. G+N+P+, G+N+P−, etc. are defined as described in “Results” section. B Total list of G+N+P+ genes that are down-regulated in Glis3KO-LID thyroid gland compared to WT-LID thyroid. C Total list of G+N+P+ genes that are up-regulated in Glis3KO-LID thyroid gland compared to WT-LID thyroid.
Additional file 7: Table S3. Lists of genes binding GLIS3, NKX2.1, PAX8, and/or FOXE1 as indicated (within 5 kb from TSS). ChIP-Seq analysis was performed with rat thyrocyte PCCl3 cells. G+N+P+, G+N+P−, etc. are defined as described in “Results” section. PAX8 data were obtained from [43].
Data Availability Statement
The PAX8 and NKX2.1 ChIP-seq data from the thyroid glands and the GLIS3-HA, NKX2.1, and FOXE1 ChIP-seq data from PCCl3 cells generated in this study were deposited in the NCBI Gene Expression Omnibus (GEO) database under accession #GSE20777. RNA-seq data from WT mice fed an ND and an LID were deposited under accession #GSE20775. GLIS3 ChIP-Seq data from mouse thyroid glands and RNA-Seq data of thyroid glands from WT and Glis3KO mice fed an LID used in this study were from GSE103297. The PAX8 ChIP-Seq data from PCCl3 cells were obtained from GSE26938.
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. Data have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession numbers GSE20777, GSE20775, GSE103297and GSE26938.