

Abstract
Faithful chromosome segregation in budding yeast requires correct positioning of the mitotic spindle along the mother to daughter cell polarity axis. When the anaphase spindle is not correctly positioned, a surveillance mechanism, named as the spindle position checkpoint (SPOC), prevents the progression out of mitosis until correct spindle positioning is achieved. How SPOC works on a molecular level is not well understood. Here we performed a genome-wide genetic screen to search for components required for SPOC. We identified the SWR1 chromatin-remodeling complex (SWR1-C) among several novel factors that are essential for SPOC integrity. Cells lacking SWR1-C were able to activate SPOC upon spindle misorientation but underwent mitotic slippage upon prolonged SPOC arrest. This mitotic slippage required the Cdc14-early anaphase release pathway and other factors including the SAGA (Spt-Ada-Gcn5 acetyltransferase) histone acetyltransferase complex, proteasome components and the mitotic cyclin-dependent kinase inhibitor Sic1. Together, our data establish a novel link between SWR1-C chromatin remodeling and robust checkpoint arrest in late anaphase.
INTRODUCTION
In budding yeast Saccharomyces cerevisiae, the mitotic spindle must align along the mother–daughter cell polarity axis to deliver one set of chromosomes to the daughter cell during mitosis. Two parallel conserved machineries, one dependent on the microtubule-associated protein Kar9 and the other on the microtubule motor Dynein, robustly orient the mitotic spindle (Li et al., 1993; Adames and Cooper, 2000; Beach et al., 2000). However, the mitotic spindle might misorient due to environmental conditions or defects in spindle positioning systems. In cells with misoriented spindles, a surveillance mechanism named the spindle position checkpoint (SPOC) halts cell cycle progression in late anaphase of mitosis (Yeh et al., 1995; Bardin et al., 2000; Bloecher et al., 2000; Pereira et al., 2000; Adames et al., 2001). The SPOC provides cells with time to correct the error in spindle positioning and assures correct segregation of the genomic DNA before the separation of mother and daughter cells, thereby preventing multiploidy and genomic instability. A SPOC-like surveillance mechanism was shown to exist in fruit fly male germline stem cells. In this system, the checkpoint arrests cells prior to mitosis in response to centrosome misorientation to promote asymmetric cell division (Cheng et al., 2008; Pereira and Yamashita, 2011).
The SPOC prevents cell cycle progression by blocking the mitotic exit network (MEN) (Caydasi et al., 2010a; Caydasi and Pereira, 2012; Weiss, 2012; Baro et al., 2017). The MEN is a GTPase-driven signaling cascade that activates Cdc14—a phosphatase that is essential for mitotic exit through inactivation of mitotic cyclin-dependent kinase (M-Cdk) (Caydasi et al., 2010a; Caydasi and Pereira, 2012; Weiss, 2012; Baro et al., 2017). Cdc14 is held inactive in the nucleolus, bound to the nucleolar resident protein, Net1 (Shou et al., 1999a). In early anaphase, Cdc14 is transiently released from the nucleolus by the Cdc-fourteen early anaphase release (FEAR) pathway (Rock and Amon, 2009). This pool of partially released Cdc14 plays an important role in spindle formation and rDNA segregation but is not sufficient to promote mitotic exit (Jaspersen and Morgan, 2000; Rock and Amon, 2009; Konig et al., 2010). For mitotic exit, Cdc14 must be fully released by the MEN in late anaphase.
The MEN is activated by the small GTPase Tem1, the activity of which is negatively regulated by the GTPase-activating protein (GAP) complex composed of Bub2 and Bfa1. MEN components including Bfa1-Bub2, Tem1, and the downstream kinases Cdc15 and Dbf2-Mob1 complex associate with the yeast microtubule-organizing center, namely the spindle pole body (SPB), throughout the cell cycle. The SPB functions as a scaffold that promotes activation of the MEN in cells with properly aligned spindles (Gruneberg et al., 2000; Visintin and Amon, 2001).
Upon spindle misalignment, the Bfa1-Bub2 GAP complex must be kept in the active form to promote GTP hydrolysis of Tem1 and hence MEN inactivation (Geymonat et al., 2002). The mother cell-enriched kinase Kin4 is a central component of SPOC that keeps the Bfa1-Bub2 GAP complex in its active form. Kin4 associates with the mother cell cortex throughout the cell cycle and SPBs in cells with misaligned spindles, where Kin4 phosphorylates Bfa1 (D’Aquino et al., 2005; Pereira and Schiebel, 2005; Maekawa et al., 2007). Kin4-phosphorylated Bfa1 forms are then recognized by the 14-3-3 family protein Bmh1. Bmh1 promotes removal of Bfa1-Bub2 GAP complexes from SPBs to prevent inactivation of Bfa1 by the polo-like kinase Cdc5 in cells with misaligned spindles (Caydasi et al., 2014), whereas PP1 (Glc7) in complex with Bud14 promotes dephosphorylation of Cdc5-phosphorylated Bfa1 (Kocakaplan et al., 2021). Cdc5 phosphorylates Bfa1 during anaphase in cells with properly aligned spindles (Hu et al., 2001). This phosphorylation reduces the activity of the Bfa1-Bub2 GAP complex in vitro and in vivo, thereby contributing to MEN activation (Geymonat et al., 2003; Caydasi and Pereira, 2012). M-Cdk was also reported to phosphorylate Bfa1 (Caydasi et al., 2017). A Cdk nonphosphorylatable mutant of Bfa1 is SPOC deficient, implying that M-Cdk keeps the Bfa1-Bub2 GAP complex active. Recently, we showed that the GSK-3 kinase Mck1 is essential to promote SPOC by keeping lower levels of the M-Cdk inhibitor (CKI) Cdc6 during mitosis (Rathi et al., 2022). The phosphorylation of Bfa1 by M-Cdk is reverted by the pool of Cdc14 phosphatase activated by the FEAR network (Caydasi et al., 2017). Kin4 prevents the inactivation of Bfa1 by FEAR-released Cdc14 by a mechanism that is currently unknown (Caydasi et al., 2017).
Although the function and regulation of Kin4 in SPOC have been more extensively studied (Chan and Amon, 2009; Caydasi et al., 2010b; Moore et al., 2010), the mechanisms by which cells initiate and maintain the SPOC upon spindle misalignment are not fully understood. Here we report the results of a genome-wide genetic screen performed to identify factors involved in SPOC. Among several novel factors we identified, we report that the SWR1 chromatin remodeling complex (SWR1-C) has a unique function in SPOC. Our data show that cells lacking SWR1-C are able to initiate the SPOC-response upon spindle misorientation; however, prolonged SPOC arrest is compromised and cells with misaligned spindles evade the mitotic arrest in spite of Kin4 activity (mitotic slippage). Interestingly, mitotic slippage observed in the absence of SWR1-C was dependent on SAGA (Spt-Ada-Gcn5 acetyltransferase) histone acetyltransferase complex, histone deacetylase Sir2, proteasome components, the m-CKI Sic1, and Cdc14-early anaphase release pathway. We propose that SWR1-C prevents premature mitotic exit upon checkpoint activation in part through counteracting Sir2 and possibly by affecting transcription of multiple factors that are not yet directly known to be involved in SPOC.
RESULTS
A genome-wide genetic screen reveals novel genes critical for SPOC integrity
Overexpression of KIN4 results in a late anaphase cell cycle arrest that mimics a constitutively active SPOC (D’Aquino et al., 2005). Thus cells overexpressing KIN4 fail to form visible colonies on agar plates (lethal phenotype). This lethality can be rescued by deletion of SPOC components that act downstream of Kin4 (Bfa1, Bub2, Bmh1) (D’Aquino et al., 2005; Pereira and Schiebel, 2005; Caydasi et al., 2014) or those that positively regulate Kin4 (PP2A-Rts1 and Elm1) (Chan and Amon, 2009; Caydasi et al., 2010a; Moore et al., 2010). To find novel SPOC components, we performed a genome-wide genetic screen using synthetic genetic array (SGA) technology (Tong et al., 2001). In this screen, we searched for gene deletions that rescue the lethality of KIN4 overexpressed from the inducible GAL1 promoter (see Figure 1A for an outline of the screen). Deletions of the known SPOC components BFA1, BUB2, RTS1, KIN4, BMH1, and MCK1 (Chan and Amon, 2009; Caydasi et al., 2010a; Moore et al., 2010; Caydasi et al., 2014; Rathi et al., 2022) were among the top 100 hits of the screen (Figure 1B and Supplemental Table S1; Data Set 1).
FIGURE 1:

Gene deletions that impair SPOC integrity. (A) Schematic representation of the SGA-based genetic screen strategy. In addition to the wild-type KIN4, the query strain carried a construct inserted in the URA3 locus for conditional KIN4 overexpression from the galactose-inducible promoter (ura3∆::URA3-GAL1-KIN4). The query strain was mated to a sporulated heterozygous diploid knockout library (goi∆::kanMX4) in which each library strain had four technical replicates spotted next to each other (2 × 2). Obtained diploids were spotted on haploid selection plates and the haploids carrying both the ura3∆::URA3-GAL1-KIN4 and the corresponding gene deletion from the library (goi∆::kanMX4) were selected. These double mutants (ura3∆::URA3-GAL1-KIN4 goi∆::kanMX4) were replicated on two types of plates with different carbon sources: glucose (no KIN4 overexpression) and raffinose/galactose (KIN4 overexpression). Plates were photographed, and colony sizes were measured in both plates to find colonies that grow under KIN4 overexpressing conditions. An example of a gene deletion with the desired phenotype is marked with a red box. (B) Plot showing colony sizes of ura3∆::URA3-GAL1-KIN4 goi∆::kanMX4. Knockout strains were ranked from better growing to poorly growing (goi∆ ranking from 1 to 4852, where goi∆ indicated the deletion of the corresponding gene of interest). The known SPOC proteins among the gene deletions with the top 100 highest colony sizes (red box) are marked in the magnification (lower plot). The dashed line indicates the threshold colony size value of 1.3, while a colony size of 1 is the median colony size on each plate. (C) Venn diagram showing the number of genes in indicated GO biological process categories. Remarkably, 70 among the top 100 hits of the screen fell into the indicated categories. (D) SPOC deficiency indexes of knockouts. Individual gene deletions were combined with the deletion of KAR9 to induce spindle misalignment. SPOC deficiency was assayed by microscopy of fixed cell populations after DNA staining; 100 cells were counted for knockout per experiment. The graph represents one of two independent experiments. The individual data values of the independent experiments are shown in Supplemental Table S2. The SPOC deficiency index of the kar9∆ was normalized to zero. Strains were considered SPOC deficient if the SPOC deficiency index was greater than 3.5 (marked by the red dashed line that is two SDs of unnormalized kar9∆ SPOC deficiency index above zero SPOC deficiency). Each bar represents a different gene deletion. Cells with normal aligned, misaligned nuclei as well as the cells with SPOC-deficient phenotypes (as depicted in E) were counted. Deletions of the known SPOC components are highlighted in green, whereas components of the SWR1-C found in the screen are highlighted in red. (E) Schematic representation of SPOC deficiency. A SPOC-deficient cell with a mispositioned anaphase spindle (indicated by two separated DNA regions in the mother cell compartment) exits mitosis and undergoes cytokinesis giving rise to cells with multiple nuclei and no nucleus. Further divisions of the same cell increase the multiple nuclei phenotype and also cause multibudded phenotypes (upper panel). SPOC-proficient cells, on the other hand, neither exit mitosis nor undergo cytokinesis as long as the spindle stays mispositioned (bottom panel). (F) Genetic and physical interactions between the proteins found to be involved in SPOC (based on experimental sources in string database). Disconnected nodes were removed. The ovals encircle the proteins that belong to the same protein complex.
The vast majority of the hits fell into Gene Ontology (GO) categories of the cell cycle, chromosome organization, transcription, and translation (Figure 1C). Several genes related to ribosome biogenesis, tRNA synthesis, and galactose metabolism were also found in the screening (Supplemental Figure S1; Supplemental Tables S1 and S2; Data Sets 1 and 2), but we excluded them from further analyses reasoning that they may interfere with KIN4 overexpression from the GAL1 promoter. To validate the hits, we individually deleted each gene in an independent strain background carrying the GAL1-KIN4 construct. Using growth assays and immunoblotting to measure Kin4 protein levels, we confirmed that deletion of 52 screen hits (genes of interest, goi∆) promoted growth upon KIN4 overexpression without affecting the extent of overexpression, whereas 8 hits including kin4∆ had reduced levels of Kin4 when grown in galactose-containing medium (Supplemental Table S2; Data Set 2; Supplemental Figure S1).
Next, we screened the hits of the SGA screen with respect to the SPOC functionality (Figure 1, D and E; Supplemental Table S2; Data Set 2). SPOC proficiency of goi∆ strains was assessed in a kar9∆ background. Kar9 is a conserved protein involved in spindle positioning (Miller and Rose, 1998). Deletion of KAR9 causes frequent spindle misalignment that allows assaying of SPOC integrity. The SPOC induces a late anaphase arrest in cells with misaligned spindles (i.e., when the mitotic spindle remained in the mother cell body). However, in the absence of SPOC, cells with misaligned spindles divide and undergo another round of budding and DNA replication. This leads to accumulation of SPOC-deficient phenotypes such as multibudded and multinucleated cells (Figure 1E). We categorized 28 goi∆ strains as SPOC deficient based on the multinucleation and multibudding phenotypes (Figure 1D). These 28 mutants with SPOC defects included deletions of genes coding for the known SPOC components Bfa1, Bub2, Bmh1, Rts1, Mck1, and Kin4 (Figure 1D), as well as genes not previously associated with SPOC. Intriguingly, many of these genes encode subunits of multiprotein complexes (Figure 1F).
SWR1-C but not INO80-C relieves the growth of KIN4 overexpressing cells
SWR1 and other subunits of SWR1-C were found in the screening as suppressors of KIN4 overexpression growth lethality (Supplemental Table S1; Data Set 1; Figure 1F). Accordingly, deletion of nonessential genes coding for SWR1-C components rescued KIN4 overexpression lethality (Figure 2 and Supplemental Figure S2). SWR1-C replaces the chromatin-bound H2A with the H2A.Z histone variant (Krogan et al., 2003; Kobor et al., 2004; Mizuguchi et al., 2004). Interestingly, HTZ1 (the gene that encodes for H2A.Z) was also found in the screen (Supplemental Table S1; Data Set 1). This suggested that deposition of H2A.Z in the nucleosomes by SWR1-C might be necessary for the late anaphase arrest induced by KIN4 overexpression. INO80-C belongs to the same nucleosome remodeler family as SWR1-C and shares components with SWR1-C (Gerhold and Gasser, 2014; Willhoft and Wigley, 2020). Disruption of genes specific to SWR1-C or INO80-C showed that only SWR1–C-related gene deletions rescued the lethality of KIN4 overexpression (Figure 2). Interestingly, among the gene deletions of different subunits of SWR1-C, yaf9∆ cells showed more pronounced growth rescue of GAL1-KIN4 overexpressing cells (Figure 2). However, Yaf9 is not an exclusive component of SWR1-C, as it also functions as part of the NuA4 acetyltransferase complex (Zhang et al., 2004). To exclude any interference by impairment of NuA4, swr1∆ and not yaf9∆ cells were further analyzed.
FIGURE 2:
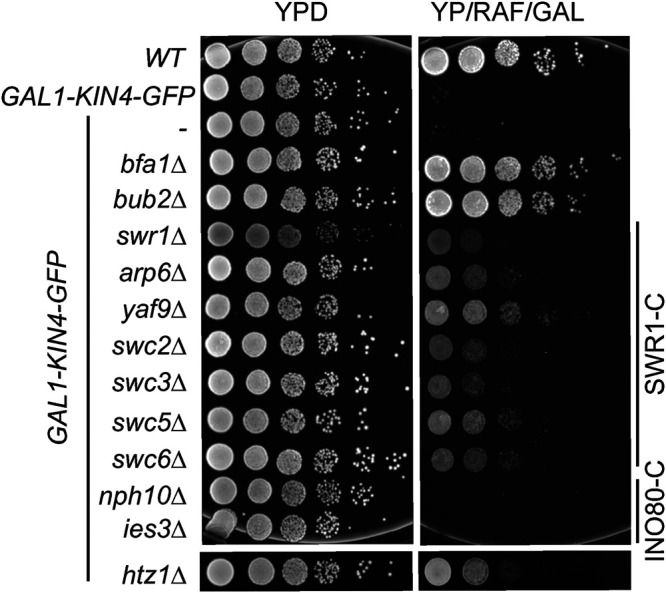
KIN4 overexpression lethality is suppressed by deletions of SWR1-C and HTZ1 but not INO80-(C) Serial dilutions of indicated strains were spotted on the agar plates that suppress (YPD) or induce (YP/RAF/GAL) KIN4 overexpression. BFA1 or BUB2 deletion reverts Kin4 overproduction lethality and was used as comparison.
SWR1-C does not promote SAC or prevent mitotic exit in MEN-compromised cells with normally aligned spindles
Known SPOC components such as Bfa1-Bub2 also function in mitotic regulation other than SPOC. We thus aimed to determine whether cells require Swr1 for such mitotic regulation. For this, we analyzed swr1∆ in two different contexts: functionality of spindle assembly checkpoint (SAC) and the ability to act as a mitotic exit inhibitor in cells with properly aligned spindles. SAC prevents anaphase onset upon failure of bipolar kinetochore-microtubule attachment (Musacchio and Salmon, 2007). The SPOC proteins Bfa1 and Bub2 but not Kin4 are required for the mitotic arrest induced by SAC (Hoyt et al., 1991; Wang and Burke, 1995; Knop et al., 1999; D’Aquino et al., 2005; Pereira and Schiebel, 2005; Maekawa et al., 2007). To investigate SWR1-C contribution to SAC, we treated cells with the microtubule depolymerizing drug nocodazole to engage this checkpoint. As expected, wild-type cells arrested as large budded cells without degrading the Separase inhibitory protein, Securin/Pds1 (Figure 3, A and B) (Musacchio and Salmon, 2007). In the absence of BUB2, Securin/Pds1 was degraded (Figure 3B, time points 75–90) and cells rebudded, indicating cell cycle progression (Figure 3A). The swr1∆ cells behaved similar to wild-type and kin4∆ cells (Figure 3, A and B). Thus SWR1-C, unlike Bfa1-Bub2, is not required for SAC.
FIGURE 3:

SWR1-C is not essential for SAC integrity. (A, B) SAC integrity of indicated cell types. Cells were released from a G1 block (t = 0) into nocodazole-containing medium. Samples were collected at indicated time points. (A) Percentages of large- and multibudded cells were scored and (B) Pds1-3HA, Clb2, Sic1 and Tubulin levels were analyzed by immunoblotting at the indicated time points. (C) Deletion of SWR1 does not rescue temperature-sensitive men-ts. Serial dilutions of SWR1 or swr1∆ cells carrying the indicated temperature-sensitive alleles of the genes coding for MEN proteins were grown on YPAD plates at indicated temperatures.
Next, we analyzed whether SWR1-C has a mitotic exit inhibitory effect in cells with normal aligned spindles. For this, we used temperature-sensitive MEN mutants (men-ts). The lack of mitotic exit inhibitors, such as Bfa1-Bub2, Bud14-Glc7, or Mck1, rescues the growth defect of men-ts mutants at their semipermissive temperature (Kocakaplan et al., 2021; Rathi et al., 2022). However, the lack of SWR1 did not rescue the growth of tem1-3, cdc15-1, mob1-67, dbf2-2, cdc14-1, or cdc14-2 (Figure 3C), leading us to conclude that the role of SWR1-C as a mitotic exit inhibitor is most likely limited to SPOC arrest.
SWR1-C does not influence Kin4 activity or Bfa1 regulation by Kin4
As deletion of SWR1 rescued the growth lethality of Kin4 overproducing cells, we reasoned that SWR1-C might be required for Kin4-dependent SPOC activation. Hallmarks of SPOC signaling are the recruitment of Kin4 to SPBs and phosphorylation of Bfa1 by Kin4 upon spindle misalignment, which subsequently promotes a decrease in SPB-bound levels of Bfa1 (Bardin et al., 2000; Pereira et al., 2000; Molk et al., 2004; D’Aquino et al., 2005; Pereira and Schiebel, 2005; Caydasi and Pereira, 2009; Monje-Casas and Amon, 2009; Caydasi and Pereira, 2012). Deletion of SWR1 influenced neither Kin4 localization (Figure 4A) nor the ability of Kin4 to phosphorylate Bfa1 in vitro (Figure 4B) or in vivo (Figure 4C). Furthermore, SPB-bound Bfa1 levels decreased in swr1∆ cells upon spindle misalignment (Figure 4D), similar to SWR1 cells (Figure 4E). These data altogether suggest that SWR1-C is not required for Kin4 activity or Kin4-dependent regulation of Bfa1 upon SPOC activation.
FIGURE 4:

Bfa1 regulation by Kin4 is not altered in swr1∆ cells. (A) The percentage of cells with Kin4 localized at the SPBs and mother cell cortex in the indicated cell types. Cells were treated by nocodazole to induce Kin4 recruitment to SPBs. rts1∆ cells served as a control in which Kin4 SPB and cortex localization diminishes; 100 cells were counted per cell type. The graph is an average of three independent experiments. Error bars show SD. (B) In vitro kinase assay of Kin4-6HA and Kin4-T209A-6HA (kinase inactive mutant) enriched from yeast cells with or without SWR1. Purified, recombinant MBP-Bfa1 was used as a substrate. Autoradiograph (32P) shows the incorporation of γ32P-ATP to MBP-Bfa1. MBP-Bfa1 levels are shown in Coomassie Brilliant Blue staining. Levels of the Kin4-6HA used in the reaction are shown in the immunoblot using anti-HA antibodies. (C) Immunoblots showing Bfa1 phosphorylation profile in the indicated cell types. In the absence of Bfa1 phosphorylation by Kin4, Bfa1 appears as slow migrating Bfa1 forms in SDS–PAGE gels (Maekawa et al., 2007). Cultures of indicated strains were arrested in G1 by alpha factor treatment (t = 0) and released in nocodazole-containing medium. Samples were collected at the depicted time points and probed with the indicated antibodies. Clb2 and Sic1 served as markers for cell cycle progression. Tubulin served as a loading control. (D, E) Representative still images from time-lapse series of Bfa1-GFP localization during anaphase spindle misalignment. Spc42-eqFP served as a SPB marker. Cell boundaries are outlined with dashed lines. Arrows point the SPBs. Scale bar: 3 µm.
SWR1-C is required for prolonged SPOC arrest
To examine the SPOC in more detail, we compared the duration of anaphase in swr1∆ kar9∆ cells upon spindle misalignment and upon correct spindle alignment through time-lapse fluorescence microscopy. A hallmark of SPOC is the long anaphase arrest of cells with misaligned spindles. The anaphase duration of cells with properly aligned spindles is normally 20 ± 3 min (Figure 5A). However, anaphase can endure for longer than 60 min in the majority of cells with misaligned spindles with an active SPOC (kar9∆ cells, Figure 5A). As previously reported, this anaphase delay does not occur in cells with misaligned spindles in the absence of Kin4 (Caydasi et al., 2010a) (Figure 5A). In this case, anaphase duration of kin4∆ kar9∆ cells with properly or misaligned spindles is equal (Figure 5A). Interestingly, the majority of swr1∆ kar9∆ cells broke their spindle despite spindle misalignment (Figure 5A). However, in contrast to kin4∆, cells lacking SWR1 could hold the anaphase arrest for a significantly longer period of time (Figure 5A, swr1∆, anaphase duration of 23 ± 3 min versus 31 ± 10 min in correctly aligned versus misaligned spindle cases, respectively). In addition, the failure of SPOC in swr1∆ kar9∆ cells was not fully penetrant, as 20% of the cells with misaligned spindles were able to stay arrested for longer than 60 min (Figure 5A). Therefore we concluded that swr1∆ kar9∆ cells are able to delay mitotic exit upon spindle mispositioning, but this delay is not maintained. Our data thus suggest a role for SWR1-C in preventing mitotic progression after SPOC engagement.
FIGURE 5:

Deletion of SWR1 causes slippage from the SPOC arrest. (A) Duration of anaphase in indicated cell types during correct spindle alignment and spindle misalignment. GFP-labeled tubulin (GFP-TUB1) was monitored by time-lapse microscopy with 1-min time resolution. The duration of anaphase was calculated as the time elapsed from the start of the fast spindle elongation phase (metaphase-anaphase transition) until spindle breakdown. N, number of cells analyzed. Asterisks indicate a significant difference according to Student’s t test (p < 0.05). Representative still images of indicated GFP-TUB1 kar9∆ cells monitored during spindle misalignment are shown on the left. Cell boundaries are outlined with dashed lines. Arrows indicate the point of spindle breakdown. Time point 1 is 1 min before the metaphase-anaphase transition, which coincides with the start of fast spindle elongation phase. Scale bar: 3 µm. (B) Representative still images from the time-lapse movies of CDC14-GFP MYO1-3mCherry bearing kar9∆ cells with misaligned spindles. Scale bar: 3 µm. The red asterisk indicates the onset of Cdc14 full release. The percentage of cells with misaligned spindles that released Cdc14-GFP fully and contracted Myo1-3mCherry is plotted. Time-lapse series of 21 cells were analyzed each in SWR1 and swr1∆. A chi-square test was performed using a significance level of 0.05. Please note that a higher percentage of kar9∆ Myo1-3mCherry cells with misaligned spindles released Cdc14 and exited mitosis in comparison to kar9∆ cells shown in A. This is most likely a consequence of Myo1 tagging, as it was not observed in cells with untagged Myo1.
We sought to understand how the absence of SWR1-C promotes slippage of cells with misaligned spindles out of the SPOC mitotic arrest. Mitotic exit and cytokinesis in budding yeast is triggered by the phosphatase Cdc14 through down-regulation of M-Cdk activity and dephosphorylation of M-Cdk targets. We thus asked whether mitotic slippage of swr1∆ cells with misaligned spindles correlates with Cdc14 release. For this, we employed fluorescence time-lapse microscopy of CDC14-GFP MYO1-3mCherry kar9∆ cells and scored the release of Cdc14-GFP from the nucleolus in cells with misaligned spindles. Myo1-3mCherry contraction served as a marker for cytokinesis. The majority of the SWR1 cells with misaligned spindles did not fully release Cdc14-GFP or contract Myo1-3mCherry (Figure 5B, SWR1), whereas a significantly greater proportion of swr1∆ cells with misaligned nuclei fully released Cdc14-GFP from the nucleolus (Figure 5B, swr1∆, 18 min, red asterisk). Thus Cdc14 full release coincides with mitotic slippage of SWR1-deleted cells. These data indicate that the MEN becomes activated in cells with misaligned spindles in the absence of SWR1-C.
The deletion of SWR1 does not promote premature FEAR release
The FEAR network promotes a partial release of Cdc14 from the nucleolus into the nucleus at the metaphase-to-anaphase transition. This pool of Cdc14 was shown to facilitate MEN activation (Jaspersen and Morgan, 2000; Konig et al., 2010; Caydasi et al., 2017), leading us to ask whether the FEAR network would be prematurely active in swr1∆ cells. To assess FEAR activity, we made use of cdc15-1 cells, in which MEN is inactivated at the restrictive temperature (37°C). MEN inactivation prevents the full release of Cdc14 immediately after FEAR and thus allows analysis of Cdc14 released by the FEAR alone. In cdc15-1 cells at restrictive temperature, FEAR-released Cdc14-GFP was frequently observed in cells with 2- to 6-µm-long spindles and not in cells with longer spindles due to the relocation of Cdc14 back to the nucleolus (Visintin et al., 2008) (Figure 6A). In cdc15-1 spo12∆ cells, the percentage of cells with FEAR-released Cdc14-GFP was markedly reduced as expected for this FEAR-less mutant (Stegmeier et al., 2002). The partial release of Cdc14-GFP was similar in SWR1 and swr1∆ cells (Figure 6A). We thus consider unlikely that Cdc14-GFP is prematurely released from the nucleolus in cells lacking SWR1. To support this conclusion, we also analyzed the spindle-associated, chromosome passenger protein Sli15. Previously, FEAR-released Cdc14 was shown to promote Sli15 spindle localization (Pereira and Schiebel, 2003). We reasoned that if SWR1 deletion caused a premature activation of the FEAR, Sli15-GFP would accumulate on spindles earlier than SWR1-bearing cells. However, this was not the case as Sli15-GFP spindle enrichment did not occur prematurely in swr1∆ cells (Figure 6B). We conclude that release of Cdc14 by the FEAR occurs at similar timing in both SWR1 and swr1∆ cells.
FIGURE 6:

Swr1 does not affect FEAR. (A) The percentage of cells with partially released Cdc14. Cultures of indicated cell types bearing mCherry-Tubulin and Cdc14-GFP were synchronized in G1 by alpha factor treatment (t = 0) at the permissive temperature of cdc15-1 (23°C) and released in alpha factor free medium at the restrictive temperature (37°C). Samples were collected every 10 min and Cdc14 release was scored as a function of spindle length. Data are an average of three independent experiments. Error bars are SD. (B) The percentage of cells with spindle-enriched Sli15-GFP. Cultures of indicated cell types bearing mCherry-Tubulin and Sli15-GFP were synchronized in G1 by alpha factor treatment (t = 0) at the permissive temperature of cdc15-1 (23°C) and released in alpha factor free medium at the restrictive temperature (37°C). Samples were collected every 10 min, and spindle-enriched Sli15 was scored as a function of spindle length. Data are an average of three independent experiments. Error bars are SD. Scale bars: 3 µm. Two-way ANOVA test was performed in A and B. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Sir2 contributes to mitotic exit in cells with misaligned spindles but not in cells overexpressing KIN4
SWR1-C functions in the DNA damage response through recruitment of Yku80 at double strand breaks to facilitate nonhomologous end-joining (van Attikum et al., 2007). We reasoned that if SWR1-C works via recruitment of Yku80, YKU80 deletion would phenocopy SWR1 deletion. However, this was not the case, as deletion of Yku80 neither rescued the growth lethality of GAL1-KIN4 overexpressing cells (Figure 7A) nor promoted SPOC deficiency in kar9∆ cells (Figure 7B).
FIGURE 7:

Sir2 contributes to SPOC deficiency of swr1∆ cells. (A) Serial dilutions of indicated strains were spotted on the agar plates that suppress (YPD) or induce (YP/RAF/GAL) KIN4 overexpression. (B) SPOC deficiency index of indicated strains. Graph shows an average of three independent experiments. A minimum of 100 cells were counted for each sample per experiment. Error bars are SD. (C) The percentage of cells with Cdc14-FEAR release. Indicated cell types were arrested in G1 using alpha factor at 23°C and released from the G1-block in alpha factor free medium at 37°C. Cells were collected every 30 min, fixed with PFA, and analyzed by microscopy. Cells with medium-to-large cells were scored based on Cdc14 nucleolar or FEAR released localization; spo12∆ cells served as a control for inactive FEAR. Scale bars: 3 µm. Graph shows an average of two experiments. A minimum of 100 cells were counted for each sample per experiment. Error bars are SD. One-way ANOVA test was performed for B and C. *p < 0.05, ***p < 0.001, ****p < 0.0001.
SWR1-C is also involved in prevention of heterochromatin spreading by antagonizing Sir-dependent silencing (Meneghini et al., 2003; Zhou et al., 2010). Accordingly, deletion of SIR2 reverses the heterochromatin spreading that takes place in SWR1–C-deficient cells (Meneghini et al., 2003; Zhou et al., 2010). We thus asked whether Swr1 works via Sir2 for its function in SPOC. We hypothesized that if SWR1-C works via antagonizing Sir2, deletion of SIR2 in swr1∆ cells would revert the swr1∆ phenotypes. Analysis of KIN4 overexpression growth lethality showed that GAL1-KIN4 swr1∆ cells behaved similarly in the presence and absence of SIR2 (Figure 7A). However, the deletion of SIR2 significantly reverted SPOC deficiency of kar9∆ swr1∆ cells (Figure 7B). Together, we concluded that SWR1 deletion rescues the lethality of KIN4 overexpression through a mechanism that does not involve Yku80 or Sir2, yet the SPOC deficiency of cells lacking Swr1 occurs in part due to the presence of Sir2.
Sir2 also functions in rDNA silencing as part of the RENT complex (Huang and Moazed, 2003) which sequesters Cdc14 in the nucleolus (Shou et al., 1999b). Considering that blockage of Cdc14 FEAR release rescues SPOC deficiency of kin4∆ (Caydasi et al., 2017) and swr1∆ cells (Figure 8C), we asked whether deletion of SIR2 inhibited FEAR release in swr1∆ cells, thereby accounting for the rescue of SPOC deficiency. We analyzed Cdc14 partial release in cdc15-1 cells where full release of Cdc14 is prevented (Figure 7C). We also analyzed spo12∆ cells as a FEAR-deficient control group. We found that unlike spo12∆, sir2∆ cells were able to release Cdc14 (Figure 7C), suggesting that Sir2 contributes to Swr1 SPOC deficiency without impinging on Cdc14-FEAR release.
FIGURE 8:

SGA screening identifies genes that are essential for the growth of swr1∆ GAL1-KIN4 cells under KIN4 overexpressing conditions. (A) Number of screen hits found within each of the indicated GO categories. (B) Physical and genetic interaction map of the hits. Circles indicate proteins with related functions. (C) Average SPOC deficiency indexes of the indicated strains. Values are an average of at least three independent experiments. Average values and SDs are listed in Data Set 6. A red line is placed at a SPOC deficiency index of 5.5, which is 2 SD below the average SPOC deficiency index of swr1∆ kar9∆. The average SPOC deficiency index of swr1∆ kar9∆ is marked with a black bar. (D) Average SPOC deficiency indexes of the indicated strains. Error bars are SD. One-way ANOVA test were performed for D. *p < 0.05.
To summarize, our results are twofold. First, SPOC deficiency of swr1∆ cells is not due to Yku80 loss of function but in part correlates with Sir2 activity. Second, Kin4 overexpressing cells may require other factors in addition to loss of SPOC functionality for their growth.
Gene expression profile of late anaphase-arrested cells in the presence and absence of SWR1-C
We hypothesized that transcription of mitotic exit-related genes could be under the control of SWR1-C. Thus we investigated how gene expression profile of late anaphase-arrested cells changes in the presence and absence of SWR1. For this, we performed a genome-wide microarray-based expression profiling. Microarray-based expression analysis of cells lacking SWR1-C was previously performed by other groups (Kobor et al., 2004; Mizuguchi et al., 2004; Morillo-Huesca et al., 2010) but not in anaphase-arrested cells with an activated SPOC. To obtain a late anaphase arrest in all cell types, we used cdc15-as (analog-sensitive allele of the MEN kinase CDC15) that blocks MEN activation upon 1NM-PP1 addition to the culture media (Supplemental Figure S3, A and B). To activate the SPOC, we made use of KIN4 overexpression (Supplemental Figure S4, A and B). We found 105 genes up-regulated and 338 genes down-regulated in swr1∆ (Supplemental Table S3, Data Set 3, and Supplemental Figure S3C); 22 of 105 up-regulated genes identified in our analysis overlapped with the findings of previous studies, whereas the overlap in the down-regulated genes was 98 of 338 (Supplemental Figure S3D). Mitosis-related GO categories were not enriched among up-regulated and down-regulated gene data sets (Supplemental Table S4; Data Set 4). Furthermore, we could not detect any known mitotic exit-related gene among differentially expressed genes in swr1∆ (Supplemental Table S3; Data Set 3). Thus we conclude that SWR1 may not control mitotic exit via transcriptional regulation of known mitotic exit-related genes.
SPOC slippage requires FEAR, proteasome, and SAGA
To determine which components contribute to both the SPOC bypass and the rescue of GAL1-KIN4 lethality by SWR1 deletion, we performed an SGA-based genome-wide screen to identify genes required for the growth rescue. We furthermore screened the hits of the genetic screen for an ability of their gene deletions to suppress SPOC deficiency of swr1∆ cells. For the SGA screen we used swr1∆ and GAL1-KIN4 swr1∆ cells. We reasoned that deletion of genes involved in SPOC bypass would cause the death of swr1∆ cells overexpressing KIN4 but would not affect the growth of swr1∆ cells (Supplemental Figure S4A). We found 69 gene deletions that caused growth defects specifically in GAL1-KIN4 swr1∆ cells but not in swr1∆ cells on galactose-containing plates (Supplemental Table S5; Data Set 5). Most of these genes were in GO categories of transcription, cytoskeletal organization, cell cycle, and cell wall organization (Figure 8A). Among them, many coded for proteins that form protein complexes or proteins participating in the same signaling pathways or cellular processes, such as the SAGA chromatin-modifying complex, the lipid-signaling PAS complex, the mannose transferase complex, the proteasome, cell wall integrity (CWI) pathway, the FEAR-network, and cytokinesis and abscission processes (Figure 8B).
In an independent strain background, we tested deletions of at least two genes from each category represented in Figure 8B. We confirmed that 90% of these gene deletions caused growth retardation in swr1∆ cells under KIN4-overexpressing conditions (Supplemental Table S6; Supplemental Figure S4B; Data Set 6). We next tested whether those gene deletions also affected SPOC in swr1∆ kar9∆ cells. Remarkably, SPOC deficiency of swr1∆ cells was completely rescued by the deletion of four genes (Supplemental Table S6; Figure 8C; Data Set 6). These were the FEAR pathway components SPO12 and SLK19, which enhance MEN activation; PRE9, which is the only nonessential 20S proteasome subunit; and the SAGA histone acetyltransferase complex component SGF73, which is involved in the regulation of RNA polymerase II (RNA PolII) transcription preinitiation (Supplemental Table S6; Figure 8C; Data Set 6) (Koutelou et al., 2010).
Different from Spo12, Slk19, Pre9, and Sgf73, deletion of the MAPK SLT2 and M-CKI SIC1 retarded formation of multinucleated and multibudded cells without completely rescuing the SPOC deficiency of swr1∆ cells (Figure 8C). Most of the gene deletions that reverted the growth phenotype of GAL1-KIN4 swr1∆ cells, however, did not revert the SPOC deficiency phenotype of swr1∆ kar9∆ cells. This implies that the late anaphase arrest in cells with correctly and misaligned spindles might have different characteristics. Interestingly, some gene deletions even caused an increment in accumulation of SPOC-deficient phenotypes. Essentially, these genes (TPM1, MDM20, ROM2) were related to actin organization and cellular polarity (Liu and Bretscher, 1989; Manning et al., 1997; Polevoda et al., 2003; Singer and Shaw, 2003), suggesting that the increased SPOC defect could stem from the inability of spindle realignment or cytokinesis.
We next asked whether the genes required for SPOC deficiency of swr1∆ cells were also required for SPOC deficiency of kin4∆ cells. In concordance with previous publications (Falk et al., 2016; Caydasi et al., 2017), deletion of FEAR network components SPO12 and SLK19 rescued SPOC deficiency of kin4∆ cells (Figure 8D). On the other hand, deletion of PRE9, SLT2, SGF73, and SIC1 did not significantly impact SPOC deficiency of kin4∆ cells despite a slight reduction due to SIC1 and SGF73 deletion (Figure 8D). These data suggest that PRE9, SLT2, SGF73, and SIC1 act by a mechanism different than the FEAR network in SPOC slippage.
Taken together, our data show that mitotic slippage of SPOC in swr1∆ cells requires the FEAR network, proteasome-dependent protein degradation, as well as RNA PolII-dependent transcription.
DISCUSSION
The SPOC exploits proteins localized to the SPBs as well as mother and daughter cell cortexes to coordinate mitotic exit with the direction of chromosome segregation. How SPOC operates on a molecular level and how it maintains a robust cell cycle arrest remains elusive. In this study, we performed a genome-wide screen that identified proteins essential for SPOC integrity. Our analysis uncovered a novel function for the chromatin-remodeling complex, SWR1-C, in preventing mitotic slippage upon prolonged SPOC arrest.
Identification of novel SPOC components
Higher levels of Kin4 inhibit cell growth due to constitutive inactivation of MEN through the Bfa1-Bub2 GAP complex (D’Aquino et al., 2005). Here we searched for gene deletions that rescued the growth defect of KIN4-overexpressing cells. Our screen was designed to identify novel genes that inhibited mitotic exit. Among them, we were particularly interested in SPOC-related genes that either directly promoted Kin4 function (e.g., Kin4 regulators or Kin4 substrates) or inhibited mitotic exit downstream or in parallel to KIN4 (e.g., inhibitors of MEN). To find SPOC regulators among those putative mitotic exit inhibitors, we performed a secondary screen using cells that lacked the spindle-positioning protein, Kar9. This screen revealed that the majority of genes identified as potential mitotic exit inhibitors, including many components of the SWR1-C, were indeed important for SPOC functionality. SWR1-C remodels the chromatin by replacing histone H2A with the histone variant H2A.Z at the chromatin-bound nucleosomes. The HTZ1, gene, which encodes for the H2A.Z histone variant, was also identified in our screen as a novel SPOC component. In this study, we investigate the function played by SWR1-C in SPOC in more detail. The function of other novel SPOC proteins will be in the scope of other studies.
SWR1-C prevents mitotic slippage during SPOC arrest
The immediate response to SPOC activation is the recruitment of Kin4 kinase to both SPBs, the phosphorylation of Bfa1 by Kin4, and the change in Bfa1 localization from asymmetric (strong at one SPB) to symmetric (weak at both SPBs). These events are essential for Tem1 inactivation that is indispensable for a late anaphase arrest (Caydasi et al., 2012). Analysis of the SPOC mechanism verified that SWR1–C-deficient cells are in fact able to initiate these SPOC responses upon spindle misorientation, yet they later release Cdc14 and constrict their actomyosin ring, indicating that cells exit mitosis and undergo cytokinesis despite Bfa1-Bub2 activation. Thus inactivation of Tem1 by Bfa1-Bub2 is essential but not sufficient to hold mitotic exit for a prolonged time. A closer look at anaphase duration in swr1∆ cells showed an anaphase arrest of 8–10 min in cells with misaligned spindles. This arrest can last for longer than 60 min in cells with a fully active SPOC, yet no arrest is seen at all in the absence of Bub2, Bfa1, or Kin4. Altogether these data led us to propose two levels of SPOC response: first, an “immediate response” that results in the inactivation of Tem1 by the core SPOC mechanism involving Bfa1-Bub2 and Kin4 and second, a “late response” that prolongs the arrest dependent on SWR1-C. Such dual-level regulation might resemble the rapid and slow responses to DNA damage. In the case of the DNA damage checkpoint, a rapid response involves degradation of the Cdk-activating phosphatase Cdc25, whereas the slow response involves transcriptional activation of targets including the CKI p21 (Shaltiel et al., 2015). Importantly, mitotic slippage that occurs in the absence of SWR1-C seems to be restricted to anaphase in cells with misaligned spindles, as deletion of SWR1 neither impaired SAC arrest nor promoted mitotic exit in MEN-compromised cells.
Genetic analysis indicated that the role of SWR1-C in DNA damage response is not relevant to our observations. Intriguingly, we found that the function of SWR1 in counteracting Sir2 to prevent heterochromatin spreading may account for SPOC slippage at least in part. In addition, our data showed that mitotic slippage that takes place in the absence of SWR1-C is dependent on the SAGA complex, which is another histone-modifying protein complex bearing histone acetyl transferase and deubiquitination functions that are important for transcriptional activation, particularly through RNA PolII dependent elongation (Koutelou et al., 2010). Chromatin is highly compacted during mitosis and most gene regulatory elements are cleared from this condensed chromosome. It was earlier thought that transcription is completely silenced during mitosis and activated during mitotic exit. However, many studies now have proven that RNA PolII remains active on the chromatin and a low-level transcription persists during mitosis in a promoter dependent manner (Liang et al., 2015; Palozola et al., 2017; Palozola et al., 2018). Owing to the role of SWR1-C in gene expression and the fact that the histone variant, Htz1 was also required for SPOC, it is tempting to speculate that SWR1–C-dependent transcriptional regulation is important for prevention of mitotic slippage during SPOC arrest. Indeed, microarray analysis of SWR1–C-dependent gene expression in a late anaphase arrest (cdc15-as) showed that transcriptional profiles of late anaphase-arrested cells differed in the presence and absence of SWR1-C. However, using these data we failed to identify individual genes or groups of genes involved in the late anaphase arrest of cells with or without spindle misalignment. More needs to be done to understand the contribution of transcriptional regulation on control of mitotic exit.
Factors required for mitotic slippage in the absence of SWR1-C
Using an unbiased genetic approach, we identified nonessential genes that contributed to mitotic slippage of SWR1–C-deficient cells with misaligned spindles (Figure 9). Most of these genes were factors already known to be involved in mitotic exit. For example, SIC1, the mitotic CKI, is important for M/G1 transition (Schwob et al., 1994). Likewise, the proteasome (Pre9 being one of the nonessential components of 20S proteasome) is required for degradation of mitotic cyclin Clb2 to achieve mitotic Cdk inactivation (Deshaies, 1997). The FEAR pathway (SPO12 and SLK19 being FEAR components), on the other hand, is crucial for priming mitotic exit at several levels, such as at the level of Bfa1-Bub2 inactivation and, Cdc15 and Dbf2/Mob1 localization at SPBs (Jaspersen and Morgan, 2000; Konig et al., 2010; Caydasi et al., 2017). Importantly, deletion of SPO12 or SLK19 reverted the SPOC deficiency of swr1∆ cells as well as kin4∆ cells (Falk et al., 2016; Caydasi et al., 2017), highlighting the role of FEAR pathway in bypassing the immediate SPOC response (Scarfone et al., 2015). Unlike the FEAR pathway, impairment of the proteasome and deletion of mitotic CKI reverted the SPOC deficiency in swr1∆ but not in kin4∆ cells. Thus mitotic slippage that occurs in the absence of SWR1-C particularly requires mitotic CKI and proteasome. Puzzling, we could not detect any difference in Sic1 or Clb2 mRNA levels during a late anaphase arrest with or without SWR1-C in our microarray analysis. We envisage that if SWR1-C directly regulates SIC1 or CLB2 it is likely not at the level of SIC1 or CLB2 transcription.
FIGURE 9:

Model for SPOC slippage. Spindle misorientation activates the SPOC kinase Kin4, which in turn keep the GAP complex Bfa1-Bub2 active. As a consequence, MEN is inhibited, and cells arrest in late anaphase until spindle misorientation is corrected. A robust late anaphase arrest requires the presence of SWR1-C to prevent mitotic slippage in SPOC-arrested cells most likely via transcriptional regulation. Mitotic slippage in the absence of SWR1-C requires Sir2, FEAR proteins Slk19 and Spo12, CWI pathway kinase Slt2, CKI Sic1, SAGA component Sgf73, and the proteosome component Pre9 as depicted. See text for details.
We found that components of the CWI pathway contributed to the SPOC deficiency of swr1∆ cells. CWI pathway regulates G1/S transition and DNA replication in response to cell wall damage (Levin, 2011; Kono et al., 2016). The CWI pathway was also shown to stabilize Sic1 (Kono et al., 2016). However, we could not detect activation of the CWI pathway in GAL1-KIN4 or GAL1-KIN4 swr1∆ cells upon induction of KIN4 overexpression (Supplemental Figure S5). How CWI might contribute to the mitotic exit of swr1∆ cells remains unclear. We furthermore found components that belong to the histone acetyl transferase and histone deubiquitylating modules (DUBms) of the SAGA complex to be crucial for mitotic exit in the absence of SWR1-C. Among those, SGF73, a component of the histone DUBm, was also necessary for mitotic slippage of swr1∆ cells with a mispositioned spindle. DUBm of SAGA is required for gene activation through RNA PolII (Frappier and Verrijzer, 2011). Thus it is tempting to speculate that genes activated by SAGA in the absence of SWR1-C might be the reason for mitotic slippage.
Chromatin remodelers and checkpoints
Chromatin remodelers implicated in checkpoint control is not limited to SWR1-C. For example, Isw2 and Ino80 chromatin remodelers facilitate S phase checkpoint deactivation by directly interacting with replication protein A (Au et al., 2011). SWI/SNF chromatin remodeling complex regulates the DNA damage checkpoint by activating the checkpoint kinase Mec1 (ATR) through interaction of the Snf2 ATPase subunit with Mec1 (Kapoor et al., 2015). In addition, Ies4 subunit of Ino80 chromatin remodeling complex is a downstream target of the Mec1/Tel1 (ATM/ATR) kinases (Morrison et al., 2007). Several other ATP-dependent chromatin remodelers are also involved in DNA damage response (Lans et al., 2012). Considering that DNA replication/damage checkpoints detect abnormalities on the DNA, it is not surprising that the chromatin remodelers participate in this process. On the other hand, data on the function of chromatin remodelers in mitotic checkpoints have so far been limited to the RSC chromatin remodeling complex. RSC chromatin remodeling complex associated with its Rsc2 subunit contributes to mitotic slippage of cells from the metaphase arrest triggered by the SAC (Rossio et al., 2010). In this case, Rsc2 physically interacts with the polo-like kinase Cdc5 and affects Net1 phosphorylation to promote Cdc14 partial release from the nucleolus during early anaphase. Thus the RSC chromatin remodeling complex promotes mitotic slippage of metaphase-arrested cells. Importantly, Rsc2 interacts with Cdc5 and hence it is likely that RSC regulates mitotic exit independently of its role in transcriptional regulation. Our finding that SWR1-C plays a critical role in preventing mitotic slippage of SPOC-arrested cells reinforces the importance of chromatin remodelers in mitotic control and opens up new insights into mitotic checkpoint regulation.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Yeast methods, strains, and growth
Yeast strains used in this study are isogenic with S288C. The strains constructed for validation of the SGA screen are indicated in Data Sets 2 and 6. Other strains are listed in Supplemental Table S7. PCR-based methods were used for gene deletions and epitope tagging (Knop et al., 1999; Janke et al., 2004). Genes of interest were expressed from their endogenous promoter unless otherwise stated. Basic yeast methods and growth media were as described (Sherman, 1991). For induction of the GAL1 promoter, yeast extract peptone medium containing raffinose (3%) and galactose (2%) (YP-Raf/Gal) was used. To synchronize the cells in the G1 phase, 10 μg/ml of synthetic alpha factor (Sigma, St. Louis, MO) were added to logarithmically growing (log-phase) cultures. For nocodazole treatment (metaphase arrest), 15 μg/ml of nocodazole (Sigma) were added to the culture media. Genetic interactions with temperature-sensitive mutants and GAL1-KIN4 strains were evaluated at a restrictive temperature and under GAL1-overexpressing conditions, respectively. Growth was assayed by performing drop tests in which serial dilutions of cultures were spotted on corresponding agar plates. Plates were incubated at an appropriate temperature for 2–3 d.
Genome-wide genetic screening
Screens were performed by sequential pinning and growth of strains as ordered colony arrays on agar plates with appropriate selective media (Baryshnikova et al., 2010) using query strains AKY1296 (Y8205 GAL1-KIN4), AKY1307 (Y8205 GAL1-KIN4 swr1∆), and AKY1346 (Y8205 swr1∆) and a heterozygous diploid yeast deletion collection (Winzeler et al., 1999) where each strain carries a deletion of a single nonessential gene. The deletion collection was pinned on agar plates in a 1536-colony format, with four technical replicates of every strain placed next to each other (2 × 2) using a ROTOR colony pinning robot (Singer Instruments, Somerset, UK). The collection was subsequently sporulated and mated with each of the query strains. The resulting diploids were sporulated and haploids carrying simultaneously the query mutations and a gene deletion allele from the deletion collection (goi∆, goi = gene of interest) were selected. All steps until here were carried out on plates with glucose as carbon source. The selected haploids were subsequently replicated either on raffinose/galactose or on glucose-containing agar media. Glucose-containing plates were photographed after 1 d of incubation at 30°C, whereas raffinose/galactose-containing plates were photographed every day up to 4 d of incubation at 30°C (d1, d2, d3, and d4 in Supplemental Table S1). Colony sizes, normalized to the median of each plate, were determined from the photographs using SGATools (Wagih et al., 2013). For each strain, mean and median colony sizes were calculated from the four technical replicates.
The cross with GAL1-KIN4 carrying strain was used to find out gene deletions that rescued KIN4 overexpression toxicity. Hits were selected employing a criteria that included colonies with “median colony size > 1.3” and “standard error of the mean < 25%” on raffinose/galactose plates (day 2) and excluded the colonies with “median colony size < 0.3” and “colony counts < 4” on glucose plates. Among such colonies, those with the top 100 highest median colony size were considered as hits.
Crosses with GAL1-KIN4 swr1∆ and swr1∆ were used to find out gene deletions that revert the growth rescue of KIN4 overexpression lethality by SWR1 deletion. To select the hits, we calculated the ratio between the median colony sizes of swr1∆ goi∆ and GAL1-KIN4 swr1∆ goi∆ on raffinose/galactose plates (day 3). Hits were considered as positives when “median colony size of swr1∆ goi∆ / GAL1-KIN4 swr1∆ goi∆ > 1,35” and “SDs < 0.2″ and “colony counts > 2.”
Analysis of checkpoint integrity
For measurement of SPOC integrity, log-phase kar9∆ cells cultured at 23°C were incubated at 30°C for 3–5 h. Cells were fixed using 70% ethanol, resuspended in phosphate-buffered saline containing 1 µg/ml 4′,6-diamino-2-phenylindole (DAPI, Sigma), and analyzed by microscopy; kar9∆ cells bearing GFP-TUB1 were fixed in 4% paraformaldehyde for 10 min at room temperature. Cells with normal and misaligned nuclei and cells with SPOC-deficient phenotypes (multiple nuclei in one cell body or single nuclei in a multibudded cell) were counted. At least 100 cells were counted per strain per experiment or time point. Analysis of each strain was repeated in three independent experiments. The SPOC deficiency index was calculated using the following formula;
SPOC deficiency index = (% SPOC-deficient phenotypes)/(% misaligned spindle) × 10.
SPOC deficiency index (normalized) = SPOC deficiency index of the kar9∆ strain bearing the indicated gene deletion – SPOC deficiency index of kar9∆ strain.
The functionality of the SAC was analyzed upon microtubule depolymerization by nocodazole treatment. Briefly, cells synchronized in G1 using alpha factor were released in nocodazole-containing media. Samples were collected for microscopy and protein extract preparation. Samples for microscopy were fixed in 70% ethanol and nuclei were stained with DAPI. The number of nuclei per cell and the budding status of at least 100 cells were recorded. Only the percentages of large budded and multibudded cells were plotted. The degradation of Pds1 was analyzed by immunoblotting.
Fluorescence microscopy
For time-lapse experiments, cells were adhered on glass-bottom dishes (MatTek, Ashland, MA) using 6% concanavalin A-Type IV (Sigma). Live-cell imaging were performed using a DeltaVision RT wide-field fluorescence imaging system (Applied Precision, Issaquah, WA) equipped with a quantifiable laser module, an OlympusIX71 microscope with plan-Apo 100× NA 1.4 oil immersion objective (Olympus, Tokyo, Japan), Photometrics CoolSnap HQ camera (Roper Scientific, Tucson, AZ), and SoftWoRx software (Applied Precision) as previously described (Caydasi and Pereira, 2009; Caydasi et al., 2014). Still images of living or fixed cells were acquired using a Zeiss Axiophot microscope equipped with a 100× NA 1.45 Plan-Fluor oil immersion objective (Zeiss, Jena, Germany), Cascade 1K CCD camera (Photometrics, Tucson, AZ), and MetaMorph software (Universal Imaging Corp., Chesterfield, PA). All images were processed in ImageJ (National Institutes of Health, Bethesda, MD), Adobe Photoshop CS3, and Adobe Illustrator CS3 (Adobe Systems, San Jose, CA). No manipulations were performed other than brightness, contrast, and color balance adjustments.
Calculation of anaphase duration
The anaphase duration of cells with correct and misaligned spindles was determined from a time-lapse series of GFP-TUB1 kar9Δ cells. Cells grown at 23°C were analyzed by live-cell imaging at 30°C for 1–1.5 h with 1-min time intervals. The time from the start of fast spindle elongation (metaphase-to-anaphase transition) until spindle breakdown was calculated as anaphase duration (Straight et al., 1997).
Protein methods
Yeast protein extracts and immunoblotting were performed as described (Janke et al., 2004). Antibodies were mouse anti-tubulin (TAT1, Sigma), mouse anti-HA (12CA5, Sigma), rabbit anti-Clb2, guinea pig anti-Sic1 (Maekawa et al., 2007), rabbit anti-Slt2/Mpk1 (SC-20168, Santa Cruz), and rabbit anti-Slt2-P (20G11, Cell Signaling). Secondary antibodies were goat anti-mouse, goat anti-rabbit, and goat anti-guinea pig IgGs coupled to horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA). In vitro kinase assays of immunoprecipitated Kin4-6HA was performed using MBP-Bfa1 purified from Escherichia coli as described previously (Maekawa et al., 2007; Geymonat et al., 2009; Caydasi et al., 2010a).
Microarray analysis of mRNA levels
The cdc15-as SWR1 GAL1-KIN4 and cdc15-as swr1∆ GAL1-KIN4 cells grown in raffinose (3%) containing medium were synchronized in G1 using alpha factor and released into 1NM-PP1 (2.5 µM) and raffinose (3%) galactose (2%) containing fresh medium. After both cultures were arrested in anaphase (percentage of cells with two DAPI staining >95%), cultures were pelleted, and pellets were washed with ice-cold dH2O and immediately frozen on dry ice. Duplicate RNA samples, extracted and purified with Qiagen RNeasy Mini kit followed by DNase I treatment, were labeled and hybridized after a single round of amplification to Affymetrix Yeast Genome 2.0 array chips essentially according to Affymetrix protocols. Microarray data analysis was performed using the limma package (Ritchie et al., 2015) implemented within the piano package (Varemo et al., 2013) in R. Robust Multichip Average was run with the affy package implemented within the piano package in R. Log2 (FoldChange) was calculated as the ratio of average log(expression) in SWR1 lacking and containing cells. Genes were considered up- or down-regulated in swr1∆ cells if Log(FoldChange) >0,5 or Log(FoldChange) ←0,5, respectively, and when p < 0.05.
GO analysis of biological process was performed using Yeastract (Monteiro et al., 2020) rank by GO function. GO biological process categories with a p < 0.05 were considered enriched when containing at least three genes.
Supplementary Material
Acknowledgments
We thank Astrid Hofmann and Dorothee Albrecht for excellent technical assistance and Elmar Schiebel for strains and access to microscopes. This work was funded by the Deutsche Forschungsgemeinschaft (DFG) Collaborative Research Center SFB1036 (Project TP21 to G.P. and Project TP10 to M.K. and A.K.). A.K.C. was supported by the German Research Council (DFG, PE1883/1-2), MSCA Individual Fellowship (796599, COHEMEX), and EMBO-IG (3918). The work of G.P. is supported by the Heisenberg program (PE1883/3), SFB873/A14 and SFB1324/B09 of the DFG.
Abbreviations used:
- CDK
cyclin dependent kinase
- CKI
cyclin dependent kinase
- CWI
Cell Wall Integrity
- FEAR
Cdc-fourteen early anaphase release
- GAP
GTPase-activating protein inhibitor
- MEN
mitotic exit network
- SAC
spindle assembly checkpoint
- SAGA
Spt-Ada-Gcn5 acetyltransferase
- SGA
synthetic genetic array
- SPOC
spindle position checkpoint
- SWR1-C
SWR1 chromatin remodeling complex.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-03-0179) on December 21, 2022.
REFERENCES
- Adames NR, Cooper JA (2000). Microtubule interactions with the cell cortex causing nuclear movements in Saccharomyces cerevisiae. J Cell Biol 149, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adames NR, Oberle JR, Cooper JA (2001). The surveillance mechanism of the spindle position checkpoint in yeast. J Cell Biol 153, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au TJ, Rodriguez J, Vincent JA, Tsukiyama T (2011). ATP-dependent chromatin remodeling factors tune S phase checkpoint activity. Mol Cell Biol 31, 4454–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin AJ, Visintin R, Amon A. (2000). A mechanism for coupling exit from mitosis to partitioning of the nucleus. Cell 102, 21–31. [DOI] [PubMed] [Google Scholar]
- Baro B, Queralt E, Monje-Casas F (2017). Regulation of mitotic exit in Saccharomyces cerevisiae. Methods Mol Biol 1505, 3–17. [DOI] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Dixon S, Vizeacoumar FJ, Myers CL, Andrews B, Boone C (2010). Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Methods Enzymol 470, 145–179. [DOI] [PubMed] [Google Scholar]
- Beach DL, Thibodeaux J, Maddox P, Yeh E, Bloom K (2000). The role of the proteins Kar9 and Myo2 in orienting the mitotic spindle of budding yeast. Curr Biol 10, 1497–1506. [DOI] [PubMed] [Google Scholar]
- Bloecher A, Venturi GM, Tatchell K (2000). Anaphase spindle position is monitored by the BUB2 checkpoint. Nat Cell Biol 2, 556–558. [DOI] [PubMed] [Google Scholar]
- Caydasi AK, Ibrahim B, Pereira G (2010a). Monitoring spindle orientation: Spindle position checkpoint in charge. Cell Div 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Khmelinskii A, Duenas-Sanchez R, Kurtulmus B, Knop M, Pereira G (2017). Temporal and compartment-specific signals coordinate mitotic exit with spindle position. Nat. Commun. 8, 14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Kurtulmus B, Orrico MI, Hofmann A, Ibrahim B, Pereira G (2010b). Elm1 kinase activates the spindle position checkpoint kinase Kin4. J Cell Biol 190, 975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Lohel M, Grunert G, Dittrich P, Pereira G, Ibrahim B. (2012). A dynamical model of the spindle position checkpoint. Mol Syst Biol 8, 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Micoogullari Y, Kurtulmus B, Palani S, Pereira G (2014). The 14-3-3 protein Bmh1 functions in the spindle position checkpoint by breaking Bfa1 asymmetry at yeast centrosomes. Mol Biol Cell 25, 2143–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Pereira G (2009). Spindle alignment regulates the dynamic association of checkpoint proteins with yeast spindle pole bodies. Dev Cell 16, 146–156. [DOI] [PubMed] [Google Scholar]
- Caydasi AK, Pereira G (2012). SPOC alert–when chromosomes get the wrong direction. Exp Cell Res 318, 1421–1427. [DOI] [PubMed] [Google Scholar]
- Chan LY, Amon A. (2009). The protein phosphatase 2A functions in the spindle position checkpoint by regulating the checkpoint kinase Kin4. Genes Dev 23, 1639–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Turkel N, Hemati N, Fuller MT, Hunt AJ, Yamashita YM (2008). Centrosome misorientation reduces stem cell division during ageing. Nature 456, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aquino KE, Monje-Casas F, Paulson J, Reiser V, Charles GM, Lai L, Shokat KM, Amon A. (2005). The protein kinase Kin4 inhibits exit from mitosis in response to spindle position defects. Mol Cell 19, 223–234. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ (1997). Phosphorylation and proteolysis: partners in the regulation of cell division in budding yeast. Curr Opin Genet Dev 7, 7–16. [DOI] [PubMed] [Google Scholar]
- Falk JE, Campbell IW, Joyce K, Whalen J, Seshan A, Amon A. (2016). LTE1 promotes exit from mitosis by multiple mechanisms. Mol Biol Cell 27, 3991–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappier L, Verrijzer CP (2011). Gene expression control by protein deubiquitinases. Curr Opin Genet Dev 21, 207–213. [DOI] [PubMed] [Google Scholar]
- Gerhold CB, Gasser SM (2014). INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends Cell Biol 24, 619–631. [DOI] [PubMed] [Google Scholar]
- Geymonat M, Spanos A, Sedgwick S (2009). Production of mitotic regulators using an autoselection system for protein expression in budding yeast. Methods Mol Biol 545, 63–80. [DOI] [PubMed] [Google Scholar]
- Geymonat M, Spanos A, Smith SJ, Wheatley E, Rittinger K, Johnston LH, Sedgwick SG (2002). Control of mitotic exit in budding yeast. In vitro regulation of Tem1 GTPase by Bub2 and Bfa1. J Biol Chem 277, 28439–28445. [DOI] [PubMed] [Google Scholar]
- Geymonat M, Spanos A, Walker PA, Johnston LH, Sedgwick SG (2003). In vitro regulation of budding yeast Bfa1/Bub2 GAP activity by Cdc5. J Biol Chem 278, 14591–14594. [DOI] [PubMed] [Google Scholar]
- Gruneberg U, Campbell K, Simpson C, Grindlay J, Schiebel E (2000). Nud1p links astral microtubule organization and the control of exit from mitosis. EMBO J 19, 6475–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT (1991). S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66, 507–517. [DOI] [PubMed] [Google Scholar]
- Hu F, Wang Y, Liu D, Li Y, Qin J, Elledge SJ (2001). Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 107, 655–665. [DOI] [PubMed] [Google Scholar]
- Huang J, Moazed D. (2003). Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes Dev 17, 2162–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Morgan DO (2000). Cdc14 activates cdc15 to promote mitotic exit in budding yeast. Curr Biol 10, 615–618. [DOI] [PubMed] [Google Scholar]
- Kapoor P, Bao Y, Xiao J, Luo J, Shen J, Persinger J, Peng G, Ranish J, Bartholomew B, Shen X (2015). Regulation of Mec1 kinase activity by the SWI/SNF chromatin remodeling complex. Genes Dev 29, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E (1999). Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15, 963–972. [DOI] [PubMed] [Google Scholar]
- Kobor MS, Venkatasubrahmanyam S, Meneghini MD, Gin JW, Jennings JL, Link AJ, Madhani HD, Rine J (2004). A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol 2, E131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocakaplan D, Karaburk H, Dilege C, Kirdok I, Bektas SN, Caydasi AK (2021). Protein phosphatase 1 in association with Bud14 inhibits mitotic exit in Saccharomyces cerevisiae. eLife 10, 72833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig C, Maekawa H, Schiebel E (2010). Mutual regulation of cyclin-dependent kinase and the mitotic exit network. J Cell Biol 188, 351–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono K, Al-Zain A, Schroeder L, Nakanishi M, Ikui AE (2016). Plasma membrane/cell wall perturbation activates a novel cell cycle checkpoint during G1 in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 113, 6910–6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutelou E, Hirsch CL, Dent SY (2010). Multiple faces of the SAGA complex. Curr Opin Cell Biol 22, 374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H, Haw RA, Pootoolal J, Tong A, Canadien V, et al. (2003). A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell 12, 1565–1576. [DOI] [PubMed] [Google Scholar]
- Lans H, Marteijn JA, Vermeulen W (2012). ATP-dependent chromatin remodeling in the DNA-damage response. Epigenet. Chromatin 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin DE (2011). Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics 189, 1145–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Yeh E, Hays T, Bloom K (1993). Disruption of mitotic spindle orientation in a yeast dynein mutant. Proc Natl Acad Sci USA 90, 10096–10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Woodfin AR, Slaughter BD, Unruh JR, Box AC, Rickels RA, Gao X, Haug JS, Jaspersen SL, Shilatifard A (2015). Mitotic Transcriptional Activation: Clearance of Actively Engaged Pol II via Transcriptional Elongation Control in Mitosis. Mol Cell 60, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HP, Bretscher A (1989). Disruption of the single tropomyosin gene in yeast results in the disappearance of actin cables from the cytoskeleton. Cell 57, 233–242. [DOI] [PubMed] [Google Scholar]
- Maekawa H, Priest C, Lechner J, Pereira G, Schiebel E (2007). The yeast centrosome translates the positional information of the anaphase spindle into a cell cycle signal. J Cell Biol 179, 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Padmanabha R, Snyder M (1997). The Rho-GEF Rom2p localizes to sites of polarized cell growth and participates in cytoskeletal functions in Saccharomyces cerevisiae. Mol Biol Cell 8, 1829–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneghini MD, Wu M, Madhani HD (2003). Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 112, 725–736. [DOI] [PubMed] [Google Scholar]
- Miller RK, Rose MD (1998). Kar9p is a novel cortical protein required for cytoplasmic microtubule orientation in yeast. J Cell Biol 140, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C (2004). ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303, 343–348. [DOI] [PubMed] [Google Scholar]
- Molk JN, Schuyler SC, Liu JY, Evans JG, Salmon ED, Pellman D, Bloom K (2004). The differential roles of budding yeast Tem1p, Cdc15p, and Bub2p protein dynamics in mitotic exit. Mol Biol Cell 15, 1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje-Casas F, Amon A. (2009). Cell polarity determinants establish asymmetry in MEN signaling. Dev Cell 16, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro PT, Oliveira J, Pais P, Antunes M, Palma M, Cavalheiro M, Galocha M, Godinho CP, Martins LC, Bourbon N, et al. (2020). YEASTRACT+: a portal for cross-species comparative genomics of transcription regulation in yeasts. Nucleic Acids Res 48, D642–D649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Chudalayandi P, Heil-Chapdelaine RA, Cooper JA (2010). The spindle position checkpoint is coordinated by the Elm1 kinase. J Cell Biol 191, 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillo-Huesca M, Clemente-Ruiz M, Andujar E, Prado F (2010). The SWR1 histone replacement complex causes genetic instability and genome-wide transcription misregulation in the absence of H2A.Z. PLoS One 5, e12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Kim JA, Person MD, Highland J, Xiao J, Wehr TS, Hensley S, Bao Y, Shen J, Collins SR, et al. (2007). Mec1/Tel1 phosphorylation of the INO80 chromatin remodeling complex influences DNA damage checkpoint responses. Cell 130, 499–511. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED (2007). The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8, 379–393. [DOI] [PubMed] [Google Scholar]
- Palozola KC, Donahue G, Liu H, Grant GR, Becker JS, Cote A, Yu H, Raj A, Zaret KS (2017). Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola KC, Liu H, Nicetto D, Zaret KS (2018). Low-Level, Global Transcription during Mitosis and Dynamic Gene Reactivation during Mitotic Exit. Cold Spring Harb Symp Quant Biol, 10.1101/sqb.2017.82.034280. [DOI] [PubMed] [Google Scholar]
- Pereira G, Hofken T, Grindlay J, Manson C, Schiebel E (2000). The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol Cell 6, 1–10. [PubMed] [Google Scholar]
- Pereira G, Schiebel E (2003). Separase regulates INCENP-Aurora B anaphase spindle function through Cdc14. Science 302, 2120–2124. [DOI] [PubMed] [Google Scholar]
- Pereira G, Schiebel E (2005). Kin4 kinase delays mitotic exit in response to spindle alignment defects. Mol Cell 19, 209–221. [DOI] [PubMed] [Google Scholar]
- Pereira G, Yamashita YM (2011). Fly meets yeast: checking the correct orientation of cell division. Trends Cell Biol, 10.1016/j.tcb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polevoda B, Cardillo TS, Doyle TC, Bedi GS, Sherman F (2003). Nat3p and Mdm20p are required for function of yeast NatB Nalpha-terminal acetyltransferase and of actin and tropomyosin. J Biol Chem 278, 30686–30697. [DOI] [PubMed] [Google Scholar]
- Rathi S, Polat I, Pereira G (2022). The budding yeast GSK-3 homologue Mck1 is an essential component of the spindle position checkpoint. Open Biol 12, 220203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JM, Amon A. (2009). The FEAR network. Curr Biol 19, R1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossio V, Galati E, Ferrari M, Pellicioli A, Sutani T, Shirahige K, Lucchini G, Piatti S (2010). The RSC chromatin-remodeling complex influences mitotic exit and adaptation to the spindle assembly checkpoint by controlling the Cdc14 phosphatase. J Cell Biol 191, 981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarfone I, Venturetti M, Hotz M, Lengefeld J, Barral Y, Piatti S (2015). Asymmetry of the budding yeast Tem1 GTPase at spindle poles is required for spindle positioning but not for mitotic exit. PLoS Genet 11, e1004938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K (1994). The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79, 233–244. [DOI] [PubMed] [Google Scholar]
- Shaltiel IA, Krenning L, Bruinsma W, Medema RH (2015). The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci 128, 607–620. [DOI] [PubMed] [Google Scholar]
- Sherman F (1991). Getting started with yeast. Methods Enzymol 194, 3–21. [DOI] [PubMed] [Google Scholar]
- Shou W, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen ZW, Jang J, Charbonneau H, Deshaies RJ (1999a). Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97, 233–244. [DOI] [PubMed] [Google Scholar]
- Shou W, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen ZW, Jang J, Shevchenko A, Charbonneau H, Deshaies RJ (1999b). Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97, 233–244. [DOI] [PubMed] [Google Scholar]
- Singer JM, Shaw JM (2003). Mdm20 protein functions with Nat3 protein to acetylate Tpm1 protein and regulate tropomyosin-actin interactions in budding yeast. Proc Natl Acad Sci USA 100, 7644–7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Visintin R, Amon A. (2002). Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell 108, 207–220. [DOI] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW (1997). Mitosis in living budding yeast: anaphase A but no metaphase plate. Science 277, 574–578. [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. (2001). Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368. [DOI] [PubMed] [Google Scholar]
- van Attikum H, Fritsch O, Gasser SM (2007). Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J 26, 4113–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varemo L, Nielsen J, Nookaew I (2013). Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res 41, 4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin C, Tomson BN, Rahal R, Paulson J, Cohen M, Taunton J, Amon A, Visintin R (2008). APC/C-Cdh1-mediated degradation of the Polo kinase Cdc5 promotes the return of Cdc14 into the nucleolus. Genes Dev 22, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Amon A. (2001). Regulation of the mitotic exit protein kinases Cdc15 and Dbf2. Mol Biol Cell 12, 2961–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O, Usaj M, Baryshnikova A, VanderSluis B, Kuzmin E, Costanzo M, Myers CL, Andrews BJ, Boone CM, Parts L (2013). SGAtools: one-stop analysis and visualization of array-based genetic interaction screens. Nucleic Acids Res 41, W591–W596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Burke DJ (1995). Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae. Mol Cell Biol 15, 6838–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss EL (2012). Mitotic exit and separation of mother and daughter cells. Genetics 192, 1165–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willhoft O, Wigley DB (2020). INO80 and SWR1 complexes: the non-identical twins of chromatin remodelling. Curr Opin Struct Biol 61, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. [DOI] [PubMed] [Google Scholar]
- Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K (1995). Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharomyces cerevisiae. J Cell Biol 130, 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Richardson DO, Roberts DN, Utley R, Erdjument-Bromage H, Tempst P, Cote J, Cairns BR (2004). The Yaf9 component of the SWR1 and NuA4 complexes is required for proper gene expression, histone H4 acetylation, and Htz1 replacement near telomeres. Mol Cell Biol 24, 9424–9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BO, Wang SS, Xu LX, Meng FL, Xuan YJ, Duan YM, Wang JY, Hu H, Dong X, Ding J, et al. (2010). SWR1 complex poises heterochromatin boundaries for antisilencing activity propagation. Mol Cell Biol 30, 2391–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.