

Abstract
Tetrahymena thermophila harbors two functionally and physically distinct nuclei within a shared cytoplasm. During vegetative growth, the “cell cycles” of the diploid micronucleus and polyploid macronucleus are offset. Micronuclear S phase initiates just before cytokinesis and is completed in daughter cells before onset of macronuclear DNA replication. Mitotic micronuclear division occurs mid–cell cycle, while macronuclear amitosis is coupled to cell division. Here we report the first RNA-seq cell cycle analysis of a binucleated ciliated protozoan. RNA was isolated across 1.5 vegetative cell cycles, starting with a macronuclear G1 population synchronized by centrifugal elutriation. Using MetaCycle, 3244 of the 26,000+ predicted genes were shown to be cell cycle regulated. Proteins present in both nuclei exhibit a single mRNA peak that always precedes their macronuclear function. Nucleus-limited genes, including nucleoporins and importins, are expressed before their respective nucleus-specific role. Cyclin D and A/B gene family members exhibit different expression patterns that suggest nucleus-restricted roles. Periodically expressed genes cluster into seven cyclic patterns. Four clusters have known PANTHER gene ontology terms associated with G1/S and G2/M phase. We propose that these clusters encode known and novel factors that coordinate micro- and macronuclear-specific events such as mitosis, amitosis, DNA replication, and cell division.
INTRODUCTION
Ciliates have provided a wealth of information on the structure and function of eukaryotic chromosomes, including DNA replication, telomeres and telomerase, and epigenetic regulation, including a noncoding RNA pathway that controls gene expression through programmed DNA elimination (Yao and Chao, 2005; Kataoka and Mochizuki, 2011; reviewed in Karrer, 2012; Bracht et al., 2013). As a prototypic ciliate, Tetrahymena thermophila harbors two functionally distinct nuclei—the micronucleus and the macronucleus—within the same cytoplasm. The diploid, germline micronucleus contains genetic material transmitted from parent to progeny during conjugation. It undergoes meiosis and mitosis; however, micronuclear chromosomes are never transcribed into mRNA. Posttranslational histone modifications package micronuclear chromosomes into constitutive heterochromatin. The cellular phenotype is conferred by the polyploid “somatic” macronucleus, where euchromatic histone modifications abound.
Upon starvation, cells of different mating types pair and produce four haploid gametic pronuclei, three of which undergo programmed nuclear death (Davis et al., 1992; Yakisich and Kapler, 2004). The surviving nucleus replicates and divides, and haploid pronuclei are reciprocally exchanged between mating partners. The resulting diploid zygotic nucleus duplicates, and one of the products differentiates into a macronucleus. During macronuclear development the genome is extensively reorganized (reviewed in Karrer, 2012). The five T. thermophila chromosomes undergo sequence-specific fragmentation, producing 180 non–ribosomal DNA (rDNA) chromosomes that are capped by telomeres and endoreplicate to ∼45C (chromosome copy number). Repetitive DNA is removed by DNA breakage and religation. The 21 kb rDNA minichromosome is amplified from 2 to 9000C. Finally, the “parental” macronucleus is destroyed and progeny propagate vegetatively (Liang et al., 2019, and references therein). Macronuclear chromosomes lack centromeres, and sister chromatids randomly assort during “amitosis,” a poorly understood process that maintains total DNA mass and copy number within a narrow range (Doerder et al., 1992; Wong et al., 2000).
To accommodate the different roles of the micro- and macronuclear chromosomes, nuclear proteins must be differentially trafficked. However, shared requirements must still be met, such as chromosome packaging into nucleosomes, DNA replication, and DNA repair. Micro- and macronuclear DNA replications utilize evolutionarily conserved replication machinery, including the origin recognition complex (ORC), the MCM2-7 replicative helicase, and other replication enzymes (Mohammad et al., 2007; Donti et al., 2009). However, micro- and macronuclei replicate at different times during the vegetative cell cycle (Allis et al., 1987). Macronuclear S phase initiates before micronucleus S, the latter of which begins before cytokinesis and is completed in daughter cells. The relative timing of nuclear division is flipped—micronuclear mitosis precedes amitotic macronuclear division (Cui and Gorovsky, 2006; Jacob et al., 2007; Cole and Sugai, 2012).
Tetrahymena’s developmental and vegetative cell cycles must employ novel regulatory pathways to coordinate the fate of chromosomes in functionally distinct nuclei. Consistent with this premise, T. thermophila encodes 34 predicted cyclins and 20 predicted cyclin-dependent kinases (CDKs) (Stover and Rice, 2011; Yan et al., 2016; Ma et al., 2020), a subset that are expressed only in mating cells (Ma et al., 2020). To gain insight into the vegetative cell cycle of Tetrahymena, we used centrifugal elutriation to profile gene expression across 1.5 vegetative cell divisions. We report the first transcriptome analysis of the vegetative cell division cycle of a binucleate ciliated protozoan and relate our finding to other eukaryotes, including yeast and mammals.
RESULTS
RNA-seq analysis across the Tetrahymena cell cycle
Previous studies indicated that centrifugal elutriation was best suited for RNA-seq analysis across the vegetative cell cycle of Tetrahymena (Liu et al., 2021, and references therein). Starvation and refeeding generates a significant lag in cell cycle progression as cells exit nutrient-deprived G0 phase (Sandoval et al., 2015). Double synchronization by sequential starvation + hydroxyurea (HU) arrest or elutriation + HU activates an unconventional DNA damage checkpoint response that includes degradation of pre–replicative complex components, increased side scatter in flow cytometry profiles, and significant compaction of the macronucleus (Lee et al., 2015; Sandoval et al., 2015). Microtubule inhibitors do not discriminate between micronuclear mitosis and macronuclear amitosis—events that occur at different stages of the vegetative cell cycle. Hence, a single round of centrifugal elutriation was used to generate a synchronized population of macronuclear G1 cells with minimal perturbation of cell physiology. This method provided sufficient cell numbers for downstream RNA-seq library preparation and analysis of cell cycle landmarks.
Owing to the complexity of the Tetrahymena cell cycle, involving two nuclei with offset S phases, and quantified level of synchrony, samples were isolated at the shortest possible intervals to maximize our ability to identify cycling transcripts. Two biological replicates were performed on samples collected every 30 min over 4 h (Ly et al., 2015). RNA was isolated for high-throughput sequencing, yielding 18 cDNA libraries generated with minimal cDNA amplification (two libraries × nine time points). With a doubling time of ∼3 h, the time points spanned two macronuclear G1 phases (Figure 1A). Flow cytometry was used to assess total DNA content (Figure 1A), and 5-ethylene-2′-deoxyuridine (EdU) labeling monitored DNA replication (Figure 1B; Supplemental Figure S1A), while 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI) fluorescence microscopy visualized micronuclear chromosome condensation, micro- and macronuclear division, and cytokinesis in individual cells (Supplemental Figure S1B). EdU labeling revealed that ∼70% of macronuclei were actively replicating in the 90 min peak population (Figure 1B; Supplemental Figure S1A) (Magiera et al., 2014). Micronuclear mitosis (peak at 120 min) (Supplemental Figure S1B) was immediately followed by micronuclear S phase (Figure 1B; Supplemental Figure S1A), which was completed in daughter cells before the onset of the second macronuclear S phase (210 min). The elutriation time course starting point (T = 0 min) contained cells that were EdU positive in either micro- or macronuclei, indicating that the initial cell population was not purely homogeneous. Micronuclear labeling decreased and macronuclear labeling increased with time in accordance with the previously reported offset nuclear S phases (Cole and Sugai, 2012, and references therein). Importantly, none of the 3216 EdU-positive cells exhibited colabeling of the micro- and macronucleus. These data argue for discrete, temporally separated S phases. Finally, macronuclear amitosis coincides with cytokinesis (both peaking around 150–180 min) (Supplemental Figure S1B).
FIGURE 1:

Validation of cell cycle synchrony by centrifugal elutriation. (A) Cells isolated at 30 min intervals after elutriation were stained with propidium iodide (PI) and analyzed by flow cytometry (adjacent lanes correspond to biological replicates). (B) EdU incorporation into micro- and macronuclei following centrifugal elutriation as determined by fluorescence microscopy of fixed cells. (C) Summary timeline for macro- and micronuclear cell cycle progression. (D) Pipeline of sample processing for RNA-seq and cytological landmark determination. (E) Heatmap of cell cycle–regulated genes. Periodically expressed genes identified by MetaCycle for genes having 1.5-fold changes in two opposite directions.
The collective data, summarized in the Figure 1C timeline, indicate that initiations of three of the four nuclear cycle events—micronuclear DNA replication, macronuclear DNA replication, and mitosis—are temporally separated, raising the possibility that cross-talk might coordinately regulate these nuclear events. In contrast, micronuclear DNA replication and macronuclear amitosis overlap. The macronuclear cell cycle consists of four phases—G1/S/G2/amitosis—while the micronuclear cell cycle has three phases—S/G2/M.
Identification of cell cycle–regulated genes
To gain insight into cell cycle control of gene expression, 18 cDNA libraries (nine per time course) were sequenced on an Illumina HiSeq 2500, resulting in ≥45 million paired end reads (43× coverage/library) with an average length of 100 base pairs (Figure 1D). The paired end reads were aligned to the T. thermophila genome (2021 version, Tetrahymena Genome Database [TGD] [http://ciliate.org/index.php/home/downloads]) (Sheng et al., 2020), and the abundance of individual transcripts was computed in each sample using the HISAT2 and StringTie (Pertea et al., 2016). In all samples ≥95.5% of the reads aligned to the macronuclear genome sequence. The reads mapped to 19,946 of the 26,259 predicted T. thermophila genes. Quantification and statistical inference of systematic changes between time points were computed by DESeq2 (Love et al., 2014). MetaCycle—an N-version programming method to explore periodic data—was used to identify cell cycle–regulated genes (Wu et al., 2016). MetaCycle implements JTK_CYCLE (JTK) and Lomb-Scargle (LS) and integrates their results. The LS method, developed by astrophysicists as a Fourier-style method for analyzing data that exhibit irregular sampling, measures the correspondence to sinusoidal curves and determines their statistical significance (Glynn et al., 2006). JTK, with its origins in statistics (Hughes et al., 2010), correlates pairs of points and then computes the significance of the correlation to that of a reference curve. Because genes with subtle changes in expression are unlikely to be biologically significant, we screened for genes with a minimum of two 1.5-fold changes in opposite directions between the two most extreme time points (p value < 0.05) using the logarithmic fold change results by DESeq2. This analysis identified 3864 candidate genes (Figure 1E; Supplemental Datafile 1). Using MetaCycle, 16% of the DESeq2 candidates were removed from further consideration. A total of 3244 genes exhibited periodic expression profiles with false discovery rate (FDR) < 0.05. The cycling genes constitute ∼11% of the total mapped genes of known or hypothetical function and ∼15% of genes expressed during the vegetative phase of the Tetrahymena life cycle.
The results of this analysis were validated by quantitative reverse transcriptase PCR (qRT-PCR) for six genes known or anticipated to be cell cycle regulated (Supplemental Figure S2). qRT-PCR was carried out with the same RNA preparations that were sequenced; it validated the RNA-Seq data for all examined samples. Some genes showed modest quantitative variation between biological replicates but displayed similar patterns of expression over the time course.
Cell cycle–regulated nucleus-specific genes of known function
The specialized roles of the micro- and macronucleus are reflected in the composition of proteins that must be selectively imported into the respective nuclei. With this in mind, we looked for evidence of cell cycle regulation at the mRNA level of nucleus-specific proteins.
Histones.
One of the most pronounced examples of cell cycle–regulated gene expression in eukaryotes is the histone gene family members (reviewed in Marzluff et al., 2008, and in MacAlpine and Almouzni, 2013). Timing and abundance are critical, as excess histones can drive genome instability (Gunjan and Verreault, 2003). In the case of Tetrahymena, micro- and macronuclear nucleosomes share common core subunits but differ in the composition of histone variants and posttranslational modifications (PTMs) that influence chromosome compaction and gene expression. The major core histones are present in both nuclei (Allis et al., 1980; Hayashi et al., 1984). RNA-seq profiles revealed that all core histone genes (HH2A.1, HH2A.2, HH2B.1, HH2B.2, HH3, HH4.1, and HH4.2) are cell cycle regulated with a single peak in mRNA abundance during macronuclear S phase (Figure 2A; 90 min).
FIGURE 2:

Cell cycle–regulated histones and chromatin modifier genes. (A) Expression profile of core histone genes. (B) Expression profile of macronuclear-specific histone variants. (C) Expression profile of the micronuclear-specific, centromere-associated histone H3 variant, CNA1. (D) Expression profiles of macro- and micronuclear-specific H1 histones (dashed line: MAC specific; dotted line: MIC specific). (E) Cell cycle–regulated histone modifiers (see the text for gene names).
Variant histones H2A.Z, H3.3, and H3.4 reside exclusively in the macronucleus (Henikoff and Smith, 2015). H2A.Z mRNA peaked during macronucleus S phase (Figure 2B; 90 min), while H3.3 and H3.4, which serve as replacement histones after transcription-associated nucleosome removal (Yu and Gorovsky, 1997; Cui et al., 2006), were constitutively expressed. Expression of the human CenpA homologue, CNA1, which functions as the micronuclear centromere-specific histone H3 variant (Cervantes et al., 2006; Cui and Gorovsky, 2006), was cell cycle regulated and peaked during micronuclear mitosis (Figure 2C; 120 min).
The micro- and macronucleus contain different histone H1 linker proteins: micronuclear linker histone MLH1 and macronuclear linker histone HHO1 (Allis et al., 1984; Wu et al., 1986; Hayashi et al., 1987). HHO1 is not essential for cell viability but is critical for full macronuclear chromatin compaction and maintenance of transcriptional regulation (Shen et al., 1995; Shen and Gorovsky, 1996). Whereas both linker histone mRNA levels are cell cycle regulated, they exhibited different expression patterns. HHO1 mRNA peaked during macronuclear S phase (Figure 2D; 90 min), and MLH1 peaked at the onset of micronuclear S phase (Figure 2D; 180 min).
Histone chaperones serve multiple roles, including deposition of histones onto newly replicated DNA, recruitment of chromatin modifiers, disassembly of nucleosomes, and replacement of damaged histones (reviewed in De Koning et al., 2007; Nabeel-Shah et al., 2021). The Tetrahymena N1/N2-related histone chaperone, NRP1 (Ttherm_01014770), is required for chromatin stability and propagation of both micro- and macronuclear chromosomes (Lian et al., 2021). NRP1 elicited a single cell cycle–regulated oscillation of mRNA, peaking before bulk histone mRNA production (30–60 min postelutriation, premacronuclear S phase; Supplemental Datafile 1). Similar results were obtained for the chromatin assembly factor 1A subunit (Ttherm_00309890) (Supplemental Datafile 1), which assembles nucleosomes onto newly synthesized DNA during S phase.
Histone-modifying enzymes.
Micro- and macronuclear histones are subjected to different PTMs (Vavra et al., 1982). Histone PTMs influence many nuclear processes, including transcription, DNA replication, and DNA repair (reviewed in Lawrence et al., 2016). The micronucleus is enriched for H3K27me3 and completely lacks the euchromatic mark H3K4me3 (Liu et al., 2007; Taverna et al., 2007). The macronucleus exclusively contains H3K27me3, H3K4me3, and H3K27me1 (Gao et al., 2013; Papazyan et al., 2014). During vegetative growth, histone H3 acetylation (Chalker et al., 2013) and deacetylation (Wiley et al., 2005) and Txr1p-catalyzed H3K27 monomethylation occur in the macronucleus (Gao et al., 2013), whereas H3K23 is trimethylated in the micronucleus (Papazyan et al., 2014).
The expressions of genes encoding five histone-modifying enzymes are cell cycle regulated. mRNA levels for the two histone acetyltransferases, HAT1 and HAT2, associated with transcription activation, peaked in early macronuclear S phase (Figure 2E; 60 min), consistent with their roles in DNA replication and repair (Masumito et al., 2005; Kurat et al., 2014; Almouzni and Cedar, 2016). By analogy, Saccharomyces cerevisiae HAT1 and HAT2 mRNAs peak in G1 and M phase, respectively (https://cyclebase.org/), and mammalian HAT1 and HAT2 mRNAs are not periodically expressed. mRNA for macronuclear-specific TXR1 peaked sharply during macronuclear G1 phase (Figure 2E; 30 min). The TXR1 homologues in Arabidopsis thaliana, ATXR5 and ATRX6, are also periodically expressed (Raynaud et al., 2006). Two of the three Drosophila Enhancer of Zeste (EZ) homologues are also cell cycle regulated in Tetrahymena. Histone methyltransferase genes EZL1, EZL2, and EZL3 are responsible for H3K27 di- and trimethylation. Whereas TXR1p is macronuclear limited, EZL2p and EZL3p are not. EZL2p mediates di- and trimethylation of H3K27 in early conjugants and vegetative growing cells (Gao et al., 2013; Zhang et al., 2013). It is required for global transcriptional silencing in the micronucleus during vegetative growth and localized repression of gene expression in the macronucleus. The role of EZL3p is less well defined. EZL2 and EZL3 mRNA levels are cell cycle regulated; they produced broad peaks that spanned late micronuclear S phase and macronuclear S phase (Figure 2E; 30–120 min). Consistent with the role of EZL1p in heterochromatin formation and DNA elimination in the developing macronucleus (Liu et al., 2007; Xu et al., 2021), EZL1 expression was low in vegetative cells and did not oscillate.
Other examined histone modification enzyme genes are not periodically expressed in Tetrahymena. Histone H3 lysine 4 (H3K4) methylation is mainly catalyzed by MLL1p and is associated with transcription activation (Cao et al., 2010). MLL1 is not periodically expressed. Similarly, genes responsible for macronuclear histone H3 demethylation (THD1, SIN3/TTHERM_00450950, RXT2/TTHERM_00992830, SAP30, PHO23/TTHERM_000046389, SAP18/TTHERM_00469050) and histone H2 monoubiquitinylation (RIN1/TTHERM_00263030; UB3 ligase) are not cell cycle regulated (Zhang et al., 2014; Nabeel-Shah et al., 2021). However, RebL1 (Ttherm_0068660)—a component of many chromatin-associated complexes—is cell cycle regulated. With its expected role in the control of macronuclear gene expression, it is worth noting that RebL1 peaked in macronuclear G1 cells (Supplemental Datafile 1; 30–60 min postelutriation). Whereas RebL1 expression is periodic, its human homologues, RBBP4 and RBBP7, are not (https://cyclebase.org/).
Condensins and cohesins.
The structural maintenance of chromosomes (SMC) complex plays a central role in chromosome segregation. Its subunits and their nuclear localization have been well studied in Tetrahymena (Uhlmann, 2016). Tetrahymena has a reduced set of SMC complexes, containing a single cohesion that resembles the meiotic cohesin of other eukaryotes, and two heterodimeric condensin I complexes that are related to condensins that mediate metaphase compaction of yeast and higher eukaryotic chromosomes (Howard-Till et al., 2019). T. thermophila condensin I complexes are targeted to either the micro- or the macronucleus and are distinguished by their kleisin/Gph subunit. Micronuclear condensins contain CPH1p and CPH2p, whereas macronuclear condensins contain CPH3p and CPH4p. CPH5p localizes to the macronucleus during development. All condensin genes are cell cycle regulated except CPH5p, which has a critical (possibly primary) role in mating cells. Consistent with previous research (Howard-Till et al., 2019), mRNA levels for the two micronuclear kleisin genes, CPH1 and CPH2, peak at 120 min, corresponding to mitosis, and their expression patterns differ from that of the macronuclear condensin subunits (Figure 3A). These data suggest that the duplicated genes may have diverged to assume nucleus-specific roles. Consistent with this idea, genes encoding subunits that are present in both the micro- and macronucleus are cell cycle regulated (Figure 3A; CPD1, CPD2, and CPG3 and SMC2 and SMC4).
FIGURE 3:
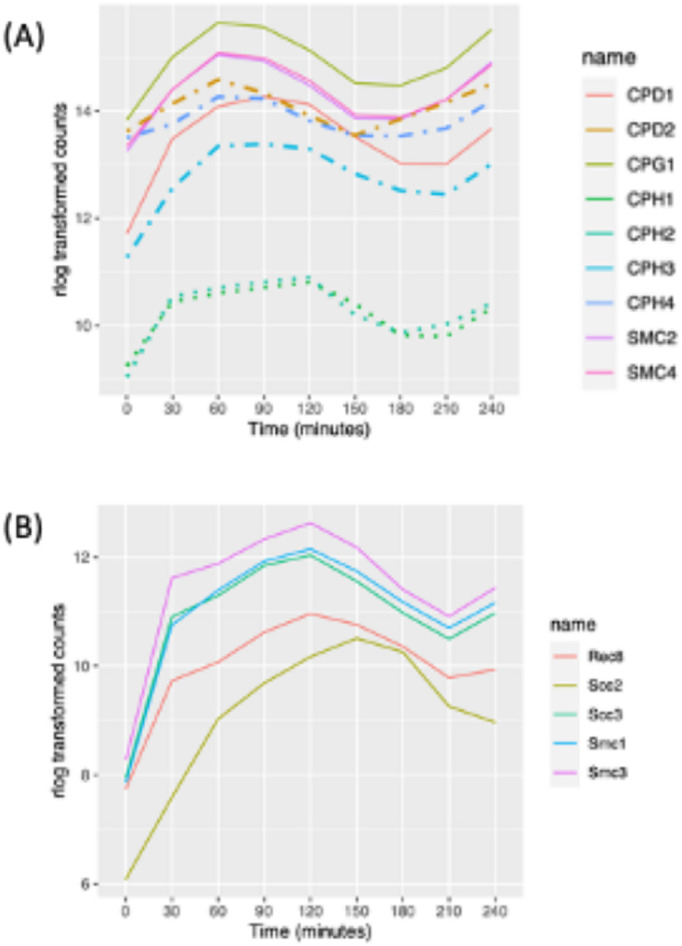
Expression profile of cohensin and condensing proteins. (A) Expression profile of cell cycle–regulated condensin genes (dashed lines: macronuclear specific; dotted lines: micronuclear specific; solid line: present in both nuclei). (B) Expression profile of cell cycle–regulated cohesin genes (all micronuclear specific).
Tetrahymena has a minimal cohesin complex consisting of SMC1p, SMC3p, SCC3p, and REC8p that localizes specifically to the micronucleus (Ali et al., 2018). This complex performs all necessary functions for mitosis and meiosis. SCC2p, which in other organisms is part of a heterodimeric complex (Scc2p/Scc4p) that helps load cohesin onto chromatin, is not required for chromosomal association of cohesin, but Tetrahymena Rec8p is hypophosphorylated in its absence (Ali et al., 2018). Consistent with the random segregation of sister chromatids in the amitotic macronucleus, cohesins are not targeted to the macronucleus. Peak mRNA levels for all of the cohesin subunits occur during micronuclear mitosis. Cohesin subunit gene expression patterns cluster closely together (Figure 3B; 120 min), except for SCC2, whose mRNA peaked slightly later (Figure 3B; 150 min).
Nuclear pore components.
A potential mechanism for the partitioning of micro- or macronuclear-limited nuclear proteins is selective nuclear import. In support of this, gene duplication and subfunctionalization has generated four micronuclear-specific, five macronuclear-specific, and one shared nuclear pore or nuclear transmembrane proteins (MicNup98Ap, MicNup153p, MicNup214p, MicPom82p; MacNup98Ap, MacNup98Bp, MacNup153p, MacNup214p, MacPom121p; shared Nup85p). Minimally, there are 16 additional shared protein subunits of micro- and macronuclear pores (Iwamoto et al., 2009, 2017). Our data revealed that the micronucleus-specific nucleoporin genes (MicNup98A, MicNup214, and MicNup153) and the micronuclear transmembrane protein, MicPom82, are cell cycle regulated (Figure 4A). MicNup153 expression peaked at micronucleus S/G2 phase, while MicPom82 and MicNup214 peaked at micronuclear S phase. MicNup98A peaked at micronucleus mitosis. In contrast, none of the macronuclear-specific or shared pore protein subunit or transmembrane protein genes were cell cycle regulated. In sharp contrast, yeast and higher eukaryotic pore proteins production is not cell cycle regulated.
FIGURE 4:

Expression profile of macro- or micronuclear-specific nucleoporin genes. (A) Cell cycle–regulated nucleoporins (dotted lines: micronuclear specific, solid lines: present in both nuclei). (B) Cell cycle–regulated expression of macronuclear- and nonnuclear-specific importin-α genes; IMA1 (macronuclear specific), IMA6, and IMA9. (C) Cell cycle–regulated expression of micronuclear importin-α genes. (D) Phylogenetic tree analysis of importin-α genes. (E) Cell cycle–regulated β-like importin proteins.
Importins and exportins.
Trafficking of proteins into the nucleus is mediated by the interaction between nuclear localization signals (NLSs) in cargo proteins and chaperones, termed importins (reviewed in Wing et al., 2022). Importins α and βform heterodimers to transport cargoes through the nuclear pore. Tetrahymena encodes 10 importinαs, one that localizes to the macronucleus (IMA1) and nine that localize to the micronucleus (Malone et al., 2008). In addition, there are two importin-related proteins of unknown function, IMA6p and IMA9p. Macronuclear-associated IMA1 was expressed at much higher levels than its micronuclear counterparts but did not meet the criteria for cell cycle regulation (Figure 4B). In contrast, six of the micronuclear importin genes, IMA3, IMA5, IMA8, IMA10, IMA12, and IMA13, are cyclically expressed. Their mRNAs exhibit different profiles that in general peak in the 30–90 min window (Figure 4C). IMA10, with peak expression at the micronuclear G2/M transition, is an essential gene that is required for chromosome segregation during mitosis and meiosis (Malone et al., 2008). Whereas IMA2 and IMA11 expression profiles met the 1.5-fold cyclic change in abundance, they were not detected by MetaCycle.
Phylogenetic tree analysis supports two importin-α gene family groupings: IMA1, IMA2, IMA3, IMA8, IMA11, and IMA13 constitute one group. Our data show that their cell cycle–regulated genes all peak during the micronuclear S/G2 window. IMA4, IMA5, IMA10, and IMA12 form the second group, and their expression peaks at micronuclear G2/M (Figure 4D) (Dereeper et al., 2008). The different importin-α expression profiles, their phylogenetic relationships, and functional data suggest that these gene family members serve nonredundant roles in protein trafficking. Finally, importin-α–related genes of unknown function, IMA6 and IMA9, are both cell cycle regulated (Figure 4E).
T. thermophila encodes 11 importin-β proteins, none of which is nucleus specific (Malone et al., 2008). IMB4 and IMB8 have a pronounced macronuclear bias. IMB2 protein accumulates in the cytoplasm in a GFP-tagged overexpression strain, while IMB6 localizes to both the micro- and macronucleus (Malone et al., 2008). Only IMB2 and IMB6 were cell cycle regulated (Figure 4E).
Identification of potential nucleus-specific cell cycle regulatory proteins
Major drivers of eukaryotic cell cycle progression include cyclins, CDKs, and E2F transcription factors. These genes are typically regulated at multiple levels, including transcription (Harashima et al., 2013; Malumbres, 2014). To identify potential micro- and macronucleus-specific cell cycle regulators, we determined the expression profiles of predicted cyclin, CDK, and E2F mRNAs and referenced them to the known function of yeast and mammalian homologues.
Tetrahymena encodes at least 34 predicted cyclin genes (Stover and Rice, 2011; Yan et al., 2016). Phylogenetic analysis suggests that 11 are cyclin D orthologues and 10 are members of the cyclin A/B family (Stover and Rice, 2011). Cyclin D regulates the G1/S transition (Grana and Reddy, 1995). Our RNA-seq analysis uncovered cell cycle regulation of four cyclin D transcripts. CYC7, CYC12, and CYC22 mRNAs peaked just before the macronuclear G1/S transition (Figure 5A; 30 min), while CYC14 mRNA was most abundant before the onset of micronuclear S phase (Figure 5A; 120 min). CYC3 and CYC26 did not exhibit a 1.5-fold change in opposite directions at two time points but met the MetaCycle criteria for oscillating genes, peaking at 180 min (micronuclear S phase). CYC9 and CYC2 mRNA levels were low and did not oscillate in vegetative growing cells, consistent with their previously defined roles during development (Yan et al., 2016; Ma et al., 2020) (Supplemental Figure S3A). The remaining cyclin D family members, CYC4, CYC13, and CYC25, were constitutively expressed during vegetative growth.
FIGURE 5:

Oscillating cyclin, CDK, and E2F mRNA abundance profiles. (A) Cell cycle–regulated expression of cyclin D gene family members. (B) Cell cycle–regulated expression of cyclin A/B genes. Solid lines: pattern 1 for cyclin A/B gene family members; dotted lines: pattern 2 for cyclin A/B gene family members. (C) Cell cycle–regulated CDK genes. (D) Cell cycle–regulated E2Fs. Solid line: potential E2F activator; dotted lines: potential E2F repressors and atypical E2F with two DNA-binding domains.
Cyclin A/B family members regulate the S/G2 or G2/M transitions (Grana and Reddy, 1995). Predicted Tetrahymena cyclin A/B gene family members exhibited two distinct oscillating patterns. mRNA levels for CYC1, CYC6, CYC20, and CYC24 peaked during macronuclear amitosis and cytokinesis, when the micronucleus is in S phase (Figure 5B; 150 and 180 min; Supplemental Figure S3B). A similar oscillation was observed for CYC18 by MetaCycle, but the 1.5-fold change threshold was not met. In contrast, the CYC15 and CYC8 (and possibly CYC10) peak levels coincided with micronuclear mitosis (Supplemental Figure S3B; 120 min). CYC11 was not cell cycle regulated, although its expression level was comparable to that of other A/B family members. As expected from previous functional studies (Yan et al., 2016), the meiosis-specific A/B cyclin gene, CYC17, was constitutively expressed at very low levels during vegetative growth.
Twenty-two putative CDK/CDK-like genes have been identified in the T. thermophila (Ma et al., 2020). Five of these genes are cell cycle regulated at the mRNA level. CDK1 mRNA was highest at 180 min (Figure 5C; late micronuclear S phase/macronuclear division/cytokinesis). CDK5, CDK10, CDK14, and CDK20 exhibited similar broad oscillating peaks (Figure 5C), and the remaining CDKs were constitutively expressed (Supplemental Figure S3C). CDK19 expression was very low in vegetative cells, consistent with its conjugation-specific role (Ma et al., 2020).
Gene predictions identified seven putative T. thermophila E2F gene family members (Zhang et al., 2017). E2F transcriptional activators drive expression of S phase–specific genes, increasing in G1 and peaking during S phase. E2F repressors have the opposite effect and peak during G2 phase (Thurlings and de Bruin, 2016). Tetrahymena E2FL1 and E2FL2 have conserved E2F/DP family winged-helix DNA-binding and CC-MB domains, features of transcriptional activators. E2FL3 and E2FL4 are most closely related to the mammalian repressors, E2F7 and E2F8. E2FL1 expression was constitutively low in vegetative growing cells, consistent with its role in meiosis (Supplemental Figure S3D) (Zhang et al., 2017). E2FL2 mRNA peaked at macronuclear G1 phase, suggesting that it is involved in activation of S phase–specific genes (Figure 5D; 30 min). The putative repressor gene E2FL3 also peaked at 30 min, while E2FL4 peaked at 150 min, corresponding to exit from macronuclear S phase. DPL1, DPL2, and DPL3 did not meet the criteria for cell cycle regulation; however, their expression profiles tracked with E2FL2 (Supplemental Figure S3D).
In mammalian cells, MuvB complexes interact with E2F4/5, DP1/2, and an Rb-like protein to generate transcriptional repressor complexes that inhibit expression of cell cycle–regulated genes (Litovchick et al., 2007; Sadasivam and DeCaprio, 2013). In contrast to the predicted Tetrahymena E2F repressors (E2FL3 and E2FL4), none of the T. thermophila paralogues and Reb1 (Rb-like)-associated proteins identified by a proteomic-based approach (Nabeel-Shah et al., 2021) is cell cycle regulated at the level of transcription (RebL1, Lin9, Lin54, Anqa1, Jinn1, Jinn2). The collective data predict that distinct master regulators control cell cycle transitions for micro- and macronuclear S phases, mitosis, and amitosis, analogous to S and M phase control in yeast and metazoa.
Periodically expressed gene clusters
Using the noise-robust soft clustering algorithm (Futschik and Carlisle, 2005), periodically expressed genes could be parsed into seven clusters, of which four have known PANTHER gene ontology (GO) biological process overrepresentation (Figure 6A). The other three clusters have unknown biological functions. The latter result is not unexpected, because only 57% of the cell cycle–regulated T. thermophila genes have assigned GO biological processes (1742 out of 3032). mRNA abundance for genes in cluster 7 was maximal during macronuclear G1 and S phase. Cluster 5 genes were maximally expressed during macronuclear S and G2 phase. Genes in cluster 1 were maximally expressed during macronuclear G2 and amitosis. Genes in cluster 2 were maximally expressed during amitosis and cytokinesis.
FIGURE 6:

Cluster and GO enrichment analysis. (A) Clusters of seven distinct periodically gene expression profiles and four subclusters (5A–D). (B) Overrepresented GO biological processes of four clusters. Overrepresented GO biological processes of subclusters within cluster 5. (C) Gene expression profiles of DNA replication protein-coding genes within cluster 7. Gene expression profile of mitotic genes within cluster 1. Gene expression profile of mitotic genes of subcluster 5C. Gene expression profile of mitotic and cell cycle checkpoint genes of subcluster 5D. (D) Coregulation analysis of the MCM6 mRNA cell cycle profile: top 10 coregulated genes.
Cluster 7.
GO overrepresentation within the 531 gene cluster 7 (macronuclear G1/S, late micronuclear S) showed a highly significant enrichment for processes linked with S phase, including DNA replication (P = 1.00E-20), DNA repair (P = 1.03E-18), regulation of the G1/S transition (P = 0.04), and histone modifiers (P = 3.36E-3) (Figure 6B; Supplemental Datafile 2). Genes in cluster 7 include proteins involved in DNA replication, such as various polymerases (POL1, POL12, POL2, POL31, POLD1), proliferating cell nuclear antigen (PCNA1), and other proteins necessary to initiate DNA synthesis (MCM2-6, CDT1, DNA Replication Factor 1 [RFA1], CDC45, and RFC1-5) (Figure 6C). DNA repair and DNA damage recognition factors known to cooperate in DNA replication were also identified, including RAD complex components (RAD50, RAD51, RAD54), MCM8, MCM9, BRCA1, and BRCT2. Importantly, genes that regulate or drive the G1/S transition, including cyclin D and E2F homologues (CYC12, CYC22, E2FL2, and E2FL4), reside in cluster 7. CDC6 and ORC1 displayed two 1.5-fold changes at two opposite directions but failed to be detected by MetaCycle; their expression level peaked at 30 min but did not display a clear peak at the second G1 phase. This may be due to the lack of synchrony at later time points (Supplemental Figure S4). It is worth noting that cluster 7 also contains a putative cell cycle checkpoint serine–threonine kinase, the DUN1 homologue (TTHERM_000851660), which is required for transient G2/M arrest after DNA damage, CDC14, which is required for mitotic exit, and the non-SMC mitotic condensation complex subunit, CPD2. Mitosis-related genes in this cluster may function in the G2/M phase transition of the germline micronucleus.
Cluster 5.
Cluster 5 contains 284 genes, whose expression peaks during macronuclear G2/micronuclear mitosis, with GO association enrichments that include the mitotic cell cycle process (P = 1.96E-07), mitotic nuclear division (P = 1.90E-07, chromosome segregation (P = 1.56E-08), chromosome condensation (P = 2.37E-07), telomere maintenance (P = 5.32E-05), DNA repair (P = 2.03E-07), and chromatin organization (P = 8.90E-06) (Figure 6B, top panel). Subclustering allowed more specific cell cycle phase associations (Figure 6B, bottom panel; Supplemental Datafile 3).
Genes in subcluster 5A peak at early macronuclear S phase (60 min) and are enriched for biological processes such as telomere maintenance (P = 2.37E-06) and nucleic acid metabolic process (P = 7.94E-07) (Supplemental Datafile 4). Subcluster 5B contains genes whose expression peaks during macronuclear G1 phase (30 min). The associated GO term is DNA repair (P = 5.32E-07) (Supplemental Datafile 5).
Subcluster 5C consists of genes that are up-regulated at late macronuclear S phase/micronuclear mitosis (90 min) and whose GO associations are enriched for the terms chromosome condensation (P = 7.44E-07), chromatin organization (P = 2.32E-06), mitotic cell cycle process (P = 3.68E-03), and mitotic nuclear division (P = 3.59E-03) (Figure 6B; Supplemental Datafile 6). The two macronucleus-specific condensin genes (CPH3 and CPH4) and the other two subunits of the condensin complex (CPD1 and CPG1) (Howard-Till et al., 2019) are in cluster 5C (Figure 6C). The PANTHER annotated pathway of mitotic cell cycle process and mitotic nuclear division are due to those four genes. Subcluster 5C also contains several histone genes in the chromatin organization pathway (HHF2, HTA1, HTA2, HTA3, HTB1, HTB2).
Subcluster 5D contains genes with high expression levels through micronuclear G2 phase and mitosis that are down-regulated at amitosis/cytokinesis. This group is enriched for biological pathways such as cell cycle checkpoint (P = 4.49E-03), chromosome condensation (P = 2.64E-02), mitotic nuclear division (P = 2.31E-04), and mitotic cell cycle process (P = 3.47E-04) (Figure 6B, bottom panel; Supplemental Datafile 7). The two micronuclear-specific condensin genes (CPH1 and CPH2) are in subcluster 5D. Cell cycle checkpoint genes in subcluster 5D include MUS81/RAD17 (TTHERM_000191179) and MAD3 (TTHERM_00393260), a subunit of the spindle assembly checkpoint complex (Figure 6C). Other genes in chromosome segregation include epsilon-tubulin (ETU1) and gamma-tubulin (GTU1). Epsilon-tubulin localizes to the pericentriolar material and is required for centriole duplication and microtubule organization. Its recruitment to the new centriole can occur only after exit from S phase (Chang and Stearns, 2000; Chang et al., 2003).
Cluster 1.
The 342 genes in cluster 1 (micronuclear mitosis, macronuclear G2) were highly enriched for mitosis-related biological processes such as mitotic cell cycle process (P = 2.95E-06), mitotic nuclear division (P = 1.48E-06), spindle organization (P = 2.96E−05), and microtubule-based processes (P = 2.98E-06). The mitosis-related biological process GO term-chromosome segregation has a higher P value (P = 2.25E-02), partly because several cohesin genes are not annotated in PANTHER (SMC1, SMC3, SCC3, and REC8) (Figure 6B; Supplemental Datafile 8). Spindle organization includes the mitotic-spindle organizing protein (Mzt1), the putative kinetochore protein (Nuf2p), the putative microtubule-associated protein (Stu2p), the putative microtubule-binding proteins (Bim1), and the hypothetical protein (TTHERM_00493000); and several genes encoding the gamma-tubulin complex (GCP2, GCP4, GCP5) (Figure 5C). Predicted mitotic genes, such as CDC31, TUB4, and TID, are also in cluster 1. Delta-tubulin 1 (DTU1), which is found in association with the centrioles and associating with only the older of the centrosomes in a newly duplicated pair, is also in this cluster (Chang and Stearns, 2000). This group includes cyclin A/B (CYC8) and cyclin D (CYC14) (Figure 5, A and B) and all genes encoding the minimal cohesin complex components (SMC1, SMC3, SCC3, and REC8) (Figure 3B).
Cluster 2.
This group contains 564 genes whose expression peaked at 150 min (macronuclear amitosis/cytokinesis). GO terms are associated with microtubule-based process (P = 6.42E-08), cytoskeleton organization (P = 1.94E-06), cellular component biogenesis (P = 4.9E-02), and centriole duplication (P = 3.33E-02) (Figure 6B; Supplemental Datafile 9). Three genes in centriole duplication from PANTHER are CPAP1, TTHERM_00532450 (centrosome protein putative), and TTHERM_01113110 (DHHC zinc finger protein). Sister Chromatid Cohesion protein 2 (SCC2), the tubulin ligases, TTLL1, TTLL2, and TTL10, components of the anaphase-promoting complex (APC1, APC2, and APC10), and two cell cycle regulators, CYC15 and E2FL3, are in this cluster. These data suggest a degree of functional conservation between mitosis and amitosis.
Coexpression profiling identifies function-related gene networks
Genes with similar expression patterns during the cell cycle frequently participate in the same pathway. Weighted gene coexpression network analysis (WGCNA) is a systems biology method for describing the correlation patterns among genes across RNA-seq samples (Zhang and Horvath, 2005; Langfelder and Horvath, 2008). Coexpression networks have been found useful for describing the pairwise relationships among genes. We identified the top 10 coexpression profiles for each cell cycle–regulated gene using WGCNA (see Supplemental Datafile 10). The results can be used for data exploration or gene screening of a given pathway. To illustrate the utility of coexpression, we used MCM6 as our query. This gene is coregulated with other genes involved in the initiation of DNA replication and replication fork progression in other eukaryotes (Cho et al., 1998; Menges et al., 2003; Grant et al., 2013) (Supplemental Figure S4). This survey identified other subunits of the MCM complex (MCM2, MCM3, MCM4, MCM5, MCM6) and the replication polymerase (POL2, POL12, DPB2). It also identified a putative MCM gene family member that was not previously annotated (TTHERM_000011759) (Figure 6D). The two remaining “top 10” coregulated genes have unknown functions; one of them contains a serine/threonine kinase domain. Strong coexpression correlations were observed in particular for genes involved in other aspects of DNA metabolism, as well as nucleocytoplasmic scaffolding proteins.
DISCUSSION
The prototypic eukaryotic cell cycle is composed of four phases, two gap phases devoted to cell growth, separated by S and M phases, which are respectively devoted to replication of the nuclear genome and segregation of chromosomes to daughter cells before cell division. Decades of research in yeast and mammalian cells have uncovered the many layers of regulation needed to ensure the fidelity of cell cycle transitions and ensuing stage-specific processes (i.e., DNA replication, chromosome segregation, cell division). They include cell cycle–regulated transcription, translational control of mRNAs, activation of proteins by phosphorylation, and ubiquitin-mediated protein degradation (Stumpf et al., 2013; reviewed in Benanti, 2016; Blank et al., 2017; reviewed in Zou and Lin, 2021; Clemm von Hohenberg et al., 2022). Posttranslational protein modification and turnover are critical for generating sharp cell cycle transitions. However, stage-specific transcription is the most common mechanism for producing the regulatory, structural, and enzymatic proteins required to duplicate and transmit nuclear and organellar components to daughter cells (reviewed in Haase and Wittenberg, 2014).
The reported number of cell cycle–regulated transcripts in S. cerevisiae varies between 600 and 1000. Human transcriptome data place this number between 1000 and 1900 (Granovskaia et al., 2010, and references therein; Dominguez et al., 2016; Giotti et al., 2017, and references therein). The increased number of cell cycle–regulated genes in metazoan reflects the diversity in cell type–specific triggers as well as organismal complexity for the coordination of multicellular tissue homeostasis.
As an early branch in the eukaryotic lineage, ciliated protozoa have evolved a truly unique system for partitioning the chromosomal functions for gene expression and sexual gene transmission into two nuclei with nonoverlapping functions—the micronucleus and macronucleus. The only known function of the micronucleus is transmission of chromosomes from parent to progeny during conjugation. Meanwhile, the macronucleus is solely dedicated to gene expression. One consequence of the separate micro- and macronuclear functions is the creation of a complex vegetative cell cycle in which both nuclei replicate and partition their respective chromosomes at different times. This arrangement poses a challenge for proteins that perform the same function at two different stages of the cell cycle. Our molecular and cytological data support a model for coordinated signaling between the micro- and macronucleus. First, cross-talk between micro- and macronuclei is consistent with our observation that none of the 3000+ EdU-positive cells that we scored incorporated label into both the micro- and macronucleus. Second, differences in cell cycle regulation of cyclin D family members argue that different licensing factors regulate entry into the two respective S phases. This would assure that the macronucleus (pure euchromatin) initiates DNA replication first and that micronuclear S phase (pure heterochromatin) does not begin DNA synthesis until cells exit macronuclear S phase. The same holds for chromosome segregation and nuclear division; however, the order is reversed: the micronucleus undergoes mitosis before amitotic macronuclear division (Figure 1C). Third, mRNA abundance for nucleus-specific pore proteins and importins is cell cycle regulated. This additional layer of regulation could play a role in the temporal separation of micro- and macronuclear DNA replication and mitosis/amitosis, which are likely to require shared and nucleus-specific proteins. We speculate that posttranslational modifications, such as those that occur in histones H3 and H4, could regulate the differential trafficking of proteins that are targeted to both nuclei.
Our RNA-seq analysis of the T. thermophila vegetative cell cycle is the first such analysis in a binucleated species. In total, steady state mRNA levels of more than 3000 genes were shown to be under cell cycle control. Sixteen percent of vegetative expressed genes exhibited cyclic oscillations that peaked at different stages of the cell cycle. This percentage is higher than that in S. cerevisiae, A. thaliana, and human cells analyzed by microarray or RNA-seq (range 5–10%). Our RNA-seq data support the idea that gene duplication and divergence plays a significant role in regulating the binucleate cell cycle. Differential regulation of gene family members could be exploited to identify micro- and macronuclear separation of function. Because many mitotic genes in yeast, higher eukaryotes, and Tetrahymena are cell cycle regulated (Cho et al., 1998; Spellman et al., 1998; Rustici et al., 2004; Giotti et al., 2017; this study), it is possible that some of these proteins would play a role amitotic chromosome transmission.
Proteomic studies previously demonstrated that two of the four condensins localize to the macronucleus (Figure 2E; CPH3, CPH4). In contrast, none of the cohesins is targeted to the amitotic macronucleus (Howard-Till et al., 2019). All cohesin subunit genes and the micronuclear condensins, CPH1 and CPH2, are cell cycle regulated, peaking before micronuclear mitosis (Figure 3, A and B). Meanwhile, expression of macronuclear condensin mRNA levels peak just before amitosis (Figure 3A). Gene expression cluster networks that reach their maximum after micronuclear mitosis may reveal components of the amitotic transmission machinery. Weighted gene coexpression network analysis can similarly be used to identify candidate genes for functional analysis.
Cyclin-CDK complexes are the master regulators of the eukaryotic cell cycle (Grana and Reddy, 1995). Gene duplication and divergence form the basis for regulated entry and exit from different stages of the mammalian cell cycle (reviewed in Martinez and Malumbres, 2020). Tetrahymena encodes 34 putative cyclin genes, a subset that are exclusively expressed during conjugation (Stover and Rice, 2011; Xu et al., 2016; Yan et al., 2016; Ma et al., 2020). In keeping with the known role of cyclin D, our data suggest that different paralogues regulate entry into macro- and micronuclear S phases. Of the 11 predicted cyclin D genes, CYC7, CYC12, and CYC22 expression peaks at the macronuclear G1/S transition, while CYC14 peaks before the onset of micronuclear S phase. Cyclin A/B family members regulate the S/G2 and G2/M transitions. Two A/B family members peak before mitotic micronuclear division—CYC15 and CYC8 (possibly CYC10). Four cyclin A/B genes (CYC1, CYC6, CYC20, and CYC24) peak later in the cell cycle, suggesting that they may regulate macronuclear amitosis. Reverse genetic methodologies and the ability to generate micronuclear/macronuclear heterokaryons (Turkewitz et al., 2002) provide opportunities to determine the functional roles of vegetative cyclins, similar to prior work on ORC and the ATR checkpoint kinase (Smith et al., 2004; Yakisich et al., 2006; Lee et al., 2015).
Our RNA-seq data identify another rich area for pursuit—histone synthesis and posttranslational modification. Although the major core histone proteins (H2A, H2B, H3, H4) are present in micro- and macronuclei, their genes produce a single increase in cell cycle–regulated mRNA abundance that peaks before macronuclear S phase, when the demand for these proteins is greatest (Figure 2, A–D). From a supply perspective, two waves of gene expression may be unnecessary. However, core histones synthesis is tightly linked to demand (Marzluff et al., 2008). If histones were produced and imported into both nuclei concurrently, supply would not be linked to demand with respect to micronuclear DNA replication. Histones protein synthesis could be under translational control, as has been shown by ribo-seq for lipogenic enzymes in yeast (Blank et al., 2017). Another nuanced mechanism would be to regulate protein transport. Nucleus-specific targeting could be controlled by euchromatic and heterochromatic posttranslational modification of histone subunits or the synthesis of nucleus-specific importins or pore proteins.
In contrast to core histone subunits, the centromeric histone H3 variant, CNA1, and micro- and macronuclear-specific H1 linker histones (MLH1 and HHO1, respectively), the macronuclear H2A.Z histone variant and macronucleus-specific chromatin modifiers (TXR1, HAT1, HAT2, EZL2, and EZL3) peak with the onset of their respective nuclear S phase. This is consistent with temporal linkage between transcription to supply and demand—an overriding feature of eukaryotic cell cycle control (reviewed in Haase and Wittenberg, 2014).
A powerful application of transcriptomics is the elucidation of genetic interaction and functional networks. We employed two strategies to our analysis of cell cycle–regulated genes. The first approach involved noise-robust soft clustering analysis—an unsupervised learning technique designed to reveal structures hidden in large time-course gene expression data sets to identify periodically expressed gene clusters. The cluster approach identified seven expression patterns that account for the 3032 cell cycle–regulated genes identified by MetaCycle. The second approach—WGCNA—uses a different algorithm to find clusters by describing the correlation patterns between genes.
Owing to the relatively poor annotation of the 26,000+ predicted T. thermophila genes, only four of the seven clusters were populated with genes with assigned GO terms. Of note, there was significant overlap for GO terms in clusters 1 and 2 for microtubule-based processes that occur at different stages of the cell cycle, suggesting that mitosis and amitosis might utilize related machinery to transmit chromosomes to daughter nuclei. Unexpectedly, GO terms for centriole replication and cellular component biogenesis were enriched at the end of the cell cycle when macronuclear amitosis occurs. Delving deeper into the clustered genes through WGCNA coexpression to study genes of unknown function could provide new insights into mechanisms that are conserved throughout evolution, as well as novel mechanisms that have evolved to solve species-specific problems, such as the maintenance of genome balance in the absence of mitosis, the temporal licensing of DNA replication in binucleate species, and the epigenetic control of replication initiation in euchromatic and heterochromatic chromosomes with nearly identical unique sequence composition.
Finally, complementary high-throughput approaches can build off of our RNA-seq analysis of the Tetrahymena vegetative cell cycle. Gro-seq could be used to further illuminate transcriptional regulation, and ribo-seq could be used to study translational control. Furthermore, mass spectroscopy and phosphoproteomics across the cell cycle could be used to probe the temporal fate of proteins in purified micro- and macronuclei.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Synchronization by centrifugal elutriation
CU428 cells (1.5 l) were grown to ∼1 × 105 cells/ml in 2% PPYS/1000U/ml pen-strep (Sigma Aldrich) at 30°C. Elutriation was used to obtain a synchronized G1 cell population as previously described (Liu et al., 2021). Cells were subsequently collected at 30 min intervals spanning 1.5 cell cycle, 240 min). For each time point, 20 ml of cells was collected; 5 ml for flow cytometry and DAPI analysis and 15 ml for RNA isolation. Centrifuged cell pellets were washed in 10 mM Tris, pH 7.4, before flow cytometry and RNA isolation. Two biological replicates were used to generate 18 cDNA libraries. Standard RNA-seq was chosen over single-cell RNA-seq due to the need to establish robust reference gene expression patterns and biomarkers to infer position in the cell cycle.
Flow cytometry, EdU labeling, and DAPI imaging
Cells were prepared for flow cytometry as previously described (Sandoval et al., 2015). CU428 cells were synchronized by centrifugal elutriation as described above. Ten minutes before each time point, cells (2 ml) were labeled for 20 min with 25 µM EdU (Life Technologies), washed twice with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 5 min, rinsed twice with PBS, and stored at 4°C. To visualize EdU incorporation into nascent DNA strands, 100 µl samples were spotted onto coverslips and then let dry for 2 h. Cells were briefly permeabilized with 0.005% Triton X-100, rinsed with PBS, and incubated for 30 min with the Click-iT EdU Alexa Fluor 594 Imaging Kit according to manufacturer protocol (Thermo Fisher). Stained coverslips were washed once with PBS, counterstained with Hoechst (Life Technologies), mounted in Vectashield mounting media (Vector Laboratories), and imaged by fluorescence microscopy. DAPI fluorescence microscopy was used to determine the percentage of cells in micronuclear mitosis, macronucleus amitosis, and cytokinesis, as previously described (Lee et al., 2015).
RNA isolation, library preparation, and RNA-seq analysis
Cell pellets were resuspended in 1 ml of RNAlater and placed at 4°C overnight. RNA was prepared using the Qiagen RNeasy kit with additional on-column DNase digestion. Total RNA (10 µg) was converted into cDNA using an Illumina RNA-seq Prep Kit. The experiment was performed in duplicate, resulting in 18 cDNA libraries. cDNA libraries spanning 1.5 cell cycles and bar-coded samples were subjected to high-throughput sequencing on an Illumina-HiSeq 2500v4 platform (35 M paired end reads/library, two biological replicates per time point). All programs in the bioinformatics workflow were provided through open source software. Adapters and low-quality read sequences were removed by Trimmomatic (Bolger et al., 2014) (https://hpc.nih.gov/apps/trimmomatic.html). Paired reads with concordant mappings were aligned to the T. thermophila macronuclear genome (SB210, 2021 version, TGD) by HISAT2, and transcript abundance in each sample was computed using StringTie (Pertea et al., 2016) (https://anaconda.org/soil/stringtie). Quantification and statistical inference of changes between time points were computed by DESeq2 (Love et al., 2014) (https://bioconductor.org/packages/release/bioc/html/DESeq2.html). Plotting was based on regularized log (rlog)-transformed DESeq2 data. This gene clustering–based algorithm was chosen over a principal component analysis (PCA) approach due to its superior performance for data analysis for high read depths (reviewed in Menon, 2018), the conventional preference for DESeq2 for RNA-seq data analysis, and compatibility with MetaCycle pattern recognition analysis.
Identification of cell cycle–regulated genes
MetaCycle was used to identify cell cycle–regulated genes (Wu et al., 2016) (https://github.com/gangwug/MetaCycle). MetaCycle implements JTK_CYCLE (JTK) and Lomb-Scargle (LS) and integrates their results. First, genes with two 1.5-fold changes in opposite directions between two time points were identified (p value < 0.05) using the logarithmic fold changes with DESeq2. A total of 3825 filtered genes with normalized counts from DESeq2 in both biological duplicates were identified and input into MetaCycle. MetaCycle::meta2d had minimum and maximum period lengths set as 120 and 240 min, respectively. A total of 3032 genes were detected with an FDR < 0.05. The 0 min time point was excluded from the MetaCycle analysis due to the transient effect of elutriation on expression of many metabolic genes. LS and JTK algorithms’ performance on peaked data was much lower (Deckard et al., 2013).
Cluster analysis
Cluster analysis was performed with noise-robust soft clustering (Futschik and Carlisle, 2005). Soft clustering is more noise robust and generates accessible internal cluster structures. It was implemented using the fuzzy c-means algorithm. Regularized log (rlog)-transformed data from DESeq2 were used as input. Optimized FCM (fuzzy c-means) parameter m = 1.37114 was used.
Analysis of gene functions
A GO overrepresentation test of gene clusters was done using PANTHER (Protein Analysis Through Evolutionary Relationships), which contains the complete sets of protein-coding genes and reports orthologues and paralogues (GO database released 2016-08-22) (Mi et al., 2016). Clustering results were input into PANTHER. A dotplot comparing pathways of clusters was generated by ggplot2 in R. Duplicate biological processes having the same genes and similar process names were removed when generating graphs. Additional gene annotation and ontology information was found using TGD (www.ciliate.org).
Coexpression profiling
The R package “WGCNA” (Zhang and Horvath, 2005) was used to perform WGCNA (https://anaconda.org/bioconda/r-wgcna). RNA-seq data were properly preprocessed using DESeq2 to generate vsd values for each sample as recommended. Before construction of the adjacency matrix, a soft threshold (β) was set by inspection of plots generated after calling the function pickSoftThreshold. The soft threshold was set to 6, where the scale-free topology (SFT) index as a function of the soft threshold reached saturation. Modules were generated after calling the function blockwiseModules. Arguments of this function were kept as default. Lists of top 10 coexpressed genes were generated for all cell cycle–regulated genes.
Data access.
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE123456. All relevant data are within this paper and its Supporting Information files.
Acknowledgments
This work was supported by National Science Foundation grant MCB-0132675 and a Tom and Jean McMullin Endowed Professorship to G.M.K.
Abbreviations used:
- C
chromosome copy number
- CDK
cyclin-dependent kinase
- DAPI
4’,6-diamidino-2-phenylindole dihydrochloride
- EdU
5-ethylene-2’deoxyuridine
- FRD
false discovery rate
- GO
gene ontology
- ORC
origin recognition complex
- PCR
polymerase chain reaction.
Footnotes
REFERENCES
- Ali EI, Loidl J, Howard-Till RA (2018). A streamlined cohesin apparatus is sufficient for mitosis and meiosis in the protist Tetrahymena. Chromosoma 127, 421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Allen RL, Wiggins JC, Chicoine LG, Richman R (1984). Proteolytic processing of h1-like histones in chromatin: a physiologically and developmentally regulated event in Tetrahymena micronuclei. J Cell Biol 99, 1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Colavito-Shepanski M, Gorovsky MA (1987). Scheduled and unscheduled DNA synthesis during development in conjugating Tetrahymena. Dev Biol 124, 469–480. [DOI] [PubMed] [Google Scholar]
- Allis CD, Glover CV, Bowen JK, Gorovsky MA (1980). Histone variants specific to the transcriptionally active, amitotically dividing macronucleus of the unicellular eucaryote, Tetrahymena thermophila. Cell 20, 609–617. [DOI] [PubMed] [Google Scholar]
- Almouzni G, Cedar H (2016). Maintenance of epigenetic information. Cold Spring Harb Perspect Biol 8, a019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benanti JA (2016). Create, activate, destroy, repeat: Cdk1 controls proliferation by limiting transcription factor activity. Curr Genet 62, 271–276. [DOI] [PubMed] [Google Scholar]
- Blank HM, Perez R, He C, Maitra N, Metz R, Hill J, Lin Y, Johnson CD, Bankaitis VA, Kennedy BK, et al. (2017). Translational control of lipogenic enzymes in the cell cycle of synchronous, growing yeast cells. EMBO J 36, 487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracht JR, Fang W, Goldman AD, Dolzhenko E, Stein EM, Landweber LF (2013). Genomes on the edge: programmed genome instability in ciliates. Cell 152, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Chen Y, Cierpicki T, Liu Y, Basrur V, Lei M, Dou Y (2010). An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS One 5, e14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes MD, Xi X, Vermaak D, Yao MC, Malik HS (2006). The CNA1 histone of the ciliate Tetrahymena thermophila is essential for chromosome segregation in the germline micronucleus. Mol Biol Cell 17, 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Meyer E, Mochizuki K (2013). Epigenetics of ciliates. Cold Spring Harb Perspect Biol 5, a017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P, Giddings TH Jr, Winey M, Stearns T (2003). Epsilon-tubulin is required for centriole duplication and microtubule organization. Nat Cell Biol 5, 71–76. [DOI] [PubMed] [Google Scholar]
- Chang P, Stearns T (2000). Delta-tubulin and epsilon-tubulin: two new human centrosomal tubulins reveal new aspects of centrosome structure and function. Nat Cell Biol 2, 30–35. [DOI] [PubMed] [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, Davis RW (1998). A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell 2, 65–73. [DOI] [PubMed] [Google Scholar]
- Clemm von Hohenberg K, Müller S, Schleich S, Meister M, Bohlen J, Hofmann TG, Teleman AA (2022). Cyclin B/CDK1 and cyclin A/CDK2 phosphorylate DENR to promote mitotic protein translation and faithful cell division. Nat Commun 13, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole E, Sugai T (2012). Developmental progression of Tetrahymena through the cell cycle and conjugation. Methods Cell Biol 109, 177–236. [DOI] [PubMed] [Google Scholar]
- Cui B, Gorovsky MA (2006). Centromeric histone H3 is essential for vegetative cell division and for DNA elimination during conjugation in Tetrahymena thermophila. Mol Cell Biol 26, 4499–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B, Liu Y, Gorovsky MA (2006). Deposition and function of histone H3 variants in Tetrahymena thermophila. Mol Cell Biol 26, 7719–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MC, Ward JG, Herrick G, Allis CD (1992). Programmed nuclear death: apoptotic-like degradation of specific nuclei in conjugating Tetrahymena. Dev Biol 154, 419–432. [DOI] [PubMed] [Google Scholar]
- Deckard A, Anafi RC, Hogenesch JB, Haase SB, Harer J (2013). Design and analysis of large-scale biological rhythm studies: a comparison of algorithms for detecting periodic signals in biological data. Bioinformatics 29, 3174–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koning L, Corpet A, Haber JE, Almouzni G (2007). Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol 14, 997–1007. [DOI] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36(Web Server issue), W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerder FP, Deak JC, Lief JH (1992). Rate of phenotypic assortment in Tetrahymena thermophila. Dev Genet 13, 126–132. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tsai YH, Gomez N, Jha DK, Davis I, Wang Z (2016). A high-resolution transcriptome map of cell cycle reveals novel connections between periodic genes and cancer. Cell Res 26, 946–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donti TR, Datta S, Sandoval PY, Kapler GM (2009). Differential targeting of tetrahymena ORC to ribosomal DNA and non-rDNA replication origins. EMBO J 28, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futschik ME, Carlisle B (2005). Noise-robust soft clustering of gene expression time-course data. J Bioinform Comput Biol 3, 965–988. [DOI] [PubMed] [Google Scholar]
- Gao S, Xiong J, Zhang C, Berquist BR, Yang R, Zhao M, Molascon AJ, Kwiatkowski SY, Yuan D, Qin Z, et al. (2013). Impaired replication elongation in Tetrahymena mutants deficient in histone H3 Lys 27 monomethylation. Genes Dev 27, 1662–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giotti B, Joshi A, Freeman TC (2017). Meta-analysis reveals conserved cell cycle transcriptional network across multiple human cell types. BMC Genomics 18, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn EF, Chen J, Mushegian AR (2006). Detecting periodic patterns in unevenly spaced gene expression time series using Lomb-Scargle periodograms. Bioinformatics 22, 310–316. [DOI] [PubMed] [Google Scholar]
- Grana X, Reddy EP (1995). Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 11, 211–219. [PubMed] [Google Scholar]
- Granovskaia MV, Jensen LJ, Ritchie ME, Toedling J, Ning Y, Bork P, Huber W, Steinmetz LM (2010). High-resolution transcription atlas of the mitotic cell cycle in budding yeast. Genome Biol 11, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GD, Brooks L 3rd, Zhang X, Mahoney JM, Martyanov V, Wood TA, Sherlock G, Cheng C, Whitfield ML (2013). Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol Biol Cell 24, 3634–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunjan A, Verreault A (2003). A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 115, 537–549. [DOI] [PubMed] [Google Scholar]
- Haase SB, Wittenberg C (2014). Topology and control of the cell-cycle-regulated transcriptional circuitry. Genetics 196, 65–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harashima H, Dissmeyer N, Schnittger A (2013). Cell cycle control across the eukaryotic kingdom. Trends Cell Biol 23, 345–356. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hayashi H, Fusauchi Y, Iwai K (1984). Tetrahymena histone H3. Purification and two variant sequences. J Biochem 95, 1741–1749. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hayashi H, Iwai K (1987). Tetrahymena histone H1. Isolation and amino acid sequence lacking the central hydrophobic domain conserved in other H1 histones. J Biochem 102, 369–376. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Smith MM (2015). Histone variants and epigenetics. Cold Spring Harb Perspect Biol 7, a019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard-Till R, Tian M, Loidl J (2019). A specialized condensin complex participates in somatic nuclear maturation in Tetrahymena thermophila. Mol Biol Cell 30, 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, Kornacker K (2010). JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms 25, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M, Mori C, Kojidani T, Bunai F, Hori T, Fukagawa T, Hiraoka Y, Haraguchi T (2009). Two distinct repeat sequences of Nup98 nucleoporins characterize dual nuclei in the binucleated ciliate tetrahymena. Curr Biol 19, 843–847. [DOI] [PubMed] [Google Scholar]
- Iwamoto M, Osakada H, Mori C, Fukuda Y, Nagao K, Obuse C, Hiraoka Y, Haraguchi T (2017). Compositionally distinct nuclear pore complexes of functionally distinct dimorphic nuclei in the ciliate Tetrahymena. J Cell Sci 130, 1822–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob NK, Lescasse R, Linger BR, Price CM (2007). Tetrahymena POT1a regulates telomere length and prevents activation of a cell cycle checkpoint. Mol Cell Biol 27, 1592–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrer KM (2012). Nuclear dualism. Methods Cell Biol 109, 29–52. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Mochizuki K (2011). Programmed DNA elimination in Tetrahymena: a small RNA-mediated genome surveillance mechanism. Adv Exp Med Biol 722, 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Lambert JP, Petschnigg J, Friesen H, Pawson T, Rosebrock A, Gingras AC, Fillingham J, Andrews B (2014). Cell cycle-regulated oscillator coordinates core histone gene transcription through histone acetylation. Proc Natl Acad Sci USA 111, 14124–14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Daujat S, Schneider R (2016). Lateral thinking: how histone modifications regulate gene expression. Trends Genet 32, 42–56. [DOI] [PubMed] [Google Scholar]
- Lee PH, Meng X, Kapler GM (2015). Developmental regulation of the Tetrahymena thermophila origin recognition complex. PLoS Genet 11, e1004875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Y, Hao H, Xu J, Bo T, Liang A, Wang W (2021). The histone chaperone Nrp1 is required for chromatin stability and nuclear division in Tetrahymena thermophila. Epigenetics Chromatin 14, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang HX, Xu J, Wang W (2019). Ran1 is essential for parental macronuclear import of apoptosis-inducing factor and programmed nuclear death in Tetrahymena thermophila. FEBS J 286, 913–929. [DOI] [PubMed] [Google Scholar]
- Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA (2011). DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 25, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Nan B, Niu J, Kapler GM, Gao S (2021). An optimized and versatile counter-flow centrifugal elutriation workflow to obtain synchronized eukaryotic cells. Front Cell Dev Biol 9, 664418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, Allis CD (2007). RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev 21, 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly T, Endo A, Lamond AI (2015). Proteomic analysis of the response to cell cycle arrests in human myeloid leukemia cells. eLife 4, e04534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Yan G, Han X, Zhang J, Xiong J, Miao W (2020). Sexual cell cycle initiation is regulated by CDK19 and CYC9 in Tetrahymena thermophila. J Cell Sci 133, jcs235721. [DOI] [PubMed] [Google Scholar]
- MacAlpine DM, Almouzni G (2013). Chromatin and DNA replication. Cold Spring Harb Perspect Biol 5, a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Gueydon E, Schwob E (2014). DNA replication and spindle checkpoints cooperate during S phase to delay mitosis and preserve genome integrity. J Cell Biol 204, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Falkowska KA, Li AY, Galanti SE, Kanuru RC, LaMont EG, Mazzarella KC, Micev AJ, Osman MM, Piotrowski NK, et al. (2008). Nucleus-specific importin alpha proteins and nucleoporins regulate protein import and nuclear division in the binucleate Tetrahymena thermophila. Eukaryot Cell 7, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M (2014). Cyclin-dependent kinases. Genome Biol 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Alonso D, Malumbres M (2020). Mammalian cell cycle cyclins. Semin Cell Dev Biol 107, 28–35. [DOI] [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, Duronio RJ (2008). Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet 9, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto H, Hawke D, Kobayashi R, Verreault A (2005). A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436, 294–298. [DOI] [PubMed] [Google Scholar]
- Menges M, Hennig L, Gruissem W, Murray JA (2003). Genome-wide gene expression in an Arabidopsis cell suspension. Plant Mol Biol 53, 423–442. [DOI] [PubMed] [Google Scholar]
- Menon V (2018). Clustering single cells: a review of approaches on high-and low-depth single-cell RNA-seq data. Brief Funct Genomics 17, 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD (2016). PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44, D336–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad MM, Donti TR, Sebastian Yakisich J, Smith AG, Kapler GM (2007). Tetrahymena ORC contains a ribosomal RNA fragment that participates in rDNA origin recognition. EMBO J 26, 5048–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeel-Shah S, Garg J, Saettone A, Ashraf K, Lee H, Wahab S, Ahmed N, Fine J, Derynck J, Pu S, et al. (2021). Functional characterization of RebL1 highlights the evolutionary conservation of oncogenic activities of the RBBP4/7 orthologue in Tetrahymena thermophila. Nucleic Acids Res 49, 6196–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazyan R, Voronina E, Chapman JR, Luperchio TR, Gilbert TM, Meier E, Mackintosh SG, Shabanowitz J, Tackett AJ, Reddy KL, et al. (2014). Methylation of histone H3K23 blocks DNA damage in pericentric heterochromatin during meiosis. eLife 3, e02996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud C, Sozzani R, Glab N, Domenichini S, Perennes C, Cella R, Kondorosi E, Bergounioux C (2006). Two cell-cycle regulated SET-domain proteins interact with proliferating cell nuclear antigen (PCNA) in Arabidopsis. Plant J 47, 395–407. [DOI] [PubMed] [Google Scholar]
- Rustici G, Mata J, Kivinen K, Lio P, Penkett CJ, Burns G, Hayles J, Brazma A, Nurse P, Bahler J (2004). Periodic gene expression program of the fission yeast cell cycle. Nat Genet 36, 809–817. [DOI] [PubMed] [Google Scholar]
- Sadasivam S, DeCaprio JA (2013). The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 13, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval PY, Lee PH, Meng X, Kapler GM (2015). Checkpoint activation of an unconventional DNA replication program in tetrahymena. PLoS Genet 11, e1005405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Gorovsky MA (1996). Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell 86, 475–483. [DOI] [PubMed] [Google Scholar]
- Shen X, Yu L, Weir JW, Gorovsky MA (1995). Linker histones are not essential and affect chromatin condensation in vivo. Cell 82, 47–56. [DOI] [PubMed] [Google Scholar]
- Sheng Y, Duan L, Cheng T, Qiao Y, Stover NA, Gao S (2020). The completed macronuclear genome of a model ciliate Tetrahymena thermophila and its application in genome scrambling and copy number analyses. Sci China Life Sci 63, 1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Yakisich JS, Kapler GM, Cole ES, Romero DP (2004). A beta-tubulin mutation selectively uncouples nuclear division and cytokinesis in Tetrahymena thermophila. Eukaryot Cell 3, 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B (1998). Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover NA, Rice JD (2011). Distinct cyclin genes define each stage of ciliate conjugation. Cell Cycle 10, 1699–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D (2013). The translational landscape of the mammalian cell cycle. Mol Cell 52, 574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Ueberheide BM, Liu Y, Tackett AJ, Diaz RL, Shabanowitz J, Chait BT, Hunt DF, Allis CD (2007). Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc Natl Acad Sci USA 104, 2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurlings I, de Bruin A (2016). E2F transcription factors control the roller coaster ride of cell cycle gene expression. Methods Mol Biol 1342, 71–88. [DOI] [PubMed] [Google Scholar]
- Turkewitz AP, Orias E, Kapler G (2002). Functional genomics: the coming of age for Tetrahymena thermophila. Trends Genet 18, 35–40. [DOI] [PubMed] [Google Scholar]
- Uhlmann F (2016). SMC complexes: from DNA to chromosomes. Nat Rev Mol Cell Biol 17, 399–412. [DOI] [PubMed] [Google Scholar]
- Vavra KJ, Allis CD, Gorovsky MA (1982). Regulation of histone acetylation in Tetrahymena macro- and micronuclei. J Biol Chem 257, 2591–2598. [PubMed] [Google Scholar]
- Wiley EA, Myers T, Parker K, Braun T, Yao MC (2005). Class I histone deacetylase Thd1p affects nuclear integrity in Tetrahymena thermophila. Eukaryot Cell 4, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing CE, Fung HYJ, Chook YM (2022). Karyopherin-mediated nucleocytoplasmic transport. Nat Rev Mol Cell Biol 23, 307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong L, Klionsky L, Wickert S, Merriam V, Orias E, Hamilton EP (2000). Autonomously replicating macronuclear DNA pieces are the physical basis of genetic coassortment groups in Tetrahymena thermophila. Genetics 155, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Anafi RC, Hughes ME, Kornacker K, Hogenesch JB (2016). MetaCycle: an integrated R package to evaluate periodicity in large scale data. Bioinformatics 32, 3351–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Allis CD, Richman R, Cook RG, Gorovsky MA (1986). An intervening sequence in an unusual histone H1 gene of Tetrahymena thermophila. Proc Natl Acad Sci USA 83, 8674–8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhao X, Mao F, Basrur V, Ueberheide B, Chait BT, Allis CD, Taverna SD, Gao S, Wang W, Liu Y (2021). A Polycomb repressive complex is required for RNAi-mediated heterochromatin formation and dynamic distribution of nuclear bodies. Nucleic Acids Res 49, 5407–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wang R, Ghanam AR, Yan G, Miao W, Song X (2016). The key role of CYC2 during meiosis in Tetrahymena thermophila. Protein Cell 7, 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakisich JS, Kapler GM (2004). The effect of phosphoinositide 3-kinase inhibitors on programmed nuclear degradation in Tetrahymena and fate of surviving nuclei. Cell Death Differ 11, 1146–1149. [DOI] [PubMed] [Google Scholar]
- Yakisich JS, Sandoval PY, Morrison TL, Kapler GM (2006). TIF1 activates the intra-S-phase checkpoint response in the diploid micronucleus and amitotic polyploid macronucleus of Tetrahymena. Mol Biol Cell 17, 5185–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan GX, Dang H, Tian M, Zhang J, Shodhan A, Ning YZ, Xiong J, Miao W (2016). Cyc17, a meiosis-specific cyclin, is essential for anaphase initiation and chromosome segregation in Tetrahymena thermophila. Cell Cycle 15, 1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MC, Chao JL (2005). RNA-guided DNA deletion in Tetrahymena: an RNAi-based mechanism for programmed genome rearrangements. Annu Rev Genet 39, 537–559. [DOI] [PubMed] [Google Scholar]
- Yu L, Gorovsky MA (1997). Constitutive expression, not a particular primary sequence, is the important feature of the H3 replacement variant hv2 in Tetrahymena thermophila. Mol Cell Biol 17, 6303–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
- Zhang C, Gao S, Molascon AJ, Wang Z, Gorovsky MA, Liu Y, Andrews PC (2014). Bioinformatic and proteomic analysis of bulk histones reveals PTM crosstalk and chromatin features. J Proteome Res 13, 3330–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Molascon AJ, Gao S, Liu Y, Andrews PC (2013). Quantitative proteomics reveals that the specific methyltransferases Txr1p and Ezl2p differentially affect the mono-, di- and trimethylation states of histone H3 lysine 27 (H3K27). Mol Cell Proteomics 12, 1678–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tian M, Yan GX, Shodhan A, Miao W (2017). E2fl1 is a meiosis-specific transcription factor in the protist Tetrahymena thermophila. Cell Cycle 16, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou T, Lin Z (2021). The involvement of ubiquitination machinery in cell cycle regulation and cancer progression. Int J Mol Sci 22, 5754. [DOI] [PMC free article] [PubMed] [Google Scholar]