

Abstract
We detail the development of a next-generation Streptococcus pneumoniae liposomal encapsulation of polysaccharides (LEPS) vaccine, with design characteristics geared toward best-in-class efficacy. The first generation LEPS vaccine, which contained 20 encapsulated pneumococcal capsular polysaccharides (CPSs) and two surface-displayed virulence-associated proteins (GlpO and PncO), enabling prophylactic potency against 70+ serotypes of Streptococcus pneumoniae (the causative agent of pneumococcal disease), was rationally redesigned for advanced clinical readiness and best-in-class coverage. In doing so, the virulent-specific GlpO protein antigen was removed from the final formulation due to off-target immunogenicity toward bacterial species within the human microbiome, while directed protection was maintained by increasing the dose of PncO from 17 to 68 μg. LEPS formulation parameters also readily facilitated an increase in CPS valency (to a total of 24) and systematic variation in protein–liposome attachment mechanisms in anticipation of clinical translation. An additional safety assessment study demonstrated that LEPS does not exhibit appreciable toxicological effects even when administered at ten times the effective dose. In summary, this new design offers the broadest, safest, and most-complete protection while maintaining desirable glycoconjugate-like features, positioning the LEPS vaccine platform for clinical success and a global health impact.
Keywords: pneumococcal disease, biofilm, liposome, serotype, translation
Graphical Abstract
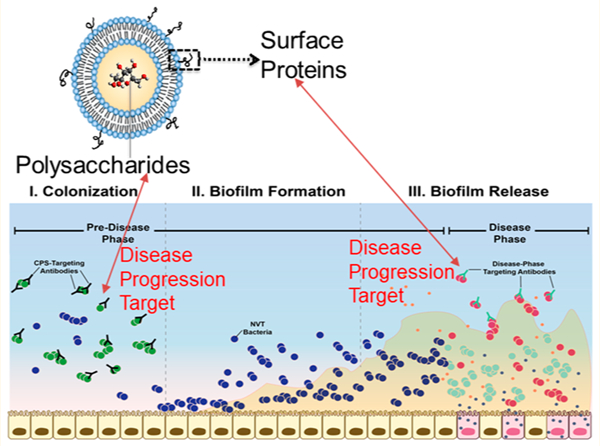
Pneumococcal disease, resulting from Streptococcus pneumoniae virulence, represents one of the most substantial bacterial diseases across the globe, causing an estimated 500 000 deaths of children under the age of five per year.1 Within the United States alone, pneumococcal pneumonia is responsible for approximately 400 000 hospitalizations annually and represents an economic burden in the billions of dollars.2–4 Two classes of licensed pneumococcal vaccines, both comprised of bacterial surface capsular polysaccharides (CPSs), which define the S. pneumoniae serotypes most commonly isolated with invasive disease, have been introduced to combat this challenge with variable success. The most successful class utilizes polysaccharides conjugated to a carrier protein, termed a pneumococcal conjugate vaccine (PCV) or glycoconjugate vaccine. Glycoconjugates are preferred over free CPSs [represented most prominently by Pneumovax 23 (PPSV23) (Merck)], as the covalent attachment of CPS to an immunogenic carrier protein elicits antibody class switching from immunoglobulin M (IgM) to immunoglobulin G (IgG), strengthens immunological memory,5 and demonstrates effectiveness in protecting young children.6
In high-income countries, such as the United States, vaccination regimens with PCVs have reduced the incidence of invasive pneumococcal disease (IPD) by greater than 90% in children under 5 and 62% across the population as a whole.7,8 These benefits result from an anticolonization immune response to the most invasive S. pneumoniae serotypes. Immunization against the CPSs of S. pneumoniae therefore provides an effective means of preventing colonization by serotypes that cause the majority of IPD.9
However, despite the benefits of implementing PCVs in vaccination schedules, disease caused by nonvaccine-type (NVT) serotypes remains a pressing challenge. Current PCVs prevent colonization for only a fraction of S. pneumoniae serotypes (13 out of 90+), while simultaneously opening an anatomical niche for NVT serotypes to colonize as asymptomatic biofilms within the nasopharynx (NP).10–13 One notable example was the marked increase in the 19A serotype following the introduction of PCV7 (a PCV containing seven CPSs but not the one specific for 19A).10–13 Though some emerging serotypes may demonstrate a lower propensity to cause invasive disease,9,14 external stimuli such as influenza infections can trigger S. pneumoniae to disseminate from normally benign nasopharyngeal biofilms and cause disease (henceforth referred to as breakthrough disease).15–18 Furthermore, capsule-switching events can enable more virulent serotypes to escape vaccination altogether.10–12,19–22 As a result of niche replacement by NVT serotypes and capsule switching, serotypes not included in Prevnar 13 (PCV13; extended to 13 CPSs) are responsible for nearly 75% of IPD in the United States.23 Consequently, providing broad-spectrum immunization against both S. pneumoniae colonization and breakthrough disease, especially with the threat of emerging NVT serotypes, remains a significant challenge to current vaccine paradigms.
In prior work, we leveraged transcriptional analysis to identify two highly conserved proteins (GlpO and PncO) demonstrating promise as vaccine antigens against virulent S. pneumoniae (i.e., bacteria triggered for release from nasopharyngeal biofilms).24,25 Covaccination with these proteins provided 100% protection in sepsis and pneumonia challenge models without disturbing initial bacterial colonization.24 A vaccine formulation featuring the encapsulation of pneumococcal CPSs within protein-decorated liposomes mediated the same antibody class switching associated with PCVs and generated similar protection results.26 This approach, coined “liposomal encapsulation of polysaccharides” (LEPS), yielded a liposomal vaccine formulation containing 20 encapsulated CPSs (1, 2, 3, 4, 5, 6A, 6B, 7F, 8, 9N, 9V, 12F, 14, 17F, 18C, 19A, 19F, 20, 22F, 23F; to prevent colonization of invasive serotypes) surface-decorated with GlpO and PncO (to prompt a PCV antibody class switching event and prevent breakthrough disease with NVT serotypes). During this study, the liposome surface was decorated with GlpO and/or PncO via noncovalent binding between a modified lipid (DOGS-NTA-Ni) and a modified protein (histidine tag).
The LEPS(20V)/GlpO+PncO formulation demonstrated noninferior immunogenicity to Pfizer’s PCV13 (the current best-in-class commercial PCV) for the 13 overlapping serotypes with coverage extended to an additional seven serotypes (2, 8, 9N, 12F, 17F, 20, and 22F). Efficacy was demonstrated against all 20 serotypes using accepted preclinical animal challenge models (sepsis and pneumonia) and an opsonophagocytic activity (OPA) assay, the established clinical correlate of protection. Furthermore, a modified OPA assay was developed using biofilm-released (BFR) bacteria mimicking the virulent pneumococcal phenotype to demonstrate serum-antibody-mediated opsonization against a total of 70 serotypes. These results represent the most comprehensive coverage of S. pneumoniae in a vaccine to date.26 However, critical questions remained regarding the ultimate potential of the LEPS vaccine as a clinical asset.
In this study, we developed a next-generation LEPS formulation to ameliorate safety concerns and establish a best-in-class pneumococcal vaccine. Potential off-target reactogenicity associated with the GlpO protein antigen was addressed as were alternative protein–liposome attachment strategies (streptavidin–biotin binding)27 to extend our concept of noncovalent mimicking of PCVs. We then assessed the most comprehensive LEPS design to date (including 24 CPSs) using a single-dose escalation study. In summary, the successful outcomes reported herein represent a significant step in the development of a universal pneumococcal disease vaccine.
RESULTS AND DISCUSSION
Risk Assessment for Off-Target Immunogenicity Associated with GlpO.
Although no negative toxicological scoring was observed within initial immunization studies,24,26,28 risk of off-target effects associated with inclusion of GlpO in the LEPS vaccine were identified through a protein sequence homology search (i.e., BLASTp; >50% homology threshold) conducted for the GlpO and PncO proteins against human commensal bacteria. While no hits were identified for PncO, a substantial number of microflora protein targets were found to possess >50% homology to GlpO (Supplementary Table 1). From the subset of bacteria identified, ten species, representing variable degrees of GlpO homology and physiological microbiota locations, were selected for further experimental analysis.
An immune-absorbance assay was first used to determine whether serum antibodies from mice vaccinated with S. pneumoniae GlpO demonstrate off-target bacterial binding. Antibody binding to bacterial species was observed relative to a sham vaccination (Figure 1a). Furthermore, antibody binding for several species was comparable to results obtained for BFR S. pneumoniae EF3030 (serotype 19F), which is the direct target of the GlpO vaccine. When the immune-absorbance assay results were evaluated in the context of protein homology (Supplementary Figure 1), a significant positive correlation (R2 = 0.858) was observed between strength of binding and sequence homology. Given the observed immune-absorbance results, an OPA assay was performed using four representative microbiota bacterial species to determine whether the resulting antibody binding also promoted OPA. As shown in Figure 1b, significant killing activity was observed for each bacterial species compared to BFR EF3030. Combined, these results suggest that vaccination with GlpO could impact the human microbiome.
Figure 1.
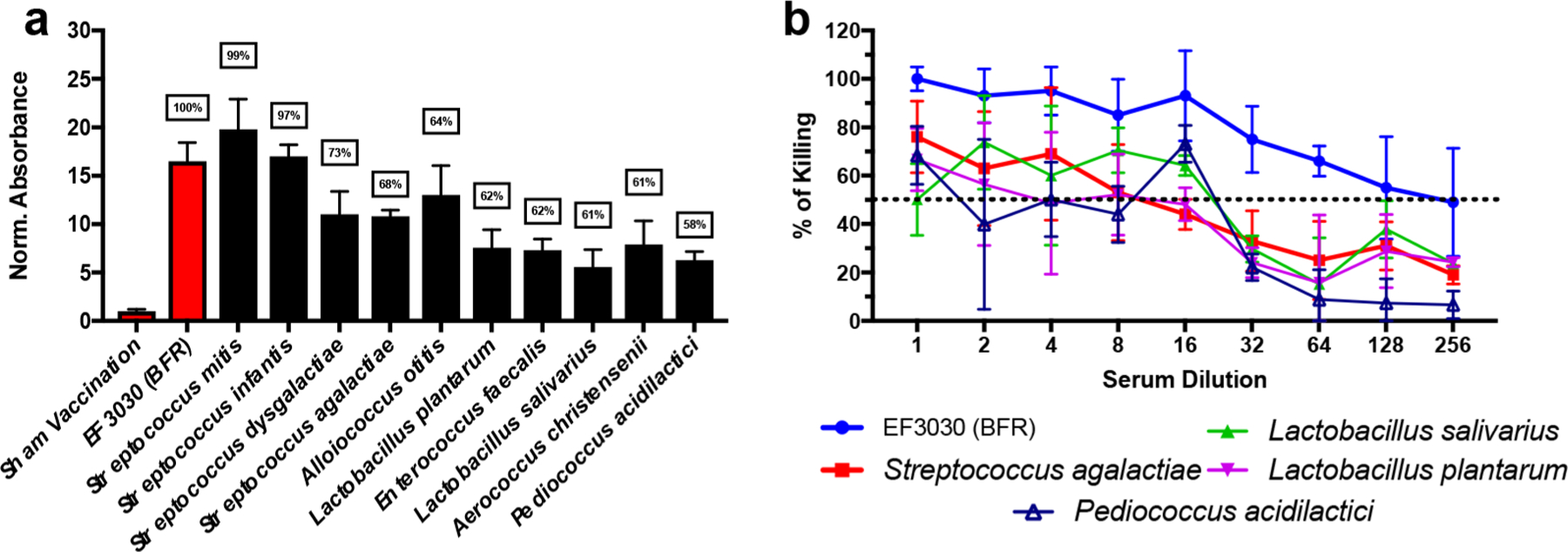
Vaccination with GlpO demonstrates a risk for potential off-target immunogenicity. (a) Normalized absorbance relative to sham vaccination for each respective bacterial species; boxed numbers represent homology shared with predicted protein epitopes of GlpO. (b) OPA assay results for BFR S. pneumoniae EF3030, S. agalactiae, L. salivarius, L. platarum, and P. acidilactici. The dashed line represents 50% killing of bacteria. Error bars represent the 95% confidence interval of four technical replicates.
GlpO-associated off-target reactivity represents a substantial concern since our initial studies demonstrated that vaccination with PncO alone could not provide 100% protection against breakthrough pneumococcal disease.24 Although we only evaluated a subset of the bacterial species with GlpO homologues, we were concerned due to the potential for cross reactivity against microflora not associated with the upper respiratory tract (e.g., intestinal microflora). Disruption of the beneficial human gut microbiome has been shown to have harmful repercussions on human health (e.g., nutrition).29 Furthermore, previous studies have demonstrated that the microbiome plays an important role in enhancing vaccine efficacy (e.g., oral rotavirus vaccine).30,31 Consequently, vaccination of infants with GlpO could result in potentially severe side effects and reduce efficacy for other routine pediatric vaccines. We therefore sought to improve the efficacy and immunogenicity of a LEPS vaccine formulated with PncO alone to justify removal of GlpO.
Protein Antigen Dose Escalation for the LEPS(20V) Formulation.
In an effort to establish dose limits and assess individual protein antigen effectiveness, we conducted immunogenicity and OPA assessment of the 20-valent LEPS (LEPS(20V)) vaccine formulation with varying doses of protein. The experimental plan would also test the potential of increased PncO levels to compensate for the absence of GlpO in vaccines designed to eliminate potential off-targets effects on the microbiome.
To formulate the LEPS vaccine, CPSs for each of the 20 serotypes were individually encapsulated within a liposome composed of DOPC/DOPG/DOGS-NTA-Ni/cholesterol/DSPE-PEG2000. All 20 variations of the LEPS particle were individually surface-decorated with His-tagged PncO at various protein concentrations. Attachment of PncO to liposomes was achieved through the noncovalent binding between the His tag and the DOGS-NTA-Ni lipid. Any unbound protein was removed using dialysis, and the final LEPS particles were combined to form LEPS(20V)/PncO.
Vaccine efficacy was determined using a BFR pneumonia challenge model spanning six bacterial serotypes (9N [covered by current vaccines and LEPS(20V)] and 6C, 15C, 16F, 23A, and 35B [NVTs]). PCV13, PPSV23, and a LEPS formulation containing CRM197 were included as benchmarks. As shown in Figure 2, vaccination with GlpO, PncO, and GlpO+PncO substantially improved survival for all tested serotypes compared to PCV13 and PPSV23. Furthermore, vaccination with GlpO and/or PncO in LEPS(20V) improved survival across all serotypes compared to LEPS(20V) formulated with CRM197. This indicates that protection against BFR NVT bacteria is due to inclusion of the pneumococcal breakthrough virulence proteins. Although incomplete protection was observed for GlpO or PncO against the 6C, 15C, 16F, and 23A serotypes at the initial dose of 34 μg, increasing the dose restored 100% protection comparable to vaccine formulations combining the proteins. There is also a trend of improved protection of mice subjects when using PncO relative to GlpO (6C [64 μg dose], 15C [34 μg dose], 16F [34 and 68 μg doses], and 23A [34 and 68 μg doses]).
Figure 2.
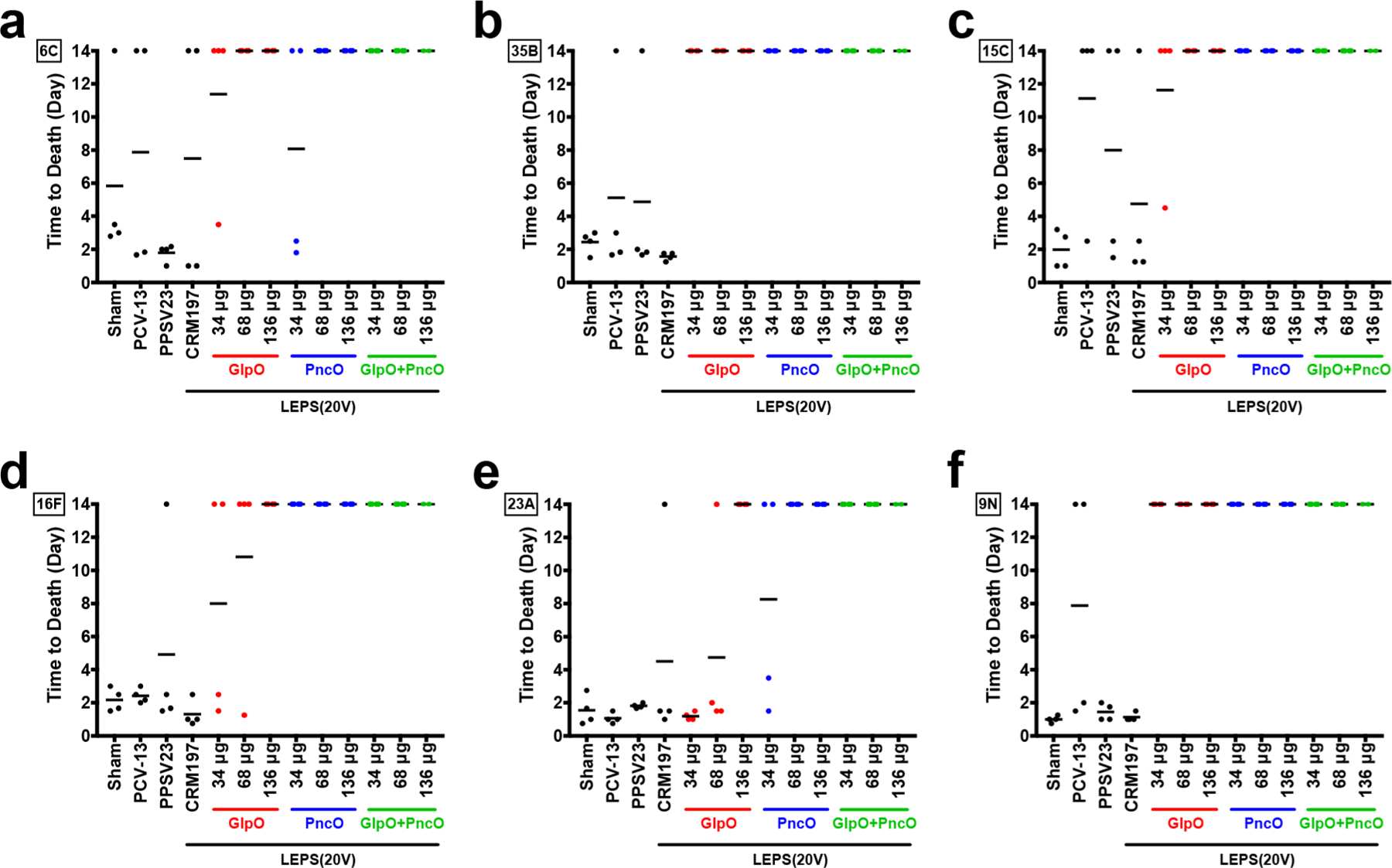
Time to death in BFR pneumonia challenge model. Serotypes tested included 6C (a), 35B (b), 15C (c), 16F (d), 23A (e), and 9N (f). Four CD-1 mice were vaccinated with a sham control, PCV13, PPSV23, and LEPS(20V) with CRM197, GlpO, PncO, and GlpO+PncO. Doses represent micrograms of each protein (34, 68, or 136 μg). Each dot represents an individual mouse, and black lines represent the average time to death.
To evaluate immunogenicity, the production and neutralization activity of antibodies resulting from vaccination were measured. When quantifying antibody titers across doses for GlpO (Supplementary Figure 2), PncO (Figure 3a–c), and GlpO+PncO (Supplementary Figure 3), production of antibodies across all CPS and protein antigens was observed. Neutralization activity of elicited antibodies was then measured using OPA assays with either planktonic or BFR S. pneumoniae serotypes (Supplementary Tables 2 and 3). Interestingly, we observed that increasing the PncO dose from 34 to 68 μg improved relative antibody titers for all 20 CPS (Supplementary Table 8) and improved OPA for 14/20 serotypes (Supplementary Table 9), even though CPS content remained constant. Although this phenomenon seems unintuitive, it is possible that increased localization of protein to the liposome surface simultaneously increases immunogenicity toward both the liposomal surface and its cargo. For example, a prior study observed that adsorption of hemagglutinin (HA) to liposomes increased immunogenicity relative to encapsulated HA.32 In addition, coadministration of Haemophilus influenza and meningococcal C vaccines containing the same carrier protein enhanced immunogenicity toward the attached sugar.33,34 However, further doubling the PncO dose to 136 μg decreased both relative antibody titers and OPA assay performance (Supplementary Tables 8 and 9). It is likely that this further increase in PncO dose suppresses antibody production against the enclosed CPS, which is consistent with prior observations for increased carrier content.35,36 The results indicate that increasing surface localization of PncO to intermediate concentrations can increase CPS immunogenicity and vaccine efficacy while enabling a single virulence-specific protein antigen to prevent breakthrough pneumococcal disease.
Figure 3.
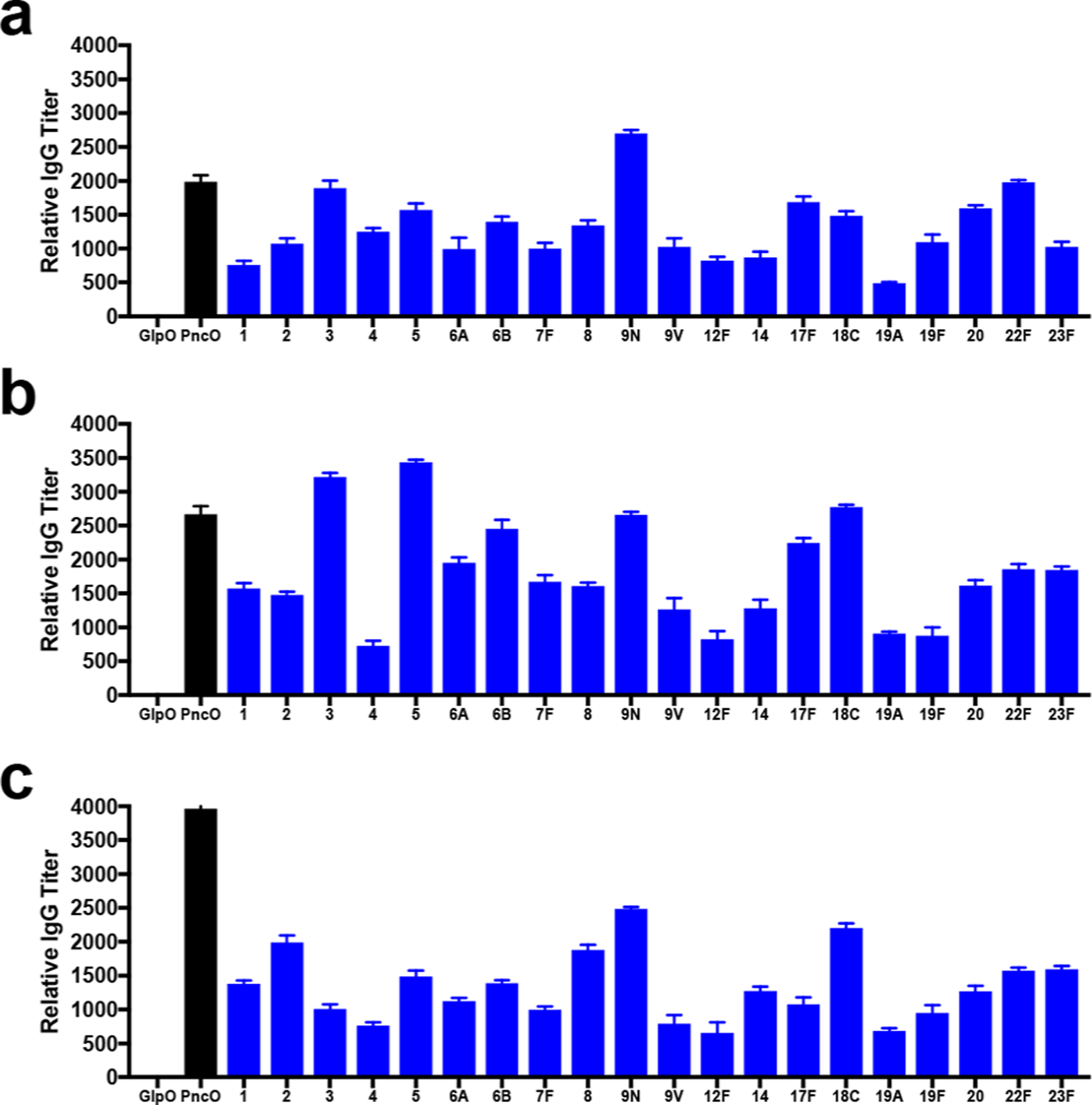
Relative antibody titers for protein and CPS antigens in the LEPS(20V)/PncO dose escalation study. CD-1 mice were vaccinated subcutaneously with LEPS(20V)/PncO at 34 μg (a), 68 μg (b), and 136 μg (c) of PncO. Error bars represent the 95% confidence interval from four biological replicates.
PncO Dose Escalation in an IAV Pneumonia Challenge Model.
Due to pending removal of GlpO from a next-generation LEPS vaccine, we conducted an additional confirmation of PncO efficacy as a stand-alone protein antigen using an influenza A virus (IAV) pneumonia challenge model (which triggers in vivo biofilm release of virulent pneumococci). Mice colonized with EF3030 (19F) S. pneumoniae were inoculated with IAV to promote bacterial dissemination from the nasopharynx biofilm into the blood and lungs. As shown in Figure 4, increasing the immunization dose of PncO decreased the bacterial load across each location. Furthermore, increasing PncO doses provided more rapid clearance of bacteria from all four anatomical sites. For example, reduction of CFU in the NP below a log10 of three was observed after 2, 4, 9, and 12 days for PncO doses of 136, 68, 34, and 17 μg, respectively. PncO doses above 68 μg typically prevented dissemination of bacteria into the lungs and blood, with only a marginal improvement observed when the dose was increased from 68 to 136 μg. Regardless, this study further demonstrates that increasing the dose of PncO beyond the initial 17 μg level reduces S. pneumoniae propagation and dissemination.
Figure 4.
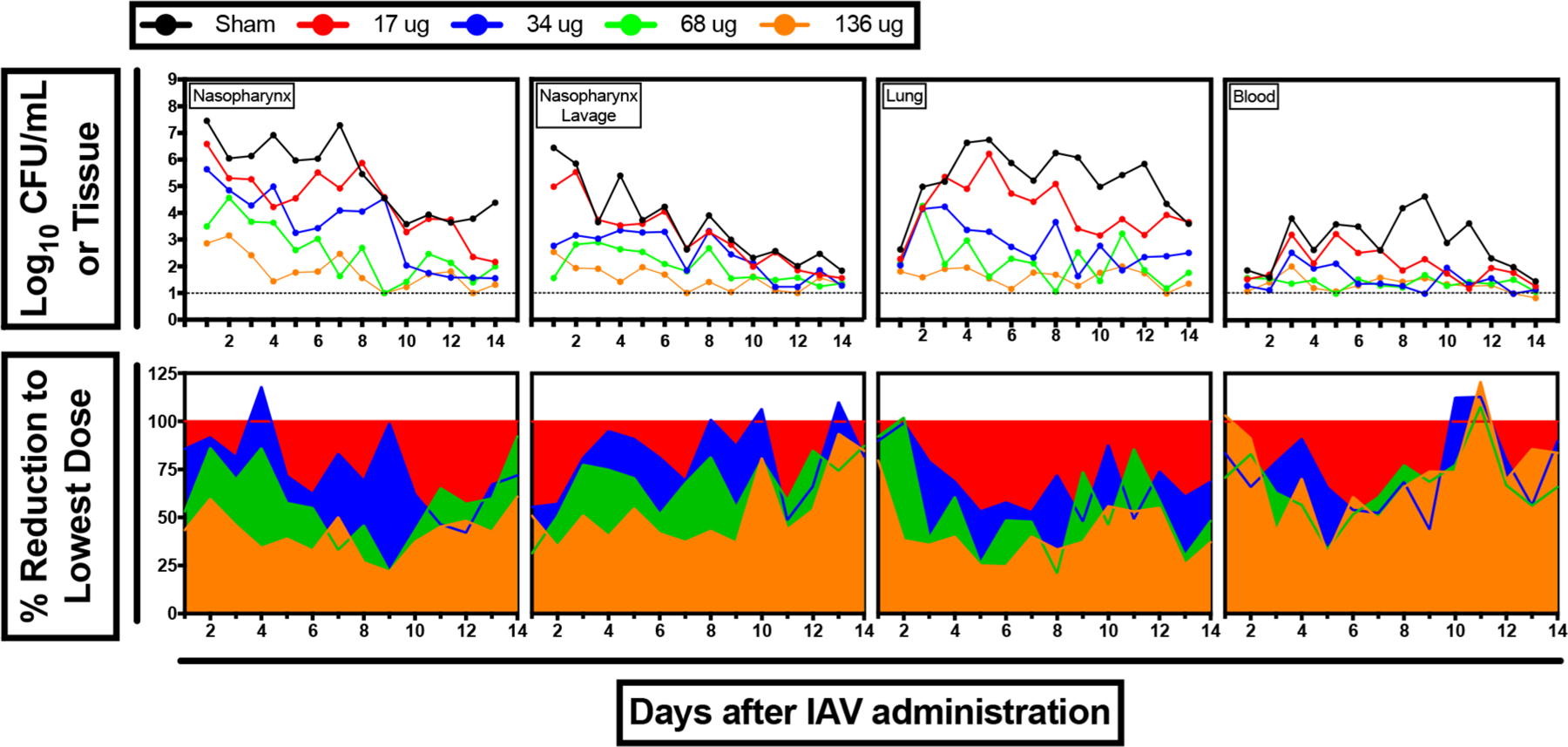
IAV pneumonia challenge for PncO dose escalation. (a) Bacterial load in various physiological locations. (b) Effect of vaccination on bacterial burden (i.e., percent reduction) relative to 17 μg dose across physiological location.
Building a 24-Valent LEPS Vaccine.
To further establish the general scaling flexibility of the LEPS vaccine platform, four new serotype CPSs (10A, 11A, 15B, and 33F) were introduced to the 20-valent formulation and tested for subsequent immunogenicity and efficacy. These serotypes were selected to encompass all remaining serotypes included within currently licensed vaccines. Immunogenicity was evaluated initially for each CPS separately as shown in Figure 5a,b. Potent antibody production and IgM to IgG class switching were achieved with each CPS, a key hallmark of PCV immune activation and consistent with our previous studies using the LEPS platform.26
Figure 5.
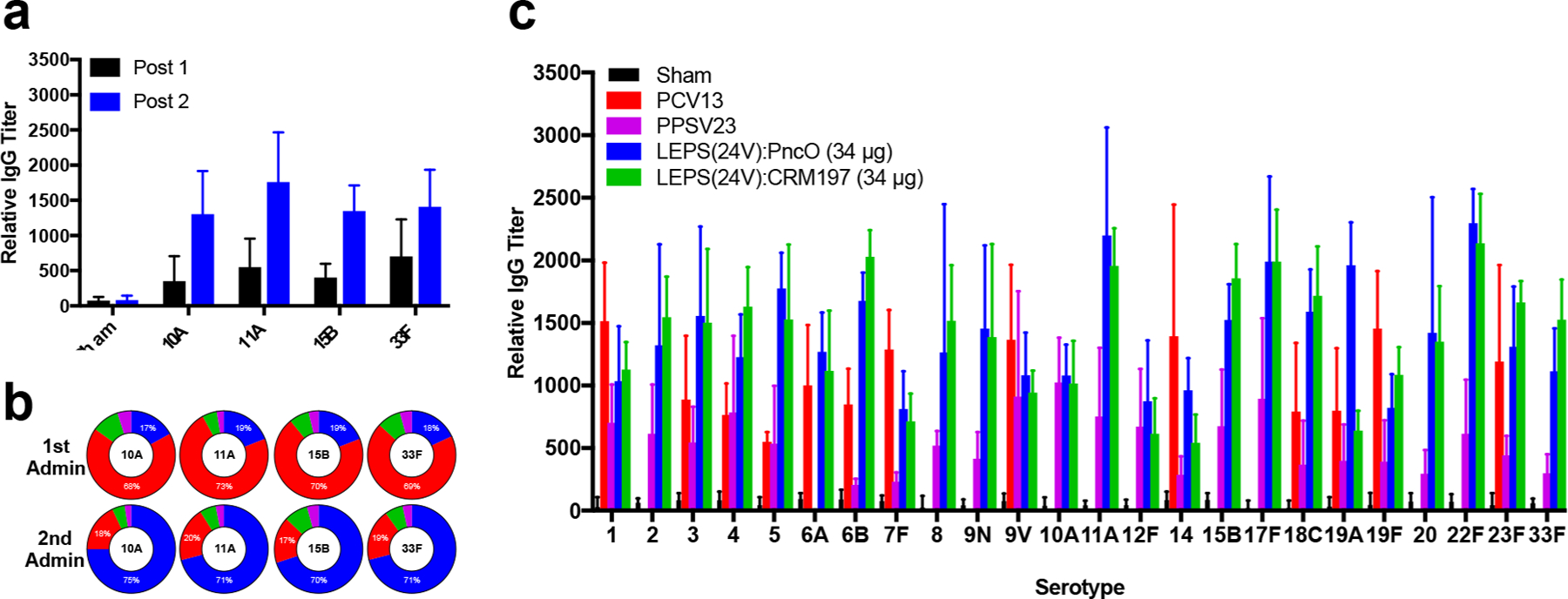
Expansion of LEPS vaccine to include 10A, 11A, 15B, and 33F CPS. (a) Relative antibody titers for each CPS post-dose 1 and 2 administration. (b) Composition of CPS antibody pool segmented by IgG (blue), IgM (red), IgA (green), and IgE (purple) post-vaccination. (c) Relative antibody titers resulting from sham, PCV13, PPSV23, LEPS(24V)/PncO, and LEPS(24V)/CRM197. Error bars represent the 95% confidence interval of four biological replicates.
Following individual testing, the four new CPS liposomes were combined with the LEPS(20V) formulation to create a 24-valent vaccine (LEPS(24V)). Relative antibody titers across all 24 serotypes were quantified and benchmarked to current licensed vaccines (i.e., PCV13 and PPSV23). As shown in Figure 5c, LEPS(24V) formulated with either PncO or CRM197 produced significant titers of CPS antibodies for all serotypes with comparable performance to PCV13 and PPSV23. Both the CRM197 and PncO LEPS(24V) formulations generated OPA titers comparable to PCV13 across all shared and unshared serotypes (Supplementary Table 4). These results indicate that LEPS provides a generalizable and scalable process for assembling a CPS-based vaccine against S. pneumoniae.
Developing Alternative Protein–Liposome Attachment Strategies.
A key feature of the LEPS platform is the noncovalent surface localization of his-tagged proteins (by using lipids containing nickel-NTA) and the subsequent simplification of final vaccine assembly relative to the covalent methods used in current commercial PCVs.26 Only 3.8 μg of nickel or cobalt is delivered per dose (daily parenteral dose limits are 22 and 5 μg for nickel and cobalt, respectively),37 and we have not observed any adverse reactions during immunization studies to date. However, potential toxicity and/or adverse immunogenicity concerns are associated with heavy metals such as nickel and his-tagged chelation components.38 Thus, to address such concerns prior to clinical translation, we assessed alternative noncovalent attachment methods for carrier or antigen proteins. Initial alterations included variations in the chelation metal (nickel [Ni2+] vs cobalt [Co2+]). In addition, two biotin–streptavidin systems were evaluated through a biotinylated lipid (DSPE-PEG-biotin) incorporated into the liposome. In one system, biotinylated liposomes were incubated with streptavidin and biotinylated CRM197 to create a biotin–streptavidin–biotin (BSB) linkage. In the alternative system, biotinylated liposomes were incubated with streptavidin-linked PspA (pneumococcal surface protein A, an additional S. pneumoniae protein antigen39) to form a biotin–streptavidin (BS) linkage.
An electrophoretic mobility shift assay (EMSA) was utilized to determine the fraction of protein bound to the surface of the liposomes. As shown in Figure 6a, all liposomal variants provided at least 50% protein binding, with both the BS and BSB strategies demonstrating comparable or enhanced binding relative to metal chelation options across all incubation times. Interestingly, when these formulations were utilized to deliver the 19F CPS, surface protein binding efficiency served as a general proxy for immunogenicity. Following immunization of mice, the degree of CPS antibody class switching (Figure 6b) and titer (Figure 6c) correlated with protein binding efficiency for each formulation. Specifically, the BS formulation outperformed metal-NTA samples in both antibody titer and antibody class switching. Furthermore, these trends extended to neutralization activity as measured by OPA (Figure 6d). Finally, all formulations were characterized using dynamic light scattering to estimate liposome size distribution and surface charge. As shown in Supplementary Table 5, properties across liposomal formulations were similar. However, the inclusion of proteins and polysaccharides increased liposome size, and the inclusion of proteins resulted in an increased negative charge at the liposome surface. The combined results highlight the flexibility of the LEPS platform to retain efficacy and structural features across formulation variations designed to facilitate clinical translation and utility.
Figure 6.
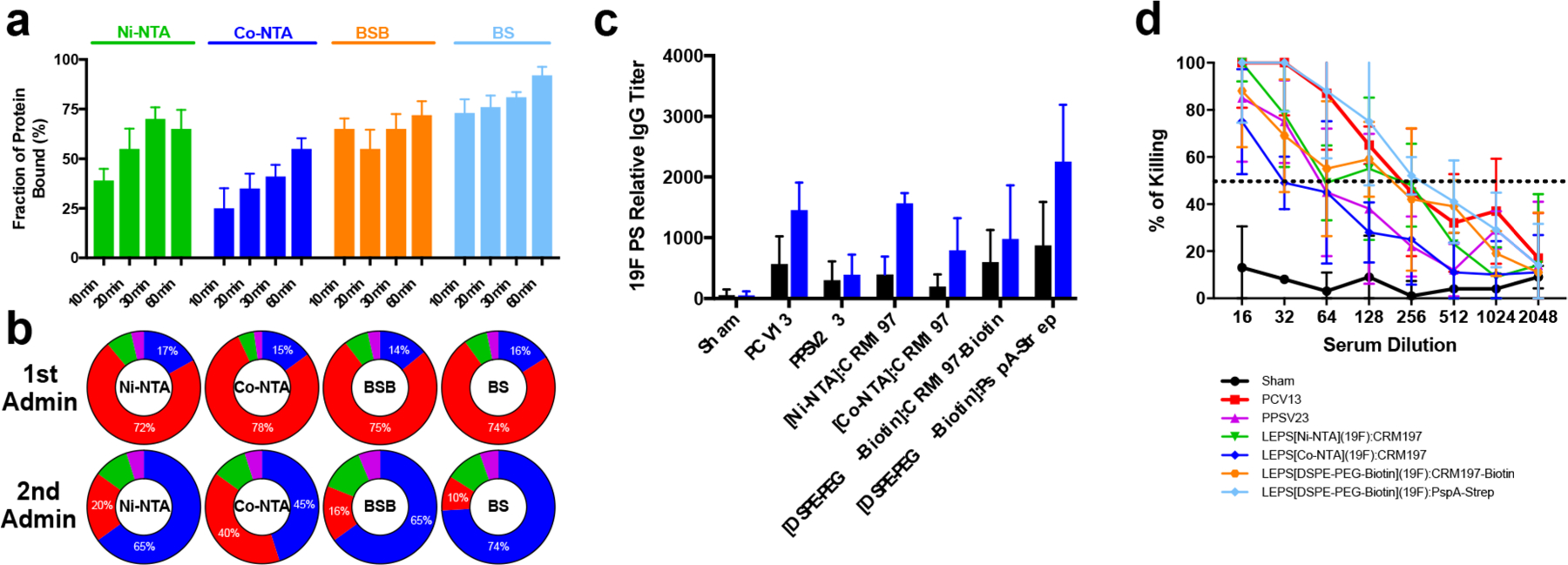
Characterization of alternative protein–liposome attachment strategies. (a) Fraction of protein bound to liposomes as measured by an EMSA assay using Ni-NTA, Co-NTA, BSB, and BS attachment. Incubations were conducted for 10, 20, 30, or 60 min. Error bars represent the standard deviation of three technical replicates. (b) Composition of CPS antibody pool segmented by IgG (blue), IgM (red), IgA (green), and IgE (purple) post-vaccination. (c) Relative antibody titer for alternative protein–liposome attachment formulations relative to PCV13 and PPSV23 following first (black) and second (blue) administrations. Error bars represent the standard deviation of four biological replicates. (d) OPA assay using alternative protein–liposome attachment formulations; dashed line represents 50% killing of bacteria. Error bars represent the 95% confidence interval of four replicates.
Single-Dose Assessment of LEPS Acute Toxicity.
We next performed a single-dose escalation study in CD-1 mice to evaluate acute toxicity and identify a maximum tolerated dose (MTD) for the LEPS vaccine. Animals were vaccinated with an initial treatment dose of 34 μg of protein and 2.2 μg of each CPS (except 6B [4.4 μg]) as well as 5× and 10× doses. As shown in Supplementary Table 6, both male and female mice tolerated up to 10× the treatment doses of protein and CPS in LEPS.
During this study, hematological effects associated with vaccination were determined by measuring metabolite and leukocyte concentrations in blood (Tables 1 and 2). The blood levels of various chemicals were measured to evaluate hepatotoxicity [alanine aminotransferase (ALAT), alkaline phosphatase (AP), aspartate aminotransferase (ASAT)], nephrotoxicity (creatine, urea), anemia (hemoglobin, hematocrite), hyperlipidemia (cholesterol, triglycerides), and electrolyte disturbances (sodium, potassium) associated with vaccination. As shown in Table 1, the levels of various chemicals in the blood were mostly similar across all doses. Although the concentration of sodium was statistically different across different groups or relative to the sham control, there was no discernible pattern in the concentrations measured. It was also observed that the concentration of different leukocytes was similar across all groups (Table 2). Although a slight increase in lymphocytes associated with increasing vaccine dose was observed, suggesting a potential immune response, the differences were not statistically significant. Despite observation of mild reactogenecity at the 10× dose upon vaccination, it was limited to initial soreness and irritation that did not cause prolonged distress. The results demonstrate that the LEPS vaccine does not produce pronounced acute toxicity in animals.
Table 1.
Concentration for Metabolites in Blood for CD-1 Mice
| sham |
alum/PncO |
LEPS(20V)/CRM197 |
|||||
|---|---|---|---|---|---|---|---|
| molecule | – | 1× | 5× | 10× | 1× | 5× | 10× |
| Male CD-1 Mice (N = 5) | |||||||
| ALAT (U/L) | 36.0 ± 5.0 | 36.7 ± 4.9 | 35.6 ± 4.2 | 34.9 ± 6.3 | 36.7 ± 4.3 | 36.7 ± 6.1 | 34.9 ± 4.7 |
| AP (U/L) | 140.0 ± 30.0 | 141.4 ± 36.3 | 134.4 ± 27.9 | 135.8 ± 30.6 | 145.6 ± 27.6 | 147.0 ± 24.0 | 133.0 ± 30.9 |
| ASAT (μmol/L) | 70.0 ± 14.0 | 71.4 ± 12.0 | 69.3 ± 10.9 | 70.7 ± 16.7 | 70.0 ± 16.8 | 70.0 ± 17.2 | 68.6 ± 10.9 |
| bilirubine (mmol/L) | <15 | <15 | <15 | <15 | <15 | <15 | <15 |
| calcium (mmol/L) | 2.5 ± 0.0 | 2.6 ± 0.1 | 2.5 ± 0.1 | 2.5 ± 0.0 | 2.5 ± 0.0 | 2.5 ± 0.1 | 2.4 ± 0.1 |
| cholesterol (mmol/L) | 3.7 ± 0.8 | 3.8 ± 0.7 | 3.8 ± 0.9 | 3.7 ± 0.9 | 3.9 ± 0.9 | 3.8 ± 1.0 | 3.8 ± 0.7 |
| creatinine (μmol/L) | 27.0 ± 2.1 | 28.4 ± 2.1 | 26.5 ± 2.2 | 25.7 ± 2.2 | 27.0 ± 2.3 | 25.9 ± 2.5 | 25.7 ± 1.6 |
| glucose (mmol/L) | 10.8 ± 1.1 | 11.3 ± 1.2 | 10.3 ± 1.3 | 10.3 ± 1.2 | 11.3 ± 1.0 | 10.5 ± 1.0 | 11.1 ± 1.1 |
| hematocrite (L/L) | 0.4 ± 0.0 | 0.4 ± 0.0 | 0.4 ± 0.0 | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.4 ± 0.0 |
| hemoglobin (mmol/L) | 8.0 ± 0.5 | 7.7 ± 0.6 | 7.9 ± 0.5 | 8.2 ± 0.5 | 7.6 ± 0.5 | 8.3 ± 0.6 | 7.9 ± 0.5 |
| LD (U/L) | 455.0 ± 144.0 | 464.1 ± 122.4 | 464.1 ± 133.9 | 450.5 ± 159.8 | 459.6 ± 175.7 | 468.7 ± 169.9 | 459.6 ± 147.2 |
| potassium (mmol/L) | 4.1 ± 0.2 | 4.3 ± 0.1 | 4.3 ± 0.2 | 4.0 ± 0.1 | 3.9 ± 0.2 | 4.2 ± 0.2 | 4.2 ± 0.2 |
| phosphate (mmol/L) | 2.2 ± 0.3 | 2.1 ± 0.3 | 2.1 ± 0.3 | 2.2 ± 0.2 | 2.1 ± 0.2 | 2.1 ± 0.3 | 2.2 ± 0.4 |
| sodium (mmol/L) | 150.0 ± 2.0 | 157.5 ± 2.3 | 151.5 ± 1.5 | 147.0 ± 1.9 | 148.0 ± 2.0 | 151.0 ± 2.0 | 147.0 ± 1.5 |
| triglyceride (mmol/L) | 1.5 ± 0.4 | 1.4 ± 0.5 | 1.4 ± 0.5 | 1.4 ± 0.5 | 1.5 ± 0.4 | 1.5 ± 0.4 | 1.4 ± 0.5 |
| urea (mmol/L) | 7.8 ± 0.8 | 8.1 ± 1.0 | 7.6 ± 0.7 | 8.1 ± 0.8 | 7.6 ± 1.0 | 7.9 ± 0.6 | 8.1 ± 0.6 |
| Female CD-1 Mice (N = 5) | |||||||
| ALAT (U/L) | 3.4 ± 8.0 | 34.0 ± 6.6 | 34.7 ± 8.2 | 34.7 ± 7.5 | 34.0 ± 7.9 | 33.0 ± 8.6 | 35.7 ± 8.2 |
| AP (U/L) | 144.0 ± 31.0 | 148.3 ± 38.4 | 146.9 ± 28.8 | 139.7 ± 31.3 | 139.7 ± 30.4 | 146.9 ± 35.0 | 146.9 ± 28.8 |
| ASAT (μmol/L) | 78.0 ± 14.0 | 78.0 ± 12.6 | 78.0 ± 10.9 | 74.1 ± 16.4 | 78.8 ± 16.2 | 76.4 ± 15.7 | 79.6 ± 15.8 |
| bilirubine (mmol/L) | <15 | <15 | <15 | <15 | <15 | <15 | <15 |
| calcium (mmol/L) | 2.5 ± 0.0 | 2.4 ± 0.0 | 2.4 ± 0.0 | 2.5 ± 0.0 | 2.4 ± 0.0 | 2.4 ± 0.0 | 2.4 ± 0.0 |
| cholesterol (mmol/L) | 3.0 ± 0.5 | 3.0 ± 0.4 | 2.9 ± 0.6 | 3.0 ± 0.5 | 3.0 ± 0.6 | 3.0 ± 0.5 | 3.0 ± 0.5 |
| creatinine (μmol/L) | 30.0 ± 2.0 | 28.8 ± 1.8 | 28.5 ± 1.8 | 31.5 ± 2.0 | 29.1 ± 2.2 | 31.5 ± 2.0 | 28.8 ± 2.4 |
| glucose (mmol/L) | 8.4 ± 0.9 | 8.8 ± 1.0 | 8.8 ± 1.1 | 8.7 ± 1.0 | 8.7 ± 1.0 | 8.1 ± 1.0 | 8.8 ± 0.7 |
| hematocrite (L/L) | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.4 ± 0.0 | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.4 ± 0.0 | 0.4 ± 0.0 |
| hemoglobin (mmol/L) | 8.6 ± 0.2 | 8.8 ± 0.2 | 8.5 ± 0.2 | 8.6 ± 0.2 | 9.0 ± 0.2 | 8.7 ± 0.2 | 9.0 ± 0.2 |
| LD (U/L) | 678.0 ± 100 | 698.3 ± 124.0 | 691.6 ± 76.0 | 684.8 ± 114.0 | 705.1 ± 77.0 | 698.3 ± 81.0 | 664.4 ± 108.0 |
| potassium (mmol/L) | 3.8 ± 0.2 | 3.6 ± 0.2 | 3.7 ± 0.2 | 3.8 ± 0.2 | 3.6 ± 0.2 | 3.8 ± 0.2 | 3.8 ± 0.2 |
| phosphate (mmol/L) | 2.6 ± 0.2 | 2.7 ± 0.2 | 2.5 ± 0.2 | 2.6 ± 0.1 | 2.7 ± 0.2 | 2.5 ± 0.2 | 2.5 ± 0.2 |
| sodium (mmol/L) | 153.0 ± 2.0 | 146.9 ± 2.4 | 146.9 ± 1.5 | 157.6 ± 1.7 | 148.4 ± 2.2 | 151.5 ± 2.0 | 160.7 ± 2.0 |
| triglyceride (mmol/L) | 1.0 ± 0.3 | 1.0 ± 0.2 | 1.0 ± 0.2 | 1.0 ± 0.3 | 1.0 ± 0.3 | 1.0 ± 0.2 | 1.0 ± 0.3 |
| urea (mmol/L) | 7.1 ± 1.3 | 7.3 ± 1.5 | 6.9 ± 1.6 | 7.3 ± 1.6 | 7.2 ± 1.0 | 7.5 ± 1.3 | 7.3 ± 1.4 |
Table 2.
Hematological Assessment of Leukocytes in CD-1 Mice
| sham |
alum/PncO |
LEPS(20V)/CRM197 |
|||||
|---|---|---|---|---|---|---|---|
| blood cell | – | 1× | 5× | 10× | 1× | 5× | 10× |
| Male CD-1 Mice (N = 5) | |||||||
| basophiles | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 |
| eosinophiles | 3.1 ± 2.2 | 3.2 ± 2.0 | 2.8 ± 2.1 | 2.9 ± 2.2 | 2.9 ± 2.5 | 3.1 ± 2.5 | 3.0 ± 2.5 |
| lymphocytes | 79.1 ± 5.8 | 80.8 ± 2.7 | 81.4 ± 2.4 | 82.4 ± 3.8 | 80.4 ± 1.4 | 80.7 ± 3.4 | 82.3 ± 4.0 |
| monocytes | 8.7 ± 5.4 | 8.0 ± 5.2 | 8.3 ± 5.6 | 7.4 ± 2.5 | 8.5 ± 4.4 | 8.3 ± 4.1 | 7.8 ± 6.2 |
| neutrophiles | 8.0 ± 2.9 | 8.0 ± 3.3 | 7.3 ± 2.5 | 7.2 ± 2.3 | 8.4 ± 3.0 | 7.8 ± 3.4 | 7.3 ± 3.5 |
| erythrocytes | 7.5 ± 0.3 | 7.2 ± 0.3 | 7.1 ± 0.2 | 7.9 ± 0.3 | 7.9 ± 0.3 | 7.7 ± 0.2 | 7.4 ± 0.2 |
| leukocytes | 6.1 ± 1.3 | 6.7 ± 1.1 | 6.8 ± 1.0 | 6.5 ± 1.2 | 6.1 ± 1.0 | 6.2 ± 1.3 | 6.9 ± 1.0 |
| thrombocytes | 1100.0 ± 150.0 | 1122.0 ± 160.5 | 1089.0 ± 126.0 | 1056.0 ± 153.0 | 1155.0 ± 124.5 | 1078.0 ± 156.0 | 1100.0 ± 147.0 |
| Female CD-1 Mice (N = 5) | |||||||
| basophiles | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| eosinophiles | 5.1 ± 2.3 | 5.3 ± 2.8 | 5.3 ± 2.5 | 5.4 ± 2.7 | 5.3 ± 1.9 | 5.2 ± 2.3 | 5.3 ± 2.8 |
| lymphocytes | 75.4 ± 5.1 | 78.2 ± 4.7 | 80.7 ± 5.0 | 78.9 ± 5.4 | 80.7 ± 3.9 | 76.2 ± 4.1 | 79.9 ± 6.2 |
| monocytes | 6.5 ± 2.7 | 6.6 ± 3.2 | 6.0 ± 2.1 | 6.7 ± 3.1 | 6.6 ± 2.8 | 6.4 ± 3.2 | 6.4 ± 3.1 |
| neutrophiles | 13.1 ± 4.3 | 10.0 ± 4.2 | 8.1 ± 4.0 | 9.2 ± 3.5 | 7.9 ± 4.1 | 12.6 ± 5.2 | 9.1 ± 4.2 |
| erythrocytes | 8.1 ± 0.3 | 8.1 ± 0.2 | 8.1 ± 0.2 | 7.9 ± 0.2 | 7.7 ± 0.3 | 8.5 ± 0.2 | 8.3 ± 0.3 |
| leukocytes | 4.2 ± 0.9 | 4.5 ± 0.7 | 4.5 ± 1.0 | 4.7 ± 1.0 | 4.5 ± 0.9 | 4.4 ± 0.7 | 4.2 ± 0.9 |
| thrombocytes | 936.0 ± 225.0 | 964.1 ± 265.5 | 982.8 ± 258.8 | 907.9 ± 229.5 | 973.4 ± 200.2 | 898.6 ± 177.8 | 945.4 ± 240.8 |
In conclusion, we have redesigned the LEPS formulation in preparation for clinical readiness by focusing on the PncO antigen, optimizing PncO dose, varying protein surface attachment mechanisms, and expanding CPS valency to 24 serotypes. These developments simplify not only vaccine clinical and regulatory strategies but also manufacturing by reducing the number of required antigen proteins. We also validated the importance of protein and CPS colocalization in LEPS by demonstrating a relationship between increased surface protein content and CPS immunogenicity, noting a correlation between immune response and liposome–protein surface attachment efficiency. In total, these results offer a next-generation LEPS vaccine capable of surpassing effectiveness of current polysaccharide-conjugate options.
MATERIALS AND METHODS
Ethics Statement.
This study was conducted in strict accordance with the guidelines in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). All protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo, Buffalo, NY, and all bacterial inoculations and treatments were performed under conditions designed to minimize any potential suffering of the animals.
Bacterial and Cellular Strains.
S. pneumoniae strains in this study included >70 serotypes,26 with S. pneumoniae EF3030 (serotype 19F) serving as a control in OPA and immunosorbent studies. Bacterial species listed in Supplementary Table 1 were used in microflora studies which included OPA and immunosorbent assays. NCI-H292 epithelial cells [CRL1849, American Type Culture Collection (ATCC)] were utilized to support biofilm growth. Human HL-60 cells (CCL-240, ATCC) were used in OPA assays.
Homology Search Description.
Protein homologue conservation in commensal bacterial species (Supplementary Table 1) was calculated by accounting for gap and mismatches in amino acid sequences, available in BLAST (http://blast.ncbi.nlm.nih.gov/). Surface accessibility was established using the InterPro database (http://www.ebi.ac.uk/interpro/), and epitope regions were predicted with the bepipred linear epitope prediction method using IEDB Analysis Resource (http://tools.immuneepitope.org/bcell/). The reference sequence used for this study was from S. pneumoniae D39 (National Center for Biotechnology Information Genome accession #CP000410.1).
Bacterial Growth and Seeding. The S. pneumoniae strains used in this study were initially grown on Todd-Hewitt agar plates supplemented with 0.5% yeast extract and 5% sheep blood (v/v) and were incubated overnight at 37 °C. Single colonies were transferred to 5 mL of Todd-Hewitt broth containing 0.5% yeast extract and were incubated at 37 °C to an OD600 of 0.6. For biofilm formation, bacteria were then diluted 1:10 in chemically defined medium (CDM) to an OD600 of approximately 0.5 mL, which was then added to each well of a 24-well plate containing a layer of fixed H292 cells. Alternatively, cultured S. pneumoniae were collected by centrifugation, washed once with and resuspended in PBS, and quantified by OD600 measurement for experiments requiring planktonic cells.
Bacteria utilized for the GplO off-target immunogenicity assessment were cultured following ATCC recommendations except Aerococcus christensenii. Streptococcus mitis, Streptococcus infantis, and Streptococcus agalactiae were cultured using brain heart influsion broth (ATCC Medium 44). Streptococcus dysgalactiae (ATCC Medium 18: trypticase Soy broth), Alloiococcus otitis (ATCC Medium 243: Mycoplasma medium), Lactobacillus salivarius (ATCC Medium 78: Lactobacillus medium), Lactobacillus plantarum (ATCC Medium 416: Lactobacilli MRS Agar/Broth), and Pediococcus acidilactici (ATCC Medium 416: Lactobacilli MRS Agar/Broth) were grown using the media indicated. All bacteria were cultured under aerobic conditions and at 37 °C with Streptococcus mitis, Lactobacillus plantarum, and Pediococcus acidilactici grown in an atmosphere containing 5% CO2. Aerococcus christensenii was cultured at 36 °C with 6% CO2 in sheep blood broth.40
H292 Cell Growth and Fixation.
NCI-H292 epithelial cells were first cultured in RPMI 1640 with the addition of fetal bovine serum (FBS) in T75 flasks at 37 °C and 5% CO2. Upon reaching 100% confluency, media was removed and cells were incubated with trypsin-EDTA solution until the cell layer was dispersed. Cells were then collected via centrifugation, diluted 1:8 in RPMI 1640 with FBS, transferred to 24-well plates, and grown as described above until reaching 100% confluency. After which, cells were washed three times with PBS, prefixed in 4% buffered paraformaldehyde at 34 °C for 48 h, and washed three additional times with PBS.
Biofilm Growth and Release.
CDM-grown pneumococci were seeded onto fixed H292 cells and incubated at 34 °C for 48 h. Throughout the biofilm growth phase, the medium was changed every 12 h. To promote biofilm-release of pneumococci, biofilms were exposed to heat (38.5 °C) for 4 h. Released cells were then collected by centrifugation, washed once with and resuspended in PBS, and quantified by OD600 measurement. Biofilm-released cells were used in specified pneumococcal disease pneumonia models, immune-absorbance assays, and OPA assays.
Protein Production and Purification.
All proteins (CRM197, GlpO, PncO, PspA) containing polyhistidine or streptavidin tags were produced in Escherichia coli BL21(DE3). Bacterial strains were inoculated into 3 mL of lysogeny broth (LB) and grown overnight at 37 °C. Bacteria were then transferred into 1 L of LB media, grown at 22 °C to an OD600 of 0.4 to 0.6, induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), and cultured overnight at 22 °C. Proteins were purified using a French press to disrupt the cells and passing cell lysate over a fast protein liquid chromatography column (GE Healthcare HisTrap HP, 1 × 1 mL). The final protein concentration was measured using the Pierce Micro BCA Protein Assay kit.
LEPS Formulation and Assembly.
LEPS carriers, which had a total lipid mass of 500 μg, were composed of (1) DOPC/DOPG/DOGS-NTA-Ni/cholesterol/DSPE-PEG2000, (2) DOPC/DOPG/DOGS-NTA-Co/cholesterol/DSPE-PEG2000, or (3) DOPC/DOPG/DOGS/cholesterol/DSPE-PEG-biotin at a molar ratio of 3:3:1:4:0.1. Lipids were dissolved in chloroform, sonicated for 1 min, evaporated to form a film using a rotary evaporator, rehydrated using a phosphate-buffered saline (PBS) solution containing a single capsular polysaccharide antigen to form the liposomal carrier, and passed through a hand-held extruder (10–12 times) with a pore size of 200 nm. Under standard conditions, individually prepared LEPS samples, each containing a different CPS, were incubated with protein antigens (CRM197, GlpO, PncO) containing a polyhistidine tag for 30 min at 4 °C to facilitate surface binding. When testing alternative surface attachment strategies (Figure 6), his-tagged proteins were incubated with liposomes containing DOGS-Ni-NTA or DOGS-Co-NTA for 10, 20, 30, or 60 min at 4 °C. Biotinylated protein antigens were first generated using an Amine Biotinylation Reagents Kit (ThermoFisher Scientific) prior to incubation with streptavidin (Sigma-Aldrich) at a 1:1 ratio for 10, 20, 30, or 60 min at 4 °C in solution with liposomes containing DSPE-PEG-biotin. Streptavidin-labeled proteins were separately incubated for 10, 20, 30, or 60 min at 4 °C with DSPE-PEG-biotin containing liposomes. For comparative surface attachment LEPS studies, protein levels were 34 μg. Unbound proteins or unencapsulated CPSs were removed via overnight dialysis in PBS at 4 °C. Individual samples were then combined to form the final 20- or 24-valent LEPS formulations. When testing alternative protein attachment LEPS formulations in antibody and OPA assays, liposomes formed using 30 min incubations were utilized.
Electrophoretic Mobility Shift Assay (EMSA).
EMSA experiments were performed with 2.5 μg of protein incubated with 50 μg of liposomes, followed by electrophoresis in a 0.75% agarose gel with 50 V applied for 90 min and imaging with an IVIS Lumina II system. This approach was utilized to measure the percent of protein bound to liposomes for the Ni-NTA, Co-NTA, biotin–streptavidin, and biotin–streptavidin–biotin LEPS systems.
Animal Immunization and Collection of Serum.
Outbred 6-week-old male and female CD-1 mice were used in immunization experiments. Mice were immunized via subcutaneous injection (200 μL). However, during toxicological assessments, mice were immunized intramuscularly. The background solution used for the formulations was PBS (“sham” negative controls were background solutions administered without antigen components). When protein antigens were used in isolation, administration levels were either 17 μg (Figure 1) or indicated (Figure 4), and both situations included coadministration of alum (as aluminum phosphate from Sigma-Aldrich; added according to the manufacturer’s instructions). Protein antigen concentrations were escalated with the following concentrations: 34, 68, and 136 μg. When combined, GlpO and PncO were administered at concentrations of 68, 136, and 272 μg. As positive controls, mice were also immunized with Prevnar 13 (PCV13) or Pneumovax 23 (PPSV23) as described previously.26 On day 14 postimmunization, mice were boosted with the same formulations. On days 14 and 28 post-immunization, serum samples were collected from the mice via retro-orbital bleeding for antibody and OPA analysis; if not indicated, antibody titers represent those from day 28 serum samples.
Measurement of Antibody Titers and Classes.
Antigen antibody titer analysis was conducted as described previously,28 with the method extended to include 20–24 CPSs from associated serotypes.
OPA Assay.
Extending upon a previous protocol,41 human HL-60 cells were differentiated with dimethlyformamide to quantify antibody-mediated opsonophagocytosis and killing of S. pneumoniae (both planktonic and BFR) and commensal bacteria (Figure 1b) exposed to dilutions of sera collected from immunized mice subjects to identify the 50% killing end point (quantified by CFU counts). HL-60 cells and pneumococci were incubated for 75 min.
Pneumococcal Challenge Models.
BFR and IAV pneumonia studies were conducted as described previously.24,26 Briefly, for BFR pneumonia models, mice were challenged with 1 × 106 CFU of pneumococci strains through intranasal (with isoflurane) administration. In IAV pneumonia models, mice were colonized by administering with 1 × 106 CFU of mouse-passaged planktonic S. pneumoniae EF3030 intranasally, without isoflurane. Next, to mimic influenza-induced pneumonia, pneumococci colonization was followed by intranasal inoculation with 40 plaque-forming units of IAV. The mouse-adapted IAV strain A/PR/8/34 (H1N1) (ATCC VR-95) was used, and titers were determined by plaque assays. Mice were monitored every 4 h for signs of morbidity (huddling, ruffled fur, lethargy, and abdominal surface temperature). Mice found to be moribund were euthanized via CO2 asphyxiation and cervical dislocation.
Bacterial Load Assessment.
At 48 h after infection or upon becoming moribund, mice were euthanized (as described previously), and a combination of nasopharynx tissue, nasopharyngeal lavage fluid, lung, and blood samples was collected and assessed for bacterial burden.42 Briefly, tissue and anatomical samples, lavage fluid, and blood were homogenized (on a setting of 10 for 30 s or until homogenized completely; Tissue-Tearor, BioSpec Products Inc.) to ensure dissociation of bacterial aggregates and then serially diluted on tryptic soy and 5% blood agar plates before enumeration.
Immune-Absorbance Assay.
Serum from GlpO-vaccinated mice was diluted at ratios of 1:10, 1:100, and 1:1000 and incubated in 96-well plates with the bacterial strains utilized in the study to quantify off-target GlpO effects (Supplementary Table 1) with 105 bacterial cells per well at 37 °C for 3 h. The plate was then blocked with 3% bovine serum albumin for 1 h at 22 °C. After binding, cells were washed with PBS and incubated with the secondary antibody (antimouse IgG) conjugated with alkaline phosphatase for 2 h at 37 °C.28 Reporter antibodies unbound to serum antibodies were removed through washing the cells with PBS. p-Nitro-phenylphosphate was then added to develop the signal, and the reaction was quenched using 0.75 M NaOH. The signal was detected using a plate reader spectrophotometer at an absorbance of 405 nm.
Toxicology Study Design.
Outbred 6-week male and female CD-1 mice were obtained from Charles River. Mice were injected intramuscularly with LEPS(20V)/CRM197 or alum/PncO where both CRM and PncO were formulated at 1×, 5×, and 10× of the initial dose (34 μg). Blood samples were collected via retro orbital bleeding approximately 6 h before and 2 days after immunization and subjected to MASCOT hematology profiling (Drew Scientific) according to the manufacturer’s protocol. For 2 weeks after immunization, mice were monitored for weight and behavioral changes. Mice were sacrificed 14 days after immunization via CO2 asphyxiation followed by cervical dislocation, and vital organs were harvested and weighed.
Statistical Analysis.
Statistical analysis was performed using the GraphPad Prism software (version 6.0h.283; GraphPad Software Inc., La Jolla, CA) to determine the 95% confidence intervals of all experimental samples and controls. Experimental sample numbers are provided in specific figure captions.
Supplementary Material
ACKNOWLEDGMENTS
The authors recognize support from NIH awards AI088485 and AI117309 (B.A.P.) and a SUNY-Buffalo Schomburg fellowship (C.H.J.). The authors also thank David Briles (University of Alabama at Birmingham) for plasmid pUAB055 (containing pspA) and Alejandro Hochkoeppler (University of Bologna) for the CRM197 genetic construct.
ABBREVIATIONS
- LEPS
liposomal encapsulation of polysaccharides
- CPS
capsular polysaccharide
- PCV
pneumococcal conjugate vaccine
- IPD
invasive pneumococcal disease
- NVT
non-vaccine-type
- NP
nasopharynx
- OPA
opsonophagocytic activity
- BFR
biofilm-released
- IAV
influenza A virus
- BSB
biotin–streptavidin–biotin
- BS
biotin–streptavidin
- MTD
maximum tolerated dose
- ALAT
alanine aminotransferase
- AP
alkaline phosphatase
- ASAT
aspartate aminotransferase
- LD
lactate dehydrogenase
- PCV13
Prevnar 13
- PPSV23
Pneumovax 23
Footnotes
The authors declare the following competing financial interest(s): C.H.J., A.B.H., and B.A.P. are cofounders of Abcombi Biosciences Inc., a company focused on vaccine design.
REFERENCES
- (1).O’Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, and Cherian T (2009) Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374 (9693), 893–902. [DOI] [PubMed] [Google Scholar]
- (2).Huang SS, Johnson KM, Ray GT, Wroe P, Lieu TA, Moore MR, Zell ER, Linder JA, Grijalva CG, Metlay JP, and Finkelstein JA (2011) Healthcare utilization and cost of pneumococcal disease in the United States. Vaccine 29 (18), 3398–3412. [DOI] [PubMed] [Google Scholar]
- (3).Centers for Disease Control and Prevention (2015) Pneumococcal Disease. In Epidemiology and Prevention of Vaccine-Preventable Diseases (Hamborsky J, Kroger A, and Wolfe C, Eds.) 13th ed., Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- (4).Weycker D, Strutton D, Edelsberg J, Sato R, and Jackson LA (2010) Clinical and economic burden of pneumococcal disease in older US adults. Vaccine 28 (31), 4955–4960. [DOI] [PubMed] [Google Scholar]
- (5).Thanawastien A, Cartee RT, Griffin TJ, Killeen KP, and Mekalanos JJ (2015) Conjugate-like immunogens produced as protein capsular matrix vaccines. Proc. Natl. Acad. Sci. U. S. A 112 (10), E1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Weintraub A (2003) Immunology of bacterial polysaccharide antigens. Carbohydr. Res 338 (23), 2539–47. [DOI] [PubMed] [Google Scholar]
- (7).CDC (2000) Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Streptococcus pneumoniae, 1999, Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- (8).CDC (2015) Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Streptococcus pneumoniae, 2015, Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- (9).Brueggemann AB, Peto TEA, Crook DW, Butler JC, Kristinsson KG, and Spratt BG (2004) Temporal and Geographic Stability of the Serogroup-Specific Invasive Disease Potential of Streptococcus pneumoniae in Children. J. Infect. Dis 190 (7), 1203–1211. [DOI] [PubMed] [Google Scholar]
- (10).Golubchik T, Brueggemann AB, Street T, Gertz RE, Spencer CCA, Ho T, Giannoulatou E, Link-Gelles R, Harding RM, Beall B, Peto TEA, Moore MR, Donnelly P, Crook DW, and Bowden R (2012) Pneumococcal genome sequencing tracks a vaccine escape variant formed through a multi-fragment recombination event. Nat. Genet 44 (3), 352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, and Bentley SD (2011) Rapid Pneumococcal Evolution in Response to Clinical Interventions. Science 331 (6016), 430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Brueggemann AB, Pai R, Crook DW, and Beall B (2007) Vaccine Escape Recombinants Emerge after Pneumococcal Vaccination in the United States. PLoS Pathog 3 (11), e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pai R, Moore MR, Pilishvili T, Gertz RE, Whitney CG, and Beall B (2005) Active Bacterial Core Surveillance Team, Postvaccine Genetic Structure of Streptococcus pneumoniae Serotype 19A from Children in the United States. J. Infect. Dis 192 (11), 1988–1995. [DOI] [PubMed] [Google Scholar]
- (14).Sleeman KL, Griffiths D, Shackley F, Diggle L, Gupta S, Maiden MC, Moxon ER, Crook DW, and Peto TEA (2006) Capsular Serotype–Specific Attack Rates and Duration of Carriage of Streptococcus pneumoniae in a Population of Children. J. Infect. Dis 194 (5), 682–688. [DOI] [PubMed] [Google Scholar]
- (15).Klugman KP, Chien Y-W, and Madhi SA (2009) Pneumococcal pneumonia and influenza: A deadly combination. Vaccine 27 (Supplement 3), C9–C14. [DOI] [PubMed] [Google Scholar]
- (16).McCullers JA, and Rehg JE (2002) Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J. Infect. Dis 186 (3), 341–50. [DOI] [PubMed] [Google Scholar]
- (17).Pettigrew MM, Marks LR, Kong Y, Gent JF, Roche-Hakansson H, and Hakansson AP (2014) Dynamic Changes in the Streptococcus pneumoniae Transcriptome during Transition from Biofilm Formation to Invasive Disease upon Influenza A Virus Infection. Infect. Immun 82 (11), 4607–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Marks LR, Davidson BA, Knight PR, and Hakansson AP (2013) Interkingdom Signaling Induces Streptococcus pneumoniae Biofilm Dispersion and Transition from Asymptomatic Colonization to Disease. mBio 4 (4), e00438–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wyres KL, Lambertsen LM, Croucher NJ, McGee L, von Gottberg A, Liñares J, Jacobs MR, Kristinsson KG, Beall BW, Klugman KP, Parkhill J, Hakenbeck R, Bentley SD, and Brueggemann AB (2013) Pneumococcal Capsular Switching: A Historical Perspective. J. Infect. Dis 207 (3), 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Coffey TJ, Daniels M, Enright MC, and Spratt BG (1999) Serotype 14 variants of the Spanish penicillin-resistant serotype 9V clone of Streptococcus pneumoniae arose by large recombinational replacements of the cpsA-pbp1a region. Microbiology (London, U. K.) 145 (8), 2023–2031. [DOI] [PubMed] [Google Scholar]
- (21).McEllistrem MC, Noller AC, Visweswaran S, Adams JM, and Harrison LH (2004) Serotype 14 variants of the France 9V(–3) clone from Baltimore, Maryland, can be differentiated by the cpsB gene. J. Clin. Microbiol 42 (1), 250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Albarracín Orio AG, Cortes PR, Tregnaghi M, Piñas GE, Argentinean Network Pneumococcus Study Group, and Echenique JR (2008) A new serotype 14 variant of the pneumococcal Spain9V-3 international clone detected in the central region of Argentina. J. Med. Microbiol 57 (8), 992–999. [DOI] [PubMed] [Google Scholar]
- (23).CDC (2015) Trends by Serotype Group, 1998–2015, Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- (24).Li Y, Hill A, Beitelshees M, Shao S, Lovell JF, Davidson BA, Knight PR, Hakansson AP, Pfeifer BA, and Jones CH (2016) Directed Vaccination Against Pneumococcal Disease. Proc. Natl. Acad. Sci. U. S. A 113 (25), 6898–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Beitelshees M, Li Y, and Pfeifer BA (2016) Enhancing Vaccine Effectiveness with Delivery Technology. Curr. Opin. Biotechnol 42, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jones CH, Zhang G, Nayerhoda R, Beitelshees M, Hill A, Rostami P, Li Y, Davidson BA, Knight P, and Pfeifer BA (2017) Comprehensive vaccine design for commensal disease progression. Sci. Adv 3 (10), e1701797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang F, Lu Y-J, and Malley R (2013) Multiple antigenpresenting system (MAPS) to induce comprehensive B- and T-cell immunity. Proc. Natl. Acad. Sci. U. S. A 110 (33), 13564–13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li Y, Beitelshees M, Fang L, Hill A, Ahmadi MK, Chen M, Davidson BA, Knight P, Smith RJ, Andreadis ST, Hakansson AP, Jones CH, and Pfeifer BA (2016) In situ Pneumococcal Vaccine Production and Delivery Through a Hybrid Biological-Biomaterial Vector. Sci. Adv 2 (7), e1600264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Institute of Medicine (2013) The National Academies Collection: Reports funded by National Institutes of Health. In The Human Microbiome, Diet, and Health: Workshop Summary, National Academies Press (US) National Academy of Sciences, Washington, D.C. [Google Scholar]
- (30).Harris VC, Armah G, Fuentes S, Korpela KE, Parashar U, Victor JC, Tate J, de Weerth C, Giaquinto C, Wiersinga WJ, Lewis KDC, and de Vos WM (2017) Significant Correlation Between the Infant Gut Microbiome and Rotavirus Vaccine Response in Rural Ghana. J. Infect. Dis 215 (1), 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jamieson AM (2015) Influence of the microbiome on response to vaccination. Hum. Vaccines Immunother 11 (9), 2329–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Barnier-Quer C, Elsharkawy A, Romeijn S, Kros A, and Jiskoot W (2013) Adjuvant effect of cationic liposomes for subunit influenza vaccine: influence of antigen loading method, cholesterol and immune modulators. Pharmaceutics 5 (3), 392–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dagan R, Poolman J, and Siegrist C-A (2010) Glycoconjugate vaccines and immune interference: A review. Vaccine 28 (34), 5513–5523. [DOI] [PubMed] [Google Scholar]
- (34).Pöllabauer EM, Petermann R, and Ehrlich HJ (2009) The influence of carrier protein on the immunogenicity of simultaneously administered conjugate vaccines in infants. Vaccine 27 (11), 1674–1679. [DOI] [PubMed] [Google Scholar]
- (35).Dagan R, Goldblatt D, Maleckar JR, Yaïch M, and Eskola J (2004) Reduction of Antibody Response to an 11-Valent Pneumococcal Vaccine Coadministered with a Vaccine Containing Acellular Pertussis Components. Infect. Immun 72 (9), 5383–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dagan R, Eskola J, Leclerc C, and Leroy O (1998) Reduced Response to Multiple Vaccines Sharing Common Protein Epitopes That Are Administered Simultaneously to Infants. Infect. Immun 66 (5), 2093–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).U. S. Food and Drug Administration, Center for Drug Evaluation and Research, and Center for Biologics Evaluation and Research (2015) Q3D Elemental Impurities Guidance for Industry, US Department of Health and Human Services, Washington, D.C. [Google Scholar]
- (38).Rappuoli R, and Bagnoli F (2011) Vaccine design: innovative approaches and novel strategies, Horizon Scientific Press, Poole, UK. [Google Scholar]
- (39).Shaper M, Hollingshead SK, Benjamin WH, and Briles DE (2004) PspA Protects Streptococcus pneumoniae from Killing by Apolactoferrin, and Antibody to PspA Enhances Killing of Pneumococci by Apolactoferrin. Infect. Immun 72 (9), 5031–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jose A, Cunha BA, Klein NC, and Schoch PE (2014) Aerococcus christensenii native aortic valve subacute bacterial endocarditis (SBE) presenting as culture negative endocarditis (CNE) mimicking marantic endocarditis. Heart Lung 43 (2), 161–163. [DOI] [PubMed] [Google Scholar]
- (41).Romero-Steiner S, Frasch CE, Carlone G, Fleck RA, Goldblatt D, and Nahm MH (2006) Use of Opsonophagocytosis for Serological Evaluation of Pneumococcal Vaccines. Clin. Vaccine Immunol 13 (2), 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tyx RE, Roche-Hakansson H, and Hakansson AP (2011) Role of Dihydrolipoamide Dehydrogenase in Regulation of Raffinose Transport in Streptococcus pneumoniae. J. Bacteriol 193 (14), 3512–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.