

Abstract
Inflammation following joint trauma contributes to cartilage degradation and progression of post traumatic osteoarthritis (PTOA). Therefore, drug delivery vehicles that deliver effective anti-inflammatory treatments have the potential to prevent PTOA. We have developed solid and hollow, thermoresponsive nanoparticles for the controlled release of our anti-inflammatory MK2-inhibiting (MK2i) peptide for intra-articular injection to halt inflammation that contributes to the advancement of PTOA. This system exploits the thermosensitive characteristic of N-isopropyl acrylamide (NIPAm) to transition phases when passing through its lower critical solution temperature (LCST). The nanoparticles (NPs) swell below the LCST and constrict above it.
Non-crosslinked poly(NIPAm) (pNIPAm), held above its LCST, formed hydrophobic cores around which shells composed of NIPAm, degradable crosslinker N, N′-bis (acryloyl) cystamine (BAC), sulfated 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS), and acrylic acid (AAc) were polymerized. Removal of the non-cross-linked pNIPAm cores via diffusion produced thermosensitive, degradable nanoparticles with low density, or hollow, cores. The data presented here revealed low-density, termed hollow, nanoparticles (hNPs) load and release significantly more MK2i than solid nanoparticles (sNPs). Furthermore, drug loading below the LCST of NIPAm results in roughly 2.5 times more therapeutic encapsulation compared to loading particles in their constricted state. Hollow nanoparticles increase drug loading compared to solid nanoparticles, are taken up into chondrocytes within 24 h, cleared from the cells within 6 days, significantly decrease the secretion of the proinflammatory cytokine IL-6, and, via intra-articular injection, are successfully delivered into the joint space of rats. The peptide loaded nanoparticles provide a reproducible platform for intra-articular delivery of therapeutics.
Keywords: N-isopropylacrylamide, Core-Shell nanoparticle, Thermosensitive, Cell-penetrating peptide, Inflammation, Osteoarthritis, Intra-articular
1. Introduction
Osteoarthritis (OA) is the most prevalent joint disease worldwide and causes significant pain, disability, and economic loss. OA affects more than 50 million people accounting for $185.5 billion in healthcare costs annually in the U.S. alone [1,2]. OA is characterized by inflammation of the synovial joint, including the synovial tissue, and degradation of extracellular matrix (ECM) molecules of articular cartilage resulting in the breakdown of the cartilage itself. Injury to articular cartilage triggers the release of inflammatory cytokines (e.g. IL-6, TNF- α) that increase the secretion of catabolic enzymes by chondrocytes [2]. The enzymatic fragments of degraded articular cartilage further stimulate inflammation resulting in the upregulation of cytokines and matrix metalloproteases (MMPs), thus generating a damaging cyclic process perpetuating OA [2]. Suppressing inflammation within the synovial joint may halt the vicious cascade that perpetuates cartilage matrix breakdown and the progression of OA following joint trauma.
Current non-surgical OA treatments, including non-steroidal anti-inflammatory drugs (NSAIDs), opioids, and intra-articular (IA) injections of viscosupplements and corticosteroids, which are primarily palliativeand are meant to control pain rather than prevent further joint degeneration. However, overuse of NSAIDs can lead to increased gastrointestinal [3,4], cardiovascular [5], and kidney complications [6], and opioids can have severe adverse side effects, primarily addiction [7]. Corticosteroids are the most prominent IA drug used for treating OA, but may promote chondrocyte destruction and increase the necessity for joint replacement and the likelihood for surgical intervention [7]. Therefore, there is a need for a safe, targeted therapeutic that can slow or stop the progression of PTOA.
Within the inflammatory pathway, mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 (MK2) is activated by p38 MAPK during OA and stabilizes the mRNA encoding pro-inflammatory cytokines (i.e. IL-1β, IL-6, and TNF-α) [8,9]. Our lab has developed a series of MK2 inhibiting (MK2i) cell-penetrating peptides (CPPs) that suppress MK2 phosphorylation, thus suppressing pro-inflammatory cytokine production; two variants of MK2i peptides, YARAAARQARAKALARQLGVAA (YARA) and KAFAKLAARLYR-KALARQLGVAA (KAFAK), differ in sequence only in their CPP region, however differ tremendously in their kinase inhibition selectivity profiles [10,11]. Despite their therapeutic value, both MK2i peptides are highly susceptible to enzymatic degradation within the extracellular space. To overcome enzymatic degradation of the MK2i peptide and prolong sustained delivery time, thermoresponsive, anionic NPs were synthesized and investigated for MK2i peptide loading, release, and subsequent in vitro bioactivity [12,13].
Our previous two generations of thermoresponsive NPs were either termed “solid” nanoparticles (sNPs) or “hollow” nanoparticles (hNPs) with the latter synthesized as core-shell particles followed by core removal that required disulfide bond reduction leaving a nondegradable shell. Limitations in peptide loading and release from the previous nanoparticle generations led us to modify synthesis protocols and to alter the crosslinking strategy to improve core removal, peptide loading and release, and to support particle degradation in vitro and in vivo.14–16 Here, the cores are synthesized with a high initiator to monomer ratio to limit polymer molecular weight and potentially enhance removal of the core from the fully formed core-shell nanoparticle. In addition, shells, formed directly around the cores, contained a labile disulfide crosslink, N,N′-bis(acryloyl) cystamine (BAC), to facilitate particle degradation and controlled release of the loaded peptide. Below the LCST, the nanoparticles swelled to aid peptide diffusion into the particle as well as support the formation of ionic bonds between the peptide and particle. The particle shell contained degradable crosslinker BAC as well as negative-acrylamido-2-methyl-1-propane-sulfonic acid (AMPS) to interact ionically with the cationic MK2i peptide, and acrylic acid (AAc) for future particle modification. These nanoparticles presented here will be used to encapsulate and deliver MK2i anti-inflammatory peptide into the joint space.
In previous generations of nanoparticles, YARA retention within the particle was unsuccessful, limiting the usefulness of the delivery system, while KAFAK was retained and used as the MK2i [14–16]. However, YARA has been found to be more specific to MK2 inhibition [10,11,17], making it desirable to deliver this version of the MK2 inhibitor peptide. The nanoparticles presented here allows for YARA retention within monodispersed, thermoresponsive, degradable NPs allowing for its sustained release into inflamed chondrocytes. In this work, we seek to understand how crosslink density effected particle degradation and YARA loading and release. Fluorophore labeling of the core polymer, via co-polymerization with vinyl-fluorophore monomers, allowed quantification of core removal using flow cytometry as well as nanoparticle visualization. Fluorescent labeling also allowed the use of intravital imaging to track particle retention within the synovial joint of rats. The body of results presented here support the hypothesis that lowering the crosslink density within the hollow poly(NIPAm-co-AMPSAAc-BAC) nanoparticles will increase YARA uptake into the particles. Further, the incorporation of the degradable crosslinker, BAC, will allow therapeutic release and NP clearance from the cell, thus supporting suppression of the production of the inflammatory cytokine interleukin 6 (IL-6) in chondrocytes treated with YARA-loaded, lower crosslink density hollow nanoparticles as compared to YARA-loaded crosslink density matched solid nanoparticles, see Scheme 1.
Scheme 1.
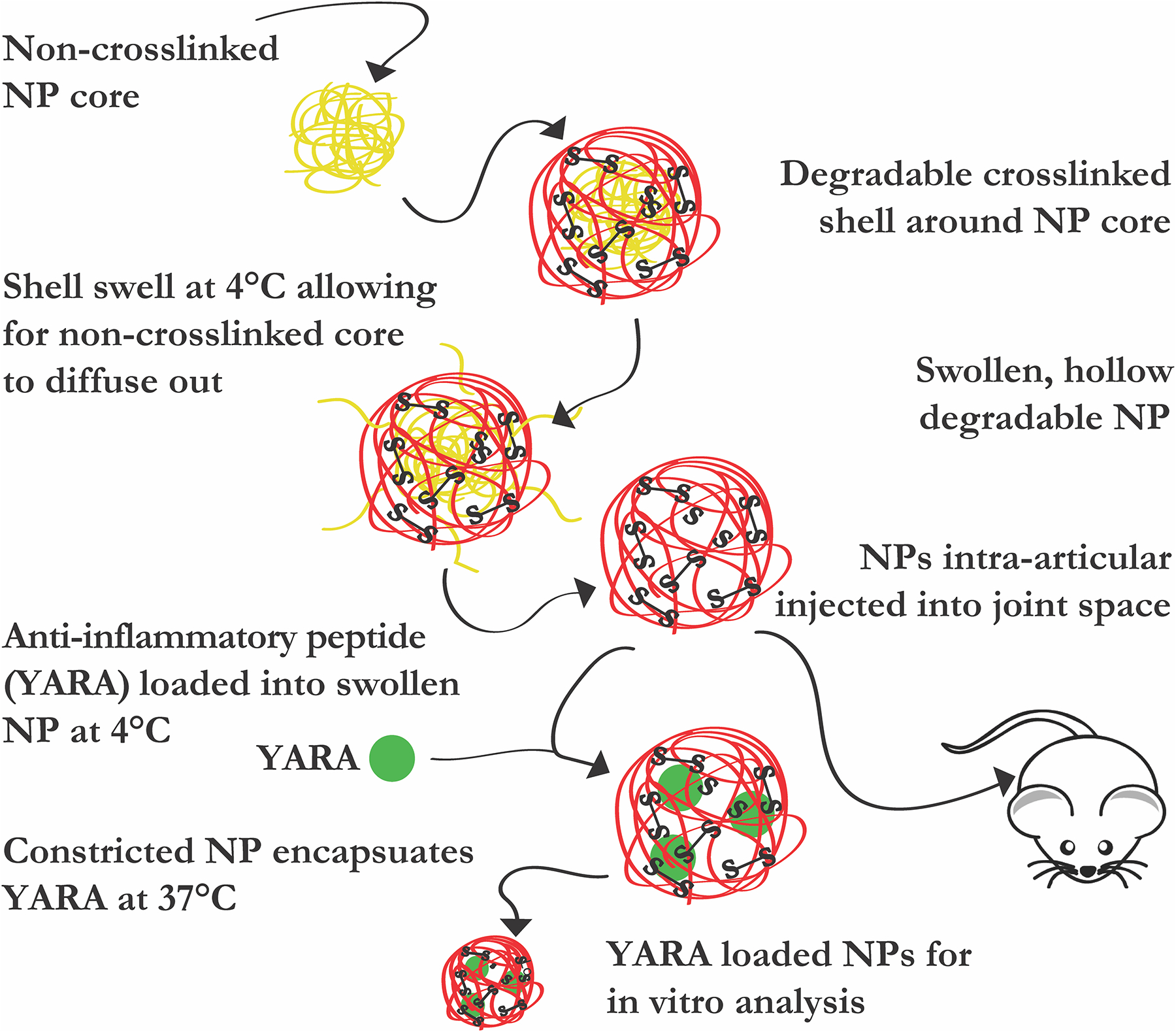
Graphical representation of reported studies. Diffusion of pNIPAm chains diffuse out of poly(NIPAm-co-AMPS-AAc-BAC) shell to generate hollow, thermoresponsive, degradable nanoparticle to load anti-inflammatory YARA for in vitro studies and in vivo delivery.
2. Materials and methods
2.1. Cell culture
Fetal bovine knees were purchased from Animal Technologies (Tyler, TX) and primary chondrocytes were harvested 24 h after slaughter as previously described [13]. Briefly, cartilage slices, 150–200 μm thick, were shaved from the load-bearing femoral condyle and washed three times with 1× PBS. In 2.5 ml increments, shaved cartilage was added to 25 ml of Dulbecco’s Modified Eagle Media (FBS DMEM)/F12 containing 0.1% bovine serum albumin, 0.2% w/v collagenase P, 100 units/ml penicillin, 100 μg streptomycin, and 3% FBS), then incubated at 37 °C for 2 h. Released chondrocytes were filtered through sterile 70 μm cell strainer and centrifuged at 1000 RPM for 5 min. The supernatant was removed and the pellet re-suspended in 10 ml of 10% FBS DMEM/F12. The centrifugation and resuspension were repeated three times. Chondrocytes were counted and plated at 20,000 cells/cm2 at 37 °C and 5% CO2 in a humidified incubator. Initial media was changed after 24 h. All chondrocytes were used between passage 2 and 6.
2.2. Materials
N-isopropyl acrylamide (NIPAm, ≥98%), N,N′-Bis(acryloyl) cystamine (99%, BAC), N, N′-methylene-bis-diacrylaminde (MBA), sodium dodecyl sulfate (SDS; 20% w/v in water), 2-acrylamido-2-methyl-1-propanesulfonic acid (99%, AMPS), dithiothreitol (98%, DTT), fluorescein o-acrylate (98%, FL), rhodamine B isothiocyanate (98%, RBITC), ethanol (99.5%, EtOH), N-diisopropylethylamine (99%, DIPEA), potassium persulfate (99%, KPS), trifluoroacetic acid (TFA), and dimethyl sulfoxide (DMSO) were acquired from Sigma Aldrich (St. Louis, MO). Dimethylformamide (DMF), dichloromethane (DCM), acetonitrile (ACN), trifluoroacetic acid (TFA), triisopropylsilane (TIPS), and phenol were purchased from Thermo Fisher (Waltham, MA). Dialysis membrane tubing was purchased from Spectrum Laboratories (Dominguez, CA). NIPAm and BAC were stored under nitrogen at 4 °C and −20 °C, respectively. AMPS was stored at room temperature in a desiccator. All water used in synthesis, dialysis, and testing was treated by a Millipore milliQ system (Billerica, MA; 18.2 MΩ cm resistivity).
2.3. Nanoparticle synthesis
Solid Particles:
Solid NPs (sNPs) were synthesized via precipitation reaction by dissolving 794.7 mg NIPAm, 78.0 mg AMPS, 48.2 mg BAC, 4.81 μl AAc, and 164 μl of a 20% SDS solution in 5 ml milliQ water in the reaction flask under a nitrogen blanket. After 15 min, 33.7 mg KPS dissolved in 2 ml milliQ water was injected into the reaction flask, still under nitrogen blanket, and refluxed at 70 °C for 4 h. sNPs were allowed to equilibrate to room temperature before purification.
Hollow Particles:
The nanoparticle (NP) core-shell complex was polymerized via precipitation reaction. The NP cores were synthesized by dissolving 394.5 mg NIPAm in 3 ml milliQ water in a scintillation vial and injecting it into a 100 ml three-neck flask under reflux and a nitrogen blanket with 35 ml milliQ water and 164 μl of a 20% SDS solution at 70 °C. Following a 15 min equilibration time, 67.4 mg KPS dissolved in 2 ml milliQ water was injected into the reaction flask to initiate the core polymerization, which continued for 2 h. NP cores were exposed to atmospheric oxygen for 45 min to terminate free-radical polymerization followed by a 15 min nitrogen purge. The NP shells were polymerized around the cores by injection of 794.7 mg NIPAm, 78.0 mg AMPS, 48.2 mg BAC, 4.81 μl AAc, and 164 μl 20% SDS dissolved in 5 ml milliQ water into the reaction flask. After 15 min, 33.7 mg KPS dissolved in 2 ml milliQ water was injected into the reaction flask, still under nitrogen blanket, and the mixture was refluxed at 70 °C for 4 h. Following polymerization, pre-dialysis NP (pd-NP) core-shell solution was allowed to equilibrate to room temperature before purification via transverse flow filtration (TFF).
To remove core polymer by diffusion, half of each lyophilized, TFF-purified pd-NP batch was dissolved in 50 ml of milliQ water and dialyzed in 10 kDa dialysis tubing (Spectrum Laboratories, Dominguez, CA) at 4 °C for 14 days; milliQ water was changed daily. Following dialysis, the now hollow NPs (hNPs) were frozen and lyophilized. Each batch was placed in opaque coverings during dialysis and lyophilization to prevent photobleaching.
Varying Crosslink Density:
sNP and hNP batches with differing crosslink density were used for drug loading and release studies. Varying amounts of BAC were added to the NP reaction mixture [24.1 mg BAC (0.5× NPs), 48.2 mg BAC (1× NPs), and 96.4 mg BAC (2× NPs)] for polymerization into the shells and solid particles (nomenclature in Table 1) via respective methods for sNP and hNP listed above to investigate the role of crosslink density on therapeutic loading and release.
Table 1.
Nomenclature for various nanoparticle batches.
| Pre-Dialysis (Cores In) | Post-Dialysis (Cores Out) | |
|---|---|---|
| Unlabeled Core + Unlabeled Shell | pd-NP | hNP |
| FL Core + Unlabeled Shell | pd-NPcFL | hNPcFL |
| Unlabeled Core + RBITC Shell | pd-NPsRBITC | hNPsRBITC |
| FL Core + RBITC Shell | pd-NPcFLsRBITC | hNPcFLsRBITC |
| Solid | Hollow | |
| 24.1 mg BAC | 0.5× sNP | 0.5× hNP |
| 48.2 mg BAC | 1× sNP | 1× hNP |
| 96.4 mg BAC | 2× sNP | 2× hNP |
| RBITC Labeled Shell | ||
| Degradable Fluorescent | hNPsRBITC-BAC | |
| Non-Degradable Fluorescent | sNPsRBITC-MBA |
Fluorophore Incorporation:
For FL-core NP batches, 0.1 mol% FL dissolved in 1 ml DMSO was injected after initial NIPAm and SDS injection, then core polymerization was initiated, creating the fluorescently-labeled co-poly(NIPAm-FL) core. For RBITC-shell NP batches, 0.1 mol% RBITC dissolved in 1 ml DMSO was injected following NIPAm, AMPS, BAC, AAc, and SDS addition and before shell polymerization initiation, resulting in fluorescently-labeled co-poly(NIPAm-AMPS-AAc-BAC-RBITC) shells formed around the core. Each NP batch was synthesized three times for experimental replicated, and tested three times for technical replicates. All nomenclature for the various batches is shown in Table 1.
Non-degradable Fluorescent Particles:
Non-degradable particles were synthesized via the same fluorophore incorporation protocol as described above, except using non-degradable crosslinker N, N′-methylenediacrylaminde (MBA) instead of BAC to generate non-degradable copoly(NIPAm-AMPS-AAc-MBA-RBITC) (sNPsRBITC-MBA) particles.
2.4. Nanoparticle purification
All sNP batches were purified using tangential flow filtration (TFF), KR2i from Spectrum Laboratories (Dominguez, CA), equipped with 10 kDa filter. sNPs and pd-NPs were filtered against 18.2 MΩ·cm resistivity until 100 ml of permeate was collected. Following purification, respective particle batches were frozen, lyophilized, and stored at room temperature. During all times, NPs were covered to prevent photobleaching.
2.5. Nanoparticle characterization
During the 45 min oxygen purge following core polymerization, 1 ml of reaction solution was analyzed using dynamic light scattering (DLS) on Nano-ZS90 Zetasizer (Malvern, Westborough, MA) at 70 °C to size the cores.
Following purification and lyophilization, sNP and pd-NP batches were respectively dissolved at 1 mg/ml in milliQ water, and subjected to temperature sweeps from 18.0 °C – 42.0 °C, in 1.5 °C increments, equilibrating for 3 min between each step, and measuring three times per step using DLS. The same procedure was followed for hNPs after dialysis to obtain their physical characteristics. Zeta (ζ)-potential was obtained on a Nano-ZS90 Zetasizer at 1 mg/ml sample concentration in milliQ water at 25.0 °C and 42.0 °C using folded capillary cells. All temperature trends and ζ-potential measurements were run in experimental and technical triplicate.
2.6. Flow cytometry
Confirmation of core removal was performed using pd-NPs with cores labeled with 0.1 mol% FL. Pd-NPs and hNPs were analyzed using Attune NxT Flow Cytometry (Thermofisher Scientific, Rockford, IL) to quantify the absorbance of the NPs before and after dialysis across all batches. The Attune flow cytometer with YL2 and BL1 lasers set to 225 mV assessed pd-NP and hNP fluorescence. Samples of each pd-NP and hNP batch were prepared at 1 mg/ml. The Attune flow cytometer pulled 150 μl of each solution at 150 μl/min and record the fluorescence; two rinses between samples and 1 ml of focusing fluid were flushed through to prevent potential contamination. Flow cytometry data was analyzed using FlowJo software.
2.7. Drug loading and release
One mg of each sNP and hNP batch were dissolved with 2 mg YARA in 1 ml ethanol (EtOH), incubated for 24 h at 4 °C or 42 °C for drug loading. Following incubation, loaded sNPs/hNPs were centrifuged at 17,000 g for 90 min and 500 μL of the supernatant was collected, centrifuged again for 90 min at 17,000 g, then 300 μL of that supernatant was collected for post-load analysis to quantify remaining unloaded peptide using a C18 reverse phase column and high-performance liquid chromatography (HPLC). Respectively, 1 ml of milliQ water was added to loaded sNP and hNP to resuspend them prior to freezing and lyophilizing.
Drug release was measured by dissolving loaded sNPs and hNPs, respectively, at 1 mg/ml in 1× PBS and incubating in a shaker at 37 °C and 200 RPM. For all time points, the solution was centrifuged for 10 min at 17,000 g, 500 μL of solution was removed and replaced with 500 μL of fresh 1× PBS, and frozen. Drug release was quantified using reverse phase HPLC. Briefly, 300 μl ml supernatant from respective time-points was thawed and analyzed for peptide content.
2.8. Nanoparticle degradation
In vitro degradation using glutathione:
1× hNPs were dissolved at 500 μg/ml in solutions of: 10 mM glutathione (GSH) pH 5.2, 0.01 mM GSH, pH 7.2, and ultrapure water pH 7.2 to mimic intracellular (intra) and extracellular (extra) GSH concentrations, respectively, with water serving as the control. Dynamic light scattering measurements were taken using a Nano-ZS90 Zetasizer (Malvern, Westborough, MA) at 37 °C from samples collected at Day 0, 1, 3, 5, 7, and 9 to obtain the respective diameter, PDI, and particle size distribution.
TEM:
One mg of 0.5×, 1×, or 2× hNPs was dissolved in 1× PBS or 1 mM DTT, and mixed at 37 °C at 200 RPM. Daily, the solutions were centrifuged at 17,000 g for 10 min, 0.5 ml of the supernatant was removed, and then 0.5 ml of fresh solution was added. Ten μl of each suspended NP treatment was adsorbed onto 400 mesh copper grids for 10 min, then excess solvent was removed. Mesh grids were stained with 2% urinal acylate for 10 s and immediately blotted to remove excess solution. Samples were then analyzed using transmission electron microscopy (TEM) (F.E.I. Company, Hillsboro, OR) at the UC Davis core facility.
2.9. Peptide synthesis and purification
YARAAARQARAKALARQLGVAA (YARA) was synthesized using a CEM Liberty Blue Peptide Synthesizer (Matthews, NC). Briefly, FMOC protected L-amino acids were individually dissolved in synthesis grade DMF to yield 0.2 M solutions. Rink-Amide (Sigma Aldrich, St. Louis, MO) resin was added to the reaction vessel of the CEM Liberty Blue peptide synthesizer. Synthesis occurred at 90 °C for 4–30 min per amino acid, time varying for each amino acid. YARA was cleaved from the Rink-Amide resin using 2 ml of a cleavage cocktail (4.4 ml TFA, 0.25 ml phenol, 0.25 ml milliQ water, and 0.10 ml TIPS) for 3 h, precipitated with 0 °C diethyl ether, centrifuged at 1000 g for 5 min four times, and dried overnight at room temperature. YARA was purified using reverse phase fast-protein liquid chromatography (FPLC). Quantification of molecular weight was assessed using Matrix Assisted Laser Desorption/Ionization – Time of Flight (MALDI-TOF) mass spectroscopy.
2.10. In vitro nanoparticle uptake, clearance, and imaging
Chondrocytes were seeded at 30,000 cells/cm2 into 24-well IBIDI cell culture plate and incubated for 24 h at 37 °C. Media was removed and frozen for later analysis, then cells were washed three times with Hank’s Balanced Salt Solution (HBSS) followed by addition of 400 μl of: media (n = 6), 400 μl of media containing 2 mg/ml of degradable hNPsRBITC-BAC (n = 6), or 2 mg/ml of non-degradable sNPsRBITC-MBA (n = 6) to the respective wells and cells were incubated for 24 h at 37 °C. Following incubation, media was aliquoted and frozen, then all wells were washed three times with HBSS then 400 μl of 75 nM Lysotracker Blue DND-22 in media (ThermoFisher Waltham, MA) was added to each well and incubated for 45 min at 37 °C under low light conditions. Each well was washed with HBSS and imaged. Following imaging, 400 μl of fresh FBS DMEM media replaced HBSS. Brightfield and fluorescent images were collected daily for 8 days; Lysotracker Blue DND-22 was only imaged after day 1. For all images, wells were washed three times and imaged in HBSS, then HBSS was replaced with fresh FBS DMEM media. All confocal images, brightfield, Lysotracker Blue DND-22 (excitation: 373 nm, emission: 422 nm), and RBITC (excitation: 570 nm, emission: 623 nm), were taken using FV3000 Confocal Laser Scanning Microscope (Olympus, Tokyo, Japan) at 60× magnification. The fluorescence of the aliquoted media was analyzed using Spectramax M5 to quantify of RBITC in media (excitation: 570 nm, emission: 623 nm).
2.11. Nanoparticle cytotoxicity
CellTiter Aqueous One assay was used to assess the cytotoxicity of all YARA loaded nanoparticles. Briefly, the respective batches of sNPs and hNPs were loaded as described previously. Chondrocytes seeded at 20,000 cells/cm2 into a 96-well plate were incubated for 24 h at 37 °C at 5% CO2. Nanoparticles were added at a concentration of 2, 4. 6, 8, 10, and 12 mg/ml for a total volume of 150 μl to each well, n = 4 per treatment; cells treated only with FBS DMEM media served as the control and were incubated for 24 h. Each well was washed three times with PBS, and 100 μl of fresh FBS DMEM media plus 20 μl of CellTiter Aqueous One (Promega, Madison, WI) added to each well and incubated for 3 h at 37 °C. Absorbance of each well was measured using a Spectramax M5 plate reader according to manufacturer’s protocol.
2.12. In vitro inflammatory stimulation and cytokine analysis
Chondrocytes were seeded at 20,000 cells/cm2 within a 96-well plate and then incubated for 48 h at 37 °C in FBS DMEM media. Following incubation, media was stored, wells were washed three times with 1× PBS, and on Day 0 inflammation was stimulated with 1 ng/ml of IL-1β, as described by Pratta et al. [18] On Day 2, media was removed and stored, then wells were washed three times with 1× PBS, and 150 μl of: 250 μM of YARA-loaded nanoparticles (n = 6) in media plus 1 ng/ml of IL-1β, 250 μM of free YARA in media plus 1 ng/ml IL-1β (n = 6), 1 ng/ml media plus IL-1β alone (n = 6), or media alone (n = 6) was added to respective wells. On days 2 and 4, media was collected, wells were washed three times with 1× PBS, and fresh media containing 1 ng/ml IL-1β was added to each well with the exception of the unstimulated controls. Media was tested for IL-6 concentration using a Bovine IL-6 Duoset enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol. Proceeding each step, each well was washed three times with buffer (0.05% Tween 20 in PBS), unless noted. Briefly, the capture antibody was coated onto clear, polystyrene high-bind 96-well plates and incubated for 24 h at room temperature. Then incubated with the Reagent Diluent (5% Tween 20 in PBS) for 2 h at room temperature. Next, 100 μl of sample/standard curve was added to respective wells and incubated for 2 h. Next, the detection antibody, diluted to working concentration, was added and incubated for 2 h at room temperature. Streptavidin-HRP, at working dilution, was added and incubated for 20 min in the dark at room temperature. Then, a substrate solution, at working concentration, was added, incubated in the dark for 20 min at room temperature followed by the direct addition of stop solution to the substrate solution, and the absorbance of each well was read at 490, 540, and 570 nm using Spectramax M5.
2.13. In vivo nanoparticle retention
Following acclimation, 10-week old Fisher 344 rats purchased from Envigo (Huntingdon, UK) were anesthetized with isoflurane and hair was removed from both rat knees. Next, 150 μl of 2.0 mg/ml hNPsRBITC in PBS (n = 5) or 150 μl PBS alone for negative control (n = 3) was injected into their left joint space, with the right joint serving as the untreated control. Rats were imaged and fluorescence intensity measured using an In Vivo Image System (IVIS) at the UC Davis Center for Molecular and Genomic Imaging (CMGI) at 557 nm excitation and 623 nm emission. IVIS was used to examine the fluorescence of hNPsRBITC and to quantify the presence of the fluorescent hNPsRBITC within rat joints with timepoints taken: immediately prior to injection, immediately after injection, daily for seven days, and immediately following dissection. Rats were imaged daily for 7 days then sacrificed using CO2 euthanasia. The hind limbs were dissected and imaged again to verify successful injection into the joint space. Total radiance emission (TRE) fluorescence was collected and analyzed.
2.14. Statistical analysis
Paired student’s t-test was used to analyze significant difference between particle diameter for the various batches listed in Table 1, Supplemental Table 2, see Supplemental Fig. 1. Paired student’s t-test was also used to assess the statistical difference between daily TRE measured in PBS and hNPsRBITC treated knees in vivo; p < 0.05. Twoway multi-comparison ANOVA was used to access differences in treatments for in vitro IL-6 knockdown, p < 0.05, as well as the TRE in vivo significant difference pre- and post-injection. Data is expressed as mean values ± standard deviation unless otherwise noted. Each batch of NPs was synthesized in experimental triplicate and each experimental run in technical triplicate, unless otherwise stated.
3. Results & discussion
3.1. Nanoparticle synthesis & characterization
Poly(NIPAm) (pNIPAm) has an LCST of approximately 32 °C, where it transitions between a hydrophilic (swollen) phase below its LCST, and a hydrophobic (collapsed) phase above its LCST. Wide-spread investigation of pNIPAm and its derivatives in biomedical applications include: drug delivery triggered by reactive oxygen species [19], tumor-cell imaging [20], gram positive bacteria detection [21], controlled release of biomolecules [22], and delivery of macromolecular therapeutics [23,24], and are due in large part to the tunability of the LCST near physiological temperature. Numerous micro- and nanoparticles exploit the thermoresponsive behavior of pNIPAm to form core-shell particles with metallic [25–31], inorganic [32–36], or polymer cores [14,37–41] and polymeric shells. Of the mentioned thermoresponsive particles, only those synthesized around inorganic cores (IC) or polymer cores (PC) form hollow particles, with a majority of the hollow particles being derived from those with inorganic (e.g. silica) cores. However, current core removal processes require the use of solvents; ICs were degraded using 0.05 M NaOH [32,35] or hydrofluoric acid [33,34], and PCs were degraded with either: 1 mM DTT [14], chloroform [38], tetrahydrofuran (THF) [40], or etched out using sodium carbonate [39]. These core degradation methods introduce additional solvents necessitating further purification and have the potential to modify the particle. Rather than use additional solvents, here we exploited the LCST of pNIPAm to generated non-crosslinked, removable pNIPAm cores, around which a crosslinked poly(NIPAm-co-AMPS-AAc-BAC) shell was polymerized to create particles with low-polymer density centers, termed hollow NP (hNP), for loading and release of cationic CPPs.
Polymerized non-crosslinked pNIPAm readily forms 50–60 nm particle cores and poly(NIPAm-FL) chains form 80–100 nm cores above their LCST (70 °C) and dissociate when temperature is dropped to below the LCST, Supplemental Table 1. Polymerizing poly(NIPAm-co-AMPSAAc-BAC) or fluorescently-labeled poly(NIPAM-co-AMPS-AAc-BACRBITC) shells around these cores at 70 °C allowed for formation of stable particles from which the non-crosslinked core can be removed via diffusion.
All pre-dialyzed and hollow nanoparticle batches (nomenclature is listed in Table 1) were sized using DLS and ζ-potential was evaluated to investigate the effect of fluorophore incorporation into the polymer backbone on size and the charge at the surface of the nanoparticles, Supplemental Table 1. Previously, fluorescein O-acrylate (FL) was polymerized into crosslinked poly(NIPAm-co-BAC-FL) cores to track core removal, and was also polymerized into the shell of primarily pNIPAm particles to track endocytosis into macrophages or bovine chondrocytes, but this study did not show the effect of FL on particle size [14–16,37]. Here, FL was polymerized into the core to verify core-shell formation and core removal. DLS measurements show that FL incorporation caused a 63.26% increase in core diameter, Supplemental Table 1. After shell polymerization around the FL core, pd-NPcFL were 49.22% and 55.77% larger than pd-NP at 18.0 °C and 42.0 °C respectively, while hNPcFL exhibited a 49.60% at 18.0 °C and 47.56% at 42.0 °C increase compared to hNP, Fig. 1 and Supplemental Table 1. This suggests that FL either affects particle size sterically or through altered hydrophobic interactions of the pNIPAm chains. RBITC was polymerized into the shells to validate shell formation around FL-labeled cores, rather than simply new NP formation, and caused the size of both pd-NPsRBITCs and hNPsRBITCs to increase as shown in Fig. 1 and Supplemental Table 1; when compared to unlabeled pd-NPs, the pd-NPsRBITCs had a 32.21% at 18.0 °C and 31.02% at 42.0 °C increase in size and the hNPsRBITC increased 30.62% at 18.0 °C and 28.48% at 42.0 °C compared to hNP. The dual labeled pd-NPcFLsRBITC/hNPcFLsRBITC batch followed the same trend, increasing both core size and shell size, but not altering the pd-NPcFLsRBITC nor hNPcFLsRBITC thermosensitive behavior, Fig. 1 and Supplemental Table 1. While the fluorophores did increase the relative size of the core, shell, and core-shell complex, these data serve as excellent indicators of both core removal and core-shell particle formation.
Fig. 1.
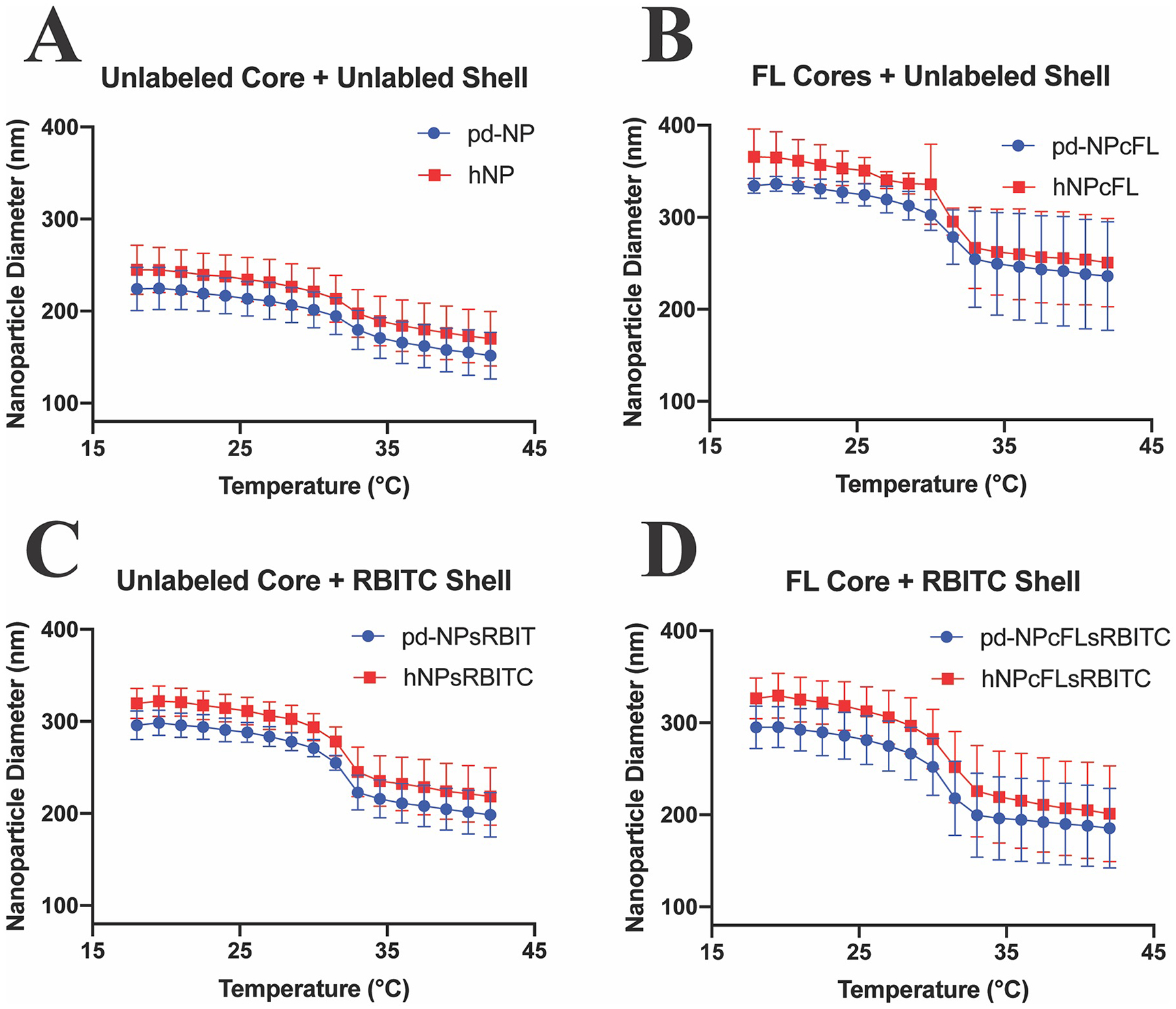
DLS hydrodynamic diameter temperature sweep from 18.0–42.0 °C of pre-dialyzed (blue circles) and hollow (red squares) nanoparticle batches. Each batch was synthesized three times, and each synthesis tested three times. A) Unlabeled Core + Unlabeled Shell; B) FL-labeled Core + Unlabeled Shell; C) Unlabeled Core + RBITC-labeled Shell; D) FL-labeled Core + RBITC Labeled Shell. Statistical size difference between the various nanoparticle batches is displayed in Supplemental Fig. 1A.
Interestingly, all hNP batches were slightly larger than their pd-NP and sNP counterparts. This is believed to be due to the non-crosslinked chains from the core diffusing out of the particles and leaving increased space for shell polymer chain rearrangement, which perhaps increases chain mobility and supports particle swelling. The characteristics match previously published work where the hNPs were shown to be larger than sNP counterparts [14,32,35,39,42]. A majority of the hNPs reported had a diameter greater than 300 nm [14,32,34,35,37,38,42], and particles larger than 200 nm have been shown to initiate an inflammatory response [43], thus our goal was to develop a method to repeatedly produce particles that maintained a size below 200 nm when delivered at 37 °C.
Particles with ζ-potential values between −20 to −30 mV are considered moderately stable and − 30 mV or lower are highly stable [44]. All pd-NPs, hNPs, and sNPs presented here were colloidally stable with ζ-potential −25 to −35 mV, except for pd-NPcFLsRBITC. pd-NPcFLsRBITC exhibited a ζ-potential of −4.51 ± 0.63 mV and was considered unstable [44]. Surprisingly, hNPcFLsRBITC was colloidally stable with a ζ-potential of −24.54 ± 9.42 mV, suggesting the interaction between FL and RBITC fluorophores partially masked the negative charge from the sulfate groups at the particle surface and the removal of the pNIPAm-FL core restored the surface presentation of sulfate groups. All hNPs had a more negative ζ-potential than their pre-dialyzed counterparts, further substantiating the claim that the shell polymer chains are better able to rearrange, resulting in increased surface charge following core removal. The size, polydispersity, and ζ-potential of the various NPs are listed in Supplemental Table 1 and Supplemental Table 2 above and below their LCST at 18.0 °C and 42.0 °C. Importantly, the increased negative ζ-potential of the particles presented here, as compared to previous versions reported by our laboratory, has improved colloidal stability and facilitated improved loading of CPP, particularly the highly specific MK2 inhibitor peptide YARA. Statistical size difference between the various nanoparticle batches is displayed in Supplemental Fig. 1A.
Further attesting to the robust synthesis method, the polydispersity (PDI) of the particles was found to be low. The PDI recorded by DLS is a measure of size uniformity, and a PDI < 0.1 is considered to indicate that a particle population is monodisperse [45]. All particle batches presented here were shown to be monodisperse, Supplemental Table 1. Overall, the synthesis methods was shown to produce particles of repeatable and uniform size [14,38].
3.2. Dialysis of nanoparticles and core removal quantification using flow cytometry
Following particle synthesis, suspending the pre-dialyzed core-shell particles in an aqueous environment at 4 °C supported particle swelling, which in turn allowed the non-crosslinked pNIPAm chains within the core to diffuse from the particle to generate hNPs. Removal of the non-crosslinked core was quantified using: DLS, TEM, and, for the first time to our knowledge, flow cytometry.
For analysis by flow cytometry, particles were maintained below the LCST, in their swollen state, to ensure that they were larger than 200 nm (the minimum size detectable by the flow cytometer). The data presented in Supplemental Fig. 2 shows the emergence of a secondary, FL-negative population in hNPcFL compared to the FL-positive pd-NPcFL batch as a result of the core polymer chains diffusing through the crosslinked shell, albeit not completely. At moderate to low monomer to initiator ratio, NIPAm is known to self-crosslink, however here a significantly higher monomer to initiator ratio was used limiting self-crosslinking of NIPAm [46]. The reason full core removal is not observed is likely due to a combination of: some NIPAm self-crosslinking occurring, physical entanglement between the non-crosslinked poly (NIPAm-co-FL) chains and crosslinked poly(NIPAm-co-AMPS-AAc-BAC) shell, and steric hinderance. However, there is a clear emergence of a secondary FL-negative population demonstrating that the majority of poly(NIPAm-co-FL) diffuses from the core-shell particle complex, Supplemental Fig. 2. Additional analysis via flow cytometry, Supplemental Fig. 2, showed pd-NPsRBITC and hNPsRBITC were unaffected by the dialysis, indicating dialysis does not affect the particle shell. Flow cytometry results confirmed the majority of core removal was achieved after 14 days of dialysis.
3.3. Core-shell nanoparticle formation – confirmed with flow cytometry
To verify the intended shell formed around the existing cores rather than forming new particles separate from the cores, RBITC-labeled shells were polymerized around FL-labeled cores (pd-NPcFLsRBITC). The data in Fig. 2 shows the existence of minor particle populations that only contain FL (core) or RBITC (new particles), but a major core-shell complex population existed within pd-NPcFLsRBITC batch (count 44,387) compared to unlabeled particle batch (count 1292) confirming a relatively pure population of core-shell particles for further study. Further, confirmation that the core polymer was removed during the 14 days of dialysis support the use of unlabeled particles for evaluating drug release and in vivo function as ultimately, unlabeled particles would be used for in vivo drug delivery.
Fig. 2.
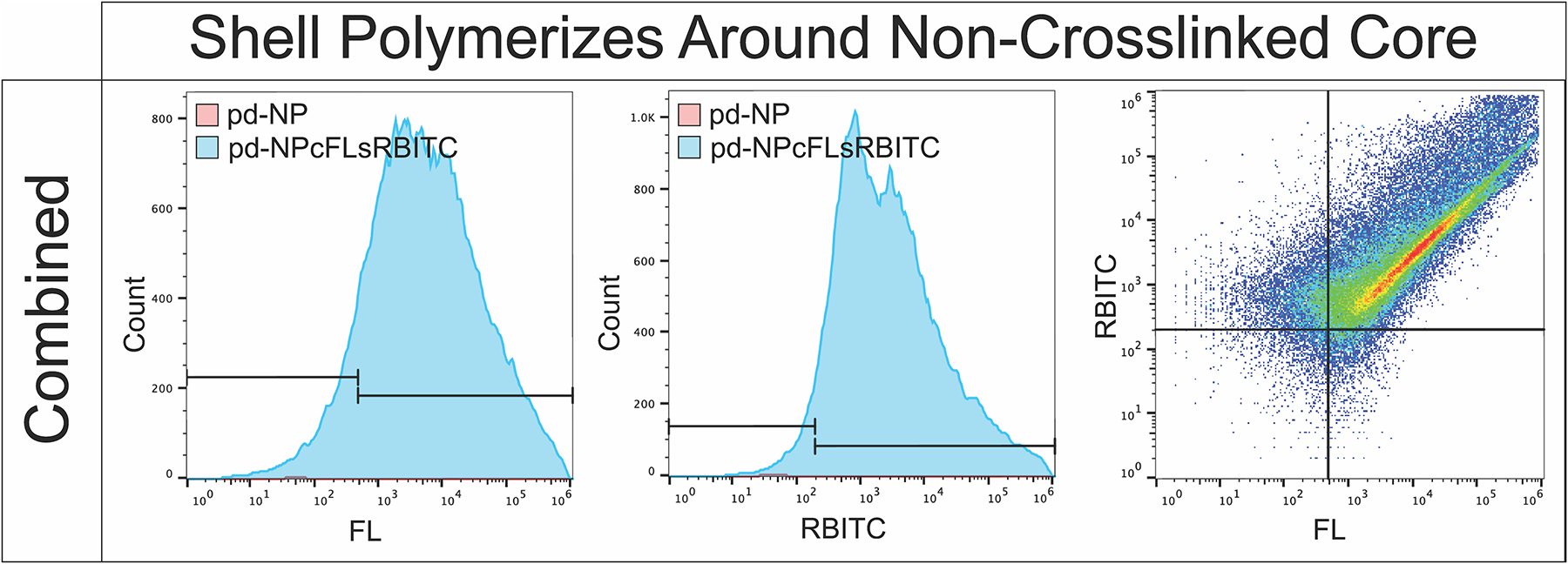
Flow cytometry confirms RBITC labeled crosslinked shells primarily polymerize around FL-labeled non-crosslinked cores forming a single nanoparticle core-shell complex, rather than two distinct particles.
3.4. Drug loading & release from nanoparticles of varying crosslink density
Unlabeled hNPs and sNPs were synthesized with various amounts of BAC (0.5× NPs, 1× NPs, and 2× NPs) to assess the benefits of hollow vs. solid nanoparticles as well as the effects of crosslink density on particle swelling and drug loading and release, nomenclature shown in Table 1. The size of each sNP, pd-NP, and hNP batch of varying crosslink density and physical characteristics (ζ-potential, and PDI data) are shown in Fig. 3, Supplemental Fig. 3, and Supplemental Table 2, respectively, with the statistical size differences between sNPs and hNPs displayed in Supplemental Fig. 1B.
Fig. 3.
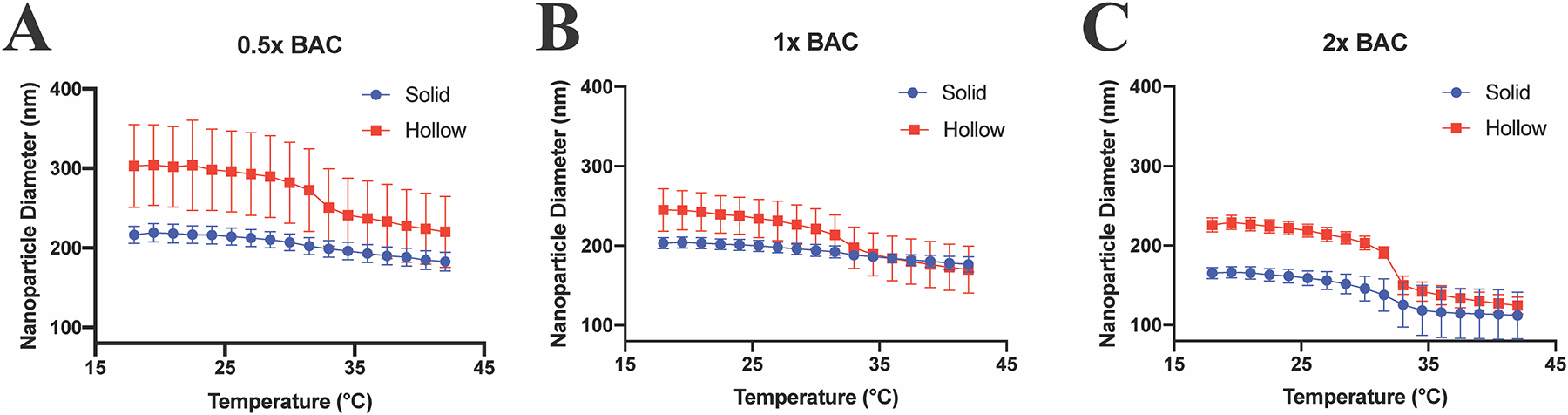
Lowering crosslink density increases swelling difference between solid (blue circles) and hollow (red squares) nanoparticles. DLS hydrodynamic diameter temperature sweep from 18.0–42.0 °C of solid (blue) and hollow (red) nanoparticle with varying crosslink density. A) 24.1 mg BAC (0.5× NPs); B) 48.2 mg BAC (1× NPs); C) 96.4 mg BAC (2× NPs). Statistical size difference between the various nanoparticle batches is displayed in Supplemental Fig. 1B.
As expected, at lower crosslink density hNPs show increased overall diameter compared to their pd-NP counterparts and as compared to corresponding sNPs for all batches. The 0.5× particles exhibited the greatest increase in diameter with hNPs being 18.73% at 18.0 °C and 19.91% at 42.0 °C larger than 0.5× pd-NPs. There was an evident, but lesser, size difference in the 1× particles, with 1× hNPs being 9.32% at 18.0 °C and 12.16% at 42 °C larger than 1× pd-NPs. Noticeably, the 2× pd-NP and hNP exhibited similar sizes across the temperature profile, with only 0.32% difference at 18.0 °C, and 2.36% difference at 42.0 °C respectively; showing that increasing the crosslink density affects NP swelling, Supplemental Fig. 3 and Supplemental Table 2. Comparing sNPs and hNPs, the 0.5× hNPs were 40.09% at 18.0 °C and 20.41% at 42.0 °C larger than 0.5× sNPs; 1× hNPs were 20.47% at 18.0 °C larger than sNPs, but were statistically identical at 42.0 °C; 2× hNPs were 36.67% at 18.0 °C and 11.26% at 42.0 °C larger than 2× sNP counter parts, Fig. 3 and Supplemental Table 2, and statistics displayed in Supplemental Fig. 1B.
While the authors are unaware of studies investigating drug loading and crosslink density on hollow, thermosensitive nanoparticles, Bartlett et al. found increasing crosslink density for poly(NIPAm)-based particles decreased the size of the particle uniformly both above and below the LCST of pNIPAm, agreeing with the findings presented here. However, their lower crosslink density poly(NIPAm)-based particles had a more positive ζ-potential and thus a lower charge to volume ratio, which Bartlett et al. believed caused lower drug loading. However, here the 2× hNPs have a more positive ζ-potential and load more peptide than the 2× sNPs, showing drug loading isn’t solely dependent on particle charge, Supplemental Table 2.
These data support the theory presented by Bartlett et al. that increased drug loading is a result of increased crosslink density at relatively low overall crosslink densities, as shown in Fig. 3 and Supplemental Table 2, with the 0.5× loading the least amount of peptide. However, these data contradict that same theory as 1× NPs load more than their respective 2× NP counterparts suggesting that crosslink density, which is inversely related to polymer mesh size, can retard peptide loading by decreasing mesh size to a point where it interferes with peptide diffusion into the particles.
Hypothetically, the sNPs and hNPs should load more YARA when swollen at 4 °C, than when collapsed at 42 °C, with hNPs loading more YARA than sNPs due to removal of the unsulfated core and decreased polymer density of the hNPs. To test this, 1000 μg NPs and 2000 μg YARA were dissolved in 1 ml EtOH at either 4 °C or 42 °C. Below the LCST of pNIPAm at 4 °C, 0.5× sNPs and hNPs loaded 14.3% and 19.2% of YARA respectively, while only loading 0.0% and 5.2% above its LCST at 42 °C, respectively, Fig. 4, Supplemental Fig. 4, and Supplemental Table 2. Interestingly, the 1× hNPs loaded the most YARA above and below the LCST when compared to 0.5 and 2× hNP and sNP; loading 57.4% at 4 °C and 24.6% at 42 °C. The 1× sNPs also loaded more YARA below the LCST, 25.3% at 4 °C and 8.7% at 42° as compared to 0.5× and 2× sNP. Consistent with 1× hNPs, the 2× hNPs, both above and below the LCST, loaded more YARA than the 2× sNPs. The 2× hNPs loaded 42.1% below the LCST and 18.1% above their LCST, while 2× sNPs loaded 11.1% below the LCST and 8.7% above the LCST, Fig. 4, Supplemental Fig. 4, and Supplemental Table 2. The results also suggest that while mesh size is important, temperature, which will affect the strength of the ionic bonds between particle and peptide, also plays a role in peptide loading; at low temperatures the expanded particles that support rapid peptide diffusion into the particle also support stronger polymer-peptide interactions and high loading. Previous studies only examined drug loading into the reported particles [14,39] or pNIPAm gels [47] below the LCST of pNIPAM, when the particles were swollen. Here we confirmed the hypothesis that more peptide is loaded into the nanoparticle when loaded below the LCST of NIPAm. However, these results also suggest the void space created by the removal of the core influences the amount of drug that is loaded into the particle, as confirmed by 0.5× and 1× hNPs loading more than their sNP counterparts. Of the six batches analyzed for drug loading, 1× hNPs loaded the most YARA below the LCST, Fig. 4, Supplemental Fig. 4A, and Supplemental Table 2. In summary, crosslink density, the thermoresponsive property of NIPAm, and loading temperature all play a role in drug loading.
Fig. 4.
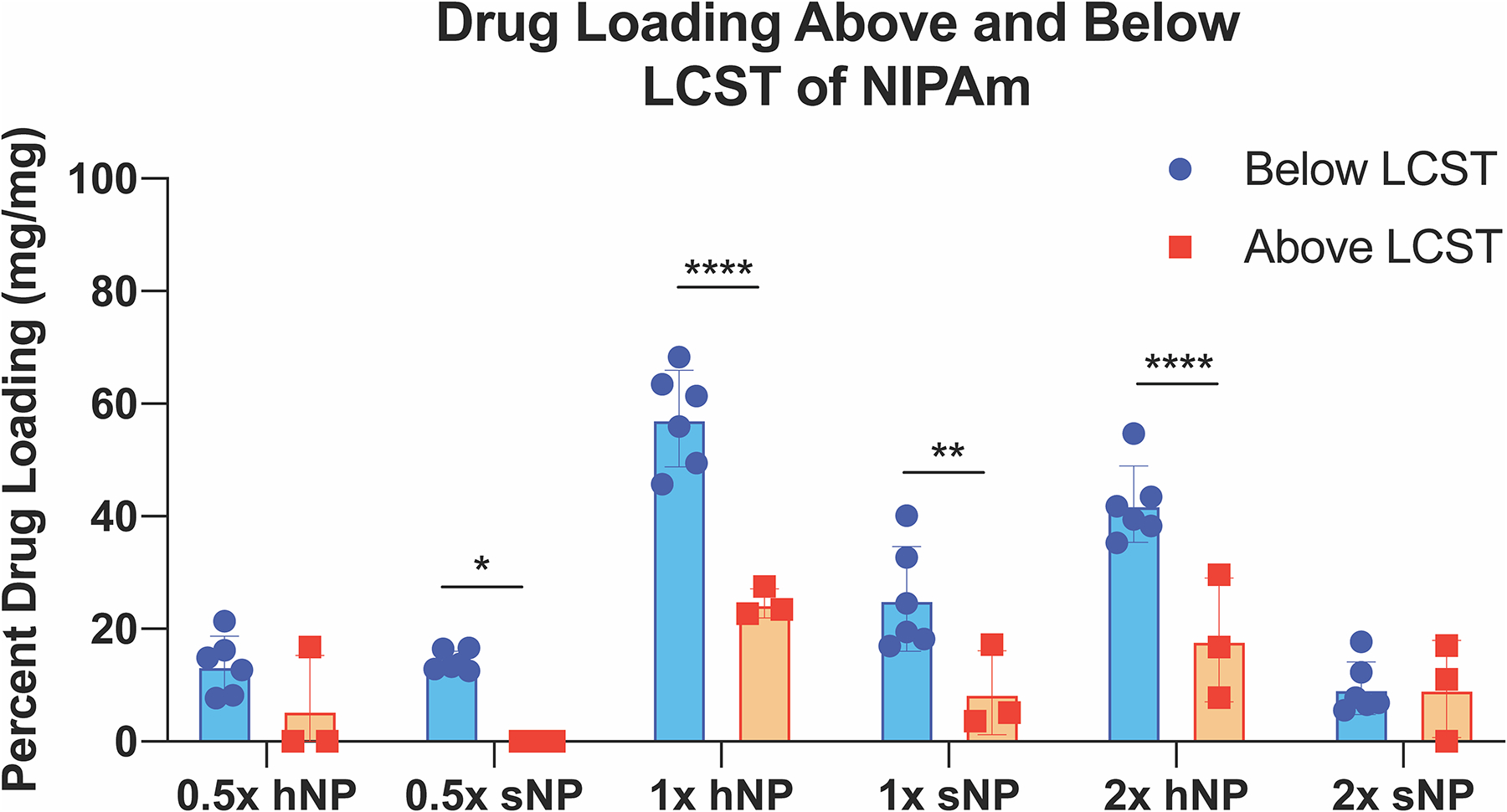
YARA loading into the primarily pNIPAm nanoparticles is dependent on temperature and crosslink density with 1× hNPs loading the most YARA above and below the LCST of NIPAm. Stats: * = p < .05, ** = p < .01, *** = p < .0001. Statistical differences between YARA loading between batches loaded at the same temperature shown in Supplemental Fig. 4.
Previous studies suggested that a portion of the loaded peptide became permanently entrapped in collapsed particles [14–16], and it was theorized that the peptide was stuck within the non-degradable high polymer density core. For this research, we hypothesized that lower density hNPs would support greater peptide release than sNPs. These data presented here support this hypothesis where all hNP batches release more YARA than the respective sNP counterparts, Fig. 5. Notably, increasing crosslink density decreases the difference between hNP and sNP release, shown in Fig. 5. The 0.5× hNPs release a higher percent of loaded peptide at any point compared to 0.5× sNPs; releasing 77.6% at the end of 5 days whereas sNPs release 43.5%. The 1× hNPs also continuously released more YARA than 1× sNPs, releasing 49.9% and 30.8%, respectively. The 2× particles showed the smallest difference in release when comparing hNPs to sNPs, releasing 48.2% and 40.5%, respectively.
Fig. 5.
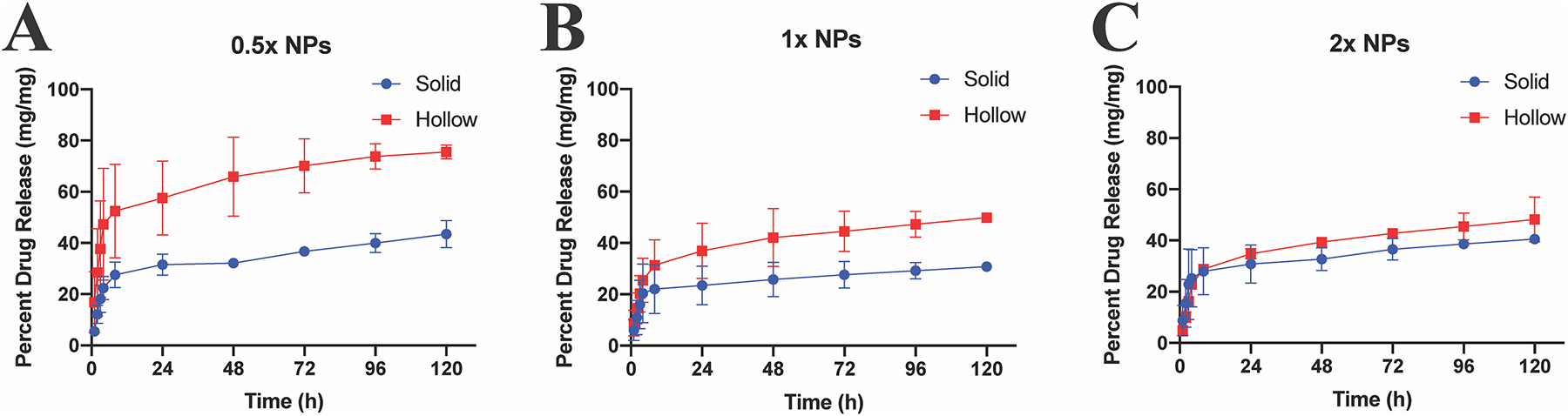
Sustained release of YARA from solid (blue circles) and hollow (red squares) nanoparticles of varying crosslink density at 37 °C at 200 RPM for 120 h. While controlled release of YARA is observed at all crosslink densities studied, the amount released at any timepoint is affected by crosslink density.
All of the described sNPs and hNPs show continual sustained release at the conclusion of 5 days, but begin to plateau around day 3. However, as also seen previously, not all of the peptide is released at this timepoint. A limitation of this in vitro study is that the particles do not degrade in PBS, Supplemental Fig. 5, and therefore it is likely that total YARA release will occur within the cells as the NPs degrade completely via reduction of the disulfide bonds. In comparison to previous studies showing CPP release from thermosensitive particles, KAFAK-hNPs loaded 470 ± 18 mg of peptide and released 53% over 4 days, with 89% release happening within the first 12 h [14], while here the 1× hNPs loaded 1147 ± 172 mg of YARA, and released roughly 24% within the first 12 h, and 49.9% after 5 days. While previous hNPs were unable to encapsulate YARA, the comparisons between 1× hNP + YARA and previous 1× hNP + KAFAK [14], demonstrates an improved sustained release of a more specific MK2i CPP. Thus, the particles presented here appear to be preferable in situations where sustained, rather than a more burst-type release is necessary.
3.5. Degradation, cytotoxicity, uptake, and clearance of nanoparticles
Dithiothreitol (DTT) is a synthetic analog of glutathione (GSH) found naturally within the cytoplasm of cells [48,49]. Glutathione reduces disulfide bonds and these nanoparticles exploit this ability for their breakdown and clearance from the cell. DTT was used in vitro to assess NP degradation. Each of the 0.5× hNP, 1× hNP, and 2× hNP batches show degradation in the presence of DTT as compared to PBS, Supplemental Fig. 5. The 0.5× hNPs degraded fully after 2 days, while the 1× and 2× hNPs had not fully degraded after 4 days, and exhibited different morphology and were present at a lower concentration, suggesting that some particles degraded more quickly than others, as compared to particles incubated in PBS alone.
Next, GSH was used to mimic the intracellular and extracellular environment to which nanoparticle are expected to be exposed, and degradation of the particles was assessed. The endosomal/lysosomal concentration of glutathione (GSH) is 10 mM and the pH 5, while the extracellular concentration of GSH is 0.01 mM at pH 7.2. The hNPs dissolved in 10 mM GSH show a significant increase in particle diameter (300% at Day 9) and particle distribution, Supplemental Fig. 6A/C, due to the disulfide bond being cleaved within the particle shell. Conversely, the diameter, distribution, and PDI of hNPs dissolved in extracellular concentrations of GSH and water remain relatively unchanged, Supplemental Fig. 6A/D/E, show the intracellular environment supports degradation via the cleavage of the disulfide bond within the BAC crosslinker. To the authors’ knowledge, these fully degradable particles are unique among hollow pNIPAm particles. However, two studies using solid particles examined degradation [16,39], both of which exploited reducible disulfide crosslinkers. Complete degradation of pNIPAM-based particles is expected to be critical for complete drug release and possibly also for in vivo clearance of the particles or degraded polymer chains.
The in vitro cytotoxicity of 0.5×, 1×, and 2× sNPs and hNPs was accessed utilizing bovine chondrocytes. Shown in Supplemental Fig. 7, all sNPs and hNPs treatments were nontoxic compared to untreated control chondrocytes. This mirrors previous work were pNIPAm-incorporated particles were non-cytotoxic to chondrocytes [16], macrophages [14,15], human hepatocellular carcinoma (HepG2) cells [30], and alveolar basal epithelial cells [50].
In order to knockdown the inflammatory response, YARA must be taken up into chondrocytes prior to its degradation. To evaluate uptake via endocytosis, RBITC-labeled nanoparticles were synthesized using degradable hNPsRBITC crosslinked with BAC (hNPsRBITC-BAC) and compared to non-degradable N, N′-methylenediacrylaminde (MBA) crosslinker (sNPsRBITC-MBA). The hNPsRBITC-BAC and sNPsRBITC-MBA were respectively incubated with chondrocytes and compared to media control, Fig. 6. As the endosome becomes more acidic, the environment is conducive to disulfide bonds cleavage [51] and, in conjunction with glutathione, is believed to be responsible for the degradation of the nanoparticles. The majority of the hNPsRBITC-BAC particles were cleared from chondrocytes by Day 5, while the non-degradable sNPsRBITC-MBA particles were observable within the cell for the duration of the 8 day experiment suggesting that loss of fluorescence in cells treated with degradable particles is not due entirely to fluorescence quenching, or loss from oxidation, but from polymer removal from the cells. All images from Day 1 to 8 are shown in Supplemental Fig. 8. Additionally, the media was analyzed for RBITC; fluorescence was detected in the media from cells treated with hNPsRBITC-BAC only through day 5, while the media obtained from cells incubated with sNPsRBITC-MBA had no detectable RBITC following initial incubation, Supplemental Fig. 9. The data here agrees with previous work demonstrating that pNIPAm particles were endocytosed into chondrocytes [14–16], but shows the degradation/clearance of the BAC crosslinked particles from the cells. The chondrocyte data in conjunction with TEM data shows nanoparticle degradation as well as clearance from chondrocytes.
Fig. 6.
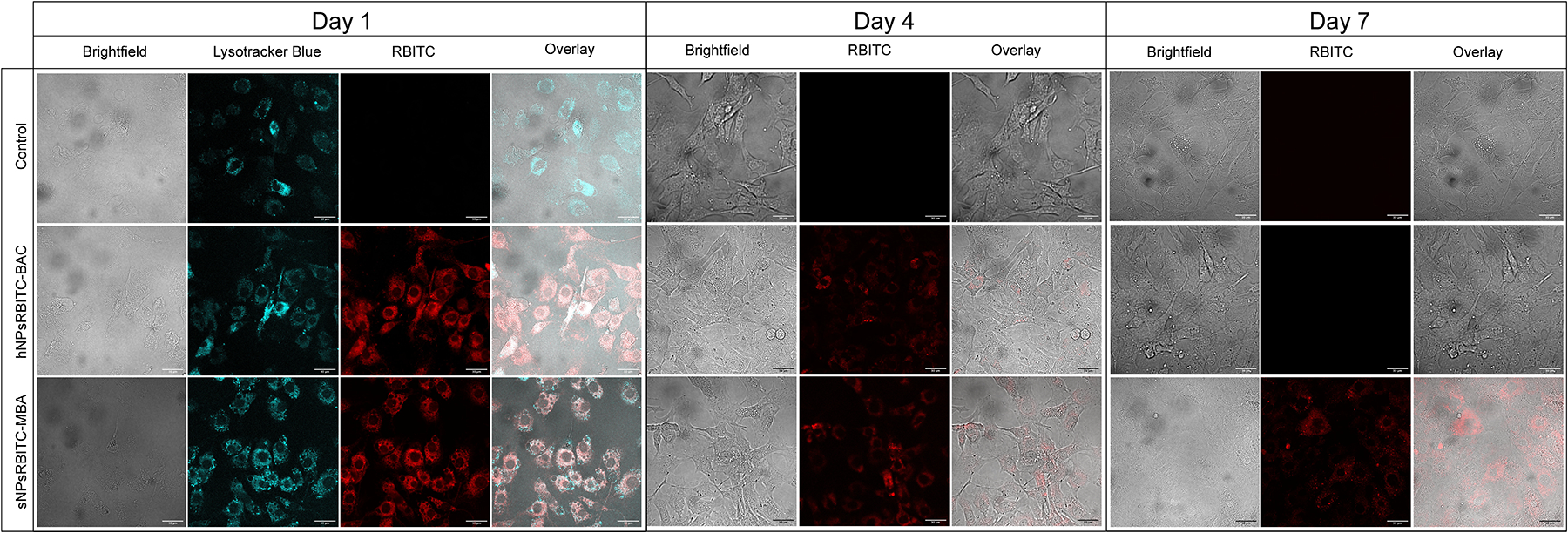
Chondrocytes endocytose hNPsRBITC-BAC and sNPsRBITC-MBA into the cytoplasm of the cells and BAC crosslinked particles were degraded/cleared from cells within 7 days. Daily images from day 1 through 8 shown in Supplemental Fig. 8. Scale bar: 30 μm.
3.6. Inflammation inhibition in bovine chondrocytes
IL-6 is an inflammatory cytokine that leads to the progression of OA. There are currently therapies for the prevention of IL-6 secretion, but they lack of specificity – knocking down other essential biological pathways [52]. Previously, a series of MK2i peptides were developed that are capable of knocking down proinflammatory cytokine production, including IL-6. While studies have examined the non-specific MK2i variant KAFAK encapsulation within particles, only two of the studies investigated the KAFAK-loaded particles with bovine chondrocytes [13,16]. Other studies either did not examine loaded-particle effect in cells [32,37,40], or investigated the effects on monocytes and macrophage cell lines [12,14,15]. Here we examine the specific peptide MK2i variant YARA, which leaves a multitude of crucial pathways unaltered [10]. Chondrocytes were stimulated with IL-1β to induce inflammation as measured by IL-6 production, Fig. 7A, or left unstimulated to serve as a baseline, Fig. 7B. Following stimulation, chondrocytes were further stimulated with IL-1β (untreated control), Fig. 7A, or treated with additional IL-1β plus 250 μM free-YARA, Fig. 7C, or IL-1β plus NPs containing 250 μM YARA to analyze the suppression of IL-6 secretion induced by delivery of the YARA peptide Fig. 7D–F.
Fig. 7.
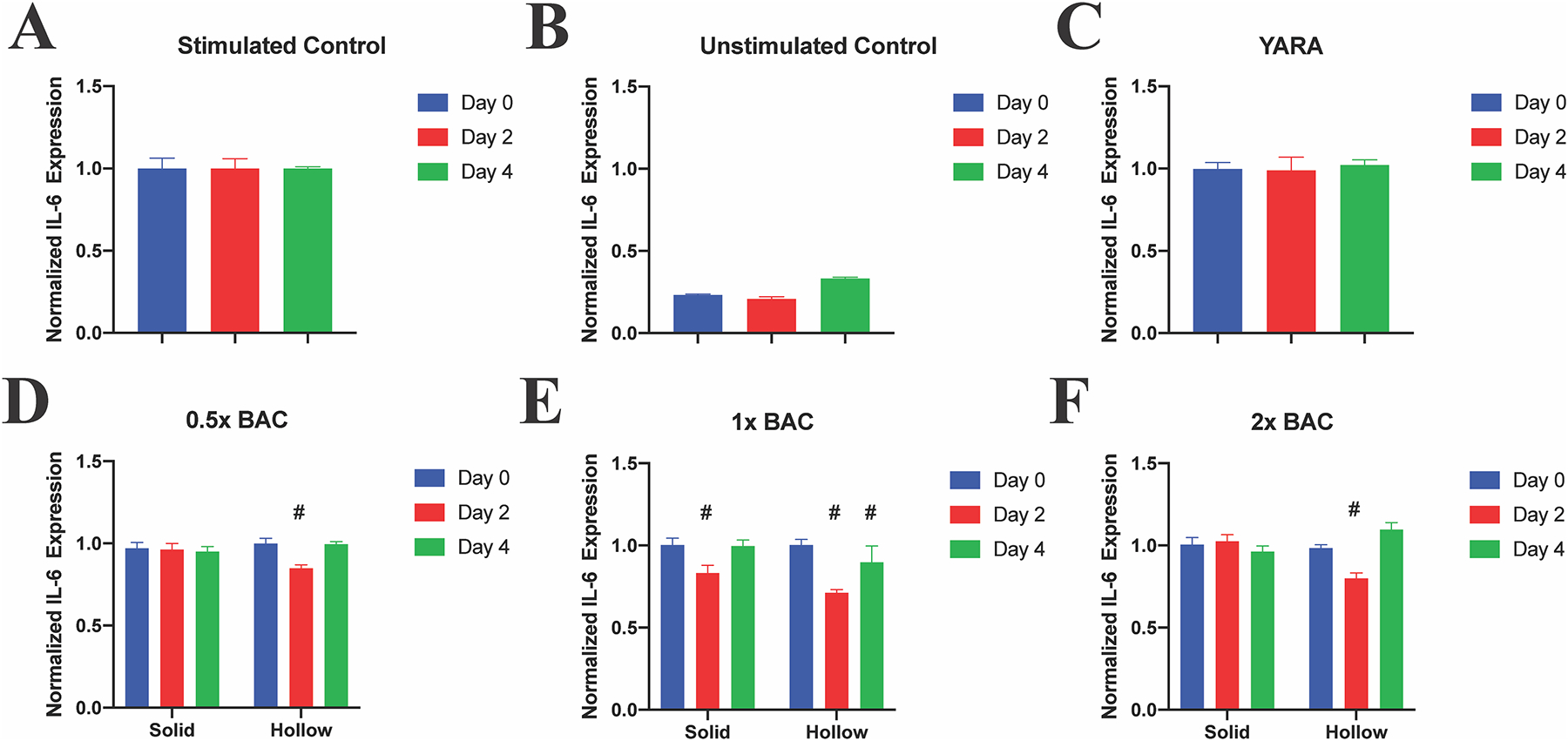
IL-1β-stimulated chondrocytes treated with sNP + YARA, hNP + YARA, or free YARA once on Day 2. All treatments significantly reduce IL-6 expression, with 1× hNPs reducing expression until Day 4, while free YARA did not alter IL-6 expression. Stats: # represents statistical significance compared to Day 0.
IL-6 production was detected in all control and treatment groups, Supplemental Fig. 10, and analyzed by normalizing, by day, to the unstimulated control of the same day and using a 2-way multi-comparison ANOVA. Notably, free YARA did not have a significant effect in reducing IL-6 expression (Fig. 7C), likely due in part to proteolytic degradation and in part due to impaired caveolae-induced endocytosis of YARA when cells are cultured on stiff polystyrene culture plates, demonstrating the advantage of using nanoparticle carriers. Of the six different nanoparticle treatments, only 1× hNP significantly reduced IL-6 production 2 and 4 days after a single treatment on Day 0, Fig. 7E. The continual IL-6 knockdown is believed to be attributed to the sustained release of YARA from 1× hNPs shown in Fig. 5 and degradation of the particle, Fig. 6 and Supplemental Fig. 5, potentially allowing for full drug release. Combining increased drug release and slow particle degradation allows for prolonged peptide activity.
A single treatment of YARA-loaded NPs was used to fully assess the release of YARA into the cell. While these data presented does not show clinical relevance for IL-6 knockdown, it provides a basis for further studies. These data show 1× hNPs allow for the most YARA loading compared to the five other presented NP batches, in addition to being the only treatment to show significant IL-6 knockdown 4 days after the initial, single treatment. Future studies will build upon these data to optimize the dose and frequency of treatments to match unstimulated chondrocyte IL-6 secretion levels.
3.7. Intra-Articular (IA) delivery of nanoparticles into rats joint space
IA injections support effective delivery with limited potential for systemic side-effects [53,54]. Numerous studies are underway involving IA injection, with a few focusing on micro- and nanoparticle treatments specifically for OA [53,54]. To assess the IA retention time of our particles, rats were injected with hNPsRBITC (n = 5) or 1× PBS (n = 3) as control. The rats injected with hNPsRBITC had 1004.72% increase in total radiant efficiency (TRE) following injection compared to the TRE prior to injection, where the PBS injected rats only saw a 9.04% TRE increase, Fig. 8A, showing that increased TRE is due to RBITC within hNPsRBITC and there for a successful injection. The region of interest (ROI) was analyzed for all rats and all time points, Fig. 8B, to quantify hNPsRBITC retention time. Analysis of the ROI over time demonstrated that the particles remain in the joint space for up to 7 days. The rat’s hind limbs were dissected following sacrifice and imaged again to further confirmed the successful injection of hNPsRBITCs into the joint space, Fig. 8C. Previous studies examined IA delivery of unbound therapeutics into the synovial space and showed retention time was less than 72 h [55,56]. However, in 2014 Morgen et al. found that the use of cationic solid dextran-based nanoparticles allowed for 70% retention of particles within the joint after 7 days [57], showing the benefit of nanocarriers and the potential correlation between particle charge and retention time within the joint space as both their cationic particles and these anionic particles remain in the joint for up to 7 days.
Fig. 8.
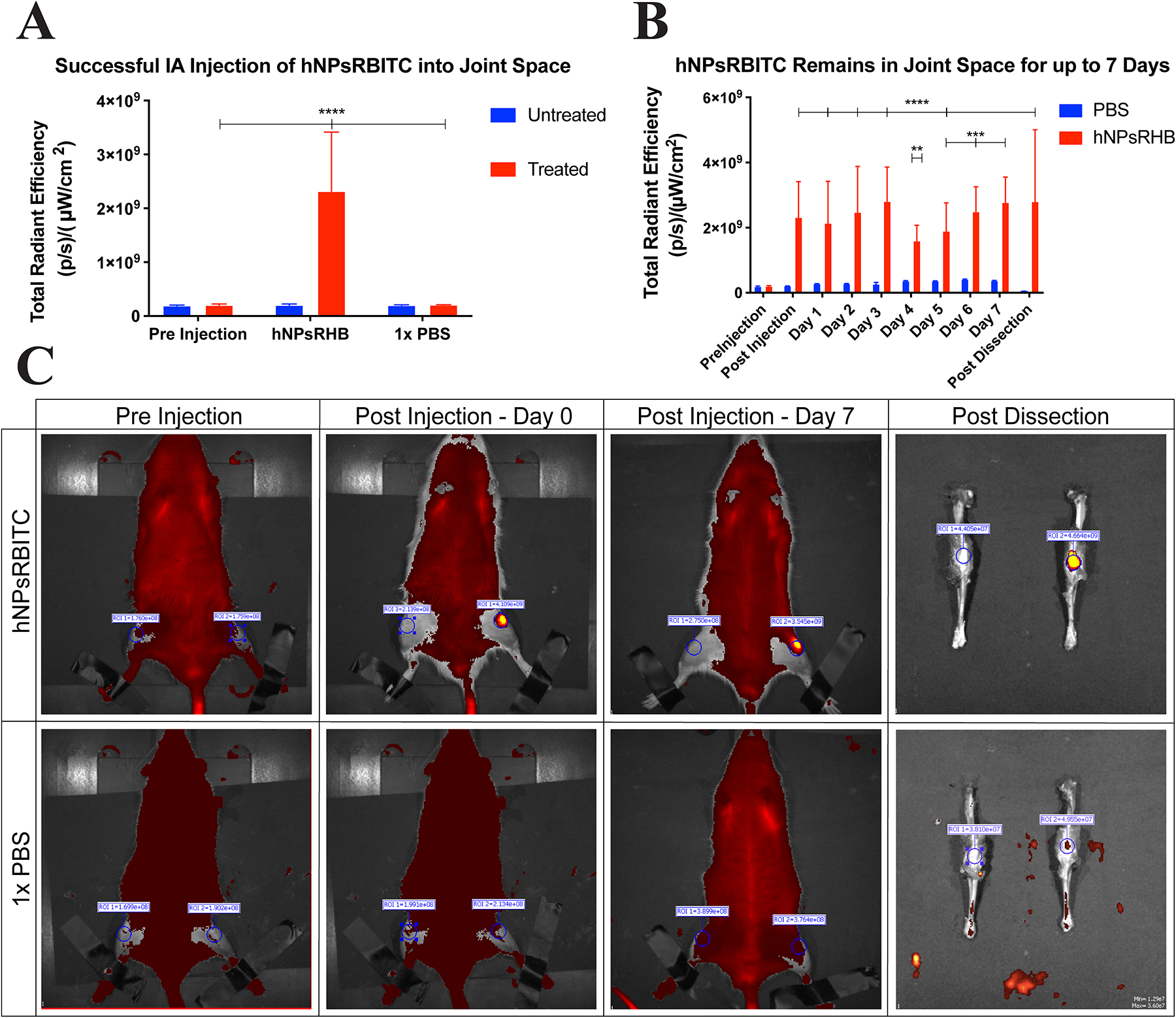
hNPsRBITC was successfully injected into the intra-articular joint space of rats and remained within the joint for 7 days. n = 5 for hNPsRBITC injected rats, n = 3 for PBS injected rats. A) TRE comparison pre and post intra-articular injection of hNPsRBITC and PBS into rats; B) Daily TRE of rat joints; C) IVIS images of rats pre- and post-injection as well as post dissection.
4. Conclusion
This work demonstrates the uptake and release of MK2 inhibiting peptide YARA from various nanoparticle systems to knock down the inflammatory cytokine IL-6 in stimulated bovine chondrocytes. The 1× hNPs presented here significantly improve upon previous work to produce monodisperse solid and hollow nanoparticles with repeatable ζ-potential, drug loading, and release. The benefit of loading low crosslink density poly(NIPAm-co-AMPS-AAc-BAC) nanoparticles below their LCST to increase drug loading is noted here as well. In addition, cationic CPP showed continual released up to 5 days. Importantly, the in vitro experiments here suggest that hNPsRBITC were cleared from chondrocytes approximately 5 days after treatment, and can suppress IL-6 production for 4 days. Finally, the hNPsRBITC particles were successfully delivered in vivo into the joint space, via intra-articular injection, and remain within the joint space for up to 7 days in agreement with previously IA delivery of charge particles [57]. Together, this body of data shows the promise of hollow, thermoresponsive poly(NIPAm-co-AMPS-AAc-BAC) nanoparticles loaded with cationic MK2i peptide YARA.
Supplementary Material
Acknowledgments
We wish to thank Dr. Vasilios Morikis for his training and assistance in our flow cytometry work and Vanessa Dartora for her training with our HPLC drug release. Additionally, we would like to thank the National Center for Advancing Translational Sciences for their funding and training.
Funding
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health, United States of America under award number R01AR065398 and the National Center for Advancing Translational Sciences of the National Institutes of Health, United States of America, through grant number UL1 TR001860 and linked award TL1 TR001861. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Declaration of Competing Interest
The authors declare the following competing financial interest(s): Moerae Matrix, Inc. has a worldwide exclusive license to the MK2 inhibitor peptide. A. Panitch owns greater than 5% of Moerae Matrix, Inc.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jconrel.2020.04.007.
References
- [1].Hootman JM, Helmick CG, Barbour KE, Theis KA, Boring MA, Updated projected prevalence of self-reported doctor-diagnosed arthritis and arthritis-attributable activity limitation among US adults, 2015–2040, Arthritis Rheum. 68 (7) (2016) 1582–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Klatt AR, Paul-Klausch B, Klinger C, Kühn C, Renno JH, Banerjee M, et al. , A critical role for collagen II in cartilage matrix degradation: collagen II induces proinflammatory cytokines and MMPS in primary human chondrocytes, J. Orthop. Res 27 (1) (2009) 65–70. [DOI] [PubMed] [Google Scholar]
- [3].Rostom A, Dube C, Ga W, Tugwell P, Welch V, Jolicoeur E, et al. , Prevention of NSAID-Induced Gastroduodenal Ulcers (Review), 4 (2011). [DOI] [PubMed] [Google Scholar]
- [4].Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, et al. , Vascular and upper gastrointestinal eff ects of non-steroidal anti-infl ammatory drugs: meta-analyses of individual, Lancet 382 (9894) (2013) 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gislason GH, Rasmussen J, Abildstrom S, ASchramm T, Hansen M, Fosbøl E, et al. , Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure, Arch. Intern. Med 169 (2) (2009) 141–149. [DOI] [PubMed] [Google Scholar]
- [6].Lee A, Mg C, Jc C, Jf K, Jp K, Effects of Nonsteroidal Anti-Inflammatory Drugs On Postoperative Renal Function In Adults With Normal Renal Function (Review), 2 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mora JC, Przkora R, Cruz-almeida Y, Knee Osteoarthritis: Pathophysiology and Current Treatment Modalities, (2018), pp. 2189–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jones SW, Brockbank SMV, Clements KM, Le Good N, Campbell D, Read SJ, et al. , Mitogen-activated protein kinase-activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology, Osteoarthr. Cartil 17 (1) (2009) 124–131. Available from: 10.1016/j.joca.2008.05.001. [DOI] [PubMed] [Google Scholar]
- [9].Philp AM, Davis ET, Jones SW, Developing anti-inflammatory therapeutics for patients with osteoarthritis, Rheumatol. (United Kingdom) 56 (6) (2017) 869–881. [DOI] [PubMed] [Google Scholar]
- [10].Ward B, Seal BL, Brophy CM, Panitch A, Design of a bioactive cell-penetrating peptide: when a transduction domain does more than transduce, J. Pept. Sci 15 (10) (2009) 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brugnano JL, Chan BK, Seal BL, Panitch A, Cell-penetrating peptides can confer biological function: regulation of inflammatory cytokines in human monocytes by MK2 inhibitor peptides, J. Control. Release 155 (2) (2011) 128–133. Available from: 10.1016/j.jconrel.2011.05.007. [DOI] [PubMed] [Google Scholar]
- [12].Bartlett RL, Panitch A, Thermosensitive nanoparticles with pH-triggered degradation and release of anti-inflammatory cell-penetrating peptides, Biomacromolecules 13 (8) (2012) 2578–2584. [DOI] [PubMed] [Google Scholar]
- [13].Bartlett RL, Sharma S, Panitch A, Cell-penetrating peptides released from thermosensitive nanoparticles suppress pro-inflammatory cytokine response by specifically targeting inflamed cartilage explants, Nanomed. Nanotechnol. Biol. Med 9 (3) (2013) 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McMasters J, Poh S, Lin JB, Panitch A, Delivery of anti-inflammatory peptides from hollow PEGylated poly(NIPAM) nanoparticles reduces inflammation in an ex vivo osteoarthritis model, J. Control. Release 258 (March) (2017) 161–170. Available from: 10.1016/j.jconrel.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Poh S, Lin JB, Panitch A, Release of anti-inflammatory peptides from Thermosensitive nanoparticles with degradable cross-links suppresses pro-inflammatory cytokine production, Biomacromolecules 16 (4) (2015) 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lin JB, Poh S, Panitch A, Controlled release of anti-inflammatory peptides from reducible thermosensitive nanoparticles suppresses cartilage inflammation, Nanomed. Nanotechnol. Biol. Med 12 (2016) 2095–2100. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1549963416300570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ward BC, Kavalukas S, Brugnano J, Barbul A, Panitch A, Peptide inhibitors of MK2 show promise for inhibition of abdominal adhesions, J. Surg. Res 169 (1) (2012) 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pratta MA, Yao W, Decicco C, Tortorella MD, Liu RQ, Copeland RA, et al. ,Aggrecan protects cartilage collagen from proteolytic cleavage, J. Biol. Chem 278 (46) (2003) 45539–45545. [DOI] [PubMed] [Google Scholar]
- [19].Gupta MK, Martin JR, Werfel TA, Shen T, Page JM, Duvall CL, Cell Protective, ABC Triblock Polymer-Based Thermoresponsive Hydrogels with ROS-Triggered Degradation and Drug Release, (2014). [DOI] [PubMed] [Google Scholar]
- [20].Online VA, Wang H, Ke F, Mararenko A, Wei Z, Banerjee P, et al. , Responsive polymer – fl uorescent carbon nanoparticle hybrid nanogels for optical, (2014), pp. 7443–7452. [DOI] [PubMed] [Google Scholar]
- [21].Swift T, Katsikogianni M, Hoskins R, Teratarantorn P, Douglas I, MacNeil S, et al. , Highly-branched poly(N-isopropyl acrylamide) functionalised with pendant Nile red and chain end vancomycin for the detection of gram-positive bacteria, Acta Biomater. 87 (2019) 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chien-Chi Lin, Anseth KS, PEG hydrogels for the controlled release of biomolecules in regenerative medicine Chien-chi, Pharm. Res 26 (3) (2009) 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang S, Langer R, Traverso G, Nanoparticulate drug delivery systems targeting inflammation for treatment of inflammatory bowel disease, Nano Today 16 (2017) 82–96. Available from: 10.1016/j.nantod.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Karg M, Pich A, Hellweg T, Hoare T, Lyon LA, Crassous JJ, et al. , Nanogels and microgels: from model colloids to applications, recent developments, and future trends, Langmuir 35 (19) (2019) 6231–6255. [DOI] [PubMed] [Google Scholar]
- [25].Liu G, Wang D, Zhou F, Liu W, Electrostatic self-assembly of au nanoparticles onto Thermosensitive magnetic core-shell microgels for thermally Tunable and magnetically recyclable catalysis, Small 11 (23) (2015) 2807–2816. [DOI] [PubMed] [Google Scholar]
- [26].Reimhult E, Schroffenegger M, Lassenberger A, Design principles for thermo-responsive core-shell nanoparticles: controlling thermal transitions by brush morphology, Langmuir 35 (22) (2019) 7092–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kakwere H, Leal MP, Materia ME, Curcio A, Guardia P, Niculaes D, et al. , Functionalization of strongly interacting magnetic nanocubes with (thermo)responsive coating and their application in hyperthermia and heat-triggered drug delivery, ACS Appl. Mater. Interfaces 7 (19) (2015) 10132–10145. [DOI] [PubMed] [Google Scholar]
- [28].Hannecart A, Stanicki D, Vander Elst L, Muller RN, Lecommandoux S, Thévenot J, et al. , Nano-thermometers with thermo-sensitive polymer grafted USPIOs behaving as positive contrast agents in low-field MRI, Nanoscale 7 (8) (2015) 3754–3767. [DOI] [PubMed] [Google Scholar]
- [29].Shen S, Ding B, Zhang S, Qi X, Wang K, Tian J, et al. , Near-infrared light-responsive nanoparticles with thermosensitive yolk-shell structure for multimodal imaging and chemo-photothermal therapy of tumor, Nanomed. Nanotechnol. Biol. Med 13 (5) (2017) 1607–1616. Available from: 10.1016/j.nano.2017.02.014. [DOI] [PubMed] [Google Scholar]
- [30].Xiao Q, Li Y, Li F, Zhang M, Zhang Z, Lin H, Rational design of a thermalresponsive-polymer-switchable FRET system for enhancing the temperature sensitivity of upconversion nanophosphors, Nanoscale 6 (17) (2014) 10179–10186. [DOI] [PubMed] [Google Scholar]
- [31].Schroffenegger M, Reimhult E, Thermoresponsive core-shell nanoparticles: does core size matter? Materials (Basel) 11 (9) (2018) 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dubbert J, Nothdurft K, Karg M, Richtering W, Core-shell-shell and hollow double-shell microgels with advanced temperature responsiveness, Macromol. Rapid Commun 36 (2) (2015) 159–164. [DOI] [PubMed] [Google Scholar]
- [33].Liu T, Zhang W, Song T, Yang X, Li C, Hollow double-layered polymer micro-spheres with pH and thermo-responsive properties as nitric oxide-releasing reservoirs, Polym. Chem 6 (17) (2015) 3305–3314. [Google Scholar]
- [34].Li C, Ma Y, Niu H, Zhang H, Hydrophilic hollow molecularly imprinted polymer microparticles with photo- and Thermoresponsive template binding and release properties in aqueous media, ACS Appl. Mater. Interfaces 7 (49) (2015) 27340–27350. [DOI] [PubMed] [Google Scholar]
- [35].Schmid AJ, Dubbert J, Rudov AA, Pedersen JS, Lindner P, Karg M, et al. , Multi-shell hollow nanogels with responsive shell permeability, Sci. Rep 6 (March) (2016) 1–13. Available from: 10.1038/srep22736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Su Y, Ojo OF, Tsengam IKM, He J, McPherson GL, John VT, et al. , Thermoresponsive coatings on hollow particles with mesoporous shells serve as stimuli-responsive gates to species encapsulation and release, Langmuir 34 (48) (2018) 14608–14616. [DOI] [PubMed] [Google Scholar]
- [37].Kitayama Y, Yoshikawa K, Takeuchi T, Efficient pathway for preparing hollow particles: site-specific crosslinking of spherical polymer particles with Photoresponsive groups that play a dual role in Shell crosslinking and Core shielding, Langmuir 32 (36) (2016) 9245–9253. [DOI] [PubMed] [Google Scholar]
- [38].Naseem K, Begum R, Wu W, Irfan A, Farooqi ZH, Advancement in multi-functional Poly(styrene)-Poly(N-isopropylacrylamide) based core–shell microgels and their applications, Polym. Rev 58 (2) (2018) 288–325. Available from 10.1080/15583724.2017.1423326. [DOI] [Google Scholar]
- [39].Hadipour Moghaddam SP, Yazdimamaghani M, Ghandehari H, Glutathione-sensitive hollow mesoporous silica nanoparticles for controlled drug delivery, J. Control. Release 282 (January) (2018) 62–75. Available from 10.1016/j.jconrel.2018.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chu F, Polzer F, Severin N, Lu Y, Ott A, Rabe JP, et al. , Thermosensitive hollow Janus dumbbells, Colloid Polym. Sci 292 (8) (2014) 1785–1793. [Google Scholar]
- [41].Brändel T, Dirksen M, Hellweg T, Tuning the swelling properties of smart multiresponsive core-shell microgels by copolymerization, Polymers (Basel) 11 (8) (2019) 1269. Available from: https://www.mdpi.com/2073-4360/11/8/1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Herman ES, Exploring Complex Interactions Within Microgels and Microgel Assemblies, (2014) (December). [Google Scholar]
- [43].García-Couce J, Almirall A, Fuentes G, Kaijzel E, Chan A, Cruz LJ, Targeting polymeric nanobiomaterials as a platform for cartilage tissue engineering, Curr. Pharm. Des 25 (2019) 1–18. Available from: http://www.eurekaselect.com/173321/article. [DOI] [PubMed] [Google Scholar]
- [44].Bhattacharjee S, DLS and zeta potential - what they are and what they are not? J. Control. Release 235 (2016) 337–351. Available from: 10.1016/j.jconrel.2016.06.017. [DOI] [PubMed] [Google Scholar]
- [45].Bhattacharjee S, DLS and zeta potential - What they are and what they are not? J. Control. Release 235 (2016) 337–351. [DOI] [PubMed] [Google Scholar]
- [46].Gao J, Frisken BJ, Influence of reaction conditions on the synthesis of self-cross-linked N-isopropylacrylamide microgels, Langmuir 19 (13) (2003) 5217–5222. [DOI] [PubMed] [Google Scholar]
- [47].Cao M, Wang Y, Hu X, Gong H, Li R, Cox H, et al. , Reversible thermoresponsive peptide-PNIPAM hydrogels for controlled drug delivery, Biomacromolecules (2019), 10.1021/acs.biomac.9b01009 acs.biomac.9b01009. Available from:. [DOI] [PubMed] [Google Scholar]
- [48].Estrela JM, Ortega A, Obrador E, Glutathione in cancer biology and therapy, Critical Reviews in Clinical Laboratory Sciences, 43 2006, pp. 143–181. [DOI] [PubMed] [Google Scholar]
- [49].Griffith OW, Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine, Anal. Biochem 106 (1) (1980) 207–212. [DOI] [PubMed] [Google Scholar]
- [50].Chen J, Wu M, Veroniaina H, Mukhopadhyay S, Poly(N-isopropylacrylamide) derived nanogels demonstrated thermosensitive self-assembly and GSH-triggered drug release for efficient tumor therapy, Polym. Chem (2019) 3–6. [Google Scholar]
- [51].Arunachalam B, Phan UT, Geuze HJ, Cresswell P, Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT), Proc. Natl. Acad. Sci. U. S. A 97 (2) (2000) 745–750. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10639150%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC15401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jones SA, Scheller J, Rose-john S, Yamamoto N, Fuller G, Kakehi T, Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling, Cell 121 (9) (2011) 3375–3383. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21881215%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3163962%5Cnhttp://dl.acm.org/citation.cfm?id=1358628.1358957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Brown S, Kumar S, Sharma B, Intra-articular targeting of nanomaterials for the treatment of osteoarthritis, Acta Biomater. 93 (2019) 239–257. Available from 10.1016/j.actbio.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Maudens P, Jordan O, Allémann E, Recent advances in intra-articular drug delivery systems for osteoarthritis therapy, Drug Discov. Today 23 (10) (2018) 1761–1775. Available from 10.1016/j.drudis.2018.05.023. [DOI] [PubMed] [Google Scholar]
- [55].Owen S, Francis H, Roberts M, Disappearance kinetics of solutes from synovial fluid after intra- articular injection, Br. J. Clin. Pharmacol 38 (4) (1994) 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gerwin N, Hops C, Lucke A, Intraarticular drug delivery in osteoarthritis, Adv. Drug Deliv. Rev 58 (2) (2006) 226–242. [DOI] [PubMed] [Google Scholar]
- [57].Morgen M, Tung D, Boras B, Miller W, Malfait AM, Tortorella M, Nanoparticles for improved local retention after intra-articular injection into the knee joint, Pharm. Res 30 (1) (2013) 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.