

Abstract
Alzheimer’s disease (AD) patients suffer progressive cerebral atrophy before dementia onset. However, the region-specific atrophic processes and the influences of age and apolipoprotein E (APOE) on atrophic trajectory are still unclear. By mapping the region-specific nonlinear atrophic trajectory of whole cerebrum from amnestic mild cognitive impairment (aMCI) to AD based on longitudinal structural magnetic resonance imaging data from Alzheimer’s disease Neuroimaging Initiative (ADNI) database, we unraveled a quadratic accelerated atrophic trajectory of 68 cerebral regions from aMCI to AD, especially in the superior temporal pole, caudate, and hippocampus. Besides, interaction analyses demonstrated that APOE ε4 carriers had faster atrophic rates than noncarriers in 8 regions, including the caudate, hippocampus, insula, etc.; younger patients progressed faster than older patients in 32 regions, especially for the superior temporal pole, hippocampus, and superior temporal gyrus; and 15 regions demonstrated complex interaction among age, APOE, and disease progression, including the caudate, hippocampus, etc. (P < 0.05/68, Bonferroni correction). Finally, Cox proportional hazards regression model based on the identified region-specific biomarkers could effectively predict the time to AD conversion within 10 years. In summary, cerebral atrophic trajectory mapping could help a comprehensive understanding of AD development and offer potential biomarkers for predicting AD conversion.
Keywords: Alzheimer’s disease, mild cognitive impairment, atrophic trajectory, apolipoprotein E, Aging
Introduction
Alzheimer’s disease (AD) is a serious neurodegenerative disease that progressively impairs the multidimensional cognitive functions of the elderly. Although mounting clinical studies have shown that it is difficult to cure dementia based on current medical technology, early intervention can effectively slow down AD conversion (Norton et al. 2014; Siemers et al. 2016; Kivipelto et al. 2018). Thus, identifying the high-probable AD patients before severe cognitive impairment is crucial for early prevention. Amnestic mild cognitive impairment (aMCI) is considered as a prodromal stage of AD, and most of the elderly with aMCI eventually progress to dementia at an annual rate of about 10–15% (Petersen et al. 2001; Levey et al. 2006; Risacher et al. 2009). Current neuroimaging research on the longitudinal trajectories of brain structural and functional changes in aMCI has led to a deeper understanding of AD development and offered potential biomarkers for the early prediction and prevention of AD (Jack et al. 2010; Delor et al. 2013; Jack Jr and Holtzman 2013; Lindemer et al. 2015; Guerrero et al. 2016; Corriveau-Lecavalier et al. 2019; Wang et al. 2020).
AD patients suffered severe gray matter atrophy in widespread brain regions, including the medial temporal lobes (MTL), posterior cingulate cortex (PCC), and so on (Karas et al. 2004; Whitwell et al. 2008; Pini et al. 2016). Detectable gray matter atrophy has been identified up to ten years before clinical AD, even when mild cognitive decline is absent (Tondelli et al. 2012). Longitudinal trajectory analyses also demonstrated progressive atrophy during the process of AD conversion in several brain regions, such as the hippocampus, amygdala, and entorhinal cortex (Younes et al. 2014; Pegueroles et al. 2017; Younes et al. 2019). These studies highlighted the potentials of brain morphometric measures in monitoring disease progression and early predicting AD conversion. However, most prior studies delineating atrophic trajectory from aMCI to AD focused on only limited brain regions (Jack Jr et al. 2008; An et al. 2016; Bilgel and Jedynak 2019). Furthermore, many longitudinal studies only explored the linear atrophic progress with AD progression because of limited follow-up time points; little is known about the nonlinear atrophic trajectory from aMCI to AD (Jack Jr et al. 2008; Lo et al. 2011; Corriveau-Lecavalier et al. 2019; Dicks et al. 2019). Thus, this study focused on portraying the nonlinear longitudinal atrophic trajectories of the whole cerebrum from aMCI to AD.
It is well known that AD conversion could be regulated by several risk factors, such as the ε4 variants of the apolipoprotein E (APOE) and aging (Armstrong 2019), which raises questions about whether and how these risk factors affect brain atrophic trajectory during AD development. APOE ε4 is the most influential genetic risk factor for AD (Hsiung et al. 2004). Early studies had found that APOE ε4 carriers had severer gray matter loss than noncarriers in the AD, aMCI, and even cognitive-normal adults (Spampinato et al. 2011; Taylor et al. 2014; Susanto et al. 2015; Nao et al. 2017). Moreover, aging is another strong risk factor for AD and brain atrophy. Strong evidence supports an increased risk of developing AD increases with aging (Guerreiro and Bras 2015). However, it is interesting to note that more severe brain atrophy was found in younger AD patients, including the MTL and inferior parietal lobe (Fan et al. 2012); longitudinal studies have also shown that early-onset AD patients suffered more rapid cortical atrophy than late-onset AD (Cho et al. 2013). Hence, we hypothesized that APOE polymorphism and aging could modulate the atrophic trajectory of the cerebral subregions during AD conversion.
This study aimed to map the atrophic trajectory of the whole cerebrum from early aMCI to AD onset. Based on the longitudinal structural magnetic resonance imaging (sMRI) data from Alzheimer’s Disease Neuroimaging Initiative (ADNI) datasets, a linear mixed-effects (LME) model was introduced to identify brain regions whose atrophic trajectories following aging are faster for aMCI patients who finally progressed to AD relative to cognitively normal (CN). Then we delineated the atrophic trajectories of these brain regions as a function of time-to-conversion and explored the regulation effects of APOE ε4 and aging risk factors on the atrophic trajectories. Finally, based on the identified brain volumetric features, we introduced a Cox proportional hazards regression model to test the potentials of these identified volumetric features in predicting the conversion of AD. The pipeline of this study is shown in Fig. 1.
Fig. 1.

A schematic summary of the study design. Abbreviations: AAL = Automated Anatomical Labeling; ADAS13 = Alzheimer’s Disease Assessment Scale Cognitive-13; ADNI = Alzheimer’s Disease Neuroimaging Initiative; aMCI_C = amnestic mild cognitive impairment converters; APOE = apolipoprotein E; AUC = area under curve of receiver operating characteristic curve; CDRSB = Clinical Dementia Rating Scale Sum of Boxes; CN = cognitively normal; GMV = gray matter volume; MMSE = Mini-Mental State Examination; RAVLT = Rey Auditory Verbal Learning Test; sMRI = structural magnetic resonance imaging.
Materials and methods
Participants
Datasets used in this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) project (http://adni.loni.usc.edu). We downloaded the longitudinal clinical and sMRI data of ADNI 1/GO/2/3 datasets from the Image Data Archive at the Laboratory of Neuro Imaging (IDA, https://ida.loni.usc.edu/login.jsp). We first enrolled the aMCI and CN subjects based on the reported diagnosis of the ADNI database according to the published criteria (Petersen et al. 2010). Second, we determined the stage of dementia during the follow-up based on the CDRSB scores (O'Bryant et al. 2008): CDRSB scores = 0 for normal cognitive stage, CDRSB scores from 0.5 to 4 for mild cognitive impairment (MCI) stage, and CDRSB scores ≥4.5 for dementia stage. For CN controls, their cognition must maintain normal (CDRSB = 0) at all follow-up times. The aMCI converters (aMCI_C) patients should start with MCI (4 ≥ CDRSB ≥0.5) and finally progress to AD (CDRSB ≥4.5) during follow-up. The stable aMCI (aMCI_S) should start with MCI but not progress to dementia or revert to CN during follow-up (4 ≥ CDRSB ≥0.5). Based on the above inclusion criteria, we initially enrolled 278 CN, 281 aMCI_C, and 290 aMCI_S subjects. After a series of quality control steps (see Supplementary Fig. 1), the longitudinal sMRI data of 232 CN subjects (1265 sMRI scans), 276 aMCI_C patients (1709 sMRI scans), and 279 aMCI_S patients (1669 sMRI scans) were finally recruited in the formal analyses. Each subject had at least two follow-up sMRI scans. We additional enrolled 24 qualified subjects (including 189 sMRI scans) who developed from CN to AD during follow-up as an independent dataset to test the performance of the prediction model.
The sMRI data were acquired using 3D T1-weighted (T1W) magnetization prepared rapid acquisition gradient-echo (MPRAGE) imaging sequences. The detailed acquisition information was available at the website: http://adni.loni.usc.edu/methods/mri-tool/mri-analysis/. Besides, several cognitive measurements were included in this study: the Alzheimer’s Disease Assessment Scale Cognitive-13 (ADAS13) and CDRSB were recorded as the measures of dementia severity (O'Bryant et al. 2008; Yagi et al. 2019); the Mini-Mental State Examination (MMSE) was recorded as a measure of cognitive impairment (Folstein et al. 1975); the Rey Auditory Verbal Learning Test immediate recall (RAVLTimm) was recorded as a measure of memory function (Moradi et al. 2017).
Data preprocessing and gray matter volume quantification
All T1W sMRI data were processed using a longitudinal pipeline based on CAT12 tool (http://www.neuro.uni-jena.de/cat12-html/cat.html) and SPM12 (https://www.fil.ion.ucl.ac.uk/spm/software/spm12/). For each subject, the sMRI data of all runs were firstly rigidly co-registered with the baseline images and averaged to generate the subject-specific average T1W images. Then the average T1W images were segmented and normalized into Montreal Neurological Institute (MNI) space using DARTEL algorithm (Ashburner 2007). For each run, the rigidly aligned T1W images were segmented into different tissue components, such as the gray matter (GM), white matter (WM), and cerebrospinal fluid (CSF). Then the GM component of each run was warped into the MNI space using the subject’s deformation field map generated at the prior step and modulated by the Jacobian determinants, termed absolute gray matter volume (GMV).
The Automated Anatomical Labeling (AAL) atlas and Automated Anatomical Labeling 3 (AAL3) atlas were used to parcel the whole cerebrum into 90 and 140 cortical and subcortical regions, respectively; and the average GMV of each cerebral region of each subject scan was extracted (Tzourio-Mazoyer et al. 2002; Rolls et al. 2020). Finally, the intracranial volume (ICV) (sum of GM, WM, and CSF volumes) was also calculated for further statistics. In contrast to the old version of the AAL atlas, AAL3 had finer-grained subdivisions for the anterior cingulate cortex, orbitofrontal cortex, thalamus, and basal ganglia. The results of the two atlases were cross-validated in the following statistics.
Statistical models for trajectory mapping
A LME model was introduced to delineate the longitudinal atrophic trajectories of the cerebrum during aMCI progressed into AD. LME model allows for modeling both fixed and random effects, and it adapts to irregular follow-up intervals with missing time points (Bernal-Rusiel et al. 2013). In addition, we added the linear and quadratic terms to model the nonlinear atrophic trajectories of the cerebral subregions during AD conversion. Subject-specific random intercepts and slopes terms were considered as random effects to account for the between-subject variance. Site-specific random intercepts term was also modeled to remove the between-site heterogeneity. Besides, ICV, sex, and education were modeled as fixed nuisance covariates. We estimated the effects of linear and quadratic terms based on a joint (or multiple) hypothesis testing using the F-Statistic (Blackwell 2008; Griffiths and Hill 2021). Joint hypothesis testing incorporates multiple null hypotheses into a single test (for example, we hypothesized that all the linear terms and quadratic interaction terms are zero), which will return an F-statistic and P-value for this joint null hypothesis. The LME was performed using MATLAB 2019 (https://www.mathworks.com/). In the present study, several LME models were constructed according to different purposes as described in the following:
Model 1: Effect of group on the cerebral atrophic trajectories with aging
To identify cerebral subregions whose atrophic trajectories were specific for aMCI_C progression rather than normal aging, we first modeled the atrophic trajectories during aging for aMCI_C, CN, and their interactions on the atrophic process. Then, we estimated the total linear and quadratic interaction effects between the age and group based on the joint hypothesis testing using the F-Statistic. We also separately estimated the linear and quadratic age main effects for aMCI_C and CN using joint hypothesis testing. A Bonferroni correction method was used to correct multiple comparisons (P < 0.05/90, Bonferroni correction). The model was described in equation (1):
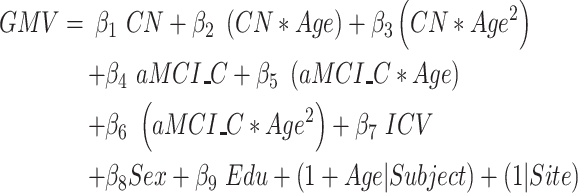 |
(1) |
Age and Age2 represent the linear and quadratic terms of age, respectively.
Model 2: Effect of APOE on the cerebral atrophic trajectories with aging in aMCI_C
This model evaluated the influence of APOE polymorphism on the atrophic trajectories with aging for aMCI_C patients in brain regions identified by Model 1. This model was similar to Model 1, except that APOE was used to substitute the group factor (joint hypothesis testing, P < 0.05/68, Bonferroni correction, equation (2)).
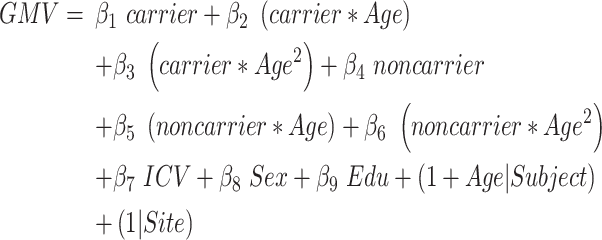 |
(2) |
Age and Age2 represent the linear and quadratic terms of age, respectively.
Model 3: Atrophic trajectories from aMCI to AD
This model portrayed the quadratic atrophic trajectories of all identified conversion-specific cerebral subregions as a function of time-to-conversion in aMCI_C patients. Time-to-conversion (or termed Time in the equations) is defined as the duration (months) between the time point of a certain sMRI scan and the time point at AD conversion with a negative sign representing time before conversion and positive representing time after conversion. The time point at AD conversion is defined as the first date that an aMCI_C patient reached CDRSB scores equal to or higher than 4.5. This model included the linear and quadratic fixed effects of time-to-conversion, and age and age2 were considered as nuisance covariates to control the influence of age on the estimation of the atrophic trajectory (joint hypothesis testing, P < 0.05/68, Bonferroni correction, equation (3)).
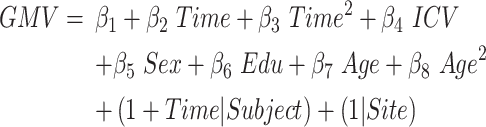 |
(3) |
Time and Time2 represent the linear and quadratic terms of time to AD conversion.
Model 4: Effect of APOE on the atrophic trajectories from aMCI to AD
This model evaluated the influence of APOE polymorphism on the quadratic atrophic trajectories from aMCI to AD. In the first model, we sub-grouped the aMCI_C patients into APOE ε4 carriers versus noncarriers; then we estimated the total linear and quadratic interaction effects between the APOE ε4 carry status and time-to-conversion (joint hypothesis testing, P < 0.05/68, Bonferroni correction, equation (4)):
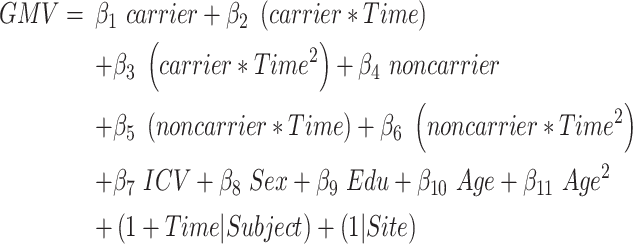 |
(4) |
In the second model, we additionally sub-grouped the aMCI_C patients into five groups with diverse APOE genotypes (ε4/ε4, ε3/ε4, ε2/ε4, ε3/ε3, and ε2/ε3); then we remodeled the interaction effects between the APOE genotypes and time-to-conversion (joint hypothesis testing, P < 0.05/68, Bonferroni correction, equation (5)):
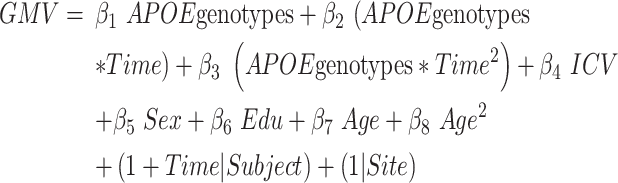 |
(5) |
Model 5: Effect of age on the atrophic trajectories from aMCI to AD
This model evaluated the influence of age on the quadratic atrophic trajectories from aMCI to AD. We estimated the total linear and quadratic interaction effects between the age and time-to-conversion (joint hypothesis testing, P < 0.05/68, Bonferroni correction, equation (6)). Moreover, the annual atrophic rate was calculated for patients with age at 60 and 80 years, respectively.
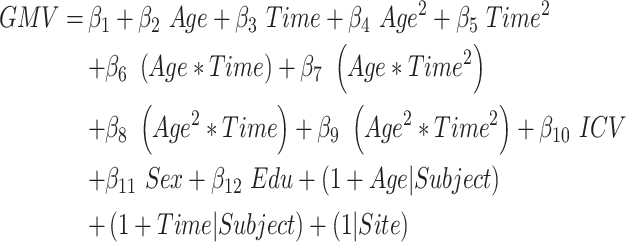 |
(6) |
Model 6: Interaction of age and APOE on the atrophic trajectories from aMCI to AD
This model evaluated the interaction between age and APOE on the quadratic atrophic trajectories from aMCI to AD. The total linear and quadratic interaction effects of the three factors (age, APOE and time-to-conversion) were estimated (joint hypothesis testing, P < 0.05/68, Bonferroni correction, equation (7)). In addition, the annual atrophic rate was calculated for different age subgroups of APOE carriers and noncarriers, respectively.
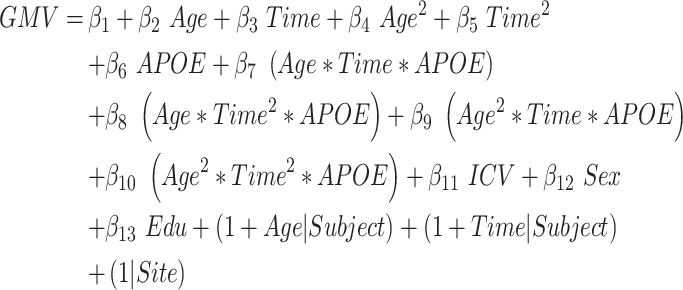 |
(7) |
Model 7: Cognitive decline trajectories from aMCI to AD
This model evaluated the quadratic cognitive decline trajectories as a function of time-to-conversion in aMCI_C and had the same independent variables with equation (3) (joint hypothesis testing, P < 0.05/4, Bonferroni correction). Cognitive measures included ADAS13, CDRSB, MMSE, RAVLTimm (equation (8)).
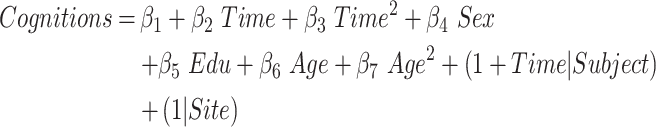 |
(8) |
Model 8: Correlation between cognitive decline and GMV atrophy
This model evaluated the quadratic association between GMV atrophy and cognitive decline. This model included the linear and quadratic terms of cognition as fixed effects (joint hypothesis testing, P < 0.05/68, Bonferroni correction). An example of MMSE was shown in equation (9).
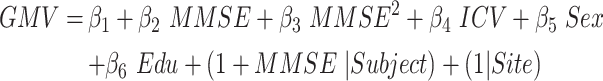 |
(9) |
In addition, the annual atrophic rate was calculated within one year, five years, and ten years before conversion using equation (10):
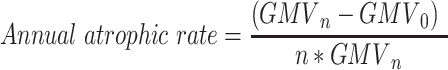 |
(10) |
In which n represents the number of years before AD conversion.
Statistical analyses for demographic data
The demographic data were analyzed using the Statistical Package for the Social Sciences version 26.0 (SPSS). A one-way analysis of variance (ANOVA) (three groups or higher) or two-sample t-test (two groups) was used to compare differences in continuous variables such as age, educational years, follow-up times and duration, cognitive scores between the CN, aMCI_C and aMCI_S groups (P < 0.05). In addition, the chi-square test was used to compare differences in categorical variables such as gender and APOE ε4 status (P < 0.05).
Predicting AD conversion during follow up
A Cox proportional hazards regression model was introduced to test the potential of identified AD conversion-related volumetric features in predicting the time to dementia conversion. All the involved aMCI subjects (including aMCI_C and aMCI_S) were included in the survival analysis. We excluded the sMRI follow-up scans after AD conversion for aMCI_C. The time when aMCI_C patients first converted into AD was regarded as the end event in the survival model. The survival time was defined as years from the first sMRI scan to AD conversion (aMCI_C) or follow-up censoring (aMCI_C or aMCI_S). We first fitted a Cox proportional hazards regression model with the GMV of the 68 cerebral subregions as features and end event (conversion or not conversion) at certain follow-up time points as labels. In addition, to evaluate the predictive value of each of the 68 cerebral subregions, we additionally fitted the Cox proportional hazards regression models with the GMV of a single cerebral region as the feature one-by-one. To evaluate how well these features could discriminate between converters and non-converters, we calculated time-dependent areas under the receiver operating characteristic (ROC) curve (AUC) at each follow-up and the overall average AUC across all follow-ups. The GMV was adjusted for gender, educational years, and site effects using linear regression before survival analysis.
Subject-wise cross-validation was adopted during model training to avoid overestimating the predictive ability (Cudeck and Browne 1983). First, the total aMCI patients were randomly separated into five subsets with approximately equal sample sizes, in which four subsets were used for training, and the remaining one subset was used to test the performance. Considering the randomness of subgrouping, we randomly shuffled the aMCI patients five times and averaged the performances of the 5 times as the final outcomes. Besides, an independent testing dataset including 24 qualified subjects who developed from CN to AD during follow-up was also enrolled to test the performance of the prediction model. A permutation method was used to test the significance of the predictive performance. Specifically, we randomly shuffled the end event (conversion or not conversion) of the training dataset and trained a pseudo-survival model, which was then used to predict the event of the testing dataset to obtain the null AUC. The above step was repeated 1000 times to obtain a null distribution for AUCs. The significance of performance was determined by judging how likely the actual AUC falls into the null distribution (P < 0.05). The survival analysis was carried out using the scikit-survival package (version 0.15.1) of Python.
Result
Demographic findings
The demographic characteristics and cognitive performance of the study population are presented in Table 1. One-way ANOVA demonstrated no statistical difference in age at the first sMRI scan (F = 0.086, P = 0.918) and follow-up duration (F = 2.388, P = 0.093) among the CN, aMCI_C, and aMCI_S. Post hoc analysis demonstrated no statistical difference in gender, education, and the number of sMRI scans per subject between aMCI_C and aMCI_S patients (P > 0.05). The aMCI_C patients had a higher ratio of males (Chi-square test: χ2 = 4.64, P = 0.031), lower education (two-sample t-test: t = 2.64, P = 0.009), a higher ratio of APOE ε4 carriers (Chi-square test: χ2 = 70.64, P < 0.001), and worse cognitive functions at the first sMRI scan (two-sample t-test: all P < 0.001) than the CN. Additionally, within the aMCI_C patients, 183 APOE ε4 carriers (containing at least one ε4 allele copy) and 93 noncarriers were identified (Supplementary Table 1), including five APOE genotypes: ε4/ε4 (46 cases), ε3/ε4 (129 cases), ε2/ε4 (8 cases), ε3/ε3 (84 cases), and ε2/ε3 genotypes (9 cases) (Supplementary Table 2). All aMCI_C patients had 1165 sMRI scans before AD conversion, of which 235 cases had 544 sMRI scans during the dementia stage (Supplementary Table 3). Two-sample t-test found that APOE ε4 carriers were younger than the noncarriers (t = −3.05, P = 0.003), and RAVLTimm was worse for APOE ε4 carriers than noncarriers (t = −2.31, P = 0.022) (Supplementary Table 1).
Table 1.
Main demographic and clinical characteristics of the participants.
| Characteristic | CN | aMCI_C | aMCI_S | Statistics | P values | CN vs. aMCI_C P values | aMCI_C vs. aMCI_S P values | Validation (CN to AD) |
|---|---|---|---|---|---|---|---|---|
| Number of subjects | 232 | 276 | 279 | – | – | – | – | 24 |
| Total no. of sMRI scans | 1265 | 1709 | 1669 | – | – | – | – | 189 |
| Age at first sMRI scan (years) | 73.87 ± 6.12 | 74.12 ± 6.80 | 73.99 ± 7.56 | F = 0.086 | 0.918 | – | – | 75.68 ± 4.39 |
| Gender (males/females) | 114/118 | 162/114 | 164/115 | χ 2 = 6.118 | 0.047 | 0.031 | 0.984 | 8/16 |
| Education (years) | 16.66 ± 2.52 | 16.04 ± 2.73 | 15.79 ± 2.92 | F = 6.584 | 0.001 | 0.009 | 0.303 | 15.71 ± 2.68 |
| APOE ε4 (carriers/noncarriers)a | 67/165 | 183/93 | 120/159 | χ 2 = 73.652 | <0.001 | <0.001 | <0.001 | 10/14 |
| No. sMRI scans per subject | 5.45 ± 2.43 | 6.19 ± 2.25 | 5.98 ± 1.74 | F = 7.844 | <0.001 | <0.001 | 0.219 | 7.88 ± 2.67 |
| Follow-up duration (years) | 4.20 ± 2.98 | 3.86 ± 2.32 | 3.72 ± 2.24 | F = 2.388 | 0.093 | – | – | 7.08 ± 3.25 |
| First sMRI scan | ||||||||
| ADAS13 | 9.32 ± 4.55 | 20.91 ± 6.74 | 15.09 ± 5.25 | F = 260.22 | <0.001 | <0.001 | <0.001 | 9.74 ± 3.98 |
| CDRSB | 0.00 ± 0.00 | 1.97 ± 0.94 | 1.26 ± 0.63 | F = 548.72 | <0.001 | <0.001 | <0.001 | 0.00 ± 0.00 |
| MMSE | 29.06 ± 1.17 | 26.91 ± 1.87 | 27.81 ± 1.79 | F = 103.31 | <0.001 | <0.001 | <0.001 | 29.42 ± 0.95 |
| RAVLT immediate | 45.01 ± 10.10 | 29.28 ± 8.71 | 35.07 ± 9.88 | F = 168.89 | <0.001 | <0.001 | <0.001 | 43.00 ± 9.43 |
a APOE ε4 carriers indicate subjects having one or two copies of ε4 alleles. Continuous variables are shown as “mean ± standard deviation.” P values in bold indicate significant differences between groups.
Cerebral atrophic trajectories as a function of age in CN and aMCI_C groups
Among the 90 cerebral subregions in AAL atlas, LME identified 68 regions whose quadratic atrophic trajectories with aging in aMCI_C were faster than the CN, including the bilateral hippocampus, parahippocampal gyrus (PHG), superior temporal pole (STP), insula, and so on, indicating that these 68 subregions were specific indicators for aMCI-to-AD conversion (joint hypothesis testing, P < 0.05/90, Fig. 2A-D). Sixty of the 68 cerebral subregions could be verified by AAL3 atlas using a strict threshold (P < 0.05/90); the remaining 8 subregions can be validated by AAL3 atlas using a looser threshold (P values: from 5.59e−4 to 5.08e−2). Moreover, AAL3 atlas additionally identified four subregions whose atrophic trajectories with aging in aMCI_C were faster than the CN, including the left olfactory cortex, left calcarine fissure, right superior occipital gyrus, and right supramarginal gyrus (P < 0.05/90), and 3 of them can be validated by AAL atlas using a looser threshold (P values: from 2.13e−3 to 5.26e−3) except for the left olfactory cortex (P = 0.193) (Fig. 3, Supplementary Table 4). An example of the atrophic trajectories as a function of the age of the bilateral hippocampus for the aMCI_C and CN was shown in Fig. 2E (left: F = 105.89, P = 3.80e−45, right: F = 116.01, P = 3.09e−49). In the aMCI_C, AAL atlas further identified that APOE ε4 carriers had faster atrophy in 45 of the 68 cerebral subregions than the noncarriers, of which 42 cerebral regions can be verified by AAL3 atlas (Supplementary Fig. 2, Supplementary Fig. 3, and Supplementary Table 5, P < 0.05/68).
Fig. 2.

Cerebral longitudinal atrophic trajectories following aging in CN and aMCI_C groups. (A) The ordered -log10(P) values of cerebral regions with significantly accelerated cerebral atrophy following age in aMCI_C relative to CN. A linear mixed-effects model was used to estimate the quadratic cerebral atrophic differences between the groups (P < 0.05/90, Bonferroni correction). Asterisk (*) indicates cerebral regions that the AAL3 atlas can validate. (B) The cortical maps of the group differences in quadratic cerebral atrophy as a function of aging. Color bar represents the -log10(P) values. Dark blue represents non-significant. (C) and (D) represents -log10(P) values of the quadratic cerebral atrophy as a function of aging in CN and aMCI_C, respectively. (E) Age-related atrophy changes in the hippocampus. Solid red (CN) and blue (aMCI_C) lines represent the fitted GMV change as a function of aging. The full names of the brain regions are shown in Supplementary Table 4. Abbreviations: aMCI_C = amnestic mild cognitive impairment converters; CN = cognitively normal; GMV = gray matter volume; L = left; R = right; y = year.
Fig. 3.

The cerebral subregions with faster quadratic atrophy as a function of aging in aMCI_C groups compared to CN identified by AAL and AAL3 atlas. (A) The identified 68 cerebral regions by AAL atlas. (B) The identified 64 cerebral regions by AAL3 atlas. (C) Overlaps of the identified cerebral regions between AAL and AAL3 atlas (P < 0.05/90, Bonferroni correction). Abbreviations: aMCI = amnestic mild cognitive impairment; AAL = Automated Anatomical Labeling; CN = cognitively normal; L = left; R = right.
Cerebral atrophic trajectories from aMCI to AD
Based on the identified 68 AAL cerebral regions that showed faster atrophy during aging in aMCI_C, we further fitted the quadratic atrophic trajectories of these regions as a function of time-to-conversion. We found all of these 68 cerebral regions showed quadratic atrophy as a function of time-to-conversion, especially in the bilateral STP, caudate, hippocampus, and pallidum, and all the positive findings revealed by the AAL atlas could be verified by AAL3 atlas (Fig. 4A and B, Supplementary Fig. 4 and Supplementary Table 6, joint hypothesis testing, P < 0.05/68). One example of the bilateral hippocampal atrophic trajectories during AD conversion was shown in Fig. 4C (left: F = 167.48, P = 4.02e−67, right: F = 140.76, P = 2.69e−57). The annual atrophic rates of these regions were highest within one year before conversion, followed by five years, and the lowest for ten years, suggesting accelerated atrophy during conversion from aMCI to AD. The top 5 highest annual atrophic rate was shown in the right pallidum (11.89%), left pallidum (8.96%), left STP (4.25%), left hippocampus (4.18%), and right caudate (4.14%) within one year before AD conversion (Fig. 4D).
Fig. 4.

Cerebral longitudinal atrophic trajectories from aMCI to AD. (A) The ordered -log10(P) values of cerebral regions with significantly accelerated cerebral atrophy from aMCI to AD. A linear mixed-effects model was used to estimate the quadratic cerebral atrophic trajectories as a function of time to AD conversion (P < 0.05/68, Bonferroni correction). Asterisk (*) indicates cerebral regions that the AAL3 atlas can validate. (B) The quadratic cerebral atrophic maps as a function of time to AD conversion. The color bar represents the -log10(P) values. Dark blue represents non-significant. (C) The atrophic trajectories of the hippocampus during the conversion. Solid blue lines represent the fitted hippocampal volume as a function of time to AD conversion. (D) The maps of average annual atrophic rate within 1 year, 5, and 10 years prior to AD conversion. The full names of the brain regions are shown in Supplementary Table 6. Abbreviations: L = left; m = month; R = right.
Effects of APOE and aging on the cerebral atrophic trajectories from aMCI to AD
LME fitting based on the AAL atlas further identified eight cerebral regions whose atrophic trajectories following time-to-conversion were regulated by APOE mutation, including the bilateral caudate and hippocampus, right insula and olfactory gyrus, and left Heschel gyrus and superior temporal gyrus (STG) (Fig. 5A, joint hypothesis testing, P < 0.05/68). Six of the 8 cerebral subregions can be verified by AAL3 atlas using a strict threshold (P < 0.05/68), and the other two brain regions can be validated by AAL3 atlas using a looser threshold (left Heschel gyrus: P = 4.82e−3, left STG: P = 2.08e−2). Besides, the left middle temporal gyrus was additionally identified by the AAL3 atlas (P < 0.05/68) (Supplementary Fig. 5, Supplementary Table 7). Post hoc analysis demonstrated that APOE ε4 carriers suffered faster accelerated quadratic atrophy than the noncarriers in these areas (Fig. 5B and C, P < 0.05/8). The atrophic trajectories of the hippocampus from aMCI to AD for the ε4 carrier and noncarrier patients were shown in Fig. 5D (left: F = 8.58, P = 1.97e−4, right: F = 11.91, P = 7.34e−6). The further inter-genotype comparison demonstrated that APOE ε4 carriers (ε4/ε4, ε3/ε4, and ε2/ε4) encountered the fastest accelerated quadratic atrophy, followed by ε3 homozygous, and ε2/ε3 carriers slowed down the atrophic trajectories (Fig. 5D, Supplementary Fig. 6).
Fig. 5.

Effects of APOE ε4 on the cerebral longitudinal atrophic trajectories from aMCI to AD. (A) The cortical maps of difference in cerebral longitudinal atrophic trajectories between APOE ε4 carriers and noncarriers. A linear mixed-effects model was used to estimate the effect of APOE polymorphism on the quadratic cerebral atrophic trajectories as a function of time to AD conversion (Fig. < 0.05/68, Bonferroni correction). The color bar represents the -log10(P) values. Dark blue represents non-significant. Panels (B) and (C) represent the quadratic cerebral atrophic maps as a function of time to AD conversion in APOE ε4 carriers and noncarriers, respectively. (D) The atrophic trajectories of the hippocampus during the conversion in APOE ε4 carriers and noncarriers (left panels) and in five APOE genotypes (right panels). Abbreviations: APOE = apolipoprotein E; L = left; m = month; R = right.
In addition, the AAL atlas identified a significant regulation of aging on the atrophic trajectories in 32 cerebral regions following time to AD conversion (Fig. 6A and B, joint hypothesis testing, P < 0.05/68), especially for bilateral STP, STG, and right hippocampus, in which 28 subregions can be verified by AAL3 atlas using a strict threshold (P < 0.05/68), and the remaining 4 can be verified using a loose threshold (P values from 9.48e−4 to 7.07e−3). Two cerebral subregions (right superior frontal gyrus and left Rolandic operculum) were additionally identified by the AAL3 atlas (Supplementary Fig. 7, Supplementary Table 8, P < 0.05/68). For example, younger patients had faster accelerated atrophy in the bilateral hippocampus than those older (Fig. 6C, left: F = 8.25, P = 1.38e−6, right: F = 12.55, P = 4.62e−10). Besides, the annual atrophic rates of younger patients appeared more rapid than older patients, and the greatest atrophic rate was observed within one year before conversion, with about 5.55% in the left PCC at 60 years old, while only 2.47% at 80 years old (Fig. 6D).
Fig. 6.

Effects of age on the cerebral longitudinal atrophic trajectories from aMCI to AD. Panels (A) and (B) represent the cerebral regions with significant Age×Time interaction effects on cerebral atrophy. A linear mixed-effects model was used to estimate the quadratic interaction between age and time to AD conversion on GMV (P < 0.05/68, Bonferroni correction). Asterisk (*) indicates cerebral regions that the AAL3 atlas can validate. (C) The quadratic atrophic trajectories of the hippocampus as a function of time to AD conversion at an interval of five years between 60 and 80 age. (D) The maps of average annual atrophic rate within 1 year, 5 years, and 10 years prior to AD conversion at age 60 (left panel) and 80 (right panel) years. The full names of the brain regions are shown in Supplementary Table 8. Abbreviations: GMV = gray matter volume; L = left; R = right; y = year.
Finally, a complex interaction between APOE gene risk and aging on the atrophic trajectories following AD conversion was found in 15 cerebral regions by AAL atlas (Fig. 7A and B, joint hypothesis testing, P < 0.05/68), in which 10 subregions can be verified by AAL3 atlas using a strict threshold (P < 0.05/68), and the remaining 5 can be verified using a loose threshold (P values from 1.21e−3 to 9.70e−3). Besides, the right lingual gyrus was additionally identified by AAL3 atlas (Supplementary Fig. 8, Supplementary Table 9, P < 0.05/68). Post hoc analysis demonstrated the regulation effects of aging on the atrophic trajectories of these areas following time to AD conversion were only evident in APOE ε4 carriers while not in noncarriers (Fig. 7C and D and Supplementary Fig. 9, P < 0.05/15). The atrophic trajectories of the hippocampus at different ages for the APOE ε4 carriers and noncarriers were presented in Fig. 7E (left: F = 5.42, P = 2.44e−4, right: F = 7.27, P = 8.35e−6).
Fig. 7.

Effects of APOE ε4 and age on the cerebral longitudinal atrophic trajectories from aMCI to AD. Panels (A) and (B) represent the cerebral regions with significant APOE×Age×Time interaction effects on the cerebral atrophy, respectively. A linear mixed-effects model was used to estimate the quadratic interaction between APOE, age, and time to AD conversion on GMV (P < 0.05/68, Bonferroni correction). Asterisk (*) indicates cerebral regions that the AAL3 atlas can validate. (C) and (D) represent the effect of aging on the quadratic cerebral atrophic trajectories as a function of time to AD conversion in APOE ε4 carriers and noncarriers, respectively. (E) The effects of APOE polymorphism and aging on the quadratic atrophic trajectories of the hippocampus as a function of time to AD conversion. The full names of the brain regions are shown in Supplementary Table 9. Abbreviations: APOE = apolipoprotein E; GMV = gray matter volume; L = left; R = right; y = year.
The trajectory of cognitive changes from aMCI to AD and associations with cerebral atrophy
Quadratic LME model further identified significantly accelerated cognitive decline following the development of AD (joint hypothesis testing, ADAS13: F = 289.21, P = 1.12e−107; CDRSB: F = 584.92, P = 4.60e−191; MMSE: F = 264.30, P = 7.68e−100; RAVLTimm: F = 142.79, P = 1.12e−57). The cognitive decline became more severe when the onset time was closer and worsen after dementia (Fig. 8A). Finally, based on the AAL atlas, worse cognitive impairment was correlated with greater atrophy of the bilateral hippocampus (Fig. 8B) and other cerebral regions such as the STP and insula, which could be verified by AAL3 atlas (Supplementary Table 10, P < 0.05, Bonferroni correction).
Fig. 8.

The association between cognitive decline and hippocampal volume from aMCI to AD. (A) the quadratic cognitive decline trajectories as a function of time to AD conversion. A linear mixed-effects model was used to estimate the quadratic changes of cognitive functions as a function of time to AD conversion. (B) The quadratic association between hippocampal volume and cognitive functions. Abbreviations: ADAS13 = Alzheimer’s Disease Assessment Scale Cognitive-13; CDRSB = Clinical Dementia Rating Scale Sum of Boxes; lHip = left hippocampus; m = month; MMSE = Mini-Mental State Examination; RAVLTimm = Rey Auditory Verbal Learning Test immediate recall; rHip = right hippocampus.
Performance of AD conversion prediction
Using the Cox proportional hazards regression model based on 68 cerebral subregions GMV features, the permutation test demonstrated significantly higher time-dependent AUCs at each follow-up point (from 0.688 to 0.773) and average AUC (0.714) compared to the null AUCs distribution for the aMCI testing dataset (P < 0.001) (Fig. 9A). Furthermore, on an independent testing dataset with 24 participants who developed from CN to AD during follow-up, the time-dependent AUCs at each follow-up point (from 0.800 to 0.880) and average AUC (0.814) were also significantly higher than the null distribution (permutation test, P < 0.001) (Fig. 9B), indicating that these AD conversion-related cerebral atrophic features could reliably predict the time to AD conversion.
Fig. 9.

The time-dependent prediction results through Cox proportional hazards regression model. (A) Left panel shows the time-dependent AUCs of survival model based on all 68 GMV features for the aMCI testing subjects. Black line and shades represent the true AUCs and their 95% CI for the 5 randomizations during cross-validation, and gray line and shades represent the pseudo-AUCs and 95% CI for the null distribution by 1000 permutations. The right panel shows the histogram of the null distribution of average AUC. (B) Time-dependent AUCs and average AUC of survival model based on all 68 GMV features for the independent testing cohort from CN to AD conversion. (C) Average AUC of the survival models based on the single GMV feature of each cerebral subregion for the aMCI testing subjects. Asterisk (*) indicates statistically significant based on the permutation test (P < 0.05). Abbreviations: AUC = area under curve of receiver operating characteristic curve; CI = confidence interval; GMV = gray matter volume; sMRI = structural magnetic resonance imaging.
We further evaluate the predictive value of the Cox proportional hazards regression model based on a single cerebral GMV feature. Permutation test identified 62 out of the 68 cerebral subregions whose GMV feature could significantly predict the time to AD conversion (P < 0.001). The cerebral subregions with the top 5 average AUC included the left amygdala (average AUC = 0.745), left middle temporal gyrus (average AUC = 0.736), right inferior temporal gyrus (average AUC = 0.730), left inferior temporal gyrus (average AUC = 0.728), and left hippocampus (average AUC = 0.726) (Fig. 9C and Supplementary Table 11).
Discussion
In this study, a major aim was to map the cerebral longitudinal atrophic trajectories during the conversion from early aMCI to AD. Linear mixed-effects model unraveled that most cerebral regions suffered quadratically accelerated atrophy during the progression from early aMCI to AD, which can be verified in both AAL and AAL3 atlas. Furthermore, APOE ε4 carriers with younger age demonstrated faster atrophic rates than the noncarriers with older age. Finally, the Cox proportional hazards regression model using the GMV of the identified cerebral regions could effectively predict the time to dementia conversion within 10 years. Our study demonstrated that cerebral atrophic trajectory mapping helps a comprehensive understanding of AD development and offers potential biomarkers for early predicting AD conversion.
Early studies had reported progressively whole-brain shrinkage (Jack Jr et al. 2008), MTL atrophy (Younes et al. 2014; Lindemer et al. 2015; Pegueroles et al. 2017; Younes et al. 2019), white matter degeneration (Lindemer et al. 2015), and ventricular expansion (Jack Jr et al. 2008) when individuals progressed from aMCI to AD. Our findings agreed with these findings. Moreover, except for the MTL atrophy, we unraveled that most other cerebral regions suffered obvious GMV loss during the AD conversion, especially areas outside the MTL, such as the superior temporal pole and gyrus, insula, striatum, and cingulate cortex. These cerebral regions are of particular interest because they are affected very early in AD progression. Our findings were in line with early neuropathological studies showing that neurofibrillary degeneration gradually spreads from the MTL to the anterior temporal cortex, polymodal association areas (prefrontal, parietal inferior, temporal superior), and finally to all neocortical areas in aging and Alzheimer’s disease (Delacourte et al. 1999). A study also reported that fibrillar amyloid-β plaque deposition was first observed in the striatum of AD at 17 years before symptom onset (Rodriguez-Vieitez et al. 2016).
In comparison with early studies focusing on the linear changes of GMV over time-to-AD conversion (Lo et al. 2011; Corriveau-Lecavalier et al. 2019), our models further emphasized the quadratic GMV atrophy of these regions: slightly atrophy during the early aMCI, accelerated atrophy when closer to AD conversion, and acceleration after conversion. We found that the top 5 highest annual atrophic rate was shown in the right pallidum (11.89%), left pallidum (8.96%), left superior temporal pole (4.25%), left hippocampus (4.18%), and right caudate (4.14%) within one year before AD conversion. These findings were in line with previous reports of progressive atrophy of the MTL (Younes et al. 2014; Bilgel and Jedynak 2019) and superior temporal cortex (Risacher et al. 2010; Phillips et al. 2019; Contador et al. 2021) during the progression of AD. However, there were inconsistent reports on the atrophy of subcortical structures in AD, especially for the pallidum. Several neuroimaging studies had reported unchanged pallidus volume in the AD (Yi et al. 2016) or aMCI (Roh et al. 2011; Benzinger et al. 2013). In contrast, mild atrophy in striatal structures (including the caudate, pallidum, and putamen) was also detected in MCI and AD (Tang et al. 2014). Furthermore, increased amyloid deposition has been reported in these striatal structures in pre-symptomatic autosomal dominant AD mutation carriers (Benzinger et al. 2013), abnormal insula-pallidus connectivity was reported in early-stage AD (Chen et al. 2013), and the GMV of the pallidum was associated with the short-term memory in MCI patients (Valdés Hernández et al. 2020). These studies supported our findings of accelerated atrophy of the striatal structures (pallidus, caudate, and putamen) during aMCI progressed into AD. It should be noted that the identified annual atrophic rate of bilateral pallidus was extraordinarily double higher than that of hippocampus within one year before AD conversion. A possible reason is that the segmentation of gray matter near the pallidus is not quite accurate based on the 3D T1W MPRAGE sMRI images because this sequence is not good enough to visualize the contours of some subcortical structures, especially for the pallidum and thalamus (Visser et al. 2016). It can also partially interpret the negative findings for the thalamic subdivisions defined by AAL3. Moreover, we also validated that the cerebral atrophic trajectories were closely associated with accelerated cognitive decline during the conversion from aMCI to AD (Dicks et al. 2019). Thus, the unraveled cerebral atrophic trajectories from aMCI to AD in the present study provides potential neuroimaging biomarkers for monitoring and predicting AD progression and highlights the importance of early intervention before severe atrophy (Whitwell 2010; Counts et al. 2017).
We also found that APOE and aging selectively modulated the atrophic trajectories of specific cerebral regions. First, we found that the ε4 alleles could speed up the atrophy during conversion from aMCI to AD and during aging progress in several brain regions such as caudate, hippocampus and insula. Furthermore, we also found that APOE ε4 carriers (ε4/ε4, ε3/ε4 and ε2/ε4) encountered the fastest accelerated quadratic atrophy, followed by ε3 homozygous, and slowest for ε2/ε3 carriers in the hippocampus, superior temporal gyrus, insula and caudate. These findings were consistent with earlier studies showing more severe atrophy of multiple brain regions in APOE ε4 carriers of AD (Hashimoto et al. 2001; Spampinato et al. 2011), progressive aMCI patients (Spampinato et al. 2011; Susanto et al. 2015), and even cognitive-normal adults (Taylor et al. 2014; Nao et al. 2017) than the noncarriers. A recent study reported that APOE ε4 carriers demonstrated faster atrophic rates of the whole brain, hippocampus, entorhinal cortex and middle temporal gyrus after transitioning to dementia but not during aMCI (Chen et al. 2021). In contrast, we further unraveled that the APOE regulation effects occur before AD conversion, supported by early studies showing the impact of APOE ε4 on the brain atrophy of aMCI (Spampinato et al. 2011; Susanto et al. 2015), and cognitive-normal adults (Taylor et al. 2014; Nao et al. 2017). Thus, our findings further verify the evidence that APOE ε4 could accelerate cerebral atrophy and AD progression, while ε2 genotype could slow down the cerebral atrophy and AD conversion (Salvadó et al. 2021).
On the influence of age on atrophic progression, we found younger patients atrophied faster than older patients during AD conversion in 32 cerebral regions, particularly in STP, STG, and hippocampus. The annual atrophic rates of these regions decreased with aging and increased with close to AD conversion, indicating that patients with younger age and closer to AD onset demonstrate a higher atrophic rate of cerebral regions. These results were consistent with prior findings showing more severe brain atrophy in the MTL for younger aMCI and AD patients (Fan et al. 2012; Fiford et al. 2018), and more rapid cortical thinning in early-onset AD patients than late-onset AD (Cho et al. 2013). The difference with previous studies was that we evaluated the interaction between aging and time to AD onset on the cerebral atrophy, which provided more direct evidence about how aging regulates the progress of AD conversion.
Furthermore, we examined whether aging regulation on cerebral atrophic trajectories from aMCI to AD is still affected by APOE risk. Our results showed that aging exerted a strong region-specific regulation on cerebral atrophy during AD conversion only for APOE ε4 carriers rather than the noncarriers. A few studies described the effects of APOE genotype in combination with age on brain atrophy (Lind et al. 2006; Cacciaglia et al. 2018; Martí-Juan et al. 2021). For example, Martí-Juan et al. reported a nonlinear interaction between APOE ε4 allele load and aging in the hippocampal surface of cognitively intact individuals (Martí-Juan et al. 2021). We found that not only the hippocampus but also many other cerebral regions demonstrate strong Aging×APOE interactions on atrophic trajectories. One example is the primary auditory cortex. Recent studies have shown significantly gray matter atrophy (Ren et al. 2018), white matter integrity impairment (Ma et al. 2016) and decreased GABA (Gao et al. 2015) in the primary auditory cortex of the presbycusis elderly. Presbycusis has been indicated as an independent risk factor for mild cognitive decline and AD (Panza et al. 2015) and APOE ε4 allele is the common strong risk factor for presbycusis and AD (Kurniawan et al. 2012). Thus, the strong Aging×APOE interactions on Heschel’s atrophic trajectories might indicate that Heschel’s atrophy is a potential early indicator of AD conversion for APOE ε4 carriers with younger age. In summary, these findings indicated that early intervention is particularly crucial for younger subjects accompanied with APOE ε4 alleles and cerebral atrophy to prevent cognitive worsening (Bernick et al. 2012).
Identifying AD conversion-related markers is critical for monitoring the disease progression and predicting AD conversion (Davatzikos et al. 2009; Misra et al. 2009; Davatzikos et al. 2011). Based on the identified cerebral atrophic features, we further tried to test their potential in predicting the risk of aMCI converted to AD at a certain follow-up time. It is still a challenge in predicting whether and when an individual will convert to AD. Early studies have tried to predict AD conversion by classifying between aMCI-to-AD converters and non-converters with an accuracy of about 66–81% (see review by Arbabshirani et al. (Arbabshirani et al. 2017) for details). However, the definition of the aMCI converters and non-converters has technical flaws because non-converters may be finally converted into AD if their follow-up durations are long enough. Furthermore, this strategy omits the time factor (how long the individual will convert) and thus has limited clinical value. Recently, some studies have tried to predict the time when individuals will convert to AD based on the follow-up neuroimaging data and achieved exciting results (Thung et al. 2018; Vogel et al. 2018; Bilgel and Jedynak 2019; Lloret et al. 2019; Nakagawa et al. 2020). For example, Bilgel et al. trained a linear regression model to predict AD dementia onset age using a quantitative template created from several neuroimaging, CSF, and cognitive biomarkers. They achieved a mean error of fewer than 1.5 years (Bilgel and Jedynak 2019). Instead, we introduced a survival model termed Cox proportional hazards regression model in the present study. This model is commonly applied in clinical prediction, which can provide not only the time when an aMCI patient will convert to AD (as the example by Bilgel et al.) but also the conversion risk (or probability) at this time point (Del Valle et al. 2020). Based on the identified AD conversion-related 68 cerebral subregions GMV features, our model achieved a relative high prediction performance in both cross-validation aMCI testing cohorts (time-dependent AUCs: 0.688–0.773, average AUC: 0.714) and in an independent testing cohort who develop from CN to AD (time-dependent AUCs: 0.800–0.880, average 0.814). Our findings were consistent with a recent study showing good performance in predicting the time to AD conversion using a deep learning-based survival model (Nakagawa et al. 2020). Furthermore, we also evaluated the predictive potential of the GMV feature of a single cerebral region, and found that 62/68 regions could significantly predict the time to AD conversion with the top 5 valued subregions including the left amygdala, left middle temporal gyrus, right inferior temporal gyrus, left inferior temporal gyrus, and left hippocampus. The results were consistent with early reported showing the potentials of biomarkers derived from these regions in predicting the progression to AD (Desikan et al. 2010; Tang et al. 2014; Li et al. 2020). Thus, our findings suggested that the GMV of the identified cerebral subregions derived from the cerebral atrophic trajectory mapping could be used as potential biomarkers to predict the time to AD conversion; moreover, we provided the prediction power of each cerebral GMV biomarker for future validation and clinical application.
There are some limitations in the present study. First, as discussed above, on the quantification of subcortical volumes, the 3D T1W MPRAGE may not be optimal in segmenting the subcortical structures, especially for the thalamus and pallidum. Therefore, a multimodal strategy (incorporating T1-, T2-, PD-, and susceptibility-weighted images) is preferred to precisely segment the subcortical structures and quantify their volume and validate our findings in the subcortical structures in the future (Plassard et al. 2019). Second, the cerebral atrophic trajectory from aMCI to AD delineated in the present study should be validated by other longitudinal datasets with different races and data collection strategies.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the staff of the Tianjin Key Laboratory of Functional Imaging and the General Hospital of Tianjin Medical University for technical and administrative support, commentary, and advice.
Contributor Information
Xiaotong Wei, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Xiaotong Du, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Yingying Xie, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Xinjun Suo, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Xiaoxi He, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Hao Ding, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China; School of Medical Imaging, Tianjin Medical University, Tianjin 300070, China.
Yu Zhang, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Yi Ji, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Chao Chai, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Meng Liang, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China; School of Medical Imaging, Tianjin Medical University, Tianjin 300070, China.
Chunshui Yu, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China; School of Medical Imaging, Tianjin Medical University, Tianjin 300070, China.
Yong Liu, School of Artificial Intelligence, Beijing University of Posts and Telecommunications, Beijing 100876, China.
Wen Qin, Department of Radiology, Tianjin Medical University General Hospital, Tianjin 300052, China; Tianjin Key Lab of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China.
Funding
This work was supported by the Natural Science Foundation of China (81971599, 81771818, 82030053, 81601473, 81571659, 81971694, and 81425013), National Key Research and Development Program of China (2018YFC1314300 and 2017YFC0909200), Tianjin Natural Science Foundation (19JCYBJC25100 and 17JCYBJC29200), Tianjin Key Technology R&D Program (17ZXMFSY00090), the Science & Technology Development Fund of Tianjin Education Commission for Higher Education (2018KJ082), the China Postdoctoral Science Foundation (2017 M611175) and the Tianjin Key Project for Chronic Diseases Prevention (17ZXMFSY00070). In addition, data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012).
Author Contributions
X.W., W.Q. & Y.L. designed research; X.W., X.D., Y.X., X.S., X.H., H.D., Y.Z., Y.J. & C.C. implemented the methods; X.W., X.D., Y.X. & X.S. analyzed data; W.Q., Y.L., M.L. & C.Y. provided guidance and advice. X.W. & W.Q. contributed to the writing of the manuscript.
Conflict of interest statement
The authors declare no conflict of interest.
References
- An H, Son SJ, Cho S, Cho EY, Choi B, Kim SY, Alzheimer's Disease Neuroimaging I . Large intracranial volume accelerates conversion to dementia in males and APOE4 non-carriers with mild cognitive impairment. Int Psychogeriatr. 2016:28:769–778. [DOI] [PubMed] [Google Scholar]
- Arbabshirani MR, Plis S, Sui J, Calhoun VD. Single subject prediction of brain disorders in neuroimaging: Promises and pitfalls. NeuroImage. 2017:145:137–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RA. Risk factors for Alzheimer's disease. Folia Neuropathol. 2019:57:87–105. [DOI] [PubMed] [Google Scholar]
- Ashburner J. A fast diffeomorphic image registration algorithm. NeuroImage. 2007:38:95–113. [DOI] [PubMed] [Google Scholar]
- Benzinger TL, Blazey T, Jack CR Jr, Koeppe RA, Su Y, Xiong C, Raichle ME, Snyder AZ, Ances BM, Bateman RJ, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A. 2013:110:E4502–E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal-Rusiel JL, Greve DN, Reuter M, Fischl B, Sabuncu MR. Statistical analysis of longitudinal neuroimage data with Linear Mixed Effects models. NeuroImage. 2013:66:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernick C, Cummings J, Raman R, Sun X, Aisen P. Age and rate of cognitive decline in Alzheimer disease: implications for clinical trials. Arch Neurol. 2012:69:901–905. [DOI] [PubMed] [Google Scholar]
- Bilgel M, Jedynak BM. Predicting time to dementia using a quantitative template of disease progression. Alzheimers Dement (Amst). 2019:11:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell MJMBR. Multiple hypothesis testing: The F-test; 2008. pp. 1–7
- Cacciaglia R, Molinuevo JL, Falcón C, Brugulat-Serrat A, Sánchez-Benavides G, Gramunt N, Esteller M, Morán S, Minguillón C, Fauria K, et al. Effects of APOE-ε4 allele load on brain morphology in a cohort of middle-aged healthy individuals with enriched genetic risk for Alzheimer's disease. Alzheimers Dement. 2018:14:902–912. [DOI] [PubMed] [Google Scholar]
- Chen G, Zhang HY, Xie C, Chen G, Zhang ZJ, Teng GJ, Li SJ. Modular reorganization of brain resting state networks and its independent validation in Alzheimer's disease patients. Front Hum Neurosci. 2013:7:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XR, Shao Y, Sadowski MJ. Segmented Linear Mixed Model Analysis Reveals Association of the APOEɛ4 Allele with Faster Rate of Alzheimer's Disease Dementia Progression. J Alzheimers Dis. 2021:82:921–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Jeon S, Kang SJ, Lee JM, Lee JH, Kim GH, Shin JS, Kim CH, Noh Y, Im K, et al. Longitudinal changes of cortical thickness in early- versus late-onset Alzheimer's disease. Neurobiol Aging. 2013:34:1921.e9–1921.e15. [DOI] [PubMed] [Google Scholar]
- Contador J, Perez-Millan A, Tort-Merino A, Balasa M, Falgas N, Olives J, Castellvi M, Borrego-Ecija S, Bosch B, Fernandez-Villullas G, et al. Longitudinal brain atrophy and CSF biomarkers in early-onset Alzheimer's disease. NeuroImage Clinical. 2021:32:102804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau-Lecavalier N, Mellah S, Clement F, Belleville S. Evidence of parietal hyperactivation in individuals with mild cognitive impairment who progressed to dementia: A longitudinal fMRI study. Neuroimage Clin. 2019:24:101958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Ikonomovic MD, Mercado N, Vega IE, Mufson EJ. Biomarkers for the Early Detection and Progression of Alzheimer's Disease. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2017:14:35–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudeck R, Browne MW. Cross-Validation Of Covariance Structures. Multivar Behav Res. 1983:18:147–167. [DOI] [PubMed] [Google Scholar]
- Davatzikos C, Xu F, An Y, Fan Y, Resnick SM. Longitudinal progression of Alzheimer's-like patterns of atrophy in normal older adults: the SPARE-AD index. Brain. 2009:132:2026–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davatzikos C, Bhatt P, Shaw LM, Batmanghelich KN, Trojanowski JQ. Prediction of MCI to AD conversion, via MRI, CSF biomarkers, and pattern classification. Neurobiol Aging. 2011:32:2322.e2319–2322.e2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, Lavin Y, Swartz TH, Madduri D, Stock A, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020:26:1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacourte A, David JP, Sergeant N, Buee L, Wattez A, Vermersch P, Ghozali F, Fallet-Bianco C, Pasquier F, Lebert F, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease. Neurology. 1999:52:1158–1165. [DOI] [PubMed] [Google Scholar]
- Delor I, Charoin JE, Gieschke R, Retout S, Jacqmin P. Modeling Alzheimer's Disease Progression Using Disease Onset Time and Disease Trajectory Concepts Applied to CDR-SOB Scores From ADNI. CPT Pharmacometrics Syst Pharmacol. 2013:2:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Cabral HJ, Settecase F, Hess CP, Dillon WP, Glastonbury CM, Weiner MW, Schmansky NJ, Salat DH, Fischl B, et al. Automated MRI measures predict progression to Alzheimer's disease. Neurobiol Aging. 2010:31:1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicks E, Vermunt L, van der Flier WM, Visser PJ, Barkhof F, Scheltens P, Tijms BM, Alzheimer's Disease Neuroimaging I . Modeling grey matter atrophy as a function of time, aging or cognitive decline show different anatomical patterns in Alzheimer's disease. Neuroimage Clin. 2019:22:101786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Holland D, Desikan RS, Dale AM, McEvoy LK. Rates of Decline in Alzheimer Disease Decrease with Age. PLoS One. 2012:7:e42325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiford CM, Ridgway GR, Cash DM, Modat M, Nicholas J, Manning EN, Malone IB, Biessels GJ, Ourselin S, Carmichael OT, et al. Patterns of progressive atrophy vary with age in Alzheimer's disease patients. Neurobiol Aging. 2018:63:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975:12:189–198. [DOI] [PubMed] [Google Scholar]
- Gao F, Wang G, Ma W, Ren F, Li M, Dong Y, Liu C, Liu B, Bai X, Zhao B, et al. Decreased auditory GABA+ concentrations in presbycusis demonstrated by edited magnetic resonance spectroscopy. NeuroImage. 2015:106:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths WE, Hill RCJTAS. On the Power of the F-test for Hypotheses in a Linear Model. 2021:1–7.
- Guerreiro R, Bras J. The age factor in Alzheimer's disease. Genome Med. 2015:7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero R, Schmidt-Richberg A, Ledig C, Tong T, Wolz R, Rueckert D, Alzheimer's Disease Neuroimaging I . Instantiated mixed effects modeling of Alzheimer's disease markers. NeuroImage. 2016:142:113–125. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Yasuda M, Tanimukai S, Matsui M, Hirono N, Kazui H, Mori E. Apolipoprotein E epsilon 4 and the pattern of regional brain atrophy in Alzheimer's disease. Neurology. 2001:57:1461–1466. [DOI] [PubMed] [Google Scholar]
- Hsiung GY, Sadovnick AD, Feldman H. Apolipoprotein E epsilon4 genotype as a risk factor for cognitive decline and dementia: data from the Canadian Study of Health and Aging. CMAJ. 2004:171:863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron. 2013:80:1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Weigand SD, Shiung MM, Przybelski SA, O'Brien PC, Gunter JL, Knopman DS, Boeve BF, Smith GE, Petersen RC. Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology. 2008:70:1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. The Lancet Neurology. 2010:9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas GB, Scheltens P, Rombouts SA, Visser PJ, van Schijndel RA, Fox NC, Barkhof F. Global and local gray matter loss in mild cognitive impairment and Alzheimer's disease. NeuroImage. 2004:23:708–716. [DOI] [PubMed] [Google Scholar]
- Kivipelto M, Mangialasche F, Ngandu T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol. 2018:14:653–666. [DOI] [PubMed] [Google Scholar]
- Kurniawan C, Westendorp RG, de Craen AJ, Gussekloo J, de Laat J, van Exel E. Gene dose of apolipoprotein E and age-related hearing loss. Neurobiol Aging. 2012:33:2230.e7–2230.e12. [DOI] [PubMed] [Google Scholar]
- Levey A, Lah J, Goldstein F, Steenland K, Bliwise D. Mild cognitive impairment: an opportunity to identify patients at high risk for progression to Alzheimer's disease. Clin Ther. 2006:28:991–1001. [DOI] [PubMed] [Google Scholar]
- Li B, Zhang M, Riphagen J, Morrison Yochim K, Li B, Liu J, Salat DH. Prediction of clinical and biomarker conformed Alzheimer's disease and mild cognitive impairment from multi-feature brain structural MRI using age-correction from a large independent lifespan sample. Neuroimage Clin. 2020:28:102387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind J, Larsson A, Persson J, Ingvar M, Nilsson LG, Bäckman L, Adolfsson R, Cruts M, Sleegers K, Van Broeckhoven C, et al. Reduced hippocampal volume in non-demented carriers of the apolipoprotein E epsilon4: relation to chronological age and recognition memory. Neurosci Lett. 2006:396:23–27. [DOI] [PubMed] [Google Scholar]
- Lindemer ER, Salat DH, Smith EE, Nguyen K, Fischl B, Greve DN, Alzheimer's Disease Neuroimaging I . White matter signal abnormality quality differentiates mild cognitive impairment that converts to Alzheimer's disease from nonconverters. Neurobiol Aging. 2015:36:2447–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret A, Esteve D, Lloret MA, Cervera-Ferri A, Lopez B, Nepomuceno M, Monllor P. When Does Alzheimer's Disease Really Start? The Role of Biomarkers. Int J Mol Sci. 2019:20:5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo RY, Hubbard AE, Shaw LM, Trojanowski JQ, Petersen RC, Aisen PS, Weiner MW, Jagust WJ. Longitudinal change of biomarkers in cognitive decline. Arch Neurol. 2011:68:1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Li M, Gao F, Zhang X, Shi L, Yu L, Zhao B, Chen W, Wang G, Wang X. DTI Analysis of Presbycusis Using Voxel-Based Analysis. AJNR Am J Neuroradiol. 2016:37:2110–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí-Juan G, Sanroma-Guell G, Cacciaglia R, Falcon C, Operto G, Molinuevo JL, González Ballester M, Gispert JD, Piella G. Nonlinear interaction between APOE ε4 allele load and age in the hippocampal surface of cognitively intact individuals. Hum Brain Mapp. 2021:42:47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra C, Fan Y, Davatzikos C. Baseline and longitudinal patterns of brain atrophy in MCI patients, and their use in prediction of short-term conversion to AD: results from ADNI. NeuroImage. 2009:44:1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi E, Hallikainen I, Hänninen T, Tohka J. Rey's Auditory Verbal Learning Test scores can be predicted from whole brain MRI in Alzheimer's disease. NeuroImage: Clinical. 2017:13:415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Ishida M, Naito J, Nagai A, Yamaguchi S, Onoda K. Prediction of conversion to Alzheimer's disease using deep survival analysis of MRI images. Brain communications. 2020:2:fcaa057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nao J, Sun H, Wang Q, Ma S, Zhang S, Dong X, Ma Y, Wang X, Zheng D. Adverse Effects of the Apolipoprotein E epsilon4 Allele on Episodic Memory, Task Switching and Gray Matter Volume in Healthy Young Adults. Front Hum Neurosci. 2017:11:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer's disease: an analysis of population-based data. Lancet Neurol. 2014:13:788–794. [DOI] [PubMed] [Google Scholar]
- O'Bryant SE, Waring SC, Cullum CM, Hall J, Lacritz L, Massman PJ, Lupo PJ, Reisch JS, Doody R, Texas Alzheimer's Research C . Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer's research consortium study. Arch Neurol. 2008:65:1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, Solfrizzi V, Logroscino G. Age-related hearing impairment-a risk factor and frailty marker for dementia and AD. Nat Rev Neurol. 2015:11:166–175. [DOI] [PubMed] [Google Scholar]
- Pegueroles J, Vilaplana E, Montal V, Sampedro F, Alcolea D, Carmona-Iragui M, Clarimon J, Blesa R, Lleo A, Fortea J, et al. Longitudinal brain structural changes in preclinical Alzheimer's disease. Alzheimers Dement. 2017:13:499–509. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Doody R, Kurz A, Mohs RC, Morris JC, Rabins PV, Ritchie K, Rossor M, Thal L, Winblad B. Current concepts in mild cognitive impairment. Arch Neurol. 2001:58:1985–1992. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR Jr, Jagust WJ, Shaw LM, Toga AW, et al. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010:74:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JS, Da Re F, Irwin DJ, McMillan CT, Vaishnavi SN, Xie SX, Lee EB, Cook PA, Gee JC, Shaw LM, et al. Longitudinal progression of grey matter atrophy in non-amnestic Alzheimer's disease. Brain. 2019:142:1701–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pini L, Pievani M, Bocchetta M, Altomare D, Bosco P, Cavedo E, Galluzzi S, Marizzoni M, Frisoni GB. Brain atrophy in Alzheimer's Disease and aging. Ageing Res Rev. 2016:30:25–48. [DOI] [PubMed] [Google Scholar]
- Plassard AJ, Bao S, D'Haese PF, Pallavaram S, Claassen DO, Dawant BM, Landman BA. Multi-modal imaging with specialized sequences improves accuracy of the automated subcortical grey matter segmentation. Magn Reson Imaging. 2019:61:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren F, Ma W, Li M, Sun H, Xin Q, Zong W, Chen W, Wang G, Gao F, Zhao B. Gray Matter Atrophy Is Associated With Cognitive Impairment in Patients With Presbycusis: A Comprehensive Morphometric Study. Front Neurosci. 2018:12:744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA, McDonald BC. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr Alzheimer Res. 2009:6:347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risacher SL, Shen L, West JD, Kim S, McDonald BC, Beckett LA, Harvey DJ, Jack CR Jr, Weiner MW, Saykin AJ, et al. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging. 2010:31:1401–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Schöll M, Chiotis K, Thordardottir S, Graff C, Wall A, et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer's disease. Brain. 2016:139:922–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, Qiu A, Seo SW, Soon HW, Kim JH, Kim GH, Kim MJ, Lee JM, Na DL. Volume reduction in subcortical regions according to severity of Alzheimer's disease. J Neurol. 2011:258:1013–1020. [DOI] [PubMed] [Google Scholar]
- Rolls ET, Huang CC, Lin CP, Feng J, Joliot M. Automated anatomical labelling atlas 3. NeuroImage. 2020:206:116189. [DOI] [PubMed] [Google Scholar]
- Salvadó G, Ferreira D, Operto G, Cumplido-Mayoral I, Arenaza-Urquijo EM, Cacciaglia R, Falcon C, Vilor-Tejedor N, Minguillon C, Groot C, et al. The protective gene dose effect of the APOE ε2 allele on gray matter volume in cognitively unimpaired individuals. Alzheimers Dement. 2021. 10.1002/alz.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement. 2016:12:110–120. [DOI] [PubMed] [Google Scholar]
- Spampinato MV, Rumboldt Z, Hosker RJ, Mintzer JE, Alzheimer's Disease Neuroimaging I . Apolipoprotein E and gray matter volume loss in patients with mild cognitive impairment and Alzheimer disease. Radiology. 2011:258:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susanto TA, Pua EP, Zhou J, Alzheimer's Disease Neuroimaging I . Cognition, brain atrophy, and cerebrospinal fluid biomarkers changes from preclinical to dementia stage of Alzheimer's disease and the influence of apolipoprotein e. J Alzheimers Dis. 2015:45:253–268. [DOI] [PubMed] [Google Scholar]
- Tang X, Holland D, Dale AM, Younes L, Miller MI, Alzheimer's Disease Neuroimaging I . Shape abnormalities of subcortical and ventricular structures in mild cognitive impairment and Alzheimer's disease: detecting, quantifying, and predicting. Hum Brain Mapp. 2014:35:3701–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JL, Scanlon BK, Farrell M, Hernandez B, Adamson MM, Ashford JW, Noda A, Murphy GM Jr, Weiner MW. APOE-epsilon4 and aging of medial temporal lobe gray matter in healthy adults older than 50 years. Neurobiol Aging. 2014:35:2479–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thung KH, Yap PT, Adeli E, Lee SW, Shen D. Conversion and time-to-conversion predictions of mild cognitive impairment using low-rank affinity pursuit denoising and matrix completion. Med Image Anal. 2018:45:68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondelli M, Wilcock GK, Nichelli P, De Jager CA, Jenkinson M, Zamboni G. Structural MRI changes detectable up to ten years before clinical Alzheimer's disease. Neurobiol Aging. 2012:33:825.e25–825.e36. [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage. 2002:15:273–289. [DOI] [PubMed] [Google Scholar]
- Valdés Hernández MC, Clark R, Wang SH, Guazzo F, Calia C, Pattan V, Starr J, Della Sala S, Parra MA. The striatum, the hippocampus, and short-term memory binding: Volumetric analysis of the subcortical grey matter's role in mild cognitive impairment. Neuroimage Clin. 2020:25:102158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser E, Keuken MC, Douaud G, Gaura V, Bachoud-Levi AC, Remy P, Forstmann BU, Jenkinson M. Automatic segmentation of the striatum and globus pallidus using MIST: Multimodal Image Segmentation Tool. NeuroImage. 2016:125:479–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JW, Vachon-Presseau E, Pichet Binette A, Tam A, Orban P, La Joie R, Savard M, Picard C, Poirier J, Bellec P, et al. Brain properties predict proximity to symptom onset in sporadic Alzheimer's disease. Brain. 2018:141:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HF, Shen XN, Li JQ, Suckling J, Tan CC, Wang YJ, Feng L, Zhang C, Tan L, Dong Q, et al. Clinical and biomarker trajectories in sporadic Alzheimer's disease: A longitudinal study. Alzheimers Dement (Amst). 2020:12:e12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL. Progression of atrophy in Alzheimer's disease and related disorders. Neurotox Res. 2010:18:339–346. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Shiung MM, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR Jr. MRI patterns of atrophy associated with progression to AD in amnestic mild cognitive impairment. Neurology. 2008:70:512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Kanekiyo M, Ito J, Ihara R, Suzuki K, Iwata A, Iwatsubo T, Aoshima K, Alzheimer's Disease Neuroimaging I, Japanese Alzheimer's Disease Neuroimaging I . Identification of prognostic factors to predict cognitive decline of patients with early Alzheimer's disease in the Japanese Alzheimer's Disease Neuroimaging Initiative study. Alzheimers Dement (N Y). 2019:5:364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi HA, Möller C, Dieleman N, Bouwman FH, Barkhof F, Scheltens P, van der Flier WM, Vrenken H. Relation between subcortical grey matter atrophy and conversion from mild cognitive impairment to Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2016:87:425–432. [DOI] [PubMed] [Google Scholar]
- Younes L, Albert M, Miller MI, Team BR . Inferring changepoint times of medial temporal lobe morphometric change in preclinical Alzheimer's disease. Neuroimage Clin. 2014:5:178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes L, Albert M, Moghekar A, Soldan A, Pettigrew C, Miller MI. Identifying Changepoints in Biomarkers During the Preclinical Phase of Alzheimer's Disease. Front Aging Neurosci. 2019:11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.