

Abstract
RNA is challenging to target with bioactive small molecules, particularly those of low molecular weight that bind with sufficient affinity and specificity. In this report, we developed a platform to address this challenge, affording a novel bioactive interaction. An RNA-focused small-molecule fragment collection (n = 2500) was constructed by analyzing features in all publicly reported compounds that bind RNA, the largest collection of RNA-focused fragments to date. The RNA-binding landscape for each fragment was studied by using a library-versus-library selection with an RNA library displaying a discrete structural element, probing over 12.8 million interactions, the greatest number of interactions between fragments and biomolecules probed experimentally. Mining of this dataset across the human transcriptome defined a drug-like fragment that potently and specifically targeted the microRNA-372 hairpin precursor, inhibiting its processing into the mature, functional microRNA and alleviating invasive and proliferative oncogenic phenotypes in gastric cancer cells. Importantly, this fragment has favorable properties, including an affinity for the RNA target of 300 ± 130 nM, a molecular weight of 273 Da, and quantitative estimate of drug-likeness (QED) score of 0.8. (For comparison, the mean QED of oral medicines is 0.6 ± 0.2). Thus, these studies demonstrate that a low-molecular weight, fragment-like compound can specifically and potently modulate RNA targets.
Graphical Abstract
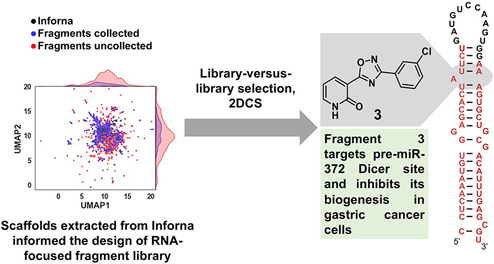
Introduction
Targeting RNAs and affecting their functions could transform medicine and chemical biology. ENCODE and other projects suggest that 1% of our genome encodes protein while as much as 80% is transcribed into RNA. (1) Other studies have suggested that there are as many as 100-fold more potential RNA targets than protein targets. (2,3) Like proteins, much of the function of RNA is due to its structure, providing an opportunity to affect disease-causing RNAs with structure-binding small molecules.
Various approaches have been implemented to identify and optimize RNA structure-binding ligands, including screening of small-molecule collections, (4−7) structure-based drug design, (8−10) and sequence-based design. (11) In the latter approach, the composite of RNA folds present in the transcriptome are mined for overlap with structural elements selected by small-molecule ligands, defined by using a library-versus-library selection called two-dimensional combinatorial screening (2DCS). (11) Collectively, this lead identification strategy, named Inforna, rationally defines novel targets for small molecules agnostically.
We have previously defined the preferred RNA 3D folds of many small molecules and enhanced their affinities and potencies by tethering two small molecules together such that they bind two adjacent structural elements in the same RNA target simultaneously. (12) Doing so, however, increases the modality’s molecular weight. Indeed, the assumption from previous studies is that fragments bind their targets with modest affinities (μM to mM) and must be lead-optimized for bioactivity. (13) To study the limits of physicochemical properties of compounds that bind RNA and to test the assertion that fragment-like compounds must be lead optimized for bioactivity, a 240-member RNA-focused fragment-like library was constructed (molecular weight ≤ 300 Da), selected by possessing chemical features that are found in known RNA binders. These compounds were then tested for their RNA-binding capacity in a massively parallel format to define their binding landscapes. Importantly, by mining these interactions to identify RNAs that have targetable folds in the human transcriptome, we find that a fragment, without lead optimization, can be a potent and specific RNA-targeted small molecule.
Results
Design of an RNA-Focused Fragment Library
The chemical features of all publicly available RNA binders in Inforna, both those identified by 2DCS and those reported in the literature by other laboratories, were computationally analyzed using Scaffold hopper, (14) affording 71 scaffolds privileged for binding RNA. The mostly highly enriched chemical scaffolds include pyrimidine, triazole, thiazole, furan, thiophene, benzimidazole, and quinoline (Figure 1A). We then searched ChemDiv’s fragment library (n = 11,788) for these chemical scaffolds, yielding 2500 RNA-focused fragments. The structures of all RNA-focused fragments as well as the cheminformatic analysis thereof is provided in the Supporting Information.
Figure 1.
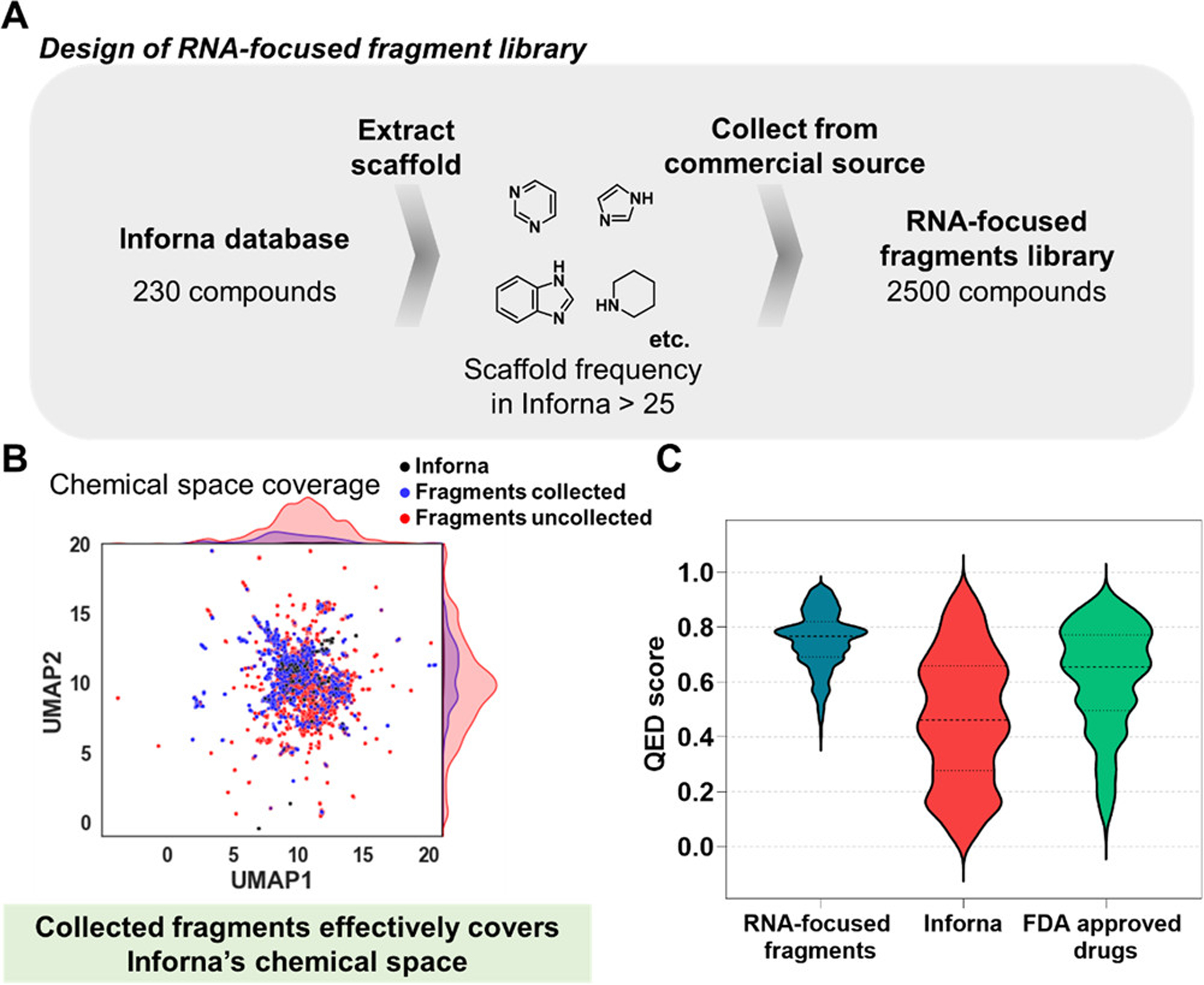
Design of an RNA-focused fragment library informed by known RNA-binding molecules. (A) Scaffolds enriched in the Inforna database were extracted by Scaffold hopper (14) and used in the design of an RNA-focused fragment library (n = 2500). See the Supporting Information for all structures and informatic analysis. (B) All fragments and Inforna compounds were embedded in two dimensions as UMAP1 and UMAP2 to visualize the chemical space that they cover. (C) Violin plot analysis comparing the distribution of quantitative estimate of drug-likeness (QED) score for RNA-focused fragment library (blue), Inforna (red), and 771 FDA-approved drugs that are orally bioavailable (green).
To visualize the chemical space the selected fragments share with Inforna, we generated uniform manifold approximation and projection (UMAP) plots by using Morgan fingerprints (1024 bit vector, radius 2; Figure 1B and Figure S1). (15,16) Approximately 30% of Inforna compounds (68 of 230 compounds) and 29% of RNA-focused fragments (737 of 2500 compounds) were distributed in the region of UMAP1 = 7 to 11 and UMAP2 = 10 to 13; likewise, 13% of both collections were distributed in the region of UMAP1 = 10 to 14 and UMAP2 = 6 to 10 (30 of 230 Inforna compounds; 335 of 2500 RNA-focused fragments). Notably, 22% of ChemDiv’s fragment library (2593 of 11,788 compounds) were distributed in this same region. There were no fragments available in the region covering UMAP1 = 11.8 to 12.9, UMAP2 = 12.7 to 13.3, where 24% of Inforna compounds were distributed. Likewise, ~50% of the selected fragments lie outside the UMAP regions covered by Inforna small molecules, indicating that the RNA-focused fragment library is also diverse. Collectively, this analysis suggests that RNA-focused fragments were efficiently selected from ChemDiv’s fragment library to cover Inforna’s chemical space while maintaining chemical diversity.
Next, the drug-likeness of the RNA-focused library fragments was assessed using their physicochemical properties. According to a guideline named ‘Rule of Three’, fragments are recommended to meet the following criteria: (a) molecular weight ≤ 300 Da, (b) logP ≤ 3, (c) number of hydrogen bond acceptors (HBAs) ≤ 3, and (d) number of hydrogen bond donors (HBDs) ≤ 3. (17) All members of the RNA-focused fragment library are within the guidelines for molecular weight (Figure S2A). Although 80% of fragments in the library deviate from the “Rule of Three” guidelines for the number of HBAs, 98% and 82% of fragments meet the guidelines for the number of HBDs and logP, respectively (Figure S2B–D).
The library was then evaluated based on its quantitative estimate of drug-likeness (QED) score, which can be calculated from eight parameters (molecular weight, octanol–water partition coefficient (AlogP), HBA, HBD, polar surface area (PSA), number of rotatable bonds, number of aromatic rings, and number of structural alerts). QED scores ranges from 0 to 1, with 0 being unfavorable and 1 desirable for orally available drugs. (18) The mean QED for Inforna compounds was 0.47 ± 0.23, which was lower than that of 771 FDA approved orally available drugs (mean = 0.6 ± 0.2; Figure 1C). (18) In contrast, the mean QED for the RNA-focused fragments was 0.75 ± 0.10, indicating that this library is much more drug-like in terms of physicochemical properties. Overall, our analyses show that the RNA-focused fragment library possesses improved physicochemical properties as a small-molecule drug library while sharing similar structural features with RNA-binding compounds.
Selection of the RNA Motifs that Bind Fragments in a Massively Parallel Library-Versus-Library Format
The binding landscapes of these RNA-focused fragments were defined using our selection platform, 2DCS, where the RNA structures preferred by members of a small-molecule library are selected on a microarray surface. For these studies, the RNA structures were selected from a library of RNAs displaying randomized regions in the patterns of a 3 × 3 nucleotide internal loop [3 × 3 internal loop library (ILL)] or a 3 × 2 nucleotide internal loop (3 × 2 ILL). The randomized region in each RNA was embedded in a hairpin cassette, allowing amplification by reverse transcription followed by polymerase chain reaction (RT-PCR). The 3 × 3 ILL encodes for 4096 unique RNA folds, while the 3 × 2 ILL encodes for 1024 (Figure S3). Thus, 12,800,000 million RNA fold-fragment interactions were probed.
We first studied the general RNA-binding capacity of the fragments by absorbing them onto an agarose-coated microarray (a method named AbsorbArray (19)) and incubating with each radioactively labeled ILL individually in the absence of competing oligonucleotides. Of the 2500 fragments, 12 bound to the 3 × 3 ILL (hit rate of 0.48%), while seven bound to the 3 × 2 ILL (hit rate of 0.28%) (Figure S4). Of these, four fragments (1, 2, 3, and 4) overlapped between the two ILLs (Figure S4). The identified binding fragments comprise various chemotypes, such as benzimidazole, pyridine, thiazole, and triazole, which are known to bind RNA, (20,21) as well as novel chemotypes within the local chemical space screened, such as the five-membered heterocycle cores, pyrazole, isoxazole, and oxadiazole. The physicochemical properties of fragments that bound RNA were compared to the RNA-focused fragment library and Inforna. As expected, properties of the hit fragments and the RNA-focused fragment library are similar, while significant differences were observed between the hit fragments and small molecules in Inforna (Figure S4 and Table S1). The average molecular weight for the hit fragments was 240 ± 66 Da, which, unsurprisingly, is similar to the RNA-focused fragment library (250 ± 40 Da) and less than that of Inforna small molecules (457 ± 204 Da). Other differences between the fragments with RNA-binding capacity and Inforna compounds were observed, for example, 3.3 ± 1 HBAs for the hit fragments vs 6 ± 1 HBAs for Inforna small molecules; and 2 ± 1 HBDs for the hit fragments vs 5 ± 1 HBDs for Inforna compounds. Interestingly, the average number of HBA for the fragment hits was the same as that of the entire RNA-focused fragment library (3.3 ± 1); however, the hit fragments had more HBDs (2 ± 1 vs 1 ± 1).
To identify fragments that specifically bind to structured RNA folds, not general RNA-binding capacity, and to define their binding landscapes, 2DCS selections were performed in the presence of excess competitor oligonucleotides. These competitor oligonucleotides mimic the regions common to all members of the library (12) and also include fully paired DNA duplexes [d(AT11) and d(GC)11] and tRNA (Figure S3). These highly stringent conditions restrict fragment binding to the randomized regions of the RNA fold library and eliminate non-specific nucleic acid binders. Of the 12 fragments that bound to the 3 × 3 ILL and the seven fragments that bound the 3 × 2 ILL in the primary screen, only three (1, 2, and 3; the same for both RNA libraries) bound to 3 × 3 ILL and 3 × 2 ILL under the stringent conditions of the 2DCS selection (Figure 2A and Figure S5). These three compounds are structurally different than those present in Inforna as their average Tanimoto scores are <0.4 when compared to the known RNA binders (Figure S6).
Figure 2.
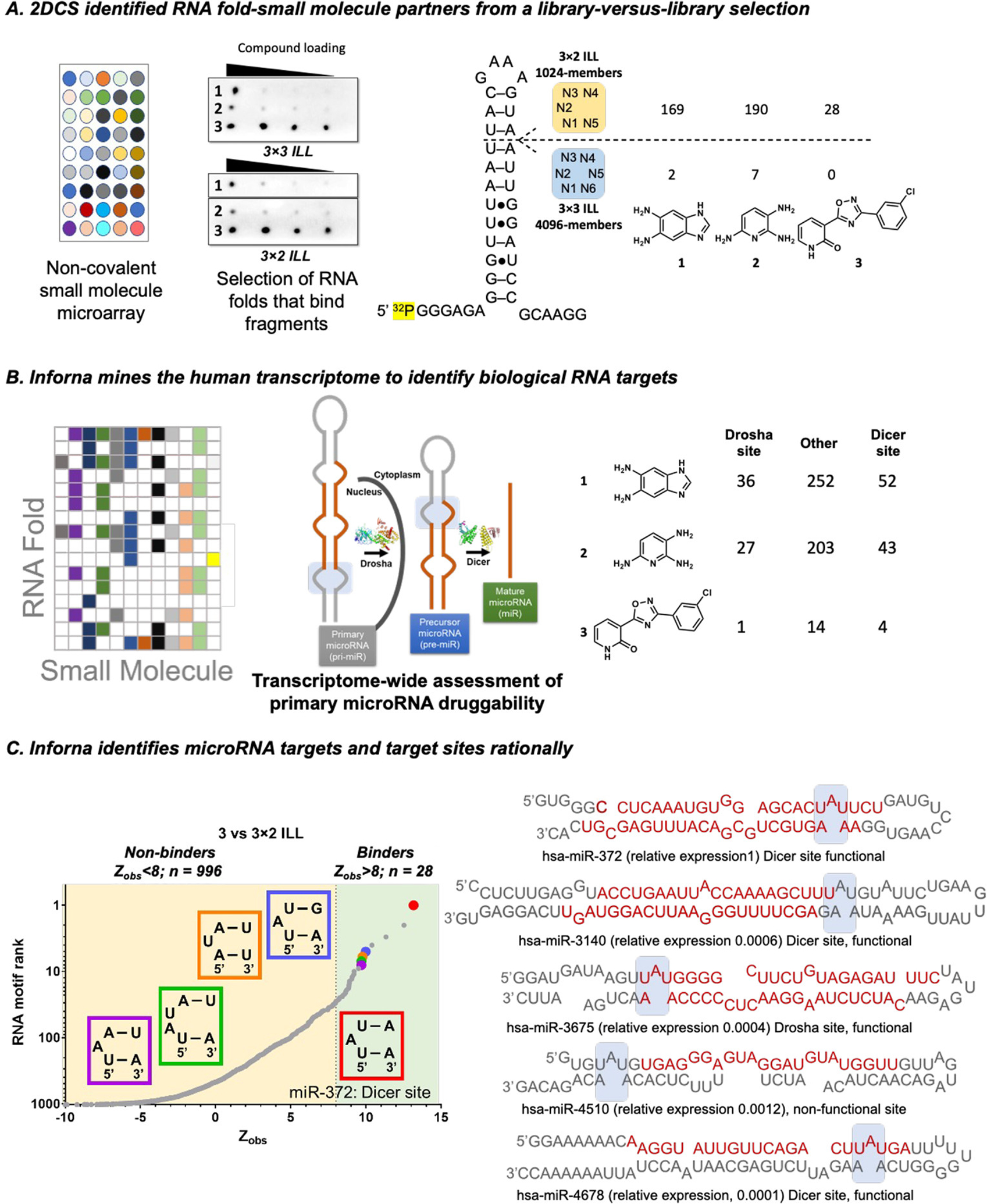
RNA targets of fragments, as selected by 2DCS. (A) The fragments were studied for binding RNA libraries that display 3 × 3 or 3 × 2 nucleotide internal loops (3 × 3 ILL and 3 × 2 ILL, respectively) using AbsorbArray, where the compounds are non-covalently immobilized on agarose coated glass slide. Three fragments (1, 2, and 3) bind RNAs specifically as determined by a secondary selection in the presence of competitor oligonucleotides (2DCS) (left). RNA targets of compounds were identified from sequencing analysis. The number of RNA targets yielded for each compound for individual RNA ILLs when a cutoff of Zobs > 8 was employed (right). (B) Scheme for RNA mining using Inforna. Inforna mines all human miRNA precursors and identifies the RNA target that has a small molecule-binding motif present in its secondary structure. (C) Inforna mining further studies whether the RNA motif is present in a functional (Dicer or Drosha) site or non-functional site of the pri- and pre-miRNAs. Fragment 3 is predicted to bind 5′UAU/3′A_A with the highest Zobs (left). Secondary structures of all miRNAs containing the A bulge (5′UAU/3′A_A) (right). Among them, only four miRNAs (miR-372, miR-3140, miR-3675, and miR-4678) harbor the A bulge in a functional site. However, the expression of miR-3140, miR-3675, and miR-4678 is >100-fold lower than miR-372 in AGS cells.
The RNA folds selected by each fragment were deconvoluted by RNA sequencing (RNA-seq). Binding landscapes were defined by calculating the statistical enrichment of each RNA fold in the RNA-seq data after 2DCS selection as compared to the fold’s frequency in the starting ILL, a method dubbed high-throughput structure–activity relationships through sequencing (HiT-StARTS). (22) HiT-StARTS uses a pooled population comparison to calculate statistical significance, reported as Zobs. We have previously shown that Zobs is correlated with affinity and that Zobs > 8 affords high-affinity, selective binding, determined by empirical data. (22) Using this cut-off, fragment 1 binds 169 RNA structures in 3 × 2 ILL and two RNA structures in 3 × 3 ILL; 2 binds 190 RNAs from 3 × 2 ILL and seven from 3 × 3 ILL; and 3 binds 28 structures from 3 × 2 ILL and none in 3 × 3 ILL (Figure 2B). These data suggest that 1 and 2 bind more RNA motifs than 3 in both the libraries tested and hence is a more selective fragment.
We generated a graphical representation, or LOGO, (23) of the RNA folds preferred by and discriminated against by each fragment. Here, we analyzed the sequences for each library individually, either the RNAs with Zobs values in the top 0.5% (enriched) or the lowest 0.5% Zobs (discriminated against). (23) A LOGO comprises a stack of letters at each position, and the relative sizes of the letters indicate the frequency of each nucleotide in the sequences, reported in bits. In general, fragment 1 and fragment 2 prefer and discriminate against similar nucleotides in the randomized region of the ILLs, while differences are observed for fragment 3 (Figures S7 and S8). Fragment 3 has clear nucleotide preferences in both types of internal loops. For the 3 × 3 ILL, pyrimidine-rich loops are preferred, particularly at positions N1, N2, N3, and N6. Likewise, fragments 2 and 3 also prefer pyrimidine-rich loops, but the preferences are perhaps not as strong (Figure S7A). In a similar vein, the most discriminated against nucleotides by 3 are at positions N1–N3, U, C, and G, respectively (Figure S7B). For the 3 × 2 ILL, fragment 3 also has clear preferences, A at positions N1 and N3 and U at N2 (Figure S8A). Adenosine residues are also preferred by 1 and 2 at N1, N3, and N4. Interestingly, the LOGO analyses for 1 and 2 indicate that N1 and N5 likely form a base pair, therefore forming 2 × 1, rather than 3 × 2, nucleotide internal loops (Figure S8A). Fragments 1 and 2 discriminate against G and U nucleotides, which are positionally dependent (Figure S8B). Again, the pattern observed for 3 is different, with G and C discriminated against at N1 and G and A at N4 (Figure S8B).
Informatic Mining of RNA Fold-Fragment Interactions across the Transcriptome Identified a Bioactive Interaction between 3 and Oncogenic pre-miR-372
We next searched for the RNA folds preferred by each fragment in all human microRNAs (miRNAs), or the miRnome. MiRNAs are small non-coding RNAs that regulate gene expression by binding to mRNAs with complementary 3′ untranslated regions (UTRs). (24,25) They are transcribed as primary transcripts (pri-miRNAs) that are processed step-wise by Drosha and Dicer to generate precursor (pre-) and mature miRNAs, respectively (Figure 2B). Targets were prioritized as follows: (i) disease-association, (ii) location of the binding site in a Drosha or Dicer processing site, and (iii) Zobs (Figure 2B). Based on these criteria, 1 is predicted to bind functional site of five RNA targets (pre-miR-18a, pre-miR-18b, and pre-miR-107/103 as well as pri-miR-9–3 and pri-miR-27a), and 2 is predicted to bind the functional sites of four RNA targets (pre-miR-18a and pre-miR-18b as well as pri-miR-9–3 and pri-miR-27a) (Figure S9). Interestingly, the most optimal match was between 3 and the 3D structure present in the Dicer processing site of pre-miR-372, an A bulge closed by two UA base pairs, 5′UAU/3′A_A. Indeed, the Zobs for 5′UAU/3′A_A was the highest among all folds in the 3 × 2 ILL and is the only disease-associated RNA 3D fold predicted to bind 3 (Figure 2C). This A bulge is present in four other miRNAs, miR-3140, miR-3675, miR-4510, and miR-4678. Of these, 3 is expected to affect the biogenesis of miR-3140, miR-3675, and miR-4678, as the 5′UAU/3′A_A is harbored in a processing (functional) site (Figure 2C). (26) None of these miRNAs is associated with disease nor are they highly expressed in the gastric cancer cell line (AGS) used in this study vide infra (Figure 2C and Figure S10).
Previous studies have shown that miR-372 is tumorigenic and upregulated in various cancers including gastric cancer, (4,27) breast cancer, (28) and gliomas. (29,30) In particular, the gastric cancer cell line, AGS, expresses high levels of miR-372, which represses the translation of large tumor suppressor kinase 2 (LATS2) mRNA and thereby induces cellular proliferation as an oncogenic phenotype (Figure 3A). (27) The Duca lab has identified multimodal small molecules (8) and functionalized polyamines (4) as ligands that bind the precursor miR-372 and inhibit its biogenesis. However, no reports of bioactive fragments targeting RNA in general or miR-372 have been reported. We therefore characterized the 3–miR-372 interaction in vitro and then its inhibition of miR-372 biogenesis, de-repression of LATS2, and reversal of proliferation in AGS gastric cancer cells.
Figure 3.
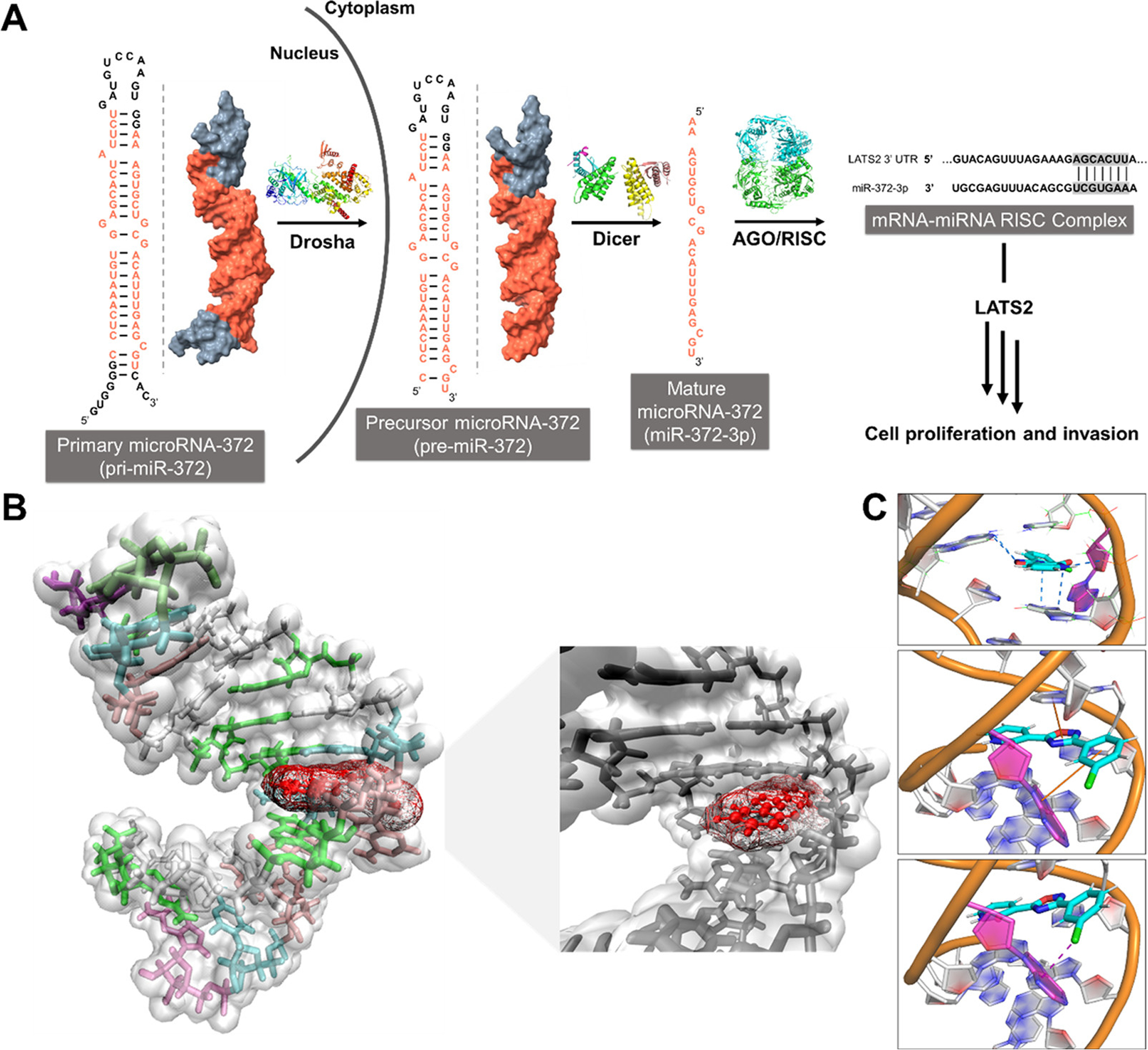
Schematic of the biogenesis of miR-372 and a model of fragment 3 bound to pre-miR-372’s Dicer processing site. (A) Biogenesis of miR-372 and its downstream biology. (B) Model of the interaction of 3 with the A bulge present in pre-miR-372’s Dicer processing site, determined by docking and molecular dynamics simulations. (C) Network of hydrogen bonds (top), stacking interactions (middle), and a halogen bond (bottom) stabilize the interaction of 3 within the binding pocket.
To study the binding affinity and the specificity of 3 for the miR-372 Dicer site, we measured the change in its fluorescence polarization as a function of RNA concentration. Indeed, 3 bound a model of the Dicer site avidly with a Kd of 1.7 ± 0.6 μM, with no saturable binding observed to an RNA in which the A bulge is mutated to an AU base pair (Figure S11). The fragment bound pre-miR-372 with a Kd of 300 ± 140 nM, likewise with no saturable binding to a pre-miR-372 mutant where the A bulge was mutated to an AU base pair (Figure S12). We next studied the ability of fragment 3 to inhibit Dicer processing of pre-miR-372 in vitro. Indeed, 3 inhibited Dicer processing of pre-miR-372 (Figure S13A) but, in accord with its lack of affinity for the pre-miR-372 mutant (A bulge converted to an AU pair), had no effect on the processing of the mutant (Figure S13B,C). Collectively, these data support the hypothesis that the fragment binds to the three-dimensional structure in a biologically active target selected by 2DCS and identified by Inforna.
To elucidate the driving forces of the interaction of fragment 3 with miR-372’s A-bulge, we used a combination of docking and molecular dynamics (MD) simulations (Figure 3B). A network of hydrogen bonds form between the compound and the A bulge’s closing base pairs as well as a backbone hydrogen bond and stacking interactions with A-bulge and the uracil of the closing base pair. Interestingly, a halogen bond between Cl atom of 3 and the A-bulge also stabilizes the bound state of this compound (Figure 3C).
To assess the inhibitory effect of 3 on miR-372 processing in cells, we measured levels of pre- and mature miR-372 upon fragment treatment by real-time quantitative polymerase chain reaction (RT-qPCR). A dose-dependent reduction of mature miR-372 was observed, with an IC50 of ~1 μM (Figure 4A). Fragment 3 boosted abundance of pre-miR-372 in a dose-dependent manner, with a significant increase observed at both 1 μM (~30%, P = 0.013) and 10 μM (~70%, P < 0.0001), in accordance with its putative mode of action, inhibition of Dicer processing (Figure 4B). To assess the selectivity of the fragment, we studied the effect of 3 (1 μM) across the AGS miRnome (n = 379 miRNAs expressed in AGS cells). A statistically significant effect was only observed on miR-372 abundance (P < 0.01) (Figure 4C).
Figure 4.
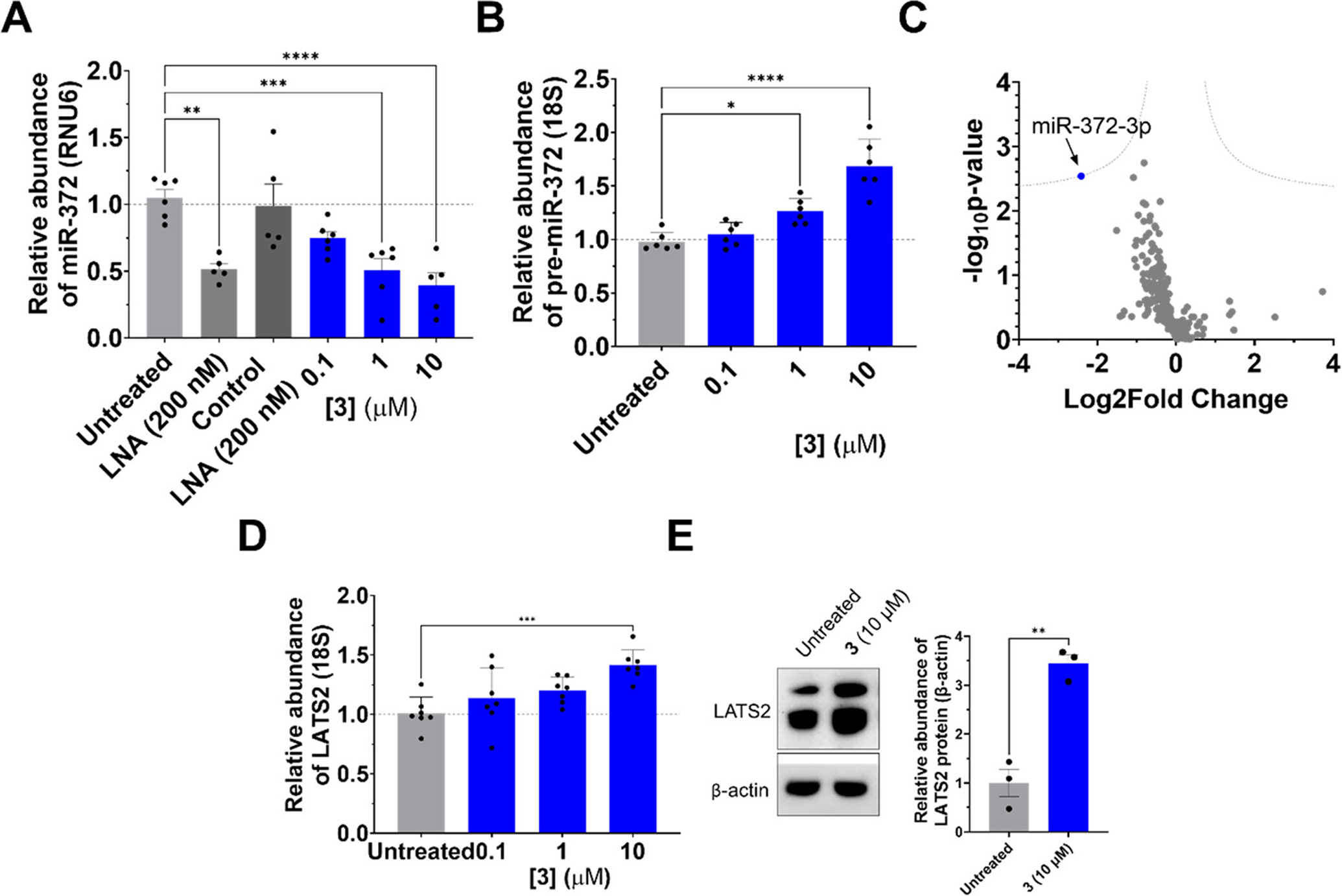
Fragment 3 inhibits biogenesis of miR-372 in human AGS gastric cancer cells. (A) Abundance of mature miR-372 upon treatment with 3, as measured by RT-qPCR (n = 6). (B) Abundance of pre-miR-372 upon treatment with 3, as measured by RT-qPCR (n = 6). (C) miRnome-wide profiling of AGS cells treated with 3 (1 μM, n = 3). Dotted lines represent a false discovery rate (FDR) of 1% and variance of S0 (0.1). (D) De-repression of miR-372’s downstream target LATS2 upon treatment with 3, as measured by RT-qPCR (n = 6). (E) Levels of LATS2 protein upon treatment with 3 (10 μM), as measured by Western blotting (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 as determined by one-way ANOVA compared to untreated samples. Data are reported as the mean ± SEM for all panels.
As aforementioned, aberrant expression of miR-372 in AGS cells enhances proliferation via direct repression of LATS2. (8,27) The effect of 3 on LATS2 mRNA and protein levels were therefore measured by RT-qPCR and Western blotting, respectively. A dose-dependent increase in LATS2 mRNA levels was observed, with a statistically significant effect at 10 μM (~40%, P = 0.004) (Figure 4D). LATS2 protein levels were also boosted upon treatment with 3, with a 3-fold increase observed at 10 μM (P = 0.002) (Figure 4E).
Since 3 de-repressed LATS2 protein levels, we next studied whether abrogating this circuit was sufficient to reverse oncogenic cellular phenotypes driven by miR-372. (8,27) Proliferation was assessed using a colorimetric MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. Indeed, 3 dose-dependently decreased AGS cell proliferation (Figure 5A), mirroring the concentrations required to de-repress LATS2 mRNA and protein levels (Figure 4D,E). As reduced LATS2 protein abundance has also been associated with an invasive phenotype in gliomas through the Hippo signaling pathway, (31,32) we also studied the effect of 3 on the invasion of AGS cells. The fragment (1 μM) significantly decreased the number of invasive cells upon treatment (~40%, P = 0.02) (Figure 5B,C).
Figure 5.
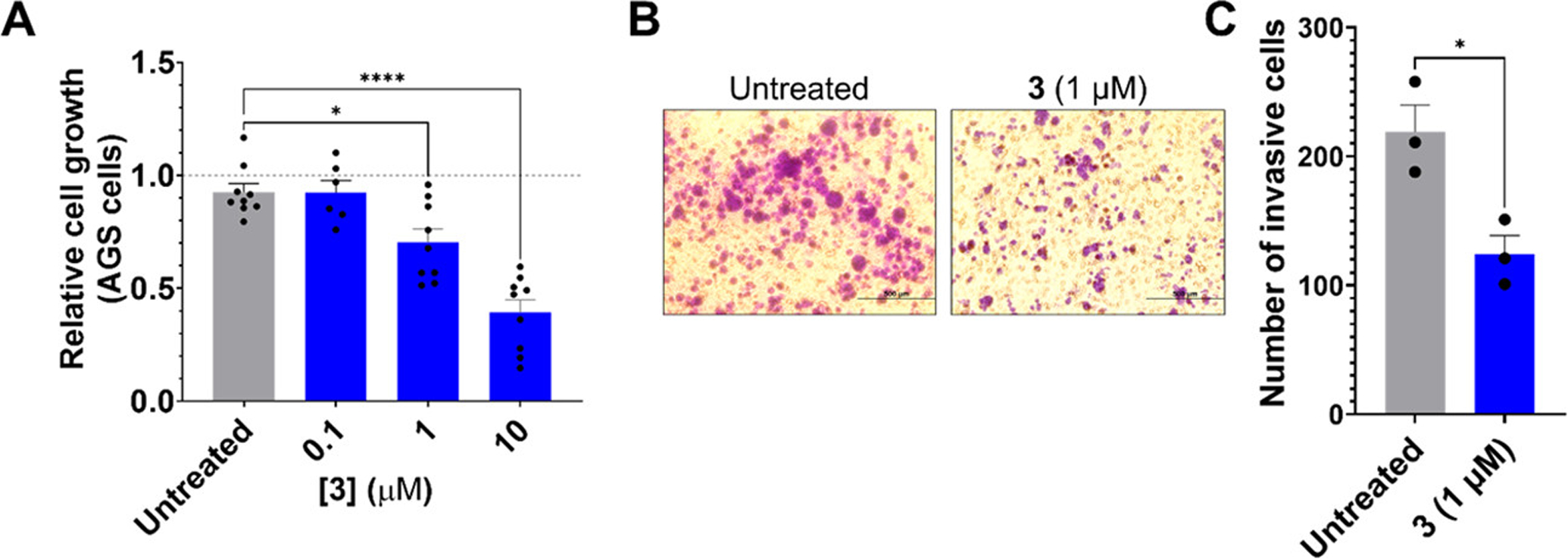
Fragment 3 reduces proliferative and invasive phenotypes in AGS gastric cancer cells. (A) Effect of 3 on proliferation of AGS cells, as determined by MTS assay (n = 9). (B) AGS cell invasion, as studied by Boyden chamber assay, upon treatment with 1 μM of 3. (C) Quantification of the number of invasive cells measured in B. *P < 0.05 and ****P < 0.0001 as determined by one-way ANOVA compared to untreated samples for panel A and by a two-tailed Student’s t test for panel C. Data are reported as means ± SEM for all panels.
To provide evidence that 3 reduced proliferation via the miR-372–LATS2 circuit, we knocked down LATS2 mRNA with an siRNA. Upon transfection of AGS cells with a LATS2-targeting siRNA, LATS2 mRNA levels were reduced by 80 ± 1% (P = 0.0002), while a scrambled, control siRNA had no effect (Figure 6A). AGS cells transfected with the scrambled control siRNA and treated with 3 (10 μM) reduced proliferation (~60%, P < 0.0001; Figure 6B), as expected. In contrast, no significant change in proliferation was observed for cells transfected with LATS2 siRNA and treated with 3 (Figure 6B), as the downstream target is no longer present. Together, these data suggest that fragment 3 reverses a proliferative phenotype via the miR-372–LATS2 circuit.
Figure 6.
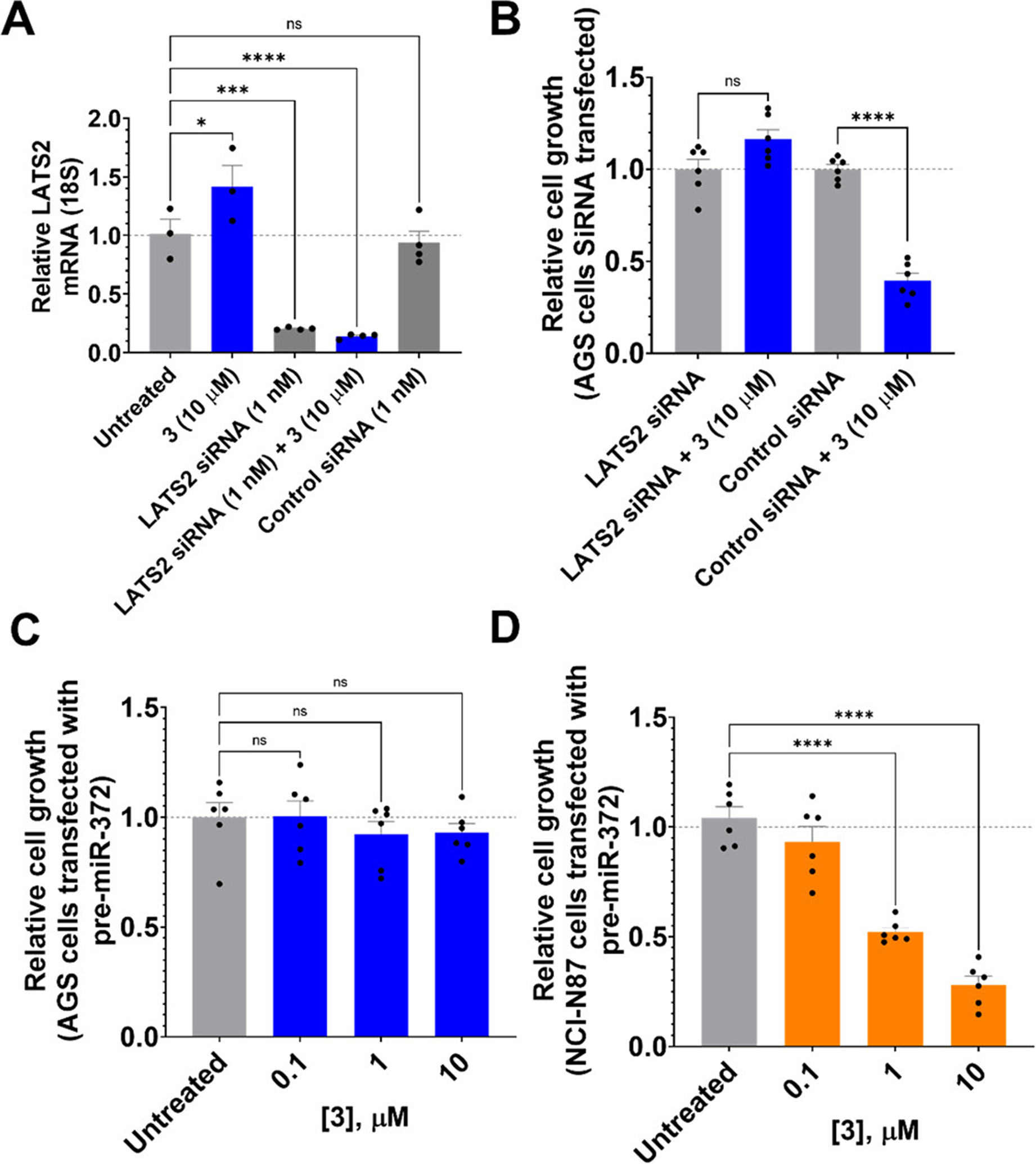
Fragment 3 reduces cell proliferation via the miR-372–LATS2 circuit. (A) siRNA knockdown of LATS2 in the presence and absence of 3, as measured by RT-qPCR. (B) Effect of 3 on cell proliferation of AGS cells upon siRNA knockdown of LATS2, as determined by MTS assay. (C) Effect of fragment 3 on the proliferation of AGS cells forced to overexpress pre-miR-372 (n = 6). Overexpression of the target leads to ablation of the compound’s effect. (D) Effect of 3 on proliferation of NCI-N87 cells forced to express pre-miR-372 (n = 6). *P < 0.05, ***P < 0.001, and ****P < 0.0001 as determined by one-way ANOVA compared to untreated samples. Data are reported as the mean ± SEM for all panels.
To further assess the selectivity of fragment 3 on the miR-372-associated phenotype, AGS cells were transfected with pre-miR-372 or the pre-miR-372 mutant in which the A bulge at the Dicer processing site was mutated to the AU base pair. Forced expression of both pre-miRNAs further enhanced the proliferative phenotype of AGS cells (Figure S14A,B) and reduced the potency of 3 (Figure 6C and Figure S14C), as expected based on increasing abundance of the target.
As an additional control, we studied the effect of fragment 3 on a gastric cancer cell line that does not aberrantly express miR-372. The gastric carcinoma cell line NCI-N87 has 16-fold lower expression of miR-372 than AGS cells have. (4) Fragment 3 had no effect on the proliferation of NCI-N87 cells at concentrations active in AGS cells (Figure S15A). Because the proliferation of NCI-N87 cells was unaffected by 3 treatment, we studied if forced expression of pre-miR-372 or the pre-miR-372 mutant could induce a proliferative phenotype in NCI-N87 cells. Indeed, forced expression of both RNAs enhanced proliferation (Figure S15B,C), but proliferation induced only by forced expression of WT pre-miR-372 could be rescued by 3 (Figure 6D and Figure S15D). These data confirm that 3 binding to the A bulge at the Dicer processing site of pre-miR-372 is critical in regulating the proliferative phenotype, further validating the inhibition of the miR-372–LATS2 circuit by compound 3.
Discussion
One of the challenges in the RNA-targeting field is overcoming the perception that compounds that affect disease pathways do not possess favorable physicochemical properties as compared to known orally bioavailable small-molecule medicines. In this study, we identified and characterized RNA-focused small-molecule fragments that bind RNA folds in a massively parallel selection format, assessing 12.8 million binding events. The results defined fragments that bind RNA structures and the cellular targets that house them. This fully described fragment collection can be used to define compounds that bind to a variety of additional RNA targets. By using a library-versus-library selection coupled with informatics, we have found that a member of this collection (3, MW = 273 Da; Kd = 300 nM) bound oncogenic pre-miR-372 and inhibited its processing in cells, affecting a miR-372-driven proliferative and invasive phenotype in a gastric cancer cell line.
As there is limited information known on the properties of compounds that can affect RNA targets, we are cautious to assign broad ranging properties to bioactive ligands at this early stage of development. However, here, we have shown that compounds of low molecular weight and, hence fragment-like characteristics, should be considered when identifying bioactive chemical matter that binds RNA, affording mechanistically defined small molecules that selectively affect RNA-mediated pathways. It will be interesting to study whether fragments outside the heterocycle space in particular or the currently known RNA-binding space in general have binding capacity and if these interactions can be detected by the methods established herein or others. (33−35) Interestingly, some of the fragment hits observed from chemical cross-linking and isolation by pull-down fragment mapping (Chem-CLIP-Frag-Map) suggest that non-heterocyclic compounds also bind RNAs. (36,37)
Importantly, compound 3 has a QED score of 0.8, which is higher than most RNA-binding compounds identified so far and is comparable with the QED scores of orally bioavailable FDA-approved small-molecule drugs. When the QED score and the binding affinity of all previously reported RNA-targeting bioactive small molecules are compared, (38) only four have QED scores of >0.6 and binding affinities of <1000 nM─branaplam (in clinical trial for the treatment of spinal muscular atrophy), (39) ribocil (an antibacterial that targets a riboswitch), (40) a small-molecule inhibitor of miR-544 biogenesis, (41) and 3 from this study (Figure S16 and Table S2). Interestingly, branaplam and the risdiplam (FDA-approved for the treatment of SMA) derivative SMN-C5 (42) are not RNA-only binders and act to stabilize a ternary complex with a pre-mRNA and the U1 small nuclear ribonucleoprotein particle (snRNP) complex. Importantly, SMN-C5, (39,42) branaplam, (39) ribocil, (40) and the antibacterial linezolid that targets 23S rRNA (43,44) were identified from phenotypic screens, and their mechanisms of action were elucidated later and linked to RNA binding. (39,40,42,45) Therefore, our target-agnostic approach that yielded 3 shows that bioactive drug-like ligands can be identified by using Inforna and 2DCS selection platforms to purposefully target RNA.
Analyses of QED and affinity should be evaluated with caution for a variety of reasons, however. For example, the affinity of RNA-binding compounds can be affected by ionic strength as well as other factors. Perhaps, a more informative analysis would evaluate QED and bioactivity, which could be complicated by differences in the RNA-targeted small molecules’ modes of action and if the small molecules inhibit the RNA target substoichiometrically, for example, degraders. (46)
Previous strategies adopted to study fragment binding of RNA include NMR spectroscopy, (47) mass spectrometry (MS), (33) equilibrium dialysis, (34) Chem-CLIP-Frag-Map, (36,37) and selective 2′ hydroxyl acylation analyzed by primer extension (SHAPE) probing (35) (Figure 7). For each approach, the initial fragment hit had low affinity for the RNA target─high μM to mM─which required further optimization to obtain higher-affinity, selective binders (Table S3). Additionally, these strategies were implemented for a specific RNA target, (33−36,47) with the exception of transcriptome-wide Chem-CLIP-Frag-Map, (37) although such target-agnostic studies require modification of the fragment library with photo-cross-linking elements. (36,37) A recent report from our laboratory showed that DNA-encoded library (DEL) screening could rapidly screen diverse chemical matter for RNA-binding capacity, with subsequent molecular fingerprints generated by 2DCS. (48) Akin to the study herein, these fingerprints were mined to identify targetable structures within the human miRnome, affording an inhibitor of pri-miR-27a that had two binding sites for the small molecule. (48)
Figure 7.

Summary of fragment-based ligand identification strategies for RNA. Previously reported methods for identification of fragment binders for RNA, including NMR spectroscopy, (47) mass spectrometry (MS), (33) equilibrium dialysis, (34) Chem-CLIP-Frag-Map, (36,37) and SHAPE probing, (35) compared to this work. Representative examples of fragment hits identified from each method and their affinities for the RNA target are provided.
Importantly, these studies show that fragments that share physicochemical properties with known RNA binders can have sufficient affinity and selectivity to affect its biological function in cells. Further, biological activity can occur at reasonable concentrations for the molecules to serve as chemical probes or lead medicines.
Conclusions
A bioactive, 273 Da molecular weight fragment, 3, was obtained from the 2DCS selection of an RNA-focused fragment library. The molecule binds to the Dicer processing site of pre-miR-372 with nM affinity and selectively inhibited the miRNA’s biogenesis at low μM concentrations in cancer cells. The fragment has limited off-targets across the miRnome and specifically reverses miR-372-driven proliferative and invasive phenotypes in a gastric cancer cells. Our study sets the stage for fragment-based lead optimization efforts to target RNA broadly and affect disease-associated phenotypes by using chemical matter with physicochemical properties that suggest oral bioavailability. These and other fragment-based drug discovery approaches to target RNA (9,33−37,47,49−53) will accelerate the discovery of small molecules that bind RNA and affect its function.
Supplementary Material
Acknowledgments
The authors thank Jessica L. Childs-Disney for her help in writing the manuscript. This work was supported by the US National Cancer Institute (R01 CA249180A to M.D.D.)
Footnotes
The authors declare the following competing financial interest(s): M.D.D. is a founder of Expansion Therapeutics.
Supporting Information
- Supporting figures, experimental methods, synthetic methods and characterization, and computational methods (PDF).
- List of compounds used in this study: (i) RNA-focused fragments selected from ChemDiv’s fragment library, (ii) remaining fragments not selected from ChemDiv’s fragment library, and (iii) Inforna compounds; SMILES code, UMAP parameters, rule of three (RO3) properties, and QED values (XLSX).
REFERENCES
- 1.ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489 (7414), 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sztuba-Solinska J; Chavez-Calvillo G; Cline SE, Unveiling the druggable RNA targets and small molecule therapeutics. Bioorg. Med. Chem 2019, 27 (10), 2149–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu A-M; Choi YH; Tu M-J, RNA drugs and RNA targets for small molecules: principles, progress, and challenges. Pharmacol. Rev 2020, 72 (4), 862–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staedel C; Tran TPA; Giraud J; Darfeuille F; Di Giorgio A; Tourasse NJ; Salin F; Uriac P; Duca M, Modulation of oncogenic miRNA biogenesis using functionalized polyamines. Sci. Rep 2018, 8 (1), 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peacock H; Suga H, Discovery of de novo macrocyclic peptides by messenger RNA display. Trends Pharmacol. Sci 2021, 42 (5), 385–397. [DOI] [PubMed] [Google Scholar]
- 6.Bugaut A; Rodriguez R; Kumari S; Hsu STD; Balasubramanian S, Small molecule-mediated inhibition of translation by targeting a native RNA G-quadruplex. Org. Biomol. Chem 2010, 8 (12), 2771–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donlic A; Morgan BS; Xu JL; Liu A; Roble C Jr; Hargrove AE, Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold. Angew. Chem. Int. Ed. Engl 2018, 57 (40), 13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vo DD; Staedel C; Zehnacker L; Benhida R; Darfeuille F; Duca M, Targeting the production of oncogenic microRNAs with multimodal synthetic small molecules. ACS Chem. Biol 2014, 9 (3), 711–721. [DOI] [PubMed] [Google Scholar]
- 9.Binas O; de Jesus V; Landgraf T; Völklein AE; Martins J; Hymon D; Kaur Bains J; Berg H; Biedenbänder T; Fürtig B; Lakshmi Gande S; Niesteruk A; Oxenfarth A; Shahin Qureshi N; Schamber T; Schnieders R; Tröster A; Wacker A; Wirmer-Bartoschek J; Wirtz Martin MA; Stirnal E; Azzaoui K; Richter C; Sreeramulu S; José Blommers MJ; Schwalbe H, 19F NMR-based fragment screening for 14 different biologically active RNAs and 10 DNA and protein counter-screens. Chembiochem 2021, 22 (2), 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connelly CM; Numata T; Boer RE; Moon MH; Sinniah RS; Barchi JJ; Ferré-D’Amaré AR; Schneekloth JS, Synthetic ligands for PreQ1 riboswitches provide structural and mechanistic insights into targeting RNA tertiary structure. Nat. Commun 2019, 10 (1), 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velagapudi SP; Gallo SM; Disney MD, Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol 2014, 10 (4), 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Disney MD, Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 2019, 141 (17), 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lundquist KP; Panchal V; Gotfredsen CH; Brenk R; Clausen MH, Fragment-based drug discovery for RNA targets. ChemMedChem 2021. 16, 2588. [DOI] [PubMed] [Google Scholar]
- 14.Scaffold hopper http://tripod.nih.gov/ (accessed 2020-10-12).
- 15.McInnes L; Healy J; Saul N; Großberger L, UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw 2018, 3 (29), 861. [Google Scholar]
- 16.Rugard M; Jaylet T; Taboureau O; Tromelin A; Audouze K, Smell compounds classification using UMAP to increase knowledge of odors and molecular structures linkages. PLoS One 2021, 16 (5), e0252486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Congreve M; Carr R; Murray C; Jhoti H, A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8 (19), 876–877. [DOI] [PubMed] [Google Scholar]
- 18.Bickerton GR; Paolini GV; Besnard J; Muresan S; Hopkins AL, Quantifying the chemical beauty of drugs. Nat. Chem 2012, 4 (2), 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Velagapudi SP; Costales MG; Vummidi BR; Nakai Y; Angelbello AJ; Tran T; Haniff HS; Matsumoto Y; Wang ZF; Chatterjee AK; Childs-Disney JL; Disney MD, Approved anti-cancer drugs target oncogenic non-coding RNAs. Cell Chem. Biol 2018, 25 (9), 1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Childs-Disney JL; Tran T; Vummidi BR; Velagapudi SP; Haniff HS; Matsumoto Y; Crynen G; Southern MR; Biswas A; Wang ZF; Tellinghuisen TL; Disney MD, A massively parallel selection of small molecule-RNA motif binding partners informs design of an antiviral from sequence. Chem 2018, 4 (10), 2384–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haniff HS; Knerr L; Liu X; Crynen G; Boström J; Abegg D; Adibekian A; Lekah E; Wang KW; Cameron MD; Yildirim I; Lemurell M; Disney MD, Design of a small molecule that stimulates vascular endothelial growth factor A enabled by screening RNA fold–small molecule interactions. Nat. Chem 2020, 12 (10), 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velagapudi SP; Luo Y; Tran T; Haniff HS; Nakai Y; Fallahi M; Martinez GJ; Childs-Disney JL; Disney MD, Defining RNA-small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent. Sci 2017, 3 (3), 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider TD; Stephens RM, Sequence LOGOs: a new way to display consensus sequences. Nucleic Acids Res 1990, 18 (20), 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambros V; Bartel B; Bartel DP; Burge CB; Carrington JC; Chen X; Dreyfuss G; Eddy SR; Griffiths-Jones S; Marshall M; Matzke M; Ruvkun G; Tuschl T, A uniform system for microRNA annotation. RNA 2003, 9 (3), 277–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fire A; Xu S; Montgomery MK; Kostas SA; Driver SE; Mello CC, Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391 (6669), 806–811. [DOI] [PubMed] [Google Scholar]
- 26.Costales MG; Haga CL; Velagapudi SP; Childs-Disney JL; Phinney DG; Disney MD, Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J. Am. Chem. Soc 2017, 139 (9), 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho WJ; Shin JM; Kim JS; Lee MR; Hong KS; Lee JH; Koo KH; Park JW; Kim KS, miR-372 regulates cell cycle and apoptosis of AGS human gastric cancer cell line through direct regulation of LATS2. Mol. Cells 2009, 28 (6), 521–527. [DOI] [PubMed] [Google Scholar]
- 28.Cheng X; Chen J; Huang Z, miR-372 promotes breast cancer cell proliferation by directly targeting LATS2. Exp. Ther. Med 2018, 15 (3), 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X; Hao B; Han G; Liu Y; Dai D; Li Y; Wu X; Zhou X; Yue Z; Wang L; Cao Y; Liu J, miR-372 regulates glioma cell proliferation and invasion by directly targeting PHLPP2. J. Cell. Biochem 2015, 116 (2), 225–232. [DOI] [PubMed] [Google Scholar]
- 30.Xia L; Nie D; Wang G; Sun C; Chen G, FER1L4/miR-372/E2F1 works as a ceRNA system to regulate the proliferation and cell cycle of glioma cells. J. Cell. Mol. Med 2019, 23 (5), 3224–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo C; Liang C; Yang J; Hu H; Fan B; Liu X, LATS2 inhibits cell proliferation and metastasis through the Hippo signaling pathway in glioma. Oncol. Rep 2019, 41 (5), 2753–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y; Geng D; Zhang Y; Zhao M; Wang Y; Jiang Y; Yu R; Zhou X, LATS2 inhibits malignant behaviors of glioma cells via inactivating YAP. J. Mol. Neurosci 2019, 68 (1), 38–48. [DOI] [PubMed] [Google Scholar]
- 33.Swayze EE; Jefferson EA; Sannes-Lowery KA; Blyn LB; Risen LM; Arakawa S; Osgood SA; Hofstadler SA; Griffey RH, SAR by MS: A ligand based technique for drug lead discovery against structured RNA targets. J. Med. Chem 2002, 45 (18), 3816–3819. [DOI] [PubMed] [Google Scholar]
- 34.Cressina E; Chen L; Abell C; Leeper FJ; Smith AG, Fragment screening against the thiamine pyrophosphate riboswitchthiM. Chem. Sci 2011, 2 (1), 157–165. [Google Scholar]
- 35.Zeller Meredith J; Favorov O; Li K; Nuthanakanti A; Hussein D; Michaud A; Lafontaine Daniel A; Busan S; Serganov A; Aubé J; Weeks Kevin M, SHAPE-enabled fragment-based ligand discovery for RNA. Proc. Natl. Acad. Sci. U. S. A 2022, 119 (20), e2122660119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suresh BM; Li W; Zhang P; Wang KW; Yildirim I; Parker CG; Disney MD, A general fragment-based approach to identify and optimize bioactive ligands targeting RNA. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (52), 33197–33203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong Y; Gibaut QMR; Rouse W; Childs-Disney JL; Suresh BM; Abegg D; Choudhary S; Akahori Y; Adibekian A; Moss WN; Disney MD, Transcriptome-wide mapping of small-molecule RNA-binding sites in cells informs an isoform-specific degrader of QSOX1 mRNA. J. Am. Chem. Soc 2022, 144 (26), 11620–11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warner KD; Hajdin CE; Weeks KM, Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov 2018, 17 (8), 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palacino J; Swalley SE; Song C; Cheung AK; Shu L; Zhang X; Van Hoosear M; Shin Y; Chin DN; Keller CG; Beibel M; Renaud NA; Smith TM; Salcius M; Shi X; Hild M; Servais R; Jain M; Deng L; Bullock C; McLellan M; Schuierer S; Murphy L; Blommers MJ; Blaustein C; Berenshteyn F; Lacoste A; Thomas JR; Roma G; Michaud GA; Tseng BS; Porter JA; Myer VE; Tallarico JA; Hamann LG; Curtis D; Fishman MC; Dietrich WF; Dales NA; Sivasankaran R, SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol 2015, 11 (7), 511–517. [DOI] [PubMed] [Google Scholar]
- 40.Howe JA; Wang H; Fischmann TO; Balibar CJ; Xiao L; Galgoci AM; Malinverni JC; Mayhood T; Villafania A; Nahvi A; Murgolo N; Barbieri CM; Mann PA; Carr D; Xia E; Zuck P; Riley D; Painter RE; Walker SS; Sherborne B; de Jesus R; Pan W; Plotkin MA; Wu J; Rindgen D; Cummings J; Garlisi CG; Zhang R; Sheth PR; Gill CJ; Tang H; Roemer T, Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526 (7575), 672–677. [DOI] [PubMed] [Google Scholar]
- 41.Haga CL; Velagapudi SP; Strivelli JR; Yang WY; Disney MD; Phinney DG, Small molecule inhibition of miR-544 biogenesis disrupts adaptive responses to hypoxia by modulating ATM-mTOR signaling. ACS Chem. Biol 2015, 10 (10), 2267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sivaramakrishnan M; McCarthy KD; Campagne S; Huber S; Meier S; Augustin A; Heckel T; Meistermann H; Hug MN; Birrer P; Moursy A; Khawaja S; Schmucki R; Berntenis N; Giroud N; Golling S; Tzouros M; Banfai B; Duran-Pacheco G; Lamerz J; Hsiu Liu Y; Luebbers T; Ratni H; Ebeling M; Cléry A; Paushkin S; Krainer AR; Allain FHT; Metzger F, Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat. Commun 2017, 8 (1), 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palacino J; Swalley SE; Song C; Cheung AK; Shu L; Zhang X; Van Hoosear M; Shin Y; Chin DN; Keller CG; Beibel M; Renaud NA; Smith TM; Salcius M; Shi X; Hild M; Servais R; Jain M; Deng L; Bullock C; McLellan M; Schuierer S; Murphy L; Blommers MJJ; Blaustein C; Berenshteyn F; Lacoste A; Thomas JR; Roma G; Michaud GA; Tseng BS; Porter JA; Myer VE; Tallarico JA; Hamann LG; Curtis D; Fishman MC; Dietrich WF; Dales NA; Sivasankaran R, SMN2 splice modulators enhance U1–pre-mRNA association and rescue SMA mice. Nat. Chem. Biol 2015, 11 (7), 511–517. [DOI] [PubMed] [Google Scholar]
- 44.Brickner SJ; Hutchinson DK; Barbachyn MR; Manninen PR; Ulanowicz DA; Garmon SA; Grega KC; Hendges SK; Toops DS; Ford CW; Zurenko GE, Synthesis and antibacterial activity of U-100592 and U-100766, two oxazolidinone antibacterial agents for the potential treatment of multidrug-resistant gram-positive bacterial infections. J. Med. Chem 1996, 39 (3), 673–679. [DOI] [PubMed] [Google Scholar]
- 45.Moellering RC, Linezolid: the first oxazolidinone antimicrobial. Ann. Intern. Med 2003, 138 (2), 135–142. [DOI] [PubMed] [Google Scholar]
- 46.Kloss P; Xiong L Fau - Shinabarger DL; Shinabarger Dl Fau - Mankin AS; Mankin AS, Resistance mutations in 23 S rRNA identify the site of action of the protein synthesis inhibitor linezolid in the ribosomal peptidyl transferase center. J. Mol. Biol 1999, 294 (1), 93–101. [DOI] [PubMed] [Google Scholar]
- 47.Childs-Disney JL; Yang X; Gibaut QMR; Tong Y; Batey RT; Disney MD, Targeting RNA structures with small molecules. Nat. Rev. Drug Discov 2022. 10.1038/s41573-022-00521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sreeramulu S; Richter C; Berg H; Wirtz Martin MA; Ceylan B; Matzel T; Adam J; Altincekic N; Azzaoui K; Bains JK; Blommers MJJ; Ferner J; Fürtig B; Göbel M; Grün JT; Hengesbach M; Hohmann KF; Hymon D; Knezic B; Martins JN; Mertinkus KR; Niesteruk A; Peter SA; Pyper DJ; Qureshi NS; Scheffer U; Schlundt A; Schnieders R; Stirnal E; Sudakov A; Tröster A; Vögele J; Wacker A; Weigand JE; Wirmer-Bartoschek J; Wöhnert J; Schwalbe H, Exploring the druggability of conserved RNA regulatory elements in the SARS-CoV-2 genome. Angew. Chem. Int. Ed. Engl 2021, 60 (35), 19191–19200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benhamou RI; Suresh BM; Tong Y; Cochrane WG; Cavett V; Vezina-Dawod S; Abegg D; Childs-Disney JL; Adibekian A; Paegel BM; Disney MD, DNA-encoded library-versus-RNA-encoded library selection enables design of an oncogenic non-coding RNA inhibitor. Proc. Natl. Acad. Sci. U. S. A 2021, 119, e2114971119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warner Katherine D.; Homan P; Weeks Kevin M.; Smith Alison G.; Abell C; Ferré-D’Amaré Adrian R., Validating fragment-based drug discovery for biological RNAs: lead fragments bind and remodel the TPP riboswitch specifically. Chem. Biol 2014, 21 (5), 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daldrop P; Reyes Francis E.; Robinson David A.; Hammond Colin M.; Lilley David M.; Batey Robert T.; Brenk R, Novel ligands for a purine riboswitch discovered by RNA-ligand docking. Chem. Biol 2011, 18 (3), 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeiger M; Stark S; Kalden E; Ackermann B; Ferner J; Scheffer U; Shoja-Bazargani F; Erdel V; Schwalbe H; Göbel MW, Fragment based search for small molecule inhibitors of HIV-1 Tat-TAR. Bioorg. Med. Chem. Lett 2014, 24 (24), 5576–5580. [DOI] [PubMed] [Google Scholar]
- 53.Tam B; Sherf D; Cohen S; Eisdorfer SA; Perez M; Soffer A; Vilenchik D; Akabayov SR; Wagner G; Akabayov B, Discovery of small-molecule inhibitors targeting the ribosomal peptidyl transferase center (PTC) of M. tuberculosis. Chem. Sci 2019, 10 (38), 8764–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee M-K; Bottini A; Kim M; Bardaro MF; Zhang Z; Pellecchia M; Choi B-S; Varani G, A novel small-molecule binds to the influenza A virus RNA promoter and inhibits viral replication. Chem. Commun 2014, 50 (3), 368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.