

Abstract
A cyclin-dependent kinase (CDK) inhibitor, p57Kip2, is an important molecule involved in bone development; p57Kip2-deficient (p57-/-) mice display neonatal lethality resulting from abnormal bone formation and cleft palate. The modulator 1α,25-dihydroxyvitamin D3 (l,25-(OH)2VD3) has shown the potential to suppress the proliferation and induce the differentiation of normal and tumor cells. The current study assessed the role of p57Kip2 in the 1,25-(OH)2VD3-regulated differentiation of osteoblasts because p57Kip2 is associated with the vitamin D receptor (VDR). Additionally, 1,25-(OH)2VD3 treatment increased p57KIP2 expression and induced the colocalization of p57KIP2 with VDR in the osteoblast nucleus. Primary p57-/- osteoblasts exhibited higher proliferation rates with Cdk activation than p57+/+ cells. A lower level of nodule mineralization was observed in p57-/- osteoblasts than in p57+/+ cells. In p57+/+ osteoblasts, 1,25-(OH)2VD3 upregulated the p57Kip2 and opn mRNA expression levels, while the opn expression levels were significantly decreased in p57-/- cells. The osteoclastogenesis assay performed using bone marrow cocultured with 1,25-(OH)2VD3-treated osteoblasts revealed a decreased efficiency of 1,25-(OH)2VD3-stimulated osteoclastogenesis in p57-/- cells. Based on these results, p57Kip2 might function as a mediator of 1,25-(OH)2VD3 signaling, thereby enabling sufficient VDR activation for osteoblast maturation.
Introduction
Treatment with 1α,25-dihydroxyvitamin D3 (l,25-(OH)2D3) suppresses the proliferation and induces the differentiation of osteoblastic cell lines. The induced growth inhibition was accompanied by a blockade of the transition from the G1 to S phase of the cell cycle [1]. We are interested in the regulatory role of l,25(OH)2D3 in cell growth and differentiation. The cyclin-dependent kinase inhibitor (CDKI) p57Kip2 shares homology with the Cip/Kip family molecules p21Cip1 and p27Kip1 in the N-terminal domain (CDK inhibitory domain); additionally, CDKIs bind a variety of cyclin-CDK complexes and inhibit their kinase activities in vitro [2, 3]. Transfection of p57Kip2 into Saos-2 osteosarcoma cells induced arrest at G1 phase through a mechanism that does not appear to require Rb or p53 [3]. Several reports have shown that SAOS2 cells were defective for p53 and Rb (Hinds et al. (1992), van der Heudel and Harlow [4, 5]). Decreased p57Kip2 expression levels have been detected in several types of tumors [6–9]; consequently, p57Kip2 might function as a tumor suppressor.
Mice deficient in the p57Kip2 gene exhibit defective endochondral bone formation. Most p57Kip2-deficient mice die shortly after birth as a result of severe cleft palate. In studies using mice lacking Cip/Kip family CDKIs (p21Cip1, p27Kip1 and p57Kip2), only p57Kip2-deficient mice exhibited developmental abnormalities, including several defects attributed to abnormal bone formation [10–12]. Based on these results, p57Kip2 might function as a cellular mediator of bone formation.
Bone resorption by osteoclasts is an important event in calcium homeostasis and bone metabolism. The maturation of osteoblasts involves mineralization and the upregulation of osteoclastogenic genes, and 1,25-(OH)2VD3, which is the most active metabolite of vitamin D3, is a hormone required for osteoblast function [13–17]. This hormone exerts a positive effect on bone mass when administered in vivo. Osteoblasts express nuclear vitamin D3 receptors (VDRs) and show increased expression of osteopontin (opn) and rankl after treatment with 1,25-(OH)2VD3. Osteoblasts are considered the major target of 1,25-(OH)2VD3 in bone [15,18]. Through interactions with nuclear VDRs, 1,25-(OH)2VD3 inhibits osteoblastic cell proliferation and upregulates the expression of VDR-dependent genes, including opn [19] and rankl [20], which increase the functional activity of mature osteoblasts to induce osteoclastogenesis.
The current study assessed the role of p57Kip2 in the 1,25-(OH)2VD3-regulated differentiation of osteoblasts because p57Kip2, but not other Cip/Kip molecules, was associated with VDR. In osteoblasts, VDR-dependent genes, including opn and rankl, were upregulated by VD3-VDR, and p57-/- osteoblasts did not show upregulation of these genes. Because significantly higher levels of an osteoclastogenesis inhibitor, osteoprotegerin, were detected in p57-/- osteoblasts than p57+/+ cells, p57Kip2 may suppress osteoprotegerin (opg) expression in osteoblasts. Based on these results, we hypothesized that p57Kip2 might be a mediator of 1,25-(OH)2VD3 signaling.
Materials and methods
Mice
Experimental animals carrying a targeted mutation in the p57Kip2 loci were supplied by F. Hoffmann-La Roche, Ltd (Basel, Switzerland) [12]. The current study was approved by the Animal Use and Care Committee of Hoshi University. Mouse genotypes were determined by PCR. The p57Kip2 locus is imprinted and expressed from the maternally derived allele. All experiments were conducted in accordance with the approval of the Hoshi University Animal Care and Use Committee (certificate number 20–070).
Histology and immunohistochemistry
Dissected tissues were fixed with 4% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4). For the histological analysis, fixed samples were demineralized for 2 days in 10% formic acid, dehydrated with ascending concentrations of ethanol, cleared in xylene and embedded in paraffin wax. Embedded samples were sectioned at 5 μm; each section was retrieved from the water bath. For tartrate-resistant acid phosphatase (TRAP) enzyme histochemistry, the sections were deparaffinized, followed by immersion in 50 ml of an aqueous solution containing 5 mg of naphthol AS-BI phosphate (Sigma, St. Louis, MO), 25 mg of red violet LB salt (Sigma) and 100 mM L-(+) tartaric acid (0.76 g; Sigma) diluted in 0.1 M sodium acetate buffer (pH 5.4) for 15 min at 37°C. For OPN immunohistochemistry, dewaxed sections were treated with 0.1% hydrogen peroxidase for 20 min to inhibit endogenous peroxidases; subsequently, dewaxed samples were pre-incubated with 1% bovine serum albumin in phosphate-buffered saline (pH 7.2; BSA-PBS) for 30 min at room temperature. Antisera against OPN (LSL Co., Tokyo, Japan) diluted 1:3000 were applied to the sections overnight at 4°C, after which the specimens were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Chemicon International Inc., Temecula, CA). Immunoreactions were visualized with diaminobenzidine as a substrate prior to observation under a light microscope. These sections were lightly counterstained with methyl green.
Preparation of primary mouse osteoblasts
Primary cultures of calvarial osteoblasts were prepared using the sequential collagenase/dispase digestion method [21]. Briefly, calvaria were removed from newborn offspring derived from p57+/- males and p57+/- females; the neonates were denuded of soft tissue and digested with 1 mg/ml collagenase and 2 mg/ml dispase for 15 min at 37°C in PBS with gentle agitation. The procedure was performed twice; cells from the second digestion were harvested and grown to confluence in α-MEM supplemented with 1% penicillin-streptomycin (Wako Pure Chemicals, Osaka, Japan) and 10% FCS.
Immunoblot analysis
Cells were plated in 6-well plastic dishes and cultured with α-MEM containing 10% FCS, 5 mM β-glycerophosphate, 50 mg/ml ascorbic acid and antibiotics. The cells were treated with 1,25-(OH)2VD3 (10 nM) for 24 h prior to collection for protein assays. Cells were rinsed twice with ice-cold phosphate-buffered saline and lysed with 180 μl of Nonidet P-40 lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10 mM NaF, 5 mM EDTA, 5 mM EGTA, 2 mM sodium vanadate, 0.5% sodium deoxycholate, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 2 mg/ml aprotinin and 0.1% Nonidet P-40]; subsequently, the lysates were cleared by centrifugation at 15,000 × g for 5 min at 4°C. For immunoblot analyses, samples were separated on 9 or 12.5% SDS-PAGE gels. Immunoblotting was performed with an enhanced chemiluminescence detection system (GE Healthcare Life Sciences, Uppsala, Sweden). Anti-VDR and anti-β-Actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A rabbit antibody against p57Kip2 was raised against the peptides corresponding to the C-terminus of the p57Kip2 protein.
Immunoprecipitation for the detection of the endogenous p57Kip2-VDR complex
Cells were plated in 60-mm plastic dishes and cultured with α-MEM containing 10% FCS, 5 mM β-glycerophosphate, 50 mg/ml ascorbic acid and antibiotics. The cells were treated with 1,25-(OH)2VD3 (10 nM) for 24 h prior to collection for protein assays. Cells were rinsed twice with ice-cold phosphate-buffered saline and lysed with 500 μl of RIPA buffer (1 mM sodium vanadate, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM phenylmethylsulfonyl fluoride and 2 mg/ml aprotinin in phosphate-buffered saline); subsequently, the whole lysates were cleared by centrifugation at 15,000 × g for 5 min at 4°C. For VDR immunoprecipitation, the lysates (200 μg protein) were mixed with anti-VDR antibodies (Santa Cruz, CA). The immunocomplexes were precipitated with Protein A/G-Sepharose beads (GE Healthcare Life Sciences); subsequently, the pellets were washed six times with ice-cold RIPA buffer. The precipitates were separated on 9% SDS-PAGE gels. Immunoblotting for the detection of both p57Kip2 and VDR was performed with an enhanced chemiluminescence detection system (GE Healthcare Life Sciences, Uppsala, Sweden). Anti-p57Kip2 and anti-VDR antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Immunofluorescence staining
Primary mouse osteoblasts were treated with 10 nM 1,25(OH)-VD3 for 24 h. The cells were fixed with paraformaldehyde and incubated with the following primary antibodies: mouse anti-VDR (D-6: Santa Cruz Biotechnology) and rabbit anti-p57Kip2 (EP2515Y, Abcam). After washing, the cells were incubated with the following secondary antibodies: Alexa Fluor 647-goat anti-mouse IgG and Alexa Fluor 488-goat anti-rabbit IgG (Jackson Immuno Reasearch). The cells were mounted with mounting medium containing DAPI to label the nuclei. Stained cells were detected by confocal microscopy (FV1200, Olympus); blue staining indicated DAPI-stained nuclei, red staining indicated VDR, and green staining indicated p57Kip2. The scale bar represents 100 μm.
Nodule mineralization
Osteoblasts were plated in 12-well multiplates at a density of 2.5 × 104 cells/well and grown to confluence for 10 days. Media were then replaced with mineralizing media (α-MEM supplemented with antibiotics, 10% FCS, 10 μM β-glycerophosphate, 100 μg/ml ascorbic acid and 100 nM dexamethasone). Following an additional 3 weeks of culture [22], mineralization was detected with the von Kossa staining method. The von Kossa-stained area was measured with the application MetaMorph.
Quantitative RT-PCR
The expression levels of the murine p57Kip2 and opn mRNAs in mouse primary osteoblasts were evaluated with quantitative RT-PCR (qRT-PCR) using a Prism 7000 System (Applied Biosystems, Foster City, CA) and SYBR Green I fluorescence as previously described [23–25]. The expression levels of the human p57Kip2 and opn mRNAs in SaOS2 cells were also assessed using qRT-PCR. The cDNA templates were synthesized from 1 μg of total RNA using the First Strand cDNA Synthesis Kit (GE Healthcare Life Sciences). The relative levels of the mouse and human p57Kip2 and opn mRNAs, which were normalized to the reference gene hypoxanthine-guanine phosphoribosyl transferase (hprt1), were determined using the comparative Ct (cycles at threshold fluorescence) method. All experiments were independently repeated three times, i.e., each experiment was performed in triplicate. The sequences of the PCR primers are as follows: mouse p57Kip2 (forward 5’-AACCGCTGGGACTTCAACTTC-3’, reverse 5’-AGACTCGCTGTCCACCTCCAT-3’), mouse opn (forward 5’-CCCTCGATGTCATCCCTGTT-3’, reverse 5’-CTGCCCTTTCCGTTGTTGTC-3’), mouse hprt1 (forward 5’-TGGGAGGCCATCACATTGT-3’, reverse 5’-AGCAGGTCAGCAAAGAACTTATAGC-3’), mouse rankl (forward 5’-CCAGCATCAAAATCCCAAGTTC-3’, reverse 5’-TGCCCGACCAGTTTTTCG-3’), mouse opg (forward 5’-GCCTGGGACCAAAGTGAATG-3’, reverse 5’-CTTGTGAGCTGTGTCTCCGTTT-3’), human p57KIP2 (forward 5’-AGTCCCTCGACGGCCTCGAG-3’, reverse 5’-CGGGACCGGGACACTAGGCA-3’), and human opn (forward 5’-ATGAGCATTCCGATGTGATTG-3’, reverse 5’-TGTGGAATTCACGGCTGA-3’).
Constructs and transfection
Expression plasmids for amino terminally HA-tagged VDR (HA-VDR) were constructed by ligating the cDNA fragments into the FLAG-pcDNA3.1(-) vector. For transfection, the human osteosarcoma cell line SaOS2 and its tetracycline (tet)-off p57Kip2 stable transfectant were used. Cells (5 × 105) were grown in 100-mm culture dishes; Lipofectamine (Invitrogen) was employed according to the manufacturer’s instructions. Cells were harvested or analyzed after 48 h. Immunoblotting and immunoprecipitation were performed as described previously [26]. For immunoprecipitation, the cell lysates were mixed with anti-FLAG M2 affinity gel (Sigma); FLAG-tagged proteins were eluted with the 3 × FLAG peptide after washing. The eluate was subjected to immunodetection utilizing anti-FLAG, anti-VDR and anti-p57Kip2 antibodies.
Statistical analysis
All data were analyzed with Student’s t-test and two-way ANOVA. Differences were considered statistically significant when p < 0.01.
Results
The VD3-dependent interaction between VDR and p57Kip2
We hypothesized that the 1,25-(OH)2VD3 (VD3) activity in osteoblasts might depend on the p57Kip2 protein. In the immunoprecipitation analysis of the primary mouse osteoblasts, p57Kip2 was detected in the complex precipitated with anti-VDR antibodies in the VD3-stimulated osteoblasts (Fig 1A). VD3 stimuli also induced the colocalization of p57Kip2 with VDR in the nuclei of the primary mouse osteoblasts (Fig 1B).
Fig 1. The association of p57Kip2 with VDR is dependent on 1,25-(OH)2VD3.

A. The association of p57Kip2 with VDR is dependent on 1,25-(OH)2VD3 in primary p57+/+ osteoblasts. Top photo: immunoprecipitation (IP) was performed with an anti-VDR antibody, and immunoblotting (IB) was conducted with an anti-p57Kip2 antibody. Bottom: immunoprecipitation and immunoblotting were performed with an anti-VDR antibody. B. Immunofluorescence staining of primary mouse osteoblasts that were treated with 10 nM 1,25-(OH)2VD3 for 24 h. Blue indicates DAPI-stained nuclei, red indicates VDR, and green indicates p57Kip2. The scale bar represents 100 μm.
p57Kip2 enhances the transcriptional activities of VDR
Next, we used SaOS2 cells characterized by tet-off regulation of p57Kip2 to assess the effects of p57Kip2 on VDR activation. In humans and rodents, the expression of opn transcripts depends on the formation of the 1,25-(OH)2VD3-VDR complex at the VDR response element (VDRE) [27, 28]. In the absence of tet, the expression of both the p57Kip2 and opn mRNAs was upregulated (Fig 2A and 2B). In the presence of tet, p57Kip2 transcripts were also downregulated by the tet-off system (Fig 2A). Because opn transcripts were simultaneously decreased in the presence of tet, p57Kip2 expression increased the levels of the opn mRNA (Fig 2B). We performed a luciferase assay employing a reporter plasmid with the opn 5’ flanking region-luciferase cDNA to investigate the role of p57Kip2 in the activation of the VDRE. Following cotransfection of the reporter plasmid and VDR expression plasmids into SaOS2 tet-off p57Kip2-stable cells, luciferase activity increased by two-fold upon the addition of 1,25-(OH)2VD3. Our SaOS2 tet-off p57Kip2-stable cells may have expressed p57Kip2 at sufficient levels to activate the opn 5’ flanking region when the cells were cultured in medium lacking tet. The p57Kip2-on cells might have expressed p57Kip2 at excessively high levels that were unable to increase the activity of the opn 5’ flanking region through VDR expression, and the opn 5’ flanking region was strongly activated by 1,25-(OH)2VD3 (Fig 2D). Under the tet-off condition (basal levels of p57Kip2), VDR expression was sufficient for opn promoter activation by 1,25-(OH)2VD3; furthermore, the induction of both VDR and p57Kip2 expression increased the activation of the promoter after the 1,25-(OH)2VD3 treatment (Fig 2C). Thus, p57Kip2 activated the opn 5’ region, and the coexistence of p57Kip2 and VDR increased the 1,25-(OH)2VD3-induced activation of opn expression. Osteogenic homeostasis mediated by 1,25-(OH)2VD3 might be sufficient in osteoblasts expressing both p57Kip2 and VDR.
Fig 2. p57Kip2 Upregulates the VD3-dependent expression of opn transcripts.

Expression levels of both p57Kip2 (A) and opn (B) depended on the presence of tetracycline in the SaOS2 tet-off p57Kip2 transfectant. An asterisk indicates statistical significance: #, p < 0.01. (C) The activity of the opn promoter was estimated in SaOS2 tet-off p57Kip2-transfected cells. In the SaOS2 tet-off p57Kip2 transfectant, p57Kip2 expression (- tetracycline) upregulated opn promoter activity independently of 1,25-(OH)2VD3. Under equal transfection conditions, an asterisk indicates a statistically significant difference between 0.1% ethanol and 1,25-(OH)2VD3: *, p < 0.01. Under equal culture conditions, a # symbol indicates a statistically significant difference among transfection conditions: #, p < 0.01. The open columns represent the vehicle (0.1% ethanol: EtOH), whereas the closed columns represent 10 nM 1,25-(OH)2VD3.
Effects of the ablation of p57Kip2 in primary cultured osteoblasts
We prepared primary calvarial osteoblasts harvested from neonates to examine the role of p57Kip2 in osteoblast maturation in detail. The primary p57-/- osteoblasts displayed a higher proliferation rate than wild-type (p57+/+) cells (S Fig 1 in S1 File). The continuous induction of differentiation in the confluent osteoblasts was observed in an in vitro culture for three weeks. After extended culture, p57-/- osteoblasts exhibited lower levels of mineralization than p57+/+ cells. The levels of p57Kip2 transcripts and proteins in cultured primary mouse osteoblasts were assessed after treatment with 1,25-(OH)2VD3. The qRT-PCR analysis revealed the upregulation of the p57Kip2 transcript in 1,25-(OH)2VD3-treated mouse osteoblasts (Fig 3A). Concomitant with the increase in the levels of p57Kip2 transcripts, substantially increased levels of the p57Kip2 protein were detected using immunoblotting (Fig 3B). These findings were consistent with a previous report noting the stabilization of p57Kip2 protein in rat osteoblasts in the presence of 1,25-(OH)2VD3 [29]. Subsequently, we hypothesized that 1,25-(OH)2VD3 activity in osteoblasts might depend on the levels of the p57Kip2 protein. An analysis of opn mRNA levels in p57+/+ osteoblasts in the early phase of mineralization revealed an upregulation of opn transcripts, which was increased two-fold by 1,25-(OH)2VD3. In p57-/- cells, the induction of differentiation did not lead to increased opn expression; however, the 1,25-(OH)2VD3 treatment induced opn expression (Fig 3C). Thus, the expression of opn transcripts in osteoblasts might depend on VDR and p57Kip2. Nodule mineralization was observed during the extended culture of both p57-/- and p57+/+ cells; furthermore, the mineralized nodules in p57-/- cells were significantly smaller than those in p57+/+ cells (Fig 3D). Additionally, the area of the mineralized nodules was reduced by approximately 30% (Fig 3D and 3E). Primary p57+/+ osteoblasts expressed opn mRNA at higher levels than p57-/- cells, and the differences were more obvious in confluent osteoblasts stimulated with mineralization medium (Fig 3F). Immunohistochemistry findings demonstrated lower levels of Opn-expressing osteoblasts in the bone medulla of p57-/- neonates than in p57+/+ neonates (S Fig 2 in S1 File). Based on these results, we hypothesized that p57Kip2 might be necessary to ensure the sufficient bioactivity of 1,25-(OH)2VD3 during osteoblastic maturation.
Fig 3. Effects of the ablation of p57Kip2 on primary cultured osteoblasts.
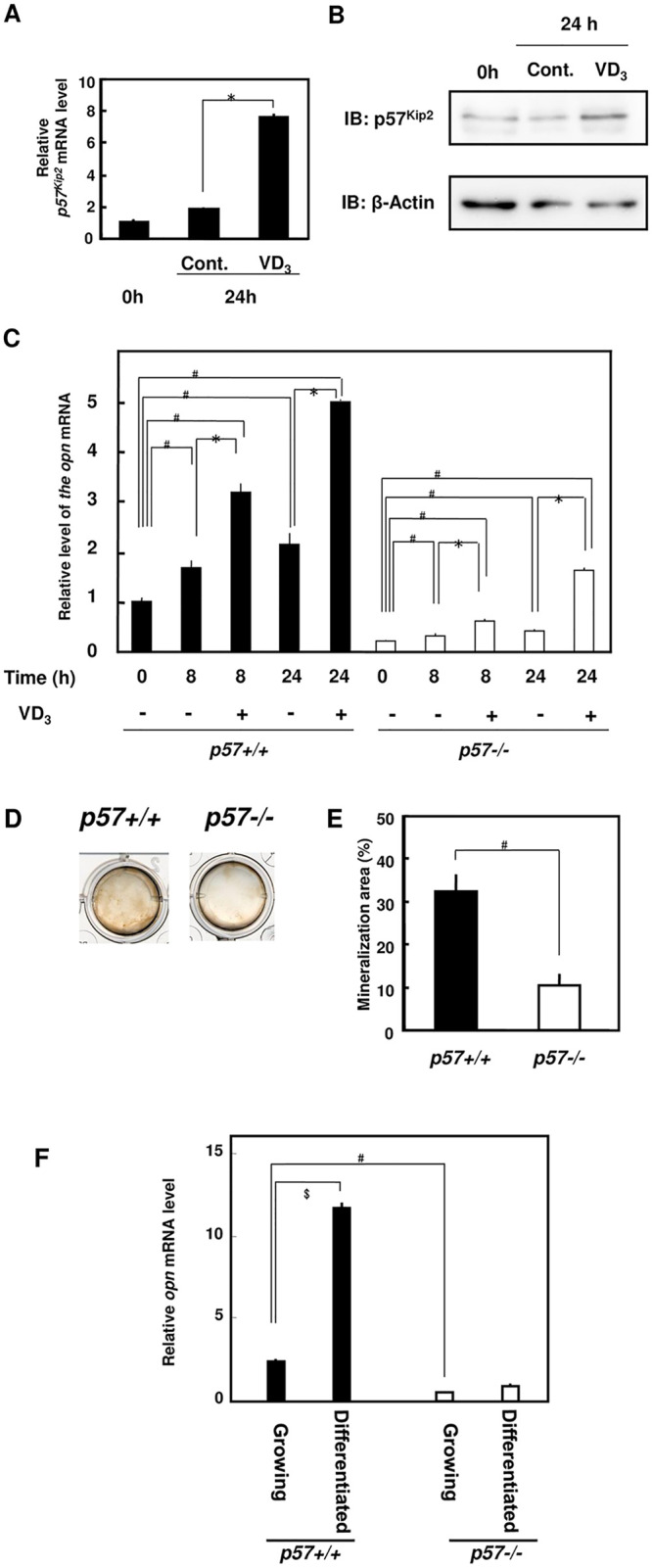
(A) Results of the quantitative RT-PCR analysis of p57Kip2 transcripts. The results were normalized to hprt levels. An asterisk indicates a statistically significant difference: *, p < 0.01. (B) Immunoblot showing the levels of p57Kip2. In A and B, cells were treated with the vehicle (0.1% ethanol: Cont.) or 10 nM 1,25-(OH)2VD3 (VD3) for 24 h. (C) The effect of 10 nM 1,25-(OH)2VD3 on the expression of the osteopontin mRNA was analyzed using quantitative RT-PCR. The results were normalized to the hprt mRNA. Under equal conditions, an asterisk indicates a statistically significant difference between 0.1% ethanol and 1,25-(OH)2VD3: *, p < 0.01. The # symbol indicates a statistically significant difference between p57+/+ and p57-/- cells: #, p < 0.01. (D) Images of von Kossa staining of nodule mineralization in extended-culture osteoblastic cells. Primary osteoblasts were plated in 12-well multiplates, grown to confluence and incubated for 21 days with ascorbic acid, β-glycerophosphate and dexamethasone. (D) Photo of von Kossa-stained cells. (E) Graph of the analysis of the cells shown in (D) [mineralization area (%)]. An asterisk indicates a statistically significant difference: *, p < 0.01. (F) The expression of the osteopontin mRNA in p57+/+ and p57-/- osteoblasts was analyzed using quantitative RT-PCR. Cells grown to 70% confluence were “growing cells”, and mineralized confluent cells were “differentiated cells”. The results were normalized to the hprt mRNA. The # symbol indicates a statistically significant difference between p57+/+ and p57-/- cells: #, p < 0.01. The $ symbol indicates a statistically significant difference between growing and differentiated cells: $, p < 0.01.
p57-/- osteoblasts exhibit defects in osteoclastogenesis
We hypothesized that p57Kip2 might play a role in the 1,25-(OH)2VD3-induced osteoclastogenesis of osteoblasts. We cocultured 1,25-(OH)2VD3-treated osteoblasts with bone marrow cells to identify the roles of p57Kip2 in the osteoclastogenic activity of osteoblasts. Notably, 1,25-(OH)2VD3 stimulated osteoclastogenesis in p57+/+ primary osteoblasts more effectively than in p57-/- osteoblasts (S Fig 3 in S1 File). Based on these findings, p57Kip2 might be necessary to maintain proper mineralization and p57Kip2 might promote VD3 signaling. Receptor activator of NF-kappa B ligand (Rankl) is a membrane-bound signal transducer responsible for the differentiation and maintenance of osteoclasts. In p57+/+ osteoblasts, rankl transcripts were upregulated 4.7-fold after 1,25-(OH)2VD3-treatment (72 h) (Fig 4A). Opg, also known as an osteoclastogenesis inhibitory factor, functions as a decoy receptor for Rank to obstruct Rankl-Rank signaling and inhibit osteoclastogenesis. The expression of opg transcripts was significantly increased in p57-/- osteoblasts (Fig 4B). Rankl expression might depend on p57Kip2 to some extent, and opg expression might be suppressed by p57Kip2. Thus, the defects in p57-/- osteoblasts might result from disturbances in rankl and opg expression levels. Additionally, 1,25-(OH)2VD3 upregulated rankl expression by 4.7-fold in p57+/+ cells and 1.4-fold in p57-/- cells (Fig 4A). The expression of rankl is regulated by VD3-VDR activation [30–32]. The p57Kip2 deficiency reduced cellular responses to 1,25-(OH)2VD3. In contrast to rankl, opg expression was not altered by 1,25-(OH)2VD3 in both p57+/+ and p57-/- cells (Fig 4B). The ratio of rankl/opg expression may be useful as an indicator of the osteoclastogenic activity of osteoblasts [32, 33]. As shown in Fig 4D, p57+/+ cells displayed higher rankl/opg ratios than p57-/- cells. Treatment with 1,25-(OH)2VD3 significantly increased the ratio in p57+/+ cells (Fig 4C). From these results, we concluded that the ablation of p57Kip2 upregulated opg, suppressed 1,25-(OH)2VD3-dependent rankl expression, and resulted in defects in osteoclastogenic activities in osteoblasts.
Fig 4. VD3-induced rankl expression in osteoblasts depended on p57Kip2.

(A) Quantitative RT-PCR analysis of the levels of the rankl mRNA in osteoblasts treated with 0.1% ethanol (-) or 10 nM 1,25-(OH)2VD3 (+) for 72 h. Levels of the rankl mRNA were normalized to the mRNA levels of the constitutive gene hprt1. (B) Quantitative RT-PCR analysis of the expression of the opg mRNA in osteoblasts treated with 0.1% ethanol (-) or 10 nM 1,25-(OH)2VD3 (+) for 72 h. Levels of the opg mRNA were normalized to the mRNA levels of the constitutive gene hprt1. (C) The relative ratio of the rankl mRNA/opg mRNA was calculated and compared between p57+/+ and p57-/- osteoblasts. In the graphs, relative levels in p57+/+ cells were estimated. The # symbol indicates a statistically significant difference between p57+/+ and p57-/- cells: #, p < 0.01. The asterisk indicates a statistically significant difference between 0.1% ethanol (-) and 10 nM 1,25-(OH)2VD3 (+): *, p < 0.01.
Discussion
As shown in the current study, p57Kip2 specifically interacted with VDR and was required for osteoclastogenesis activities in osteoblasts. Studies using knockout mice indicated that p57Kip2 functions mainly as a CDKI in mouse osteoblasts, because the lack of genes encoding other Cip/Kip family molecules, including both p21Cip1 and p27Kip1, did not result in abnormal bone formation, which was observed in p57-/- mice [11–13].
The degradation of p57Kip2 via the proteasome pathway inhibits osteoblast maturation [34–37]. Thus, the expression of p57Kip2 is necessary for osteoblast maturation. Following the evaluation of a novel ubiquitin ligase, FBL12, which is involved in TGF-1β-induced degradation of p57Kip2, Kim et al. [36] noted that p57Kip2 overexpression promotes the differentiation of primary osteoblasts. VD3 increased p57Kip2 levels in mouse osteoblasts. We revealed the VD3-dependent upregulation of p57Kip2 in mouse osteoblasts, and an increase in levels of the p57Kip2 protein might also be associated with an increase its stabilization.
In our study, VDR was specifically associated with the CDKI domain of p57Kip2. Cip/Kip molecules contain a characteristic CDKI domain (Fig 5). The p57Kip2 CDKI domain contains the specific hydrophilic AELNAEDQN peptide and hydrophobic PLRGPGRLQ peptide. The 3D structure of p57Kip2 has not yet been reported, but these p57Kip2-specific peptides might be involved in the interaction with VDR. As shown in the study by Valcheva et al. [38], G0-synchronized primary VDR-deficient vascular smooth muscle cells express p57Kip2 at higher levels than wild-type cells. These findings prompted us to investigate the regulation of p57Kip2 levels by the VDR complex. The LBD of VDR was responsible for the interaction with p57Kip2. This peptide of VDR contains the common LBD for other nuclear receptors, including PXR, LXR, FXR THR and RXR. According to our results, p57Kip2 might interact with various nuclear receptors to regulate their activity. Joseph B et al. reported that p57Kip2 cooperated with Nurr1, the same nuclear receptor as VDR, and activated transcriptional activity at its transcription factor binding site (NBRE). Therefore, we investigated whether p57Kip2 also cooperated with VDR to activate transcriptional activity at the transcription factor binding site (VDRE) [39]. The coexistence of p57 and VDR facilitated 1,25-(OH)2VD3-dependent VDR activation, and p57 functioned as a cofactor of VDR.
Fig 5. CDKI domains of Cip/Kip family molecules.

Characteristic CDKI domains were compared among mouse (Mus musculus) p57Kip2, human (Homo sapiens) p57Kip2, p27Kip1 and p21Cip1. Identical amino acid residues in 3 of 4 peptides are enclosed with a red line. A blue line surrounds p57Kip2-specific peptides, which were shared by mouse and human sequences.
As shown in Fig 4, p57Kip2 regulated opg expression in mouse osteoblasts, in contrast to opn and rankl. The osteoclastogenic activities of osteoblasts depend on Rankl and Opg. Rankl is a membrane-bound signal transducer responsible for the differentiation and maintenance of osteoclasts; in addition, Rankl promotes osteoclast differentiation. In conjunction with the differences in rankl mRNA levels, p57-/- osteoblasts were inferior to p57+/+ cells in terms of osteoclast induction activity. As shown in our current study, p57Kip2 regulated opg expression, and the ablation of p57Kip2 significantly upregulated the expression of the opg mRNA. Opg is the decoy receptor for Rankl to prevent osteoclastogenesis. Defects in osteoclastogenesis due to a lack of p57Kip2 might result from both an increase in opg expression and the downregulation of rankl. Several studies have described the regulation of opg expression in osteoclastogenesis [20, 27, 40–42], but no information is available on opg downregulation. When opg was discovered by Yasuda et al. [41], most researchers believed that opg expression was vitamin D-dependent. However, Nakamichi et al. [43] reported that rankl mRNA expression was VDR dependent but opg expression in osteoblast-specific KO mice did not change after treatment with 1, 25 vitamin D3. These results suggested that opg expression might not be VDR dependent. Recently, cancer cells were shown to release more Opg than normal cells [44–48]. Because p57Kip2 is a tumor suppressor protein, p57Kip2-deficient cancer cells may express and release Opg.
The levels of the opn mRNA in osteoblasts are consistent with the in vivo osteomalacia-like phenotype; however, in vitro, p57-/- osteoblasts cultured with maturation medium produced fewer mineralized nodules than p57+/+ cells. The expression of the opn mRNA was also upregulated by 1,25-(OH)2VD3-VDR activity, similar to rankl. Kitazawa et al. [30–32] previously reported that 1,25-(OH)2VD3 enhances osteoclastogenesis via the transactivation of the rankl gene in osteoblasts through the VDRE in both humans and mice. Urano et al. [29] observed the upregulation of p57Kip2 transcripts and increased levels of the p57Kip2 protein in rat osteoblasts (p57+/+) cultured in the presence of 1,25-(OH)2VD3. We expected that sufficient VDR activation by 1,25-(OH)2VD3 activity might be mediated by p57Kip2 during osteoblast maturation. An assessment of the SaOS2 tet-off p57Kip2 stable cell line yielded data consistent with our expectations, at least in terms of the activation of the opn promoter containing the VDRE. Of course, our results might be partially attributed to the interaction of p57Kip2 and VDR in osteoblasts and might depend on the other pathway via CDK activation.
In the present study p57Kip2 formed complexes with VDR and function as a cell cycle regulator and a mediator of the 1,25-(OH)2VD3-induced transcriptional activation of osteoblast genes in mineralizing osteoblastic cells. These data identified possible roles for p57Kip2 in regulating osteoblast differentiation and bone metabolism.
Supporting information
(PDF)
(PPTX)
Acknowledgments
We would like to thank T Usui for providing the SaOS2 tet-off p57Kip2 cells. We would also like to thank T Tomuro, A Nara, Y Takahashi, R Oikawa, Dr. A Karakawa, Dr. Y Nakamichi, Prof. S Inoue, Prof. H Itabe and Prof. M Tomita for their support of this study. In addition, we wish to thank Prof. E Abe for critically reading the manuscript.
Data Availability
All relevant data are within the manuscript and its Supporting information files.
Funding Statement
This work was supported by JSPS KAKENHI Grant Numbers JP15K07950 (KT), JP20K09474(HA), JP20K07789 (TU), JP19H01068 (KA), JP18H05215 (KIN), andJP19K07091 (NH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Eelen G, Verlinden L, Van Camp M, Van Hummelen P, Marchal K, De Moor B, et al. The effects of 1α,25-dihydroxyvitamin D3 on the expression of DNA replication genes. J Bone Miner Res. 2004;19: 133–146. [DOI] [PubMed] [Google Scholar]
- 2.Lee MH, Reynisdottir I, Massague J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995;9: 639–649. [DOI] [PubMed] [Google Scholar]
- 3.Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, et al. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9: 650–662. [DOI] [PubMed] [Google Scholar]
- 4.Hinds P.W., Mittnacht S., Dulic V., Arnold A., Reed S.I., and Weinberg R.A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992; 70: 993–1006. doi: 10.1016/0092-8674(92)90249-c [DOI] [PubMed] [Google Scholar]
- 5.van der Heuvel S. and Harlow E. Distinct roles for cyclin dependent kinases in cell cycle control. Science, 1993; 26: 2050–2054. [DOI] [PubMed] [Google Scholar]
- 6.Pateras IS, Apostolopoulou K, Niforou K, Kotsinas A, Gorgoulis VG. p57KIP2: "Kip"ing the cell under control. Mol Cancer Res. 2009;7: 1902–1919. [DOI] [PubMed] [Google Scholar]
- 7.Borriello A, Caldarelli I, Bencivenga D, Criscuolo M, Cucciolla V, Tramontano A, et al. p57Kip2 and cancer: time for a critical appraisal. Mol Cancer Res. 2011;9: 1269–1284. [DOI] [PubMed] [Google Scholar]
- 8.Kavanagh E, Joseph B. The hallmarks of CDKN1C (p57, KIP2) in cancer. Biochim Biophys Acta. 2011;1816: 50–56. doi: 10.1016/j.bbcan.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 9.Jayapal SR, Kaldis P. p57Kip2 regulates T-cell development and lymphoma. Blood. 2014;123: 3370–3371. [DOI] [PubMed] [Google Scholar]
- 10.Zhang P, Liégeois NJ, Wong C, Finegold M, Hou H, Thompson JC, et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith–Wiedemann syndrome. Nature. 1997;387: 151–158. [DOI] [PubMed] [Google Scholar]
- 11.Yan Y, Frisen J, Lee MH, Massague J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997;11: 973–983. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi K, Nakayama K, Nakayama K. Mice lacking a CDK inhibitor, p57Kip2, exhibit skeletal abnormalities and growth retardation. J Biochem. 2000;127: 73–83. [DOI] [PubMed] [Google Scholar]
- 13.Gallagher JC, Riggs BL, Eisman J, Hamstra A, Arnaud SB, DeLuca HF. Intestinal calcium absorption and serum vitamin D metabolites in normal subjects and osteoporotic patients: effect of age and dietary calcium. J Clin Investig. 1979;64: 729–736. doi: 10.1172/JCI109516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lian JB, Stein GS. Transcriptional control of vitamin D-regulated proteins. J Cell Biochem. 1992;49: 37–45. doi: 10.1002/jcb.240490108 [DOI] [PubMed] [Google Scholar]
- 15.Kato S, Suzawa M, Takada I, Takeyama K, Yanagizawa J, Fujiki R, et al. The function of nuclear receptors in bone tissues. J Bone Miner Metab. 2003;21: 323–336. doi: 10.1007/s00774-003-0453-3 [DOI] [PubMed] [Google Scholar]
- 16.Suda T, Ueno Y, Fujii K, Shinki T. Vitamin D and bone. J Cell Biochem. 2003;88: 259–266. doi: 10.1002/jcb.10331 [DOI] [PubMed] [Google Scholar]
- 17.Meyer MB, Benkusky NA, Lee C-H, Pike JW. Genomic determinants of gene regulation by 1,25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. J Biol Chem. 2014;289: 19539–19554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haussler MR, Haussler CA, Jurutka PW, Thompson PD, Hsieh JC, Remus LS, et al. The vitamin D hormone and its nuclear receptor: molecular actions and disease states. J Endocrinol. 1997;154: S57–SS73. [PubMed] [Google Scholar]
- 19.Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF, Denhardt DT. Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc Natl Acad Sci U S A. 1990;87: 9995–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95: 3597–3602. doi: 10.1073/pnas.95.7.3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKee MD, Nanci A. Osteopontin: an interfacial extracellular matrix protein in mineralized tissues. Connect Tissue Res. 1996;35: 197–205. doi: 10.3109/03008209609029192 [DOI] [PubMed] [Google Scholar]
- 22.Marzia M, Sims NA, Voit S, Migliaccio S, Taranta A, Bernardini S, et al. Decreased c-Src expression enhances osteoblast differentiation and bone formation. J Cell Biol. 2000; 151: 311–320. doi: 10.1083/jcb.151.2.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urano T, Narusawa K, Shiraki M, Usui T, Sasaki N, Hosoi T, et al. Association of a single nucleotide polymorphism in the WISP1 gene with spinal osteoarthritis in postmenopausal Japanese women. J Bone Miner Metab. 2007;25: 253–258. doi: 10.1007/s00774-007-0757-9 [DOI] [PubMed] [Google Scholar]
- 24.Urano T, Usui T, Takeda S, Ikeda K, Okada A, Ishida Y, et al. TRIM44 interacts with and stabilizes terf, a TRIM ubiquitin E3 ligase. Biochem Biophys Res Commun. 2009;383: 263–268. doi: 10.1016/j.bbrc.2009.04.010 [DOI] [PubMed] [Google Scholar]
- 25.Urano T, Shiraki M, Yagi H, Ito M, Sasaki N, Sato M, et al. GPR98/Gpr98 gene is involved in the regulation of human and mouse bone mineral density. J Clin Endocrinol Metab 2012; 97: E565–574. doi: 10.1210/jc.2011-2393 [DOI] [PubMed] [Google Scholar]
- 26.Hofbauer LC, Heufelder AE. Osteoprotegerin and its cognate ligand: a new paradigm of osteoclastogenesis. Eur J Endocrinol. 1998;139: 152–154. doi: 10.1530/eje.0.1390152 [DOI] [PubMed] [Google Scholar]
- 27.Koszewski NJ, Reinhardt TA, Horst RL. Vitamin D receptor interactions with the murine osteopontin response element. J Steroid Biochem Mol Biol. 1996;59: 377–388. doi: 10.1016/s0960-0760(96)00127-6 [DOI] [PubMed] [Google Scholar]
- 28.Staal A, van Wijnen AJ, Birkenhäger JC, Pols HA, Prahl J, DeLuca H, et al. Distinct conformations of vitamin D receptor/retinoid X receptor-alpha heterodimers are specified by dinucleotide differences in the vitamin D-responsive elements of the osteocalcin and osteopontin genes. Mol Endocrinol. 1996;10: 1444–1456. doi: 10.1210/mend.10.11.8923469 [DOI] [PubMed] [Google Scholar]
- 29.Urano T, Hosoi T, Shiraki M, Toyoshima H, Ouchi Y, Inoue S. Possible involvement of the p57Kip2 gene in bone metabolism. Biochem Biophys Res Commun. 2000;269: 422–426. [DOI] [PubMed] [Google Scholar]
- 30.Kitazawa S, Kajimoto K, Kondo T, Kitazawa R. Vitamin D3 supports osteoclastogenesis via functional vitamin D response element of human RANKL gene promoter. J Cell Biochem. 2003;89: 771–777. [DOI] [PubMed] [Google Scholar]
- 31.Kitazawa R, Kitazawa S, Maeda S. Promoter structure of mouse RANKL/TRANCE/OPGL/ODF gene. Biochim Biophys Acta. 1999;1445: 134–141. doi: 10.1016/s0167-4781(99)00032-9 [DOI] [PubMed] [Google Scholar]
- 32.Kitazawa R, Kitazawa S. Vitamin D3 augments osteoclastogenesis via vitamin D-responsive element of mouse RANKL gene promoter. Biochem Biophys Res Commun. 2002;290: 650–655. [DOI] [PubMed] [Google Scholar]
- 33.Thomas GP, Baker SU, Eisman JA, Gardiner EM. Changing RANKL/OPG mRNA expression in differentiating murine primary osteoblasts. J Endocrinol. 2001;170: 451–460. doi: 10.1677/joe.0.1700451 [DOI] [PubMed] [Google Scholar]
- 34.Urano T, Yashiroda H, Muraoka M, Tanaka K, Hosoi T, Inoue S, et al. p57Kip2 is degraded through the proteasome in osteoblasts stimulated to proliferation by transforming growth factor β1. J Biol Chem. 1999;274: 12197–12200. [DOI] [PubMed] [Google Scholar]
- 35.Nishimori S, Tanaka Y, Chiba T, Fujii M, Imamura T, Miyazono K, et al. Smad-mediated transcription is required for transforming growth factor-β1-induced p57Kip2 proteolysis in osteoblastic cells. J Biol Chem. 2001;276: 10700–10705. [DOI] [PubMed] [Google Scholar]
- 36.Kim M, Nakamoto T, Nishimori S, Tanaka K, Chiba T. A new ubiquitin ligase involved in p57KIP2 proteolysis regulates osteoblast cell differentiation. EMBO Rep. 2008;9: 878–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, et al. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci U S A. 2003;100: 10231–10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valcheva P, Cardus A, Panizo S, Parisi E, Bozic M, Lopez Novoa JM, et al. Lack of vitamin D receptor causes stress-induced premature senescence in vascular smooth muscle cells through enhanced local angiotensin-II signals. Atherosclerosis. 2014;235: 247–255. doi: 10.1016/j.atherosclerosis.2014.05.911 [DOI] [PubMed] [Google Scholar]
- 39.Joseph B, Wallén-Mackenzie A, Benoit G, Murata T, Joodmardi E, Okret S, et al. p57(Kip2) cooperates with Nurr1 in developing dopamine cells. Proc Natl Acad Sci U S A. 2003; 100: p15619–15624. doi: 10.1073/pnas.2635658100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247: 610–615. doi: 10.1006/bbrc.1998.8697 [DOI] [PubMed] [Google Scholar]
- 41.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837 [DOI] [PubMed] [Google Scholar]
- 42.Hakeda Y, Kobayashi Y, Yamaguchi K, Yasuda H, Tsuda E, Higashio K, et al. Osteoclastogenesis inhibitory factor (OCIF) directly inhibits bone-resorbing activity of isolated mature osteoclasts. Biochem Biophys Res Commun. 1998;251: 796–801. doi: 10.1006/bbrc.1998.9523 [DOI] [PubMed] [Google Scholar]
- 43.Nakamichi Y, Udagawa N, Horibe K, Mizoguchi T, Yamamoto Y, Nakamura T, et al. VDR in Osteoblast-Lineage Cells Primarily Mediates Vitamin D Treatment-Induced Increase in Bone Mass by Suppressing Bone Resorption. J Bone Miner Res. 2017; 32:1297–1308. doi: 10.1002/jbmr.3096 [DOI] [PubMed] [Google Scholar]
- 44.Weichhaus M, Segaran P, Renaud A, Geerts D, Connelly L. Osteoprotegerin expression in triple-negative breast cancer cells promotes metastasis. Cancer Med. 2014;3: 1112–1125. doi: 10.1002/cam4.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goswami S, Sharma-Walia N. Osteoprotegerin secreted by inflammatory and invasive breast cancer cells induces aneuploidy, cell proliferation and angiogenesis. BMC Cancer. 2015;15: 935. doi: 10.1186/s12885-015-1837-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fisher JL, Thomas-Mudge RJ, Elliott J, Hards DK, Sims NA, Slavin J, et al. Osteoprotegerin overexpression by breast cancer cells enhances orthotopic and osseous tumor growth and contrasts with that delivered therapeutically. Cancer Res. 2006;66: 3620–3628. doi: 10.1158/0008-5472.CAN-05-3119 [DOI] [PubMed] [Google Scholar]
- 47.Pettersen I, Bakkelund W, Smedsrød B, Sveinbjørnsson B. Osteoprotegerin is expressed in colon carcinoma cells. Anticancer Res. 2005;25: 3809–3816. [PubMed] [Google Scholar]
- 48.Corey E, Brown LG, Kiefer JA, Quinn JE, Pitts TEM, Blair JM, et al. Osteoprotegerin in prostate cancer bone metastasis. Cancer Res. 2005;65: 1710–1718. doi: 10.1158/0008-5472.CAN-04-2033 [DOI] [PubMed] [Google Scholar]