

Abstract
Genome editing holds great potential for cancer treatment due to the ability to precisely inactivate or repair cancer-related genes. However, delivery of CRISPR/Cas to solid tumors for efficient cancer therapy remains challenging. Here, we targeted tumor tissue mechanics via a multiplexed dendrimer lipid nanoparticle (LNP) approach involving co-delivery of focal adhesion kinase (FAK) siRNA, Cas9 mRNA, and sgRNA (siFAK+CRISPR-LNPs) to enable tumor delivery and enhance gene editing efficacy. We show that gene editing was enhanced >10-fold in tumor spheroids due to increased cellular uptake and tumor penetration of nanoparticles mediated by FAK-knockdown. siFAK+CRISPR-PD-L1-LNPs reduced extracellular matrix stiffness and efficiently disrupted PD-L1 expression by CRISPR/Cas gene editing, which significantly inhibited tumor growth and metastasis in four mouse models of cancer. Overall, we provide evidence that modulating the stiffness of tumor tissue can enhance gene editing in tumors, which offers a new strategy for synergistic LNPs and other nanoparticle systems to treat cancer using gene editing.
In vivo inactivation or repair of cancer-related genes using the robust and programmable clustered regularly interspaced short palindromic repeat (CRISPR) associated protein (CRISPR/Cas) system1–10 represents an exciting approach for cancer treatment11. However, CRISPR-based therapies for solid tumors face critical obstacles. The first is cancer’s limitless replicative potential, a cancer hallmark12, that results in an expansive tumor burden whereby editing a small number of cells would not be able to reverse disease symptoms11, 13. The second is the uniquely stiff and fibrotic stroma of the tumor microenvironment. The physically dense tumor microenvironment thus acts as a barrier to efficient tumor therapy, blocking nanoparticle uptake into tumors, access to enough cells to overcome replicative potential, and immune cell infiltration to tumor tissue14. Therefore, we developed a multiplexed nanoparticle siRNA + Cas9 mRNA + sgRNA approach to decrease tumor mechanics and ECM stiffness, increase nanoparticle endocytosis and tissue penetration, and reduce the therapeutic modification threshold to allow gene editing therapy to provide significant survival benefit in genetically engineered mice harboring aggressive tumors.
Among properties of the tumor microenvironment, increased stiffness results from abundant extracellular matrix (ECM) that enhance intrinsic mechanical properties15. Cell-induced deformation is a particular process that ECM undergoes in tumors. Cancer and stromal cells can exert considerable actomyosin-generated forces on the ECM, which contribute to increased ECM stiffness16, 17. These “inside-out” transmitted tensile forces are primarily mediated by integrin-dependent adhesions of attached cells, in a process involving focal adhesion kinase (FAK) activation18. Therefore, targeting FAK in tumor tissue can modulate the mechanical properties of tumor cells, as well as stromal cells and the tumor ECM. In addition, inhibition of FAK activity regulates the tumor immunoenvironment leading to elevated CD8+ cytotoxic T cells infiltration14, 19. However, infiltrated T cells will be inhibited by PD-L1 overexpression on tumor cells, which acts as an inhibitor of T cell responses through sending a critical “don’t find me” signal to the immune system20. This genetic alteration of PD-L1 in cancer cells represents an immune checkpoint blockade of cancer immunotherapy21. Taken together, these features of the tumor microenvironment, stiff ECM and PD-L1 overexpression, present an opportunity for CRISPR-mediated disruption of PD-L1 expression in solid tumors for efficient cancer therapy if the aforementioned challenges can be overcome.
Herein, we developed a multiplexed nanoparticle approach to reduce tumor stiffness and enhance CRISPR gene editing efficiency in tumor tissues for improved therapeutic effect. We co-packaged siRNA (anti-FAK), mRNA (Cas9), and targeted sgRNA into self-assembled lipid nanoparticles (LNPs) to resolve two critical barriers to cancer therapy (stiff ECM and PD-L1 overexpression) (Scheme 1). We found this approach successfully inhibited FAK activity and triggered an intriguing enhancement phenomenon in multiple tumor cells that aided siFAK+CRISPR-LNP access to enough cells to overcome replicative potential to enhance the delivery and overall gene editing >10-fold. We revealed that this gene editing enhancement induced by delivery of siFAK decreased membrane tension regulated by the contraction force of tumor cells and thus increased LNP endocytosis and tumor penetration. Particular focus was placed on ovarian cancer as a representative cancer type that often involves metastasis, and liver cancer as a representative cancer type that involves fibrosis and ECM barriers. Through modulating mechanical properties of tumor cells and breaking down the ECM in vivo, siFAK+CRISPR-LNPs decreased potential metastasis in a mouse metastasis model of ovarian cancer, improved cancer therapy in a tumor xenograft mouse model, and dramatically extended survival in an aggressive MYC-driven transgenic mouse model of liver cancer. Regulating the mechanical properties of tumor cells/ECM for enhancing the genetic suppression in tumor tissues provides an innovative strategy for treating cancer using CRISPR. We anticipate that this general approach could further synergize with additional types of cancer therapeutics in the future.
Scheme 1 ǀ. Schematic illustration for targeting the mechanical properties of tumors to open a double checkpoint blockade of cancer (stiff ECM plus immunosuppression) to enable cancer therapy.

Proposed model for how dendrimer lipid nanoparticles (LNPs) encapsulating FAK siRNA, Cas9 mRNA, and targeted sgRNAs could exhibit enhanced penetration into tumors with increased gene editing of PD-L1 for improved cancer therapy.
siFAK+CRISPR-LNPs enhance gene editing in vitro via modulation of tumor tensile force
To examine the hypothesis that FAK silencing would improve CRISPR gene editing, we first had to develop a nanoparticle system capable of delivering three types of nucleic acids in one nanoparticle: FAK siRNA (siFAK), Cas9 mRNA, and sgRNA. We constructed self-assembled lipid nanoparticles (siFAK+CRISPR-LNPs) (Fig. 1a) that utilized 5A2-SC8 as the ionizable amino lipid dendrimer to bind negatively charged RNAs and facilitate endosomal escape after cellular uptake due to charge acquisition at endosomal pH. 5A2-SC8 was selected based on our prior work delivering siRNA and mRNA to the liver22–24. To further develop LNPs for co-encapsulation of three nucleic acids of different physiochemical properties,9, 24–30 we optimized the mass ratio of 5A2-SC8 : RNA and LNP molecular composition with respect to other lipids (5A2-SC8 : cholesterol : DOPE : DMG-PEG2000 : DSPE-PEG2000 = 15:30:15:2:1 (mol)). This led to targeted LNPs capable of triple RNA loading with high RNA encapsulation efficiency (Supplemental Fig. 1a). Optimized LNPs were ~ 120 nm in size and ~10 mV in average surface charge, which can remain stable in serum-containing medium (5% w/v) (Supplemental Fig. 1b–c) without obvious changes in size over 72 h incubation (Supplemental Fig. 1d). We note CRISPR was deployed using Cas9 mRNA because this approach results in transient Cas9 expression, which can minimize off-target effects without risk of integration, as compared to viral or pDNA delivery approaches1.
Fig. 1 ǀ. FAK knockdown enhances LNP-mediated mRNA delivery and CRISPR gene editing.

a, Schematic illustration showing triple loading of FAK siRNA, Cas9 mRNA, and sgRNA into 5A2-SC8 LNPs. b, RT-qPCR quantification of time-dependent FAK expression in cells treated with siFAK+CRISPR-PD-L1-LNPs. n=4 biologically independent samples. Error bars represent mean ± s.d. c, Representative western blot analysis of FAK expression in IGROV1 cells treated with PBS, siCtrl+CRISPR-PD-L1-LNPs, and siFAK+CRISPR-PD-L1-LNPs for 12 h and 24 h. d, and e, Representative fluorescence microscopy images (d) and quantification of GFP fluorescence intensity (e) of HeLa-GFP cells treated with PBS, siCtrl+CRISPR-GFP-LNPs, and siFAK+CRISPR-GFP-LNPs for 48 h. Scale bar = 20 μm. **P < 0.01 determined by two-tailed t-test. n=3 biologically independent samples. Error bars represent mean ± s.d.. f and g, Representative T7E1 assay for siFAK+CRISPR-LNPs-mediated cleavage enhancement of GFP in HeLa-GFP cells (f) and PD-L1 in IGROV1 cells (g). HeLa-GFP cells: total RNA: 2.67 μg/mL, Cas 9 mRNA: sgRNA: siRNA = 2:0.25:2). IGROV1 cells: total RNA: 1.25 μg/mL, Cas 9 mRNA: sgRNA: siRNA = 2:1:2). h, Representative immunofluorescence of PD-L1 in IGROV1 cells after 48 h treatment with PBS, siCtrl+CRISPR-PD-L1-LNPs, and siFAK+CRISPR-PD-L1-LNPs. (Total RNA: 1.25 μg/mL, Cas 9 mRNA: sgRNA: siRNA = 2:1:2). PD-L1 (Red), Nuclei (DAPI, Blue). Scale bar = 10 μm. i, T7E1 mismatch cleavage (top) and quantification of integrated optical density (IOD) (bottom) for siFAK+CRISPR-GFP-LNPs-mediated gene editing enhancement of GFP in HeLa-GFP cell tumor spheroids cultured on the different stiffness substrates (~100 Pa and ~300 Pa). The concentration of total RNA for HeLa-GFP tumor spheroids was 2.67 μg/mL, Cas 9 mRNA: sgRNA: siRNA = 2:0.25:2. n=3 biologically independent samples. Error bars represent mean ± s.d.. ****P < 0.0001 analyzed two-tailed t-test. Data was analyzed using Image J. j and k, Representative images (j) and quantification of fluorescence intensity (k) of the penetration of nanoparticles in the IGROV1 tumor spheroids treated with siCtrl+Cy5-mRNA-LNPs and siFAK+Cy5-mRNA-LNPs after 48 h incubation. Scale bar = 50 μm. l and m, mCherry expression (l) and deep distribution (m) in tumor spheroids treated with PBS, siCtrl+mRNA-LNPs, and siFAK+mRNA-LNPs. Scale bar = 100 μm.
We then delivered siFAK+CRISPR-LNPs specifically targeting GFP into human HeLa cells stably expressing green fluorescent protein (HeLa-GFP) and assessed efficacy of gene silencing and gene editing. siRNA-mediated gene silencing successfully inhibited FAK expression (Fig. 1b and c). Maximal knockdown (~80%) was quantified from 4 h to 72 h post-administration of siFAK+CRISPR-LNPs (Fig. 1b–c). As expected, GFP expression was inhibited through administration of all LNPs containing Cas9 mRNA and sgGFP (siFAK+CRISPR-GFP-LNPs). Intriguingly, GFP expression was reduced more in HeLa-GFP cells treated with siFAK+CRISPR-GFP-LNPs compared to cells treated with siCtrl+CRISPR-GFP-LNPs (in which luciferase siRNA served as control) (Fig. 1d and e). T7 endonuclease 1 (T7E1) mutation detection assay results revealed that the frequency of indels in GFP DNA were higher in cells treated with siFAK+CRISPR-GFP-LNPs than in cells treated with siCtrl+CRISPR-GFP-LNPs (Fig. 1f). These data indicated that multiplexed LNPs successfully delivered all three types of nucleic acids to human cancer cell lines to achieve highly efficient gene silencing and genome editing. An intriguing phenomenon was also revealed where FAK knockdown significantly enhanced gene editing of GFP. The results were confirmed through administration of mCherry mRNA and luciferase mRNA in multiple tumor cell types (Supplemental Fig. 2a–f). The enhancement of mRNA delivery and protein expression was time- and siRNA-concentration-dependent irrespective of the ratio of loaded cargoes (Supplemental Fig. 2b, c, 3a–b). Based on the higher delivery efficacy of simultaneous co-delivery of siRNA and mRNA in vivo than sequential delivery using multiple LNPs (Supplemental Fig. 4–8), we utilized the simultaneous delivery approach for further studies. Next, to confirm that delivery enhancement was regulated by FAK knockdown, we inactivated FAK using a small molecule inhibitor in cells co-administered with siCtrl+mRNA-LNPs. Consistent with the siFAK co-delivery results, mRNA delivery efficacy and protein expression were also enhanced in cells treated with an FAK inhibitor drug (Supplemental Fig. 9). Furthermore, we delivered siFAK+CRISPR-LNPs to human ovarian cancer cells (IGROV1) specifically targeting PD-L1 (siFAK+CRISPR-PD-L1-LNPs) to examine the gene editing efficiency. Insertions and deletions (indels) at the PD-L1 locus were also significantly increased with FAK knockdown (Fig. 1g), leading to distinctly reduced PD-L1 protein expression (Fig. 1h). Thus, we concluded that suppression of FAK improves nanoparticle-based CRISPR gene editing efficacy in multiple tumor cell lines.
Next, we examined delivery in 3D multicellular spheroids that have been shown to recapitulate critical in vivo physiologic tumor parameters31. siFAK+CRISPR-GFP-LNPs were able to achieve genome editing in spheroids, where DNA cleavage was 7-fold higher than siCtrl+CRISPR-GFP-LNPs (Fig. 1i). We further delivered reporter mCherry mRNA and Cy5-labelled mRNA to examine both efficacy and spatial LNP delivery. siFAK+mRNA-LNPs penetrated throughout the entire spheroid within 4 hours as tracked by Cy5 fluorescence (Fig. 1j–k and Supplemental Fig. 10), which led to mCherry expression following mRNA translation throughout the entire spheroid (Fig. 1l and m). In contrast, siCtrl+mRNA-LNPs were unable to penetrate the center of the spheroids and only delivered mRNA to the periphery (Fig. 1j–m and Supplemental Fig. 11). These results suggested that FAK knockdown can overcome the physical barriers of tumor spheroids and distinctly increase RNA delivery.
To investigate the mechanism for how FAK knockdown enhances gene editing efficacy of siFAK+CRISPR-LNPs, we first quantified the cellular uptake of LNPs using confocal microscopy and flow cytometry. Comparing Cy5-labeled siCtrl+mRNA-LNPs and Cy5-labeled siFAK+mRNA-LNPs treated cells, higher cellular uptake was observed in the siFAK group (Fig. 2a–b). Next, we used various small molecules to inhibit distinct cellular uptake pathways. The results indicated that FAK silencing increased cellular uptake of LNPs mainly through modulating clathrin- and caveolae-dependent endocytosis pathways (Fig. 2c and Supplemental Fig. 12). Membrane invagination and endocytosis are the major steps for these two pathways, involving membrane tension regulated by tensile forces generated by actin filament (F-actin) and the actomyosin network. With this insight, we then examined dynamic changes of F-actin/stress fibers and the actomyosin network distribution (Fig. 2d–e and Supplemental Fig. 13). Compared with the strong stress F-actin and rich actomyosin network in cells treated with siCtrl+CRISPR-LNPs, both stress F-actin and actomyosin network were decreased, indicating that the contraction force of cells treated with siFAK+CRISPR-LNPs was distinctly decreased. The decreased contraction was also examined using a collagen-based contraction assay32 (Supplemental Fig. 14). Interestingly, in cells treated with siFAK+CRISPR-LNPs, the actomyosin network majorly accumulated at the peripheral area of cells with distinct decrease of F-actin stress fibers and alignment (Fig. 2d–e), which might induce membrane invagination, which can also decrease the membrane tension because membranes flatten at high tension and invaginate at low tension33, 34. We further stained the cell membrane and noticed that the membrane was deformed and endocytosed after administration of siFAK+CRISPR-LNPs, which was time-, and siFAK-concentration dependent (Fig. 2f–g and Supplemental Fig. 15a–b). In addition, decreased Yes-associated protein (YAP) expression and localization of YAP to the nucleus further showed that cellular contraction was decreased by FAK knockdown35 (Supplemental Fig. 16a–c). Combined with the endocytosis of cell membrane-protein integrin β1 in IGROV1 and HepG2 cells treated with siFAK+CRISPR-LNPs (Supplemental Fig. 17 and 18a–c), we concluded that siFAK+CRISPR-LNPs reduce the contraction force to enhance the cellular endocytosis and penetration of nanoparticles.
Fig. 2 ǀ. FAK-knockdown enhances the endocytosis of siFAK+CRISPR-LNPs through dynamic alteration of the contraction force and cell membrane tension.

a and b, Representative confocal images (a) and flow cytometry quantification (b) of time-dependent cellular uptake of siCtrl+cy5-mRNA-LNPs and siFAK+cy5-mRNA-LNPs in IGROV1 cells. LNPs (Cy5, Red), cytoskeleton (phalloidin-iFluor 488, Green), and nucleus (DAPI, Blue). Scale bar = 20 μm. n=3 biologically independent samples. Error bars represent mean ± s.d.. c, Quantification of the cellular uptake of LNPs when using small molecule inhibitors of various cellular uptake pathways. Cellular uptake was quantified through observing the Cy5-labled-mRNA-LNPs in cells. n=3 biologically independent samples. Error bars represent mean ± s.d.. ns, no significant difference, ***P < 0.001 determined by two-tailed t-test. The results were analyzed using Image J. d, Representative confocal images of IGROV1 cells treated with siCtrl+CRISPR-LNPs and siFAK+CRISPR-LNPs at different time points. F-actin (Red), P-myosin II (P-myo.II, Green), Nuclei (DAPI, Blue). Scale bar = 20 μm. e, Quantification of the distribution of F-actin and P-myo.II on the white arrow lines in the confocal images (d). f, Schematic illustration FAK-knockdown induced reduction of cellular contraction force and membrane tension under siFAK+CRISPR-LNPs treatment. g, Representative membrane invagination and endocytosis regulated by administration of siFAK+cy5-mRNA-LNPs. Scale bar = 10 μm. Membrane, yellow; Nuclei, blue; siFAK+Cy5-mRNA-LNPs, red.
To further demonstrate that decreasing mechanical properties of cells could enhance gene editing, we modulated the cell stiffness by controlling the substrate stiffness using different concentrations of matrigel matrix36 (~20 mg/mL, ~300 Pa; and ~10 mg/mL, ~100 Pa)37. We found that mRNA delivery and gene editing efficacy were significantly enhanced through decreasing the mechanical properties of the tumor tissue regulated by soft substrates through administration of reporter mRNA, siFAK+CRISPR-LNPs targeting luciferase in HeLa-Luc, and targeting GFP in HeLa-GFP tumor spheroids (Fig. 1i, Supplemental Fig. 19a–d). Notably, a >12-fold enhancement of CRISPR/Cas-mediated gene editing in the stiffer tumor spheroids was quantified following administration of siFAK+CRISPR-GFP-LNPs (Fig. 1i), indicating that FAK knockdown enhances gene editing in complex tumor models.
siFAK+CRISPR-PD-L1-LNPs inhibit xenograft tumor growth in vivo
FAK is overexpressed in several advanced-stage solid cancers, especially ovarian cancer38, which increases the contraction of tumor cells and stiffness of ECM39. Therefore, to evaluate gene editing and antitumor efficacy of siFAK+CRISPR-PD-L1-LNPs, C57BL/6 mice bearing ID8-Luc xenograft tumors were used to perform the following experiments through local administration of siFAK+CRISPR-PD-L1-LNPs and other controls (PBS, empty LNPs, siCtrl+CRISPR-PD-L1-LNPs, siFAK+CRISPR-Ctrl-LNPs) (Supplemental Fig. 20a). We proceeded to (i) examine LNP distribution and penetration; (ii) measure protein expression following translation of the delivered mRNA; (iii) investigate the ECM and the infiltration of immune cells in tumor microenvironment; (iv) and compare the tumor growth in mice after 30-day treatment. Deep LNP penetration and enhanced mRNA translation to protein expression in tumor tissues were observed for siFAK+Cy5-mRNA-LNPs and siFAK+mCherry mRNA-LNPs (Fig. 3a–b and Supplemental Fig. 20b). We studied changes in tumor stiffness through examining compressive modulus of tumor tissue, deposition and crosslinking of collagen I, and other proteins associated with cell mechanics (YAP, P-myosin II, and integrin β1) (Fig. 3c–d, and Supplemental Fig. 21–22). We found that tumor stiffness decreased in the tumor microenvironment of mice treated with siFAK+CRISPR-PD-L1-LNPs and siFAK+CRISPR-Ctrl-LNPs, leading to a less aggressive tumor phenotype evidenced by decreased P-myosin II, integrin β1, and collagen I fibers (Fig. 3c–d and Supplemental Fig. 20c, 21, and 22). Moreover, the infiltration of immune cells, especially T cells, in the tumor tissue were also increased after administration siFAK+CRISPR-PD-L1-LNPs (Supplemental Fig. 23). The combination of a reduction in ECM stiffness regulated by gene silencing of FAK, and enhanced gene editing of PD-L1 should inhibit tumor growth (Fig. 3e–f). The average tumor volume of mice treated with siFAK+CRISPR-PD-L1-LNPs was ~16 mm3 which was smaller than the tumor volume of mice treated with PBS (~200 mm3), empty LNPs (~180 mm3), siCtrl+CRISPR-PD-L1-LNPs (~90 mm3), and siFAK+CRISPR-Ctrl-LNPs (~65 mm3) (Fig. 3e–f). Meanwhile, there was no significant acute toxicity to mice following administration of siFAK+CRISPR-PD-L1-LNPs (Supplemental Fig. 24). Taken together, we concluded that regulating the ECM stiffness through FAK knockdown is beneficial for gene editing of PD-L1, which in turn significantly inhibits tumor growth. Furthermore, using a mouse metastasis model of ovarian cancer, we demonstrated that siFAK+CRISPR-PD-L1-dLNPs could decrease ovarian cancer metastasis due to the combination of decreasing cellular adhesion by FAK knockdown and improved the gene editing efficacy of PD-L1 (Supplemental Fig. 25a–e).
Fig. 3 ǀ. siFAK+CRISPR-PD-L1-LNPs targeted tumor stiffness and PD-L1 to inhibit xenograft tumor growth.
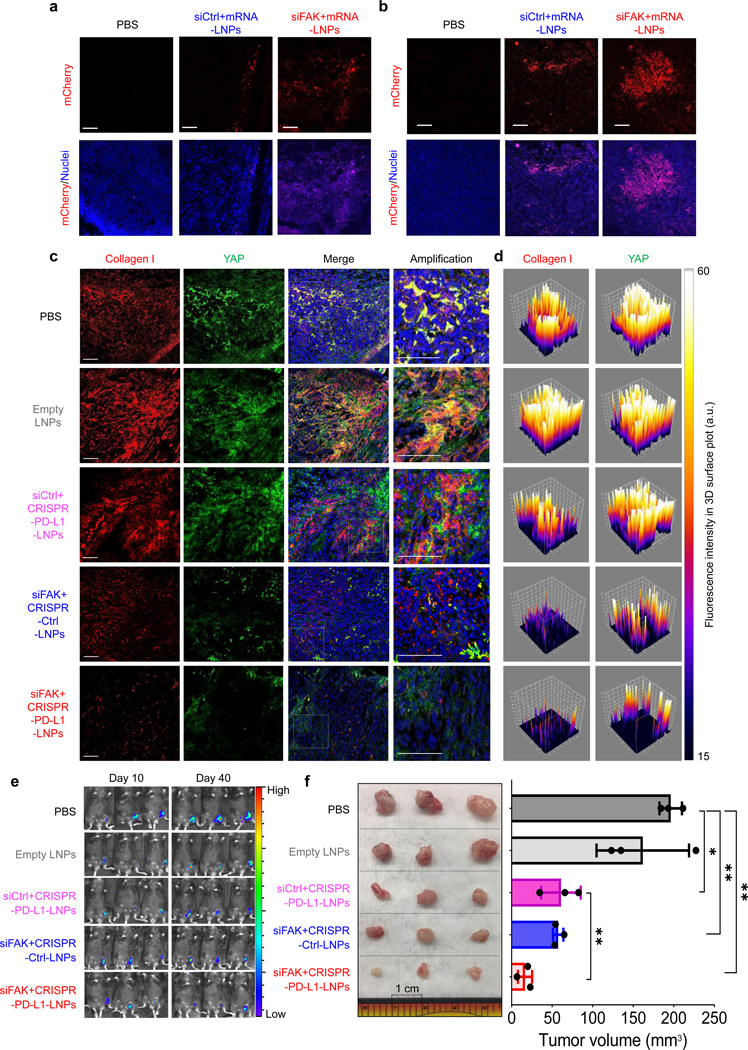
a and b, Representative confocal images of mCherry expression and distribution at the edge (a) and center (b) of fixed tumor tissues after local administration of PBS, siCtrl+mRNA-LNPs, and siFAK+mRNA-LNPs. Scale bar = 50 μm. Nuclei (blue), mCherry (red). c and d, Representative 3D construction of immunofluorescence (c) and 3D surface plot of quantification (d) of collagen I and YAP in fixed tumor tissues after 30-day therapy of mice by weekly local injection of PBS, empty LNPs, siCtrl+CRISPR-PD-L1-LNPs, siFAK+CRISPR-Ctrl-LNPs, and siFAK+CRISPR-PD-L1-LNPs. Collagen I (Red), YAP (Green), nuclei (Blue). Scale bar = 100 μm. e, Bioluminescence imaging of whole animals bearing ID8-Luc xenograft tumors treated with PBS, empty LNPs, siCtrl+CRISPR-PD-L1-LNPs, siFAK+CRISPR-Ctrl-LNPs, and siFAK+CRISPR-PD-L1-LNPs at day 40. n = 3 mice per group. f, Excised tumor (left) and tumor size (right) show the in vivo therapeutic efficacy of siFAK+CRISPR-PD-L1-LNPs and the other control groups. Tumor size measurement began on day 10 and continued every 3 days. n = 3 mice per group. Error bars represent the mean ± s.d., *P <0.05, **P <0.01, ***P <0.001, was determined by one-way ANOVA with multiple comparison test.
siFAK+CRISPR-LNPs enhance gene editing in a genetically engineered liver cancer model
To further evaluate the effects of combined FAK silencing and enhanced gene editing, we examined antitumor efficacy in aggressive, genetically engineered mouse model (GEMM) of liver cancer harboring a tetracycline (tet)-repressible human MYC transgene (tet-MYC)40. Upon removal of doxycycline (dox), rapid tumor growth leads to death within 60 days without treatment. Moreover, high levels of fibrosis with collagen deposition have been frequently detected in liver cancer models that involve MYC overexpression41, leading to stiffer tumors embedded in normal tissue which influences treatment responses and hinders nanoparticle uptake42, 43. Here, we followed the treatment regimen outlined in Fig. 4a to test whether enhancing the gene editing through reducing the tumor mechanics can improve therapeutic outcomes. Examining the livers at day 35, 45, and 55 (Fig. 4b–c and Supplemental Fig. 26a–c, 27a, and b), we found that siFAK+CRISPR-PD-L1-LNPs and siFAK+CRISPR-Ctrl-LNPs treated mice had markedly reduced levels of collagen fibrosis (Fig. 4b–c), indicating that reduction of FAK expression in cancer cells decreased tumor tissue stiffness, which was also confirmed by reduced YAP, P-myosin II, and integrin β1 in tumors (Fig 4b–c and Supplemental Fig. 27a–b). The high efficacy of mRNA delivery was further demonstrated using Cy5-labeled mCherry- and luciferase-mRNA (siFAK+mRNA-LNPs) (Supplemental Fig. 28a–b).
Fig. 4 ǀ. siFAK+CRISPR-LNPs enabled enhancement of gene editing though decreasing tumor stiffness in an aggressive, genetically engineered liver cancer model.
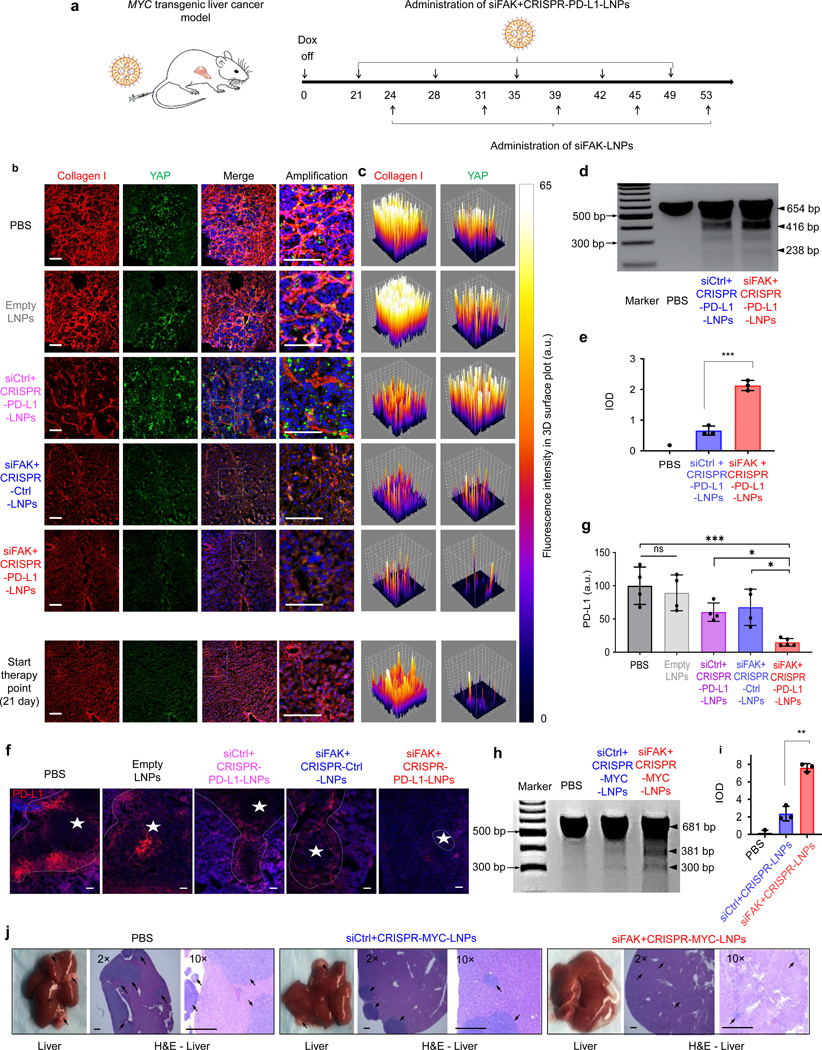
a, Schematic illustration of administration regimen for systemic therapy in the MYC-driven liver cancer mouse model. b and c, Representative 3D construction of immunofluorescence (b) and 3D surface plot of quantification (c) of collagen I and YAP in the tumor during the therapy process (day 45). Collagen I (Red), YAP (Green), nuclei (Blue). Scale bar = 100 μm. d and e, Representative T7E1 results (d) and quantification of IOD (e) for siFAK+CRISPR-PD-L1-LNPs-mediated cleavage enhancement of PD-L1 in the tumor compared with that of siCtrl+CRISPR-PD-L1-LNPs. n=3 biologically independent samples. Error bars represent mean ± s.d.. ***P <0.001 determined by two-tailed t-test. f, Representative PD-L1 expression in liver tissues scanned using confocal imaging and captured under two channels: nuclei (DAPI, blue), PD-L1 (red). Scale bar =100 μm. g, Quantification of PD-L1 expression using Image J. n=4 biologically independent samples. Error bars represent mean ± s.d.. *P <0.05, ***P <0.001 determined by were determined by one-way ANOVA with multiple comparison test. h and i, Representative T7E1 results (h) and quantification of IOD (i) for siFAK+CRISPR-MYC-LNPs-mediated cleavage enhancement of MYC in the tumor compared with that of siCtrl+CRISPR-MYC-LNPs with sgRNA targeting MYC. n=3 biologically independent samples. Error bars represent mean ± s.d.. **P <0.01 determined by two-tailed t-test. j, Representative liver images and H&E staining of liver sections of mice following systemic administration of PBS, siCtrl+CRISPR-MYC-LNPs, and siFAK+CRISPR-MYC-LNPs. 3 mg/kg total RNA administered; Cas9 mRNA (1.0 mg/kg): sgRNA (0.5 mg/kg): siRNA (1.5 mg/kg) = 2:1:3 (wt)s. 5A2-SC8: RNA = 10:1 (wt). The mice were injected starting on day 21. The liver was examined on day 31. Arrows showed the tumors in the liver. Scale bar = 500 μm.
We next investigated CRISPR/Cas-mediated gene editing in MYC mice following i.v. injection of PBS, siFAK+CRISPR-PD-L1-LNPs, and siCtrl+CRISPR-PD-L1-LNPs. The results showed that DNA cleavage efficacy was significantly enhanced by FAK knockdown, leading to a distinct decrease of PD-L1 expression in the tumor tissue (Fig. 4d–g). Significant enhancement of gene editing in vivo was also confirmed through examining gene editing efficacy of siFAK+CRISPR-MYC-LNPs specifically targeting MYC through quantifying indels and tumor growth inhibition in the liver (Fig. 4h–j).
Successful LNP delivery of siFAK, Cas9 mRNA, and sgPD-L1 leads to a reduction of PD-L1 expression and ECM stiffness, which will enhance anti-tumor immune response mediated by increase of immune cell infiltration (Fig. 5a). We examined the infiltration of CD8+ T cells44, and macrophages present in the tumor microenvironment45, 46, which are crucial to inhibit tumor growth. As expected, higher numbers of CD8+ T cells and macrophages infiltrated into the tumors of mice treated with siFAK+CRISPR-PD-L1-LNPs compared with mice treated with siCtrl+CRISPR-PD-L1-LNPs and siFAK+CRISPR-Ctrl-LNPs. There were few CD8+ T cells in the tumors of mice treated with PBS and empty LNPs at days 35 and 55 (Fig. 5b–d and Supplemental Fig. 29). This enhancement of CD8+ T cell and macrophage infiltration within the tumor could be representative of host immune reactions against cancer cell growth and were most significantly associated with a better survival45, 47.
Fig. 5 ǀ. Systemic administration of siFAK+CRISPR-PD-L1-LNPs significantly extended survival of mice bearing aggressive, MYC-driven cancer.

a, Proposed model for how decreasing ECM stiffness could enhance gene editing to thereby increase immune cells infiltration. b and c, Representative IHC for CD8+ T cells (b) and macrophage cells (c) infiltration in the tumor (day 55). Scale bar = 100 μm. d, Quantification of infiltration of macrophage cells and CD8+ T cells from confocal images. Error bars represent mean ± s.d. n=3 mice with 2 tissue sections per mouse. ns, no significant difference, **P <0.01, ***P <0.001, ****P <0.0001 determined by one-way ANOVA with multiple comparison test. e, and f, Representative whole body (e) and liver (f) images of mice treated with PBS, empty LNPs, siCtrl+CRISPR-PD-L1-LNPs, siFAK+CRISPR-Ctrl-LNPs, and siFAK+CRISPR-PD-L1-LNPs (55 days). g, H&E staining of liver sections of mice treated with PBS, empty LNPs, siCtrl+CRISPR-PD-L1-LNPs, siFAK+CRISPR-Ctrl-LNPs, and siFAK+CRISPR-PD-L1-LNPs at day 35 (14 days after treatment initiation). The arrows in the images indicate tumors. Scale bar = 500 μm. h and i, Abdominal circumference measurements (therapy began on day 21) (h) and survival curves (i) of mice show the in vivo therapeutic efficacy of siFAK+CRISPR-PD-L1-LNPs as compared to the other control groups (PBS, empty LNPs, siCtrl+CRISPR-PD-L1-LNPs, and siFAK+CRISPR-Ctrl-LNPs, (n = 6 mice each group). Error bars represent mean ± s.d. (h) 3 mg/kg total RNA injection; Cas9 mRNA (1.0 mg/kg): sgRNA (PD-L1, 0.5 mg/kg); siRNA (1.5 mg/kg) = 2:1:3 (wt). 5A2-SC8: RNA = 10:1 (wt).
siFAK+CRISPR-PD-L1-LNPs extend survival of mice bearing MYC-driven cancer
Building on these results, we further investigated tumor growth and survival of MYC mice following longer term treatments with PBS, empty LNPs, siCtrl+CRISPR-PD-L1-LNPs, siFAK+CRISPR-Ctrl-LNPs, and siFAK+CRISPR-PD-L1-LNPs with sgRNA specifically targeting PD-L1. We started a therapeutic regimen by weekly i.v. injection of siFAK+CRISPR-PD-L1-LNPs. Due to the short duration of siRNA-mediated gene silencing22, 48–50, we administered siFAK-LNPs one more time per week. At day 55, the abdomens of mice that received siFAK+CRISPR-PD-L1-LNPs were similar in circumference to normal, wild type mice and much smaller than abdomens of MYC mice treated with PBS (Fig. 5e), indicating that tumor growth was suppressed. Decreased tumor growth in mice treated with siFAK+CRISPR-PD-L1-LNPs was also confirmed through analysis of liver photographs and H&E staining (Fig. 5f–g and Supplemental Fig. 30a–b). Liver tumor growth in mice treated with siFAK+CRISPR-Ctrl-LNPs and siCtrl+CRISPR-PD-L1-LNPs was also slower than that of mice treated with PBS and empty LNPs at day 35. Among all groups, mice treated with siFAK+CRISPR-PD-L1-LNPs clearly had the smallest tumors at day 55 (Supplemental Fig. 30b), demonstrating that FAK silencing to improve gene editing enhanced overall cancer therapy.
The curve of abdominal circumference of mice treated with only gene editing by siCtrl+CRISPR-PD-L1-LNPs showed a plateau from day 21 to 45 (Fig. 5h), suggesting that gene editing of PD-L1 had therapeutic efficacy at the early time points when tumors were smaller and LNP uptake and penetration were less hindered by ECM (low collagen I deposition at 35 day) (Supplemental Fig. 26b–c). After 45 days, the abdominal circumference curve of mice treated with siCtrl+CRISPR-PD-L1-LNPs showed steep increase. Considering that collagen I deposition and crosslinking was significantly increased in tumors at 45 and 55 days (Fig. 4b–c and Supplemental Fig. 26a–c), we hypothesized that high ECM stiffness in the tumor acted as a barrier to siCtrl+CRISPR-PD-L1-LNP uptake and penetration. After 45 days, gene editing therapeutic efficacy was distinctly decreased. The abdominal circumference curve rose slowly in the groups treated by siFAK+CRISPR-Ctrl-LNPs and siFAK+CRISPR-PD-L1-LNPs, indicating that FAK knockdown inhibited tumor growth. In turn, the enhancement of LNP delivery due to FAK silencing reduced the ECM and thus increasing gene editing of tumor cells mediated by CRISPR further increasing the therapeutic efficacy (Fig. 5e–g). Therefore, administration of siFAK+CRISPR-PD-L1-LNPs significantly extended the survival of mice (>100 days) in comparison to the PBS treated group (60 days) (Fig. 5i). There was a significant increase in survival compared to mice treated with siCtrl+CRISPR-PD-L1-LNPs (~67 days), which is approximately the effect measured previously for delivery of Let-7g22 and siRNA against Anilin48. Enhanced tumor therapy through gene editing increased by FAK knockdown was also demonstrated using small molecule inhibitors of FAK and CRISPR-LNPs targeting KRAS (a RAS human oncogene51) in A549 cells in vitro and in tumor xenografts (Supplemental Fig. 31a–h). This general multiplexed approach may therefore be applicable to a variety of oncogenic targets in the future. In addition, in vitro and in vivo safety studies revealed no off-target editing and high tolerability (Supplemental Fig. 32a–d and 33a–c). Overall, we concluded that LNPs inhibited the rapid tumor growth and enhanced gene editing of PD-L1 to improve immunotherapeutic efficacy in multiple difficult-to-treat cancer models.
Conclusion
Development of effective cancer therapeutics requires precise targeting of specific genes via approaches that can simultaneously overcome cancer hallmarks, including a dense tumor microenvironment11. Although CRISPR offers a new approach for accurate therapeutic intervention, including permanent inactivation of PD-L1, this approach is currently hindered by the physically dense tumor microenvironment that blocks nanoparticle uptake and immune cell infiltration. We overcame this challenge by developing multiplexed LNPs designed to deliver siFAK to break down the ECM, Cas9 mRNA to deploy Cas protein, and targeted sgRNA to knockout cancer targets. We found that FAK inhibition decreased the contractile force and membrane tension properties of tumor cells and ECM stiffness, which significantly enhanced CRISPR gene editing in tumor cells in vitro and in vivo by increasing LNP endocytosis and tumor penetration. This approach aided access to enough cells to overcome replicative potential and enhanced overall gene editing >10-fold. siFAK+CRISPR-PD-L1-LNP therapy was tested in four mouse models of human cancer, showing that this approach can inhibit metastasis and tumor growth. Administration of siFAK+CRISPR-PD-L1-LNPs reduce PD-L1 expression and ECM stiffness, which will increase immune cell infiltration for immunotherapy to significantly extend survival of mice bearing MYC-driven HCC. We envision that this multiplexed, generalizable strategy can be used for a variety of cancer targets in various cancer types, offering a new approach for treating cancer using CRISPR gene editing.
Supplementary Material
Acknowledgments
D.J.S. acknowledges support from the Cancer Prevention and Research Institute of Texas (CPRIT) (RP190251), the National Institutes of Health (NIH) (R01 EB025192-01A1, R01 CA269787-01), American Cancer Society (ACS) (RSG-17-012-01), and the Cystic Fibrosis Foundation (CFF) (SIEGWA18XX0). H.Z. acknowledges support from the NIH (R01 DK111588, R01 DK125396, R01 CA251928), the Moody Medical Research Institute, and an Emerging Leader Award from The Mark Foundation for Cancer Research (#21-003-ELA). We also acknowledge the UTSW Tissue Resource (National Cancer Institute (5P30CA142543)) and the Moody Foundation Flow Cytometry Facility. T.W. acknowledges a CPRIT Training Grant (RP160157). L.T.J. acknowledges the Pharma Foundation. We are also very grateful to Professor Zong Sheng Guo (University of Pittsburgh) for sharing ID8-Luc cells with us.
Footnotes
Competing interests
D.J.S. is a co-founder and consultant of ReCode Therapeutics, which has licensed intellectual property from UT Southwestern. H.Z. has a sponsored research agreement with Alnylam Pharmaceuticals, consults for Flagship Pioneering, and serves on the SAB of Ubiquitix. H.Z.’s interests are not directly related to the contents of this paper.
Methods
Please see Supplementary Information for full methods.
Data Availability.
All data that support the plots within this paper and other findings of this study are shown in the figures and available from the corresponding author upon reasonable request.
References
- 1.Wang HX et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: Challenges and opportunities for nonviral delivery. Chem. Rev 117, 9874–9906 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Jinek M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mali P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xue W. et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin H. et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 32, 551–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maddalo D. et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long CZ et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller JB et al. Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. 56, 1059–1063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei T. et al. Delivery of tissue-targeted scalpels: Opportunities and challenges for in vivo CRISPR/Cas-based genome editing. ACS Nano 14, 9243–9262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang CH, Lee KC & Doudna JA Applications of CRISPR-Cas enzymes in cancer therapeutics and detection. Trends Cancer 4, 499–512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D. & Weinberg RA Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Cox DB, Platt RJ & Zhang F. Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang H. et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 22, 851–860 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohammadi H. & Sahai E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 20, 766–774 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Humphrey JD, Dufresne ER & Schwartz MA Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lampi MC & Reinhart-King CA Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci. Trans. Med. 10, eaao0475 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Seong J, Wang N. & Wang Y. Mechanotransduction at focal adhesions: from physiology to cancer development. J. Cell. Mol. Med. 17, 597–604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serrels A. et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 163, 160–173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casey SC et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Topalian SL, Drake CG & Pardoll DM Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 24, 207–212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou K. et al. Modular degradable dendrimers enable small RNAs to extend survival in an aggressive liver cancer model. Proc. Natl. Acad. Sci. USA 113, 520–525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng Q. et al. Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater. 30, e1805308 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Cheng Q. et al. Selective ORgan Targeting (SORT) nanoparticles for tissue specific mRNA delivery and CRISPR/Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ball RL, Hajj KA, Vizelman J, Bajaj P. & Whitehead KA Lipid nanoparticle formulations for enhanced co-delivery of siRNA and mRNA. Nano Lett. 18, 3814–3822 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Patel S. et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 11, 983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abumanhal-Masarweh H. et al. Tailoring the lipid composition of nanoparticles modulates their cellular uptake and affects the viability of triple negative breast cancer cells. J. Control. Release 307, 331–341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei T, Cheng Q, Min Y-L, Olson EN & Siegwart DJ Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 11, 3232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S. et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat. Mater. 20, 701–710 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SM et al. A systematic study of unsaturation in lipid nanoparticles leads to improved mRNA transfection in vivo. Angew. Chem. Int. Ed. 60, 5848–5853 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta G, Hsiao AY, Ingram M, Luker GD & Takayama S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control. Release 164, 192–204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laklai H. et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 22, 497–505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaksonen M. & Roux A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 19, 313–326 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Echarri A. & Del Pozo MA Caveolae–mechanosensitive membrane invaginations linked to actin filaments. J. Cell Sci. 128, 2747–2758 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Dupont S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Kraning-Rush CM, Califano JP & Reinhart-King CA Cellular traction stresses increase with increasing metastatic potential. Plos One 7, e32572 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaudhuri O. et al. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 13, 970–978 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Hoadley KA et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stokes JB et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 10, 2135–2145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shachaf CM et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431, 1112–1117 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Zheng K, Cubero FJ & Nevzorova YA c-MYC making liver sick: Role of c-MYC in hepatic cell function, homeostasis and disease. Genes 8, 123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egeblad M, Rasch MG & Weaver VM Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 22, 697–706 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paszek MJ et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Fourcade J. et al. CD8+ T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 72, 887–896 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schreiber RD, Old LJ & Smyth MJ Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Sica A. & Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naito Y. et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 58, 3491–3494 (1998). [PubMed] [Google Scholar]
- 48.Zhang S. et al. Knockdown of anillin actin binding protein blocks cytokinesis in hepatocytes and reduces liver tumor development in mice without affecting regeneration. Gastroenterology 154, 1421–1434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller JB & Siegwart DJ Design of synthetic materials for intracellular delivery of RNAs: From siRNA-mediated gene silencing to CRISPR/Cas gene editing. Nano Res. 11, 5310–5337 (2018). [Google Scholar]
- 50.Wu SY, Lopez-Berestein G, Calin GA & Sood AK RNAi therapies: Drugging the undruggable. Sci. Trans. Med. 6, 240–247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cox AD & Der CJ Ras history: The saga continues. Small GTPases 1, 2–27 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the plots within this paper and other findings of this study are shown in the figures and available from the corresponding author upon reasonable request.