

Abstract
Limb-girdle muscular dystrophy type R25 (LGMDR25) is caused by recessive mutations in BVES encoding a cAMP-binding protein, characterized by progressive muscular dystrophy with deteriorating muscle function and impaired cardiac conduction in patients. There is currently no therapeutic treatment for LGMDR25 patients. Here we report the efficacy and safety of recombinant adeno-associated virus 9 (AAV9)-mediated systemic delivery of human BVES driven by a muscle-specific promoter MHCK7 (AAV9.BVES) in BVES-knockout (BVES-KO) mice. AAV9.BVES efficiently transduced the cardiac and skeletal muscle tissues when intraperitoneally injected into neonatal BVES-KO mice. AAV9.BVES dramatically improved body weight gain, muscle mass, muscle strength, and exercise performance in BVES-KO mice regardless of sex. AAV9.BVES also significantly ameliorated the histopathological features of muscular dystrophy. The heart rate reduction was also normalized in BVES-KO mice under exercise-induced stress following systemic AAV9.BVES delivery. Moreover, intravenous AAV9.BVES administration into adult BVES-KO mice after the disease onset also resulted in substantial improvement in body weight, muscle mass, muscle contractility, and stress-induced heart rhythm abnormality. No obvious toxicity was detected. Taken together, these results provide the proof-of-concept evidence to support the AAV9.BVES gene therapy for LGMDR25.
Keywords: AAV, adeno-associated virus, BVES, cardiac arrhythmia, gene therapy, LGMDR25, limb-girdle muscular dystrophy, POPDC1
Graphical abstract
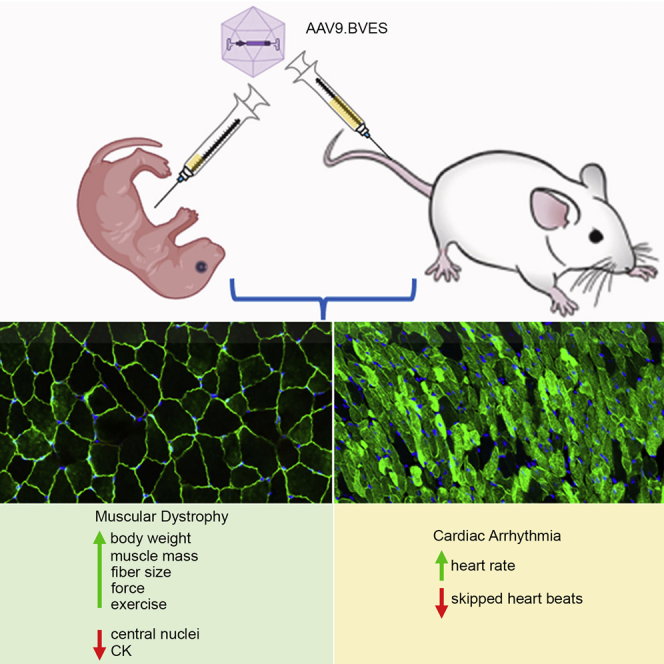
Systemic administration of AAV9.BVES at the neonatal or adult stage improves body weight gain, muscle mass, muscle strength, exercise performance, and histopathological features of muscular dystrophy without obvious toxicity in a mouse model of LGMDR25. The heart rate reduction is also normalized in BVES-KO mice following systemic AAV9.BVES delivery.
Introduction
Limb-girdle muscular dystrophies (LGMDs) are a highly heterogeneous group of muscular disorders that are characterized by progressive muscle weakness and wasting in legs and arms with some affected by heart dysfunction. Genetic defects in the blood vessel epicardial substance (BVES) gene (also known as Popeye domain containing protein 1 or POPDC1) were recently identified to cause autosomal recessive limb-girdle muscular dystrophy type X or R25 (LGMD2X or LGMDR25).1 LGMDR25 patients showed elevated serum creatine kinase (CK) and progressive proximal limb muscle weakness and atrophy2 with some eventually becoming wheelchair bound.1 Heart rhythm abnormalities including arrhythmia,3,4 atrioventricular (AV) block,3,4 and sinus bradycardia5 have also been recorded in LGMDR25 patients. The precise prevalence of LGMDR25 is unknown, but an increasing number of patients carrying BVES mutations have recently been identified.2,4,6,7 To date, there are no treatments available for LGMDR25 patients.
BVES encodes a cAMP-binding transmembrane protein, belonging to the Popeye domain containing (POPDC) protein family, which contains two other members, POPDC2 and POPDC3.8 All POPDC proteins are highly expressed in striated muscles.9 Interestingly, patients carrying mutations in BVES often display a loss of membrane localization of POPDC2,2 a risk protein of cardiac arrhythmia.10 BVES has been implicated in various cellular processes, including protein trafficking,3 myoblast differentiation,11 cell migration,12 cell proliferation,13 and signaling cascades.14 Consistent with patient studies, BVES-deficient zebrafish displayed muscular dystrophy as well as AV block in heart,3 and BVES ablated mice displayed the defect of muscle regeneration upon cardiotoxin (CTX) challenge in skeletal muscle and stress-induced bradycardia.5,15 We recently established a BVES-knockout (BVES-KO) mouse model with all coding exons deleted, which developed muscular dystrophy and atrophy characterized by decreased fiber size and increased central nucleation (H.L. and R.H., unpublished data). This model faithfully recapitulated the pathological features of LGMDR25 patients, supporting its use for preclinical translational studies.
AAV-mediated gene therapy has been widely exploited for various genetic diseases, including muscular dystrophies,16 due to the broad tissue tropism and safety in humans.15,16 In particular, AAV gene therapy has recently received Food and Drug Administration approval for hereditary retinal dystrophy (Luxturna)17 and spinal muscular atrophy (Zolgensma).18 AAV9 has been shown to be highly efficient at transducing major tissues, including the liver, heart, skeletal muscle, and central nervous system.19,20 Here, we explored the feasibility to treat LGMDR25 by AAV9-mediated systemic delivery of a BVES transgene using BVES-KO mice. Our results showed that both neonatal and adult administration of AAV9.BVES can dramatically improve the function and pathology of BVES-KO mice, without notable serious adverse events. This study highlights the great promise of AAV9.BVES gene therapy for LGMDR25.
Results
AAV9.BVES transduces skeletal muscle and heart efficiently in BVES-KO mice
To generate AAV9.BVES, we constructed an AAV transfer plasmid by placing the human BVES cDNA fused with 3x HA tag under the control of a skeletal and cardiac muscle-specific promoter MHCK721 (Figure 1A). We first performed a dosage escalating study with intraperitoneal injection of AAV9.BVES (from 1E13 to 1E14 vg/kg) into postnatal day 3 (P3) BVES-KO pups. Immunofluorescence examination showed a dose-dependent transduction in skeletal muscle and heart when killed at 1 month of age, with almost complete transduction in skeletal muscle and ∼70% in cardiomyocytes upon 1E14 vg/kg AAV9.BVES administration (Figure 1B). Western blot analysis confirmed the dose-dependent increase of BVES transgene expression in skeletal muscle with both anti-HA and anti-BVES antibodies (Figures 1C and 1D). Because a substantial amount of cardiomyocytes were not transduced at the highest dose (1E14 vg/kg), we further increased the dose of AAV9.BVES to 2E14 vg/kg in our following experiments.
Figure 1.

Dose-dependent AAV9.BVES transduction in skeletal muscle and heart of BVES-KO mice
(A) Schematic diagram of AAV9.BVES vector. (B) Immunofluorescence staining with anti-HA antibody of gastrocnemius muscle and heart cross-sections derived from 1-month-old BVES-KO mice treated with different doses of AAV9.BVES. Scale bar, 100 μm. (C and D) Western blot analysis of gastrocnemius muscle from 1-month-old BVES-KO mice treated with different doses of AAV9.BVES. Gastrocnemius muscles from 1-month-old WT and untreated BVES-KO mice were used as controls.
To test if early intervention with AAV9.BVES can prevent the disease development in BVES-KO mice, we administered AAV9.BVES (2E14 vg/kg) into P3 BVES-KO pups (22 male and eight female) through intraperitoneal injection. One injected mouse was killed at 3 months of age to verify the expression of the BVES transgene by immunofluorescence staining with the anti-HA antibody. As shown in Figures 2A and S1, BVES was mainly expressed in various skeletal muscle fibers including gastrocnemius (GA), quadriceps (QU), tibial anterior (TA), diaphragm (DIA), and soleus (SOL). It was also highly expressed in the heart muscles (Figure S2). There was no positive staining in the BVES-KO mouse tissues without AAV9.BVES injection, confirming the specificity of the anti-HA antibody (Figures 2A and S2). BVES was hardly detectable in the other major organs, such as liver, lung, kidney, and colon (Figure S2), suggesting that the combination of AAV9 delivery and the muscle-specific MHCK7 promoter is highly efficient in restricting the expression of BVES transgene in striated muscle fibers. A few cells in the liver tissue showed positive staining, likely due to leaky expression from the MHCK7 promoter as reported before.21 The tissue distribution pattern of the BVES transgene is similar to that of the endogenous BVES (Figure S3). Both endogenous and transgenic BVES were mainly observed at the sarcolemma of skeletal muscle fibers (Figures 2A and S4). Some intercellular staining was also evident, particularly in cardiomyocytes (Figure S2 and S4).
Figure 2.

Human BVES was specifically and efficiently expressed in the striated muscles following intraperitoneal injection of AAV9.BVES in neonatal male BVES-KO mice
(A) BVES was widely expressed in various skeletal muscles of BVES-KO mice following AAV9.BVES administration. Scale bar, 100 μm. (B–D) Western blot showed BVES was highly expressed in GA muscle (B) and heart (C) in BVES-KO mice with neonatal injection of AAV9.BVES. Arrows indicate the specific bands of BVES. (D) Densitometry quantification of the western blot data. ∗∗p < 0.01; ∗∗∗p < 0.001 (unpaired, two-tailed Student’s t test). Data are mean ± SEM
Western blot analysis further confirmed the transgene expression in skeletal muscle (Figure 2B) and heart (Figure 2C) of BVES-KO mice treated with AAV9.BVES when killed at 9 months of age. Endogenous BVES was detected as three major bands in skeletal muscle and two major bands in heart samples, consistent with previous reports that BVES may exist as monomer, dimer, and glycosylated forms.11,22,23 The BVES antibody also detected some non-specific bands as they appeared in both wild-type (WT) and BVES-KO samples. The BVES transgene was strongly expressed in both the skeletal muscle and heart of BVES-KO mice treated with AAV9.BVES as detected by the antibodies against BVES or the HA tag (Figures 2B and 2C). Densitometry analysis showed that the expression of the BVES transgene was expressed at ∼20-fold of WT levels in both GA and heart muscles of BVES-KO mice treated with AAV9.BVES (Figure 2D).
Neonatal delivery of AAV9.BVES normalizes muscle function and exercise performance in BVES-KO mice
To examine whether neonatal administration of AAV9.BVES can prevent the disease development, the BVES-KO mice treated with or without AAV9.BVES at P3 were monitored monthly for body weight gain. As compared with age-matched WT males, the male BVES-KO mice showed significantly reduced body weight gain, particularly after 3 months of age (Figure 3A). AAV9.BVES greatly improved the body weight gain in BVES-KO mice, and there was no significant difference in body weight between the treated BVES-KO and WT mice at 8 months of age (30.0 ± 1.5 g versus 30.1 ± 1.8 g, p = 1.0). When killed at 9 months of age, the AAV9.BVES-treated BVES-KO mice were not visually different from the WT mice, whereas the untreated BVES-KO mice looked much smaller than the WT controls (Figure 3B). Similarly, AAV9.BVES-treated female BVES-KO mice also showed significant improvement in body weight gain, similar to their WT counterparts (Figures S5A and S5B).
Figure 3.

Systemic neonatal administration of AAV9.BVES normalized muscle function and mass in male BVES-KO mice
(A) Monthly body weight measurements of WT and BVES-KO mice with or without AAV9.BVES. (B) Representative photographs of WT and BVES-KO mice with or without AAV9.BVES at 9 months of age. (C) Tetanic torque measurements of the posterior compartment muscles of WT and BVES-KO mice with or without AAV9.BVES at 5 months of age. (D and E) Running time to exhaustion (D) and total running distance (E) of WT and BVES-KO mice with or without AAV9.BVES at 6 months of age on a 15° uphill treadmill. ∗∗p < 0.01; ∗∗∗∗p < 0.0001; ns, not significant. (F) Representative images of dissected skeletal muscles from male WT and BVES-KO mice with or without AAV9.BVES. (G and H) The net mass of GA (G) and QU (H) in 9-month-old male mice with the designated genotype. (I and J) The relative mass of GA (I) or QU (J) muscle normalized to the body weight in 9-month-old male mice with the designated genotype. ∗∗p < 0.01; ∗∗∗∗p < 0.0001; ns, not significant (one-way ANOVA with Turkey’s multiple comparisons test). Data are mean ± SEM.
To test if AAV9.BVES treatment improves muscle function, we first measured the muscle contractility using an in vivo muscle test system. Maximum plantarflexion tetanic torque was measured during supramaximal electric stimulation of the tibial nerve at 150 Hz. As compared with age-matched WT mice, the BVES-KO mice at 5 months of age produced significantly reduced torque while AAV9.BVES treatment fully normalized the contractile strength in the KO mice (Figure 3C). Similar improvement in the contractile strength was also detected in AAV9.BVES-treated female KO mice (Figure S5C). Next, we subjected the mice to uphill (15°) treadmill running until exhaustion. Both the time to exhaustion (Figure 3D) and the total running distance (Figure 3E) were significantly decreased in male BVES-KO mice as compared with WT; however, AAV9.BVES treatment fully normalized both parameters in male BVES-KO mice. Similarly, AAV9.BVES significantly improved exercise performance in female BVES-KO mice (Figures S5D and S5E). These studies showed that neonatal administration of AAV9.BVES completely restored body weight gain, contractile strength, and exercise performance in BVES-KO mice. Consistent with the reduced body weight, we found that the muscle mass was dramatically reduced in GA and QU muscles of BVES-KO mice, which was significantly improved by AAV9.BVES treatment (Figures 3F–3J).
Adult administration of AAV9.BVES also significantly improves muscle function and exercise performance in BVES-KO mice
To further study the translational potential of AAV9.BVES gene therapy, we examined whether systemic administration of AAV9.BVES (2E14 vg/kg) into adult BVES-KO mice after disease onset can show beneficial effects on muscle function and exercise performance. As our study revealed that BVES-KO mice displayed the retarded body weight gain starting at around 3 months of age (see Figure 3), we treated the 4-month-old BVES-KO mice with AAV9.BVES through tail vein injection and measured the body weight, force contractility, exercise performance, and muscle mass as described above. Western blot analysis confirmed the transgene expression in skeletal muscle and heart of BVES-KO mice treated with AAV9.BVES when killed at 9 months of age (Figures S6A–S6C). While the untreated BVES-KO mice failed to gain weight, AAV9.BVES-treated BVES-KO animals showed a steady increase in their body weight within 18 weeks after treatment, although still lighter than their age- and gender-matched WT controls (Figure 4A). At 6 weeks after AAV9.BVES treatment, we started to observe significant differences in the body weight between treated and untreated BVES-KO mice (Figure 4A). The AAV9.BVES-treated BVES-KO mice were visually larger than the untreated BVES-KO animals (Figure 4B). At 2 months after AAV9.BVES treatment, the contractile strength was significantly increased as compared with the untreated controls (Figure 4C). Moreover, AAV9.BVES treatment in BVES-KO mice restored their exercise performance to the WT levels (Figures 4D and 4E). At 9 months of age, we killed the mice to study the impact of AAV9.BVES treatment on muscle mass. As shown in Figures 4F–4J, AAV9.BVES treatment significantly increased the mass of GA and QU muscles.
Figure 4.

Systemic adult administration of AAV9.BVES improved muscle function and mass in male BVES-KO mice
(A) Biweekly body weight measurements of WT and BVES-KO mice with or without AAV9.BVES. (B) Representative photographs of WT and BVES-KO mice with or without AAV9.BVES at 9 months of age. (C) Tetanic torque measurements of the posterior compartment muscles of WT and BVES-KO mice with or without AAV9.BVES at 6 months of age. (D and E) Running time to exhaustion (D) and total running distance (E) of WT and BVES-KO mice with or without AAV9.BVES at 6 months of age on a 15° uphill treadmill. (F) Representative images of dissected skeletal muscles from male WT and BVES-KO mice with or without AAV9.BVES. (G and H) The net mass of GA (G) and QU (H) in 9-month-old male mice with the designated genotype. (I, J) The relative mass of GA (I) or QU (J) muscle normalized to the body weight in 9-month-old male mice with the designated genotype. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns, not significant (one-way ANOVA with Turkey’s multiple comparisons test). Data are mean ± SEM.
Together, these results showed that AAV9.BVES gene transfer into adult BVES-KO mice can also significantly improve muscle function, exercise performance, and muscle mass even after disease onset.
AAV9.BVES gene therapy improves muscle pathology
Like the patients with LGMDR25, BVES-KO mice exhibited muscular dystrophy and atrophy as evidenced by elevated serum CK levels, significant fiber size variability, centrally nucleated muscle fibers (CNFs), and smaller muscle mass. Either adult or neonatal administration of single-dose AAV9.BVES significantly normalized the serum CK levels (Figure S7). H&E staining showed that a considerable number of CNFs and necrotic fibers were present in BVES-KO GA muscles and that systemic AAV9.BVES administration at either neonatal or adult stage ameliorated these pathological alterations in the BVES-KO mice (Figure 5A). To quantify the muscle size and CNF percentages, we performed immunofluorescence staining with antibodies against dystrophin, myosin heavy chain 2B (MyHC-IIB), and DAPI (Figure 5B). The type IIB fibers had a higher percentage of CNFs than the non-IIB fibers (11.9% ± 2.9% versus 7.5% ± 2.9%) in male KO GA muscles at 9 months of age (Figures 5C and S8A). Either neonatal or adult administration of AAV9.BVES significantly reduced the percentage of CNFs (3.0% ± 1.0% and 7.4% ± 1.3% in MyHC-IIB+ fibers, respectively) (Figure 5C) and increased the percentage of MyHC-IIB+ muscle fibers with larger area (Figures 5D and 5E), with neonatal injection showing higher improvement. The relative number of MyHC-IIB+ muscle fibers within the GA muscles were not significantly different among the WT, KO, and treatment groups (Figure S8B). Examination of other major skeletal muscles showed similar improvement (Figures S9 and S10A–S10C). Interestingly, the QU and TA muscles had higher percentage of CNFs as compared with SOL and DIA in BVES-KO mice (Figures S9 and S10), consistent with our observation that MyHC-IIB+ fibers were more severely affected. Neonatal treatment with AAV9.BVES dramatically reduced the percentage of CNFs in BVES-KO skeletal muscles, while adult administration also showed a similar trend of improvement (Figure S10B). The AAV9.BVES treatment also increased muscle fiber size in QU and TA muscles of BVES-KO mice (Figure S10C).
Figure 5.

Systemic AAV9.BVES gene delivery improved the pathohistological defects in male BVES-KO mice
(A) H&E staining of GA muscle sections in WT and BVES-KO mice with or without AAV9.BVES at 9 months of age. Scale bar, 100 μm. (B) Immunostaining of MyHC-IIB and dystrophin (Dys) in GA muscles from WT and BVES-KO mice with or without AAV9.BVES. Scale bar, 100 μm. (C) Quantification of CNFs in MyHC-IIB muscle fibers from GA muscles of WT and BVES-KO mice with or without AAV9.BVES at 9 months of age. (D) The fiber size distribution of MyHC-IIB muscle fibers from GA muscles from WT and BVES-KO mice with or without AAV9.BVES at 9 months of age. (E) Mean CSA of MyHC-IIB muscle fibers from GA muscles from WT and BVES-KO mice with or without AAV9.BVES at 9 months of age. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001; ns, not significant (two-way ANOVA with Turkey’s post-tests). Data are mean ± SEM.
AAV9.BVES gene transfer improves heart rhythm
Previous studies showed that patients with LGMDR25 presented arrythmia, AV block, and sinus bradycardia.2,3,4,7 Similarly, the BVES-KO mice displayed lower heart rate and severe repetitive drops of the heart rate during and after swimming stress.5 To examine whether AAV9.BVES gene transfer can correct the heart rate defect, we monitored male and female mice by electrocardiogram (ECG). The P wave and QRST wave were not significantly different among the mice in different groups except that the RR interval was increased in the BVES-KO mice (Tables S1, S2, and S3). At the rest condition, 10 of 21 untreated male BVES-KO mice showed delayed or skipped beats, whereas the frequency of ECG abnormality was greatly reduced in AAV9.BVES-treated groups (neonatal, two out of 10; adult, zero out of seven) (Figures 6A–6C and Table S4). Under the rest condition, the average heart rate of untreated male BVES-KO mice was significantly lower than that of WT (Figures 6B and 6C). We then subjected the animals to an episode of 10-min uphill treadmill running, immediately followed by ECG recording. Skipped heart beats were again observed primarily in untreated BVES-KO mice (Figure 6D and Table S4). The mice in all groups showed an increase in heart rate following the treadmill running with the BVES-KO mice still displaying reduced heart rate as compared with the WT animals (Figures 6E and 6F). AAV9.BVES treatment at either neonatal or adult stage normalized the heart rate of BVES-KO mice in response to exercise-induced stress (Figures 6E and 6F). AAV9.BVES showed similar benefits for improving ECG in female BVES-KO mice where abnormal heart beats were observed in nine out of 15 mice (Figures S11A–S11D). Moreover, we measured the cardiac function by echocardiography. We found no significant differences in ejection fraction or other parameters among the animals at 9 months of age, regardless of genotype or treatment (Figure S12 and Table S5).
Figure 6.

Systemic AAV9.BVES gene delivery normalized ECG abnormalities in male BVES-KO mice
(A and D) Representative ECG data and quantification of skipped heart beats of the male WT and BVES-KO mice with or without neonatal or adult administration of AAV9.BVES before (A) and after (D) treadmill running. (B and E) Heart rate (before or after exercise) of the male WT and BVES-KO mice with or without neonatal administration of AAV9.BVES. (C and F) Heart rate (before or after exercise) of the male WT and BVES-KO mice with or without adult administration of AAV9.BVES. ∗p < 0.05, ∗∗p < 0.01; ns, not significant (one-way ANOVA with Turkey’s multiple comparisons test). Data are mean ± SEM.
AAV9.BVES has minimal toxicity in BVES-KO mice
To examine the safety profile of AAV9.BVES, we measured the serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) for liver toxicity, and blood urea nitrogen (BUN) for kidney toxicity. The BVES-KO mice showed elevated AST (Figure 7A) and normal ALT (Figure 7B) levels as compared with the WT animals, which was not unexpected, as increases in these enzymes were frequently observed in muscular dystrophy patients and animals.19 Consistent with the observation that AAV.BVES treatment improved the muscular dystrophy phenotype in BVES-KO mice, AAV9.BVES treatment also normalized the serum levels of AST (Figure 7A). Measurement of BUN did not show significant changes in the treated or untreated BVES-KO mice (Figure 7C). H&E staining showed no detectable alterations in livers of BVES-KO mice treated with or without AAV9.BVES (Figure 7D). These results indicate that systemic delivery of AAV9.BVES did not induce obvious liver or kidney toxicity in BVES-KO mice.
Figure 7.

The safety profile of AAV9.BVES therapy in male BVES-KO mice
(A–C) Measurements of serum AST (A), ALT (B), and BUN (C) of WT and BVES-KO mice with or without neonatal or adult administration of AAV9.BVES at 9 months of age. ∗p < 0.05; ∗∗p < 0.01; ∗∗p < 0.001; ns, not significant (two-way ANOVA with Turkey’s post-tests). Data are mean ± SEM. (D) H&E staining of liver sections from WT and BVES-KO mice with or without neonatal or adult administration of AAV9.BVES at 9 months of age. Scale bar, 100 μm.
Discussion
In this study, we evaluated the efficacy and safety of recombinant AAV carrying the human BVES transgene as a promising gene therapy for LGMDR25. We have used the AAV9 serotype and muscle-specific promoter to better target skeletal and cardiac muscles. Our data showed that systemic administration of a single dose of AAV9.BVES before or after disease onset ameliorated the muscular dystrophy and heart rhythm abnormality without overt adverse effects in BVES-KO mice. These findings provided a foundation for translating AAV-mediated BVES gene transfer to LGMDR25 patients.
Our recent study characterizing the phenotype of BVES-KO mice showed that this mouse model accurately recapitulates the clinical pathological features of LGMDR25 (H.L. and R.H., unpublished data), supporting its use for preclinical translational studies. Like LGMDR25 patients, the BVES-KO mice developed muscular dystrophy, muscular atrophy, and cardiac conduction defect. Thus, targeting both the skeletal and cardiac muscles must be considered for the long-term treatment of this disease. The use of AAV9 and the skeletal and cardiac muscle-specific promoter MHCK7 allows for enhanced transgene expression in skeletal and cardiac muscles with little off-target tissue expression following systemic delivery. We demonstrated that systemic delivery of AAV9.BVES leads to nearly complete transduction and restoration of BVES expression in various limb skeletal muscles, diaphragm, and cardiac muscle with little detectable expression in liver, lung, kidney, or colon. This was accompanied by a remarkable improvement in body weight gain, muscle mass, muscle contractility, exercise performance, muscle histopathology, and ECG regularity. Of note, the neonatal intervention appeared to be more effective, as evidenced by higher body weight, lower CNFs, and better force production and exercise capacity, when compared with treatment at a later stage of the disease in BVES-KO mice. Similar observations were found in AAV9-FKRP gene therapy for LGMD2I.24 Our observations showed that the type IIB fibers were more severely affected in BVES-KO mice. Although myosin heavy chain isoform IIX, rather than IIB, is expressed in human skeletal muscle, type IIX and IIB are very similar in their contractile properties and oxidative capacity.25 It is plausible to assume that type IIX fibers in LGMDR25 patients are likely more severely affected. Future examination of patient biopsies will be needed to confirm this, and the mechanisms underlying the fiber type-associated impact of BVES deficiency will be explored.
We observed a high transgene expression with the MHCK7 promoter. The expression of BVES protein following AAV9.BVES delivery was estimated to be around 20-fold of WT levels, consistent with previous reports showing the strong activity of the MHCK7 promoter.21 This high level of BVES expression did not show any signs of toxicity during the 9 months of study. This contrasts with previous studies of some other LGMD genes, such as dysferlin, calpain3, and caveolin-3, for which the overexpression can cause overt pathology in skeletal muscle and/or heart.26,27,28,29,30,31,32 Although no overt toxicity associated with high-level BVES overexpression was observed within the 9-month study, future studies will be needed to explore the longer-term effects. It also would be interesting to test a weaker skeletal and cardiac muscle-specific promoter in the future to further minimize the potential overexpression-related side effects. Moreover, recently engineered AAV capsids with improved muscle and heart transduction efficiency, such as the AAVMYO33 and MyoAAV,34 may further lower the required dose and thus improve the safety profile for BVES gene therapy.
Gene replacement therapy has been increasingly used to treat various forms of muscular dystrophies, including Duchenne muscular dystrophy (DMD) and LGMDs.16 Several preclinical studies showed a very promising perspective, such as rAAV6-microdystrophin for DMD,35 rAAVrh74.MHCK7.hSGCA for LGMD2D,36 scAAV.MHCK7.hSGCB for LGMD2E,37 AAV8-desm-hSGCG for LGMD2C,38 and AAV9-FKRP for LGMD2I,24,39 and some of these have moved into clinical trials, including SGT-001(AAV-microdystrophin), SRP-9003 (rAAVrh74.MHCK7.hSGCB), and SRP9004 (rAAVrh74.MHCK7.hSGCA) (https://clinicaltrials.gov/). Our study places LGMDR25 onto the growing list of AAV gene therapies. So far, nine pathogenic mutations in BVES have been reported in LGMDR25 patients, including S201F,3 R88Ter,4,40 Del56 V217-K272,4 R143Ter,2 S263Ter,6 M1G,4 Q153Ter,7 I193S,7 and P134L.41 In addition, the decreased expression of BVES caused by R129W mutation in human has been linked to the Sporadic Tetralogy of Fallot,42,43 the most common type of congenital heart disease. The AAV9.BVES gene therapy may offer a mutation-independent approach for the treatment of the diseases caused by BVES mutations.
Materials and methods
Mice
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of The Ohio State University. C57BL/6N mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The BVES-KO mice (C57BL/6N-Bvestm1.1(KOMP)Vlcg/MbpMmucd) were obtained from Mutant Mouse Resource & Research Centers, UC Davis, and maintained in our barrier facility. In BVES-KO mice, all coding exons were deleted via the Cre-mediated recombination. Identification of the mutant mice was performed by PCR genotyping of genomic DNA prepared from ear clips with the following primers: KO-F, 5′-ACTTGCTTTAAAAAACCTCCCACA, KO-R, 5′-AGTCACTAGCAAGAGATCTGCACCC; WT-F, 5′-AAGTGCTGGGATTAAAGGTGTGTGC; WT-R, 5′-AAGGACACATCACAGCTTCAGG. The WT and KO allele would produce a 164-base pair (bp) and 771-bp band, respectively.
Systemic administration of AAV9.BVES in BVES-KO mice
AAV9.BVES viral particles were produced, purified and tittered at Andelyn Biosciences (Columbus, OH, USA) as previously described.19 Titers are expressed as DNase-resistant particles per milliliter (DRP/mL) and AAV9 titers used for the in vivo studies were 4 × 1013 DRP/mL. The male or female BVES-KO mice at postnatal day 3 were administered with AAV9.BVES viral particles (∼2 × 1014 vg/kg) via intraperitoneal injection. For adult administration, the male BVES-KO mice at 4 months of age were injected with AAV9.BVES viral particles (2 × 1014 vg/kg) via tail vein.
Treadmill running
Total time and distance of running to exhaustion were performed as previously described.44 Briefly, the randomized mice were first acclimated to the treadmill (LE8710MTS; Harvard Apparatus) for 2 consecutive days at low speed (10 cm/s) for 5 min. At the third day, mice were placed on the uphill (15°) treadmill with the following parameters: initial speed 10 cm/s, increased every 4 min by 5 cm/s. Mice were considered to be exhausted when the animal’s hindlimbs remained on the electric grid for more than 10 s. Time and distance were automatically collected via the software SeDaCOM (Harvard Apparatus).
Force measurement
The muscle contractility was measured as previously described19 using an in vivo muscle test system equipped with software DMC LabBook (Aurora Scientific Inc). Mice were anesthetized with 3% (w/v) isoflurane and anesthesia was maintained by 2% isoflurane (w/v) during muscle contractility measurement. The tetanic torques from the posterior compartment muscles (including gastrocnemius, soleus, and plantaris muscles) of mice were measured by electric stimulation of the sciatic nerve.
ECG recording
The ECG was measured using IX-BIO4 (iWorx Systems). C57BL/6J and BVES-KO mice with or without AAV9.BVES were anesthetized with 2% (w/v) isoflurane, and ECG was recorded for about 50 s under anesthesia before and immediately after 10-min treadmill running (5° uphill). The data were analyzed using the software Labscribe (iWorx Systems). Briefly, all skipped heart beats (defined as a prolongation of the RR interval ≥1.5 times the previous RR interval) were manually counted during the entire recording session.
Echocardiography recording
C57BL/6J and BVES-KO mice with or without AAV9.BVES were anesthetized with 1% to 2% isoflurane, and echocardiography was measured using a Vevo 2100 Ultrasound system. Echocardiography data were analyzed using VevoLab software to determine the left ventricle ejection fraction.
Immunofluorescence staining
Cryosections (10 μm) of mouse tissues were prepared using Cryostat 1905 (Leica Biosystems, Deer Park, IL). The slides were fixed with 4% paraformaldehyde for 10 min at room temperature. After washing with PBS three times (5 min each), the slides were blocked with 5% BSA plus 5% normal serum for 1 h. The slides were then incubated with primary antibodies against HA (1:1000, #3724, Cell Signaling Technology, Danvers, MA), Dystrophin (E2660, 1:500, Spring Bioscience, Pleasanton, CA), rabbit polyclonal anti-BVES (1:100, HPA014788, Sigma-Aldrich, St. Louis, MO), or MyHC-IIB (1:50, 10F5, deposited to the Developmental Studies Hybridoma Bank by Lucas, C.) at 4°C overnight. Following extensive wash with PBS, the slides were incubated with the corresponding secondary antibodies Alexa Fluor 488 goat anti-rabbit immunoglobulin (Ig)G (A-11034, 1:400; Invitrogen, Carlsbad, CA), Alexa Fluor 568 donkey anti-rabbit IgG (A-10042, 1:400; Invitrogen), or Alexa Fluor 594 goat anti-mouse IgM (A-21044, 1:400; Invitrogen) for 1 h at room temperature. Finally, the slides were sealed with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratory, Burlingame, CA). The images were taken with a Zeiss 780 confocal microscope (Carl Zeiss Microscopy, White Plains, NY) or Nikon Ti-E fluorescence microscope, magnification ×200 (Nikon, Melville, NY). Fiber size and CNF quantification were carried out using Myosight45 with manual calibration on the cross-sections of various skeletal muscles.
H&E staining
Ten microns of frozen muscle and liver sections were fixed in 10% formaldehyde for 5 min at room temperature and then proceeded to the standard protocol of H&E staining. All images were taken under a Nikon Ti-E fluorescence microscope, magnification ×200.
Western blot
The mouse tissues were lysed with cold RIPA buffer supplemented with protease inhibitors (S8830; Sigma-Aldrich) as well as PhosSTOP (PHOSS-RO; Roche, Basel, Switzerland), and the extracted proteins were quantified by DC Protein Assay Reagent (BioRad, Hercules, CA). The extracted protein samples were separated by stain-free SDS-PAGE gels (4%–15%; BioRad) and transferred onto nitrocellulose membranes (0.45 μm). Primary antibodies used include the rabbit polyclonal anti-BVES (1:1,000, HPA014788; Sigma-Aldrich), anti-HA (1:1,000, #3724; Cell Signaling Technology, Danvers, MA) and anti-GAPDH (1:2,000, MAB374; Cell Signaling Technology). Secondary horseradish peroxidase-conjugated goat anti-mouse (1:4,000) and goat anti-rabbit (1:4,000) antibodies were obtained from Cell Signaling Technology. The membranes were developed using ECL western blot substrate (Pierce Biotechnology, Rockford, IL) and scanned by ChemiDoc XRS + system (BioRad). Western blots were quantified using ImageJ.
Serological analysis
Serum samples collected from the mice were stored at −80°C for the biochemical assays. Measurements of CK (326-10; Sekisui Diagnostics), AST (ab105135; Abcam), ALT (ab105134; Abcam) and BUN (EIABUN; Thermo Fisher) were performed according to the manufacturer’s protocols.
Data analysis and statistics
The data were expressed as mean ± SEM, analyzed with GraphPad Prism 8.0.1 software (San Diego, CA, USA) and final figures were assembled with Adobe Photoshop 2020. Statistical differences were determined by two-tailed unpaired Student’s t test for two groups and one-way or two-way ANOVA with Turkey’s post-tests for multiple group comparisons using Prism 8.0.1 with the assumption of Gaussian distribution of residuals. A p < 0.05 was considered to be significant.
Acknowledgments
The authors thank all lab members for their constructive comments and suggestions. R.H. is supported by US National Institutes of Health grants (R01HL116546, R01HL159900, R01AR070752), a Parent Project Muscular Dystrophy award, and Miao family gift. R.H. has received commercial research support from Stealth Biotherapeutics. K.I.S. is supported by R01-HL138738, and K.M.P is supported by F31-HL152648-01A1.
Author contributions
R.H. conceived the study and wrote the manuscript. H.L. performed the experiments and participated in drafting the manuscript. E.H. assisted in tissue collection, and K.P. assisted in echocardiography measurements. All authors contributed to the final version of the manuscript.
Declaration of interests
R.H. and H.L. have submitted a patent application based on the results reported in this manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.11.012.
Supplemental information
References
- 1.Taghizadeh E., Rezaee M., Barreto G.E., Sahebkar A. Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: a review. J. Cell. Physiol. 2019;234:7874–7884. doi: 10.1002/jcp.27907. [DOI] [PubMed] [Google Scholar]
- 2.Beecher G., Tang C., Liewluck T. Severe adolescent-onset limb-girdle muscular dystrophy due to a novel homozygous nonsense BVES variant. J. Neurol. Sci. 2021;420:117259. doi: 10.1016/j.jns.2020.117259. [DOI] [PubMed] [Google Scholar]
- 3.Schindler R.F.R., Scotton C., Zhang J., Passarelli C., Ortiz-Bonnin B., Simrick S., Schwerte T., Poon K.L., Fang M., Rinné S., et al. POPDC1(S201F) causes muscular dystrophy and arrhythmia by affecting protein trafficking. J. Clin. Invest. 2016;126:239–253. doi: 10.1172/JCI79562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Ridder W., Nelson I., Asselbergh B., De Paepe B., Beuvin M., Ben Yaou R., Masson C., Boland A., Deleuze J.F., Maisonobe T., et al. Muscular dystrophy with arrhythmia caused by loss-of-function mutations in BVES. Neurol. Genet. 2019;5:e321. doi: 10.1212/NXG.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Froese A., Breher S.S., Waldeyer C., Schindler R.F.R., Nikolaev V.O., Rinné S., Wischmeyer E., Schlueter J., Becher J., Simrick S., et al. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Invest. 2012;122:1119–1130. doi: 10.1172/JCI59410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Indrawati L.A., Iida A., Tanaka Y., Honma Y., Mizoguchi K., Yamaguchi T., Ikawa M., Hayashi S., Noguchi S., Nishino I. Two Japanese LGMDR25 patients with a biallelic recurrent nonsense variant of BVES. Neuromuscul. Disord. 2020;30:674–679. doi: 10.1016/j.nmd.2020.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Gangfuß A., Hentschel A., Heil L., Gonzalez M., Schönecker A., Depienne C., Nishimura A., Zengeler D., Kohlschmidt N., Sickmann A., et al. Proteomic and morphological insights and clinical presentation of two young patients with novel mutations of BVES (POPDC1) Mol. Genet. Metab. 2022;136:226–237. doi: 10.1016/j.ymgme.2022.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Amunjela J.N., Swan A.H., Brand T. The role of the popeye domain containing gene family in organ homeostasis. Cells. 2019;8:1594. doi: 10.3390/cells8121594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrée B., Hillemann T., Kessler-Icekson G., Schmitt-John T., Jockusch H., Arnold H.H., Brand T. Isolation and characterization of the novel popeye gene family expressed in skeletal muscle and heart. Dev. Biol. 2000;223:371–382. doi: 10.1006/dbio.2000.9751. [DOI] [PubMed] [Google Scholar]
- 10.Rinné S., Ortiz-Bonnin B., Stallmeyer B., Kiper A.K., Fortmüller L., Schindler R.F.R., Herbort-Brand U., Kabir N.S., Dittmann S., Friedrich C., et al. POPDC2 a novel susceptibility gene for conduction disorders. J. Mol. Cell. Cardiol. 2020;145:74–83. doi: 10.1016/j.yjmcc.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Li H., Xu L., Gao Y., Zuo Y., Yang Z., Zhao L., Chen Z., Guo S., Han R. BVES is a novel interactor of ANO5 and regulates myoblast differentiation. Cell Biosci. 2021;11:222. doi: 10.1186/s13578-021-00735-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith T.K., Hager H.A., Francis R., Kilkenny D.M., Lo C.W., Bader D.M. Bves directly interacts with GEFT, and controls cell shape and movement through regulation of Rac1/Cdc42 activity. Proc. Natl. Acad. Sci. USA. 2008;105:8298–8303. doi: 10.1073/pnas.0802345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amunjela J.N., Tucker S.J. Dysregulation of POPDC1 promotes breast cancer cell migration and proliferation. Biosci. Rep. 2017;37 doi: 10.1042/BSR20171039. BSR20171039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams C.S., Zhang B., Smith J.J., Jayagopal A., Barrett C.W., Pino C., Russ P., Presley S.H., Peng D., Rosenblatt D.O., et al. BVES regulates EMT in human corneal and colon cancer cells and is silenced via promoter methylation in human colorectal carcinoma. J. Clin. Invest. 2011;121:4056–4069. doi: 10.1172/JCI44228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin A.Y., Wang L.H. Molecular therapies for muscular dystrophies. Curr. Treat. Options Neurol. 2018;20:27. doi: 10.1007/s11940-018-0509-2. [DOI] [PubMed] [Google Scholar]
- 16.Crudele J.M., Chamberlain J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019;28:R102–R107. doi: 10.1093/hmg/ddz128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Day J.W., Finkel R.S., Chiriboga C.A., Connolly A.M., Crawford T.O., Darras B.T., Iannaccone S.T., Kuntz N.L., Peña L.D.M., Shieh P.B., et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:284–293. doi: 10.1016/S1474-4422(21)00001-6. [DOI] [PubMed] [Google Scholar]
- 19.Xu L., Zhang C., Li H., Wang P., Gao Y., Mokadam N.A., Ma J., Arnold W.D., Han R. Efficient precise in vivo base editing in adult dystrophic mice. Nat. Commun. 2021;12:3719. doi: 10.1038/s41467-021-23996-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 21.Salva M.Z., Himeda C.L., Tai P.W., Nishiuchi E., Gregorevic P., Allen J.M., Finn E.E., Nguyen Q.G., Blankinship M.J., Meuse L., et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 22.Kawaguchi M., Hager H.A., Wada A., Koyama T., Chang M.S., Bader D.M. Identification of a novel intracellular interaction domain essential for Bves function. PLoS One. 2008;3:e2261. doi: 10.1371/journal.pone.0002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knight R.F., Bader D.M., Backstrom J.R. Membrane topology of Bves/Pop1A, a cell adhesion molecule that displays dynamic changes in cellular distribution during development. J. Biol. Chem. 2003;278:32872–32879. doi: 10.1074/jbc.M301961200. [DOI] [PubMed] [Google Scholar]
- 24.Vannoy C.H., Xiao W., Lu P., Xiao X., Lu Q.L. Efficacy of gene therapy is dependent on disease progression in dystrophic mice with mutations in the FKRP gene. Mol. Ther. Methods Clin. Dev. 2017;5:31–42. doi: 10.1016/j.omtm.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori K. Maintenance of skeletal muscle to counteract sarcopenia in patients with advanced chronic kidney disease and especially those undergoing hemodialysis. Nutrients. 2021;13:1538. doi: 10.3390/nu13051538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glover L.E., Newton K., Krishnan G., Bronson R., Boyle A., Krivickas L.S., Brown R.H., Jr. Dysferlin overexpression in skeletal muscle produces a progressive myopathy. Ann. Neurol. 2010;67:384–393. doi: 10.1002/ana.21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han R., Frett E.M., Levy J.R., Rader E.P., Lueck J.D., Bansal D., Moore S.A., Ng R., Beltrán-Valero de Bernabé D., Faulkner J.A., Campbell K.P. Genetic ablation of complement C3 attenuates muscle pathology in dysferlin-deficient mice. J. Clin. Invest. 2010;120:4366–4374. doi: 10.1172/JCI42390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z., Fang Q., Du T., Chen G., Wang Y., Wang D.W. Cardiac-specific caveolin-3 overexpression prevents post-myocardial infarction ventricular arrhythmias by inhibiting ryanodine receptor-2 hyperphosphorylation. Cardiology. 2020;145:136–147. doi: 10.1159/000505316. [DOI] [PubMed] [Google Scholar]
- 29.Galbiati F., Volonte D., Chu J.B., Li M., Fine S.W., Fu M., Bermudez J., Pedemonte M., Weidenheim K.M., Pestell R.G., et al. Transgenic overexpression of caveolin-3 in skeletal muscle fibers induces a Duchenne-like muscular dystrophy phenotype. Proc. Natl. Acad. Sci. USA. 2000;97:9689–9694. doi: 10.1073/pnas.160249097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aravamudan B., Volonte D., Ramani R., Gursoy E., Lisanti M.P., London B., Galbiati F. Transgenic overexpression of caveolin-3 in the heart induces a cardiomyopathic phenotype. Hum. Mol. Genet. 2003;12:2777–2788. doi: 10.1093/hmg/ddg313. [DOI] [PubMed] [Google Scholar]
- 31.Roudaut C., Le Roy F., Suel L., Poupiot J., Charton K., Bartoli M., Richard I. Restriction of calpain3 expression to the skeletal muscle prevents cardiac toxicity and corrects pathology in a murine model of limb-girdle muscular dystrophy. Circulation. 2013;128:1094–1104. doi: 10.1161/CIRCULATIONAHA.113.001340. [DOI] [PubMed] [Google Scholar]
- 32.Lostal W., Roudaut C., Faivre M., Charton K., Suel L., Bourg N., Best H., Smith J.E., Gohlke J., Corre G., et al. Titin splicing regulates cardiotoxicity associated with calpain 3 gene therapy for limb-girdle muscular dystrophy type 2A. Sci. Transl. Med. 2019;11:eaat6072. doi: 10.1126/scitranslmed.aat6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinmann J., Weis S., Sippel J., Tulalamba W., Remes A., El Andari J., Herrmann A.K., Pham Q.H., Borowski C., Hille S., et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020;11:5432. doi: 10.1038/s41467-020-19230-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tabebordbar M., Lagerborg K.A., Stanton A., King E.M., Ye S., Tellez L., Krunnfusz A., Tavakoli S., Widrick J.J., Messemer K.A., et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell. 2021;184:4919–4938.e22. doi: 10.1016/j.cell.2021.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gregorevic P., Allen J.M., Minami E., Blankinship M.J., Haraguchi M., Meuse L., Finn E., Adams M.E., Froehner S.C., Murry C.E., Chamberlain J.S. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffin D.A., Pozsgai E.R., Heller K.N., Potter R.A., Peterson E.L., Rodino-Klapac L.R. Preclinical systemic delivery of adeno-associated alpha-sarcoglycan gene transfer for limb-girdle muscular dystrophy. Hum. Gene Ther. 2021;32:390–404. doi: 10.1089/hum.2019.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pozsgai E.R., Griffin D.A., Heller K.N., Mendell J.R., Rodino-Klapac L.R. Systemic AAV-mediated beta-sarcoglycan delivery targeting cardiac and skeletal muscle ameliorates histological and functional deficits in LGMD2E mice. Mol. Ther. 2017;25:855–869. doi: 10.1016/j.ymthe.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Israeli D., Cosette J., Corre G., Amor F., Poupiot J., Stockholm D., Montus M., Gjata B., Richard I. An AAV-SGCG dose-response study in a gamma-sarcoglycanopathy mouse model in the context of mechanical stress. Mol. Ther. Methods Clin. Dev. 2019;13:494–502. doi: 10.1016/j.omtm.2019.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vannoy C.H., Leroy V., Lu Q.L. Dose-dependent effects of FKRP gene-replacement therapy on functional rescue and longevity in dystrophic mice. Mol. Ther. Methods Clin. Dev. 2018;11:106–120. doi: 10.1016/j.omtm.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng N., Gao M., Jiao K., Yue D., Xu Y., Zhao C., Lu J., Zhu W. Early respiratory muscle involvement in LGMDR25: a case report. Neuromuscul. Disord. 2022;32:692–696. doi: 10.1016/j.nmd.2022.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Mahmood A., Samad A., Shah A.A., Wadood A., Alkathiri A., Alshehri M.A., Alam M.Z., Hussain T., He P., Umair M. A novel biallelic variant in the popeye domain-containing protein 1 (POPDC1) underlies limb girdle muscle dystrophy type 25. Clin. Genet. 2022 doi: 10.1111/cge.14238. [DOI] [PubMed] [Google Scholar]
- 42.Shi Y., Li Y., Wang Y., Zhuang J., Wang H., Hu M., Mo X., Yue S., Chen Y., Fan X., et al. The functional polymorphism R129W in the BVES gene is associated with sporadic tetralogy of fallot in the han Chinese population. Genet. Test. Mol. Biomarkers. 2019;23:601–609. doi: 10.1089/gtmb.2019.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y., Li Y., Wang Y., Zhu P., Chen Y., Wang H., Yue S., Xia X., Chen J., Jiang Z., et al. BVES downregulation in non-syndromic tetralogy of fallot is associated with ventricular outflow tract stenosis. Sci. Rep. 2020;10:14167. doi: 10.1038/s41598-020-70806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knudsen N.H., Stanya K.J., Hyde A.L., Chalom M.M., Alexander R.K., Liou Y.H., Starost K.A., Gangl M.R., Jacobi D., Liu S., et al. Interleukin-13 drives metabolic conditioning of muscle to endurance exercise. Science. 2020;368:eaat3987. doi: 10.1126/science.aat3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Babcock L.W., Hanna A.D., Agha N.H., Hamilton S.L. MyoSight-semi-automated image analysis of skeletal muscle cross sections. Skelet. Muscle. 2020;10:33. doi: 10.1186/s13395-020-00250-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.