

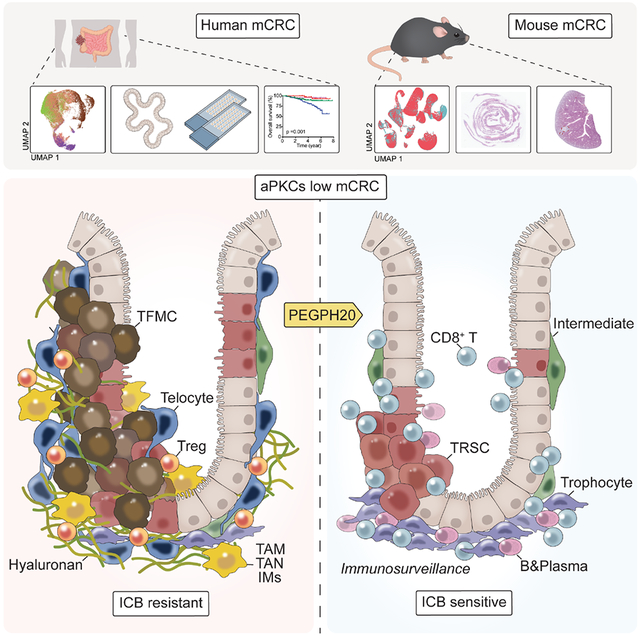
SUMMARY
Mesenchymal colorectal cancer (mCRC) is microsatellite stable (MSS), highly desmoplastic, with CD8+ T cells excluded to the stromal periphery, resistant to immunotherapy, and is driven by low levels of the atypical PKCs (aPKCs) in the intestinal epithelium. We show here that a salient feature of these tumors is the accumulation of hyaluronan (HA), which along with reduced aPKC levels, predict poor survival. HA promotes epithelial heterogeneity and the emergence of a tumor fetal metaplastic cell population (TFMC) endowed with invasive cancer features through a network of interactions with activated fibroblasts. TFMCs are sensitive to HA deposition, and their metaplastic markers have prognostic value. We demonstrate that in vivo HA degradation with a clinical dose of hyaluronidase impairs mCRC tumorigenesis and liver metastasis and enables immune checkpoint blockade therapy by promoting the recruitment of B and CD8+ T cells, including a proportion with resident memory features, and blocking immunosuppression.
Graphical Abstract
eTOC
Mesenchymal colorectal cancer (mCRC) is a poor prognosis, highly desmoplastic, and immune-excluded tumor. Martinez-Ordoñez et al. demonstrate that the synthesis of hyaluronan driven by a deficiency in the atypical PKCs in epithelial cells is a critical event in mCRC by promoting stroma crosstalks with invasive fetal metaplastic cells and immunosuppression.
INTRODUCTION
Colorectal cancer (CRC) is a major cause of cancer mortality worldwide.1 Despite improvements in systemic treatment, the metastatic disease shows a dismal 5-year survival rate of only 12%-14%.2 CRCs can develop from any region of the colon or the rectum. However, it has become increasingly clear that primary tumor locations show distinct pathophysiological and clinical characteristics.3,4 Thus, in the distal (left-sided) colon, tumors typically initiate from tubular adenomas, whereas in the proximal (right-sided) colon, tumors initiate from sessile serrated lesions (SSL). These SSL-derived proximal CRCs can evolve through a transcriptional pathway termed consensus molecular subtype (CMS) 1, are often associated with BRAF mutations, are characterized by microsatellite instability (MSI-H) with a CpG island methylator phenotype, are immune active, present with a good prognosis, and are relatively sensitive to immune checkpoint blockade therapy (ICB).5,6 In contrast, distal CRCs are microsatellite stable (MSS), often chromosomal instable, belonging to the CMS2/3 transcriptional category, are initiated by mutations in APC, TP53, KRAS, and the TGFβ pathway, have an intermediate prognosis, and account for about 50% of all CRCs.7,8 The transcriptional classification of CRC tumors also revealed a CMS4-enriched subtype, which accounts for about 35% of all CRCs and displays the highest risk of distant relapse and the worst prognosis.9,10 CMS4 tumors could evolve from SSL or tubular adenomas, are MSS, and resistant to ICB therapy.11 In contrast to CMS2/3 tumors, which are immunologically deserted, CMS4 tumors are immune excluded with CD8+ T cells accumulating in the stromal periphery and absent in the cancer epithelial core.12-14
Other distinctive features of the CMS4-enriched tumors are their high content in activated cancer-associated fibroblasts (CAFs) and the acquisition of a mesenchymal phenotype that predicts adverse outcomes in CRC patients better than the presence of prevalent mutations.7,9,11 This agrees with the increasingly accepted notion that an activated desmoplastic stroma drives the resistance of CRC cells to conventional and targeted therapies.15 However, the molecular and cellular mechanisms that drive mesenchymal tumorigenesis are still far from clear. Given the relatively scarce information on the fundamental players controlling the development of this type of intestinal tumor, it is critical to find upstream regulators of these pathways beyond the known mutated tumor suppressors and oncogenes already identified in cancers from CMS2/3 distal tubular lesions or CMS1 proximal SSL. In this regard, we have recently reported that low levels of the atypical PKCs (aPKCs; PKCζ and PKCλ/ι) are drivers for the initiation and progression of mesenchymal CRC (mCRC), as well as for their resistance to ICB.13,16 Thus, the simultaneous inactivation of both aPKCs in the mouse intestinal epithelium results in the spontaneous generation of aggressive MSS mesenchymal intestinal tumors with serrated and signet ring carcinoma features and a reactive desmoplastic and immunosuppressive tumor microenvironment (TME).13,15 aPKC-deficient tumors excluded CD8+ T cells to the stromal periphery but were infiltrated with myeloid-derived suppressor cells (MDSCs) expressing PD-L1 but were resistant to anti-PD-L1 treatment.13
Because mCRC tumors are populated by CAFs and enriched in a TGFβ signaling gene expression signature, several studies explored the possibility of treating these tumors by inhibiting TGFβ function. Surprisingly, this approach is not sufficient to reduce the tumor load or to reactivate the immune system, although it decreases the desmoplastic reaction and enables response to anti-PD-L1 therapy.13,17,18 Therefore, it could be argued that targeting CAFs, while inefficient as monotherapy, at least at the TGFβ level, might be a potential venue to promote immunotherapy in mCRC. However, the design of therapeutics aimed at selectively de-reprograming CAFs is extremely challenging due to the heterogeneity of the stromal fibroblasts in several cancers, including CRC, and a limited understanding of the master regulators of the different CAF subtypes.
The difficulty of targeting CAFs impelled the search for alternative ways to modulate the tumor stroma. Thus, several laboratories have focused their interest on targeting not the cellular components but different molecules of the TME extracellular matrix, like collagens and the glycosaminoglycan hyaluronan (HA).19-22 The rationale behind this approach is that reducing the extracellular matrix barrier purportedly protecting the tumor would result in better access to chemotherapeutics and ICB treatments. However, genetically targeting collagen I, the major fibrillar collagen type, in αSMA+ pancreatic and intestinal CAFs does not reduce but enhances tumor growth by either decreasing the barrier effect, which allows the tumor to expand free of its stromal boundaries, or by promoting the recruitment of MDSCs, which stimulates a TME conducive to immunosuppression and enhanced tumorigenesis.23,24 Therefore, a better understanding of the mesenchymal TME ecosystem will unveil the mechanistic cross-talks that define their malignancy and resistance to immunotherapy, leading to the identification of vulnerabilities to be exploited therapeutically.
Here we report a detailed characterization of the epithelial compartment and microenvironment of mCRC and its response to HA-depleting treatment.
RESULTS
Reduced expression of both aPKCs in human CRC correlates with a mesenchymal phenotype
Gene Set Enrichment Analysis (GSEA) of the CRC dataset in The Cancer Genome Atlas (TCGA) demonstrated that the transcriptome of patients with low aPKC levels (PRKCIlowPRKCZlow) was enriched in transcripts corresponding to the mesenchymal phenotype, including an activated stroma and CAF signatures, as compared to patients with high aPKC levels (PRKCIhighPRKCZhigh) (Figures 1A-1C). Stratification of TCGA patients by CMS subtypes using the CMScaller algorithm revealed that the CMS4 transcriptomic phenotype was predominant in the PRKCIlowPRKCZlow group (Figures 1D and S1A). Likewise, analysis by the recently proposed iCMS classification, based on intrinsic epithelial gene expression, demonstrated significant enrichment of PRKCIlowPRKCZlow patients in the iCMS3 subtype (Figures 1E and S1B). The PRKCIlowPRKCZlow group had a higher monocytic-myeloid abundance and an augmented expression of endothelial and activated fibroblastic signatures as compared to that of the PRKCIhighPRKCZhigh cohort, as determined by the Microenvironment Cell Populations (MCP)-counter algorithm (Figure S1C). These results demonstrated that CRC patients with low aPKC levels have a tumor microenvironment that is inflamed, highly vascularized, and rich in activated CAFs, all characteristics of the mCRC type.
Figure 1. aPKC-low levels correlate with HA-deposition and poor prognosis in human CRC.

(A) GSEA of transcriptomic data from TCGA CRC patients according to PRKCI/PRKCZ expression.
(B) GSEA plots of the indicated gene signatures from TCGA CRC patients according to PRKCI/PRKCZ expression.
(C) GSEA of transcriptomic data from TCGA CRC patients according to PRKCI/PRKCZ expression using stroma-related signatures.
(D and E) GSEA plots of the indicated gene signatures in TCGA CRC patients according to PRKCI/PRKCZ expression.
(F) Experimental design of PRKCI and PRKCZ editing in patient-derived organoids (PDOs) from CRC.
(G) Immunoblot of indicated proteins in sgPRKCI/PRKCZ and sgC PDOs (n=3 biological replicates).
(H) qPCR of HAS2 in sgPRKCI/PRKCZ and sgC PDOs. Unpaired t-test. Data shown as mean ± SEM (n=3 biological replicates). ***p < 0.001.
(I) Immunofluorescence (IF) for HA (yellow) in sgPRKCI/PRKCZ and sgC PDOs. Scale bars, 50 μm
(J) IF for aPKCs (red) and HA (green) in a human cohort of CRC samples (n=390). Scale bars, 100 μm.
(K and L) Kaplan-Meier curve for overall survival of CRC patients according to aPKCs (K) and HA (L) expression. Log-rank tests.
(M and N) Pie chart of relative distribution of CRC patients according to aPKCs/HA expression (M) and Kaplan-Meier curve for overall survival. Log-rank test (N).
(O and P) IF for aPKCs (red) and HA (green) (O) and quantification (P) in primary CRC samples and metastatic counterparts (n=21, paired). Paired t-test. ***p < 0.001. Scale bars, 100 μm.
See also Figure S1.
Low aPKC levels in CRC patients are associated with increased expression of markers of hyaluronan synthesis
The analysis of the differentially expressed genes in PRKCIlowPRKCZlow vs. PRKCIhighPRKCZhigh groups in several large datasets identified hyaluronan synthase 1 (HAS1) and 2 (HAS2) as two upregulated genes in PRKCIlowPRKCZlow patients (Figures S1D and S1E), suggesting that aPKC-deficient tumors would potentially be enriched in HA. Deletion of both aPKC genes in a human CRC patient-derived organoid showed higher levels of HAS2 and increased accumulation of HA (Figures 1F-1I), demonstrating that aPKC deficiency is sufficient to trigger the HA biosynthetic pathway in a cell-autonomous manner. Consistently, the classification of CRC patients based on a HAS score according to their HAS1, HAS2, and HAS3 levels showed a negative correlation with aPKC levels and a positive enrichment in TGFβ signaling and EMT signatures in CRC patients with a high HAS score, as compared to those with a low HAS score (Figures S1F and S1G). These findings support the notion that increased HA levels are a feature of human mCRC tumors.
Next, we analyzed the expression levels of aPKC in the tumor epithelium and HA deposition in the stroma by double immunofluorescence of a large cohort of surgically resected CRC specimens (Figure 1J). Among the 390 CRC patients in the tissue microarray (TMA), 343 with available clinical information were stratified according to aPKC/HA expression status, including 21 Stage IV patients with matched liver metastasis (Figure S1H). Kaplan-Meier curves revealed that patients with aPKC-low expression showed significantly worse overall survival than those with aPKC-high levels (Figure 1K). HA-positive patients also showed a worse prognosis than HA-negative patients (Figure 1L). Multivariate logistic regression analysis demonstrated that low aPKC expression was correlated with HA stromal deposition independent of other pathological features (Figures S1I and S1J). Survival analyses of these patients categorized into four groups based on their aPKC and HA content indicated that those who were aPKC-low and HA-positive had the worst prognosis (Figures 1M and 1N). Consistently, multivariate COX proportional hazards regression analysis demonstrated that aPKC-low/HA-positive expression was significantly associated with worse prognosis compared to aPKC high/HA-positive patients (Hazard ratio: 2.40), whereas there was no significant difference between aPKC high/HA positive patients and aPKC high/HA negative or aPKC low/HA negative patients (Figure S1K). These findings highlight HA deposition as one of the main contributors to the aggressive phenotype of aPKC-deficient tumors. All stage IV CRC patients and their matched liver metastasis studied were HA-positive, and liver metastases showed reduced expression of aPKC compared to their matched primary tumors (Figures 1O and 1P), suggesting that the loss of both aPKCs is an important event for the systemic spread of late-stage CRC.
aPKC deletion makes CRC tumors dependent on HA for enhanced malignancy
To address the potential role of HA stromal accumulation in the tumorigenesis of intestinal mesenchymal tumors in vivo, we first characterized mouse tumor organoids (MTO) with mutations in Ape, Trp53, Kras, and Tgfbr217 and with deletion of Prkci and Prkcz by CRISPR-Cas9. MTO-sgPrkci/sgPrkcz recapitulated the observed phenotype in human organoids showing in vitro upregulation of Has1, Has2, and Has3 transcripts, increased expression of HA, enrichment in EMT and TGFβ signaling signatures, and the iCMS3 patients’ epithelial signature together with a decreased expression of the iCMS2 patients' signature (Figures 2A-2E and S2A). The injection of MTO-sgPrkci/sgPrkcz into the colon submucosa of C57BL/6J mice generated larger tumors and displayed increased HA accumulation than MTO controls (Figures S2B-S2F). Next, we tested the impact of depleting HA on tumorigenesis. MTO-sgPrkci/sgPrkcz or MTO-sgC (control) were subcutaneously transplanted into syngeneic C57BL/6J mice and treated with a low dose of clinical-grade pegylated hyaluronidase (PEGPH20; 0.0375 mg/Kg), equivalent to a human dose evaluated in clinical trials, or with vehicle (Figure 2F). Tumors from MTO-sgPrkci/sgPrkcz had a more desmoplastic phenotype characterized by enhanced expression of HA, collagen, and αSMA+, which was reverted by PEGPH20 treatment (Figures 2G and 2H). MTO-sgPrkci/sgPrkcz tumors showed higher volume and weight than those from MTO-sgC but were sensitive to PEGPH20 treatment, which did not affect the growth properties of MTO-sgC tumors (Figures 2I and 2J). Consistently, transcriptomic interrogation of MTO-sgPrkci/sgPrkcz tumors treated with PEGPH20 revealed a decrease in signatures related to invasion, tumor progression, and desmoplasia, such as EMT, myogenesis, angiogenesis, TGFβ signaling, stromal activation and the CMS4 subtype (Figures 2K-2M). These data demonstrate that aPKC-deficiency switches CRC tumors to a highly mesenchymal phenotype yet confers vulnerability to HA depletion in the context of WNT-driven tumorigenesis.
Figure 2. Targeting HA disrupts the desmoplastic response and impairs mesenchymal tumorigenesis.

(A) Experimental design of Prkci and Prkcz editing in mouse tumor organoids (MTO).
(B) Immunoblot analysis of indicated proteins in MTO-sgPrkci/Prkcz and MTO-sgC (n=3 biological replicates).
(C) qPCR of indicated genes in MTO-sgPrkci/Prkcz and MTO-sgC (n=3 biological replicates). Unpaired t-test. Data shown as mean ± SEM. *p < 0.05, **p< 0.01.
(D) IF for HA (yellow) in MTO-sgPrkci/Prkcz and MTO-sgC. Scale bars, 50 μm.
(E) GSEA plots of the indicated gene set signatures for MTO-sgPrkci/Prkcz versus MTO-sgC (n=3 biological replicates).
(F-J) Subcutaneous injection of MTO-sgPrkci/Prkcz and MTO-sgC in WT mice. Mice were treated twice a week with Veh or PEGPH20 0.0375 mg/kg for 3 weeks (MTO: sgC Veh n=10, sgC PEGPH20-treated n=10, sgPrkci/Prkcz Veh n=10, and sgPrkci/Prkcz PEGPH20-treated n=8). Experimental design (F); Immunohistochemistry (IHC) for HA, Masson’s trichrome, and αSMA staining (G); quantification, one-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, (n=8), ***p < 0.001, ****p < 0.0001 (H); tumor volume, two-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, ****p < 0.0001 (I) and tumor weight, one-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, **p< 0.01 (J). Scale bars, 100 μm. Sac: sacrificed.
(K and L) GSEA from Quant-seq on MTO-sgPrkci/Prkcz PEGPH20-treated tumors (n=4) versus Veh (n=3) using compilation H (MSigDB) (K) and stroma-related signatures (L).
(M) GSEA plots of the indicated gene signatures for MTO-sgPrkci/Prkcz PEGPH20-treated tumors (n=4) versus Veh (n=3).
See also Figure S2.
HA accumulation is a critical feature of serrated mesenchymal tumors
To test whether HA is also important in serrated-originating CRC, we used an endogenous model of mesenchymal serrated MSS tumors driven by aPKC deficiency.13 Upon the inducible and simultaneous ablation of both aPKCs, organoids from inducible Prkcif/fPrkczf/f;Villin-CreER mice showed increased expression of Has1, Has2, and Has3, concomitant with enhanced HA accumulation (Figures 3A-3C). Prkcif/fPrkczf/f;Villin-Cre intestinal tissues showed a substantial deposition of HA compared to control samples (Figure 3D). The accumulation of stromal HA was detected even in the non-tumor area of the Prkcif/fPrkczf/f;Villin-Cre intestine and further increased in adenomas as they progressed from benign SSL to malignant carcinomas (Figure 3D). The stromal HA accumulation in Prkcif/fPrkczf/f;Villin-Cre tumors was eliminated by PEGPH20 treatment, accompanied by a profound remodeling of the tumor stroma, as evidenced by reduced collagen deposition and αSMA expression (Figures 3E-3G). PEGH20 treatment resulted in a significant reduction in tumor number, average size, and tumor load and lower cancer incidence in the small intestine, concomitant with fewer SSL and reduced invasive carcinomas (Figures 3H-3K, S3A, and S3B). As reported previously, this mouse model also gives rise to aggressive desmoplastic tumors in the proximal colon.13 PEGPH20 treatment reduced colon tumorigenesis in this mouse model together with stroma remodeling (Figures S3C-S3G). In keeping with these histological observations, GSEA of transcriptomic profiling of Prkcif/fPrkczf/f;Villin-Cre tumors revealed that PEGPH20 treatment resulted in a decrease in signatures related to invasion and tumor progression as well as those corresponding to stromal activation, serrated tumorigenesis and the CMS4 subtype (Figures 3L-3O).
Figure 3. Targeting HA represses mesenchymal intestinal CRC.

(A) Experimental design for tamoxifen treatment in organoids from Prkcif/fPrkczf/f;Villin-CreER mice.
(B) qPCR of indicated genes in tamoxifen- or Veh-treated organoids (n= 3 biological replicates). Unpaired t-test. Data shown as mean ± SEM. **p< 0.01, ***p < 0.001.
(C) IF for HA (yellow) of Veh****- or tamoxifen-treated organoids. Scale bars, 50 μm.
(D) IHC for HA of small intestinal sections from Prkcif/fPrkczf/f and Prkcif/fPrkczf/f;Villin-Cre mice. Scale bars, 50 μm.
(E-K) Prkcif/fPrkczf/f;Villin-Cre mice (male and female, 11-week-old) treated with Veh (n=10) or PEGPH20 (n=12), 0.0375 mg/kg for 3 weeks. Experimental design (E); IHC HA, Masson’s trichrome, and αSMA (F); staining quantification, unpaired t-test, data shown as mean ± SEM (n=5), **p< 0.01 (G); macroscopic images of small intestinal tumors (H); total tumor number, tumor size, and tumor load in small intestine, unpaired t-test and Mann-Whitney test, data shown as mean ± SEM, **p< 0.01, ***p < 0.001 (I); quantification of cancer incidence, chi-square, *p < 0.05 (J); and sessile serrated lesions (SSL) and carcinoma quantification of tumors, Mann-Whitney test, data shown as mean ± SEM, *p < 0.05, **p< 0.01 (K). Scale bars, 50 μm (F), 10 mm (H). The red arrow denotes tumors >5mm (H). Sac: sacrificed.
(L-N) GSEA of transcriptomic data from Quant-seq on PEGPH20-treated tumors versus Veh (n=3) using compilation H (MSigDB) (L), stroma-related signatures (M) and serrated-related signatures (N).
(O) GSEA plots of the indicated gene signatures for PEGPH20-treated tumors versus Veh (n=3).
See also Figure S3.
Anti-angiogenic effect of PEGPH20 on the vasculature of mCRC tumors
To define the mesenchymal phenotype at a cellular level and to understand how the extracellular accumulation of HA promotes tumorigenesis and informs the microenvironment in this type of neoplasia, we carried out a scRNA-seq analysis of Prkcif/fPrkczf/f;Villin-Cre tumors from mice treated or not with PEGPH20 (Figures 4A and 4B). Unsupervised clustering with selective markers for each population identified epithelial, stromal, and immune cells (Figures 4C and S4A-S4I). Stromal cell re-clustering and mapping of marker gene expression identified six major stromal cell types, including endothelium (Pecam1), lymphatic-endothelial cells (Lyve1), smooth muscle cells (Myh11), fibroblasts (Dcn), glial cells (Plp1), and pericytes (Rgs5) (Figures 4D, 4E, S4J and S4K). Endothelial cells were the most abundant cell population in the stroma of Prkcif/fPrkczf/f;Villin-Cre tumors (Figures 4E-4F). Analysis of the endothelial cell compartment of Pecam1+ (CD31) and Lyve1+ cells identified nine cell types (Figures 4G, 4H, S4L, and S4M), which were ascribed to the following categories: capillary (Cd36), artery (Gja4), vein (Ackr1), tip (Apln), immature (Aplnr), postcapillary (Selp), lymphatic-endothelial (Lyve1), shear-stress artery (Pi16) and proliferative (Birc5) (Figures S4L and S4M). Previous scRNAseq efforts identified tip and proliferative endothelial cells as specially enriched in the tumor endothelial cell (TEC) population, which have been proposed to be involved in tumor neo-angiogenesis.25,26 To rigorously distinguish the clusters corresponding to normal endothelial cells (NEC) from the TEC in the Prkcif/fPrkczf/f;Villin-Cre tumors, we created a TEC-specific signature by comparing tumor vs. normal endothelium from a human CRC scRNAseq dataset (GSE132465). This signature labeled Tip, Postcapillary, and Proliferating cells as TEC (Figures S4N and S4O). Also, gene signatures from lung endothelial cells supported that capillary and immature cells belong to the TEC category (Figures S4N and S4O). PEGPH20 reduced the proportion of tip cells and postcapillary and immature cells in Prkcif/fPrkczf/f;Villin-Cre tumors (Figures 4H and 4I).
Figure 4. Remodeling of the mesenchymal intestinal tumor stroma by PEGPH20 treatment.

(A) Experimental design and workflow of scRNAseq. Small intestinal tumors from Veh (n=5) and PEGPH20-treated mice (n=3) were dissected and digested into single-cell suspensions for sequencing.
(B and C) Uniform manifold approximation and projection (UMAP) of tumor cells colored by treatment (B) and by the major cellular compartments (C).
(D and E) UMAP of stromal cells colored by treatment (D) and by the major stromal cell type (E).
(F) Stromal-cell-type percentage relative to the total stromal cells count per treatment.
(G and H) UMAP of endothelial cells colored by treatment (G) and by endothelial cell type (H).
(I) Endothelial-cell-type percentage relative to the total stromal cells count per treatment.
(J and K) UMAP of fibroblast colored by treatment (J) and by fibroblast cell type (K).
(L) Violin plots for the indicated gene signatures in the fibroblast cell types. The top and bottom of the violin plots represent the minimal and maximal values, and the width is based on the kernel density estimate of the data, scaled to have the same width for all clusters. Horizontal lines represent median values. Unpaired t-test, **p< 0.01, ***p < 0.001.
(M and N) UMAP of fibroblast colored by fibroblast cell type (M) and fibroblast-cell-type percentage relative to the total fibroblast count per treatment (N).
(O) Significantly enriched extracellular matrix (ECM)-related signatures in intermediate fibroblast treated with PEGPH20 versus Veh.
(P and Q) PDGFRα staining (red) with NRG1 (green) and panCK (white), CD34 staining (green) with PDFGRα (red) and CD31 (white) (P), and quantification, unpaired t-test, data shown as mean ± SEM, **p< 0.01, ****p < 0.0001 (Q) in Veh- and PEGPH20-treated small intestine tumors (n=5).
(R and S) PDGFRα staining (red) with NRG1 (green) and panCK (white), CD34 staining (green) with PDFGRα (red) and CD31 (white) (R), and quantification, unpaired t-test and Mann-Whitney test, data shown as mean ± SEM, **p< 0.01, ****p < 0.0001 (S) in Veh- and PEGPH20-treated colon tumors (n=3). Scale bars, 100 μm.
See also Figures S4 and S5.
Next, we applied two gene signatures reflecting the transcriptional programs of tumor vessel disorganization and normalization, respectively.25 The whole endothelial compartment of Prkcif/fPrkczf/f;Villin-Cre tumors were enriched in the vessel disorganization signature, which was reversed upon PEGPH20 treatment, indicating that high HA deposition in mesenchymal tumors promotes endothelial remodeling (Figure S4P). PEGPH20 also increased the normalization signature (Figure S4P), demonstrating that HA depletion not only pruned the TECs but also normalized the endothelial landscape of the Prkcif/fPrkczf/f;Villin-Cre tumors. Further pathway analysis of the endothelium compartment of these tumors showed that PEGPH20 reduced angiogenesis, glycolysis, and hypoxia signatures (Figure S4P). In contrast, it increased the expression of signatures related to interferon (IFN) activation (Figure S4P), suggesting enhanced immunosurveillance mechanisms driven by reprogramming endothelial cells in the tumor vasculature. No changes were observed in pericyte coverage (Figures S4Q and S4R). These results demonstrate that the remodeling of the tumor endothelial vasculature is a hallmark of the high efficacy of PEGPH20 in reducing tumorigenesis.
Fibroblast heterogeneity in endogenous mesenchymal tumors
The analysis of the fibroblast compartment by re-clustering the Dcn-expressing cells identified three cell categories (Figures 4J and 4K). Previous studies classified normal intestinal fibroblasts into three cell lineages according to the expression of Pdgfra, Cd81, and Cd34. This determines a dual and compartmentalized fibroblast positioning along the crypt axis and their biological function in intestinal homeostasis.27 Thus, Pdgfrahigh fibroblasts (telocytes) are abundant in the villus region, expressing high levels of BMP ligands necessary for terminal epithelial cell differentiation.28 Conversely, PdgfralowCd81high fibroblasts (trophocytes) are CD34+, are exclusively located beneath the crypts, and express WNT pathway factors, including RSPO1 and RSPO3, all critical for adult stem cell maintenance.29,30 In line with this classification, we identified telocytes and trophocytes as components of the fibroblast compartment of Prkcif/fPrkczf/f;Villin-Cre tumors (Figures 4K, S5A-S5H). We also found that these tumors were enriched in a third fibroblast population (“intermediate”) that shares some trophocyte features, such as the expression of WNT modulators and BMP inhibitors, but also resembles telocytes in that are negative for CD81 and CD34, yet express PDGFRα, albeit at a lower level than telocytes (Figures 4K, S5A-S5H). Telocytes and intermediate fibroblasts showed enrichment of carcinoma-associated fibroblast (CAF) signatures (Figure 4L). CAFs have been previously described as inflammatory (iCAFs) or myofibroblastic (myCAFs), which are strongly activated by TGFβ.31-33 Telocytes showed higher myCAF signature expression and TGFβ response than intermediate or trophocytes (Figure 4L and S5H). Conversely, trophocytes showed a higher expression of an iCAF signature than the intermediate or the telocytes (Figure 4L and S5I-S5K). Telocytes from Prkcif/fPrkczf/f;Villin-Cre tumors express high levels of Sfrp2 and Sfrp4 (Figure S5F), critical targets of SOX2 expression in CAFs, to promote the CRC mesenchymal phenotype.12 Consistently, telocytes in these tumors displayed high expression of SOX2 targets (Figure 4L), suggesting that telocytes/myCAFs are activated and expanded through the TGFβ/SOX2 axis to drive the progression of Prkcif/fPrkczf/f;Villin-Cre tumors.
HA degradation remodels the mCRC CAF compartment
HA degradation in vivo virtually eliminated the telocyte population, the main CAF in Prkcif/fPrkczf/f;Villin-Cre tumors, and promoted the concomitant accumulation of trophocytes (Figures 4M and 4N). In contrast, the proportion of intermediate fibroblast was not affected by PEGPH20 treatment (Figures 4M and 4N). However, differential gene expression analysis showed a substantial reduction in collagens, matrix metalloproteinases, and signatures related to matrix remodeling in the intermediate fibroblasts from PEGPH20-treated mice (Figures 4O, S5K, and S5L), suggesting that the HA-rich environment is fundamental for the selective maintenance of telocytes and myCAF features. These findings were further validated by multiplex immunofluorescence. Staining of NRG1 (activated telocytes), PDGFRα (telocytes), Pan-CK (epithelial cells), and DAPI (nuclear marker) showed a strong presence of PDGFRα+/NRG1+ fibroblasts at the luminal border of Prkcif/fPrkczf/f;Villin-Cre tumors, consistent with the zonation pattern of telocytes in the normal small intestinal and colonic epithelium, that was significantly reduced by PEGPH20 treatment (Figures 4P-4S). Trophocytes identified as a PDGRα−/CD31−/CD34+ population and normally located beneath the crypts in the normal intestinal epithelium showed a broader distribution in Prkcif/fPrkczf/f;Villin-Cre tumors (Figures 4P-4S). PEGPH20 strongly increased trophocyte levels mainly at the crypt-bottom positions (Figures 4P-4S), which suggests a complete remodeling of the trophocyte population upon HA depletion.
Epithelial cancer cell hierarchical heterogeneity and dependency on HA in mCRC tumors
We next investigated whether HA depletion modulates the epithelial features of mesenchymal intestinal tumors. Epithelial cell re-clustering and mapping of the scRNAseq data from Prkcif/fPrkczf/f;Villin-Cre tumors using marker gene expression classified the non-tumor epithelial cell population of these samples as non-cycling transient amplifying (TA), cycling transient amplifying (cTA), enterocytes, goblet, Paneth, tuft, and enteroendocrine (Figures 5A, 5B, and S6A-S6C). Based on sample origin and genome-wide copy-number alteration (CNA) analysis, we classified epithelial cells as malignant or non-malignant (Figures S6D-S6F). We identified four tumor epithelial populations (Figure S6G). One of the clusters corresponds to the tumor counterpart of the cycling TA population (TcTA) (Figures 5B, S6G-S6K), which shares similarities with a recently identified cancer cell population seemingly essential for LGR5-independent tumor growth.34 Another cluster was enriched in Ly6a and Anxa10, markers of a previously reported fetal metaplastic cell (tumor fetal metaplastic cells; TFMC) (Figures 5B, S6G-S6K).35,36 A third tumor cell population was characterized by high expression of clusterin and is reminiscent of the “revival stem cell” (tumor revival stem cells; TRSC) that emerges in response to intestinal tissue damage associated with the loss of LGR5+ stem cells and activation of regenerative processes in non-tumor intestinal tissues (Figures 5B, S6G-S6K).37 A fourth cancer cell type corresponded to transformed goblet cells (TGC) and displayed some features of immature goblet cells with high levels of Ly6a, and Anxa10, a transcriptional profile characteristic of the TFMCs found in the non-goblet epithelial tumor cell compartment (Figures 5B, S6L-S6N).
Figure 5. scRNA-seq reveals a complex heterogeneity and hierarchy maintained by HA in tumor epithelial cells.

(A and B) UMAP of epithelial cells colored by treatment (A) and by epithelial cell type (B).
(C) RNA velocities visualized on the UMAP projection in (B).
(D) Violin plots for the indicated gene signatures in cycling transient amplifying (cTAs), tumor cTAs (TcTAs), tumor revival stem cells (TRSCs), and tumor fetal metaplastic cells (TFMCs). The top and bottom of the violin plots represent the minimal and maximal values, and the width is based on the kernel density estimate of the data, scaled to have the same width for all clusters. Horizontal lines represent median values. Unpaired t-test. ***p < 0.001.
(E) Scheme showing that cTAs can differentiate to TcTA and conserve epithelial cancer cell hierarchical heterogeneity.
(F) Violin plots for the indicated gene signatures in immature goblet cells, mature goblet cells, and tumor goblet cells (TGC). The top and bottom of the violin plots represent the minimal and maximal values, and the width is based on the kernel density estimate of the data, scaled to have the same width for all clusters. Horizontal lines represent median values. Unpaired t-test. *p < 0.05, ***p < 0.001.
(G) RNA velocities in tumor epithelial cells for each treatment and tumor-cell-type percentage relative to the total tumoral cells count per treatment.
(H and I) IHC for ANXA10, MUC5AC and CLU (H), and quantification, unpaired t-test, data shown as mean ± SEM, ****p < 0.0001 (I) in PEGPH20-treated tumors (n=5). Scale bars, 50 μm. ns: not significant.
(J) UMAP of goblet cells colored by goblet cell type and goblet-cell-type percentage relative to the total goblet cells count per treatment.
(K) IHC for ANXA10 with alcian blue, IF for ANXA10 (magenta) and MUC2 (green), and quantification in Veh- and PEGPH20-treated tumors (n=5). Mann-Whitney test, data shown as mean ± SEM, **p < 0.01. Scale bars, 25 μm.
(L) IF for HA (green), aPKCs (yellow), MUC5AC (cyan), and ANXA10 (magenta) in Veh- and PEGPH20-treated tumors (n=5). Scale bars, 100 μm.
(M) Kaplan-Meier curve for 8-year overall survival of TCGA CRC patients according to aPKCs/HA/ANXA10/MUC5AC expression. Log-rank test.
(N) CellphoneDB analysis of the number of ligand-receptor interactions between tumor epithelial cells and fibroblast.
(O) Anxa10 and Muc5ac mRNA levels of MTO-sgC stimulated by conditioned medium (CM) of indicated intestinal fibroblasts with or without PEGPH20 (2.5 μg/ml) for 3 days (n=3). Schematic representation and qPCR. One-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, *p < 0.05, ***p < 0.001, ****p < 0.0001.
(P) Dot plot for ligand-receptor pairs of growth factors between telocytes and tumor epithelial in Veh-treated tumors.
(Q and R) CellphoneDB analysis of the number of ligand-receptor interactions between tumor epithelial cells and fibroblast cell types (Q) and dot plot for ligand-receptor pairs of Lgr5 and Rspo co-factors between trophocyte and tumor epithelial in Veh- and PEGPH20-treated tumors (R).
See also Figures S6 and S7.
Trajectory analysis by RNA velocity established a flow from cTAs to TcTAs, which gives rise to TFMCs and TRSCs albeit some of the latter might originate directly from cTAs (Figure 5C). Transcriptomic comparison of tumor and normal intestinal epithelial cells shows that TcTAs are related to non-tumor cTAs, whereas TFMCs are more differentiated and related to enterocytes (Figures S6J). Although there is an increased expression of a BMP signaling signature in the three epithelial cell transformed populations, as compared to cTA, TFMC showed the highest BMP signaling activation while displaying reduced WNT signaling, which was maintained/enhanced in the TRSC population (Figure 5D). These data demonstrate that TFMCs are the most differentiated transformed cell populations and have features compatible with enterocyte markers (Figure S6J). The TFMC compartment is enriched in a “metaplasia” gene expression signature (Figure 5D). This is consistent with recent observations that human serrated tumors are associated with a metaplastic response whereby enterocytes acquire a fetal-gastric lineage as a cell protection mechanism against a chronic cytotoxic response.35 In our model, toxicity is most likely triggered by constitutive inflammation driven by increased epithelial apoptosis and dysfunctional Paneth cells that promote tumor initiation,13,14,38 which explains why there is a TRSC compartment in these tumors. This scenario accounts for the SSL histology characteristic of the Prkcif/fPrkczf/f;Villin-Cre adenomas, which are the precursors of the serrated carcinomas.35
Selective signaling phenotypes of mesenchymal tumor cells are maintained by stromal HA accumulation
From the signaling point of view, TcTAs are less enriched in proliferative signatures, such as “E2F_TARGETS” or “MYC_TARGETS,” than cTAs, yet these signatures were more enriched in TcTAs than in TFMC or TRSC (Figure 5D). This indicates that tumor cells are less proliferative than non-tumor cTA cells and become even less proliferative as they differentiate towards the TFMC and TRSC compartments. There is also a progressive upregulation of the EMT and TGFβ gene expression signatures going from cTAs to TcTAs, and from these to TFMC, consistent with aPKC-deficient tumor epithelial cells being more invasive yet less proliferative, with TFMC displaying the highest degree of mesenchymal activation (Figure 5D). There was also a general increase in YAP in all these transformed cells, which was highest in the TFMC population (Figure 5D). YAP has been shown to control the production of EGF family growth factors 39, suggesting that the strong ERK and EGFR activation responses previously described in Prkcif/fPrkczf/f;Villin-Cre tumors can be accounted for by the presence of this more invasive TFMC cell population.13 Tumor TRSCs were enriched in WNT signaling, suggesting that a relatively small but significant proportion of the cancer epithelial cells in Prkcif/fPrkczf/f;Villin-Cre tumors rely on WNT activation likely to function as a reserve cell type (Figure 5D). Therefore, cTAs are the most probable origin of Prkcif/fPrkczf/f;Villin-Cre tumor epithelial cells, which are hierarchically structured reflecting the non-transformed intestinal cell lineage organization but with critical alterations in tumorigenic pathways that equip the serrated tumor cells with differential signaling characteristics (Figure 5E). In the secretory compartment, TGCs showed enrichment in EMT, YAP, WNT, and metaplasia signatures, as compared to the non-tumor mature goblet population (Figures 5F and S6O), indicating that the fetal lineage transition was observed in both enterocytic and goblet populations of serrated tumors.
PEGPH20 treatment differentially affected the three non-secretory tumor cell populations (Figure 5G). Thus, while there was a limited proportional reduction of TcTAs, we found a clear reduction in the proportion of TFMCs but with a relative enrichment in the proportion of TRSCs in the persistent tumor cell population (Figure 5G). Trajectory analysis of the PEGPH20-treated condition showed a total disruption in the flow from TcTA to TFMC, while TFMC gained the ability to differentiate into TRSCs (Figure 5G). Next, we determined the localization of TFMCs (ANXA10+ and MUC5AC+) and TRSCs (CLU+) in Prkcif/fPrkczf/f;Villin-Cre tumors. Consistent with their enterocytic features and metaplastic phenotype, TFMCs were more exposed to the cancer luminal surface, whereas TRSCs were distributed at the crypt-bottom areas (Figures 5H and 5I). PEGPH20 strongly reduced the presence of TFMCs (ANXA10+ and MUC5AC+ staining), whereas that of TRSCs (CLU+ staining) was not reduced, in agreement with our scRNAseq findings (Figures 5H and 5I). MTO-sgPrkci/sgPrkcz recapitulated the observed phenotype in Prkcif/fPrkczf/f;Villin-Cre tumors showing in vitro upregulation of Anxa10, Muc5ac and Clu as compared with MTO-sgC (Figure S6P). PEGPH20 treatment reduced Anxa10 and Muc5ac expression without changes in Clu levels in MTO-sgPrkci/sgPrkcz (Figure S6P). These results support a model whereby HA depletion in Prkcif/fPrkczf/f;Villin-Cre tumors induced a lineage switch from TFMC to TRSC, suggesting that the WNT-independent TFMC population, which is likely maintained by YAP, can acquire a TRSC phenotype most probably supported by a WNT-dependent state. Regarding the goblet cell compartment, PEGPH20 treatment strongly reduced the proportion of TGCs in Prkcif/fPrkczf/f;Villin-Cre tumors, as also demonstrated by ANXA10+/Alcian Blue and ANXA10+/MUC2+ staining (Figures 5J and 5K), demonstrating that stromal HA accumulation is a crucial event for the maintenance of TGCs. Similar results in cell population changes upon PEGPH20 treatment were observed when colon tumors were analyzed (Figures S6Q and S6R).
To establish the human relevance of these findings, we have analyzed scRNA-seq data from three previously published and available human CRC datasets: GSE132465,26 GSE166555,40 and GSE17834141 for a total of 97 CRC patients. By fractionating epithelial cells from primary colorectal tumors, we found that the four representative gene signatures derived from our mouse model, namely, TcTA, TRSC, TFMC, and TGC, were highly expressed in human CRC tumors (Figures S7A-S7L). TcTA and TRSC signatures were mainly present in the iCMS2 subtype, left-side tumors, and T2-T3 stages, while TFMC and TGC were associated with the poor prognosis subtype iCMS3, right-side tumors, and the T4 stage concordant with their aggressive features found in the mouse intestine (Figure S7M). The analysis of the tumor epithelium of the CRC TMA by multiplex-Opal staining for HA, aPKC, ANXA10, and MUC5AC demonstrated that both ANXA10-high and MUC5AC-positive staining are predictors of poor prognosis, consistent with previous data showing that iCMS3 tumors have a worse prognosis and are the ones enriched in metaplasia genes 42, and with our results demonstrating that the TFMC population is enriched in iCMS3 tumors. The acquisition of metaplasia markers such as ANXA10 or MUC5AC in aPKC-low/HA-positive tumors predicted poor survival as compared to those that have not acquired these markers (Figures 5L, 5M, and S7N). Multivariate logistic regression analysis demonstrated that both ANXA10 and MUC5AC expression were significantly associated with aPKC-low and HA-positive expression independent of other pathological features (Figures S7O-S7R). These results further support the link between the fetal/metaplastic state (TFMC) and malignancy in human mCRC.
HA depletion reformats the CAF-epithelial tumor interactions
Since TFMCs colocalize with telocytes at the luminal surface of Prkcif/fPrkczf/f;Villin-Cre tumors and both populations were sensitive to PEGPH20, we posited that their interaction might be critical for the maintenance of tumor growth. Therefore, we investigated the crosstalk between fibroblasts and tumor epithelial populations by CellphoneDB analysis.43 Telocytes showed the highest interactions with tumor cells compared to those of the intermediate or trophocyte populations (Figure 5N). Consistently, conditioned media of fibroblasts isolated from Prkcif/fPrkczf/f;Villin-Cre mice (telocytes) induced the upregulation of metaplastic markers (Anxa10 and Muc5ac) in in vitro MTO cultures, which was rescued by PEGPH20 (Figure 5O). Telocytes were enriched in growth factors such as Igf1, Ereg, Fgf2/7, Nrg1, and Hgf, while their cognate receptors were broadly expressed by tumor cells (Figure 5P), which could account for the regulation of TFMCs by telocytes. We also identified Areg as a specific growth factor expressed in TFMC, interacting with Egfr and Icam1 in telocytes. Tff1, previously identified as a biomarker for SSL 44, was expressed by TFMCs and could support telocytes expressing Fgfr2 (Figure 5P). This set of interactions highlights the bi-directional telocyte-TFMC crosstalk that determines mesenchymal tumor malignancy. PEGPH20 induced a high number of communications between TRSC and trophocytes while switching off the interactions between TFMCs and telocytes (Figure 5Q). PEGPH20 treatment also enriched trophocytes in Rspo1/3, which interacted with their cognate receptor Lgr5 in the TRSC compartment (Figure 5R), in keeping with recent evidence on the maintenance by trophocytes of adult stem cell homeostasis through WNT signaling.27,45
Microenvironmental HA is critical for cancer immunosuppression in mesenchymal tumors
Our previously published data demonstrated that reduced IFN signaling, which in turn impeded CD8+ T cell-mediated immunosurveillance, was a central event for the initiation of Prkcif/fPrkczf/f;Villin-Cre tumors.13,46 PEGPH20 treatment rescued the IFN inhibition in these tumors as demonstrated by enrichment in IFN and allograft rejection signatures by GSEA of RNAseq of Prkcif/fPrkczf/f;Villin-Cre endogenous tumors and allografts from MTO-sgPrkci/sgPrkcz (Figure 6A). This enrichment in IFN upon HA depletion was more evident in the TRSCs, which was the epithelial tumor sub-population more resistant to PEGPH20 treatment (Figure 6B). These results suggest that PEGPH20 could enable ICB therapy through upregulating the IFN pathways in the “persister” epithelial cancer cells by switching them from immunoevasion to immunosurveillance mode. To explore this possibility, we determined how PEGPH20 reprogrammed the immune system of intestinal mesenchymal tumors. Unsupervised clustering of the immune component of the scRNAseq data from Prkcif/fPrkczf/f;Villin-Cre tumors identified six major cell types: tumor-associated neutrophils (TANs), tumor-associated macrophages (TAMs), dendritic cells (DCs), inflammatory monocytes (IMs), B and plasma cells, T cells, and natural killer (NK) cells (Figures 6C, S8A and S8B). Prkcif/fPrkczf/f;Villin-Cre tumors were rich in TAN, TAM, and IM, and PEGPH20 treatment reduced the levels of these myeloid cell types and increased the proportion of T and B cells (Figures 6C and 6D). Further unsupervised T and NK cell clustering identified three different CD8+ T cells and four CD4+ T cell populations, along with proliferative T cells, γδ T cells, and NK cells (Figures 6E, S8C and S8D). PEGPH20 reduced CD4+ Treg cells, which, together with the relative enrichment in CD8+ T cells, resulted in an increased CD8+:Treg ratio in their TME, a critical anti-tumorigenic state (Figure 6F). Within the CD8+ T cells, we found three functional states: CD8+ T effector memory (Tem), CD8+ T resident memory (Trm), and CD8+ T exhausted (Tex) (Figures 6E and S8D). CD8+ Tex cells showed the highest degree of dysfunctionality and exhaustion, whereas CD8+ Trm were enriched in stemness (Figure S8E). The lower levels of exhaustion markers and the expression of Gzmk suggested a pre-dysfunctional phenotype as the main characteristic of the CD8+ Tem population (Figure S8D). PEGPH20 treatment increased the proportion of CD8+ Trm and CD8+ Tex cells and resulted in the enrichment in the signatures corresponding to CD8+ T cells exhaustion, stem-like, and expansion, as indicated by the increased expression of Ccl5 (Figures 6G and 6H), suggesting strong CD8+ T cells activity in response to PEGPH20. VECTRA multiplex imaging showed a substantial accumulation of myeloid cells and CD4+ Tregs, a minimal presence of B cells, and the exclusion of CD8+ T cells from the tumoral areas in Prkcif/fPrkczf/f;Villin-Cre tumors (Figures 6I and S8F), which is consistent with the immunosuppressive environment of mCRC. PEGPH20 treatment produced a concomitant reduction of myeloid cells and CD4+ Tregs with strong recruitment of B, plasma, and CD8+ T cells to infiltrate the tumors from the endogenous and subcutaneous aPKC-deficient mouse models (Figures 6I and S8F-S8H). These results demonstrate that the immune landscape of Prkcif/fPrkczf/f;Villin-Cre tumors resembles the immunosuppressive features and immune cell composition of mCRC, which can be reprogrammed by PEGPH20 treatment.
Figure 6. HA induces immunosuppression and impairs immunosurveillance in mesenchymal intestinal tumors.

(A) GSEA of transcriptomic data from Quant-seq on Prkcif/fPrkczf/f;Villin-Cre PEGPH20-treated tumors versus Veh (n=3) (top), and MTO-sgPrkci/Prkcz PEGPH20-treated tumors (n=4) versus Veh (n=3) (bottom) using compilation H (MSigDB).
(B) Violin plots for the indicated gene signatures in TcTAs, TRSCs, TFMCs, and TGCs treated with Veh or PEGPH20. The top and bottom of the violin plots represent the minimal and maximal values, and the width is based on the kernel density estimate of the data, scaled to have the same width for all clusters. Horizontal lines represent median values. Unpaired t-test, **p < 0.01, ***p < 0.001.
(C and D) UMAP of all immune cells colored by the major immune cell type (C) and immune-cell-type percentage relative to the total immune cells count per treatment (D).
(E and F) UMAP of all T cells colored by the T cell type (E) and canonical lineage marker expression for CD8+T cell and CD4+Treg (left) showing the percentage of CD4+Treg, CD8+T, and CD8+T:CD4+Treg ratio per treatment (right) (F).
(G) T-cell-type percentage relative to the total T cells count per treatment.
(H) Violin plots for the indicated gene signatures in CD8+T cells treated with Veh or PEGPH20. The top and bottom of the violin plots represent the minimal and maximal values, and the width is based on the kernel density estimate of the data, scaled to have the same width for all clusters. Horizontal lines represent median values. Unpaired t-test, *p < 0.05, ***p < 0.001.
(I) Seven-color overlay image for the indicated protein staining in Veh- and PEGPH20-treated tumors (n=3). Scale bars, 100 μm. The white arrows denote CD4+Treg and myeloid cells; the yellow arrows mark CD8+T cells and the red arrows point to B cells.
(J) CellphoneDB analysis of ligand-receptor pairs of cytokines between telocytes, intermediate and myeloid cells in Veh- or PEGPH20-treated tumors.
(K and L) Dot plot of ligand-receptor pairs of Ccl27a/Ccl28-Ccr10 (K) and Ccl25-Ccr9 and Xcl1-Xcr1 (L) between tumor epithelial cells and immune cells in Veh- or PEGPH20-treated tumors.
(M) Predicted regulatory crosstalk between tumor epithelial cells, fibroblasts, and the immune system in Veh- or PEGPH20-treated tumors.
See also Figure S8.
The permissive immune environment of mesenchymal tumors is orchestrated by cross-compartment interactions and maintained by HA
To elucidate the underlying mechanisms of the tumor response to PEGPH20, we determined cell-specific receptor and ligand expression patterns between the three main compartments of the TME. Based on CellphoneDB analysis, the predicted cell-cell communication networks highlighted the central role of telocytes in the recruitment of TAMs, IM, and TANs to the TME driven by mesenchymal aPKC-deficient epithelial cancer cells (Figure S8I). Analysis of HA receptors showed a higher expression of Cd44 in telocytes and myeloid cells, consistent with the high sensitivity of these two populations to HA-depletion (Figure S8J). Focusing on the cytokines and their receptors that mediate myeloid cell recruitment to Prkcif/fPrkczf/f;Villin-Cre tumors, we identified stromal-immune interactions between Ccl7- and Ccl11-expressing telocytes with myeloid cells positive for their cognate receptors Ccr2/Ccr1/Ccr5, and Ccr2, respectively. Cxcl2-expressing telocytes specifically recruit neutrophils positive for Cxcr2 (Figure 6J), which can also account for the accumulation and recruitment of CD4+ Tregs that express Ccr5/Ccr2 (Figure S8K). Therefore, eliminating telocytes by HA-depletion might explain the reduction of myeloid and CD4+Tregs in mesenchymal intestinal tumors upon PEGPH20 treatment. Consistently, conditioned media from fibroblasts isolated from to Prkcif/fPrkczf/f;Villin-Cre intestinal tissue (telocytes) promoted macrophage chemotaxis, which was abolished by PEGPH20 (Figure S8L). PEGPH20 reduced the TGFβ signals provided by myeloid cells and CD4+Tregs, contributing to the low amount of activated telocytes, EMT, and invasive characteristics of TFMCs (Figure S8M and S8N). PEGPH20 also repressed the secretion of growth signals from telocytes to myeloid cells and CD4+ Tregs (Figures S8O and S8P). Spp1, previously identified as a critical marker of myeloid cells in CMS4 CRC,26 was expressed by TAMs and might also account for the maintenance of telocytes expressing its receptors, Ptger4 and Cd44, in the mesenchymal stroma of Prkcif/fPrkczf/f;Villin-Cre tumors (Figures S8O and S8P). PEGPH20 treatment eliminates the interaction between Spp1-Ptger4/Cd44 in fibroblasts and the Spp1 interactions found between telocytes and CD4+ Tregs (Figure S8P). PEGPH20 also increased the secretion of Ccl27a in TRSCs and Ccl28 in TcTAs and in the residual TFMCs to recruit B cells and that of Ccl25 in TcTAs and TFMCs to attract Ccr9-expressing T cells to the tumor epithelial compartment (Figures 6K and 6L). There was also a PEGPH20-driven interaction between Xcl1 expressed in T cells and Xcr1-expressing DCs (Figure 6L), shown to be critical for antigen presentation and a cytotoxic immune response.47,48 These results support a model whereby the elimination of telocytes by PEGPH20 alleviates the immunosuppression triggered by myeloid cells and CD4+ Tregs, which cooperates with the induction of immunosurveillance through IFN activation in the tumor epithelial cell compartment, and the release of cytokines involved in B and CD8+ T cell infiltration (Figure 6M).
HA depletion renders aPKC-deficient tumors sensitive to ICB therapy
The remodeling of the immune microenvironment by PEGPH20 suggested that it might increase responses to ICB therapy. Since draining lymph nodes of MTO-sgPrkci/sgPrkcz tumors showed a significantly increased proportion of immune cells expressing PD-L1 (Figure 7A), we tested the therapeutic potential of PEGPH20 in combination with anti-PDL1. The administration of anti-PD-L1 with PEGPH20 significantly enhanced the anti-tumor activity of PEGPH20 as determined by the reduced volume and weight of MTO-sgPrkci/sgPrkcz tumors, while anti-PD-L1 alone showed no effect (Figures 7B-7D and S9A-S9C). PEGPH20 treatment also reduced the HA content and intensity of αSMA+ stromal staining and was sufficient to bring CD8+ T and B cells to the tumor and to reduce the amount of immunosuppressive myeloid and Tregs (Figures S9D and S9E). Since the liver is the most common site for CRC metastasis, we carried out an orthotopic liver metastasis model in which MTO-sgPrkci/sgPrkcz organoids were transplanted via intrasplenic injection into syngeneic WT C57BL/6 hosts (Figure 7E). Different treatment groups were initiated two weeks after injection to allow the establishment of liver metastasis. Treatment with PEGPH20 as monotherapy was effective in reducing liver metastasis number, load, and size, as compared to vehicle, as well as that combination with anti-PD-L1 alone or plus anti-CTLA-4 further enhanced its effectiveness (Figures 7F and 7G). Liver metastases driven by MTO-sgPrkci/sgPrkcz induced a highly desmoplastic response as determined by HA and αSMA staining, which was effectively reduced by PEGPH20 treatment (Figures 7H-7K). The immunosuppressive microenvironment of these liver metastases was characterized, like in the primary tumor, by high numbers of infiltrating myeloid cells and Tregs, and the absence of B and T cells, which were all reverted by PEGPH20 with a significant decrease in myeloid and T regs and a robust infiltration inside the remaining metastasis of B and plasma cells along with infiltrating CD8+ T cells (Figures 7J-7M). These results demonstrate that PEGPH20 treatment makes aPKC-deficient tumors and their liver metastasis sensitive to anti-PD-L1 therapy.
Figure 7. The combination therapy of PEGPH20 with anti-PDL1 improves the response of mesenchymal CRC tumors.

(A) IF for PD-L1 (green) and CD45 (red) of tumor-draining lymph nodes in MTO-sgC and MTO-sgPrkci/Prkcz. Scale bars, 100 μm.
(B-D) Subcutaneous injection of MTO-sgPrkci/Prkcz in WT mice treated twice a week with PEGPH20 (0.0375 mg/kg) and αPD-L1 (5 mg/kg) for 4 weeks (n:Veh= 14, PEGPH20-treated=15 and PEGPH20-treated with αPD-L1=15). Experimental design (B); tumor volume, two-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, **p < 0.01, ****p < 0.0001 (C); tumor weight, one-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, *p < 0.05, **p < 0.01 (D). Sac: sacrificed.
(E-M) Intrasplenic injection of MTO-sgPrkci/Prkcz in WT mice. Mice were treated twice a week with Veh or PEGPH20 0.0375 mg/kg and/or αPD-L1 (5 mg/kg) and/or αCTLA-4 (100 μg/dose) for 2.5 weeks (n: Veh-treated =7, PEGPH20-treated=7, PEGPH20 and αPD-L1-treated =7, and PEGPH20 and αCTLA-4-treated=6). Experimental design (E); macroscopic images of liver metastasis tumors (F); total metastases number, tumor load, and average tumor volume, one-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (G); H&E staining and IHC for HA (H) and quantification, one-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, (n=4), ****p < 0.0001 (I); IF for αSMA (white), CD138 (green) and S100A8 (red), IF for FOXP3 (white) and CD19 (green) (J), and staining quantification, one-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, **p < 0.01, ***p < 0.001, ****p < 0.0001 (K); CD8 staining (L) and quantification, one-way ANOVA and post hoc Tukey’s test, data shown as mean ± SEM, (n=3), **p < 0.01, ***p < 0.001 (M); in liver metastases (n=5). Scale bars, 10 mm (F), 2 mm (H), 100 μm (J, L). The red line denotates liver metastases (H). Sac: sacrificed.
See also Figure S9.
DISCUSSION
The cellular and molecular interactions that define the TME of mesenchymal tumors and its potential therapeutics are poorly understood. We have recently demonstrated that reduced levels of the two aPKC isoforms correlated with features of mesenchymal tumorigenesis in human CRC, as well as that their simultaneous genetic inactivation in the intestinal epithelium is sufficient to induce this type of tumor in mice in the context of a morphologically serrated pathway driven by YAP-ERK and without mutations in BRAF or the APC/WNT cascade.13,14 Here we show that mesenchymal tumorigenesis, which is characterized in CRC by an EMT epithelium together with a desmoplastic and inflamed immune landscape with the exclusion of the CD8+ T cells to the tumor-stromal periphery, can also be induced in the context of tubular adenocarcinomas by the simultaneous inactivation of both aPKCs in organoids driven by mutations in the APC/KRAS/p53/TGFβ cassette. These observations are consistent with a large proportion of tumors harboring APC mutations that also express low aPKC levels and that show a mesenchymal phenotype. Our data reveal that a common critical feature shared by mCRCs is the accumulation of HA in their stroma. We also show that HA deposition not only contributes to higher levels of malignancy in both serrated and conventional settings but also makes these tumors vulnerable to the action of PEGPH20. The initial generation of HA can be accounted for by the cell-autonomous activation of HAS expression upon the acute simultaneous genetic inactivation of both aPKCs in the intestinal epithelium.
Treating mesenchymal tumors in mice with PEGPH20 not only results in the cell-autonomous reduction of the epithelial cell population with the most aggressive invasive features (TFMCs) but also promotes the recruitment of CD8+ T and B cells with the concomitant reduction in the infiltrating Tregs and immunosuppressive myeloid cells, which underly the reduction in tumor load. This is in contrast with the effects of inactivating the TGFβ stromal cascade, which is insufficient to reactivate the immune system or to reduce the tumor burden.13,17,18 Since other strategies to target the ECM, like the genetic inactivation of type I collagen, gave inconsistent results,23,24 PEGPH20 emerges as a potential treatment of mCRC tumors as a stroma-targeting monotherapy. The fact that PEGPH20 treatment of mCRC tumors results in the accumulation of CD8+ Trm cells explains why these PEGPH20-treated tumors further respond to anti-PD-L1 treatment, establishing HA degradation as an obligated step in the conversion of residual mCRC, PEGPH20-resistant tumor cells, from an ICB refractory state to a sensitive one.
These results should be considered in the context of previous clinical trials using PEGPH20 for the treatment of pancreatic cancer (PDAC), another type of desmoplastic neoplasia. The initial rationale was based on the assumption that this treatment would result in improved drug delivery to solid tumors and prolonged survival, as shown in preclinical models.20,21,49 Unfortunately, these clinical studies gave mixed results, and although patients showed a higher response rate in the experimental arm, there was no improvement in the duration of response or progression-free or overall survival.50-53 However, what we show here is that PEGPH20 triggers a complete remodeling of the mCRC TME, impacting not only the levels of HA but also the CAF populations and the immune compartment. A similar degree of remodeling was reportedly achieved only when PEGPH20 was used in combination with GVAX immunotherapy in PDAC preclinical models.54 A potential explanation for the discrepant findings of the lack of effect of monotherapy with PEGPH20 in PDAC as compared to the positive effects in mCRC may rely on the differences between the TME of these two types of tumors. Thus, PDAC tumors are known to be immune-deserted, characterized by a paucity of T cells in either the parenchyma or the stroma of the tumor. To make these tumors responsive to immunotherapy would require a T cell priming agent in combination with stroma-targeting. In contrast, aPKC CRC tumors are immune-excluded and, therefore, characterized by the presence of abundant immune cells that do not penetrate the parenchyma of these tumors but instead are retained in the stroma that surrounds the tumor epithelium. The recruitment of pre-existing T cells is, therefore, the rate-limiting step that is targeted by PEGPH20 to allow the cancer-immunity response in mCRC. Therefore, PEGPH20 should be considered a valid strategy to treat mCRC as monotherapy and/or as an enabler of ICB. In this regard, it is important to emphasize that the dose of PEGPH20 utilized in the PDAC clinical trials was approximately 400-fold lower than the one used in PDAC preclinical models, indicating that to achieve clinical response in PDAC patients, much higher hyaluronidase doses would be required, which will be unfeasible due to toxicity. In marked contrast, we show here that mCRC is fully sensitive to the clinical doses of PEGPH20 (0.0375 mg/Kg), which strongly suggests that perhaps PDAC was not the right type of neoplasia to be treated with PEGPH20, whereas a better response would be obtained in patients with mCRC.
Another critical question to ensure the success of PEGPH20 in the clinic is the identification of better biomarkers for patient selection. In this regard, our data show that HA expression does not predict survival in CRC patients but that low aPKC levels combined with high HA levels define the cohort of CRC patients with the worst prognosis. Evidence presented here in two mCRC mouse models establishes that low aPKC/HA-positive mCRC patients will benefit from PEGPH20-based treatment. Our analysis demonstrates that the epithelial compartment of mCRC tumors is organized, echoing the hierarchy and cell types of the normal intestine. This includes the existence of tumor progenitor-like and differentiated-like populations.55-57 Thus, according to our data, aPKC-deficient serrated tumors originate from a highly proliferative cTA population, which evolves into a tumor cycling cell type expressing a signature previously identified in Lgr5− negative CRC tumor cells.34 Therefore, our data strongly support the notion that differentiated cells retaining progenitor features can be transformed to generate a serrated mCRC. In this regard, a recent scRNAseq study in human patients established that whereas conventional adenomas originated from the expansion of adult stem cells at the bottom of the intestinal crypt, serrated adenomas emerge from a more differentiated cell state through a metaplastic process whereby intestinal cells trans-differentiate to a gastric-fetal phenotype.35 The TFMC subpopulation identified in mCRC is what accounts for the most invasive and aggressive phenotype of this type of tumor and has symbiotic crosstalk with telocytes, which is the tumor fibroblast population with the most CAF features. The fact that PEGPH20 treatment completely ablated the telocyte subpopulation contributes to the eradication of the TFMC compartment and the reformatting of the mCRC TME towards a TRSC-trophocyte-dominated condition.
TRSCs originated from the TcTA population but, in contrast to the TFMCs, retain features of adult stemness such as a heightened WNT pathway, which is reduced in the TFMC population that is instead characterized by a YAP-driven signature. TRSCs are reminiscent of the Clu+ “revival stem cells” that emerge during non-tumor intestinal injury-regeneration processes.37 This cell type is in a cooperative connection with the trophocyte fibroblasts at the bottom of the crypt through a WNT-dependent network, resembling the requirements of adult intestinal stem cells.58,59 Since the non-tumor Clu+ “revival stem cells” hold the ability to repopulate the whole normal epithelium, we posited that the TRSCs would be able to recreate the entire mCRC upon the cessation of PEGPH20 treatment. In this regard, we show here that these cells are resistant to PEGPH20 treatment in vitro and that they account for a large proportion of the whole “persister” population that is not eradicated by PEGPH20 treatment. The fact that PEGPH20 promotes the accumulation of trophocytes in the mCRC TME further helps to feed TRSCs as a possible source of therapy resistance. Our findings indicate that persistent cancer cells in PEGPH20-treated tumors are enriched in IFN signatures, which contributes to the sensitivity of these PEGPH20-treated tumors to anti-PD-L1 therapy and suggests that, although PEGPH20 treatment profoundly represses mCRC tumorigenesis, combination with ICB will help prevent cancer rebound. Therefore, PEGPH20 reformats the mCRC TME from a TFMC-telocyte-driven scenario of aggressive tumorigenesis to a TRSC-trophocyte “persister” paradigm that can be, nonetheless, ablated by ICB treatment through the upregulation of immunosurveillance IFN-driven pathways by PEGPH20.
STAR★ METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jorge Moscat (jom4010@med.cornell.edu).
Materials Availability
Cell and mouse lines generated in this study are available from the Lead Contact upon request with a completed Materials Transfer Agreement.
Data and code Availability
The Quantseq, and scRNA-seq datasets generated during this study have been deposited to the GEO repository on the NCBI website (GEO: GSE207780, GSE207778, GSE207776, and GSE207779) and are publicly available as of the date of publication.
This paper reports data derived from published and publicly available datasets GEO: GSE132465, GSE166555, GSE178341, GSE14333, and GSE39582. These datasets are located on the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-β-actin | Sigma-Aldrich | Cat# A1978, RRID: AB_476692 |
| Rabbit anti-aPKCs | Abcam | Cat# ab59364; RRID: AB_944858 |
| Rabbit anti-CD8 | Abcam | Cat# ab209775; RRID: AB_2860566 |
| Rat anti-CD8 | BD Pharmingen | Cat# 557654, RRID: AB_396769 |
| Rabbit anti-CD19 | Cell Signaling Technology | Cat# 90176S, RRID:AB_2800152 |
| Rabbit anti-MRP8 | Abcam | Cat# ab92331, RRID: AB_2050283 |
| Rabbit anti-CD138 | Thermo Fisher Scientific | Cat# PA5-16918, RRID: AB_10979011 |
| Rabbit anti-FOXP3 | Cell Signaling Technology | Cat#12653, RRID: AB_2797979 |
| Rabbit anti-PDL1 | Cell Signaling Technology | Cat# 64988, RRID: AB_2799672 |
| Rat anti-CD45 | BD Parmingen | Cat# 550539, RRID: AB_2174426 |
| Mouse anti-panCK | Thermo Fisher Scientific | Cat# MA5-13156, RRID: AB_2174426 |
| Rabbit anti-panCK | Abcam | Cat# ab9377, RRID: AB_307222 |
| Rabbit anti-Ki67 | Cell Signaling Technology | Cat#12202, RRID: AB_2620142 |
| Rabbit anti-MUC2 | Abcam | Cat# ab272692, RRID: AB_2888616 |
| Rabbit anti-MUC5AC | Cell Signaling Technology | Cat#61193, RRID: RRID:AB_2799603 |
| Rabbit anti-ANXA10 | Abcam | Cat# ab213656, RRID: RRID:AB_2921231 |
| Rabbit anti-CLU | Abcam | Cat# ab184100, RRID: AB_2892532 |
| Goat anti-CLU | R&D systems | Cat# AF2747, RRID: AB_2083314 |
| Rabbit anti-PDGFRa | Abcam | Cat# ab203491, RRID: AB_2892065 |
| Mouse anti-PDGFRb | Abcam | Cat#ab69506, RRID:AB_1269704 |
| Rabbit anti-CD34 | Abcam | Cat# ab81289, RRID: AB_1640331 |
| Mouse anti-CD31 | Thermo Fisher Scientific | Cat# RB10333P1, RRID: AB_720501 |
| Rat anti-CD31 | Dianova | Cat#DIA-310, RRID:AB_2631039 |
| Rabbit anti-NRG1 | Abcam | Cat# ab191139, RRID: RRID:AB_2921232 |
| Mouse anti-Smooth muscle actin | Dako | Cat# M0851, RRID: AB_2223500 |
| Biotinylated recombinant HA-binding protein (HTI-601) | Halozyme | Provided by Halozyme Therapeutics |
| Goat anti-Mouse IgG1, secondary, HRP | Thermo Fisher Scientific | Cat# PA1-74421, RRID: AB_10988195 |
| Goat anti-Mouse IgG, secondary, HRP | Thermo Fisher Scientific | Cat# 31430, RRID:AB_228307 |
| Goat anti-Rabbit IgG, secondary, HRP | Thermo Fisher Scientific | Cat# 31461, RRID: AB_228347 |
| Goat anti-Rat IgG, secondary, HRP | Thermo Fisher Scientific | Cat# 62-9520, RRID: AB_87993 |
| Donkey anti-Rabbit IgG, secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042, RRID: AB_2534017 |
| Goat anti-Rabbit IgG, secondary, IRDye 800 | LI-COR Biosciences | Cat# 926-32211, RRID: AB_621843 |
| Goat anti-Mouse IgG1, secondary, IRDye 800 | LI-COR Biosciences | Cat# 926-32350, RRID: AB_2782997 |
| Goat anti-Mouse IgG, secondary, IRDye 800 | LI-COR Biosciences | Cat# 926-32210, RRID: AB_621842 |
| InVivoMAb rat IgG2b isotype control | BioXCell | BE0090, RRID:AB_1107780 |
| InVivoMAb anti-mouse PD-L1 (B7-H1) | BioXCell | BE0101, RRID:AB_10949073 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | BioXCell | BP0164, RRID:AB_10949609 |
| Biological samples | ||
| Human Colorectal cancers (CRC) | Osaka City University Hospital, Osaka, JAPAN | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| HBSS (no calcium, no magnesium) | GIBCO | Cat# 14175095 |
| Dimethyl Sulfoxide | Fisher BioReagents | Cat# BP2311 |
| Chloroform | Sigma-Aldrich | Cat# 288306 |
| Methanol | Sigma-Aldrich | Cat# 1424109 |
| Tryple Express | Life Technologies | Cat# 12605-010 |
| HEPES | Gibco | Cat# 15630080 |
| PBS (no calcium, no magnesium) | Gibco | Cat# 10010-023 |
| Quick-RNA Miniprep Kit | Zymo Research | Cat# R1054 |
| RNAlater Stabilization Solution | Invitrogen | Cat# AM7021 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596018 |
| Advanced DMEM/F12 | Thermo Fisher Scientific | Cat# 12634010 |
| RPMI | Corning | Cat#15-040-CV |
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat# 35050061 |
| Penicillin-Streptomycin solution | Corning | Cat# 30-002-CI |
| Y-27632 | Tocris | Cat# 1254 |
| B27 Supplement | Thermo Fisher Scientific | Cat# 17504001 |
| B27 Supplement minus vitamin A | Thermo Fisher Scientific | Cat# 12587001 |
| N2 Supplement | Thermo Fisher Scientific | Cat# 17502048 |
| Murine EGF | Thermo Fisher Scientific | Cat# PMG8045 |
| Human EGF | Peprotech | Cat#AF-100-15 |
| Human FGF-2 | Peprotech | Cat#100-18B |
| Human IGF-1 | Biolegend | Cat#590906 |
| Recombinant human Wnt-3a | R&D systems | Cat#5036-WN/CF |
| A83-01 | Sigma-Aldrich | Cat#SML078 |
| Human [Leu15]-Gastrin I | Sigma-Aldrich | Cat# G9145 |
| Recombinant murine R-Spondin 1 | R&D systems | Cat# 3474-RS-050 |
| Recombinant murine Noggin | Peprotech | Cat# 250-38 |
| Recombinant murine M-CSF | Peprotech | Cat# 315-02 |
| Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix | Corning | Cat# 356230 |
| Liberase™ TM Research Grade | Sigma-Aldrich | Cat# 5401119001 |
| Collagenase A | Sigma-Aldrich | Cat# 10103586001 |
| Dispase II | Sigma-Aldrich | Cat# #D4693 |
| DNase I | Sigma-Aldrich | Cat# 04716728001 |
| 4-OH-tamoxifen | Millipore-Sigma | Cat# H7904 |
| TrueCut Cas9 Protein v2 | Thermo Fisher Scientific | Cat# A36498 |
| DAPI | Life Technologies | Cat# D1306 |
| Streptavidin, secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# S11226 |
| Streptavidin, secondary, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# S11223 |
| Trypan Blue Solution, 0.4% | Gibco | Cat# 15250061 |
| Critical commercial assays | ||
| Neon™ 10μl Electroporation Kit | Thermo Fisher Scientific | Cat# MPK1096 |
| Chromium Single Cell 3′ Gene Expression Solution v2 | 10X Genomics | Cat#PN-120237 |
| EasySep™ Dead Cell Removal (Annexin V) Kit | STEMCELL Technologies | Cat# 17899 |
| VECTASTAIN® Elite® ABC-HRP Kit | Vector | Cat# PK-6100 |
| Automated Opal 7-Color IHC Kit | AKOYA BIOSCIENCES | Cat# NEL821001KT |
| Manual Opal 4-Color IHC Kit | AKOYA BIOSCIENCES | Cat# NEL810001KT |
| Trichrome Stain (Masson) Kit | Sigma-Aldrich | Cat# HT15 |
| Corning Biocoat Control Inserts | Corning | Cat# 354578 |
| Alexa Fluor™ 488 Tyramide Superboost™ Kit | Thermo Fisher Scientific | Cat# B40932 |
| Deposited data | ||
| sc-RNA-seq | This study | GEO: GSE207780 |
| 3’ RNA-seq (Prkcifl/fl Prkczfl/fl Villin-Cre-Veh or -PEGPH20 treated) | This study | GEO: GSE207778 |
| 3’ RNA-seq (sgPrkci/Prkcz and sgC MTOs) | This study | GEO: GSE207776 |
| 3’ RNA-seq (sgPrkci/Prkcz MTOs-Veh or -PEGPH20 treated) | This study | GEO: GSE207779 |
| Raw Data | This study; Mendeley Data | https://doi.org/10.17632/vbzbzhrw98.1 |
| Single cell 3′ RNA sequencing from patients with CRC | Lee et al.26 | GEO: GSE132465 |
| Single cell 3′ RNA sequencing from patients with CRC | Uhlitz et al.40 | GEO: GSE166555 |
| Single cell 3′ RNA sequencing from patients with CRC | Pelka et al.41 | GEO: GSE178341 |
| Microarray from primary CRC tumors | Jorissen et al.73 | GEO: GSE14333 |
| Microarray from primary CRC tumors | Marisa et al.74 | GEO: GSE39582 |
| TCGA-COREAD | cBioportal | http://www.cbioportal.org/index.do |
| Experimental models: Cell lines | ||
| Human CRC (sgPRKCI/PRKCZ) | This study | N/A |
| Mouse tumor organoid (MTO) | Tauriello et al.17 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Prkcifl/fl Prkczfl/fl | Nakanishi et al.13 | N/A |
| C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Oligonucleotides | ||
| Real-time PCR primers | This manuscript | Table S1 |
| CRISPR guides | This manuscript | Table S2 |
| Software and algorithms | ||
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/scientificsoftware/ |
| QuPath v.0.1.3 | Queen’s University, Belfast, Northern Ireland | https://qupath.github.io |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Zen blue | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| RStudio (1.1.456) | R Core Team | https://www.r-project.org/ |
| R | R Core Team | https://www.r-project.org/ |
| GSVA; version 1.26.0 | Bioconductor | https://github.com/rcastelo/GSVA |
| GSEA (v4.1.0) | Broad Institute | http://www.broadinstitute.org/gsea/index.jsp |
| BaseSpace | Illumina | https://basespace.illumina.com/ |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| CMScaller v0.99.1 | Eide et al.72 | https://github.com/peterawe/CMScaller |
| CellPhoneDB v.2.0.0 | Efremova et al.43 | https://www.cellphonedb.org |
| GenePattern | Broad Institute | https://cloud.genepattern.org/gp/pages/login.jsf |
| Lexogen QuantSeq DE 1.3.0 | BlueBee Cloud | https://www.bluebee.com |
| Cell Ranger (v3.0) | Langmead et al.75 10X genomics | http://software.10xgenomics.com/single-cell/overview/welcome |
| Seurat 3.0 | Stuart et al.76 | https://github.com/Satijalab/seurat |
| kb_python | Bray et al.69;Melsted et al.70 | https://github.com/pachterlab/kb_python |
| scVelo package | Bergen et al.71 | https://scvelo.readthedocs.io/VelocityBasics/ |
| Harmony | Korsunsky et al.67 | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
| PhenoChart Whole Slide Viewer | AKOYA BIOSCIENCES | https://www.akoyabio.com/support/software/phenochart-whole-slide-viewer/ |
| CaseViewer | 3DHistech, Budapest, Hungary | https://www.3dhistech.com/solutions/caseviewer/ |
| Other | ||
| EVOS FL Auto Imaging System | Thermo Fisher Scientific | N/A |
| EVOS M5000 Imaging System | Thermo Fisher Scientific | N/A |
| TissueLyser II | QIAGEN | Cat# 85300 |
| NanoDrop 1000 spectrophotometer | Thermo Fisher Scientific | N/A |
| Zeiss LSM 710 NLO Confocal Microscope | Zeiss | N/A |
| Neon™ Transfection System | Thermo Fisher Scientific | Cat# MPK5000 |
| Pannoramic Scanner | 3DHistech, Budapest, Hungary | N/A |
| Vectra Polaris Automated Quantitative Pathology Multispectral Imaging System | AKOYA BIOSCIENCES | N/A |
Original raw data have been deposited in Mendeley Data (https://doi.org/10.17632/vbzbzhrw98.1).
Experimental Model and Subject Details
Mice
Animal handling and experimental procedures conformed to institutional guidelines and were approved by the Sanford-Burnham-Prebys Medical Discovery Institute Institutional Animal Care and Use Committee, and by the Weill Cornell Medicine Institutional Animal Care and Use Committee. Prkcif/fPrkczf/f;Villin-Cre, Prkcif/fPrkczf/f;Villin-Cre and Villin-CreERT2 mice were previously described.13,60,61 Prkcif/fPrkczf/f;Villin-CreERT2 mice were generated by breeding Prkcif/fPrkczf/f mice with Villin-CreERT2 (Jackson Laboratory, stock number 020282) mice. All mouse strains were generated in a C57BL/6 background and were born and maintained under pathogen-free conditions. For PEGPH20 treatment, 11-week-old Prkcif/fPrkczf/f;Villin-Cre were retro-orbitally injected twice a week for 3 weeks with a dose of 0.0375 mg/kg of PEGPH20 (Halozyme Therapeutics), until they were sacrificed. Control mice were treated with vehicle. Tumor load was calculated by multiplying tumor number and tumor diameter. For xenograft experiments, 6-7week-old male C57BL/6 were purchased from Charles River Labs (Wilmington, MA, USA). All mice were maintained on food and water ad libitum and were age-matched and co-housed for all experiments. Mice were sacrificed and small intestine, colon, tumors, or other organs were collected for analysis. All genotyping was done by PCR. Age- and sex-matched mice were used for all experiments.
Human samples
For CRC samples, a total of 343 CRC patients (male, n = 176; female, n=167) who had undergone a resection of the primary tumor at Osaka City University Hospital were analyzed. Baseline characteristics are shown in Figure S1. CRC tissues were obtained from each patient and none of the patients had undergone preoperative radiation or chemotherapy. This study was approved by the Osaka City University Ethics Committee and written informed consent was obtained from patients. De-identified human samples were sent to Weill Cornell Medicine and used for histological analyses. The study was approved by the IRB Committee of Weill Cornell Medicine.
Cell lines
Patient-derived organoids (PDOs) were isolated from patient-derived xenografts (PDXs). First, surgically resected colorectal cancer tissues were transplanted into NSG mice to generate PDXs. After engraftment, PDOs were isolated and cultured. Briefly, PDX tissues were washed vigorously with ice-cold phosphate-buffered saline (PBS) and minced into 1 mm3 fragments using surgical scalpel blades. The fragments were digested with digestion buffer containing 25 mg/ml Collagenase A (Sigma), 25 mg/ml Dispase II (Sigma), and 500 μ/ml DNase I (Sigma) for 10 min at 37C. The supernatant was filtered through a 70 μm cell strainer and centrifuged at 400G for 5 min. The pellet was embedded in Matrigel and applied onto 12-well plates as 20 μl droplets. Organoids were cultured in advanced Dulbecco’s Modified Eagle’s Medium/F12 (Thermo Fisher Scientific) supplemented with 50% Wnt-3A conditioned medium, 50 ng/ml recombinant human EGF (Peprotech), 0.25% Noggin-FC conditioned medium, 10% mouse R-spondin 1-FC conditioned medium, 500nM A83-01, 100 ng/ml recombinant human IGF-1 (BioLegend), 50 ng/ml recombinant human FGF-2 (Peprotech), 10 μM Y-27632, 1x B27 supplement (Life Technologies), 10 nM human [Leu15]-Gastrin I (Sigma), 1x Glutamax (Life Technolgies), 1x HEPES (Life Technologies), and 1x Penicillin/Streptomycin (Corning). After the first passage, Wnt-3a and R-spondin 1 were removed from the media. MTO were obtained from Dr. Eduard Batlle (Institute for Research in Biomedicine, Barcelona, Spain) and previously described 17. MTO were cultured in advanced DMEM/F12 medium supplemented with 10 mM HEPES, Glutamax, B-27 (all Life Technologies), 50 ng/ml recombinant human EGF (Peprotech). Normal intestinal organoids were cultured in Advanced DMEM/F12 containing 10 mM HEPES, 1X Glutamax, 1X N2 supplement, 1X B27 supplement, 50 ng/ml EGF, 1000 ng/ml R-spondin 1, 100 ng/ml Noggin, and 10 μM Y-27632 in an atmosphere of 95% air and 5% CO2.
Intestinal fibroblasts were isolated from Prkcif/fPrkczf/f (Fib-WT) and Prkcif/fPrkczf/f;Villin-Cre (Fib-DKO) mice and previously described.12 Cultures were tested weekly for mycoplasma contamination.
METHOD DETAILS
Colonoscopy-guided injection and Xenograft Experiments
For colonoscopy-guided orthotopic injection in the colon submucosa using MTO, a 10 μl organoid suspension of equivalent to 40-50 MTO admixed with 10% Matrigel (Corning) was directly injected into the colonic submucosa of 7 weeks old C57BL/6 mice. 4 weeks after implantation, tumors were collected and analyzed histologically. Orthotopic tumor sizes were measured by caliper after mice were sacrificed. For xenografts using MTO, a 100 μl cell suspension of equivalent to 0.5 Ö 106 cells of MTO-sgPrkci/Prkcz or MTO-sgC admixed with 30% Matrigel (Corning) was directly injected subcutaneously into the right flank of 7 weeks old C57BL/6 mice. Tumors were allowed to grow for 14 days, and mice were randomly divided to receive vehicle, n=20, or PEGPH20 (0.0375 mg/kg, 2 days each week) via retro-orbital injection, n=18. Tumors were measured twice a week. Mice were euthanized three weeks after treatment and tumors were analyzed histologically. For the combination therapy of anti-PD-L1 (Clone B7-H1, Bioxcell) antibody and PEGPH20, mice were injected with a dose of 5 mg/kg (body mass) of anti-PD-L1 antibody in combination with PEGPH20 (n=15) twice a week for 4 weeks until they were sacrificed.
Intrasplenic injection
Intrasplenic injections were used for liver colonization by introducing dissociated organoids (single cells) into the portal circulation of 10 weeks old C57BL/6 mice; MTO cells were counted and suspended in HBSS for injection, using 5 × 104 cells in 70 μl per mouse. Tumors were allowed to grow for 14 days, and mice were randomly divided to receive vehicle, n=7, or PEGPH20 (0.0375 mg/kg/, 2 days each week) via retro-orbital injection, n=7. For the combination therapy of anti-PD-L1 (Clone 10F-9G2, Bioxcell) antibody and PEGPH20, mice were injected with a dose of 5 mg/kg (body mass) of anti-PD-L1 antibody in combination with PEGPH20 (n=7) twice a week for 2.5 weeks. For the combination therapy of anti-CTLA-4 (Clone 9-D9, Bioxcell) antibody and PEGPH20, mice were injected with a dose of 100 μg of anti-CTLA-4 antibody in combination with PEGPH20 (n=6) twice a week for 2.5 weeks. Mice were euthanized 1 week after treatment. Metastasis load was calculated as the total volume of all liver metastases.
Cell Culture Experiments
All patient-derived and mouse organoid lines contain exclusively epithelial cells, with no contamination of stromal cells as verified by staining with epithelial markers. To perform PRKCI and PRKCZ editing in PDOs, single-guide RNA sequences targeting PRKCI and PRKCZ (Table S2), were purchased from Synthego and transduced into PDOs with recombinant Streptococcus pyogenes Cas9 protein (Truecut Cas9 Protein v2, Thermo. PRKCI and PRKCZ, were knocked out using the Neon Transfection System 1 (Invitrogen) following the manufacturer’s protocol and single clones were expanded and screened by protein immunoblotting. To knock out Prkcz in mouse MTO-sgPrcki,46 a 20-nucleotide single-guide RNA (sgRNA) sequence targeting mouse Prkcz was designed using the CRISPR design tool at http://crispr.mit.edu/ (Table S2). The sgRNA was cloned in the lentiCRISPR v2 vector (Addgene plasmid #52961) and transduced into MTO-sgPrcki MTO. Cells were selected with puromycin and screened for PKCζ by immunoblotting. For the deletion of Prkci and Prkcz in normal organoids, 500 nM 4-OH-tamoxifen was added the second day after organoids were passaged. For PEGPH20 treatment in vitro using MTO, 300 organoids per Matrigel dome were seeded in a p12 well plate (Thermo Fisher Scientific) and cultured as described above. PEGPH20 at 2.5 μg/ml was added every other day for 6 days for a total of three doses. For experiments with conditioned medium from intestinal fibroblasts, 1,6 x 105 fibroblasts were seeded in a 6-well plate in the presence of vehicle or 2.5 μg/ml PEGPH20 for 3 days. Supernatants were collected, centrifuged, and filtered before use.
Bone marrow macrophages differentiation and migration assay
BMDMs were prepared by centrifuging mouse leg bones in RPMI supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. After being filtered through a 70 μm nylon mesh, cells were centrifuged plated in non-treated 10 cm dishes, and incubated for 24 hr. Supernatants containing bone marrow precursors were collected, centrifuged at 1,500 rpm, and cultured in differentiation media (RPMI supplemented with 20% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 0.5 mM β-mercaptoethanol and 50 ng/ml recombinant murine MCSF (Peprotech). 2.5 × 104 BMDMs (7 days in culture) were plated in a transwell chamber (Corning Biocoat control inserts) with 8 μm membrane. BMDMs were allowed to migrate for 8 hours at 37°C, 5% CO2 in the presence of conditioned medium from intestinal fibroblasts. Cells were fixed in cold methanol and stained with crystal violet. Percentage of area was quantified using ImageJ.
Histology, Immunohistochemistry, and Immunofluorescence
Tissues from indicated mice were isolated, rinsed in ice-cold PBS, fixed in 10% neutral buffered formalin overnight at 4°C, dehydrated, and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin (H&E). For immunohistochemistry, sections were deparaffinized, rehydrated, and then treated for antigen retrieval. After blocking in Protein Block Serum-Free solutions (DAKO), tissues were incubated with primary antibody overnight at 4°C followed by incubation with biotinylated secondary antibody. Endogenous peroxidase was quenched in 3% H202 in water for 10 min at room temperature. Antibodies were visualized with avidin-biotin complex (Vectastain Elite; Vector Laboratories) using diaminobenzidine as the chromogen. Stained sections were analyzed with a Zeiss light microscope supplemented with Zen 3.3 Bule edition software. For tissue immunofluorescence, the signal was visualized using the OPAL™ 4-Color Manual IHC Kit (Akoya Biosciences, MA), which allows simultaneous detection of multiple targets in the same image. Fluorophores Opal 520, Opal 570, and TSA Plus Cyanine 5 were used, and the sections were counterstained with Spectral DAPI. For HA staining, we have used the biotinylated HA probe HTI-601 (Halozyme), but there are multiple commercial alternatives that can be used to detect HA with similar results: biotin-labeled HA-binding protein [rhAggrecan aa20-675/His (NSO/7), biotin, R&D Systems]; biotinylated hyaluronic acid binding protein [HABP – AMSBIO cat # AMS.HKD-BC41]; HABP [EMD Millipore, cat#38591]. For cell immunofluorescence, after fixation and blocking, the cells were incubated with primary antibody overnight at 4°C, followed by incubation with biotinylated secondary antibody, tyramide signal amplification, and DAPI. Coverslips were mounted and imaged under confocal microscopy.
VECTRA staining
Primary antibody dilutions were optimized individually at H220 retrieval using the Leica BondRx Automated IHC stainer and evaluated by a board-certified pathologist for specificity. Multiplexed staining was optimized and then performed using the Automated Opal 7-Color IHC Kit (NEL821001KT) from Akoya Biosciences. A library for spectral separation was generated by staining control tissue with each opal fluor conjugated to CD20. Slides were imaged in the Vectra Polaris Automated Quantitative Pathology Multispectral Imaging System. Exposure times were maximized under the constraint that no pixel saturates the detector.62,63
Tissue microarray and histological analyses
Image analysis was processed in GuPath v.0.1.3 (Gueen's University, Belfast, Northern Ireland). Scanned TMA slides were dearrayed and preprocessed as previously described.64 After dearraying, all cores were examined and those with either no tumor represented or with artefacts (tissue broken, for example) were excluded. Cell detection was determined for each core, and the total number of positive cells using DAPI staining as a cell reference was assessed. H-score, calculated based on the extent and intensity of the staining, was used for aPKCs and ANXA10 scoring. CRC patients were categorized into high and low aPKCs or ANXA10 expression groups using 90 or 60 H-score as the cut-off respectively, selected by an experienced pathologist. For HA and MUC5AC expression, CRC samples were considered positive when at least 1-30% of stromal or epithelial cells respectively were stained. For in vivo experiments, HA and Masson’s trichrome staining areas were quantified using the Color deconvolution plugin in ImageJ using H DAB or Masson’s trichrome mode respectively.
Whole-mount organoid staining
Organoid staining was performed as previously described.65 Briefly, the organoid suspension was washed once in PBS before adding 500 μl of ice-cold cell recovery solution (Corning) per well. Organoids were incubated for 30-60 minutes at 4°C on a horizontal shaker until Matrigel was dissolved. Organoids were resuspended 5-10 times transferred to a pre-coated 15 ml falcon tube (1%BSA-PBS) and centrifuged for 3 min at 70g, 4°C. The organoid pellet was resuspended in 1 ml of ice-cold PFA (4%) and organoids were fixed for 45 min. 10 ml of ice-cold PBS with 0.1% Tween-20 was added, incubated for 10 min, and centrifuged at 70g for 3 min at 4°C. The organoid pellet was resuspended in organoid wash buffer and transferred to a 24-well plate for blocking and overnight antibody incubation. Organoids were washed for 2 hours three times and then incubated overnight with secondary antibodies in the wash buffer with 1 μg/ml DAPI. The next day, the washing steps were repeated, and the organoids were mounted onto 35 mm glass-bottom dishes and imaged under confocal microscopy.
Immunoblotting Analysis
Cells for protein analysis were lysed in RIPA buffer (20 mM Tris-HCl, 37 mM NaCl2, 2 mM EDTA, 1% Triton-X, 10% glycerol, 0.1% SDS, and 0.5% sodium deoxycholate) with phosphatase and protease inhibitors. Protein concentration in lysates were determined by using Protein Assay Kit (Bio-Rad). Cell extracts were denatured, subjected to SDS-PAGE, transferred to PVDF membranes (GE Healthcare). After blocking with 5% nonfat dry milk in Tris-buffered saline and 0.1% Tween (TBS-T), the membranes were incubated with the specific antibodies (as listed in Key Resources Table) overnight at 4°C. After 2 h incubation with the appropriate fluorochrome-conjugated antibodies, the immune complexes were detected by Near-infrared fluorescence (LI-COR).
RNA Extraction and Analysis
Total RNA from cultured organoids was extracted using TRIZOL reagent (Invitrogen) and purified by the RNeasy Mini Kit (GIAGEN), followed by DNase treatment. After quantification using a Nanodrop 1000 spectrophotometer (Thermo Scientific), RNA was either processed for RNA-seq or reverse-transcribed using random primers and MultiScribe Reverse Transcriptase (Applied Biosystems). Gene expression was analyzed by amplifying 500 ng of the complementary DNA using the CFX96 Real Time PCR Detection System with SYBR Green Master Mix (BioRad) and primers described in Table S1. The amplification parameters were set at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s (40 cycles total). Gene expression values for each sample were normalized to the 18S RNA.
3’ RNA-seq Preparation and Sequencing
Total RNA was extracted using Quick-RNA MiniPrep kit (Zymo Research). Libraries were prepared from 200 ng of total RNA using the QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina from Lexogen, and optional UMIs (Vienna, Austria). Barcoded libraries were pooled, and single end sequenced (1X75) on the Illumina NextSeq 500 using the High output V2.5 kit (Illumina Inc., San Diego CA).
10x Library Preparation and Sequencing
Tumors were minced thoroughly and digested by 0.5 mg/ml Liberase TH (Sigma) for 30 minutes at 37°C. Dead cells were removed by Annexin V (STEMCELL technologies). scRNA-seq libraries were generated using the Chromium Single Cell 30 Reagent Kit v2 (10X Genomics). Cells were loaded onto the 10X Chromium Single Cell Platform (10X Genomics) at a concentration of 2,000 cells per μl (Single Cell 3’ library and Gel Bead Kit v.2) as described in the manufacturer’s protocol (10x User Guide, Revision B). Generation of gel beads in emulsion (GEMs), barcoding, GEM-RT clean-up, complementary DNA amplification and library construction were all performed as per the manufacturer’s protocol. Individual sample quality was checked using a Bioanalyzer Tapestation (Agilent). Qubit was used for library quantification before pooling. The final library pool was sequenced on an Illumina NovaSeq6000 instrument using a S1 flow cell. Average cell recovery for Prkcif/fPrkczf/f;Villin-Cre tumors was 104,954 cells with a total of 209,907 cells captured at a mean depth of 14,568 read per cell and 895 mean genes per cell.
Analysis of scRNA sequencing data
For scRNA-seq, raw sequence reads were quality-checked using FastQC software. The Cell Ranger version 2.1.1 software suite from 10X Genomics (https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest) was used to process, align, and summarize unique molecular identifier (UMI) counts against the mouse mm10 assembly reference genome analysis set, obtained from the University of California Santa Cruz (UCSC). Raw, unfiltered count matrices were imported into R for further processing. Raw UMI count matrices were filtered using the Seurat v 3.0 R package66 to remove: barcodes with very low (less than 200, empty wells) and very high (more than 3000, probably doublets) total UMI counts; matrices for which a high percentage of UMIs originated from mitochondrial features (more than 12%); and matrices for which fewer than 250 genes were expressed. Subsequently, the data were normalized using the SCTransform function, regressing out the following variables: total number of UMIs per cell and percentage of mitochondrial UMIs. Following normalization, the principal components were computed. The top principal components were identified using the ElbowPlot function and used for the UMAP dimensionality reduction. For clustering, we used Seurat in combination with Harmony software67 to correct the potential effects of technical differences between sequencing batches. The RunHarmony(), RunUMAP(), FindNeighbors(), and FindClusters() functions were run for clustering and the percentage of mitochondrial features was considered to be a source of unwanted variation and was regressed out using the Seurat package. Genes specifically expressed in each cluster were identified with the FindAllMarkers() function and the Wilcoxon test labeling the different populations using the genes differentially up-regulated in each population. For the differential gene expression analysis, the FindMarker() function with default parameters was run for comparison between populations. The cell groups were annotated based on the marker gene analysis and canonical markers from the literature (Table S3). The gene set enrichment analysis was performed using the results of the differentially expressed gene analysis between populations using Preranked GSEA analysis with default parameters (permutations=1000, enrichment score=weighted) and MSigDB H.all.v7.0.symbols (H), c2.all.v7.0.symbols (C2) and c5.all.v7.0.symbols (C5) collections. The scoring for the indicated signatures was performed using the AddModuleScore function in Seurat with default parameters. Single-cell RNA-seq data of CRC patients were obtained from GSE132465, GSE166555 and GSE178341, and processed as described above. The “TcTA”, “TRSC”, “TFMC” and “TGC” signatures (Table S3) were generated based on a list of the Top 50 upregulated genes for each tumoral population using FindAllMarkers() function in Seurat. Overlapping genes between populations were removed and mapped to human orthologues. GSE132465 was used to generate CRC TEC and hCRC CAF signatures based on a list of differentially expressed genes (upregulated genes, pdj<0.05) between tumor vs normal endothelial cells (n=84) and tumor vs normal fibroblast (n=122) respectively. Gene sets used for signature scoring are listed in Table S3 and when necessary, mapped to mouse orthologues using Ensembl BioMart. The visualization of the indicated signature score by Violin Plot and the statistical analysis (T-test) was performed using the ggplot2 package in R software.
Single-cell CNV analysis
Single-cell CNV profiles were inferred using the inferCNV R package (v1.2.1), which computes gene expression intensities across genomic positions from malignant cells as compared to a set of reference cells (goblet cells from non-tumor epithelial scRNAseq data). Input data were prepared as recommended by inferCNV authors. The algorithm was run with the following arguments: cutoff=0.1, cluster_by_groups=TRUE, denoise=TRUE, HMM = TRUE. The feature vectors from inferCNV were exported and visualized on the matching UMAP in Seurat using the add_to_seurat () function. We separated normal epithelial cells from tumor epithelial cells based on sample origin and CNVs. A small number of tumor cells clustered with normal cells and vice versa were eliminated.
CellPhoneDB analysis
CellPhoneDB was used to identify ligand-receptor interactions in scRNAseq data. After identifying different cell types in our scRNA-seq as described above, we followed recommended procedures for the preparation of input files using CellPhoneDB v.2.0.0 43. We updated the original CellPhoneDB repository with novel interactions and complexes curated from literature using ‘cellphonedb database generate’ command. All CellPhoneDB statistical analysis were performed with this updated database and percentage cell expression threshold of 5%. Cell-cell interactions heatmaps and Dot plots showing number of interactions, Log2 mean (Molecule 1, Molecule 2) and log10(p value), were generated using CellphoneDB.
scRNA-seq trajectory analysis
For RNA velocity analysis,68 loom files were generated using kb_python.69,70 The velocity index (Mus Musculus.GRCm38.98) was built using kb ref and the flag ‘lamanno’. Pseudo alignment and quantification was performed using kb count and the following parameters: technology: 10xv3 and workflow lamanno. Kb count uses bustools for quantification and return unfiltered and filtered loom files which were used for downstream analysis using scVelo.71 Briefly, genes were filtered and normalized using the filter_and_normalize function with the following parameters: min_shared_counts = 20 and n_top_genes = 2000. First and second-order moments (means and uncentered variances) were computed using the moments function, with n_pcs = 30 and n_neighbors = 30. RNA velocities were calculated with the velocity function setting mode = “stochastic”.
Bulk RNAseq analysis
For 3’RNA-Seq, read data was processed with the BlueBee Genomics Platform (BlueBee, San Mateo, CA). RNA-seq data of The Cancer Genome Atlas (TCGA) Colorectal Adenocarcinoma (Tumor Samples with mRNA data (RNA Seq V2 RSEM), 592 tumor samples from 592 patients) was downloaded through cBioportal (http://www.cbioportal.orq/index.do). GenePattern (https://genepattern.broadinstitute.org/gp/pages/index.jsf) was used to collapse gene matrix files (CollapseDataset module) or to assess the statistical significance of differential gene expression (DESeq2). Genes were sorted by log2 FC>0.3 and adj>0.05. Volcano plot representation for differentially expressed genes was generated using VolcaNoseR (https://huygens.science.uva.nl/VolcaNoseR/). Gene Set Enrichment Analysis (GSEA) was performed using GSEA 4.0 software (http://www.broadinstitute.org/gsea/index.jsp) with 1000 gene-set permutations using the gene-ranking metric t-test with the collections h.all.v6.1.symbols (H), c2.all.v6.1.symbols (C2), c5.all.v6.1.symbols (C5), or customized gene signatures (Table S3). For ASHIDA_CAF signature, differential gene expression analysis was performed as indicated above between CAF and NAF from GSE34312. Genes were sorted by log2 FC>0.5 and adj>0.05 . The classification of TCGA CRC patients based on the consensus molecular subtype7 was performed using CMScaller72 R package with default parameters (false discovery rate (FDR) = 0.5, seed = 1, RNAseq=TRUE). TCGA CRC patients were separated into two groups using the top 25 percentile and bottom 25 percentile of PRKCI and PRKCZ expression. The Microenvironment Cell Populations (MCP)-counter algorithm (webMCP-counter) was used as an independent bioinformatics tool to assess stromal cell enrichment between groups. HAS score for each patient was calculated by computing the average of HAS1, HAS2, and HAS3 expression and the gene set variation analysis (GSVA) algorithm, using standard settings. TCGA CRC patients were separated into two groups according to the median for the HAS score and compared by GSEA as described above.
Quantification and statistical analysis
All the statistical tests were justified for every figure. All samples represent biological replicates. Data are presented as the mean ± SEM. Statistical analysis was performed using GraphPad Prism 8 or R software environment (http://www.r-project.org/). Pairwise differences were measured using two-tailed independent student’s t-tests. If the data did not meet this test, a Mann-Whitney U-test was used. Statistical significance between groups of 3 or more was determined by a one-way or two-way ANOVA, followed by the Tukey’s multiple comparison test. Differences in Kaplan Meier plots were analyzed using the Log-rank test. The chi-square test or Fisher’s exact test was used to determine the significance of differences between covariates. Logistic regression analysis was employed to estimate univariate and multivariate odds ratio and 95% confidence interval (CI). Values of p < 0.05 were considered significantly different.
Supplementary Material
Table S3. Compiled gene signatures and supporting literature, related to STAR Methods.
HIGHLIGHTS.
Reduced aPKC drives HA deposition, the mCRC phenotype, and predicts poor survival
HA enhances malignancy and promotes CAF and epithelial heterogeneity in mCRC
Immunosuppression is maintained by cross-compartment interactions driven by HA
HA depletion blocks mCRC tumorigenesis and liver metastasis enabling immunotherapy
ACKNOWLEDGMENTS
Research was supported by grants by the National Cancer Institute of the National Institutes of Health under awards numbers: R01CA265892 and R01CA250025 to J.M.; R01CA246765 to M.T.D.-M; R50CA265332 to A.D. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Project support for this research was provided in part by the Center for Translational Pathology in the Department of Pathology and Laboratory Medicine, Weill Cornell Medicine. This work was funded in part thanks to a generous gift from Alan Braynin and the Torrey Coast Foundation. We thank Tavonna Bryant and Tararin Nikomborirak and the personnel of the Genomics and Optical Microscopy Cores at Weill Cornell Medicine, and of the Molecular Cytology Core at Memorial Sloan Kettering Cancer Center for technical assistance. J.M. and M.T.D.-M. are Homer T. Hirst III Professors of Oncology in Pathology.
Footnotes
DECLARATION OF INTERESTS
A.M.-O., A.D., Y.N., J.M., M.T.D.-M. are coinventors of Weill Cornell Medicine patent applications covering methods for treating mesenchymal colorectal cancer. J.M., M.T.D.-M., and D.S. are co-founders of Zelambio. All other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, and Jemal A (2020). Colorectal cancer statistics, 2020. CA Cancer J Clin 70, 145–164. 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 2.Martini G, Dienstmann R, Ros J, Baraibar I, Cuadra-Urteaga JL, Salva F, Ciardiello D, Mulet N, Argiles G, Tabernero J, and Elez E (2020). Molecular subtypes and the evolution of treatment management in metastatic colorectal cancer. Ther Adv Med Oncol 12, 1758835920936089. 10.1177/1758835920936089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huyghe JR, Harrison TA, Bien SA, Hampel H, Figueiredo JC, Schmit SL, Conti DV, Chen S, Qu C, Lin Y, et al. (2021). Genetic architectures of proximal and distal colorectal cancer are partly distinct. Gut 70, 1325–1334. 10.1136/gutjnl-2020-321534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Missiaglia E, Jacobs B, D'Ario G, Di Narzo AF, Soneson C, Budinska E, Popovici V, Vecchione L, Gerster S, Yan P, et al. (2014). Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann Oncol 25, 1995–2001. 10.1093/annonc/mdu275. [DOI] [PubMed] [Google Scholar]
- 5.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al. (2017). Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 18, 1182–1191. 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, et al. (2019). Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491. 10.1126/science.aau0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. (2015). The consensus molecular subtypes of colorectal cancer. Nat Med 21, 1350–1356. 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, and Tabernero J (2017). Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer 17, 268. 10.1038/nrc.2017.24. [DOI] [PubMed] [Google Scholar]
- 9.De Sousa EMF, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van Leersum R, Bijlsma MF, et al. (2013). Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med 19, 614–618. 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 10.Fessler E, and Medema JP (2016). Colorectal Cancer Subtypes: Developmental Origin and Microenvironmental Regulation. Trends Cancer 2, 505–518. 10.1016/j.trecan.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DV, Byrom D, et al. (2015). Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet 47, 320–329. 10.1038/ng.3225. [DOI] [PubMed] [Google Scholar]
- 12.Kasashima H, Duran A, Martinez-Ordonez A, Nakanishi Y, Kinoshita H, Linares JF, Reina-Campos M, Kudo Y, L'Hermitte A, Yashiro M, et al. (2021). Stromal SOX2 Upregulation Promotes Tumorigenesis through the Generation of a SFRP1/2-Expressing Cancer-Associated Fibroblast Population. Dev Cell 56, 95–110 e110. 10.1016/j.devcel.2020.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakanishi Y, Duran A, L'Hermitte A, Shelton PM, Nakanishi N, Reina-Campos M, Huang J, Soldevila F, Baaten BJG, Tauriello DVF, et al. (2018). Simultaneous Loss of Both Atypical Protein Kinase C Genes in the Intestinal Epithelium Drives Serrated Intestinal Cancer by Impairing Immunosurveillance. Immunity 49, 1132–1147 e1137. 10.1016/j.immuni.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakanishi Y, Diaz-Meco MT, and Moscat J (2019). Serrated Colorectal Cancer: The Road Less Travelled? Trends Cancer 5, 742–754. 10.1016/j.trecan.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moscat J, Cuervo AM, and Diaz-Meco MT (2022). Immunosurveillance, interferon, and autophagic networking in cancer: the PRKCI-ULK2 paradigm. Autophagy 18, 226–227. 10.1080/15548627.2021.1991192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reina-Campos M, Diaz-Meco MT, and Moscat J (2019). The Dual Roles of the Atypical Protein Kinase Cs in Cancer. Cancer Cell 36, 218–235. 10.1016/j.ccell.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X, et al. (2018). TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543. 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 18.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. (2013). Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120. 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Jaeghere EA, Denys HG, and De Wever O (2019). Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. Trends Cancer 5, 704–723. 10.1016/j.trecan.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, LeBleu VS, and Kalluri R (2021). Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39, 548–565 e546. 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, Filliol A, Chin L, Savage TM, Yin D, et al. (2021). Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest 131. 10.1172/JCI146987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, Geldhof V, de Rooij L, Kalucka J, Sokol L, et al. (2020). An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 37, 21–36 e13. 10.1016/j.ccell.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Lee HO, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, Vanhecke J, Verbandt S, Hong H, Min JW, et al. (2020). Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet 52, 594–603. 10.1038/s41588-020-0636-z. [DOI] [PubMed] [Google Scholar]
- 27.McCarthy N, Manieri E, Storm EE, Saadatpour A, Luoma AM, Kapoor VN, Madha S, Gaynor LT, Cox C, Keerthivasan S, et al. (2020). Distinct Mesenchymal Cell Populations Generate the Essential Intestinal BMP Signaling Gradient. Cell stem cell 26, 391–402 e395. 10.1016/j.stem.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L, Toke NH, Luo S, Vasoya RP, Fullem RL, Parthasarathy A, Perekatt AO, and Verzi MP (2019). A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat Genet 51, 777–785. 10.1038/s41588-019-0384-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KA, Kakitani M, Zhao J, Oshima T, Tang T, Binnerts M, Liu Y, Boyle B, Park E, Emtage P, et al. (2005). Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science 309, 1256–1259. 10.1126/science.1112521. [DOI] [PubMed] [Google Scholar]
- 30.Harnack C, Berger H, Antanaviciute A, Vidal R, Sauer S, Simmons A, Meyer TF, and Sigal M (2019). R-spondin 3 promotes stem cell recovery and epithelial regeneration in the colon. Nat Commun 10, 4368. 10.1038/s41467-019-12349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, Chin L, Filliol A, Wen W, Song X, et al. (2021). Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 39, 866–882 e811. 10.1016/j.ccell.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornburg M, Desbois M, Lu S, Guan Y, Lo AA, Kaufman S, Elrod A, Lotstein A, DesRochers TM, Munoz-Rodriguez JL, et al. (2021). Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 39, 928–944 e926. 10.1016/j.ccell.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 34.Morral C, Stanisavljevic J, Hernando-Momblona X, Mereu E, Alvarez-Varela A, Cortina C, Stork D, Slebe F, Turon G, Whissell G, et al. (2020). Zonation of Ribosomal DNA Transcription Defines a Stem Cell Hierarchy in Colorectal Cancer. Cell stem cell 26, 845–861 e812. 10.1016/j.stem.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen B, Scurrah CR, McKinley ET, Simmons AJ, Ramirez-Solano MA, Zhu X, Markham NO, Heiser CN, Vega PN, Rolong A, et al. (2021). Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 184, 6262–6280 e6226. 10.1016/j.cell.2021.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mustata RC, Vasile G, Fernandez-Vallone V, Strollo S, Lefort A, Libert F, Monteyne D, Perez-Morga D, Vassart G, and Garcia MI (2013). Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep 5, 421–432. 10.1016/j.celrep.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, et al. (2019). Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125. 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi Y, Reina-Campos M, Nakanishi N, Llado V, Elmen L, Peterson S, Campos A, De SK, Leitges M, Ikeuchi H, et al. (2016). Control of Paneth Cell Fate, Intestinal Inflammation, and Tumorigenesis by PKClambda/iota. Cell Rep 16, 3297–3310. 10.1016/j.celrep.2016.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, and Wrana JL (2015). Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526, 715–718. 10.1038/nature15382. [DOI] [PubMed] [Google Scholar]
- 40.Uhlitz F, Bischoff P, Peidli S, Sieber A, Trinks A, Luthen M, Obermayer B, Blanc E, Ruchiy Y, Sell T, et al. (2021). Mitogen-activated protein kinase activity drives cell trajectories in colorectal cancer. EMBO Mol Med 13, e14123. 10.15252/emmm.202114123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. (2021). Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752 e4720. 10.1016/j.cell.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joanito I, Wirapati P, Zhao N, Nawaz Z, Yeo G, Lee F, Eng CLP, Macalinao DC, Kahraman M, Srinivasan H, et al. (2022). Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat Genet 54, 963–975. 10.1038/s41588-022-01100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15, 1484–1506. 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 44.Khaidakov M, Lai KK, Roudachevski D, Sargsyan J, Goyne HE, Pai RK, Lamps LW, and Hagedorn CH (2016). Gastric Proteins MUC5AC and TFF1 as Potential Diagnostic Markers of Colonic Sessile Serrated Adenomas/Polyps. Am J Clin Pathol 146, 530–537. 10.1093/ajcp/aqw142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarthy N, Kraiczy J, and Shivdasani RA (2020). Cellular and molecular architecture of the intestinal stem cell niche. Nat Cell Biol 22, 1033–1041. 10.1038/s41556-020-0567-z. [DOI] [PubMed] [Google Scholar]
- 46.Linares JF, Zhang X, Martinez-Ordonez A, Duran A, Kinoshita H, Kasashima H, Nakanishi N, Nakanishi Y, Carelli R, Cappelli L, et al. (2021). PKClambda/iota inhibition activates an ULK2-mediated interferon response to repress tumorigenesis. Mol Cell. 10.1016/j.molcel.2021.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler S, Hutloff A, Mages HW, Ranke K, Schaefer M, et al. (2009). Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 31, 823–833. 10.1016/j.immuni.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 48.Ozga AJ, Chow MT, and Luster AD (2021). Chemokines and the immune response to cancer. Immunity 54, 859–874. 10.1016/j.immuni.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson CB, Shepard HM, O'Connor PM, Kadhim S, Jiang P, Osgood RJ, Bookbinder LH, Li X, Sugarman BJ, Connor RJ, et al. (2010). Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Molecular cancer therapeutics 9, 3052–3064. 10.1158/1535-7163.MCT-10-0470. [DOI] [PubMed] [Google Scholar]
- 50.Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, Tjulandin SA, Gladkov OA, Holcombe RF, Korn R, et al. (2016). Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin Cancer Res 22, 2848–2854. 10.1158/1078-0432.CCR-15-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, Braiteh F, Ritch PS, Zalupski MM, Bahary N, et al. (2018). HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol 36, 359–366. 10.1200/JCO.2017.74.9564. [DOI] [PubMed] [Google Scholar]
- 52.Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, Thumar J, Chiorean EG, Shields AF, Behl D, et al. (2019). Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J Clin Oncol 37, 1062–1069. 10.1200/JCO.18.01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, Hitre E, Hammel P, Hendifar AE, Bates SE, et al. (2020). Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J Clin Oncol 38, 3185–3194. 10.1200/JCO.20.00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blair AB, Kim VM, Muth ST, Saung MT, Lokker N, Blouw B, Armstrong TD, Jaffee EM, Tsujikawa T, Coussens LM, et al. (2019). Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clin Cancer Res 25, 5351–5363. 10.1158/1078-0432.CCR-18-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, Sim S, Okamoto J, Johnston DM, Qian D, et al. (2011). Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol 29, 1120–1127. 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Merlos-Suarez A, Barriga FM, Jung P, Iglesias M, Cespedes MV, Rossell D, Sevillano M, Hernando-Momblona X, da Silva-Diz V, Munoz P, et al. (2011). The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell stem cell 8, 511–524. 10.1016/j.stem.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 57.Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, Richel DJ, Stassi G, and Medema JP (2008). Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci U S A 105, 13427–13432. 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Flier LG, and Clevers H (2009). Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 71, 241–260. 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 59.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 60.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, and Gumucio DL (2002). Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 277, 33275–33283. 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 61.el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, and Robine S (2004). Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193. 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 62.Patel SS, and Rodig SJ (2020). Overview of Tissue Imaging Methods. Methods Mol Biol 2055, 455–465. 10.1007/978-1-4939-9773-2_21. [DOI] [PubMed] [Google Scholar]
- 63.Patel SS, Weirather JL, Lipschitz M, Lako A, Chen PH, Griffin GK, Armand P, Shipp MA, and Rodig SJ (2019). The microenvironmental niche in classic Hodgkin lymphoma is enriched for CTLA-4-positive T cells that are PD-1-negative. Blood 134, 2059–2069. 10.1182/blood.2019002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci Rep 7, 16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Ineveld RL, Ariese HCR, Wehrens EJ, Dekkers JF, and Rios AC (2020). Single-Cell Resolution Three-Dimensional Imaging of Intact Organoids. Journal of visualized experiments : JoVE. 10.3791/60709. [DOI] [PubMed] [Google Scholar]
- 66.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 70.Melsted P, Booeshaghi AS, Liu L, Gao F, Lu L, Min KHJ, da Veiga Beltrame E, Hjorleifsson KE, Gehring J, and Pachter L (2021). Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat Biotechnol 39, 813–818. 10.1038/s41587-021-00870-2. [DOI] [PubMed] [Google Scholar]
- 71.Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414. 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 72.Eide PW, Bruun J, Lothe RA, and Sveen A (2017). CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci Rep 7, 16618. 10.1038/s41598-017-16747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, Kerr D, Aaltonen LA, Arango D, Kruhoffer M, et al. (2009). Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin Cancer Res 15, 7642–7651. 1078–0432.CCR-09–1431 [pii] 10.1158/1078-0432.CCR-09-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, et al. (2013). Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 10, e1001453. 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Compiled gene signatures and supporting literature, related to STAR Methods.
Data Availability Statement
The Quantseq, and scRNA-seq datasets generated during this study have been deposited to the GEO repository on the NCBI website (GEO: GSE207780, GSE207778, GSE207776, and GSE207779) and are publicly available as of the date of publication.
This paper reports data derived from published and publicly available datasets GEO: GSE132465, GSE166555, GSE178341, GSE14333, and GSE39582. These datasets are located on the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-β-actin | Sigma-Aldrich | Cat# A1978, RRID: AB_476692 |
| Rabbit anti-aPKCs | Abcam | Cat# ab59364; RRID: AB_944858 |
| Rabbit anti-CD8 | Abcam | Cat# ab209775; RRID: AB_2860566 |
| Rat anti-CD8 | BD Pharmingen | Cat# 557654, RRID: AB_396769 |
| Rabbit anti-CD19 | Cell Signaling Technology | Cat# 90176S, RRID:AB_2800152 |
| Rabbit anti-MRP8 | Abcam | Cat# ab92331, RRID: AB_2050283 |
| Rabbit anti-CD138 | Thermo Fisher Scientific | Cat# PA5-16918, RRID: AB_10979011 |
| Rabbit anti-FOXP3 | Cell Signaling Technology | Cat#12653, RRID: AB_2797979 |
| Rabbit anti-PDL1 | Cell Signaling Technology | Cat# 64988, RRID: AB_2799672 |
| Rat anti-CD45 | BD Parmingen | Cat# 550539, RRID: AB_2174426 |
| Mouse anti-panCK | Thermo Fisher Scientific | Cat# MA5-13156, RRID: AB_2174426 |
| Rabbit anti-panCK | Abcam | Cat# ab9377, RRID: AB_307222 |
| Rabbit anti-Ki67 | Cell Signaling Technology | Cat#12202, RRID: AB_2620142 |
| Rabbit anti-MUC2 | Abcam | Cat# ab272692, RRID: AB_2888616 |
| Rabbit anti-MUC5AC | Cell Signaling Technology | Cat#61193, RRID: RRID:AB_2799603 |
| Rabbit anti-ANXA10 | Abcam | Cat# ab213656, RRID: RRID:AB_2921231 |
| Rabbit anti-CLU | Abcam | Cat# ab184100, RRID: AB_2892532 |
| Goat anti-CLU | R&D systems | Cat# AF2747, RRID: AB_2083314 |
| Rabbit anti-PDGFRa | Abcam | Cat# ab203491, RRID: AB_2892065 |
| Mouse anti-PDGFRb | Abcam | Cat#ab69506, RRID:AB_1269704 |
| Rabbit anti-CD34 | Abcam | Cat# ab81289, RRID: AB_1640331 |
| Mouse anti-CD31 | Thermo Fisher Scientific | Cat# RB10333P1, RRID: AB_720501 |
| Rat anti-CD31 | Dianova | Cat#DIA-310, RRID:AB_2631039 |
| Rabbit anti-NRG1 | Abcam | Cat# ab191139, RRID: RRID:AB_2921232 |
| Mouse anti-Smooth muscle actin | Dako | Cat# M0851, RRID: AB_2223500 |
| Biotinylated recombinant HA-binding protein (HTI-601) | Halozyme | Provided by Halozyme Therapeutics |
| Goat anti-Mouse IgG1, secondary, HRP | Thermo Fisher Scientific | Cat# PA1-74421, RRID: AB_10988195 |
| Goat anti-Mouse IgG, secondary, HRP | Thermo Fisher Scientific | Cat# 31430, RRID:AB_228307 |
| Goat anti-Rabbit IgG, secondary, HRP | Thermo Fisher Scientific | Cat# 31461, RRID: AB_228347 |
| Goat anti-Rat IgG, secondary, HRP | Thermo Fisher Scientific | Cat# 62-9520, RRID: AB_87993 |
| Donkey anti-Rabbit IgG, secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042, RRID: AB_2534017 |
| Goat anti-Rabbit IgG, secondary, IRDye 800 | LI-COR Biosciences | Cat# 926-32211, RRID: AB_621843 |
| Goat anti-Mouse IgG1, secondary, IRDye 800 | LI-COR Biosciences | Cat# 926-32350, RRID: AB_2782997 |
| Goat anti-Mouse IgG, secondary, IRDye 800 | LI-COR Biosciences | Cat# 926-32210, RRID: AB_621842 |
| InVivoMAb rat IgG2b isotype control | BioXCell | BE0090, RRID:AB_1107780 |
| InVivoMAb anti-mouse PD-L1 (B7-H1) | BioXCell | BE0101, RRID:AB_10949073 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | BioXCell | BP0164, RRID:AB_10949609 |
| Biological samples | ||
| Human Colorectal cancers (CRC) | Osaka City University Hospital, Osaka, JAPAN | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| HBSS (no calcium, no magnesium) | GIBCO | Cat# 14175095 |
| Dimethyl Sulfoxide | Fisher BioReagents | Cat# BP2311 |
| Chloroform | Sigma-Aldrich | Cat# 288306 |
| Methanol | Sigma-Aldrich | Cat# 1424109 |
| Tryple Express | Life Technologies | Cat# 12605-010 |
| HEPES | Gibco | Cat# 15630080 |
| PBS (no calcium, no magnesium) | Gibco | Cat# 10010-023 |
| Quick-RNA Miniprep Kit | Zymo Research | Cat# R1054 |
| RNAlater Stabilization Solution | Invitrogen | Cat# AM7021 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596018 |
| Advanced DMEM/F12 | Thermo Fisher Scientific | Cat# 12634010 |
| RPMI | Corning | Cat#15-040-CV |
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat# 35050061 |
| Penicillin-Streptomycin solution | Corning | Cat# 30-002-CI |
| Y-27632 | Tocris | Cat# 1254 |
| B27 Supplement | Thermo Fisher Scientific | Cat# 17504001 |
| B27 Supplement minus vitamin A | Thermo Fisher Scientific | Cat# 12587001 |
| N2 Supplement | Thermo Fisher Scientific | Cat# 17502048 |
| Murine EGF | Thermo Fisher Scientific | Cat# PMG8045 |
| Human EGF | Peprotech | Cat#AF-100-15 |
| Human FGF-2 | Peprotech | Cat#100-18B |
| Human IGF-1 | Biolegend | Cat#590906 |
| Recombinant human Wnt-3a | R&D systems | Cat#5036-WN/CF |
| A83-01 | Sigma-Aldrich | Cat#SML078 |
| Human [Leu15]-Gastrin I | Sigma-Aldrich | Cat# G9145 |
| Recombinant murine R-Spondin 1 | R&D systems | Cat# 3474-RS-050 |
| Recombinant murine Noggin | Peprotech | Cat# 250-38 |
| Recombinant murine M-CSF | Peprotech | Cat# 315-02 |
| Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix | Corning | Cat# 356230 |
| Liberase™ TM Research Grade | Sigma-Aldrich | Cat# 5401119001 |
| Collagenase A | Sigma-Aldrich | Cat# 10103586001 |
| Dispase II | Sigma-Aldrich | Cat# #D4693 |
| DNase I | Sigma-Aldrich | Cat# 04716728001 |
| 4-OH-tamoxifen | Millipore-Sigma | Cat# H7904 |
| TrueCut Cas9 Protein v2 | Thermo Fisher Scientific | Cat# A36498 |
| DAPI | Life Technologies | Cat# D1306 |
| Streptavidin, secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# S11226 |
| Streptavidin, secondary, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# S11223 |
| Trypan Blue Solution, 0.4% | Gibco | Cat# 15250061 |
| Critical commercial assays | ||
| Neon™ 10μl Electroporation Kit | Thermo Fisher Scientific | Cat# MPK1096 |
| Chromium Single Cell 3′ Gene Expression Solution v2 | 10X Genomics | Cat#PN-120237 |
| EasySep™ Dead Cell Removal (Annexin V) Kit | STEMCELL Technologies | Cat# 17899 |
| VECTASTAIN® Elite® ABC-HRP Kit | Vector | Cat# PK-6100 |
| Automated Opal 7-Color IHC Kit | AKOYA BIOSCIENCES | Cat# NEL821001KT |
| Manual Opal 4-Color IHC Kit | AKOYA BIOSCIENCES | Cat# NEL810001KT |
| Trichrome Stain (Masson) Kit | Sigma-Aldrich | Cat# HT15 |
| Corning Biocoat Control Inserts | Corning | Cat# 354578 |
| Alexa Fluor™ 488 Tyramide Superboost™ Kit | Thermo Fisher Scientific | Cat# B40932 |
| Deposited data | ||
| sc-RNA-seq | This study | GEO: GSE207780 |
| 3’ RNA-seq (Prkcifl/fl Prkczfl/fl Villin-Cre-Veh or -PEGPH20 treated) | This study | GEO: GSE207778 |
| 3’ RNA-seq (sgPrkci/Prkcz and sgC MTOs) | This study | GEO: GSE207776 |
| 3’ RNA-seq (sgPrkci/Prkcz MTOs-Veh or -PEGPH20 treated) | This study | GEO: GSE207779 |
| Raw Data | This study; Mendeley Data | https://doi.org/10.17632/vbzbzhrw98.1 |
| Single cell 3′ RNA sequencing from patients with CRC | Lee et al.26 | GEO: GSE132465 |
| Single cell 3′ RNA sequencing from patients with CRC | Uhlitz et al.40 | GEO: GSE166555 |
| Single cell 3′ RNA sequencing from patients with CRC | Pelka et al.41 | GEO: GSE178341 |
| Microarray from primary CRC tumors | Jorissen et al.73 | GEO: GSE14333 |
| Microarray from primary CRC tumors | Marisa et al.74 | GEO: GSE39582 |
| TCGA-COREAD | cBioportal | http://www.cbioportal.org/index.do |
| Experimental models: Cell lines | ||
| Human CRC (sgPRKCI/PRKCZ) | This study | N/A |
| Mouse tumor organoid (MTO) | Tauriello et al.17 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Prkcifl/fl Prkczfl/fl | Nakanishi et al.13 | N/A |
| C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Oligonucleotides | ||
| Real-time PCR primers | This manuscript | Table S1 |
| CRISPR guides | This manuscript | Table S2 |
| Software and algorithms | ||
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/scientificsoftware/ |
| QuPath v.0.1.3 | Queen’s University, Belfast, Northern Ireland | https://qupath.github.io |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Zen blue | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| RStudio (1.1.456) | R Core Team | https://www.r-project.org/ |
| R | R Core Team | https://www.r-project.org/ |
| GSVA; version 1.26.0 | Bioconductor | https://github.com/rcastelo/GSVA |
| GSEA (v4.1.0) | Broad Institute | http://www.broadinstitute.org/gsea/index.jsp |
| BaseSpace | Illumina | https://basespace.illumina.com/ |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| CMScaller v0.99.1 | Eide et al.72 | https://github.com/peterawe/CMScaller |
| CellPhoneDB v.2.0.0 | Efremova et al.43 | https://www.cellphonedb.org |
| GenePattern | Broad Institute | https://cloud.genepattern.org/gp/pages/login.jsf |
| Lexogen QuantSeq DE 1.3.0 | BlueBee Cloud | https://www.bluebee.com |
| Cell Ranger (v3.0) | Langmead et al.75 10X genomics | http://software.10xgenomics.com/single-cell/overview/welcome |
| Seurat 3.0 | Stuart et al.76 | https://github.com/Satijalab/seurat |
| kb_python | Bray et al.69;Melsted et al.70 | https://github.com/pachterlab/kb_python |
| scVelo package | Bergen et al.71 | https://scvelo.readthedocs.io/VelocityBasics/ |
| Harmony | Korsunsky et al.67 | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
| PhenoChart Whole Slide Viewer | AKOYA BIOSCIENCES | https://www.akoyabio.com/support/software/phenochart-whole-slide-viewer/ |
| CaseViewer | 3DHistech, Budapest, Hungary | https://www.3dhistech.com/solutions/caseviewer/ |
| Other | ||
| EVOS FL Auto Imaging System | Thermo Fisher Scientific | N/A |
| EVOS M5000 Imaging System | Thermo Fisher Scientific | N/A |
| TissueLyser II | QIAGEN | Cat# 85300 |
| NanoDrop 1000 spectrophotometer | Thermo Fisher Scientific | N/A |
| Zeiss LSM 710 NLO Confocal Microscope | Zeiss | N/A |
| Neon™ Transfection System | Thermo Fisher Scientific | Cat# MPK5000 |
| Pannoramic Scanner | 3DHistech, Budapest, Hungary | N/A |
| Vectra Polaris Automated Quantitative Pathology Multispectral Imaging System | AKOYA BIOSCIENCES | N/A |
Original raw data have been deposited in Mendeley Data (https://doi.org/10.17632/vbzbzhrw98.1).