

Abstract
The gut microbiota represents a diverse and dynamic population of microorganisms that can influence the health of the host. Increasing evidence supports the role of the gut microbiota as a key player in the pathogenesis of neurodegenerative diseases, including Alzheimer's disease (AD). Unfortunately, the mechanisms behind the interplay between gut pathogens and AD are still elusive. It is known that bacteria‐derived outer membrane vesicles (OMVs) act as natural carriers of virulence factors that are central players in the pathogenesis of the bacteria. Helicobacter pylori (H. pylori) is a common gastric pathogen and H. pylori infection has been associated with an increased risk to develop AD. Here, we are the first to shed light on the role of OMVs derived from H. pylori on the brain in healthy conditions and on disease pathology in the case of AD. Our results reveal that H. pylori OMVs can cross the biological barriers, eventually reaching the brain. Once in the brain, these OMVs are taken up by astrocytes, which induce activation of glial cells and neuronal dysfunction, ultimately leading to exacerbated amyloid‐β pathology and cognitive decline. Mechanistically, we identified a critical role for the complement component 3 (C3)‐C3a receptor (C3aR) signalling in mediating the interaction between astrocytes, microglia and neurons upon the presence of gut H. pylori OMVs. Taken together, our study reveals that H. pylori has a detrimental effect on brain functionality and accelerates AD development via OMVs and C3‐C3aR signalling.
Keywords: Alzheimer's disease, gut‐brain axis, Helicobacter pylori, bacterial extracellular vesicles (bEVs), outer membrane vesicles (OMVs), C3, complement
1. INTRODUCTION
Alzheimer's disease (AD) is a devastating age‐related neurodegenerative disorder with an alarming increasing prevalence (Cummings et al., 2021). It is already well‐known for decades that the deposition of amyloid‐beta (Aβ) protein in senile plaques outside neurons and the formation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated Tau (p‐Tau) protein inside neurons result in the loss of synapses and neurodegeneration which ultimately leads to symptoms associated with AD (DeTure and Dickson, 2019). More recently, it became increasingly clear that also inflammation, both peripheral and central, are important (and often early) events in AD progression (Tejera et al., 2019; Xie et al., 2021). Except for the recently FDA‐approved Aducanumab of which the therapeutic effect is not yet conclusively established (Lalli et al., 2021), current Alzheimer's drugs address only disease symptoms (Cummings et al., 2021). The lack of effective treatments may be attributed by the inadequate understanding of the mechanism underlying AD pathology. Over the past few decades, the three most common proposed hypotheses are the Aβ cascade, the Tau and the neuroinflammation hypothesis. However, the process inducing Aβ peptide deposit, Tau protein degeneration, neuroinflammation and ultimately neuronal cell loss, remains to be elucidated (Du et al., 2018). Due to the absence of disease‐modifying therapeutics and the disturbing multiplying prevalence worldwide, more controversial emerging hypotheses are gaining increased attention. For example, recent epidemiological and experimental evidence provides support for an infectious hypothesis proposing that pathogens (bacteria, viruses, prions, etc.) may be a common underlying cause of AD (Dominy et al., 2019; Gorle et al., 2018; Seaks and Wilcock, 2020). According to the infectious theory, pathogens or pathogen‐derived components are believed to trigger inflammation, which can lead to the accumulation of Aβ and NFTs and neuronal dysfunction, resulting in AD (Seaks and Wilcock, 2020; Xie et al., 2022b).
Outer membrane vesicles (OMVs) are nano‐sized particles surrounded by a proteolipid bilayer and are naturally released from Gram‐negative bacteria (Schwechheimer and Kuehn, 2015). They carry various biological molecules from their parental bacteria, including microbe‐associated molecular patterns (MAMPs), enzymes and toxins, that can act as virulence factors in the host (Xie et al., 2022a). OMVs have gained tremendous interest in a wide range of biomedical fields due to their inherent ability to deliver their cargo both locally and systematically to recipient cells (Xie et al., 2022a). This cell‐free intercellular communication plays an important role in the physiology and pathogenesis of the bacteria (Bielaszewska et al., 2017). For example, in mice colonized with Escherichia coli (E. coli), the E. coli OMVs were reported to spread to a wide range of host tissues, including the heart, liver, kidney, spleen, as well as the brain (Bittel et al., 2021). As OMVs contain various MAMPs, they can induce pro‐inflammatory responses in the periphery but also in the brain by crossing the blood‐brain barrier (BBB) (Finethy et al., 2017; Ha et al., 2020; Han et al., 2019). Moreover, OMVs from Porphyromonas gingivalis (P. gingivalis) and Paenalcaligenes hominis (P. hominis) cultures, isolated via protein precipitation and sucrose gradient centrifugation, respectively, were shown to be involved in the pathogenesis of AD in mice (Gong et al., 2022; Lee et al., 2020). Administration of these OMVs by oral gavage decreased the tight junctions (TJs) expression (e.g., ZO‐1, OCLN and CLDN‐5) at the BBB and entered the brain parenchyma, thereby inducing neuroinflammation, tau phosphorylation and memory dysfunction in mice (Gong et al., 2022; Lee et al., 2020). In addition, P. hominis OMVs have also been shown to penetrate the brain via the vagus nerve and cause cognitive impairment (Lee et al., 2020). However, the OMV isolation methods used in these studies may also co‐purify other components (e.g., proteins, lipids and protein aggregates), which may interfere with the interpretation of these functional studies (Xie et al., 2022a). In addition, the organic dyes (DiO and FITC) used to label OMVs may also label co‐purified components, resulting in false positive signals in OMV tracking studies (Mondal et al., 2019).
Helicobacter pylori (H. pylori) is a pathogen which infects more than half of the world's population and is associated with the development of gastroduodenal diseases. In addition, Numerous studies have shown that H. pylori infection may also increase the risk of various extragastric diseases such as cardiovascular diseases (OR = 1.58, 95% CI: 1.34–1.87) (Tong et al., 2022), hypertension (OR = 1.34, 95% CI: 1.10–1.63) (Fang et al., 2022), stroke (OR = 1.43, 95% CI:1.25–1.46) (Doheim et al., 2021) and diabetes (OR = 1.27, 95% CI: 1.11–1.45) (Mansori et al., 2020), all of which are risk factors for AD (Doulberis et al., 2018; Xie et al., 2022b). A recent meta‐analysis also identified direct evidence of a significant positive association between H. pylori infection and AD development (OR = 1.40, 95% CI: 1.12–1.76) (Fu et al., 2020). OMVs derived from H. pylori exert immunomodulatory effects by inducing the production of proinflammatory cytokines such as IL‐6 and TNF, and by promoting apoptosis of gastric epithelial cells and immune cells. These inflammatory responses may further accelerate the development of H. pylori infection (Chmiela et al., 2018; Wang et al., 2021). In addition, H. pylori OMVs are also found to induce apoptosis of human umbilical vein endothelial cells, which may promote the formation of atherosclerotic plaques (Wang et al., 2021). However, no study has attempted to determine their role in AD development and progression.
In this study, we investigated the impact of H. pylori‐derived OMVs on brain functions and AD pathology using wild‐type (WT) mice and AppNL‐G‐F AD mice. The AppNL‐G‐F strain is a second‐generation mouse model of AD in which the amyloid precursor protein (APP) is not overexpressed like in most other existing transgenic mouse models. Instead, this model uses a knock‐in approach to express human mutated APP at WT. The combined effect of three mutations associated with familial AD results in elevated levels of pathogenic Aβ (Sasaguri et al., 2017). To study the mechanism by which H. pylori OMVs affect brain functions, WT mice were used as the elevated Aβ production in AppNL‐G‐F mice already impairs brain functions (Xie et al., 2021). We reveal that H. pylori OMVs exacerbate Aβ pathology and induce cognitive impairment via regulation of glial cell activation and neuronal dysfunction. Moreover, we identify a critical role for complement component 3 (C3)‐C3a receptor (C3aR) signalling in mediating the interactions of astrocyte‐microglia‐neuron in the context of the H. pylori OMVs challenge. This study provides new crucial insights into the role of H. pylori‐derived OMVs in brain function and AD development.
2. METHODS
2.1. Key resources
Key resources were provided in Table S1.
2.2. Bacteria culture and outer membrane vesicles isolation
H. pylori 26695 (ATCC 700392) was cultured at 37°C in a microaerobic atmosphere (85% N2, 10% CO2, 5% O2). H. pylori 26695 was grown in Brucella broth (10 g meat peptone, 10 g casein peptone, 2 g yeast extract, 5 g sodium chloride, 1 g glucose, 0.1 g sodium bisulphite in 1 L) supplemented with 10% exosome‐depleted foetal bovine serum (FBS) (Characterized in Figure S1a‐d) for 72 h until the late ‐log phase was reached. Culture supernatant (2 L per batch) was harvested at an optical density at 600 nm (OD600nm) of 1.0–1.4 in an UV‐VIS spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). An OD600nm of 0.05 corresponds to an inoculum of ∼6 × 106 CFU/ml as determined by viable count in horse blood agar incubated at 37°C in the microaerophilic atmosphere for 72 h. To isolate and purify OMVs, the bacterial debris and large contaminants were removed by two centrifugation steps of (8000 × g, 4°C, 15 min). The supernatants were filtered sequentially using 0.45 and 0.22 μm membrane filters and concentrated using a 10 kDa cut‐off centrifugal filter to a volume of ∼30 ml. The purified OMVs were subsequently isolated by SEC using commercially available qEVoriginal/35 nm columns. Fractions #1‐30 of 0.5 ml were collected. Finally, fractions 7–11 were pooled and OMVs in these fractions were concentrated again using a 10 kDa cut‐off centrifugal filter until a volume of 1 ml. The number of OMV particles was measured by nanoparticle tracking analysis (NTA) using a Zetaview system (Particle Metrix, Germany). Examination of 30 μl OMV and control samples by transmission electron microscopy (TEM) did not indicate the presence of contaminating bacterial cells. The OMV protein concentrations were measured by a nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The yield of H. pylori 26695 OMVs was ∼1 mg protein (∼2 × 1012 particles) per 1 L culture supernatant. OMV samples were pooled per batch (2 L per batch) and a total of three batches were collected for this study. Each batch was characterized as shown in Figure S2(a‐d). OMV samples were aliquoted and stored in PBS at −80°C for less than 6 months. As shown in Figure S1(a‐d) and Figure S2(a‐d), there were still some residual particles in exosome‐depleted the FBS. To exclude that the observed effect was due to non‐H. pylori derived particles present in the bacterial culture medium, the OMV isolation protocol was in parallel also applied to culture medium. Briefly, same volume (i.e., 2 L per batch) of 10% exosome‐depleted FBS containing culture medium was filtered sequentially using 0.45 and 0.22 μm membrane filters. Next, a 10 kDa cut‐off centrifugal filter was used to concentrate the samples to a volume of ∼30 ml. The same SEC fractions 7–11 were then collected and concentrated again by using a 10 kDa cut‐off centrifugal filter to a volume of 1 ml. These samples were used as control in all our experiments.
2.3. TLR4 activation analysis
Lipopolysaccharide (LPS) activity levels were measured using the HEK‐Blue mTLR4 assay (InvivoGen) according to the manufacturer's protocol. Briefly, HEK‐Blue mTLR4 cells were seeded at 25,000 cells per well in a 96‐well plate in detection medium. The fractions (#1–30) from SEC were added to the reporter cell line and incubated with HEK‐Blue detection medium for 12 h, followed by absorption measurement of the culture medium at 655 nm by iMark Microplate Absorbance Reader (Bio‐Rad) and relative TLR4 activation was calculated.
2.4. Electron microscopy
Purified OMVs were visualized by negative staining TEM as described previously (Vandendriessche et al., 2021). In short, samples were spotted on a parafilm sheet. Next, formvar/C‐coated hexagonal copper grids (EMS G200H‐Cu), which were glow discharged for 10 s, were placed on top of the droplet for 1 min with the coated side of the grid down. The grids were washed five times in droplets of Milli‐Q water, stained with 1% (w/v) uranyl acetate for 10 s and air dried for 24 h before imaging. Visualization of the samples was done using a JEM 1400plus TEM (JEOL, Tokyo, Japan) operating at 80 kV.
2.5. Animals and manipulations
Wild‐type C57BL/6J, AppNL‐G‐F (carrying Arctic, Swedish, and Beyreuther/Iberian mutations) (Saito et al., 2014) and B6.Cg‐Gt(ROSA)26Sortm9(CAG‐tdTomato)Hze /J (Rosa26.tdTomato) reporter mice were bred at a conventional animal facility. Mice were kept in individually ventilated cages under a 14‐h dark/10‐h light cycle and received food and water ad libitum. Both male and female mice were used. The AppNL‐G‐F mice, C57BL/6J and Rosa26.tdTomato mice were 24–28 weeks, 12–16 weeks and 10–12 weeks of age, respectively, at the start of the experiment. The animals were randomly allocated to experimental groups. For short‐term experiments, mice were orally gavaged with H. pylori OMVs (20 μg protein, 4 × 1010 particles) or an equivalent volume of control sample alone or simultaneously i.p. injected with 0.5% DMSO/C3aRA (1 mg/kg) once per day for 5 days and sacrificed at day 7. For long‐term experiments, mice were orally gavaged with H. pylori OMVs (20 μg protein, 4 × 1010 particles) or an equivalent volume of control sample alone or simultaneously i.p. injected with 0.5% DMSO/C3aRA (1 mg/kg) three times per week (Monday, Wednesday and Friday) for 3 weeks. These mice were sacrificed at the end of treatment or cognition tests.
All animal studies were conducted in compliance with governmental and EU guidelines for the care and use of laboratory animals and were approved by the ethical committee of the Faculty of Sciences, Ghent University, Belgium (EC2021‐025 and EC2022‐109).
2.6. Cre‐recombinase loading and in vivo biodistribution analysis of H. pylori OMVs
For Cre‐recombinase loading, H. pylori OMVs and Cre‐recombinase were incubated with 0.1 mg/ml saponin at room temperature with shaking (400 rpm) for 2 h. To exclude residual Cre‐recombinase, the Cre‐recombinase‐loaded H. pylori OMV solutions were subjected to qEV isolation. The loading efficiency was validated via Cre western blot analysis (Figure S3a and b). The fractions containing OMVs were collected and concentrated using a 10‐kDa centrifugal filter. Rosa26.tdTomato mice were intragastrically administered Cre‐recombinase‐loaded H. pylori OMVs (4 × 1010 particles) once per day for 5 days. At day 7, mice were sacrificed, and the brains were collected and analysed by performing immunostaining as described in Section 2.1. The unloaded H. pylori OMVs were used as control in this experiment.
2.7. Quantification of the gastrointestinal barrier, BBB and blood‐CSF barrier permeability
Gastrointestinal barrier permeability was determined as previously described (Vandenbroucke et al., 2014). Briefly, 4 kDa FITC‐dextran was intragastrically administered 4 h before collection of blood. Plasma was isolated and gastrointestinal leakage was determined by measuring fluorescence using the FLUOstar Omega reader (BMG LABTECH) at λex/λem = 485/520 nm.
BBB and blood‐CSF barrier leakage was analysed as described previously (Vandenbroucke et al., 2012). In brief, 4 kDa FITC‐dextran was i.v. injected 15 min before CSF isolation by cisterna magna puncture. For blood‐CSF barrier analysis, 1 μl of CSF was diluted in 99 μl PBS and fluorescence was measured using the FLUOstar Omega reader (λex/λem = 485/520 nm). For the analysis of BBB integrity, mice were transcardially perfused with PBS/heparin and brain was isolated. Next, perfused brain samples were incubated in formamide overnight, samples were centrifuged for 15 min at 20,000 g in 4°C, and 100 μl supernatant was used for fluorescence measurement using the FLUOstar Omega reader (λex/λem = 485/520 nm).
2.8. Western blotting
Hippocampus and bacterial extracts were prepared in 0.5% CHAPS buffer containing complete protease inhibitor and centrifuged for 15 min at 20,000 g and 4°C, and protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Hippocampus and bacterial extraction supernatants, OMV and FBS samples were denatured in 6×Laemmli buffer, separated by SDS‐PAGE gel electrophoresis, and transferred to a nitrocellulose membrane. Following blocking with Odyssey blocking buffer (LI‐COR Biosciences), the membrane was incubated with primary antibodies, washed with PBS containing 0.1% Tween 20 and subsequently incubated with fluorophore‐ or HRP‐conjugated secondary antibodies. Protein bands were visualized by Odyssey Fc Imaging System (LI‐COR Biosciences) or WesternBright Quantum HRP substrate (advansta) in Amersham Imager 600 (GE Healthcare), and quantification was done in Image Studio (LI‐COR Biosciences) and Amersham Imager 600 integrated analysis software (GE Healthcare), respectively.
2.9. Cytokine measurements
Hippocampus and stomach lysates were prepared as described in Section 2.8 and IL‐1β, and TNF levels of plasma and tissue lysates were measured using the Bio‐Plex cytokine assay (Bio‐Rad) according to the manufacturer's instructions.
2.10. RNA extraction and RT‐qPCR analysis
Total RNA was isolated from the tissue using TRIzol reagent. After homogenizing the tissue in a tube containing zirconium oxide beads on a TissueLyser (QIAGEN), chloroform was added and the homogenate was separated into three phases by centrifugation at 20,000 g in a microcentrifuge at 4°C for 15 min. Total RNA was extracted from the upper aqueous phase using Aurum total RNA kit (Bio‐Rad) according to the manufacturer's instructions. The concentration of total RNA was determined by the Nanodrop and total RNA was reverse‐transcribed into cDNA with SensiFAST™ cDNA Synthesis Kit (Bioline). qPCR was performed on the Roche LightCycler 480 System (Applied Biosystems) using SensiFAST™ SYBR® No‐ROX Kit (Bioline). Results are given as relative expression values normalized to the geometric mean of reference genes, determined using GeNorm. The sequences of the primers are depicted in Table S2.
2.11. Aβ ELISA
Aβ was extracted and measured using a standard protocol as described previously (Steeland et al., 2018). Specifically, hippocampus tissue was homogenized in Tissue Protein Extraction Buffer containing complete protease and phosphatase inhibitors using a Precellys (Bertin Technologies) and subsequently centrifuged at 5000 g for 5 min at 4°C. Supernatant was collected and centrifuged at 4°C for 1 h at 100,000 g (TLA‐100 Rotor; Beckman Coulter). Supernatant containing soluble Aβ was collected and the pellet was further processed in GuHCl solution containing complete protease inhibitor, sonicated, vortexed, incubated for 60 min at 25°C and centrifuged at 70,000 g for 20 min at 4°C. Supernatant containing insoluble Aβ was 12 times diluted with GuHCl diluent. The levels of Aβ1‐42 and Aβ1‐40 in hippocampus lysates were determined using a sandwich ELISA assay. Briefly, the sample‐detection mixtures were added to the ELISA plate coated with anti‐Aβ1‐42 (1.5 μg/ml; JRF/cAb42/26) or anti‐Aβ1‐40 antibody (1.5 μg/ml; JRF/cAb40/28) and incubated overnight at 4°C with slow shaking. Absorption at 450 nm was measured after adding substrate solution (BD Biosciences OptEIA™) followed by stopping buffer (1 M H2SO4). The amount of Aβ was determined with GraphPad Prism 8.0 using a nonlinear regression model.
2.12. Histology
To evaluate gastric inflammation, 5 μm paraffin sections of the stomach were stained with haematoxylin (Merck) and eosin (VWR) (H&E). Scoring of immune cell infiltration in H&E‐stained stomach was done blinded. Images (20× magnification) were acquired with a BX51 discussion microscope (Olympus) using an objective lens with 3 N.A. 1.0. The images were analysed in Image J software.
2.13. Immunohistochemistry
For immunostainings on the mouse brain, the sections (one section per mouse for each staining) were cut depending on the used processing method, namely, 5 μm for paraffin, 20 μm for cryo, and 50 μm for vibratome sections. Paraffin sections were de‐paraffinized in xylene and ethanol, boiled in citrate buffer for 20 min, and followed by blocking with 5% goat serum in PBS‐T (PBS containing 0.3% Triton X‐100) solution for 1 h at room temperature. The sections were then stained with primary antibodies (anti‐GFAP, anti‐IBA1, anti‐Ki‐67, anti‐C3, anti‐C3aR, anti‐DCX, anti‐PSD‐95, anti‐SYP, anti‐NeuN, anti‐OCLN, anti‐E‐cadherin (CDH1), and anti‐RFP) in blocking buffer at 4°C overnight. After washing with PBS, sections were stained with appropriate fluorophore‐conjugated secondary antibodies in PBS or PBS containing 0.1% Triton X‐100 for 1–2 h before washing and mounting. Cryosections for anti‐ZO‐1 staining were boiled directly in citrate buffer and followed the same steps as above for paraffin staining. Vibratome sections for anti‐GFAP, anti‐IBA1, anti‐C3, anti‐CD68, anti‐PSD‐95 and anti‐SYP were treated with blocking buffer directly and followed the same steps as above for paraffin staining. A Zeiss LSM780 confocal microscope or Zeiss Axioscan Z.1 was used for imaging. Images were processed using Image J and the intensity of overlapping signals were quantified with Colocalization analysis for Image J.
2.14. Glial cell morphology quantification
To quantify the glial cell morphology, IBA1‐positive microglia and GFAP‐positive astrocytes were imaged with a 40×/63× oil objective using the confocal microscope with the z‐stack model. Images were analysed using filament tracing algorithm from Imaris software (Bitplane).
2.15. Synaptic imaging and quantification
For synaptic puncta colocalization analysis, brain paraffin sections were co‐immunostained with the anti‐SYP and anti‐PSD‐95 antibodies and imaged with the 63× oil objective with the 3× zoom using confocal microscopy. Images were processed using Image J and the number of colocalized puncta was quantified with the Synapse Counter plugin for Image J (Dzyubenko et al., 2016).
For quantification of PSD‐95 and SYP inside CD68‐positive phagosomes and IBA‐positive microglia, brain vibratome sections were co‐immunostained with anti‐IBA1, anti‐CD68, and anti‐SYP or anti‐PSD‐95 antibodies. Sections were imaged with the 40×/63× oil objective using confocal microscope with the z‐stack model. Images were analysed using the surface function from Imaris software.
2.16. Electrophysiological field recordings on acute mouse hippocampal slices
Electrophysiology was performed at the VIB‐KU Leuven Center for Brain and Disease Research Electrophysiology Expertise Unit as described previously (Largo‐Barrientos et al., 2021). Specifically, mice were anesthetized with isoflurane and brains were isolated into ice‐cold cutting solution (87 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM glucose, 25 mM NaHCO3, 0.5 mM CaCl2, 7 mM MgCl2, 75 mM sucrose, 1 mM kynurenic acid, 5 mM ascorbic acid and 3 mM pyruvic acid) at pH 7.4 and 5% CO2/95% O2. 300 μm‐thick parasagittal hippocampal sections were cut in cold cutting solution using a vibratome (VT1200 Leica), incubated at 34°C for 35 min to recover and then stored in cutting solution at room temperature. For recordings, slices were placed onto a multielectrode array (MEA 2100, Multichannel Systems) and continuously perfused with artificial cerebrospinal fluid (aCSF) solution (119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 11 mM glucose, 26 mM NaHCO3, 4 mM MgCl2 and 4 mM CaCl2) at pH 7.4 and 5% CO2/95% O2. Field excitatory post‐synaptic potentials (fEPSPs) were recorded from mossy fiber‐CA3 synapses or Schaffer collateral‐CA1 synapses by stimulating and recording from the appropriate electrodes. First, input‐output curves were established for each individual slice by applying single‐stimuli ranging from 500 to 2750 mV with 250 mV increments. Stimulus strength that corresponds to 35% of maximal response in the input‐output curve was used for the consecutive recordings. For long‐term potentiation (LTP) experiments, stable fEPSPs were recorded for 30 min to establish a baseline. Afterwards, three trains of high‐frequency stimulation (100 stimuli at 100 Hz) with 5 min intervals were applied to induce LTP and fEPSPs were recorded for 65 additional minutes. Post‐LTP fEPSPs were measured every 5 min (average of three consecutive stimulations (15 s apart)). Recordings were analysed and processed using Multi Channel Experimenter software (Multichannel Systems).
2.17. Y‐maze test
The Y‐maze test was performed with a slight modification as previously described (Saito et al., 2014). All the experiments were conducted in the light phase (9:00–18:00). Y maze apparatus (O'Hara & Co), made of grey plastic, consisted of three compartments (3 cm (W) bottom and 10 cm (W) top, 40 cm (L) and 12 cm (H)) radiating out from the centre platform (3 × 3 × 3 cm triangle), and positioned 60 cm above the floor. In this test, each mouse was placed in the centre of the maze facing toward one of the arms and then allowed to explore freely for 5 min. We recorded and analysed the spontaneous behavioural alternations of the mice using EthoVision tracking system (Noldus). A reduction in the behavioural alternations corresponds to memory impairment (Sarnyai et al., 2000).
2.18. Novel object recognition test
The NOR test was performed with a minor modification as previously described (Vandendriessche et al., 2021). All the experiments were conducted in the light phase (9:00–18:00). The mice were placed in a clear, acrylic box (40 × 40 × 40 cm). During the training trial, mice were given two sessions of 5 min; during the first session, mice were allowed to explore the arena without objects (habituation), and in the second session, two identical objects were placed at two opposite positions within the box at the same distance from the nearest corner. The mice were returned to their home cage for 15 min (short‐term memory) after the training phase. Mice were placed back in the same box for 5 min after replacing one of the familiar objects with a novel one. Exploration of the objects by the mice was defined as touching the object with their nose or mouth. All trials were videotaped and scored manually using EthoVision tracking system (Noldus). The percentage preference for the novel object was calculated using the following formula: percentage novel object preference = (novel object exploration time/(novel object exploration time + familiar object exploration time)) × 100%. A value >50% indicates preference for the novel object. To exclude the existence of olfactory cues, the box and the objects were thoroughly cleaned with 20% ethanol after each trial.
3. STATISTICAL ANALYSIS
Graphical data were analysed with GraphPad Prism 8.0 and presented as mean ± SEM. Unpaired, two‐tailed Student's t‐tests were used to determine statistical significance between two independent groups. One‐way analysis of variance (ANOVA) with Bonferroni's multiple comparison test was used to determine statistical significance among multiple groups with one independent variable. Two‐way ANOVA with Bonferroni's multiple comparison test was used to determine statistical significance among multiple groups with two independent variables. A p value of <0.05 was considered statistically significant.
4. RESULTS
4.1. Isolation and characterization of H. pylori OMVs
H. pylori OMVs were purified from bacterial cultures by a combination of ultrafiltration and size exclusion chromatography (SEC) (Figure 1a). After SEC isolation, the particle number and protein concentration of each fraction was determined by NTA and spectrophotometric analysis, respectively (Figure 1b). LPS is the major outer membrane component of Gram‐negative bacteria and one of the most abundant components of OMVs (Vanaja et al., 2016). TLR4 activation analysis demonstrated a noticeable increased LPS activity in the particle enriched fractions (Figure 1c). Next, the particle‐enriched fractions were pooled and subjected to OMV preparation by ultrafiltration using 10 kDa centrifugal filters (Figure 1a). The resulting purified H. pylori OMVs were physically intact when observed under TEM using negative staining, with an average size of 176.1 ± 4.7 nm (two peaks centred at 135 and 225 nm) in diameter as determined by NTA (Figure 1d,e). H. pylori OMV purification was further confirmed by enrichment of flagellin A (FlaA), the major component of flagellar filaments that is also present in OMVs (Zavan et al., 2019), in the purified OMV preparation and the OMV fractions #7‐11 (Figure 1f).
FIGURE 1.
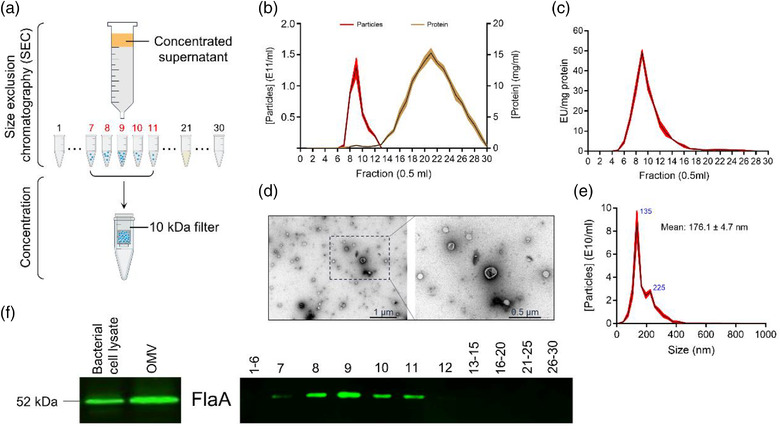
Characterization of Helicobacter pylori OMVs. (a) Graphical illustration of H. pylori OMV isolation from bacterial cultures. (b) Particle and protein concentration in the different SEC fractions (0.5 ml per fraction) were determined using nanoparticle tracking analysis (NTA; red) and Nanodrop (brown), respectively. Data show all 30 fractions. (c) Limulus amebocyte lysate (LAL) assay quantified LPS activity levels in the different SEC fractions. (d) Representative negative staining TEM images of purified H. pylori OMVs from SEC fractions #7–11. Scale bars: 1 μm (left) and 0.5 μm (right). (e) Size distributions of purified H. pylori OMVs from SEC fractions #7‐11 analysed by NTA. (f) Western blot analysis of FlaA in bacterial cell lysate and purified H. pylori OMVs from SEC fractions #7–11 (left; 10 μg total protein was loaded) and all fractions separately (right; 20 μl per fraction was loaded). The graphs are shown as mean ± SEM. Data in b, c and d are shown for at least three biological replicates.
4.2. Biodistribution of H. pylori OMVs
To examine whether gastric H. pylori‐derived OMVs can reach and deliver their content to the periphery, we loaded H. pylori OMVs with Cre‐recombinase (Figure 2a), which were consequently administered to Rosa26.tdTomato mice by oral gavage, as depicted treatment in the schedule in (Figure 2b). In this mouse model, a loxP‐flanked STOP cassette followed by a downstream red fluorescent protein variant (tdTomato) is inserted in the ROSA locus. The loxP‐flanked STOP cassette prevents transcription of the tdTomato. However, the Cre‐recombinase enzyme can delete the STOP cassette, leading to robust tdTomato expression (Madisen et al., 2010). By loading the OMVs with Cre‐recombinase enzyme, the cells to which the OMV delivered their cargo, namely, the Cre‐recombinase, will turn red due to the removal of the STOP cassette and the subsequent expression of the tdTomato fluorescent protein. Next, H. pylori OMV biodistribution and Cre‐recombinase delivery were tracked by detection of the tdTomato expression in the stomach, colon, lung, heart, liver, kidney, spleen, and brain. A clear TdTomato signal was observed in the stomach, while absent in colon sections at the end of the gastrointestinal tract. Also, spleen, lung and heart did not show a pronounced increase in TdTomato+ cells. In contrast, we did observe Cre‐mediated recombination in the liver, kidney and brain (e.g., hippocampus and cortex) (Figure 2c and Figure S4a‐f). Co‐stainings revealed that the majority of tdTomato+ cells in the brain are astrocytes as visualized using the astrocyte specific marker GFAP but also a small amount of tdTomato+ NeuN+ double‐positive neurons could be observed (Figure 2d,e and Figure S4b). In the stomach, tdTomato signal was detected in CD45+ cells (Figure S4e). These data implicate that H. pylori OMVs can travel across various biological barriers and eventually even migrate to the brain.
FIGURE 2.
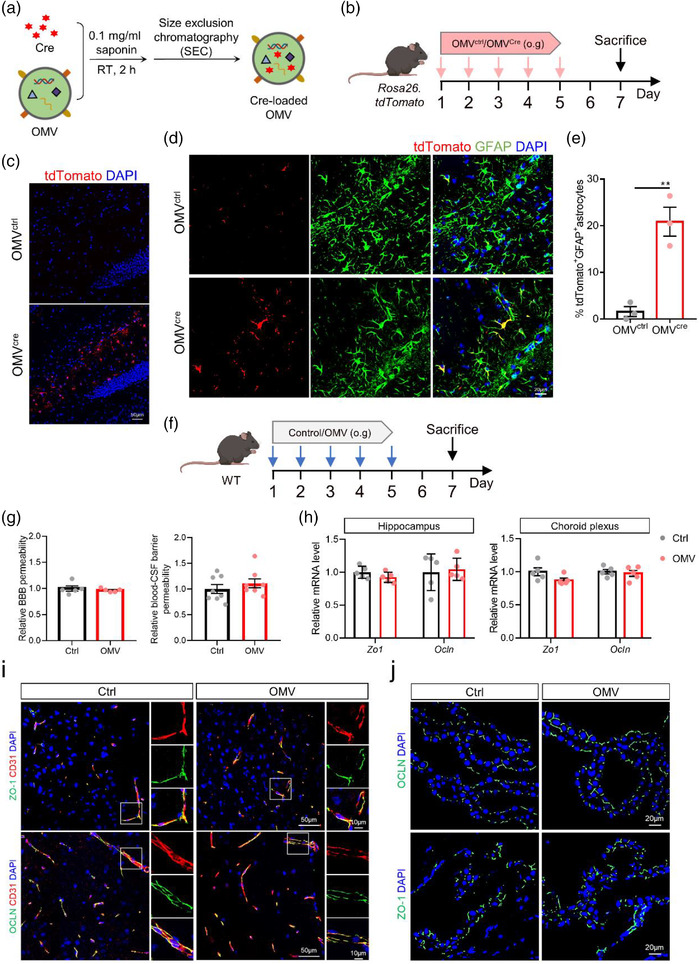
Helicobacter pylori OMVs migrate to the brain and are taken up by astrocytes. (a) Graphical illustration of saponin‐assisted Cre‐recombinase loading into H. pylori OMVs and repurification with SEC. (b) Treatment schedule of Rosa26.tdTomato mice treated with unmodified (OMVctrl) or Cre‐recombinase‐loaded (OMVcre) H. pylori OMVs. (c) Representative images of tdTomato staining in the hippocampus of Rosa26.tdTomato orally gavaged with OMVctrl and OMVCre. Scale bars, 50 μm. (d) Representative images of tdTomato and GFAP co‐staining in the hippocampus. Scale bars, 20 μm (left) and 10 μm (right). (e) Quantification of tdTomato+GFAP+ astrocytes. (f) Treatment schedule of WT mice treated with control or H. pylori OMVs. (g) Relative permeability of BBB and blood‐CSF barrier in control and OMV‐treated mice. (h) Relative gene expression of tight junctions Zo1 and Ocln in the hippocampus (left) and choroid plexus (right) in control and OMV‐treated mice. (i) Representative images of ZO‐1/CD31and OCLN/CD31 staining in the hippocampus control and OMV‐treated mice. Scale bars, 50 and 10 μm (insert). (j) Representative images of ZO‐1 and OCLN staining in choroid plexus control and OMV‐treated mice. Scale bars, 20 μm. The graphs are shown as the mean ± SEM and the datapoints are biological replicates. Images are representative for 3 (c and d) or 5 (h) biological replicates. Statistical significance was determined by two‐tailed Student's t‐test. **p < 0.01.
To investigate if the H. pylori OMV translocation to the brain is caused by OMV‐induced disruption of the gastrointestinal barrier and central nervous system (CNS) barriers, WT mice were followed the same treatment schedule and the integrity of gastrointestinal barrier, BBB and blood‐CSF barrier was analysed using a 4 kDa FITC‐conjugated dextran tracer (Figure 2f,g). The comparable FITC signal indicated that the H. pylori OMVs treatment had no apparent effect on the integrity of these barriers (Figure 2g and Figure S5a). In agreement with this, we did not observe differences in mRNA levels of the TJs Zo1 and Ocln in hippocampus and choroid plexus upon H. pylori OMV treatment (Figure 2h). Also, the protein localization of ZO‐1 and OCLN in hippocampal endothelial and choroid plexus epithelial cells showed comparable near‐continuous staining at the apical cell border upon H. pylori OMV oral gavage as seen in the control conditions (Figure 2i and j). In the stomach, we even detected higher mRNA expression Ocln and Cdh1, and increased protein expression of CDH1 in H. pylori OMVs‐treated mice compared to the control mice (Figure S5b‐d). Collectively, these results indicate that H. pylori OMVs do not disrupt the gastrointestinal and blood‐brain interfaces but may actively cross brain barriers through transcellular pathways.
4.3. H. pylori OMVs promote AD pathology in AppNL‐G‐F mice
To examine the effect of H. pylori OMVs on AD pathology, the OMVs were administered to AppNL‐G‐F mice by oral gavage following the treatment scheme shown in (Figure 3a). While OMV treatment for 1 week did not induce significant changes in Aβ pathology between H. pylori OMV‐treated and control AppNL‐G‐F mice based on the Aβ immunostaining (Figure S6a‐c), 3 weeks of H. pylori OMV treatment did have pronounced effects (Figure 3a‐c). More specifically, we observed that the amount of Aβ plaques significantly increased in the hippocampus of AppNL‐G‐F mice upon H. pylori OMV treatment compared to control treated mice (Figure 3b,c). Aβ plaque load was mostly increased in the CA1 and CA2/3 regions, but not in the dentate gyrus (DG) (Figure 3d,e). In agreement with the Aβ plaque analysis, Aβ ELISA also showed a significant increase in both soluble and insoluble Aβ1‐40 and Aβ1‐42 in the hippocampus of H. pylori OMV‐treated AppNL‐G‐F mice (Figure 3f). Additionally, morphometric analysis of Aβ stained brain sections revealed an increase in both medium (<250–600 μm2) and large (>600 μm2) plaques in the hippocampus of H. pylori OMV‐treated AppNL‐G‐F mice compared to control treated mice. In contrast, no difference in the number of small plaques (<250 μm2) could be observed (Figure 3g).
FIGURE 3.
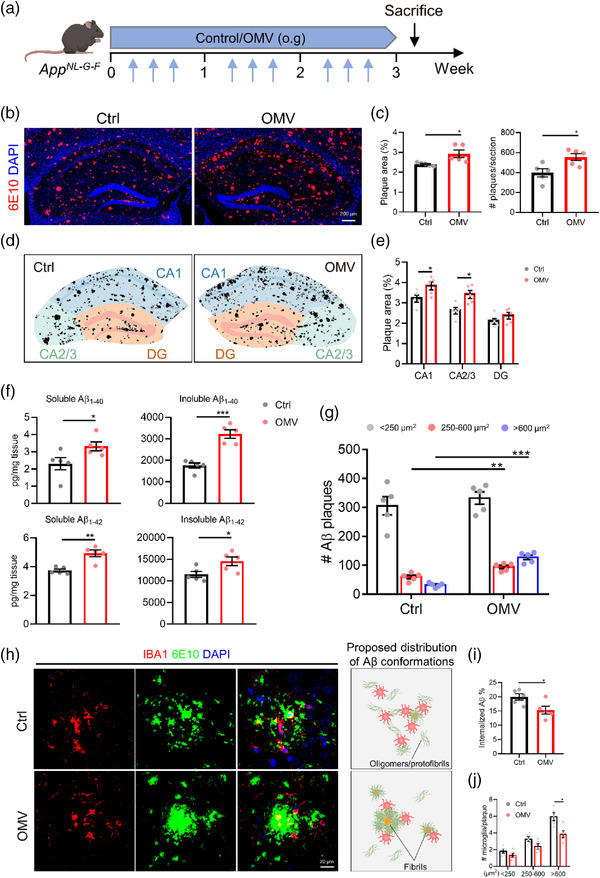
Helicobacter pylori OMVs promote AD pathology in AppNL‐G‐F mice. (a) Treatment schedule of AppNL‐G‐F mice treated with control or H. pylori OMVs. (b) Representative images of 6E10 staining in the hippocampus. Scale bars, 200 μm. (c) Quantification of Aβ plaque area (left) and number (right) in the hippocampus. (d) Corresponding masks used to quantify plaque load in subregions of the hippocampus. (e) Quantification of the plaque load in hippocampal subregions. (f) Soluble and insoluble Aβ1‐40 and Aβ1‐42 levels in the hippocampus. (g) Quantification of Aβ plaque size distribution in the hippocampus. (h) Representative images of IBA1+ microglia and 6E10 staining in the hippocampus and proposed Aβ conformations in the different plaque types. Scale bars, 20 μm. (i) Quantification of the percentage of overlay area of IBA1+ microglia and Aβ plaque. 3–5 plaques are analysed per mouse and 5 mice per group. (j) Plaques were divided into small (< 250 μm2), medium (250‐600 μm2), and large (> 600 μm2) plaques, and the number of microglia per plaque was quantified. 3–5 plaques are analysed per mouse and 5 mice per group. The graphs are shown as the mean ± SEM and the datapoints are biological replicates. Images are representative for 5 (b and h) biological replicates. Statistical significance was determined by two‐tailed Student's t‐test. *p < 0.05, **p < 0.01, ***p < 0.001.
Microglia are brain resident macrophages that play an important role in mediating Aβ degradation and clearance (Tejera et al., 2019). We observed a decrease in microglial internalized Aβ and a lower number of microglia, labelled via the microglial marker IBA1 adjacent to the large plaques in AppNL‐G‐F mice upon OMV oral gavage (Figure 3h‐j). Consistent with our Aβ size distribution analysis (Figure 3g), we observed that Aβ plaques were diffuse in AppNL‐G‐F control mice, whereas they were centrally localized in H. pylori OMV‐treated AppNL‐G‐F mice (Figure 3h). Taken together, these findings suggest that H. pylori OMV treatment accelerated the typical hallmark of AD pathology, namely, Aβ plaque formation, which might be due to impaired microglial Aβ clearance.
4.4. H. pylori OMVs activate C3‐C3aR signalling in WT and AppNL‐G‐F mice
As OMVs contain a certain amount of LPS (Vanaja et al., 2016) and as our previous study showed that LPS‐induced systemic inflammation affects brain inflammation and AD pathology (Xie et al., 2021), we next evaluated the impact of orally gavaged H. pylori OMVs on peripheral and central inflammation in WT mice (Figure S7a‐f). We observed an increased infiltration of mononuclear and polymorphonuclear cells in the stomach submucosa of H. pylori OMV‐treated mice (Figure S6a) and a significant increased gastric inflammation score based on the Updated Sydney System (Figure S7b). On the contrary, the pro‐inflammatory cytokines interleukin (IL‐)1β and tumour necrosis factor (TNF) showed no differences neither on mRNA level nor on protein level (Figure S7c and d) in the stomach. Plasma levels of IL‐1β, but not of TNF, were elevated (Figure S7e), while Il1β and Tnf gene expression in the hippocampus was not increased in H. pylori OMV‐treated mice compared to control mice (Figure S7f). These results show that H. pylori OMVs do not induce severe peripheral and central inflammation in WT mice.
H. pylori is complement sensitive and activates mainly the classic complement pathway (Berstad et al., 2001). Indeed, we found that genes involved in the classical pathway activation (C1q through C3) are significantly upregulated in WT mice treated with H. pylori OMVs while genes more specific to the lectin, alternative, or terminal pathways of the complement are not affected (Figure 4b,c and Figure S8). This was accompanied by an enhanced C3 protein expression and a higher degree of colocalization of C3 with GFAP positive cells in the hippocampus of H. pylori OMV‐treated mice compared to WT controls (Figure 4d,e). Also, western blot analysis showed an increased trend of C3 expression in the hippocampus of H. pylori OMV‐treated WT mice (Figure 4f,g).
FIGURE 4.
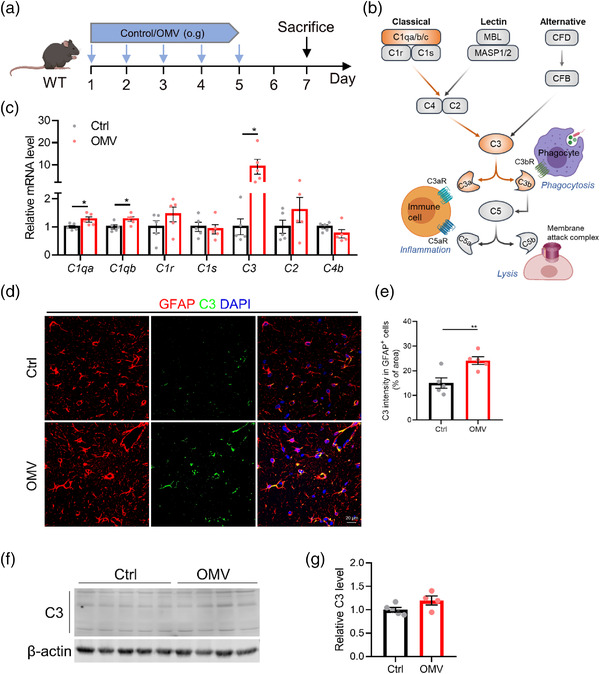
Helicobacter pylori OMVs promote C3 activation in WT mice. (a) Treatment schedule of AppNL‐G‐F mice treated with control or H. pylori OMVs. (b) Simplified schematic of the complement pathway illustrating selected proteins. Proteins involved in the classical complement pathway and upregulated by H. pylori OMVs treatment are highlighted in orange. (c) Gene expression of complement components in the hippocampus. (d) Representative images of GFAP and C3 staining in CA3 region of hippocampus. Scale bars, 20 μm. (e) Quantification of the percentage of overlay area of C3 in astrocytes. (f) Western blot analysis of C3 expression in the hippocampus. Individual lanes are biological duplicates (n = 4 or 5). (g) Quantification of the relative C3 levels in the hippocampus. The graphs are shown as the mean ± SEM and the datapoints are biological replicates. Images are representative for 5 (d) biological replicates. Statistical significance was determined by two‐tailed Student's t‐test. *p < 0.05, **p < 0.01
In contrast, C3aR expression was unaltered on protein and mRNA levels in hippocampus tissues of WT mice (Figure S9a‐c). In addition, co‐staining documented a high degree of colocalization of C3aR and IBA1 in these mice (Figure S9d,e). Next, we studied C3‐C3aR signalling in AppNL‐G‐F mice after 3 weeks of H. pylori OMVs treatment. In contrast to the WT mice, both the mRNA and protein levels of C3 showed a significant increase in the hippocampus of H. pylori OMV‐treated AppNL‐G‐F mice compared to controls as shown by qPCR and immunostaining (Figure S10a‐e), while only the protein expression level of C3aR showed a trend towards upregulation after H. pylori OMV treatment in AppNL‐G‐F mice (Figure S10f‐j). Co‐immunostaining of C3, Aβ and GFAP also showed a high degree of colocalization of C3 and Aβ/GFAP (Figure S10d). In addition, a quantitative increase of C3 intensity in this immunostaining was consistently detected in H. pylori OMV‐treated AppNL‐G‐F mice (Figure S10e). Although the H. pylori OMV treatment did not affect the C3aR expression in WT mice (Figure 4f‐j), its protein level was upregulated in H. pylori OMV‐treated AppNL‐G‐F mice (Figure S10g,h). Similarly, co‐staining C3aR and IBA1 in the hippocampus showed colocalization and an increased trend of C3aR expression in H. pylori OMV‐treated mice compared to controls (Figure S10i,j). Altogether, these results indicate H. pylori OMV treatment can induce or promote C3 activation.
4.5. H. pylori OMVs increase glial reactivity via C3‐C3aR signalling in WT mice
Astrocytes and microglia are the major glial cells in the CNS that maintain and support homeostasis of the neuronal functions (Vainchtein and Molofsky, 2020). C3‐C3aR signalling plays a prominent role in immune regulation in the CNS by mediating glial reactivity (Litvinchuk et al., 2018). Next, we assessed the effect of H. pylori OMV treatment on glial reactivity and the role of the increased C3‐C3aR signalling in this process. The WT mice were simultaneously treated with H. pylori OMVs and/or C3aRA via oral gavage and intraperitoneal injection, respectively (Figure 5a). Although no significant astrogliosis was observed in the whole hippocampus of WT mice after H. pylori OMV treatment (Figure S11a,b), a significant increase in GFAP signal was observed specifically in the hippocampal CA3 region (Figure 5b,c). Increased expression of GFAP represents astroglial activation and/or astrogliosis (Brahmachari et al., 2006). Moreover, quantitative morphometric 3D analysis of GFAP+ astrocytes in the CA3 region showed that H. pylori OMV‐treated WT mice have reactive astrocytes, characterized by significantly longer processes and increased number of segments, branching, and terminal points relative to astrocytes from control treated WT mice (Figure 5d,e). Indeed, blocking C3 signalling using C3aRA treatment did not block the H. pylori OMV effects on astrogliosis and astrocytic reactivity (Figure 5b‐e and Figure S11a,b). In parallel, we observed a significant decrease in the number of IBA+ microglia in the CA3 hippocampal region and the whole hippocampus of H. pylori OMV‐treated WT mice compared to control treated WT mice (Figure 5f,g and Figure S11c,d). However, no differences were visible in these mice on mRNA expression of microgliosis marker Aif1 or total Ki‐67+/Ki‐67+GFAP+/Ki‐67+IBA1+ cells (Figure S11e‐k). Quantitative analysis of microglial morphology showed decreased segments, branching, and terminal points in H. pylori OMV‐treated WT mice, and these phenotypic effects were blocked by C3aRA treatment (Figure 5h‐i). To further characterize the microglial activation, we performed IBA1 and CD68 co‐staining to identify CD68+ phagocytic microglia (Figure 5j). Quantification of CD68 immunoreactivity revealed that H. pylori OMV treatment in WT mice significantly increased CD68 expression in microglia and that C3aRA treatment prevented the H. pylori OMV‐induced increase (Figure 5k).
FIGURE 5.
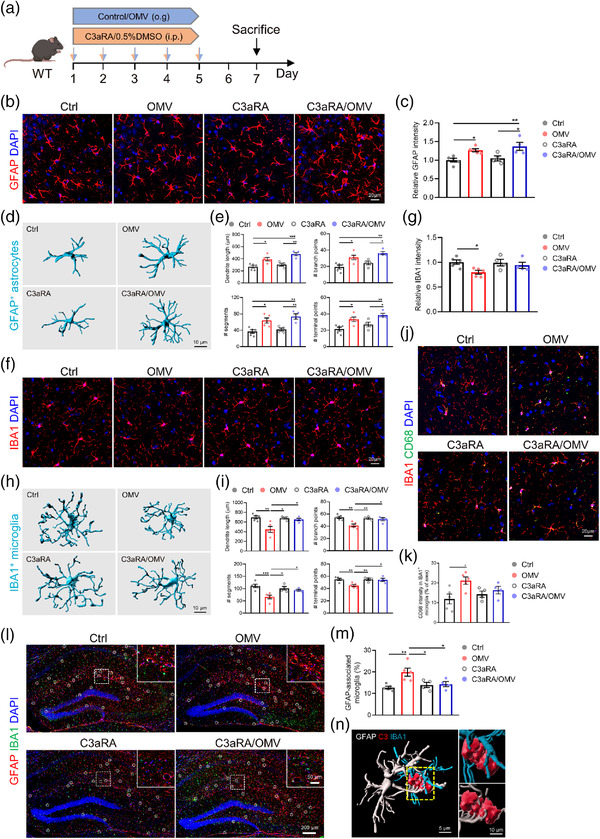
Helicobacter pylori OMVs increase glial reactivity via C3‐C3aR signalling in WT mice. (a) Treatment schedule of WT mice treated with control/H. pylori OMVs and 0.5%DMSO/C3aRA. (b) Representative images of GFAP staining in CA3 region of hippocampus. Scale bars, 20 μm. (c) Quantification of the relative GFAP intensity in CA3 region of hippocampus. (d) Imaris‐based 3D morphometric reconstruction analysis of GFAP+ astrocytes in CA3 region of hippocampus. Scale bars, 10 μm. (e) Imaris‐based quantification of cell morphology of GFAP+ astrocytes in CA3 region of hippocampus. (f) Representative images of IBA1 staining in CA3 region of hippocampus. Scale bars, 20 μm. (g) Quantification the number of IBA1+ microglia in CA3 region of hippocampus. (h) Imaris‐based 3D morphometric reconstruction analysis of IBA1+ microglia in CA3 region of hippocampus. (i) Imaris‐based quantification of cell morphology of IBA1+ microglia in CA3 region of hippocampus. Scale bars, 10 μm. (j) Representative images of IBA1 and CD68 staining in CA3 region of hippocampus. Scale bars, 20 μm. (k) Quantification of the percentage of CD68 intensity in IBA1+ microglia in CA3 region of hippocampus. (l) Representative images of GFAP and IBA1 staining in the hippocampus. Scale bars, 200 and 50 μm (insert). (m) Quantification of the percentage of astrocyte colocalized microglia. (n) Imaris‐based 3D reconstruction of the interaction between astrocytes and microglia via C3. Scale bars, 5 μm (left) and 10 μm (right). The graphs are shown as the mean ± SEM and the datapoints are biological replicates. Images are representative for 4 or 5 (b, f, j and l) biological replicates. Statistical significance was determined by two‐way ANOVA Bonferroni's multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001.
Given the specific expression of C3 and C3aR in astrocytes and microglia, respectively (Litvinchuk et al., 2018), we investigated whether there is an astrocyte‐microglia interaction via C3‐C3aR signalling after H. pylori OMV administration. We show a remarkable increase in IBA1+ microglial cells that overlap with GFAP+ astrocytes in the hippocampus of H. pylori OMV‐treated WT mice compared to the control treated mice and C3aRA/H. pylori OMV‐treated WT mice (Figure 5l and m). 3D reconstruction reveals a close spatial interaction of astrocytes, microglia and C3, suggesting the involvement of C3‐C3aR signalling in this glial cell interaction (Figure 5n).
4.6. H. pylori OMVs induce neuronal dysfunction via C3‐C3aR signalling in WT mice
We further analysed the effects of H. pylori OMV treatment on synaptic and neuronal properties and the role of C3‐C3aR signalling in this process (Figure 6a). Indeed, a reduced immunostaining intensity of presynaptic protein synaptophysin (SYP) and postsynaptic density protein 95 (PSD‐95) was observed in the CA3 area of the hippocampus of H. pylori OMV‐treated WT mice compared to WT controls (Figure 6b,c). Moreover, C3aRA treatment on H. pylori OMV‐treated WT mice resulted in a significant elevation of PSD‐95 immunoreactivity (Figure 6b,c). Similarly, high‐resolution images of co‐immunostaining of SYP and PSD‐95 revealed reduced pre‐, post‐, and double‐positive synaptic puncta in H. pylori OMV‐treated WT mice, while C3aRA treatment did prevent to a certain level (not significant trend) synapse loss (Figure 6d,e). These results were further confirmed by immunoblot analysis in the hippocampus using anti‐SYP and anti‐PSD‐95 antibodies (Figure 6f,g). These findings suggest that H. pylori OMVs are potent inducers of synaptic deficits and C3‐C3aR signalling plays a crucial role in this process.
FIGURE 6.
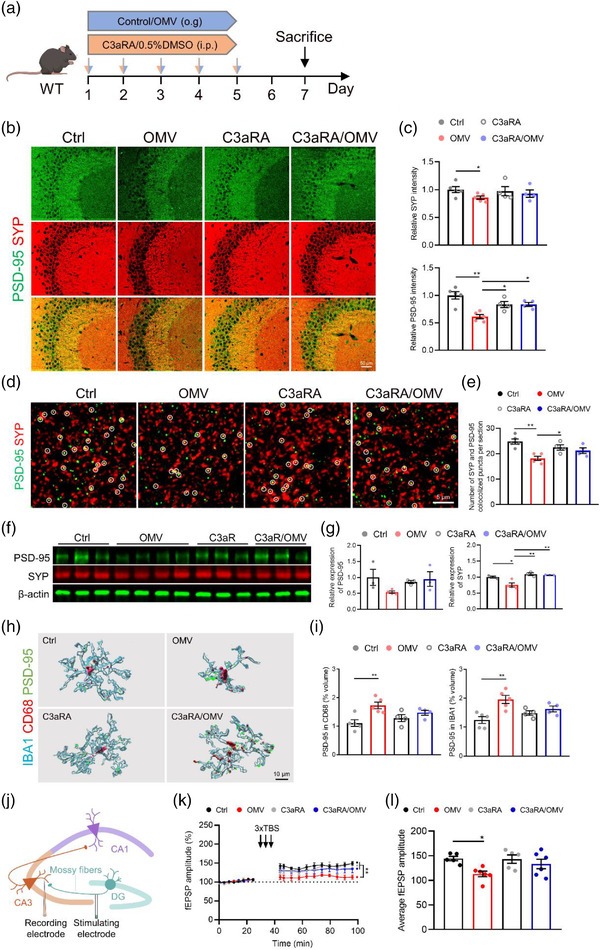
Helicobacter pylori OMVs induce synaptic deficits via C3‐C3aR signalling in WT mice. (a) Treatment schedule of WT mice treated with control/H. pylori OMVs and 0.5%DMSO/C3aRA. (b) Representative SYP and PSD‐95 co‐immunostaining in CA3 region of hippocampus. Scale bars, 50 μm. (c) Quantification of the relative intensity of SYP (upper) and PSD‐95 (lower). (d) Representative high magnification confocal images of SYP and PSD‐95 coimmunostaining in CA3 region of hippocampus of WT. Scale bars, 5 μm. (e) Quantification of the number of colocalized puncta of SYP and PSD‐95. (f) Western blot analysis of SYP and PSD‐95 in the hippocampus. Individual lanes are biological replicates (n = 3–4). (g) Quantification of relative SYP and PSD‐95 expression in the hippocampus. (h) Imaris‐based 3D reconstruction analysis of IBA1/CD68/PSD‐95 staining in CA3 region of hippocampus. Scale bars, 10 μm. (i) Quantification of the percentage of PSD‐95+ volume inside CD68+ phagosome (upper) and IBA1+microglia (lower). (j) Schematic diagram indicating the general experimental set‐up of LTP measurement. (k‐l) fEPSP amplitude recordings over time (k) and the average of the fEPSP amplitude recordings (l) after LTP induction in CA3 mossy fiber synapses. The graphs are shown as the mean ± SEM and the datapoints are biological replicates. Images are representative for 4 or 5 (b and d) biological replicates. Statistical significance was determined by two‐way ANOVA Bonferroni's multiple comparisons. *p < 0.05, **p < 0.01
As our earlier analysis showed a higher intensity of CD68+ phagocytic microglia in H. pylori OMV‐treated WT mice (Figure 5j,k), the reduced synaptic puncta were seen in H. pylori OMV‐treated WT mice may be due to increased microglial engulfment. We found that the volume of PSD‐95‐positive (Figure 6h,i) or SYP‐positive (Figure S9a,b) puncta within IBA1+ microglia are increased in H. pylori OMV‐treated WT mice and this effect was largely prevented by simultaneous C3aRA treatment. As the excessive synaptic pruning may result in imbalanced synaptic transmission and subsequent synaptic dysfunction, we evaluated the contribution of H. pylori OMVs to the synaptic function of mossy fiber‐CA3 synapses by assessing the electrophysiological properties of these synapses in acute hippocampal slices of WT mice (Figure 6j). The LTP induction was significantly decreased in H. pylori OMV‐treated WT mice compared to control treated WT mice, and this phenotype was substantially improved when combined with C3aRA treatment (Figure 6k,l and Figure S12f,g). Furthermore, we tested whether this effect could also be observed in other neuronal networks, and recorded LTP of Schaffer collateral‐CA1 synapses (Figure S12c). The LTP recordings also showed Schaffer collateral‐CA1 synapses were affected by H. pylori OMV treatment, but not as pronounced as mossy fiber‐CA3 synapses (Figure S12d‐g). When comparing the efficacy of C3aRA treatment in CA1‐ and CA3‐pathways, similar protective effects against H. pylori OMV‐induced synaptic defects could be observed (Figure 6k,l and Figure S12d‐g). These findings suggest that the detrimental effect of H. pylori OMV on synaptic plasticity is at least in part due to increased synapse elimination by activated microglia.
The abnormal activation of microglia also actively participates in the phagocytosis of live neurons (Fricker et al., 2012). In this context, we performed NeuN staining and the quantification showed reduced neuronal numbers in the CA3 region of the hippocampus in H. pylori OMV‐treated WT mice (Figure S13a,b). C3aRA treatment in H. pylori OMV‐treated WT mice resulted in a powerful blockade of neurodegeneration (Figure S13a,b). The reduced neuronal density in the hippocampus by H. pylori OMV treatment may at least partly be due to reduced proliferation of neuronal progenitor cells as demonstrated by DCX and Ki‐67 immunostaining (Figure S13c,d). In addition, incorrect microglia‐neuron interactions are known to lead to microglial phagocytosis of live neurons and excessive neuronal loss(Brown and Neher, 2014). We then quantified the CD68+ phagosome in the CA3 stratum pyramidale and revealed an increased CD68 immunoreactivity in H. pylori OMV‐treated WT mice. Again, C3aRA treatment prevented this OMV‐induced increase (Figure S13e,f).
Combined, these data demonstrate that H. pylori OMV can induce synaptic deficits and neurodegeneration, and that enhanced microglial activation phagocytosis may contribute to this process through C3‐C3aR signalling.
4.7. Blocking C3‐C3aR signalling attenuates AD pathology and memory loss induced by H. pylori OMVs in AppNL‐G‐F mice
Finally, we investigated the effect of H. pylori OMVs on memory functions and the potential of blockage of C3‐C3aR signalling in preventing memory impairments. Therefore, AppNL‐G‐F mice were simultaneously treated with H. pylori OMVs and/or C3aRA via oral gavage and intraperitoneal injection, respectively (Figure 7a). After 3 weeks of treatment, we examined the effects on cognitive function using Y‐maze and novel object recognition (NOR) tests. The H. pylori OMV‐treated AppNL‐G‐F mice showed cognitive impairment based on significant decreased spontaneous alternation in the Y‐maze task and performance to the new object in the NOR test (Figure 7b‐d). Interestingly, C3aRA treatment prevented the occurrence of cognitive impairment caused by H. pylori OMVs in AppNL‐G‐F , as shown in the NOR test, while this was less pronounced in the Y‐maze (Figure 7b‐d). Consistent with the cognitive results, H. pylori OMV treatment worsen Aβ pathology and again this effect was largely blocked by C3aRA treatment (Figure 7e‐h). In line with this, C3aRA treatment protected the microglial Aβ clearance from the influence of H. pylori OMVs, and showed the same levels of microglia internalized Aβ as AppNL‐G‐F controls (Figure 7i,j). Lastly, quantification of microglia around Aβ plaques also demonstrated that C3aRA treatment inhibited H. pylori OMV‐induced clustering decrease in AppNL‐G‐F mice compared with control AppNL‐G‐F mice (Figure 7i,k). In addition, LTP recordings revealed that Schaffer collateral‐CA1 synapses in AppNL‐G‐F mice were significantly impaired upon oral H. pylori OMV administration and this effect was blocked by C3aRA treatment (Figure S7l‐n and Figure S14a,b). Together, these findings suggest that H. pylori OMVs cause cognitive impairment in AppNL‐G‐F mice and that by blocking the C3‐C3aR signalling AD pathology and memory loss induced by H. pylori OMV can be attenuated.
FIGURE 7.
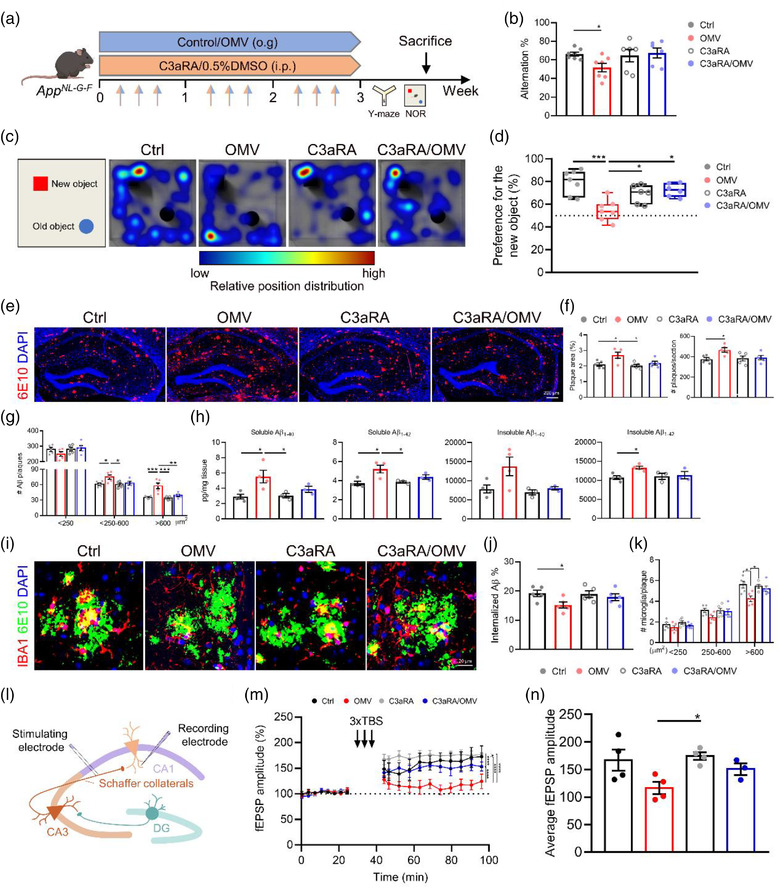
C3aR antagonist treatment blocks the effects of Helicobacter pylori OMVs on AD pathology and memory function in AppNL‐G‐F mice. (a) Treatment schedule of AppNL‐G‐F mice treated with control/H. pylori OMVs and 0.5%DMSO/C3aRA. (b) Quantification of Y‐maze spontaneous alternation. (c) Representative heat maps of tracks in NOR test. (d) The percentage of NOR discrimination. (e) Representative images of 6E10 staining in the hippocampus. Scale bars, 200 μm. (f) Quantification of the plaque area (left) and number (right) in the hippocampus. (g) Quantification of Aβ plaque size distribution in the hippocampus. (h) Soluble and insoluble Aβ1‐40 and Aβ1‐42 levels in the hippocampus. (i) Representative images of IBA1 and 6E10 staining in the hippocampus. Scale bars, 20 μm. (j) Quantification of the percentage of overlay area of microglia and Aβ plaque. (k) Plaques were divided into small (< 250 μm2), medium (250‐600 μm2), and large (> 600 μm2), the number of microglia per plaque was quantified. (l) Schematic diagram indicating the general experimental set‐up of LTP measurement in AppNL‐G‐F mice. (m‐n) fEPSP amplitude recordings over time (m) and the average of the fEPSP amplitude recordings (n) after LTP induction in CA1 Schaffer collateral synapses. The graphs are shown as the mean ± SEM and the datapoints are biological replicates. Images are representative for 5 (c, e and i) or 3–4 (m and n) biological replicates. Statistical significance was determined by two‐way ANOVA Bonferroni's multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
5. DISCUSSION
The gut microbiota is a complex community of microorganisms that resides in our gastrointestinal ecosystem and of which a disbalance not only implies various gut disorders but also CNS disorders. Growing evidence supports the concept that the gut microbiota affects host brain function including cognitive behaviour and that a microbiota dysbiosis may mediate or affect AD pathogenesis (Hu et al., 2016). However, the mechanisms of this microbiota‐gut‐brain axis are not yet fully understood.
It is well established that microbiota‐derived molecules such as LPS, peptidoglycan, and short‐chain fatty acids directly modulate neuroinflammation and Aβ accumulation (Colombo et al., 2021; Krstic et al., 2012; Xie et al., 2021). Several of these bioactive molecules are also present in microbiota‐derived OMVs, that is, nano‐sized spherical buds of the outer membrane produced by both pathogenic and non‐pathogenic Gram‐negative bacteria (Xie et al., 2022a). OMVs can transport their cargo to proximal and distal cells to exert physiological and pathological effects (Finethy et al., 2017; Ha et al., 2020; Han et al., 2019). Recently, microbial‐derived OMVs have also been demonstrated to be involved in several pathologies such as mitochondrial dysfunction (Deo et al., 2020, 2018), systemic bone loss (Kim et al., 2021), pulmonary fibrosis(Yang et al., 2019) and cognitive impairment (Durant et al., 2020; Wei et al., 2020).
H. pylori is a major human pathogen that induces inflammation of the stomach and is etiologically related to various gastric and extragastric diseases, like AD (Doulberis et al., 2018). Next to the induction of inflammation, H. pylori infection can also increase the production of AD risks, such as reactive oxygen species, lipid peroxides and homocysteine (Doulberis et al., 2018). Here, we investigated whether of H. pylori‐derived OMVs might be key players in the previously reported H. pylori effects on AD pathology. Our results indicate that OMVs can travel from the gut to the brain and eventually affect brain functions. Notably, we observed that both the gastrointestinal and brain barriers remained intact after H. pylori OMV administration, suggesting that H. pylori OMVs may enter the brain via a transcellular pathway crossing the barriers or indirectly via the vagus nerve, as was shown for P. hominis OMVs (Lee et al., 2020). In addition, we could not rule out the possibility that cargo released from disrupted OMVs might somehow reach brain barriers and eventually enter the brain. The intact barriers may limit the entry of the OMVs into the bloodstream and the brain, which may explain the lack of very pronounced peripheral and central inflammation that we observed in our study. In addition, a recent study reported that H. pylori OMVs induced approximately 5‐fold higher production of pro‐inflammatory cytokines in the spleen upon intraperitoneal injection compared to oral administration (Choi et al., 2017), indicating a positive correlation between peripheral inflammation levels and OMV amount in peripheral tissues. Taken together, peripheral inflammation is probably not the underlying mechanism by which H. pylori OMVs potentially affect brain function.
Our study reveals that H. pylori OMVs promote Aβ plaque formation in AppNL‐G‐F mice. Interestingly, the effects on Aβ pathology can only be detected after 3 weeks of oral gavage with H. pylori OMVs, suggesting that a chronic more robust H. pylori infection of the gut is needed, while a transient, acute infection does not affect the brain (Albaret et al., 2020). Of note, the Aβ plaques are most enriched in the CA1 and especially in the CA2/3 regions of the hippocampus upon H. pylori OMV administration. This difference in Aβ plaque distribution is probably due to the distribution of APP‐expressing interneurons in the different hippocampal regions as demonstrated by Rice et al. (2020). In addition to the amount of Aβ aggregation, the H. pylori OMV treatment also increased the compactness and surface area of Aβ plaques, important pathological features of AD (Yuan et al., 2016).
Astrocytes and microglia are resident cells of the CNS, and they contribute not only to the formation of the BBB but also to the homeostasis and immune functions of the CNS (Alvarez et al., 2013; Vainchtein and Molofsky, 2020). Recent evidence has shown that both cell types can take up and internalize microbial‐derived OMVs and induce an inflammatory response (Bittel et al., 2021; Wei et al., 2020). In line with this observation, we detected H. pylori OMV uptake by astrocytes and induction of activation of glial cells, reflected by the increase (astrocytes) and reduction (microglia) in dendrite length, the number of segments, branch points and terminal points, respectively. In H. pylori OMV‐treated mice, we also observed increased interaction between astrocyte‐microglia and microglia‐neuron, and ultimately the accumulation of Aβ and cognitive impairment. As the principal immune effector and phagocytic cells in CNS, microglia play an important role in Aβ clearance by a variety of phagocytic and digestive mechanisms (Tejera et al., 2019; Zuroff et al., 2017). We found that H. pylori OMV treatment resulted in a decrease in the number of Aβ phagocytosing microglia. In addition, we found that H. pylori OMV administration induced neurodegeneration and synaptic deficits, which were at least partially modulated by phagocytosis of reactive microglia. Indeed, a recent study has also shown that reactive microglia are involved in neurodegeneration and excessive synaptic pruning (Litvinchuk et al., 2018). However, we cannot exclude the additional effects of signals released by neuronal activity to instruct phagocytosis of microglia (Bartels et al., 2020). In addition, the abnormal neuronal activity can directly contribute to the Aβ aggregation as we observed in our previous study (Xie et al., 2021) and aggregation of Aβ into compact plaques in turn affects local neuronal dysregulation such as disruption of synaptic integration (Stern et al., 2004). In addition to Aβ accumulation, synapse loss is also closely associated with cognitive decline (Colom‐Cadena et al., 2020). In this respect, we speculate that the H. pylori OMV‐exacerbated Aβ pathology and synaptic dysfunction are the main contributors to the observed memory defects.
Mechanistically, we identified the C3‐C3aR signalling pathway as an important regulator in the H. pylori OMV‐mediated detrimental effects on the brain. The complement pathway is a critical regulator of innate immunity and the complement pathway molecules are expressed in the CNS where they have been implicated in AD processes (Chen et al., 2020; Hong et al., 2016). H. pylori is complement sensitive and activates mainly the classic pathway (Berstad et al., 2001). The classical complement activation requires the cleavage of the central complement factor C3 to C3a and C3b, which elicit downstream events through binding to their receptors C3aR and CR3, respectively (Stephan et al., 2012). Combining the predominant expression of astroglial C3 and microglial C3aR observed in our study and a previous study (Liddelow et al., 2017), we speculate that H. pylori OMV increases astrocyte‐microglia crosstalk via C3‐C3aR and microglia activation trigger the neuronal dysfunction, Aβ deposition and ultimately cognitive impairment. Our results are in line with previously reported that C3‐C3aR signalling is important in the astrocyte‐microglia and neuron‐immune crosstalk and that it influences network function and Aβ pathology (Lian et al., 2016, 2015). Blocking this pathway with pharmacological inhibition of C3aR curtailed H. pylori OMV induced dysfunction of glial cells (proliferation, activation and phagocytosis) and neurons (neurodegeneration and synaptic deficits), and almost completely rescued the Aβ pathology and memory loss, suggesting C3aR inhibition may represent a novel therapeutic target that may ameliorate AD pathology induced by H. pylori infection. Importantly, we observed neither the beneficial effects of C3aRA treatment alone on Aβ pathology nor cognitive functions in AppNL‐G‐F mice despite previously reported that C3aR has a detrimental effect in promoting viral‐induced synapse loss and cognitive decline (Vasek et al., 2016) and that C3aRA treatment relieves Aβ pathology (Lian et al., 2016). Whether the varied outcomes are due to the difference in mouse models, the time of treatment administration or other effects remains to be seen.
It is important to note that in addition to H. pylori, other pathogens such as periodontal bacteria (e.g., P. gingivalis and Fusobacterium nucleatum), Borrelia burgdorferi and Chlamydia pneumoniae have also been reported to be associated with the pathogenesis of AD (Vigasova et al., 2021). In addition, a recent study showed that non‐pathogenic E. coli OMVs are also able to deliver their cargo from the gut to the brain (Bittel et al., 2021), suggesting potential effects on brain functions. Our analysis was restricted to the effects of H. pylori‐derived OMV on the CNS, so it is uncertain whether these effects are specific to H. pylori OMVs or more broadly to OMVs in general. Therefore, further studies comparing OMVs from different bacteria are needed to decipher whether the observed effects are specific to H. pylori‐derived OMVs.
Taken together, we are the first to describe a novel aspect of the gut‐brain interplay, namely, that H. pylori‐derived OMVs modulate physiological functions of glial cells and neurons, and can worsen AD pathology. We further identified C3‐C3aR signalling as an essential regulator in the impact of H. pylori OMVs on brain functionality. Indeed, pharmacological inhibition of C3aR prevents H. pylori OMV induced activation of glial cells, synaptic impairment and neuronal loss, thereby rescuing Aβ pathology and memory functions. These findings not only provide new insights supporting the infectious hypothesis behind AD pathogenesis, but also identify OMVs as important players in the gut‐brain axis.
AUTHOR CONTRIBUTIONS
Junhua Xie: Conceptualization; Data curation; Investigation; Methodology; Writing—original draft. Lien Cools: Investigation; Writing—review & editing. Griet Van Imschoot: Investigation. Elien Van Wonterghem: Investigation. Marie J. Pauwels: Investigation; Writing—review & editing. Ine Vlaeminck: Investigation. Chloë De Witte: Investigation. Samir EL Andaloussi: Resources; Writing—review & editing. Keimpe Wierda: Investigation; Methodology; Writing—review & editing. Lies De Groef: Resources; Writing—review & editing. Freddy Haesebrouck: Funding acquisition; Resources; Supervision; Writing—review & editing. Lien Van Hoecke: Conceptualization; Data curation; Investigation; Methodology; Supervision; Writing—review & editing. Roosmarijn E. Vandenbroucke: Conceptualization; Data curation; Funding acquisition; Methodology, Resources; Supervision; Writing – review & editing.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
We thank M. Mercken (Johnson & Johnson Pharmaceuticals Research and Development, Beerse, Belgium) for providing the antibodies against Aβ (JRF/ABN/24, JRF/cAB40/28, and JRF/cAB42/26) for the Aβ ELISA, Femke Baeke and Riet De Rycke for performing transmission electron microscopy, Sofie De Bruyckere for technical assistance related to bacterial culture. We also want to thank the VIB Bioimaging Core for training, support and access to the instrument park. This work was supported by Ghent University Special Research fund (BOF) (BOF‐STA29‐17 and 01G03121), Research Foundation‐Flanders (FWO) (G055121N, 11M3322N, 12I3820N), Chinese Scholarship Council (CSC) (201808360194), Foundation for Alzheimer's Research Belgium (SAO‐FRA) (20190028, 20200032) and Baillet Latour Fund. The illustrations in this manuscript were partially created with BioRender.com.
Xie, J. , Cools, L. , Van Imschoot, G. , Van Wonterghem, E. , Pauwels, M. J. , Vlaeminck, I. , De Witte, C. , Andaloussi, S. E. L. , Wierda, K. , De Groef, L. , Haesebrouck, F. , Van Hoecke, L. , & Vandenbroucke, R. E. (2023). Helicobacter pylori‐derived outer membrane vesicles contribute to Alzheimer's disease pathogenesis via C3‐C3aR signalling. Journal of Extracellular Vesicles, 12, e12306. 10.1002/jev2.12306
Freddy Haesebrouck, Lien Van Hoecke and Roosmarijn E. Vandenbroucke shared senior authorship.
REFERENCES
- Albaret, G. , Sifré, E. , Floch, P. , Laye, S. , Aubert, A. , Dubus, P. , Azzi‐Martin, L. , Giese, A. , Salles, N. , Mégraud, F. , Varon, C. , Lehours, P. , & Roubaud‐Baudron, C. (2020). Alzheimer's disease and Helicobacter pylori infection: Inflammation from stomach to brain? Journal of Alzheimers Disease, 73, 801–809. [DOI] [PubMed] [Google Scholar]
- Alvarez, J. I. , Katayama, T. , & Prat, A. (2013). Glial influence on the blood brain barrier. Glia, 61, 1939–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels, T. , De Schepper, S. , & Hong, S. (2020). Microglia modulate neurodegeneration in Alzheimer's and Parkinson's diseases. Science, 370, 66–69. [DOI] [PubMed] [Google Scholar]
- Berstad, A. E. , Høgåsen, K. , Bukholm, G. , Moran, A. P. , & Brandtzaeg, P. (2001). Complement activation directly induced by Helicobacter pylori . Gastroenterology, 120, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Bielaszewska, M. , Rüter, C. , Bauwens, A. , Greune, L. , Jarosch, K.‐A. , Steil, D. , Zhang, W. , He, X. , Lloubes, R. , Fruth, A. , Kim, K. S. , Schmidt, M. A. , Dobrindt, U. , Mellmann, A. , & Karch, H. (2017). Host cell interactions of outer membrane vesicle‐associated virulence factors of enterohemorrhagic Escherichia coli O157: Intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathogens, 13, e1006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittel, M. , Reichert, P. , Sarfati, I. , Dressel, A. , Leikam, S. , Uderhardt, S. , Stolzer, I. , Phu, T. A. , Ng, M. , Vu, N. K. , Tenzer, S. , Distler, U. , Wirtz, S. , Rothhammer, V. , Neurath, M. F. , Raffai, R. L. , Günther, C. , & Momma, S. (2021). Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe‐host‐communication in vivo . Journal of Extracellular Vesicles, 10, e12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari, S. (2006). Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. Journal of Neuroscience, 26, 4930–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. C. , & Neher, J. J. (2014). Microglial phagocytosis of live neurons. Nature Reviews Neuroscience, 15, 209–216. [DOI] [PubMed] [Google Scholar]
- Chen, W.‐T. , Lu, A. , Craessaerts, K. , Pavie, B. , Sala Frigerio, C. , Corthout, N. , Qian, X. , Laláková, J. , Kühnemund, M. , Voytyuk, I. , Wolfs, L. , Mancuso, R. , Salta, E. , Balusu, S. , Snellinx, A. , Munck, S. , Jurek, A. , Fernandez Navarro, J. , Saido, T. C. , … De Strooper, B. (2020). Spatial transcriptomics and in situ sequencing to study Alzheimer's disease. Cell, 182, 976–991. [DOI] [PubMed] [Google Scholar]
- Chmiela, M. , Walczak, N. , & Rudnicka, K. (2018). Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori‐related diseases. Journal of Biomedical Science, 25, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, H. I. , Choi, J.‐. P. , Seo, J. , Kim, B. J. , Rho, M. , Han, J. K. , & Kim, J. G. (2017). Helicobacter pylori‐derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Experimental & Molecular Medicine, 49, e330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom‐Cadena, M. , Spires‐Jones, T. , Zetterberg, H. , Blennow, K. , Caggiano, A. , Dekosky, S T. , Fillit, H. , Harrison, J E. , Schneider, L. S. , Scheltens, P. , De Haan, W. , Grundman, M. , Van Dyck, C H. , Izzo, N J. , & Catalano, S M. (2020). The clinical promise of biomarkers of synapse damage or loss in Alzheimer's disease. Alzheimers Research and Therapy, 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, A. V. , Sadler, R. K. , Llovera, G. , Singh, V. , Roth, S. , Heindl, S. , Sebastian Monasor, L. , Verhoeven, A. , Peters, F. , Parhizkar, S. , Kamp, F. , Gomez De Aguero, M. , Macpherson, A. J. , Winkler, E. , Herms, J. , Benakis, C. , Dichgans, M. , Steiner, H. , Giera, M. , … Liesz, A. (2021). Microbiota‐derived short chain fatty acids modulate microglia and promote Abeta plaque deposition. Elife, 10, e59826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, J. , Lee, G. , Zhong, K. , Fonseca, J. , & Taghva, K. (2021). Alzheimer's disease drug development pipeline: 2021. Alzheimers Dement (N Y), 7, e12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo, P. , Chow, S H. , Han, M.‐. L. , Speir, M. , Huang, C. , Schittenhelm, R B. , Dhital, S. , Emery, J. , Li, J. , Kile, B T. , Vince, J E. , Lawlor, K E. , & Naderer, T. (2020). Mitochondrial dysfunction caused by outer membrane vesicles from Gram‐negative bacteria activates intrinsic apoptosis and inflammation. Nature Microbiology, 5, 1418–1427. [DOI] [PubMed] [Google Scholar]
- Deo, P. , Chow, S H. , Hay, I D. , Kleifeld, O. , Costin, A. , Elgass, K D. , Jiang, J.‐. H. , Ramm, G. , Gabriel, K. , Dougan, G. , Lithgow, T. , Heinz, E. , & Naderer, T. (2018). Outer membrane vesicles from Neisseria gonorrhoeae target PorB to mitochondria and induce apoptosis. PLoS Pathogens, 14, e1006945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deture, M A. , & Dickson, D W. (2019). The neuropathological diagnosis of Alzheimer's disease. Molecular Neurodegeneration, 14, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doheim, M. F. , Altaweel, A. A. , Elgendy, M. G. , Elshanbary, A. A. , Dibas, M. , Ali, A. A. H. A. , Dahy, T. M. , Sharaf, A. K. , & Hassan, A E. (2021). Association between Helicobacter pylori infection and stroke: a meta‐analysis of 273,135 patients. Journal of Neurology, 268, 3238–3248. [DOI] [PubMed] [Google Scholar]
- Dominy, S S. , Lynch, C. , Ermini, F. , Benedyk, M. , Marczyk, A. , Konradi, A. , Nguyen, M. , Haditsch, U. , Raha, D. , Griffin, C. , Holsinger, L J. , Arastu‐Kapur, S. , Kaba, S. , Lee, A. , Ryder, M I. , Potempa, B. , Mydel, P. , Hellvard, A. , Adamowicz, K. , … Potempa, J. (2019). Porphyromonas gingivalis in Alzheimer's disease brains: Evidence for disease causation and treatment with small‐molecule inhibitors. Science Advance, 5, eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulberis, M. , Kotronis, G. , Thomann, R. , Polyzos, S. A. , Boziki, M. , Gialamprinou, D. , Deretzi, G. , Katsinelos, P. , & Kountouras, J. (2018). Review: Impact of Helicobacter pylori on Alzheimer's disease: What do we know so far? Helicobacter, 23, e12454. [DOI] [PubMed] [Google Scholar]
- Du, X. , Wang, X. , & Geng, M. (2018). Alzheimer's disease hypothesis and related therapies. Translational Neurodegeneration, 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant, L. , Stentz, R. , Noble, A. , Brooks, J. , Gicheva, N. , Reddi, D. , O'Connor, M. J. , Hoyles, L. , McCartney, A. L. , Man, R. , Pring, E. T. , Dilke, S. , Hendy, P. , Segal, J. P. , Lim, D. N. F. , Misra, R. , Hart, A. L. , Arebi, N. , Carding, S. R. , & Knight, S. C. (2020). Bacteroides thetaiotaomicron ‐derived outer membrane vesicles promote regulatory dendritic cell responses in health but not in inflammatory bowel disease. Microbiome, 8, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzyubenko, E. , Rozenberg, A. , Hermann, D M. , & Faissner, A. (2016). Colocalization of synapse marker proteins evaluated by STED‐microscopy reveals patterns of neuronal synapse distribution in vitro . Journal of Neuroscience Methods, 273, 149–159. [DOI] [PubMed] [Google Scholar]
- Fang, Y. , Xie, H. , & Fan, C. (2022). Association of hypertension with Helicobacter pylori: A systematic review and metaanalysis. PLoS ONE, 17, e0268686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy, R. , Luoma, S. , Orench‐Rivera, N. , Feeley, E. M. , Haldar, A. K. , Yamamoto, M. , Kanneganti, T. D. , Kuehn, M. J. , & Coers, J. (2017). Inflammasome activation by bacterial outer membrane vesicles requires guanylate binding proteins. mBio, 8, e01188–e01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker, M. , Oliva‐Martín, M. J. , & Brown, G. C. (2012). Primary phagocytosis of viable neurons by microglia activated with LPS or Abeta is dependent on calreticulin/LRP phagocytic signalling. Journal of Neuroinflammation, 9, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, P. , Gao, M. , & Yung, K. K. L. (2020). Association of intestinal disorders with Parkinson's disease and Alzheimer's disease: A systematic review and meta‐analysis. ACS Chemical Neuroscience, 11, 395–405. [DOI] [PubMed] [Google Scholar]
- Gong, T. , Chen, Q. , Mao, H. , Zhang, Y. , Ren, H. , Xu, M. , Chen, H. , & Yang, D. (2022). Outer membrane vesicles of Porphyromonas gingivalis trigger NLRP3 inflammasome and induce neuroinflammation, tau phosphorylation, and memory dysfunction in mice. Frontiers in Cellular and Infection Microbiology, 12, 925435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlé, N. , Blaecher, C. , Bauwens, E. , Vandendriessche, C. , Balusu, S. , Vandewalle, J. , Van Cauwenberghe, C. , Van Wonterghem, E. , Van Imschoot, G. , Liu, C. , Ducatelle, R. , Libert, C. , Haesebrouck, F. , Smet, A. , & Vandenbroucke, R. E. (2018). The choroid plexus epithelium as a novel player in the stomach‐brain axis during Helicobacter infection. Brain, Behaviour and Immunity, 69, 35–47. [DOI] [PubMed] [Google Scholar]
- Ha, J. Y. , Choi, S.‐Y. , Lee, J. H. , Hong, S.‐H. , & Lee, H.‐J. (2020). Delivery of periodontopathogenic extracellular vesicles to brain monocytes and microglial IL‐6 promotion by RNA cargo. Frontiers in Molecular Bioscience, 7, 596366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, E.‐C. , Choi, S.‐Y. , Lee, Y. , Park, J.‐W. , Hong, S.‐H. , & Lee, H.‐J. (2019). Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF‐alpha production in human macrophages and cross the blood‐brain barrier in mice. FASEB Journal, 33, 13412–13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, S. , Beja‐Glasser, V F. , Nfonoyim, B M. , Frouin, A. , Li, S. , Ramakrishnan, S. , Merry, K M. , Shi, Q. , Rosenthal, A. , Barres, B A. , Lemere, C A. , Selkoe, D J. , & Stevens, B. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X. , Wang, T. , & Jin, F. (2016). Alzheimer's disease and gut microbiota. Science China Life Sciences, 59, 1006–1023. [DOI] [PubMed] [Google Scholar]
- Kim, H. Y. , Song, M.‐K. , Gho, Y. S. , Kim, H.‐H. , & Choi, B.‐K. (2021). Extracellular vesicles derived from the periodontal pathogen Filifactor alocis induce systemic bone loss through Toll‐like receptor 2. Journal of Extracellular Vesicles, 10, e12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic, D. , Madhusudan, A. , Doehner, J. , Vogel, P. , Notter, T. , Imhof, C. , Manalastas, A. , Hilfiker, M. , Pfister, S. , Schwerdel, C. , Riether, C. , Meyer, U. , & Knuesel, I. (2012). Systemic immune challenges trigger and drive Alzheimer‐like neuropathology in mice. Journal of Neuroinflammation, 9, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli, G. , Schott, J. M. , Hardy, J. , & De Strooper, B. (2021). Aducanumab: A new phase in therapeutic development for Alzheimer's disease? EMBO Molecular Medicine, 13, e14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Largo‐Barrientos, P. , Apóstolo, N. , Creemers, E. , Callaerts‐Vegh, Z. , Swerts, J. , Davies, C. , Mcinnes, J. , Wierda, K. , De Strooper, B. , Spires‐Jones, T. , De Wit, J. , Uytterhoeven, V. , & Verstreken, P. (2021). Lowering synaptogyrin‐3 expression rescues Tau‐induced memory defects and synaptic loss in the presence of microglial activation. Neuron, 109, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K.‐E. , Kim, J.‐K. , Han, S.‐K. , Lee, D. Y. , Lee, H.‐J. , Yim, S.‐V. , & Kim, D.‐H. (2020). The extracellular vesicle of gut microbial Paenalcaligenes hominis is a risk factor for vagus nerve‐mediated cognitive impairment. Microbiome, 8, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, H. , Litvinchuk, A. , Chiang, A C.‐A. , Aithmitti, N. , Jankowsky, J L. , & Zheng, H. (2016). Astrocyte‐microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's disease. Journal of Neuroscience, 36, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, H. , Yang, L. , Cole, A. , Sun, L. , Chiang, A. C.‐A. , Fowler, S. W. , Shim, D. J. , Rodriguez‐Rivera, J. , Taglialatela, G. , Jankowsky, J. L. , Lu, H.‐C. , & Zheng, H. (2015). NFkappaB‐activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron, 85, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , Bennett, M. L. , Münch, A. E. , Chung, W.‐S. , Peterson, T. C. , Wilton, D. K. , Frouin, A. , Napier, B. A. , Panicker, N. , Kumar, M. , Buckwalter, M. S. , Rowitch, D. H. , Dawson, V. L. , Dawson, T. M. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinchuk, A. , Wan, Y.‐W. , Swartzlander, D. B. , Chen, F. , Cole, A. , Propson, N. E. , Wang, Q. , Zhang, B. , Liu, Z. , & Zheng, H. (2018). Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer's disease. Neuron, 100, 1337–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen, L. , Zwingman, T. A. , Sunkin, S. M. , Oh, S. W. , Zariwala, H. A. , Gu, H. , Ng, L. L. , Palmiter, R. D. , Hawrylycz, M. J. , Jones, A. R. , Lein, E. S. , & Zeng, H. (2010). A robust and high‐throughput cre reporting and characterization system for the whole mouse brain. Nature Neuroscience, 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansori, K. , Moradi, Y. , Naderpour, S. , Rashti, R. , Moghaddam, A. B. , Saed, L. , & Mohammadi, H. (2020). Helicobacter pylori infection as a risk factor for diabetes: a meta‐analysis of case‐control studies. BMC Gastroenterology, 20, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal, A. , Ashiq, K. A. , Phulpagar, P. , Singh, D. K. , & Shiras, A. (2019). Effective visualization and easy tracking of extracellular vesicles in glioma cells. Biological Procedures Online, 21, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, H. C. , Marcassa, G. , Chrysidou, I. , Horré, K. , Young‐Pearse, T. L. , Müller, U. C. , Saito, T. , Saido, T. C. , Vassar, R. , De Wit, J. , & De Strooper, B. (2020). Contribution of GABAergic interneurons to amyloid‐beta plaque pathology in an APP knock‐in mouse model. Molecular Neurodegeneration, 15, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, T. , Matsuba, Y. , Mihira, N. , Takano, J. , Nilsson, P. , Itohara, S. , Iwata, N. , & Saido, T. C. (2014). Single app knock‐in mouse models of Alzheimer's disease. Nature Neuroscience, 17, 661–663. [DOI] [PubMed] [Google Scholar]
- Sarnyai, Z. , Sibille, E L. , Pavlides, C. , Fenster, R J. , Mcewen, B S. , & Tóth, M. (2000). Impaired hippocampal‐dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proceedings of the National Academy of Sciences USA, 97, 14731–14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri, H. , Nilsson, P. , Hashimoto, S. , Nagata, K. , Saito, T. , De Strooper, B. , Hardy, J. , Vassar, R. , Winblad, B. , & Saido, T. C. (2017). APP mouse models for Alzheimer's disease preclinical studies. EMBO Journal, 36, 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechheimer, C. , & Kuehn, M. J. (2015). Outer‐membrane vesicles from Gram‐negative bacteria: Biogenesis and functions. Nature Rev Microbiology, 13, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaks, C. E. , & Wilcock, D. M. (2020). Infectious hypothesis of Alzheimer disease. PLoS Pathogens, 16, e1008596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeland, S. , Gorlé, N. , Vandendriessche, C. , Balusu, S. , Brkic, M. , Van Cauwenberghe, C. , Van Imschoot, G. , Van Wonterghem, E. , De Rycke, R. , Kremer, A. , Lippens, S. , Stopa, E. , Johanson, C. E. , Libert, C. , & Vandenbroucke, R. E. (2018). Counteracting the effects of TNF receptor‐1 has therapeutic potential in Alzheimer's disease. EMBO Molecular Medicine, 10, e8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan, A. H. , Barres, B. A. , & Stevens, B. (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annual Review of Neuroscience, 35, 369–389. [DOI] [PubMed] [Google Scholar]
- Stern, E. A. (2004). Cortical synaptic integration in vivo is disrupted by amyloid‐beta plaques. Journal of Neuroscience, 24, 4535–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejera, D. , Mercan, D. , Sanchez‐Caro, J. M. , Hanan, M. , Greenberg, D. , Soreq, H. , Latz, E. , Golenbock, D. , & Heneka, M. T. (2019). Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO Journal, 38, e101064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, L. , Wang, B.‐B. , Li, F.‐H. , Lv, S.‐P. , Pan, F.‐F. , & Dong, X.‐J. (2022). An updated meta‐analysis of the relationship between Helicobacter pylori infection and the risk of coronary heart disease. Frontiers in Cardiovascular Medicine, 9, 794445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein, I. D. , & Molofsky, A. V. (2020). Astrocytes and microglia: In sickness and in health. Trends in Neuroscience, 43, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaja, S. K. , Russo, A. J. , Behl, B. , Banerjee, I. , Yankova, M. , Deshmukh, S. D. , & Rathinam, V. A. K. (2016). Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase‐11 activation. Cell, 165, 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke, R. E. , Dejonckheere, E. , Van Lint, P. , Demeestere, D. , Van Wonterghem, E. , Vanlaere, I. , Puimege, L. , Van Hauwermeiren, F. , De Rycke, R. , Mc Guire, C. , Campestre, C. , Lopez‐Otin, C. , Matthys, P. , Leclercq, G. , & Libert, C. (2012). Matrix metalloprotease 8‐dependent extracellular matrix cleavage at the blood‐CSF barrier contributes to lethality during systemic inflammatory diseases. Journal of Neuroscience, 32, 9805–9816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke, R. E. , Vanlaere, I. , Van Hauwermeiren, F. , Van Wonterghem, E. , Wilson, C. , & Libert, C. (2014). Pro‐inflammatory effects of matrix metalloproteinase 7 in acute inflammation. Mucosal Immunology, 7, 579–588. [DOI] [PubMed] [Google Scholar]
- Vandendriessche, C. , Balusu, S. , Van Cauwenberghe, C. , Brkic, M. , Pauwels, M. , Plehiers, N. , Bruggeman, A. , Dujardin, P. , Van Imschoot, G. , Van Wonterghem, E. , Hendrix, A. , Baeke, F. , De Rycke, R. , Gevaert, K. , & Vandenbroucke, R. E. (2021). Importance of extracellular vesicle secretion at the blood‐cerebrospinal fluid interface in the pathogenesis of Alzheimer's disease. Acta Neuropathologica Communications, 9, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasek, M. J. , Garber, C. , Dorsey, D. , Durrant, D. M. , Bollman, B. , Soung, A. , Yu, J. , Perez‐Torres, C. , Frouin, A. , Wilton, D. K. , Funk, K. , Demasters, B. K. , Jiang, X. , Bowen, J. R. , Mennerick, S. , Robinson, J. K. , Garbow, J. R. , Tyler, K. L. , Suthar, M. S. , … Klein, R. S. (2016). A complement‐microglial axis drives synapse loss during virus‐induced memory impairment. Nature, 534, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigasova, D. , Nemergut, M. , Liskova, B. , & Damborsky, J. (2021). Multi‐pathogen infections and Alzheimer's disease. Microbial Cell Factories, 20, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N. , Zhou, F. , Chen, C. , Luo, H. , Guo, J. , Wang, W. , Yang, J. , & Li, L. (2021). Role of outer membrane vesicles from Helicobacter pylori in atherosclerosis. Frontiers in Cell and Developmental Biology, 9, 673993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, S. , Peng, W. , Mai, Y. , Li, K. , Wei, W. , Hu, L. , Zhu, S. , Zhou, H. , Jie, W. , Wei, Z. , Kang, C. , Li, R. , Liu, Z. , Zhao, B. , & Cai, Z. (2020). Outer membrane vesicles enhance tau phosphorylation and contribute to cognitive impairment. Journal of Cellular Physiology, 235, 4843–4855. [DOI] [PubMed] [Google Scholar]
- Xie, J. , Gorlé, N. , Vandendriessche, C. , Van Imschoot, G. , Van Wonterghem, E. , Van Cauwenberghe, C. , Parthoens, E. , Van Hamme, E. , Lippens, S. , Van Hoecke, L. , & Vandenbroucke, R. E. (2021). Low‐grade peripheral inflammation affects brain pathology in the AppNL‐G‐F mouse model of Alzheimer's disease. Acta Neuropathology Communications, 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, J. , Li, Q. , Haesebrouck, F. , Van Hoecke, L. , & Vandenbroucke, R. E. (2022a). The tremendous biomedical potential of bacterial extracellular vesicles. Trends in Biotechnology, 40, 1173–1194. [DOI] [PubMed] [Google Scholar]
- Xie, J. , Van Hoecke, L. , & Vandenbroucke, R. E. (2022b). The impact of systemic inflammation on Alzheimer's disease pathology. Frontiers in Immunology, 12, 796867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, D. , Chen, X. , Wang, J. , Lou, Q. , Lou, Y. , Li, L. , Wang, H. , Chen, J. , Wu, M. , Song, X. , & Qian, Y. (2019). Dysregulated lung commensal bacteria drive interleukin‐17B production to promote pulmonary fibrosis through their outer membrane vesicles. Immunity, 50, 692–706. [DOI] [PubMed] [Google Scholar]
- Yuan, P. , Condello, C. , Keene, C. D , Wang, Y. , Bird, T. D. , Paul, S. M. , Luo, W. , Colonna, M. , Baddeley, D. , & Grutzendler, J. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron, 92, 252–264. [DOI] [PubMed] [Google Scholar]
- Zavan, L. , Bitto, N. J. , Johnston, E. L. , Greening, D. W. , & Kaparakis‐Liaskos, M. (2019). Helicobacter pylori growth stage determines the size, protein composition, and preferential cargo packaging of outer membrane vesicles. Proteomics, 19, 1970004. [DOI] [PubMed] [Google Scholar]
- Zuroff, L. , Daley, D. , Black, K. L. , & Koronyo‐Hamaoui, M. (2017). Clearance of cerebral Abeta in Alzheimer's disease: reassessing the role of microglia and monocytes. Cellular and Molecular Life Sciences, 74, 2167–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information