

Abstract
Objectives:
To identify genetic variants associated with NAS through a Genome Wide Association Study (GWAS) and estimate a Polygenic Risk Score (PRS) model for NAS.
Design:
A prospective case-control study included 476 in-utero opioid-exposed term neonates. A GWAS of 1000 Genomes-imputed genotypes was performed to identify variants associated with need for pharmacotherapy for NAS. PRS models for estimating genetic predisposition were generated via a nested cross-validation approach using 382 neonates of European ancestry. PRS predictive ability, discrimination, and calibration were assessed.
Results:
Cross-ancestry GWAS identified one intergenic locus on chromosome 7 downstream of SNX13 exhibiting genome-wide association with need for pharmacotherapy. PRS models derived from the GWAS for a subset of the European ancestry neonates reliably discriminated between need for pharmacotherapy using cis variant effect sizes within validation sets of European and African American ancestry neonates. PRS were less effective when applying variant effect sizes across datasets and in calibration analyses.
Conclusions:
GWAS has the potential to identify genetic loci associated with need for pharmacotherapy for NAS and enable development of clinically predictive PRS models. Larger GWAS with additional ancestries are needed to confirm the observed SNX13 association and the accuracy of PRS in NAS risk prediction models.
INTRODUCTION
Neonatal Abstinence Syndrome (NAS) is a constellation of signs of withdrawal in the neonate from in-utero exposure to maternal opioids.1, 2 The incidence of NAS has exponentially increased over the past decade, concurrent with the global opioid epidemic.3 NAS has a highly variable expression, making accurate prediction of the need for pharmacotherapy a significant challenge.4 Opioid exposed neonates are monitored in the hospital several days for signs indicating the need for pharmacotherapy.5–7
Despite decades of research, accurate prediction of NAS expression remains elusive. Recent studies have utilized maternal and neonatal factors associated with NAS expression and developed predictive models.8, 9 Genetic factors explain some variation and have been associated with opioid use disorder (OUD) in adults. Genetic variation in opioid receptor genes and genes involved in opioid metabolism are associated with variable response to pharmacotherapy10–14.
The objective of this study was to identify genetic variants associated with the need for pharmacotherapy of NAS through a Genome Wide Association Study (GWAS) and establish Polygenic Risk Score (PRS) models to enhance prediction of need for treatment.
METHODS
Patient population and Phenotype:
Subjects were recruited from four prospective studies - two clinical trials and two observational cohort studies.15, 16 The 8-site clinical trial compared methadone with morphine for the treatment of NAS. An observational arm of this trial included: 1) neonates whose parents consented for the trial but who did not require treatment; 2) parents who did not consent for randomization in the trial but agreed to data collection and genetic analyses.15 Additional neonates were recruited from a single center trial of sublingual buprenorphine for NAS and an observational study led by Thomas Jefferson University.16
Inclusion criteria for these studies included neonates born > 36 weeks gestational age to mothers with known OUD in medication assisted treatment programs. All neonates were monitored in hospital for a minimum 3–5 days prior to discharge home. Pharmacotherapy for NAS was based on Finnegan Scoring criteria per clinical trial and observational study protocols.15–17 Control neonates met all inclusion criteria, but did not require pharmacotherapy. Neonates were excluded if they did not meet inclusion criteria, were one of a multiple birth, had major congenital abnormalities, died or left the hospital before day 5. Institutional Review Boards at each center approved the studies and informed consent was obtained.
DNA Isolation, Genotyping and Quality Control.
Genomic DNA was extracted from buccal swabs using previously published methodologies18. All subjects were genotyped with the Infinium Omni2.5-8v1.3 array which contains probes for ~2.4 million SNPs. Subjects were assigned to ancestral groups (African American – AA, European American - EA) based on comparison to the HapMap CEU, YRI, and CHB populations using STRUCTURE and ancestral outliers were removed.19,20 SNP- and subject-level quality control (QC) were conducted using PLINK21. SNPs with missing call rate > 10% or Hardy-Weinberg p-value < 0.0001 were removed and heterozygous haploid SNPs were set to missing. Subject-level QC removed subjects with missing call rate > 10%, duplicates, first degree relatives (identity-by-descent pi-hat > 0.4), cryptically related subjects (identity-by-state distance > 0.9), and subjects with excessive homozygosity. Following genotype QC, 94 AAs (2,328,444 SNPs) and 394 EAs (2,325,421 SNPs) remained.
To expand genomic coverage, genotype imputation was performed with the Michigan Imputation Server using 1000 Genomes (1000G) Phase 3 (Version 5) reference panel.22 After removing variants with minor allele frequency (MAF) < 0.01 in the reference (1000G AFR for AAs, 1000G EUR for EAs) or study populations or with imputation Rsq < 0.8, ~13.4 million and ~8.5 million SNPs and indels remained in AAs and EAs, respectively.
Genome-wide Association Analyses.
Genome-wide variant associations with the need for pharmacotherapy were calculated separately for AA (N=94) and EA (N=382) neonates with non-missing phenotypes and covariates using RVtests software, adjusting for sex and genetic principal components as covariates.23 Results for association testing of AA and EA neonates were combined via inverse variance-weighted meta-analysis using METAL software with genomic control.24 Results were filtered to eliminate variants with MAF < 0.01 and imputation quality < 0.8. Gene associations with need for pharmacotherapy were generated from the GWAS summary statistics using MAGMA software.25
Nested Cross Validation for PRS Development.
Due to small sample size, a nested cross-validation approach was used to develop PRS models for predicting need for pharmacologic treatment (Supplemental Figure 1). Given the small number of AA neonates and the differing genetic architecture of AAs and EAs, only EA neonates were used for PRS model development. Subjects were divided into training and validation sets, with neonates from Thomas Jefferson University (N=92, ~25% of overall cohort) assigned to the validation set, establishing independent training and validation sets.
For cross validation, the training set (N=290) was divided into 5 equal sets with similar case:control ratios. Each was used as the target population in 5 separate runs of PRSice-2 utilizing the classic PRS calculation approach of clumping and thresholding (Supplemental Figure 1).26 The remaining 80% of the training set was used as the base population for PRSice-2. In preparation for running PRSice-2, genome-wide association testing was run in the base population with RVtests, adjusting for sex and genetic principal components as covariates. For the PRSice-2 analyses, the 1000 Genomes EUR group was used to improve LD estimation for clumping (MAF cutoff 0.01). Each PRSice2 analysis generated an optimal PRS model consisting of different sets of variants.
Validation of PRS Models from Cross Validation:
Results from cross validation were combined for testing in the reserved Jefferson validation set and in AA neonates using different intersections of the variant sets from the optimal PRS models. Specifically, PRS models consisting of variants present in 1, 2, 3, 4, or all 5 optimal models were tested for their discriminative and predictive abilities in the validation set. Two distinct approaches were used to assess PRS model performance, differing in the source of variant effect sizes used to calculate PRS: 1) the cis approach, which calculates PRS as the average sum of validation set effect sizes weighted by validation set allele dosages for PRS model variants, and 2) the trans approach, which calculates PRS as the average sum of training set effect sizes weighted by validation set dosages for PRS model variants. For both approaches, the dosages of variants with negative effect sizes were first inverted and the sign of the effect sizes were changed to positive. The predictive and discriminatory ability of different variant sets was assessed using ANOVA and Area Under the Receiver Operating Characteristic Curve (AUROC) statistics. The 5 PRS models were tested for their discriminative and predictive abilities in AA neonates using similar methodology.
Calibration of PRS Models:
Calibration of PRS models was performed as described previously27. Optimal PRS thresholds for discriminating need for pharmacotherapy for NAS were calculated by maximizing the Youden-Index based on kernel smoothed densities using the oc_youden_kernel function of the cutpointr package. Mean calibration of PRS models was tested by comparing the average predicted need for treatment based on the PRS threshold to the actual overall rate. PRS and thresholds were calculated using the effect sizes from the training set for the Jefferson EA validation set or the full EA set (training + validation) for AAs. Weak calibration of PRS models (not overestimating/underestimating predicted outcome) was assessed by calculating the calibration intercept and slope and examining their deviation from 0 and 1.
RESULTS:
GWAS Meta-analysis of Need for Pharmacotherapy:
Cross-ancestry GWAS meta-analysis in AA (cases = 59, controls = 35) and EA (cases = 231, controls = 151) neonates identified one intergenic locus on chromosome 7 between the LINC02889 and SNX13 genes exhibiting genome-wide significant association with need for pharmacotherapy (p = 4.22 × 10−8 for top variant rs73313786; Figure 1 – Manhattan plot; Supplemental Figure 2 – QQ plot; Supplemental Figure 3 – LocusZoom plots; Supplemental Table 1 – variants with p < 0.001). A second intergenic locus on chromosome 6 between the LOC107986667 and MEAT6 genes achieved near-genome-wide significance (p = 5.48 × 10−8 for top variant rs1566002; Supplemental Figure 4 – LocusZoom plots). Ancestry-specific GWAS revealed no genome-wide significant loci for AA (Supplemental Figure 5; Supplemental Table 2). However, the same genome-wide significant chromosome 7 intergenic locus observed in the cross-ancestry meta was observed for EA ancestry alone (p = 2.17 × 10−8 for top variant rs10277501; Supplemental Figure 6; Supplemental Table 3).
Figure 1: Manhattan Plot of Cross-ancestry GWAS of Need for Pharmacotherapy for NAS.
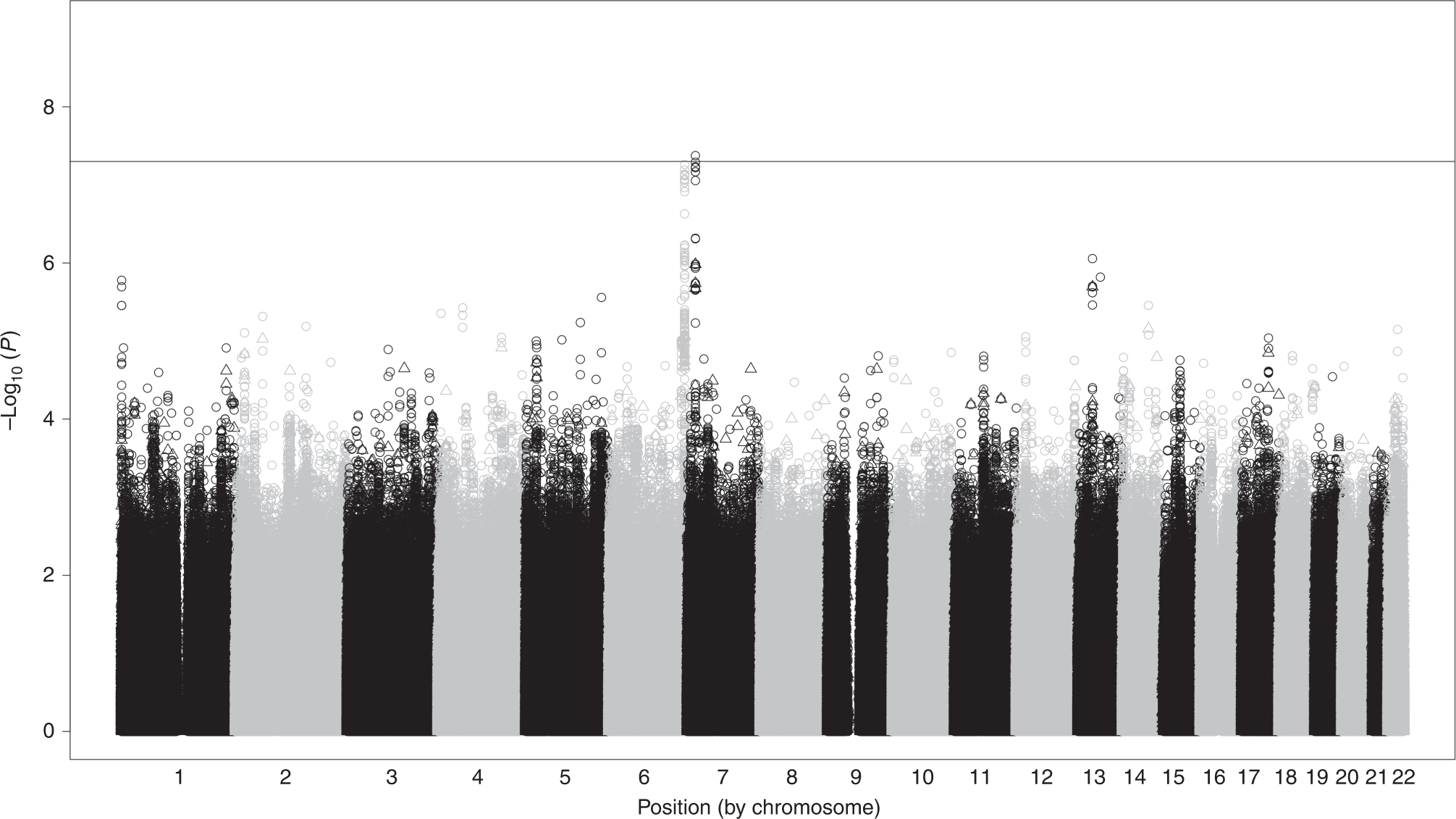
Genome-wide variant associations with need for pharmacotherapy for NAS were calculated separately for AA (N=94) and EA (N=382), adjusting for sex and genetic principal components as covariates. Association testing results for AA and EA neonates were combined via inverse variance-weighted meta-analysis with genomic control. Results were filtered to eliminate variants with MAF < 0.01 and imputation quality < 0.8. One locus on chromosome 7 downstream of SNX13 achieved genome-wide significance.
Prior studies identified 7 variants in the OPRM1, COMT, OPRD1, OPRK1, and PNOC genes associated with NAS (need for pharmacologic treatment, treatment with ≥ 2 medications, length of hospital stay).36,37 None of the previously identified variants extended to need for NAS treatment in our GWAS based on a Bonferroni-corrected p-value of 0.007 (0.05/7 SNPs) (Table 1). However, rs2614095 (intronic variant within PNOC) was nominally significant in the present study (p = 0.042) and showed the same direction of affect across studies (minor allele A being protective). Although no variants in the OPRM1 gene approached genome-wide significance, the top variant rs641457 had a beta = 0.7117 and p = 0.0037). This occurred despite different primary outcomes in the various studies (e.g., need for NAS treatment in the present study).
Table 1.
Variants previously associated with NAS severity phenotypes
| SNP | Phenotype | Gene | Minor Allele | Previous Studies | This Study (Phenotype = NAS Treatment) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | EA | AA + EA | ||||||||||
| OR | P-value | Reference | OR | P-value | OR | P-value | OR | P-value | ||||
| rs1799971 | LOS | OPRM1 | G | −6.9 | 0.02 | Wachman et al. 2015 | NA | NA | 0.806 | 0.362 | 0.806 | 0.363 |
| rs204076 | ≥ 2 Meds | OPRD1 | A | 2.8 | 0.04 | Wachman et al. 2015 | 0.630 | 0.261 | 0.785 | 0.136 | 0.766 | 0.083 |
| LOS | OPRD1 | A | 6.6 | 0.008 | Wachman et al. 2015 | |||||||
| rs2614095 | ≥ 2 Meds | PNOC | A | 0.3 | 0.01 | Wachman et al. 2015 | 0.787 | 0.566 | 0.722 | 0.043 | 0.730 | 0.042 |
| LOS | PNOC | A | −4.7 | 0.04 | Wachman et al. 2015 | |||||||
| rs351776 | ≥ 2 Meds | PNOC | A | 3.2 | 0.01 | Wachman et al. 2015 | 1.548 | 0.251 | 1.222 | 0.212 | 1.263 | 0.128 |
| LOS | PNOC | A | 4.2 | 0.05 | Wachman et al. 2015 | |||||||
| rs4732636 | NAS Treatment | PNOC | A | 2.2 | 0.05 | Wachman et al. 2015 | 0.890 | 0.796 | 1.217 | 0.258 | 1.173 | 0.342 |
| NAS Treatment | PNOC | A | 2 | 0.04 | Wachman et al. 2017 | |||||||
| ≥ 2 Meds | PNOC | A | 3.8 | 0.004 | Wachman et al. 2015 | |||||||
| LOS | PNOC | A | 5.8 | 0.01 | Wachman et al. 2015 | |||||||
| rs702764 | ≥ 2 Meds | OPRK1 | C | 4.1 | 0.003 | Wachman et al. 2015 | 1.433 | 0.327 | 1.299 | 0.210 | 1.328 | 0.133 |
| rs740603 | NAS Treatment | COMT | A | 0.4 | 0.01 | Wachman et al. 2015 | 0.767 | 0.441 | 1.063 | 0.704 | 1.008 | 0.956 |
| ≥ 2 Meds | COMT | A | 1.9 | 0.02 | Wachman et al. 2017 | |||||||
| LOS | COMT | A | 5.3 | 0.01 | Wachman et al. 2015 | |||||||
Analysis of gene-based association with the need for pharmacotherapy from the GWAS failed to identify any significant cross-ancestry or ancestry-specific associations (based on Bonferroni-corrected p-value threshold of 2.74 × 10−6 for 18,229 genes; Supplemental Tables 4–6).
PRS development for predicting the need for pharmacotherapy for NAS:
The overall sample was split into a training set (non-Jefferson samples; N=290) and a validation set (Jefferson samples; N=92). Due to small sample size, AA neonates were reserved for validation of PRS models in a different ancestry. New sets of variants for PRS calculation were established based on the number of times a variant occurred in the 5 optimal PRS models from a nested cross validation approach. This approach resulted in variant sets ranging in size from 1126 (present in all 5 optimal PRS models) to 383,754 (present in 1 of the optimal PRS models) (Supplemental Tables 7a–e).
Analysis of Discriminatory and Predictive Performance of PRS models in EA neonates.
ANOVA and AUROC on five sets of variants confirmed that all PRS models demonstrated excellent discriminatory and predictive performance for the need for pharmacotherapy in the training set (Supplemental Table 8; boxplots in Supplemental Figure 7).
All variant sets could effectively discriminate between neonates requiring pharmacotherapy and those not needing it in the EA validation set when using cis effect sizes derived from the validation set to calculate PRS, but not when using trans effect sizes from the training set (Table 2; Supplemental Figures 8 and 9). In the cis analysis, PRS models performed well, improving with increasing size of variant set (ANOVA from 1.62×10−08 for the smallest set to 4.34×10−48 for the largest; AUROCs ranging from 0.813 to 1.000). In the trans context (often used in clinical settings), performance of the PRS models was worse. No model detected significant differences between treated/untreated neonates (maximum AUROC 0.61).
Table 2.
ANOVA and AUROC analyses of PRS model performance in EA and AA validation sets
| Target Set | Effect Size Source | Variant Set | N Variants | N Samples | ANOVA P | AUROC (95% CI) |
|---|---|---|---|---|---|---|
| EA validation set | EA validation set | 1/5 CVs | 382872 | 92 | 4.34E-48 | 1 (NA, NA) |
| 2/5 CVs | 90954 | 92 | 4.93E-45 | 1 (NA, NA) | ||
| 3/5 CVs | 28013 | 92 | 7.14E-40 | 1 (NA, NA) | ||
| 4/5 CVs | 7043 | 92 | 6.64E-26 | 0.985 (0.965, 1) | ||
| 5/5 CVs | 1126 | 92 | 1.62E-08 | 0.814 (0.726, 0.902) | ||
| EA validation set | EA training set | 1/5 CVs | 382872 | 92 | 0.987 | 0.479 (0.354, 0.605) |
| 2/5 CVs | 90954 | 92 | 0.565 | 0.543 (0.418, 0.668) | ||
| 3/5 CVs | 28013 | 92 | 0.905 | 0.512 (0.391, 0.633) | ||
| 4/5 CVs | 7043 | 92 | 0.364 | 0.552 (0.432, 0.673) | ||
| 5/5 CVs | 1126 | 92 | 0.123 | 0.606 (0.485, 0.727) | ||
| AA validation set | AA validation set | 1/5 CVs | 356467 | 94 | 1.05E-38 | 0.998 (0.994, 1) |
| 2/5 CVs | 85217 | 94 | 1.58E-38 | 0.999 (0.997, 1) | ||
| 3/5 CVs | 26334 | 94 | 2.46E-30 | 0.994 (0.985, 1) | ||
| 4/5 CVs | 6721 | 94 | 2.84E-19 | 0.958 (0.923, 0.993) | ||
| 5/5 CVs | 1097 | 92 | 2.98E-06 | 0.768 (0.669, 0.868) | ||
| AA validation set | EA full set | 1/5 CVs | 356467 | 93 | 0.2 | 0.596 (0.471, 0) |
| 2/5 CVs | 85217 | 94 | 0.034 | 0.624 (0.506, 0) | ||
| 3/5 CVs | 26334 | 94 | 0.074 | 0.599 (0.48, 0) | ||
| 4/5 CVs | 6721 | 93 | 0.281 | 0.569 (0.447, 0) | ||
| 5/5 CVs | 1097 | 93 | 0.024 | 0.622 (0.502, 0) |
Analysis of Discriminatory and Predictive Performance of PRS Models in AA neonates:
PRS suffer from lack of portability across ancestries due to different linkage disequilibrium patterns and allele frequencies.28 Two approaches to assess performance in AAs were used: one where cis effect sizes from testing among AAs were used for PRS calculation and the other where trans effect sizes were derived from the full EA dataset (training + validation). All 5 PRS variant sets performed well distinguishing between treated and untreated AA neonates when AA effect sizes were used (Table 2; Supplemental Figure 10). PRS performance improved with increasing variant set size, with p-values ranging from 2.98×10−06 for the smallest set to 1.05×10−38 for the largest and AUROCs ranging from 0.768 to 0.998. Performance of the PRS models in the second approach where EA effect sizes were used for calculation of PRS (Table 2; Supplemental Figure 11) was comparable to the EA validation set (Table 2). Two variant sets resulted in significant differences between treated and untreated AA neonates with corresponding AUROCs of 0.62. Unlike the first approach, PRS performance did not improve with increasing variant size.
Calibration of PRS Models for Prediction of Need for Pharmacotherapy in EA and AA neonates.
For the EA validation set, the smallest set of variants most closely recapitulated the actual treatment rate, with others not performing as well (Table 3). For AA neonates, all variant sets resulted in a predicted treatment rate of zero based on the EA-based cut point, which likely reflects differing allele frequencies between EAs and AAs (Table 3). To assess ‘weak calibration’, the calibration intercept (target value = 0) and slope (target value = 1) were calculated for each PRS model. For EA, all PRS models had negative intercepts, indicative of overestimation of treatment risk (Table 3). The PRS models for the 2 smallest variant sets had calibration slopes > 1, suggesting that estimates of treatment risk were too moderate. Conversely, the PRS models based on the 3 largest variant sets had calibration slopes < 1, suggesting that estimates of treatment risk were too extreme (Table 3). For the AA neonates, the calibration intercepts for all models were close to the target value of 0, but the calibration slopes were all > 1 suggestive of estimates that were too moderate (Table 3).
Table 3.
PRS model calibration in EA and AA validation sets
| Target Set | Effect Size/Cutpoint Source Set | Variant Set | Optimal Cutpoint | Actual Prevalence | PRS Prevalence | Intercept | Slope |
|---|---|---|---|---|---|---|---|
| EA validation | EA training | 1/5 CVs | 0.836 | 0.587 | 0.880 | −0.489 | −0.990 |
| 2/5 CVs | 1.077 | 0.587 | 1.000 | −0.746 | −15.426 | ||
| 3/5 CVs | 1.222 | 0.587 | 0.989 | −0.902 | −1.652 | ||
| 4/5 CVs | 1.229 | 0.587 | 0.783 | −0.899 | 6.589 | ||
| 5/5 CVs | 1.352 | 0.587 | 0.630 | −1.031 | 3.958 | ||
| AA validation | EA full | 1/5 CVs | 0.725 | 0.628 | 0.000 | 0.029 | 109.181 |
| 2/5 CVs | 0.988 | 0.628 | 0.000 | −0.130 | 75.514 | ||
| 3/5 CVs | 1.071 | 0.628 | 0.000 | −0.215 | 38.727 | ||
| 4/5 CVs | 1.159 | 0.628 | 0.000 | −0.267 | 9.136 | ||
| 5/5 CVs | 1.451 | 0.628 | 0.000 | −0.436 | 6.014 |
DISCUSSION:
Being able to predict NAS expression would provide crucial information to guide treatment decisions and optimize NAS outcomes. Advances in the fields of genetics/genomics and computational biology have generated immense interest in including genomic information in predictive models. In this first-of-its-kind GWAS of neonates with in-utero opioid exposure, a genomic locus was identified upstream of the SNX13 gene that was associated with need for pharmacotherapy. PRS models that segregate neonates by their need for therapy were then developed. These findings may lead to a better understanding of NAS and provide a new tool to assist in predicting the need for pharmacotherapy of NAS. Further development of PRS in larger samples in concert with clinical data may permit a precision medicine approach.
Our multi-ancestry GWAS identified a single intergenic locus ~124 kb downstream of SNX13 with genome-wide significance with need for pharmacotherapy. SNX13 is a sorting nexin and G protein regulator involved in intracellular trafficking that has been implicated in heart failure and associated with neutrophil counts, high density lipoprotein (HDL) cholesterol level, apolipoprotein A1 level, and mean platelet volume 29–33. SNX13 is expressed in numerous tissues, including the brain, which is particularly relevant to addiction, where expression is highest in the cerebellum34. Although there is no previous evidence of SNX13 being implicated in addiction, a related sorting nexin, SNX27, has been implicated in attenuating response to cocaine in mice, possibly through regulation of neuron excitability35,36. Further studies using larger datasets are needed to establish whether the association with the locus near SNX13 is reproducible and whether SNX13 expression is regulated by the locus.
Our findings did not find a definitive association of OPRM1 with NAS pharmacotherapy by GWAS. Prior adult studies identified variants in the OPRM1 gene associated with opioid addiction37, 38. Wachman et al. described variants in OPRM1 associated with decreased length of stay (LOS) and need for pharmacotherapy13. Genes involved in the dopamine pathway (e.g., Catechol- O- Methyltransferase - COMT) and PNOC (Prepronociceptin) were also found to be associated with LOS and need for pharmacotherapy39. In a larger replication cohort of 199 mother-infant dyads, some variant associations did not meet significance threshold after correction for multiple comparison testing40. In the current study (N=476), none of the seven previously associated candidate variants were associated with need for NAS treatment after a Bonferroni correction for multiple testing. However, rs2614095, an intronic variant within PNOC, was nominally significant (p = 0.042). The minor allele A was protective in previous studies and the current study.
Recent studies have consistently noted the importance of maternal ancestry in NAS, with non-Hispanic whites displaying higher risk compared to other ancestries41,42. Our study included non-Hispanic whites and a smaller number of AAs, which allowed us to capture some of the ancestral diversity for NAS. Although there was a mixture of ancestry-specific and cross-ancestry associations, the small sample size makes it impossible to draw any definitive conclusions about the interaction of ancestry and genetic loci relative to NAS. Future studies should focus on larger cohorts with greater representation of other ancestries.
PRS holds great promise for assessing an individual’s risk of a disease or trait, how well an individual will respond to specific therapies, and other factors influenced by genetic predisposition. PRS models for predicting pharmacotherapy of NAS were developed in EAs using nested cross-validation and tested in EA and AA validation sets. Although PRS performance was consistent between EAs and AAs, it was highly dependent on the source of effect sizes for PRS calculation. PRS calculated using cis effect sizes from validation sets demonstrated excellent discrimination between neonates needing/not needing pharmacotherapy. However, PRS using trans effect sizes did not perform as well. The differences in performance likely resulted from variability in effect sizes due to small sample size or perhaps some model overfitting. The weak discrimination when using trans-effect sizes has implications for utility in clinical settings. Similarly, calibration analyses suggest that current PRS models have not been sufficiently powered to justify clinical use. However, the excellent performance of PRS models for discriminating the need for pharmacotherapy in independent cohorts using cis effect sizes suggests that these models do hold promise and could be effective with increased sample sizes and higher ancestral diversity.
While our data provide evidence supporting the potential benefit of using PRS in the management of NAS, there are some limitations. The sample size in this study was small for a GWAS, limiting the power to discover loci with small effect sizes. However, our nested cross validation approach and establishment of largely independent validation sets allowed us to make optimal use of the samples that were available. Also, the need-for-treatment phenotype relied on Finnegan Scoring criteria which can be subjective, limit phenotypic variability, and further reduce power. Unfortunately, collecting samples sufficiently powered for GWAS of NAS is extremely difficult, as evidenced by the 5 years required to assemble the cohort used in this study. Another limitation is the relatively small representation of non-EA ancestries, which is particularly problematic due to the potential for substantially different causal genetic architectures among different ancestries resulting from variation in allele effect sizes and linkage disequilibrium patterns43. In fact, recent studies have demonstrated poor trans-ancestry portability of both GWAS findings and PRS44–45. Given that the majority of genetic studies have been conducted with participants of European ancestry, the clinical applicability of these findings to individuals of non-European ancestry is limited, which exacerbates healthcare disparities46. Strengthening regulatory protections against genetic discrimination, open sharing of GWAS summary statistics, and equitable investment in global populations and ancestries are critical next steps for studies of NAS and other diseases and traits.
Supplementary Material
Impact.
Genetic associations appear to be important in neonatal abstinence syndrome
This is the first genome wide association in neonates with neonatal abstinence syndrome
Polygenic risk scores can be developed examining single nucleotide polymorphisms across the entire genome
Polygenic risk scores were higher in neonates receiving pharmacotherapy for treatment of their neonatal abstinence syndrome
Future studies with larger cohorts are needed to better delineate these genetic associations
Acknowledgements:
The authors want to thank: Adam Czynski, Erica Oliveira – Brown, Mark Hudak – Jacksonville, Jeffrey Shenberger – Baystate Medical Center, Peter Marro – Maine Medical Center, Elisha Wachman – Boston Medical Center, Barbara Engelhardt – Vanderbilt, Debra Bogen – University of Pittsburgh, and Michelle Ehrlich – Mt. Sinai
Funding Sources:
National Institute of Drug Abuse (NIDA) R01DA032889, R21DA041706-02 (JMD, BML), NIDA R01DA02976 (WK), Charles H. Hood Foundation (JMD)
Footnotes
Conflicts of Interest: None of the authors have any conflicts of interest to disclose
Informed Consent: Was required
REFERENCES:
- 1.MacMillan KD Neonatal Abstinence Syndrome: Review of Epidemiology, Care Models, and Current Understanding of Outcomes. Clin Perinatol. 46, 817–822 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Grossman M & Berkwitt A Neonatal abstinence syndrome. Semin Perinatol. 43, 173–186 (2019). [DOI] [PubMed] [Google Scholar]
- 3.U.S. Agency for Healthcare and Quality (AHRQ) Trends in neonatal abstinence syndrome births in the United States. Accessed September 30, 2021. https://www.hcup-us.ahrq.gov/reports/Trends_NeonatalAbstinenceSyndrome_Births_UnitedStates.pdf
- 4.Jansson LM & Patrick SW Neonatal Abstinence Syndrome. Pediatr Clin North Am. 66, 353–367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devlin LA & Davis JM A practical approach to neonatal opiate withdrawal syndrome. Am J Perinatol. 35, 324–330 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Patrick SW, Barfield WD, & Poindexter BB, for the COMMITTEE ON FETUS AND NEWBORN, COMMITTEE ON SUBSTANCE USE AND PREVENTION. Neonatal Opioid Withdrawal Syndrome. Pediatrics. 146, e2020029074 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Winkelman TNA, et al. Incidence and costs of neonatal abstinence syndrome among infants with Medicaid: 2004-2014. Pediatrics. 141, e20173520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh R, et al. Predictors of pharmacologic therapy for neonatal opioid withdrawal syndrome: a retrospective analysis of a statewide database. J Perinatol. 41, 1381–1388 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Patrick SW, al. Development and validation of a model to predict neonatal abstinence syndrome. J Pediatr. 229, 154–160 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kendler KS, et al. Illicit psychoactive substance use, heavy use, abuse, and dependence in a US population-based sample of male twins. Arch Gen Psychiatr. 57, 261–269 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Goldman D, Oroszi G, & Ducci F The genetics of addictions: uncovering the genes. Nat Rev Genet. 6, 521–532 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Smith AH, et al. Genome-wide association study of therapeutic opioid dosing identifies a novel locus upstream of OPRM1. Mol Psychiatry. 22, 346–352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wachman EM, et al. Association of OPRM1 and COMT single-nucleotide polymorphisms with hospital length of stay and treatment of neonatal abstinence syndrome. JAMA. 309, 1821–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wachman EM & Farrer LA The genetics and epigenetics of neonatal abstinence syndrome. Semin Fetal Neonatal Med. 2019;24:105–110 [DOI] [PubMed] [Google Scholar]
- 15.Davis JM, et al. Comparison of safety and efficacy of methadone vs morphine for treatment of neonatal abstinence syndrome: a randomized clinical trial. JAMA Pediatr. 172, 741–748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kraft WK, et al. Buprenorphine for the treatment of the neonatal abstinence syndrome. N Engl J Med. 376, 2341–2348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaltenbach K & Jones HE Neonatal abstinence syndrome: presentation and treatment considerations. J Addict Med. 10, 217–223 (2016). [DOI] [PubMed] [Google Scholar]
- 18. [Accessed Sep 30 2021]. https://www.dnagenotek.com/US/support/protocols/oragene-discover.html.
- 19.Frazer KA, et al. , for the International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 449, 851–861 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falush D, Stephens M, & Pritchard JK Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 7, 574–578 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das S, et al. Next-generation genotype imputation service and methods. Nat Genet. 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhan X, et al. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics. 32, 1423–1426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willer CJ, Li Y, & Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Leeuw C, et al. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput Biol. 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi SW & O’Reilly PF PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience. 8:giz082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Calster B, et al. On behalf of Topic Group ‘Evaluating diagnostic tests and prediction models’ of the STRATOS initiative. Calibration: the Achilles heel of predictive analytics. BMC Med. 17, 230 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin AR, et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 51, 584 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, et al. SNX13 reduction mediates heart failure through degradative sorting of apoptosis repressor with caspase recruitment domain. Nat Commun. 5, 5177 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Chen MH, et al. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell. 182, 1198–1213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vuckovic D, et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell. 182, 1214–1231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willer CJ, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 45, 1274–1283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson TG, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 17, e1003062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lonsdale J, Thomas J, Salvatore M et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munoz MB, Slesinger PA Sorting nexin 27 regulation of G protein-gated inwardly rectifying K+ channels attenuates in vivo cocaine response. Neuron. 82, 659–669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rifkin RA, Huyghe D, Li X, Parakala M, Aisenberg E, Moss SJ, Slesinger PA GIRK currents in VTA dopamine neurons control the sensitivity of mice to cocaine-induced locomotor sensitization. Proc Natl Acad Sci U S A. 115, E9479–E9488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bauer IE, Soares JC & Nielsen DA The role of opioidergic genes in the treatment outcome of drug addiction pharmacotherapy: a systematic review. Am J Addict. 24, 15–23 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Smith AH, et al. Genome-wide association study of therapeutic opioid dosing identifies a novel locus upstream of OPRM1. Mol Psychiatr. 22, 346–352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wachman EM, et al. Variations in opioid receptor genes in neonatal abstinence syndrome. Drug Alcohol Depend 155, 253–259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wachman EM, et al. Association of maternal and infant variants in PNOC and COMT genes with neonatal abstinence syndrome severity. Am J Addict. 26, 42–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parikh A, et al. Racial association and pharmacotherapy in neonatal opioid withdrawal syndrome. J Perinatol. 39, 1370–1376 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Brown SE, et al. Association of race and ethnicity with withdrawal symptoms, attrition, opioid use, and side-effects during buprenorphine therapy. J Ethn Subst Abuse. 9, 106–114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galinsky KJ, et al. Estimating cross-population genetic correlations of causal effect sizes. Genet Epidemiol. 43, 180–188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vilhjálmsson, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lam M et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 51, 1670–1678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin AR, et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.