

Summary
Cardiac fibrosis is a hallmark in late-stage familial dilated cardiomyopathy (DCM) patients, although the underlying mechanism remains elusive. Cardiac exosomes (Exos) have been reported relating to fibrosis in ischemic cardiomyopathy. Thus, we investigated whether Exos secreted from the familial DCM cardiomyocytes could promote fibrogenesis. Using human iPSCs differentiated cardiomyocytes we isolated Exos of angiotensin II stimulation conditioned media from either DCM or control (CTL) cardiomyocytes. Of interest, cultured cardiac fibroblasts had increased fibrogenesis following exposure to DCM-Exos rather than CTL-Exos. Meanwhile, injecting DCM-Exos into mouse hearts enhanced cardiac fibrosis and impaired cardiac function. Mechanistically, we identified the upregulation of miRNA-218-5p in the DCM-Exos as a critical contributor to fibrogenesis. MiRNA-218-5p activated TGF-β signaling via suppression of TNFAIP3, a master inflammation inhibitor. In conclusion, our results illustrate a profibrotic effect of cardiomyocytes-derived Exos that highlights an additional pathogenesis pathway for cardiac fibrosis in DCM.
Subject areas: Fibrosis, Cell biology
Graphical abstract
Highlights
-
•
Angiotensin II conditioned DCM cardiomyocytes secrete exosomes to promote fibrosis
-
•
Highly present of miR-218-5p in DCM exosomes mediate the profibrotic effect
-
•
TNFAIP3 plays an essential role in the miR-218-5p induced TGF-beta activation
Fibrosis; Cell biology
Introduction
Familial dilated cardiomyopathy (DCM) is the most common form of inherited cardiomyopathy, accounting for approximately 30–50% of DCM cases.1,2 It significantly contributes to heart failure in all age groups, often with onset in younger patients.3,4,5 The pathophysiology of DCM includes inadequate neurohumoral stimulation, abnormal myocyte calcium cycling, ventricular remodeling, excessive extracellular matrix (ECM) accumulation, and accelerated apoptosis.6 Previous studies have identified over 50 genes encoding cytoskeletal, sarcomeric, mitochondrial, nuclear membrane, and RNA-binding proteins that are mutated in the familial DCM.7,8 Most of these mutations are associated with sarcomeric proteins such as titin, β-myosin heavy chain, and cardiac troponin T (TNNT2), which are responsible for the generation and regulation of cardiac contraction.8 Recent studies exploring the molecular mechanisms of such DCM-associated sarcomeric protein mutations have attributed the disease pathology to reduced calcium sensitivity and epigenetic dysregulation in cardiomyocytes (CMs).9,10,11 However, the excessive cardiac fibrosis observed in late-stage DCM patients is hardly correlated with those mutations.
Preclinical and clinical studies have suggested that progressive myocardial fibrosis is a hallmark of adverse remodeling in DCM, relating to disease severity.12,13 Excessive myocardial fibrosis increases myocardial stiffness, perturbs the coordination of myocardial excitation-contraction coupling, and disrupts CM perfusion.14 Once myocardial fibrosis is present in nonischemic DCM, it does not regress in size or resolve over time.15 The dysregulated ECM is mainly caused by the activation of resident cardiac fibroblasts (CFs),16 which transforms into a myofibroblast phenotype that promotes the overproduction of collagens (type I and III), various growth factors, and cytokines.17 However, the mechanisms underlying fibroblast activation in patients with familial DCM have been elusive. Previous studies have indicated that CFs can directly sense the mechanical load through surface receptors and mechanosensitive membrane channels.18,19 Moreover, stretching of the ventricle due to dilation activates paracrine signaling from CMs to CFs.20
Exosomes are nanosized (30–200 nm) vesicles released by most cell types.21 They contain biologically active RNAs (especially miRNAs) and proteins that can be transferred to recipient cells through binding, fusion, or endocytosis.22 In the heart, endogenous Exos from different subtypes of cells acts as vehicles for intercellular communication to maintain cellular homeostasis and respond to pathological stress.23,24 Recent studies have indicated that cardiac Exos play a critical role in the pathogenesis of various cardiovascular diseases.25,26,27 For instance, Exos isolated from CMs of rats with either doxorubicin-induced cardiomyopathy or myocardial infarction can promote fibrosis.28 We, therefore, hypothesized that Exos derived from genetic mutation familial DCM CMs (DCM-Exos) are involved in cardiac fibrogenesis and pathological remodeling of the heart.
Here, we used familial DCM patient-specific iPSCs differentiated CMs (iCMs) as a model to investigate the functional role of cardiac Exos in the pathogenesis of fibrosis in DCM hearts. By comparing Exos secreted by healthy iCMs and DCM iCMs, we demonstrated that DCM-Exos increased cardiac fibrosis both in vitro and in vivo. Expression profiles showed a significantly increased level of miR-218-5p in the DCM-Exos, which provides an alternative pathway to activate TGF-β signaling in CFs. Further functional assays demonstrated that miR-218-5p suppressed TNFAIP3, a master inflammation inhibitor, which subsequently promoted fibrogenesis by activating TGF-β signaling. Together, this study elucidated a distinctive miRNA profile in familial DCM-Exos that may delineate a unique pathogenesis pathway for cardiac fibrosis in familial DCM.
Results
Characterization of cardiomyocyte-derived Exos
As shown in the schematic design (Figure 1A), we first differentiated three DCM patients’ iPSCs (Table S1) and three healthy donors’ iPSCs (CTLs) into cardiomyocytes (iCMs). The purity of the cardiomyocytes was confirmed by flow cytometry (Figure S1A). The cardiac marker cardiac troponin T (cTNT) was primarily expressed in differentiated iCMs (average cTNT+> 95%). In addition, we determined the expression of two cardiac sarcomeric isoforms, α-myosin heavy chains (Myh6) and β-myosin heavy chains (Myh7), by qRT-PCR. The ratio of Myh6 to Myh7 (Myh6/Myh7) was similar between CTL-iCMs and DCM-iCMs, indicating the similar maturation of iCMs (Figure S1B).
Figure 1.

Characterization of Exos derived from iCMs
(A) Schematic overview of the study design. Three DCM iPSC lines and three healthy control iPSC lines were differentiated into cardiomyocytes. Exosomes were isolated from ANG II conditioning media of either CTL-iCMs or DCM-iCMs and further used in experiments.
(B) Representative immunoblots detect canonical exosome markers with equal protein loading (10 μg) of cells, cell debris (Debris), or Exos fractions. The exosomal markers CD63, CD9, and Alix were presented in the Exo samples. Oppositely, the ER marker, calnexin, was only present in cells and debris but not in Exos.
(C) TEM images of CTL-Exos and DCM-Exos. Exos were visualized as cup-shaped structures with diameters <200 nm.
(D and E) Quantification of CTL- and DCM-Exos by Nanosight NTA. N = 6 CTL vs. 6 DCM; NS = nonsignificant.
Next, the Exos were isolated from the conditioning media of iCMs. During the conditioning, we added angiotensin II (ANG II) to mimic the pathological stimulation in DCM patients.29,30 The same amount of protein lysis of iCMs, cell debris, and Exo fraction was examined by immunoblots. Compared to the iCMs and cell debris, tetraspanins such as CD63 and CD9; and endosomal sorting complexes required for transport (ESCRT) related Alix were enriched in Exo samples. In contrast, the ER membrane marker, calnexin, was only expressed in the cell and cell debris fractions (Figure 1B). In addition, the Exo fraction showed typical cup-shaped vesicles with diameters <200 nm under the transmission electron microscope (TEM) (Figure 1C). These results exhibited that Exos was successfully isolated from the conditioning media of iCMs. Finally, we used the Nanosight Tracking Analysis (NAT) system (NS300) to quantify the vesicle number. The NTA analysis showed there were no significant differences between DCM-Exos and CTL-Exos regarding Exo production or size distribution (Figures 1D and 1E).
DCM-Exos upregulated fibrotic gene expression in CFs
We first labeled the CTL-Exos and DCM-Exos with PKH26 to visualize Exo uptake in cardiac fibroblasts (CFs). Consistent with previous studies,31,32 the Exos were uptaken easily by CFs. The endocytosed Exos were observed under the microscope as early as 2 h after treatment (Figure 2A). The number of uptaken Eoxs kept increasing along with an increase in incubation time (Figure S2A). There was no significant difference in uptake efficiency between CTL-Exos and DCM-Exos (Figures 2B and S2B).
Figure 2.

DCM-Exos induced up-regulation of fibrotic genes in CFs
(A) Representative fluorescent images of CTL-Exos or DCM-Exos (PKH26-labeled) treatment in CFs. Endocytosed Exos (orange) were observed in the cytoplasm of CFs (bar = 200 μm (left); 900 nm (right)).
(B) Quantifications of Exo uptake in CFs. The uptake ratio was evaluated by the number of Exo uptaken cells (orange) divided by the total number of nuclei (blue) in the image (N = 22 pics/group), NS, nonsignificant.
(C) qRT-PCR of fibrotic genes in CFs. N = 6; ∗ = p< 0.05.
(D) Representative immunoblots. The CF cell lysis of different treatment groups was probed with specific antibodies (Col1a, Col3a, and CTGF).
(E) Quantification of protein expression in immunoblots. Col1a, col3a, and CTGF expression were normalized to the loading control (GAPDH), N = 6. Data represented as mean ± SD; ∗ = p< 0.05; one-way ANOVA followed by Tukey post hoc test.
Next, CTL-Exos, DCM-Exos, and vehicle (PBS) were used to treat CFs for 72 h. Of interest, compared with the CTL-Exos, DCM-Exos significantly upregulated the expression of fibrotic markers. The RNA and protein expressions of collagen I (Col1a), collagen III (Col3a), and connective tissue growth factor (CTGF) were significantly increased in the DCM-Exos treated CFs, rather than the CTL-Exos or vehicle (Figures 2C–2E). This profibrotic effect was only observed with DCM-Exos derived from the conditioning media. The Exos (CTL and DCM) derived from basal media of iCMs or iPSCs did not increase the fibrotic markers in CFs (Figure S3).
DCM-Exos promoted cardiac fibrogenesis in mouse hearts
To confirm the profibrotic effect of DCM-Exos in vivo, we injected equal amounts of CTL-Exos, DCM-Exos, or vehicle into the wild-type CD-1 mouse hearts by the animal surgeon in the lab. CTL-Exos and DCM-Exos derived from the conditioning media of iCMs were applied in this experiment. Cardiac function was examined by echocardiography (echo) at 3-time points: day 0 (the day before injection), day 7, and day 14 post-injection (Figure 3A). At day 14, the LVEF of the DCM-Exo group was significantly reduced compared with its baseline (Figure 3B). In the CTL-Exos, and vehicle groups, there were no significant changes. Histological analysis of day14 hearts indicated increased fibrotic area in the DCM-Exo group compared with CTL-Exo (Figure 3C). The red staining, which enhanced the visualization of collagen I and III fibers, indicated the fibrosis area in sections.33 Unlike the myocardial infarction murine model,34 there was no significant increase in proliferation markers, such as Ki67, in all three groups indicating the fibrotic effects through the modulation of fibroblast-associated ECM expression rather than promoting the CF proliferation (Figure S4).
Figure 3.

Intracardial injection of DCM-Exo promoted fibrosis in mouse hearts
(A) Representative echocardiographic images. Images were taken respectively from PBS, CTL-Exo, or DCM-Exo injection groups on day 0 and day 14.
(B) LVEF was determined by echocardiography. The injection of DCM-Exo significantly decreased LVEF on day 14 (N = 8/group; ∗ = p< 0.05). Injection of PBS, or CTL-Exo resulted in nonsignificant change between day 14 and baseline.
(C) Representative pictures of picrosirius red/fast green staining in series sections of the mouse hearts. The fibrotic area (Red) was quantified by ImageJ. N = 8/group, ∗∗ = p< 0.01.
(D) Representative immunoblots of mouse hearts (PBS, CTL-Exo, or DCM-Exo treatments). Fibrotic markers, Col1a, Col3a, and CTGF were detected by specific antibodies.
(E) Quantification of fibrotic markers expression. DCM-Exos injection resulted in significant upregulation of fibrotic markers (N= 8/group). GAPDH was used as a protein loading control. Data represented as mean ± SD; ∗ = p< 0.05; one-way ANOVA followed by Tukey post hoc test.
In addition, the fibrotic markers Col1a, Col3a, and CTGF showed significant upregulation in the DCM-Exo treated hearts compared with the CTL-Exos injection hearts (Figure 3D). Together, the results showed that intramyocardial injection of DCM-Exos promoted fibrosis in the mouse heart and, meanwhile, impaired cardiac function.
MiR-218-5p played a crucial role in inducing fibrogenesis in the CFs
Previous studies have shown that miRNAs in Exos cargo were cell-specific, and disease-specific and played a critical role in the Exos-mediated effects.25,31 We hypothesized whether the highly expressed miRNAs in the DCM-Exo were responsible for the profibrotic effect. The small RNA next-generation sequencing (NGS) was performed on the total RNAs isolated from CTL-Exos and DCM-Exos (3 donors vs. 3 DCM patients). The NGS result revealed a total of 22 upregulated miRNAs (>1.5-fold, p< 0.01) in the DCM-Exos compared with the CTL-Exos (Figure 4A).
Figure 4.

MiR-218-5p played an essential role in the profibrotic effect
(A) Volcano plot of differential expression miRs in CTL-Exos and DCM-Exos. (B) miRNA screening assay. qRT-PCR results showed miR-218-5p mimics significantly upregulated expression fibrotic of markers (N= 3; one-way ANOVA; ∗ = p< 0.05).
(C) MiR-218-5p upregulated fibrotic marker in a dose-dependent manner.
(D) ANG II stimulation up-regulated miR-218-5p expression in DCM-iCMs (N= 3; ∗= p <0.05, ∗∗∗ = p< 0.001).
(E) Intracardial injection of miR-218-5p increased the fibrotic area in mouse hearts (N= 8, Student’s t test, ∗∗ = p< 0.01). Data represented as mean ± SD.
Next, we performed the miR screening assay to analyze the profibrotic effect of these highly expressed miRNAs from the DCM-Exo. Every single miRNA was transfected into the CFs separately. The expression of the fibrotic markers in the CFs was determined by qRT-PCR. The screening assay identified miR-218-5p, the second high-expression miRNA in the DCM-Exos, which resulted in a significant upregulation of fibrotic markers (Figure 4B). To further confirm this result, we transfected CFs with a different dose of miR-218-5p. Consistent with the screening assay, the expression of Col1a, Col3a, and CTGF showed a dose-dependent increase, confirming the fibrotic effect of this crucial miRNA (Figure 4C). MiR-218-5p expression in DCM-Exos was confirmed by qRT-PCR. Compared with CTL-Exo, miR-218-5p expression increased more than 6-fold in the DCM-Exos (Figure S5). We found that miR-218-5p expression in the CTL-iCMs and DCM-iCM is similar at the baseline. However, the miR-218-5p was significantly increased in DCM-iCMs with conditioning media (Figure 4D).
Lastly, we intracardially injected the miR-218-5p mimics with transfection reagent into the wild-type CD-1 mice hearts. Compared with miR scramble control (miR-CTL), miR-218 injection caused a significant increase in fibrosis area in the hearts (Figure 4E). Taken together, the results showed that miR-218-5p was responsible for the fibrotic effect mediated by DCM-Exos.
MiR-218-5p knockdown ameliorated the profibrotic effect of DCM-Exo
Opposite to the overexpression of miR-218-5p, the CFs were transfected with miR-218-5p mimics inhibitor. The presence of the miR-218-5p inhibitor significantly decreased the expression of Col1a, Col3a, and CTGF, even with further treatment of DCM-Exo (Figure 5A). On the other hand, we tested the reduction of the endogenous miR-218-5p expression by delivering a specific miRNA targeting sequence, zip-miR218 (SBI Inc.) into the DCM-iCMs. Zip-miR218 would permanently knock down miR-218-5p expression in cells by expressing a specific-designed shRNA. We transfected DCM-iCMs with either zip-miR218 or zip-GFP as a control separately. 72 h after transfection, green fluorescence in DCM-iCMs was detected in both zip-miR218 and zip-GFP, indicating the successful introduction of genes (Figure 5B).
Figure 5.

MiR-218-5p inhibition attenuated the fibrotic effect elicited by the DCM-Exos
(A) qRT-PCR of fibrotic gene expression. MiR-218-5p inhibition significantly reduced the DCM-Exo-induced fibrotic effect. One-way ANOVA followed by Tukey’s post hoc test; N= 3; ∗∗ = p< 0.01.
(B) Representative fluorescent images. iCMs were transfected by zip-GFP as control or zip-miR218. Bar = 150 μm.
(C) qRT-PCR analysis of miR-218 expression in Exos derived from zip-miR218 transfected iCMs. Student’s t test, N= 3; ∗∗ = p< 0.01.
(D) qRT-PCR analysis of fibrotic gene expression. MiR-218-5p knockdown DCM-Exos (ExoZipmiR218−GFP) significantly reduced the expression of fibrotic markers. N = 3; Student’s t test; ∗ = p< 0.05. Data represented as mean ± SD.
We isolated the Exos from the conditioning media of DCM-iCMs, which were transfected with either zip-GFP (ExoZip−GFP) or zip-miR218 (ExoZip-miR218). The qRT-PCR result confirmed that, compared with ExoZip−GFP, the expression of miR-218-5p was significantly downregulated in ExoZip-miR218 (Figure 5C). Meanwhile, compared with ExoZip−GFP, ExoZip-miR218 treatment exhibited a significant reduction of Col1a, Col3a, and CTGF in CFs (Figure 5D). Again, these results showed an essential role of miR-218-5p in regulating the profibrotic effect of DCM-Exo.
MiR-218-5p promoted fibrogenesis via activation of canonical TGF-β signaling
The TGF-β-mediated Smad signaling pathway was crucial in inducing fibrogenesis.35,36 TGF-β acted through receptors to promote Smad2/3 phosphorylation, followed by translocation of the phosphorylated Smad complex into the nucleus, where the Smads worked as transcription factors to increase the synthesis of the col I and III and thus induced fibrosis.36
Thus, we investigated whether miR-218-5p affected TGF-β signaling pathway in the CFs. MiR-218-5p mimics were transfected in the CFs. In parallel, the CFs were treated with either TGF-β1 or the TGF-β inhibitor SB525334.37 The immunoblots indicated that Smad2 phosphorylation was dramatically increased with TGF-β1 treatment, indicating activation of TGF-β/Smad signaling (Figure 6A). Oppositely, SB525334 treatment effectively reduced Smad2 phosphorylation compared with TGF-β1 treatment alone. Meanwhile, inhibition of TGF-β/Smad signaling by also halted the upregulated of fibrogenic markers induced by TGF-β1 treatment.
Figure 6.

MiR-218-5p increased Smad2 phosphorylation in CFs
(A) Immunoblots of cell lysates derived from CFs treated with TGF-β, miR-218-5p, and a TGF-β inhibitor (SB525334). Specific antibodies were used to detect Col1a, Col3a, CTGF, phospo-Smad2 (p-SMAD2), and GAPDH.
(B) Quantitation of protein expression in immunoblots. GAPDH was used as a protein loading control. TGF-β inhibitor (SB525334) significantly reduced TGF-β1-induced fibrotic effect (lane 3). MiR-218-5p increased the phosphorylation of Smad2. N= 3; one-way ANONVA followed by Tukey’s post hoc test, ∗ = p< 0.05; ∗∗ = p< 0.01; ∗∗∗ = p< 0.001. Data represented as mean ± SD.
On the other hand, without the TGF-β1 treatment mediated receptor signaling pathway, miR-218-5p overexpression led to a significant increase in the phosphorylation of Smad2 (Figure 6B; p-SMAD2, lane 4). The fibrotic markers were concurrently upregulated with the increase of Smad2 phosphorylation. Of interest, SB525334 treatment dramatically abolished the Smad2 phosphorylation and decreased the profibrotic effects of miR-218-5p. These results indicated that the profibrotic effect of miR-218-5p was through activation of the TGF-β/Smad2 signaling pathway, not independent of the presence of TGF-β1.
TNFAIP3 was an essential mediator of miR-218-5p induced fibrotic effect
MiR-218-5p was produced from two precursor genes, MIR218-1 and MIR218-2, located in the introns of SLIT2 and SLIT3. Although we showed that miR-218-5p was responsible for the fibrotic effect, the mechanism of miR-218-5p regulating fibrosis was still unknown. By interactive predictions of RNA22 38, the miR-218-5p exhibited a direct binding sequence at 5′UTR of the TNFAIP3 immediately before the start codon region (Figure 7A). In addition, TNFAIP3 was reported previously as a suppressor to the canonical TGFβ/Smad-dependent pathway.39 Thus, we first determined the expression of TNFAIP3 in miR-218-5p overexpression. The immunoblots exhibited miR-218-5p overexpression significantly decreased TNFAIP3 expression in CFs (Figure 7B).
Figure 7.

MiR-218-5p targeted to TNFAIP3
(A) Targeted sequence prediction of miR-218-5p in RNA22.
(B) Immunoblots (left) indicated TNFAIP3 expression was significantly repressed in CFs transfected with miR-218-5p.
(C) Luciferase assay of miR-218-5p and TNFNAIP3 5′UTR region. Co-transfection of miR-218-5p + WT sequence significantly reduced luciferase activity compared with the scramble CTL + WT. One-way ANOVA followed by Tukey’s post hoc test, ∗∗ = p< 0.01; NS = nonsignificant.
(D) Representative immunoblots of CFs treatment.
(E) Quantitation of expression of TNFAIP3, Col1a, Col3a, and CTGF. One-way ANOVA followed by Tukey’s post hoc test; ∗∗ = p<0.01, ∗∗∗ = p< 0.001. Data represented as mean ± SD.
Next, we co-transfected scramble control (Scr CTL) and miR-218-5p mimics into HEK293A cells with a luciferase reporter system containing either the wild-type (WT) or the mutated (Mut) sequence from 5′-UTR of TNFAIP3, including the first 25 bp of the start codon (Table S2). Decreased luciferase activity was observed in HEK293A cells co-transfected with the miR-218-5p mimic and TNFAIP3-WT (miR-218 + WT), compared with the cells co-transfected with scramble CTL and TNFAIP3-WT (Scr CTL + WT). When we transfected the mutation sequence, luciferase activity was unchanged, regardless of the presence of the miR-218-5p mimics (Figure 7C). Altogether, we identified that TNFAIP3 is a direct target of miR-218-5p in the CFs.
Moreover, the overexpression of TNFANIP3 ameliorated miR-218-5p induced fibrotic effects. As shown in immunoblots, TNFAIP3 overexpression significantly downregulated the expressions of Col1a, Col3a, and CTGF (Figures 7D and 7E). As a direct target of miR-218-5p, TNFAIP3 was essential in mediating the fibrotic effect of miR-218-5p.
Restoration of TNFAIP3 ameliorated the fibrotic effect of DCM-Exo
As a critical mediator of miR-218-5p, we investigated TNFAIP3 expression in the CTL-Exo and DCM-Exo treated CFs. As shown in the immunoblots, DCM-Exo treatment dramatically decreased TNFAIP3 expression in the CFs, like miR-218-5p overexpression (Figure 8A).
Figure 8.

Restoration of TNFAIP3 ameliorated the DCM-Exo-induced fibrotic effect
(A) Representative immunoblots of CFs treated with either CTL-Exos or DCM-Exos. DCM-Exo treatment significantly decreased TNFAIP3 expression. Student’s t test; ∗∗ = p <0.01.
(B) Representative immunoblots of TNFAIP3, Col1a, Col3a, CTGF, and GAPDH detection in CFs treated with CTL-Exos, DCM-Exos, or overexpression of TNFAIP3.
(C–F) Quantitation of protein expression. TNFAIP3 overexpression significantly reduced the expression of fibrotic markers. One-ANOVA followed by Tukey’s post hoc test; N= 3; ∗∗ = p< 0.01.
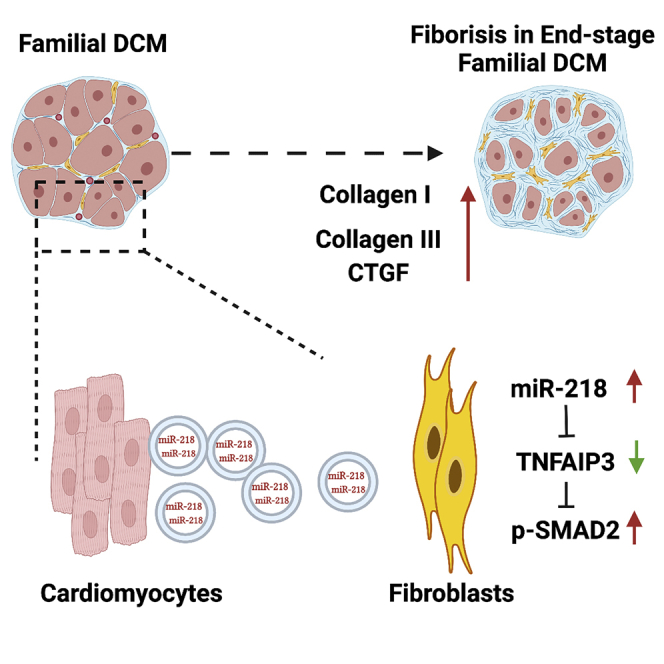
(G) Schematic picture showing the working model of our study. Data represented as mean ± SD.
Next, we overexpressed TNFAIP3 by transfecting the TNFAIP3 cDNA ORF in CFs. As shown in Figure 8B, TNFAIP3 overexpression significantly upregulated TNFAIP3 in the CFs (Figure 8C). Opposite to the DCM-Exo treatment, the expression of Col1a, Col3a, and CTGF was effectively reduced by the introduction of TNFAIP3 in CFs (Figures 8D–8F). The results confirmed that the restoration of TNFAIP3, which is an essential mediator of miR-218-5p, effectively ameliorated the fibrotic effect of DCM-Exos.
Taken together, our data indicated that miR-218-5p activated TGFβ/smad2 pathway, inducing profibrotic effects in CFs via receiving the DCM-Exo. Mechanistically, miR-218-5p repressed the TNFAIP3 expression and further regulated the fibrotic markers. In contrast, we restored the expression of TNFAIP3 could effectively block miR-218-5p and DCM-Exo induced profibrotic effect.
Discussion
Exos are nanosized vesicles containing functional biomolecules (proteins, lipids, RNA, and DNA) that can be horizontally transferred to recipient cells.40,41 Although it arouses much interest in investigating their functions in different diseases, the pathogenic nature of cardiac Exos is largely unexplored.25,31,42 Familial DCM is a genetic form of heart disease causing thin and weakened heart chambers often leading to heart failure. Besides ‘sick’ heart muscle, excessive ECM has been proven to significantly contribute to disease progression, impair contractility, and increase cardiovascular hospitalization and mortality.43,44 Cardiac fibroblasts are responsible for ECM dysregulation. However, the signals which activate the fibroblasts may also play a critical role.45,46
Here, we investigated the profibrotic effect of the Exos derived from three familial DCM patients’ iPSCs differentiated cardiomyocytes. Using iPSC-derived cardiac cells demonstrated a significant opportunity to investigate the potential functions of Exos in the heart.47,48 Compared with CTL-Exos, DCM-Exos treatment significantly increased the expression of fibrogenic genes in cardiac fibroblast cells. In vivo, injection of DCM-Exos into mouse hearts induced enlarged fibrosis and impaired cardiac function. These were not observed in hearts injected with CTL-Exos. These results suggested that Exo-mediated crosstalk between DCM cardiomyocytes and fibroblasts independently elicited a profibrotic effect. The clinical development of DCM reflects the cumulative effects of detrimental remodeling in the heart. In contrast, our in vivo experiments may only show the acute response of the heart to DCM-Exos. In the future, the long-term effect of DCM-Exos will need to be investigated in animal models.
Previous studies showed that exosomal miRNAs reflected the biology of their celltype of origin and disease status.31,48 Our small RNA sequencing results indicated differential miRNA expression between DCM-Exos and CTL-Exos. Amongst differential miRs, miR-218-5p was highly present in the DCM-Exos and critical to mediate the profibrotic effect. Overexpression of miR-218 in vitro and in vivo indicated significant upregulation of fibrotic markers (col1a, col3a, CTGF), as well as increased fibrotic area. Oppositely, the knockdown of miR-218-5p presence in the DCM-Exos ameliorated the profibrotic effect of the DCM-Exos. Furthermore, we exhibited that miR-218-5p overexpression activated canonical TGF-β signaling by increasing the phosphorylation of Smad2/3 in CFs. MiR-218-5p played an essential role as a tumor suppressor in cancers such as gastrointestinal stromal tumor, non-small cell lung cancer, and pancreatic cancer.49,50,51 In addition, miR-218-5p affected cardiac development, causing cardiac malformation, atrial hypoplasia, and ventricular septum insufficiency. Overexpression of miR-218-5p in zebrafish embryos can inhibit standard heart development.52 Inhibition of miR-218-5p was shown to be against cardiac fibrosis in rats with myocardial infarction.53 The upregulation of miR-218-5p in the DCM-Exos was consistent that Exos can facilitate their effects via the delivery of specific miR in the cargo. The mechanism of upregulated miR-218-5p in the DCM-Exos seemed to be related to the stress stimulation, although a detailed regulatory mechanism is not explored.
In the study, we further identified TNFAIP3 as the target of miR-218-5p, which regulated the profibrotic effect in the CFs. TNFAIP3 is a zinc finger (de)ubiquitinating enzyme that plays a crucial role in regulating NF-κB activity.54,55,56 Deficiency of TNFAIP3 developed severe inflammation in mice and increased the sensitivity in cells with an inflammatory stimulation.54 Although miR-218-5p was reported to directly target CTGF in the colorectal cancer,57 our results indicated that miR-218-5p overexpression significantly increased the expression of several fibrotic makers, including the CTGF (Figures 4C and 7D). These different results may be because of the different cell-type specificities between CF and colorectal cancer. On the other hand, the TNFAIP3 overexpression successfully reduced upregulated fibrotic markers, which were induced by miR-218-5p overexpression (Figure 7D), indicating the master regulatory role of TNFAIP3 in the profibrotic effect of miR-218-5p. It was notable that exposure to sustained inflammatory signaling increased the sensitivity of CFs to TGF-β stimulation and exhibited an increased profibrotic phenotypic response.58 Consistently, our results indicated that miR-218-5p activates TGF-β signaling by suppressing the inflammatory inhibitor TNFAIP3. The repression of TNFAIP3 enhanced inflammation that would further magnify fibrogenic effects in CFs.
Taken together, the results of this study explored a new mechanism of Exos-mediated profibrotic effect in familial DCM. The phenotypic fibrotic alteration in CFs was attributed to differential miRs (miR-218-5p) in the DCM-Exo cargoes. From a clinical respective, this cumulative effect of detrimental Exos was non-negligible. Compared with much evidence that cardiac Exos were responsible for protective effects,22 the uncovering of the detrimental functions of Exos may provide us with an alternative way to reduce cardiac fibrosis and manage the disease progression.
Limitations of the study
Owing to the lack of proper technique to monitor the dynamics of exosomes in cells (or in vivo), we isolated the exosomes from the conditioned media of the cultured cardiomyocytes as in most studies.59 The results of our study reflected the profibrotic nature of the DCM cardiomyocyte-derived exosome while uptake into the cardiac fibroblast of the heart. A limitation was that it was hard to mimic the dynamic process and chronic effect of the exosome uptake in vivo. Most studies of exosome biogenesis and release have mainly been performed using in vitro grown cell lines as a model.60 The accurate exosome secretion data, such as exosome secretion speed and amount in human hearts is unavailable so far. In future studies, the live cell/exosome imaging system could assist us to explore the kinetics of exosome trafficking in the hearts.61,62 In addition, cardiac fibrosis is a dynamic and complex process.63 Owing to the phenotypic plasticity, the fibroblasts could shift between activation (myofibroblast) and resting status.64,65,66 Thus, the long-term profibrotic effect of exosomes needs to be investigated in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD63 | Abcam | RRID: AB_940915 |
| Calnexin | Cell Signaling Technology | RRID: AB_2228381 |
| CD9 | Cell Signaling Technology | RRID: AB_2798139 |
| Alix | Cell Signaling Technology | RRID: AB_2299455 |
| Col1a | Cell Signaling Technology | RRID: AB_2800169 |
| Col3a | Invitrogen (M) | PA586281 |
| Col3a | Cell Signaling Technology (H) | 66887 |
| CTGF | Abcam | RRID: AB_305688 |
| P-SMAD2 | Cell Signaling Technology | RRID: AB_2798798 |
| TNFAIP3 | Cell Signaling Technology | RRID: AB_10698880 |
| GAPDH | Millipore | MAB374 |
| Bacterial and virus strains | ||
| MIRZIP-218 | System Biosciences | MZIP218 |
| MIRZIP CTL | System Biosciences | MZIP000 |
| Chemicals, peptides, and recombinant proteins | ||
| CHIR99021 | Selleckchem | 2924 |
| Wnt-C59 | Selleckchem | 7037 |
| Angiotensin II | Sigma-Aldrich | 9525 |
| TGF-beta 1 | Novus Biologicals | 7754 |
| SB525334 | Selleckchem | 1476 |
| Experimental models: Cell lines | ||
| Healthy iPSC line1 | Stanford University BioBank | 15 |
| Healthy iPSC line2 | Stanford University BioBank | 479 |
| Healthy iPSC line3 | Stanford University BioBank | 480 |
| DCM patient iPSC line1 | Stanford University BioBank | 17 |
| DCM patient iPSC line2 | Stanford University BioBank | 18 |
| DCM patient iPSC line3 | Stanford University BioBank | 19 |
| Experimental models: Organisms/Strains | ||
| Crl:CD1 | Charles River | CD-1 |
| Oligonucleotides | ||
| microRNA mimics | Horizon | C-300573 |
| 5′UTR wildtype | This paper | Genewiz |
| 5′UTR mutation | This paper | Genewiz |
| Recombinant DNA | ||
| TNFAIP3 | Origene | SC330856 |
| Software and algorithms | ||
| Prism GraphPad | Dotmatics | https://www.graphpad.com/scientific-software/prism/ |
| MicroRNA Analysis | This paper | https://doi.org/10.6084/m9.figshare.21764141 |
| RNA22 | Jefferson CMC | https://cm.jefferson.edu/rna22/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sunjay Kaushal (SKaushal@luriechildrens.org).
Materials availability
This study did not generate new unique reagents.
Method details
Cardiac fibroblast culture
Cardiac fibroblasts were purchased from Lonza (CC-2904) and cultured in the cardiac fibroblast growth medium (CC-4526). Passages 1-3 were used in this study.
iPSCs differentiation
Healthy and DCM (TNNT2R173W) iPSCs were purchased from the Stanford University Cardiovascular Institute Biobank (Table S1). iPSC lines were cultured in Essential 8 medium (Thermo Fisher Scientific; A1517001) and passaged in split ratios of 1:6. Until the cells reached ∼85% confluence (Day 0), the medium was changed to RPMI1640 (Thermo Fisher Scientific; 61870036) supplemented with B27 minus insulin (Thermo Fisher Scientific; A1895601) and 6.5 μmol/L CHIR99021 (Selleckhem; S2924). On day 2, the medium was changed to RPMI1640 supplemented with B27 minus insulin only. From day 3–5, the medium was changed to RPMI1640 supplemented with B27 minus insulin and 2 μmol/L Wnt-C59 (Selleckhem; S7037). The medium was changed on day 5 and every other day thereafter to RPMI1640 supplemented with B27 (Thermo Fisher Scientific; 17504044). Contractile CMs were noted from days 9-15. Since day 15, RPMI1640 medium without d-glucose (Thermo Fisher Scientific;11879020) was used.67,68 The selection took one-two week, and iCMs were replated into new flasks after selection. Four days after replating, the iCMs were ready for the Exos isolation.
Exo isolation by ultracentrifugation
Medium containing 5 μM ANG II was added to replated iCMs and was collected after 60 h of conditioning. Exos were isolated from the conditioned medium by differential centrifugation.69 The conditioned medium was centrifuged for 10 min at 300 × g to remove cells and then for 10 min at 2,000 × g to remove apoptotic bodies. Exos in the resulting supernatant were isolated by ultracentrifugation (100,000 × g; 75 min). Pellets were suspended in PBS for further applications.
Nanosight Tracking Analysis
Exo samples were diluted and loaded onto a Nanosight instrument (Melvin Instruments; NS300) for nanoparticle analysis according to the manufacturer’s instructions.
Transmission electron Microscopy
Exo samples were sent to Creative Bioarray, Inc. Pictures were taken using a JEOL JEM1400 transmission electron microscope (TEM).
Profibrotic assay in vitro
Cardiac fibroblasts were treated with CTL-Exo or DCM-Exo (1.5 x 109 p/ml) for 72 h, respectively. RNAs were isolated from cells after treatments. The expression of fibrotic markers were determined by qRT-PCR.
Quantitative reverse transcriptase PCR
Quantitative reverse transcriptase PCR (qRT-PCR) was performed according to the manufacturer’s instructions (Qiagen; 205,311). Each reaction was performed in triplicate using a Fast SYBR-Green reaction mix. Quantitect primer assays for each primer set were obtained from Qiagen. The primers of microRNA were purchased from Thermofisher Inc. (Waltham, MA). Cycle threshold (CT) values of the housekeeping gene were subtracted from the corresponding gene of interest. The expression 2ˆ-dCT determined the fold change of expression level for each gene. Final values were averaged, and results were represented as fold expression with the SD.
Western blotting
Protein concentrations were determined by DC kit (Bio-Rad; 5000112). Primary antibodies used were: mouse anti-human CD63 (1:1,000; ab59479; Abcam); anti-CD9 (1:1000; #13174 cell signaling); anti-HSP70 (1:1000, #4872 cell signaling); anti-Col1a (1:1,000; 91144; Cell Signaling Technology), anti-Col3a (PA5-27828; cell signaling #66887); anti-CTGF (1:1000, Abcam6992), anti-TNFAIP3 (1:1000, #5630, #4625 cell signaling); anti-pSMAD2 (1:1000, #3108 cell signaling); anti-GAPDH (MA1-16757) . Secondary antibodies used were goat anti-mouse IgG (cell signaling); goat anti-rabbit IgG (cell signaling).
Total RNA isolation
RNA was isolated from cardiac fibroblasts or mouse heart tissues.70 RNA from Exo samples was isolated using a Total Exosome RNA Isolation Kit (Invitrogen, 4478545). Generally, the cell and Exo samples were dissociated in the QIAzol Lysis Reagent. Chloroform was added into homogenized samples to extract total RNA. The affinity binding columns were used to purify the RNAs extracted from all samples.
Next-generation sequencing library generation and sequencing
Exosomal RNA samples (4 μg RNA per sample) from CTL-Exo and DCM-Exo (N = 3 vs. 3) were sent to Novogene, Inc. (Sacramento, CA) for library generation and sequencing. The sequencing libraries were generated using the NEBNext Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs, Ipswich, MA) according to the manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. Library quality was assessed before sequencing. Differentially-expressed miRNAs were identified based on a model using the negative binomial distribution. Normalized log10 miRNA expression values were used to generate a heatmap with Morpheus (Broad Institute).
Intramyocardial injection of Exos or miR-218 mimics
Animal experiments were done in accordance with the Institutional Animal Care and Use Committee of the University of Maryland Baltimore. Ten-week-old CD-1 mice (Charles River) were anesthetized during surgery. The heart was exposed via a left thoracotomy, and 5 × 1012 DCM-Exos, CTL-Exos (all Exos were isolated from ANG II conditioned cardiomyocytes), or vehicle (PBS) were injected into the myocardium on the left ventricle (LV). The miR-218-5p mimics (horizon, C300573-05) were formulated with Max-Suppressor in vivo RNALancerII, a lipid-based delivery reagent, according to the manufacturer’s instructions before injections. Transthoracic echocardiograms were acquired at 7 and 14 days after injection. Cardiac function was evaluated based on echocardiographic parameters. M-mode images of the LV in the parasternal short-axis view were obtained at the level of the papillary muscles using a high-resolution ultrasound machine (Vevo 2100: VisualSonics) equipped with a 20-MHz scan head for calculation/estimation of LV fractional shortening and ejection fraction. Data were calculated from 5 cardiac cycles according to the generally accepted formula. All mice were euthanized by day 14 after injection. Hearts were kept for histological analyses and further experiments.
Transfection of zip-miR and zip-GFP
The zip-miR128 and zip-GFP were purchased from System Bioscience. The reagents were added to the purified iCMs according to the manufacturer’s instructions.
Picrosirius red/fast green staining and fibrosis area analysis
Paraffin-embedded mouse heart tissues were sectioned and processed for picrosirius red/fast green staining.33 The heart tissue samples were rehydrated using a graded ethanol series and then immersed in PBS at room temperature. The rehydrated samples were then incubated in 0.04% picrosirius red and 0.1% fast green for 30 min, then rehydrated and mounted in DPX mounting medium. Fibrotic areas were analyzed with ImageJ software (NIH).
Luciferase reporter gene assay
Wild-type (WT) tumor necrosis factor alpha-induced protein 3 (TNFAIP3) 5ʹ-UTR and mutant-type (MUT) TNFAIP3 5ʹ-UTR sequences were synthesized by Genewiz. The miR-218-5p mimic or the miRNA scramble control was co-transfected into HEK293A cells using Lipofectamine 2000. After 48 h, luciferase activity was detected by the Dual-Luciferase Reporter Assay Kit (Promega).
Quantification and statistical analysis
Parametric data were analyzed using GraphPad Prism. Student’s t-tests were used for comparisons between the two groups. For comparing three or more groups, one-way ANOVA followed by an appropriate (Bonferroni or Tukey) posthoc test will be applied. p values <0.05 were considered statistically significant. Data are represented as mean ± SD.
Acknowledgments
We acknowledge Dr. Fern P. Finger for her assistance in preparing this manuscript. D.L. is funded by Maryland Stem Cell Research Fund grant 2021-MSCRFL-5695 and National Heart, Lung, and Blood Institute grant HL118491-06A1. S.K. is funded by grants from the National Heart, Lung, and Blood Institute grants (R01HL118491, R01 HL139060-01A1, R42HL131226-01). The authors also thank the Department of Surgery, University of Maryland School of Medicine for financial support.
Author contributions
X.F., S.S., D.L., and S.K. designed the experiments. X.F. and M.Y.A. performed cell culture and iPSC differentiation, Exo isolation, and experiments. R.M. performed flow cytometry. L.C. performed animal surgery and echocardiography analysis. T.K. constructed 5′UTR and mutations. X.F., L.C., and D.L. analyzed the data. X.F. wrote the original draft. M.G., P.S., C.H., P.Y., D.L., and S.K. edited and finalized the manuscript. All authors critically reviewed and approved the final version of the manuscript.
Declaration of interests
This article was prepared while T.K. was employed at the University of Maryland School of Medicine. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
S.K. is a founder and shareholder in Secretome Therapeutics. R.M.,S.S., M.G., and P.S. are shareholders in Secretome Therapeutics.
Published: January 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.105963.
Contributor Information
Deqiang Li, Email: dqli@som.umaryland.edu.
Sunjay Kaushal, Email: skaushal@luriechildrens.org.
Supplemental information
Data and code availability
-
•
Analysis data have been deposited at Figshare and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.McNally E.M., Golbus J.R., Puckelwartz M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elliott P., Andersson B., Arbustini E., Bilinska Z., Cecchi F., Charron P., Dubourg O., Kühl U., Maisch B., McKenna W.J., et al. Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 3.Maron B.J., Towbin J.A., Thiene G., Antzelevitch C., Corrado D., Arnett D., Moss A.J., Seidman C.E., Young J.B., et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of Care and outcomes Research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 4.Watkins H., Ashrafian H., Redwood C. Inherited cardiomyopathies. N. Engl. J. Med. 2011;364:1643–1656. doi: 10.1056/NEJMra0902923. [DOI] [PubMed] [Google Scholar]
- 5.Rath A., Weintraub R. Overview of cardiomyopathies in childhood. Front. Pediatr. 2021;9:708732. doi: 10.3389/fped.2021.708732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultheiss H.P., Fairweather D., Caforio A.L.P., Escher F., Hershberger R.E., Lipshultz S.E., Liu P.P., Matsumori A., Mazzanti A., McMurray J., Priori S.G. Dilated cardiomyopathy. Nat. Rev. Dis. Primers. 2019;5:32. doi: 10.1038/s41572-019-0084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNally E.M., Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res. 2017;121:731–748. doi: 10.1161/CIRCRESAHA.116.309396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cahill T.J., Ashrafian H., Watkins H. Genetic cardiomyopathies causing heart failure. Circ. Res. 2013;113:660–675. doi: 10.1161/CIRCRESAHA.113.300282. [DOI] [PubMed] [Google Scholar]
- 9.Robinson P., Griffiths P.J., Watkins H., Redwood C.S. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 2007;101:1266–1273. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 10.Wu H., Lee J., Vincent L.G., Wang Q., Gu M., Lan F., Churko J.M., Sallam K.I., Matsa E., Sharma A., et al. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised beta-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell. 2015;17:89–100. doi: 10.1016/j.stem.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun N., Yazawa M., Liu J., Han L., Sanchez-Freire V., Abilez O.J., Navarrete E.G., Hu S., Wang L., Lee A., et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012;4:130–147. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandawat A., Chattranukulchai P., Mandawat A., Blood A.J., Ambati S., Hayes B., Rehwald W., Kim H.W., Heitner J.F., Shah D.J., Klem I. Progression of myocardial fibrosis in nonischemic DCM and association with mortality and heart failure outcomes. JACC Cardiovasc. Imaging. 2021 doi: 10.1016/j.jcmg.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke M.A., Chang S., Wakimoto H., Gorham J.M., Conner D.A., Christodoulou D.C., Parfenov M.G., DePalma S.R., Eminaga S., Konno T., et al. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight. 2016;1:215–222. doi: 10.1172/jci.insight.86898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frangogiannis N.G. The extracellular matrix in ischemic and nonischemic heart failure. Circ. Res. 2019;125:117–146. doi: 10.1161/CIRCRESAHA.119.311148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapelko V.I. Extracellular matrix alterations in cardiomyopathy: the possible crucial role in the dilative form. Exp. Clin. Cardiol. 2001;6:41–49. [PMC free article] [PubMed] [Google Scholar]
- 16.Krenning G., Zeisberg E.M., Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freundlich B., Bomalaski J.S., Neilson E., Jimenez S.A. Regulation of fibroblast proliferation and collagen synthesis by cytokines. Immunol. Today. 1986;7:303–307. doi: 10.1016/0167-5699(86)90067-8. [DOI] [PubMed] [Google Scholar]
- 18.Perrucci G.L., Barbagallo V.A., Corlianò M., Tosi D., Santoro R., Nigro P., Poggio P., Bulfamante G., Lombardi F., Pompilio G. Integrin alphanubeta5 in vitro inhibition limits pro-fibrotic response in cardiac fibroblasts of spontaneously hypertensive rats. J. Transl. Med. 2018;16:352. doi: 10.1186/s12967-018-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross R.S., Borg T.K. Integrins and the myocardium. Circ. Res. 2001;88:1112–1119. doi: 10.1161/hh1101.091862. [DOI] [PubMed] [Google Scholar]
- 20.Koitabashi N., Danner T., Zaiman A.L., Pinto Y.M., Rowell J., Mankowski J., Zhang D., Nakamura T., Takimoto E., Kass D.A. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J. Clin. Invest. 2011;121:2301–2312. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Théry C., Zitvogel L., Amigorena S. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 22.Gartz M., Strande J.L. Examining the paracrine effects of exosomes in cardiovascular disease and repair. J. Am. Heart Assoc. 2018;7:e007954. doi: 10.1161/JAHA.117.007954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bellin G., Gardin C., Ferroni L., Chachques J.C., Rogante M., Mitrecic D., Ferrari R., Zavan B. Exosome in cardiovascular diseases: a complex world full of hope. Cells. 2019;8 doi: 10.3390/cells8020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ranjan P., Kumari R., Verma S.K. Cardiac fibroblasts and cardiac fibrosis: precise role of exosomes. Front. Cell Dev. Biol. 2019;7:318. doi: 10.3389/fcell.2019.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gartz M., Lin C.W., Sussman M.A., Lawlor M.W., Strande J.L. Duchenne muscular dystrophy (DMD) cardiomyocyte-secreted exosomes promote the pathogenesis of DMD-associated cardiomyopathy. Dis. Model. Mech. 2020;13:dmm045559. doi: 10.1242/dmm.045559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bang C., Batkai S., Dangwal S., Gupta S.K., Foinquinos A., Holzmann A., Just A., Remke J., Zimmer K., Zeug A., et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Invest. 2014;124:2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X., Sucharov J., Stauffer B.L., Miyamoto S.D., Sucharov C.C. Exosomes from pediatric dilated cardiomyopathy patients modulate a pathological response in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2017;312:H818–H826. doi: 10.1152/ajpheart.00673.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J., Yu X., Xue F., Li Y., Liu W., Zhang S. Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk. Am J. Transl. Res. 2004;10:4350–4366. [PMC free article] [PubMed] [Google Scholar]
- 29.Bleumink G.S., Schut A.F.C., Sturkenboom M.C.J.M., Deckers J.W., van Duijn C.M., Stricker B.H.C. Genetic polymorphisms and heart failure. Genet. Med. 2004;6:465–474. doi: 10.1097/01.gim.0000144061.70494.95. [DOI] [PubMed] [Google Scholar]
- 30.Kitzman D.W., Little W.C., Brubaker P.H., Anderson R.T., Hundley W.G., Marburger C.T., Brosnihan B., Morgan T.M., Stewart K.P. Pathophysiological characterization of isolated diastolic heart failure in comparison to systolic heart failure. JAMA. 2002;288:2144–2150. doi: 10.1001/jama.288.17.2144. [DOI] [PubMed] [Google Scholar]
- 31.Qiao L., Hu S., Liu S., Zhang H., Ma H., Huang K., Li Z., Su T., Vandergriff A., Tang J., et al. microRNA-21-5p dysregulation in exosomes derived from heart failure patients impairs regenerative potential. J. Clin. Invest. 2019;129:2237–2250. doi: 10.1172/JCI123135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao L., Wang L., Wei Y., Krishnamurthy P., Walcott G.P., Menasché P., Zhang J. Exosomes secreted by hiPSC-derived cardiac cells improve recovery from myocardial infarction in swine. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aay1318. eaay1318-1959. [DOI] [PubMed] [Google Scholar]
- 33.Segnani C., Ippolito C., Antonioli L., Pellegrini C., Blandizzi C., Dolfi A., Bernardini N. Histochemical detection of collagen fibers by sirius red/fast green is more sensitive than van Gieson or sirius red alone in normal and inflamed rat colon. PLoS One. 2015;10:e0144630. doi: 10.1371/journal.pone.0144630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kretzschmar K., Post Y., Bannier-Hélaouët M., Mattiotti A., Drost J., Basak O., Li V.S.W., van den Born M., Gunst Q.D., Versteeg D., et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proc. Natl. Acad. Sci. USA. 2018;115:E12245–E12254. doi: 10.1073/pnas.1805829115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khalil H., Kanisicak O., Prasad V., Correll R.N., Fu X., Schips T., Vagnozzi R.J., Liu R., Huynh T., Lee S.J., et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saadat S., Noureddini M., Mahjoubin-Tehran M., Nazemi S., Shojaie L., Aschner M., Maleki B., Abbasi-Kolli M., Rajabi Moghadam H., Alani B., Mirzaei H. Pivotal role of TGF-beta/smad signaling in cardiac fibrosis: non-coding RNAs as effectual players. Front. Cardiovasc. Med. 2020;7:588347. doi: 10.3389/fcvm.2020.588347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakao A., Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hanai J., Heldin C.H., Miyazono K., ten Dijke P. TGF-Beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miranda K.C., Huynh T., Tay Y., Ang Y.S., Tam W.L., Thomson A.M., Lim B., Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 39.Bhattacharyya S., Wang W., Graham L.V.D., Varga J. A20 suppresses canonical Smad-dependent fibroblast activation: novel function for an endogenous inflammatory modulator. Arthritis Res. Ther. 2016;18:216. doi: 10.1186/s13075-016-1118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Théry C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 41.Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 42.Jadli A.S., Parasor A., Gomes K.P., Shandilya R., Patel V.B. Exosomes in cardiovascular diseases: pathological potential of nano-messenger. Front. Cardiovasc. Med. 2021;8:767488. doi: 10.3389/fcvm.2021.767488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Assomull R.G., Prasad S.K., Lyne J., Smith G., Burman E.D., Khan M., Sheppard M.N., Poole-Wilson P.A., Pennell D.J. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J. Am. Coll. Cardiol. 2006;48:1977–1985. doi: 10.1016/j.jacc.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 44.Gulati A., Jabbour A., Ismail T.F., Guha K., Khwaja J., Raza S., Morarji K., Brown T.D.H., Ismail N.A., Dweck M.R., et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. 2013;309:896–908. doi: 10.1001/jama.2013.1363. [DOI] [PubMed] [Google Scholar]
- 45.Pellman J., Zhang J., Sheikh F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: mechanisms and model systems. J. Mol. Cell. Cardiol. 2016;94:22–31. doi: 10.1016/j.yjmcc.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hall C., Gehmlich K., Denning C., Pavlovic D. Complex relationship between cardiac fibroblasts and cardiomyocytes in Health and disease. J. Am. Heart Assoc. 2021;10:e019338. doi: 10.1161/JAHA.120.019338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung J.H., Fu X., Yang P.C. Exosomes generated from iPSC-derivatives: new direction for stem cell therapy in human heart diseases. Circ. Res. 2017;120:407–417. doi: 10.1161/CIRCRESAHA.116.309307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandy M., Rhee J.W., Ozen M.O., Williams D.R., Pepic L., Liu C., Zhang H., Malisa J., Lau E., Demirci U., Wu J.C. Atlas of exosomal microRNAs secreted from human iPSC-derived cardiac cell types. Circulation. 2020;142:1794–1796. doi: 10.1161/CIRCULATIONAHA.120.048364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C., Ge S., Hu C., Yang N., Zhang J. MiRNA-218, a new regulator of HMGB1, suppresses cell migration and invasion in non-small cell lung cancer. Acta Biochim. Biophys. Sin. 2013;45:1055–1061. doi: 10.1093/abbs/gmt109. [DOI] [PubMed] [Google Scholar]
- 50.He H., Hao S.J., Yao L., Yang F., Di Y., Li J., Jiang Y.J., Jin C., Fu D.L. MicroRNA-218 inhibits cell invasion and migration of pancreatic cancer via regulating ROBO1. Cancer Biol. Ther. 2014;15:1333–1339. doi: 10.4161/cbt.29706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tu L., Wang M., Zhao W.Y., Zhang Z.Z., Tang D.F., Zhang Y.Q., Cao H., Zhang Z.G. miRNA-218-loaded carboxymethyl chitosan-Tocopherol nanoparticle to suppress the proliferation of gastrointestinal stromal tumor growth. Mater. Sci. Eng. C Mater. Biol. Appl. 2017;72:177–184. doi: 10.1016/j.msec.2016.10.052. [DOI] [PubMed] [Google Scholar]
- 52.Chiavacci E., Dolfi L., Verduci L., Meghini F., Gestri G., Evangelista A.M.M., Wilson S.W., Cremisi F., Pitto L. MicroRNA 218 mediates the effects of Tbx5a over-expression on zebrafish heart development. PLoS One. 2012;7:e50536. doi: 10.1371/journal.pone.0050536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qian L., Pan S., Shi L., Zhou Y., Sun L., Wan Z., Ding Y., Qian J. Downregulation of microRNA-218 is cardioprotective against cardiac fibrosis and cardiac function impairment in myocardial infarction by binding to MITF. Aging (Albany NY) 2019;11:5368–5388. doi: 10.18632/aging.102112. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Lee E.G., Boone D.L., Chai S., Libby S.L., Chien M., Lodolce J.P., Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dixit V., Mak T.W. NF-kappaB signaling. Many roads lead to madrid. Cell. 2002;111:615–619. doi: 10.1016/s0092-8674(02)01166-2. [DOI] [PubMed] [Google Scholar]
- 56.Wertz I.E., O'Rourke K.M., Zhou H., Eby M., Aravind L., Seshagiri S., Wu P., Wiesmann C., Baker R., Boone D.L., et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 57.Lun W., Wu X., Deng Q., Zhi F. MiR-218 regulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via targeting CTGF. Cancer Cell Int. 2018;18:83. doi: 10.1186/s12935-018-0575-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang W., Chancey A.L., Tzeng H.P., Zhou Z., Lavine K.J., Gao F., Sivasubramanian N., Barger P.M., Mann D.L. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation. 2011;124:2106–2116. doi: 10.1161/CIRCULATIONAHA.111.052399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verweij F.J., Bebelman M.P., Jimenez C.R., Garcia-Vallejo J.J., Janssen H., Neefjes J., Knol J.C., de Goeij-de Haas R., Piersma S.R., Baglio S.R., et al. Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J. Cell Biol. 2018;217:1129–1142. doi: 10.1083/jcb.201703206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hessvik N.P., Llorente A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018;75:193–208. doi: 10.1007/s00018-017-2595-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sung B.H., von Lersner A., Guerrero J., Krystofiak E.S., Inman D., Pelletier R., Zijlstra A., Ponik S.M., Weaver A.M. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat. Commun. 2020;11:2092. doi: 10.1038/s41467-020-15747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Betzer O., Barnoy E., Sadan T., Elbaz I., Braverman C., Liu Z., Popovtzer R. Advances in imaging strategies for in vivo tracking of exosomes. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020;12:e1594. doi: 10.1002/wnan.1594. [DOI] [PubMed] [Google Scholar]
- 63.González A., López B., Ravassa S., San José G., Díez J. The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim. Biophys. Acta. Mol. Cell Res. 2019;1866:1421–1432. doi: 10.1016/j.bbamcr.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 64.Souders C.A., Bowers S.L.K., Baudino T.A. Cardiac fibroblast: the renaissance cell. Circ. Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Travers J.G., Kamal F.A., Robbins J., Yutzey K.E., Blaxall B.C. Cardiac fibrosis: the fibroblast awakens. Circ. Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gibb A.A., Lazaropoulos M.P., Elrod J.W. Myofibroblasts and fibrosis: mitochondrial and metabolic control of cellular differentiation. Circ. Res. 2020;127:427–447. doi: 10.1161/CIRCRESAHA.120.316958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun N., Panetta N.J., Gupta D.M., Wilson K.D., Lee A., Jia F., Hu S., Cherry A.M., Robbins R.C., Longaker M.T., Wu J.C. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad. Sci. USA. 2009;106:15720–15725. doi: 10.1073/pnas.0908450106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burridge P.W., Matsa E., Shukla P., Lin Z.C., Churko J.M., Ebert A.D., Lan F., Diecke S., Huber B., Mordwinkin N.M., et al. Chemically defined generation of human cardiomyocytes. Nat. Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lobb R.J., Becker M., Wen S.W., Wong C.S.F., Wiegmans A.P., Leimgruber A., Möller A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles. 2015;4:27031. doi: 10.3402/jev.v4.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chomczynski P., Mackey K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 1995;19:942–945. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Analysis data have been deposited at Figshare and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.