

Abstract
The invasion and establishment of An. stephensi mosquitoes in the Horn of Africa represents a significant regional threat, which may jeopardise malaria control, particularly in urban areas which were formally free from disease transmission. Novel vector surveillance methods are urgently needed, both agnostic to mosquito larval morphology, and simple to implement at the sampling stage. Using new multiplex TaqMan assays, specifically targeting An. stephensi and Ae. aegypti, we validated the use of environmental DNA (eDNA) for simultaneous vector detection in shared artificial breeding sites. Study findings demonstrated that An. stephensi and Ae. aegypti eDNA deposited by as few as one second instar larva in 1L of water was detectable. Characterization of molecular insecticide resistance mechanisms, using novel amplicon-sequencing panels for both vector species, was possible from eDNA shed by as few as 16–32 s instar larvae in 50 ml of water. An. stephensi eDNA, derived from emergent pupae for 24 h, was remarkably stable, and still detectable ~ 2 weeks later. eDNA surveillance has the potential to be implemented in local endemic communities and at points of country entry, to monitor the spread of invasive vector species. Further studies are required to validate the feasibility of this technique under field conditions.
Subject terms: Genetics, Molecular biology, Diseases
Introduction
Vector-borne diseases (VBDs) account for 17% of all infectious diseases worldwide and are responsible for more than 700,000 deaths annually1. In sub-Saharan Africa, malaria, transmitted by Anopheles mosquitoes, remains the leading cause of morbidity and mortality (247 million new cases and 619,000 deaths in 2021); despite the widespread deployment of effective vector control measures, especially the use of insecticide-treated nets (ITNs) and indoor residual spraying (IRS), which have averted more than 1.5 billion cases and 7.6 million malaria-related deaths since 20002. Concurrently, approximately 51 million individuals are infected with lymphatic filariasis3, a severe cause of disability and stigma, and many arthropod-borne viruses (arboviruses) including, dengue, chikungunya, Rift Valley fever, yellow fever, Zika, o’nyong’nyong and West Nile, circulate unabated among humans, wildlife and livestock across the continent4,5. Over 27,000 cases of viruses transmitted by Aedes mosquitoes have been reported in West Africa since 2007, but seroprevalence surveys, indicative of prior disease exposure, suggest this is a gross underestimation of the actual arboviral disease burden6. While there is growing recognition that parts of Africa are at serious risk for arbovirus outbreaks, the understanding of the current distribution of these diseases is inadequate, as regional VBD surveillance is focused primarily on malaria, detection is usually reactive in response to outbreaks7, and robust evidence for efficacious control tools targeting arbovirus vectors is lacking8.
Anopheles stephensi is a highly competent malaria vector whose distribution until 2011 encompassed the Indian subcontinent, parts of South-East Asia and the Arabian Peninsula. Recently it has become an invasive vector species in the horn of Africa, recognised by the WHO as a “biological threat”9. An. stephensi was first reported in Djibouti in 201210 and since then this species has spread to neighbouring Ethiopia (2016)11,12, the Republic of Sudan (2019)13,14, Somalia (2019)15, Yemen (2021)15 and most recently Nigeria (2020)15. The invasion of An. stephensi in Djibouti was accompanied by an increase in malaria cases from 25 in 2012 to 14,810 in 201716, with outbreaks reported in Djibouti City, including historically malaria-free areas that were considered unsuitable for anopheline breeding sites10. Unlike An. arabiensis, the main malaria vector species in this region, An. stephensi often breeds in urban areas in man-made water containers, buckets, discarded tyres, and water storage tanks for domestic use and construction. These are sites that may not be under routine surveillance by the National Malaria Control Programme (NMCP); such larval habitats are often used by Aedes species, vectors responsible for dengue outbreaks and transmission of other arboviruses, including yellow fever, in Ethiopia17,18. An. stephensi and Ae. aegypti have already been observed in the same larval habitats in eastern Ethiopia19. Given the propensity of the former vector species to colonize urban environments quickly, breeding mainly in man-made water containers, mathematical models estimate over 126 million people in cities across Africa are at risk of malaria transmitted by An. stephensi20 and that annual Plasmodium falciparum cases could increase by 50% if no vector control interventions are implemented21.
As the spread of An. stephensi poses a significant problem to urban populations in Africa, the WHO recommends targeted surveillance to monitor existing populations, dispersal dynamics and to characterize their insecticide resistance profiles to inform prospective vector control strategies22. Moreover, dengue has been recognised as a significant public health threat in Ethiopia, where a need for improved vector surveillance and control has been recognised17. Novel vector surveillance methods are urgently required, which demand little prior knowledge of mosquito larval morphology and are quick and easy to implement at the sampling stage, with the ability to simultaneously detect the presence of multiple species of interest. Recent advances in molecular techniques are providing new opportunities for surveillance and prevention of VBDs, which can be used to better target vector control strategies. Detection of the traces of genetic material that organisms leave behind in their environment is one possible approach. Environmental DNA (eDNA) found in collected samples may allow us to indirectly confirm the presence of a species of interest and its abundance even when there are no obvious signs of the organism being present23,24. eDNA can be extracted from environmental samples, such as water from potential mosquito larval habitats, and can be detected using sensitive molecular techniques such as qPCR. eDNA decay and degradation are environmentally-dependent and due to its nature, detection of eDNA is an indicator of recent presence of the species of interest25. This approach can be especially useful for detection of invasive species, including mosquitoes24,26,27.
This study assessed the suitability of using eDNA for simultaneous detection of An. stephensi and Ae. aegypti from the same larval habitats and characterization of their molecular insecticide resistance mechanisms, under controlled laboratory conditions.
Results
PCR primer testing and validation
To enable simultaneous detection of An. stephensi and Ae. aegypti eDNA sampled from shared breeding sites, we designed a multiplex TaqMan assay based on single-nucleotide polymorphisms (SNPs) in COX1. Initially the analytical sensitivity, linearity, and dynamic range of the qPCR assay for eDNA detection was estimated using serial dilutions of An. stephensi and Ae. aegypti gDNA derived from colony strains, tested in technical triplicate. Both qPCR assays produced good linearity (R2 = 0.99 for both An. stephensi and Ae. aegypti assays) and efficiencies of 90.01% and 94.62%, respectively (Supplementary Fig. S1A,B). The LODs (limits of detection) and LOQs (limits of quantification) were determined to be 0.0000154 copies per reaction for individual detection of An. stephensi and Ae. aegypti. Next, the analytical sensitivity, linearity and dynamic range of the multiplex qPCR assay was evaluated using An. stephensi and Ae. aegypti gDNA serially diluted in equal proportions. Similarly, the multiplex assay was highly efficient (102.2% and 104.9% for An. stephensi and Ae. aegypti, respectively) with good linearities (R2 = 0.99 for simultaneous detection of An. stephensi and Ae. aegypti gDNA). The LOD/LOQs were determined to be 0.0000154 copies/reaction for simultaneous detection of An. stephensi and Ae. aegypti (Supplementary Fig. S1C).
The An. stephensi and Ae. aegypti probes were highly specific to gDNA from each species; no amplification was detected with gDNA from other sympatric or medically important vector species: An. arabiensis, An. gambiae s.s., An. funestus s.s. or Culex quinquefasciatus. All qPCR data are reported in Supplementary File S1.
Detection of An. stephensi and Ae. aegypti eDNA in artificial breeding sites
To investigate the sensitivity of An. stephensi and Ae. aegypti eDNA detection, artificial breeding sites of 50 ml or 1L of water were simulated with known numbers of second instar larvae (Fig. 1).
Figure 1.

Design of experiments to investigate the impact of different environmental conditions on An. stephensi and Ae. aegypti eDNA detection. Numbers of second instar larvae are denoted on each artificial breeding site. Experiment 1 investigated eDNA detection of different densities of An. stephensi and Ae. aegypti in 50 ml artificial breeding sites. Experiment 2 investigated eDNA detection of different densities of An. stephensi and Ae. aegypti in 1L artificial breeding sites, including different ratios of An. stephensi:Ae. aegypti co-habiting the same breeding site. Experiment 3 investigated the rate of eDNA degradation. An. stephensi pupae were left in each breeding site for 24 h, prior to manual removal of all emerged adults, remaining pupae, and pupal skins; eDNA detection was performed for the following 14 days. Figure created with BioRender.com.
In 50 ml artificial breeding sites (Fig. 1, Experiment 1), a dose response was evident for detection levels of both vector species; with increasing densities of larvae, cycle threshold (Ct) values began to decrease significantly (Fig. 2). The average Ct values for An. stephensi for each larval density were 1 larva: 31.89 [95% confidence interval (CI): 31.35–32.42]; 2 larvae: 27.36 [95% CI 24.72–29.99]; 4 larvae: 26.74 [95% CI 25.75–27.73]; 8 larvae: 25.42 [95% CI 23.77–27.07]; 16 larvae: 21.96 [95% CI 21.09–22.83]; 32 larvae: 21.01 [95% CI 20.09–21.92] (Fig. 2A). The average Ct values for Ae. aegypti for each larval density were 1 larva: 33.66 [95% CI 31.15–36.16]; 2 larvae: 29.11 [95% CI 27.19–31.03]; 4 larvae: 29.43 [95% CI 28.54–30.32]; 8 larvae: 27.85 [95% CI 26.0–29.70]; 16 larvae: 26.53 [95% CI 24.93–28.12]; 32 larvae: 25.31 [95% CI 23.32–27.30] (Fig. 2B).
Figure 2.

Taqman qPCR detection of An. stephensi (A) and Ae. aegypti (B) from 50 ml breeding sites. qPCR detection for all extractions were run in technical triplicate. Conditions sharing a superscript do not differ significantly (Dunn’s multiple comparisons test, p > 0.05). Error bars indicate 95% confidence intervals (CIs).
Similarly, a dose response was detectable for both vector species in 1L artificial breeding sites (Fig. 1, Experiment 2) with increasing larval densities; unsurprisingly, by comparison to the 50 ml breeding sites, Ct values were generally higher at each larval density (Fig. 3A,B). The average Ct values for An. stephensi for each larval density were 1 larva: 37.82 [95% CI 36.16–39.47]; 2 larvae: 34.90 [95% CI 33.27–36.53]; 4 larvae: 33.06 [95% CI 32.14–33.98]; 8 larvae: 25.32 [95% CI 23.44–27.21]; 16 larvae: 21.71 [95% CI 20.08–23.33]; 32 larvae: 21.86 [95% CI 20.95–22.78] (Fig. 3A). The average Ct values for Ae. aegypti for each larval density were 1 larva: 37.70 [95% CI 36.72–38.68]; 2 larvae: 34.91 [95% CI 33.18–36.65]; 4 larvae: 32.68 [95% CI 32.20–33.16]; 8 larvae: 30.57 [95% CI 30.26–30.89]; 16 larvae: 28.80 [95% CI 28.47–29.12]; 32 larvae: 28.54 [95% CI 27.58–29.50] (Fig. 3B).
Figure 3.

Taqman qPCR detection of An. stephensi (A), Ae. aegypti (B) or both species in equal (C) or 4:40 (D) proportions from 1L breeding sites. qPCR detection for all extractions were run in technical triplicate. Conditions sharing a superscript do not differ significantly (Dunn’s multiple comparisons test, p > 0.05). Error bars indicate 95% confidence intervals (CIs).
In 1L artificial breeding sites with mixed vector species in equal proportions (Fig. 1, Experiment 2), Ct values were generally lower than those detected in 1L breeding sites containing individual species at lower larval densities (Fig. 3C). The average Ct values for An. stephensi in mixed breeding sites at each larval density were: 2 larvae: 30.79 [95% CI 30.22–31.35]; 4 larvae: 30.65 [95% CI 30.44–30.86]; 8 larvae: 31.57 [95% CI 31.17–31.96]; 16 larvae: 31.84 [95% CI 29.53–34.15]; 32 larvae: 27.71 [95% CI 27.35–28.07] (Fig. 3C). The average Ct values for Ae. aegypti in mixed breeding sites at each larval density were: 2 larvae: 32.29 [95% CI 31.88–32.71]; 4 larvae: 29.01 [95% CI 28.71–29.31]; 8 larvae: 36.63 [95% CI 35.16–38.10]; 16 larvae: 28.18 [95% CI 27.78–28.58]; 32 larvae: 27.88 [95% CI 27.51–28.26] (Fig. 3C).
In 1L artificial breeding sites with mixed vector species at a 4:40 ratio (An. stephensi:Ae. aegypti), both species were still detectable, with higher average Ct values for the minority species (An. stephensi); the average Ct value for An. stephensi was 37.20 [95% CI 36.43–37.97] and for Ae. aegypti was 29.72 [95% CI 29.08–30.36] (Fig. 3D).
Rate of An. stephensi eDNA degradation in artificial breeding sites
A longitudinal cohort of 1L artificial breeding sites were monitored for 2 weeks, following the emergence of 32 An. stephensi pupae, to observe the rate of eDNA degradation. A negative dose response was detectable with increasing Ct values over time (Fig. 4).The average Ct values for An. stephensi eDNA at each time point were 24 h: 28.90 [95% CI 27.47–30.32]; 48 h: 28.31 [95% CI 27.99–28.63]; 72 h: 28.97 [95% CI 28.53–29.41]; 96 h: 29.72 [95% CI 29.45–29.99]; 120 h: 29.48 [95% CI 28.92–30.04]; 144 h: 31.19 [95% CI 30.49–31.88]; 168 h: 31.38 [95% CI 31.05–31.71]; 192 h: 31.56 [95% CI 31.17–31.94]; 216 h: 32.82 [95% CI 32.30–33.34]; 240 h: 32.57 [95% CI 31.93–33.20]; 264 h: 33.77 [95% CI 33.24–34.31]; 288 h: 36.96 [95% CI 36.43–37.50] (Fig. 4). An. stephensi eDNA was undetectable by qPCR after 312 h (13 days).
Figure 4.
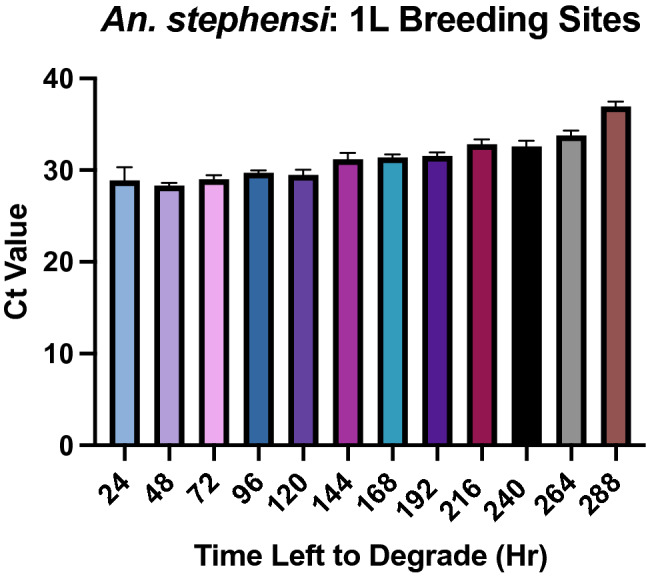
Rate of An. stephensi eDNA degradation over 12 days. qPCR detection for all extractions were run in technical triplicate. Error bars indicate 95% confidence intervals (CIs).
Detection of insecticide resistance genes from eDNA in artificial breeding sites
Characterisation of published insecticide resistance mechanisms in An. stephensi using amplicon-sequencing, was undertaken with eDNA extracted from 50 ml artificial breeding sites. Amplicons covering the voltage-gated sodium channel (vgsc), glutathione-s-transferase 2 (GSTe2) and acetylcholinesterase-1 (ace-I) genes, which contain genetic variants associated with insecticide resistance, were tested28.
Of the 6 eDNA samples tested (containing 1, 2, 4, 8, 16, and 32 larvae), no PCR amplicons were generated for either eDNA-L1 or -L2 samples. Average read depth per amplicon for eDNA-L4, -L8, -L16 and -L32 is presented in Table 1; eDNA-L32 was the only sample that produced all 7 amplicons when imaged on an agarose gel. The ACE-1_II amplicon for eDNA-L4 was the only incidence of a sample which produced an average coverage too low for SNP calling (> 50). Primer pairs for VGSC II, III and IV amplicons proved the least sensitive, with only eDNA-L32 yielding positive results (Table 1).
Table 1.
Coverage for An. stephensi eDNA-L4, -L8, -L16, and -L32 insecticide resistance amplicon targets.
| Chr | Start | End | Target Gene | Amplicon | L4 | L8 | L16 | L32 |
|---|---|---|---|---|---|---|---|---|
| 2 | 60,913,833 | 60,914,331 | ace-1 | ACE-1_I | 4.12 | 43.42 | 67.90 | 836.42 |
| 2 | 60,913,236 | 60,913,796 | ace-1 | ACE-1_II | 7021.93 | 3620.82 | 3163.21 | 2547.49 |
| 3 | 42,830,105 | 42,830,628 | vgsc | VGSCI | 58.99 | 253.52 | 12.91 | 1032.54 |
| 3 | 42,817,282 | 42,817,823 | vgsc | VGSCII | 2980.09 | 1117.36 | 1067.52 | 685.12 |
| 3 | 42,809,619 | 42,810,112 | vgsc | VGSCIII | 0.00 | 0.00 | 0.00 | 140.11 |
| 3 | 42,808,876 | 42,809,271 | vgsc | VGSCIV | 0.00 | 0.00 | 0.00 | 248.51 |
| 3 | 70,580,282 | 70,580,771 | GSTe2 | GSTe2 | 0.00 | 0.00 | 0.00 | 281.57 |
Chromosome number (chr), start and end of the target amplicon, the target gene, amplicon name and coverage are displayed.
A total of 40 variants were identified, accounting for 33 single nucleotide polymorphisms (SNPs) and 7 insertions or deletions (indels). Only one missense mutation was identified in the Ace-1 gene in amino acid 177; this SNP was present in the eDNA-L8 and eDNA-L16 samples, with eDNA-L8 genotyped as heterozygous and eDNA-L16 as the homozygous alternate. This SNP has been previously reported in both colony and wild-caught samples28. No SNPs associated with insecticide resistance were identified.
Similarly, eDNA from Ae. aegypti 50 ml artificial breeding sites containing 4 and 8 larvae did not amplify using amplicon-seq primers, targeting the vgsc and ace-1 genes29. PCR reactions using eDNA from Ae. aegypti artificial breeding sites containing 16 larvae (eDNA-L16) were able to amplify 5 targets: ace-1 and four amplicons for the vgsc (DomainIVS6, DomainIV, Domain IIIExon36 and DomainIIIExon35; Table 2).
Table 2.
Coverage for Ae. aegypti eDNA-L16 insecticide resistance amplicon targets.
| Chr | Start | End | Target Gene | Amplicon | L16 |
|---|---|---|---|---|---|
| 3 | 161,500,008.00 | 161,500,475.00 | ace1 | Ace1 | 24,756.60 |
| 3 | 315,931,391.00 | 315,931,846.00 | vgsc | DomainIVS6 | 32,260.40 |
| 3 | 315,931,739.00 | 315,932,233.00 | vgsc | DomainIV | 6567.40 |
| 3 | 315,938,703.00 | 315,939,191.00 | vgsc | DomainIIIExon36 | 1391.40 |
| 3 | 315,939,789.00 | 315,939,789.00 | vgsc | DomainIIIExon33_34 | 0.00 |
| 3 | 315,983,627.00 | 315,984,124.00 | vgsc | DomainII | 0.00 |
| 3 | 315,983,774.00 | 315,984,267.00 | vgsc | DomainIIExon26 | 0.00 |
| 3 | 315,998,366.00 | 315,998,809.00 | vgsc | DomainIIS4 | 0.00 |
| 3 | 316,080,615.00 | 316,081,078.00 | vgsc | DomainI | 6.30 |
| 3 | 315,938,919.00 | 315,939,398.00 | vgsc | DomainIIIExon35 | 790.40 |
Chromosome number (chr) start and end of the target amplicon, the target gene, amplicon name and coverage are displayed.
Analysis of the DNA sequences obtained for the eDNA-L16 sample did not show any mutations. The sample coverage for each target amplicon is displayed in Table 2 and showed very good coverage ranging from 790.4 to 32,260.40-fold. Five targets, however, did not amplify or showed coverage below the cut-off of 50 (DomainIIIExon33_34, DomainII, DomainIIExon26, DomainIIS4 and DomainI). Differences in gene amplification may reflect differences in relative primer efficiency.
Discussion
The invasion and establishment of An. stephensi in the Horn of Africa represents an imminent and significant regional threat, which may jeopardise malaria control, particularly in urban areas which were formally free from disease transmission20,21. To develop novel methods of vector surveillance, this study evaluated the feasibility of using eDNA for simultaneous detection of An. stephensi and Ae. aegypti, which have both been observed co-habiting in natural breeding sites in Ethiopia19. Study findings demonstrated that An. stephensi and Ae. aegypti eDNA deposited by as few as one second instar larvae in 1L of water was detectable by qPCR. In general, dose responses were evident for detection levels of both vector species, with increasing densities of larvae, resulting in significantly lower Ct values, with some degree of stochastic variation observed. This aligns with previous laboratory studies which demonstrated similar levels of detection of An. gambiae s.s. in artificial breeding sites26. Furthermore, the multiplex TaqMan assay developed in this study displayed comparable levels of detection between both vector species, when used to amplify eDNA from mixed breeding sites, in a 50:50 or 1:10 An. stephensi:Ae. aegypti ratio in 1L of water.
Anopheles breeding sites can be highly heterogenous in both their ecology and temporality, which will directly affect the sensitivity of eDNA detection. In rural environments, breeding sites commonly comprise rice paddies, ponds, puddles, village pumps and associated troughs, cesspools, water basins, agricultural trenches, dams, edges of rivers/streams, in animal/human footprints and irrigation canals and drains30–32. In urban landscapes any natural or man-made feature that collects any form of stagnant water is a potential breeding site, particularly in and around deteriorating infrastructure, such as broken water pipes, blocked drains, construction sites, lorry tyre tracks and catch pits33. An. gambiae s.l. breeding sites are frequently freshwater habitats that are small, temporary, clean and sun-exposed, although some studies suggest that these larvae can survive in dirty and polluted environments34,35; while An. funestus s.l. tend to breed in larger semi-permanent water bodies containing aquatic vegetation and algae36. In Ethiopia, An. stephensi has been found in both rural and urban areas, breeding in Aedes-type sites (e.g. artificial water containers, buckets and tanks)19. With such variation in breeding site environments, it is anticipated that the rate of eDNA persistence will differ quite considerably, with respect to water turbidity, velocity, UV exposure, salinity/pH, pollution, and co-occupancy (e.g. with other insects, mosquito species, fish or tadpoles). In our study, An. stephensi eDNA, derived from emergent pupae for 24 h, was remarkably stable, and still detectable almost two weeks later. Similarly, eDNA from 15 Ae. albopictus larvae, reared for 12 days in 500 ml water, was demonstrable up to 25 days after live material was removed27. Our study may have in fact underestimated the sensitivity of eDNA detection, by only allowing larvae to remain in artificial breeding sites for 24 h, as well as the rate of eDNA degradation, due to highly controlled insectary conditions (i.e. constant temperature, humidity and light:dark cycles).
In addition to vector species identification, this study investigated the potential to exploit eDNA for molecular insecticide resistance surveillance. Using recently developed amplicon-seq panels for An. stephensi28 and Ae. aegypti29, fragments of key insecticide resistance genes (including portions of the voltage-gated sodium channel and acetylcholinesterase) were able to be amplified from eDNA shed by as few as 16–32 s instar larvae in 50 ml of water; as expected, no insecticide resistance mutations were identified in our insecticide-susceptible insectary colonies. The detection sensitivity of insecticide resistance genes in natural breeding sites may be greater than in our laboratory study, particularly as larval densities can be higher and vector populations will deposit eDNA throughout their ~ 2 week lifecycle in breeding sites. Conventional insecticide resistance monitoring is based on larval dipping from known, productive breeding sites, rearing of vectors to the adult stage, followed by phenotypic characterization in bioassays and/or genotypic analysis using various molecular techniques37. eDNA detection offers a new avenue to explore molecular insecticide resistance at the community-level, circumventing some of the laborious sampling requirements and potentially allow wider-scale surveillance of water bodies which may be less productive sites or entirely unknown to local entomologists. This strategy could be combined with additional analytical techniques, such as high-performance liquid chromatography, to survey the extent of breeding site contamination with agricultural pesticides, a known driver of insecticide resistance selection in mosquito vector populations38,39.
Water sampling for mosquito eDNA detection represents a cheap, low-technology tool, which requires virtually no entomological skills or training, and thus has the potential to be easily integrated into citizen science initiatives. Furthermore, with regards to An. stephensi specifically, eDNA detection could be considered for surveillance of seaports in countries at greatest risk of introduction of this invasive species40. Following laboratory validation in this study, further work is required to assess the feasibility of sampling eDNA of An. stephensi/Ae. aegypti in situ, including an assessment of environmental risk factors which impact eDNA detection levels and comparison of eDNA quantification versus larval density per breeding site.
Conclusions
This study validated the use of eDNA for simultaneous detection of An. stephensi and Ae. aegypti in shared artificial breeding sites. Study findings demonstrated that An. stephensi and Ae. aegypti eDNA deposited by as few as 1 s instar larva in 1 L of water was detectable. Characterization of molecular insecticide resistance mechanisms, using novel amplicon-seq panels for both vector species, was possible from eDNA derived from as few as 16–32 s instar larvae in 50 ml of water. eDNA surveillance has the potential to be implemented in local endemic communities as part of citizen science initiatives, and/or in cargo ports at points of country entry, to monitor the spread of invasive mosquito vector species. Further studies are required to validate the feasibility of this technique under field conditions.
Materials and methods
Laboratory-reared second instar An. stephensi (SD500 strain) and Ae. aegypti (AEAE strain) larvae were used in all experiments. An. stephensi SD500 was colonised from Pakistan in 1982 and is insecticide susceptible41. Ae. aegypti AEAE was colonised from West Africa before 1926 and is insecticide susceptible (Aiyenuro, personal communication). Larvae were reared in sterile, distilled water, in plastic bowls (20 × 18 × 7 cm) under controlled insectary conditions (26–28 °C, relative humidity 70–80% and 12:12 h light:dark cycles) and fed once daily with NISHIKOI staple fish food pellets (Nishikoi, UK).
Experiment 1: 50 ml artificial breeding sites
In the first experiment, 21 different conditions were tested by adding 50 ml of sterile, distilled, water to 21 sterile 50 ml falcon tubes (Fisher Scientific, UK). We performed three biological replicates with 6 different larval densities: 1, 2, 4, 8, 16 and 32 of either An. stephensi or Ae. aegypti larvae (Fig. 1). Three negative control experimental habitats with no larvae were run in parallel for each condition. Larvae were left in each 50 ml breeding site for 24 h, prior to manual removal, using a sterile pipette.
Experiment 2: 1L artificial breeding sites
In the second experiment, 21 different conditions were tested by adding 1L of sterile, distilled water to 21 sterile plastic rectangular containers. We performed three biological replicates with 6 different larval densities: 1, 2, 4, 8, 16 and 32 larvae. Three negative control experimental habitats with no larvae were run in parallel for each condition. Larvae were left in each 1L breeding site for 24 h, prior to manual removal, using a sterile pipette. An. stephensi and Ae. aegypti were evaluated initially in separate 1L breeding sites and then in mixed breeding sites. The mixed breeding site experiments used 24 different conditions, comprising three biological replicates with 5 larval densities: 2, 4, 8, 16 and 32 larvae of An. stephensi and Ae. aegypti in equal proportions (50:50), three biological replicates with larvae of the two species in different proportions: 4:40 An. stephensi:Ae. aegypti larvae and three negative control breeding sites.
Experiment 3: eDNA degradation in artificial breeding sites
In the third experiment, the rate of eDNA degradation in artificial breeding sites was assessed. One litre of sterile, distilled water was added to 9 sterile plastic rectangular containers. We performed three biological replicates with 32 An. stephensi pupae added to each container. Three negative control experimental habitats with no pupae were run in parallel for each condition. Pupae were left in each 1L breeding site for 24 h, prior to manual removal of all emerged adults, remaining pupae, and pupal skins, using a sterile pipette. Breeding sites were then sampled daily for 14 days, following the removal of adults and pupae.
eDNA extraction
eDNA from water samples were concentrated prior to extraction. For each breeding site replicate, 15 ml of water was added to a sterile 50 ml falcon tube (Fisher Scientific, UK) and 1.5 ml of 3 M sodium acetate solution (pH 5.2) (Sigma-Aldrich, UK) was immediately added, followed by 33 ml of absolute ethanol and stored overnight at − 20°C. Samples were then centrifuged at 8000 rpm at 6 °C for 30 min. The supernatant was discarded, and the pellet was washed in 20 ml of absolute ethanol by centrifuging at 8000 rpm at 6 °C for 10 min. The supernatant was discarded, and ethanol allowed to evaporate at 65°C. The pellet was dissolved in 720 μl ATL buffer and 80 μl proteinase K (Qiagen, UK) and incubated overnight at 56 °C. eDNA was extracted using a Qiagen DNeasy 96 Blood and Tissue kit (Qiagen, UK), according to the manufacturer’s protocol, with minor modifications.
An. stephensi and Ae. aegypti eDNA PCR primer design and validation
We designed a multiplex TaqMan assay to distinguish between An. stephensi (ASTE016222) and Ae. aegypti (AAEL018662) eDNA based on a fragment of cytochrome oxidase I (COX1). Forward (5ʹ-CAGGAATTACWTTAGACCGAMTACC-3ʹ) and reverse (5ʹ-TCAAAATAARTGTTGRTATAAAATRGGGTC-3ʹ) primers and two TaqMan fluorescent-labelled probes (An. stephensi probe 5ʹ-/5HEX/ATTACTATA/ZEN/TTACTTACAGACCG/3IABkFQ/-3ʹ and Ae. aegypti probe 5ʹ-/56FAM/ATTACTATG/ZEN/TTATTAACAGACCG/3IABkFQ/-3ʹ) were designed using Geneious Prime® 2021.1.1 to amplify a 202 bp region of COXI (Fig. 5). Primers and probes were selected by reference to all available COXI sequences from NCBI GenBank (n = 4402) for Ae. aegypti, An. arabiensis, An. gambiae s.s., An. pharoensis, An. quadriannulatus, An. funestus, An. stephensi and Cx. quinquefasciatus, which were used to confirm the presence/absence of species-specific SNPs and to account for additional intra-species genetic diversity.
Figure 5.

Consensus alignment of reference COXI sequences used for primer and probe design. Positions of forward and reverse primers are highlighted by light blue asterisks. Species-specific probes are highlighted in teal, with single-nucleotide polymorphisms in colour.
Standard curves of Ct values for each probe were generated individually and then multiplexed using a ten-fold serial dilution of control An. stephensi or Ae. aegypti DNA (single extractions from SD500 and AEAE laboratory colonies, respectively) to assess PCR efficiencies. Genomic DNA concentrations were determined using the Qubit 4 fluorometer 1X dsDNA HS assay (Invitrogen, UK). Standard curve reactions were performed in a final volume of 10 μl containing 2X PrimeTime® Gene Expression Master Mix (IDT, USA), 250 nM of forward and reverse primers and 150 nM of each probe and 2 μl genomic DNA. Reactions were run on a Stratagene Mx3005P Real-Time PCR system (Agilent Technologies) at 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. All assays were run in technical triplicate alongside PCR no-template controls (NTCs).
To confirm primer and probe specificity, the multiplex assay was used to assess amplification of stock genomic DNA from other medically important sympatric vector species: An. arabiensis, An. gambiae s.s., An. funestus and Cx. quinquefasciatus.
Detection of An. stephensi and Ae. aegypti eDNA from artificial breeding sites
Following eDNA extraction, eDNA detection was performed in a final volume of 10 μl containing 2X PrimeTime® Gene Expression Master Mix (IDT, USA), 250 nM of forward and reverse primers, 150 nM of each probe and 4.2 μl eDNA. Reactions were run on a Stratagene Mx3005P Real-Time PCR system (Agilent Technologies) at 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. All assays were run in technical triplicate alongside PCR no-template controls (NTCs).
qPCR data analysis
Stratagene MxPro qPCR software (Agilent Technologies, UK) was used to produce qPCR standard curves. qPCR assay limits of detection (LOD) were determined using the “Generic qPCR LoD/LoQ calculator”42, implemented in R version 4.0.243. All other statistical analyses were conducted in GraphPad Prism v9.4.0.
Amplicon primer design and PCR amplification
To characterize molecular mechanisms of insecticide resistance in An. stephensi, briefly, three genes of interest were identified, and a total of 7 amplicon primer pairs were designed to target SNPs, previously associated with insecticide resistance28. To enable sample multiplexing, each primer had a 6 bp barcode attached to the 5ʹ end to allow for sample identification in downstream analysis. Amplicons were generated using the eDNA extracted from experiment #1 (1, 2, 4, 8, 16, and 32 larvae in 50 ml artificial breeding sites). PCR reactions contained 5X Q5 reaction buffer (New England Biolabs, UK), Q5® High-Fidelity DNA polymerase (New England Biolabs, UK), 250 nM of forward and reverse primers and 2 µl of eDNA. PCR reaction conditions were an initial 30 s denaturation at 95 °C, followed by 35 cycles of 98 °C for 10 s, 60 °C for 30 s and 72 °C for 30 s, and a final elongation step at 72 °C for 2 min. PCR products were visualized on SYBR Safe 1% agarose gels (Invitrogen, UK), prior to purification using Agencourt AMPure XP magnetic beads (Beckman Coulter, UK), using a ratio of 0.7:1 (µl of beads to DNA).
To characterize molecular mechanisms of insecticide resistance in Ae. aegypti, briefly, sequences for portions of the voltage-gated sodium channel (vgsc; AAEL023266-RL) and acetylcholinesterase-1 (Ace-1; AAEL000511-RJ) were extracted from publicly available assemblies for Ae. aegypti (LVP AGWG)29. Forward and reverse primers were designed using PrimerBLAST software to amplify regions of 450–500 bp that contain known SNP loci or regions of interest, with 6 bp inline barcodes and partial Illumina tails that allow the samples to be sequenced on an Illumina sequencing platform. PCR reactions contained 5X Q5 reaction buffer (New England Biolabs, UK), Q5® High-Fidelity DNA polymerase (New England Biolabs, UK), 250 nM of forward and reverse primers and 2 µl of eDNA extracted from experiment #1 (1, 2, 4, 8 and 16 larvae in 50 ml artificial breeding sites). PCR reaction conditions were 98 °C for 30 s, followed by 35 cycles of 98 °C for 10 s, 57 °C for 60 s and 72 °C for 90 s. PCR products were visualized on SYBR Safe 1% agarose gels (Invitrogen, UK), prior to purification using Agencourt AMPure XP magnetic beads (Beckman Coulter, UK), using a ratio of 0.7:1 (µl of beads to DNA).
Amplicon sequencing
The concentration of purified PCR products was measured using the Qubit 4 fluorometer 1X dsDNA HS assay (Invitrogen, UK) and samples with unique barcode combinations were pooled in equal concentrations to create an overall pool of 20 ng/µl in 25 µl total volume. A second PCR to insert Illumina adaptors (no further library preparation required) and amplicon sequencing was performed at Genewiz (Illumina-based Amplicon-EZ service).
Amplicon sequencing analysis
Samples were demultiplexed based on the in-line barcodes in each forward and reverse primer using an in-house python script29. Sequences were trimmed, aligned to reference (Ae. aegypti LVP AGWG for Ae. aegypti or UCI_ANSTEP_V1 assembly for An. stephensi) and data were quality checked using FastQC and Samclip. Paired-end reads were mapped against the reference sequences using the BWA-MEM algorithm. Variants were then called using three packages: Freebayes, GATK, and an in-house naive variant caller. Identified variants were then compared and normalised against each other to reduce false negatives and positives, and then filtered and annotated using BCFtools; for An. stephensi, variants were annotated using snpEFF, based on a manually built database for the UCI An. stephensi genome assembly. SNPs were then manually filtered to ensure that they appeared in more than one sample, with an allele depth of > 50.
Supplementary Information
Acknowledgements
This project was funded by UK aid from the UK government [Health Research Programme Consortia (RPCs): RAFT (Resilience against Future Threats through Vector Control), PO8615]; however, the views expressed do not necessarily reflect the UK government’s official policies. Study funding was also provided by a Wellcome Trust/Royal Society Sir Henry Dale Fellowship awarded to TW (101285/Z/13/Z): https://wellcome.org and https://royalsociety.org.
Author contributions
M.K., S.C., and L.A.M. designed the study. M.K. led the entomological experiments with support from N.M.P. and B.P. L.A.M. led the qPCR analysis. H.A.P., M.O.C., E.C. and S.C. performed the amplicon sequencing. T.W. provided laboratory resources. J.P. and T.G.C. performed the bioinformatics analysis. S.C. and J.L. provided project oversight and funding. M.K., H.A.P., M.O.C. and L.A.M. drafted the manuscript, which was revised by co-authors. All authors read and approved the final manuscript.
Data availability
The qPCR datasets generated during this study are contained within Supplementary File S1. Sequence data generated by this study will be available at Sequence Read Archive (SRA) BioProject PRJEB56471 (accession numbers: ERS13525860-ERS13525863).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mojca Kristan, Email: mojca.kristan@lshtm.ac.uk.
Louisa A. Messenger, Email: louisa.messenger@lshtm.ac.uk
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-29657-y.
References
- 1.Global vector control response 2017–2030. Global vector control response51 (2017).
- 2.Organization WH. World Malaria Report 2021. World Health Organization; 2022. [Google Scholar]
- 3.Collaborators, L. B. o. D. N. T. D. The global distribution of lymphatic filariasis, 2000–18: A geospatial analysis. Lancet Glob. Health8, e1186-e1194 (2020). [DOI] [PMC free article] [PubMed]
- 4.Mordecai EA, Ryan SJ, Caldwell JM, Shah MM, LaBeaud AD. Climate change could shift disease burden from malaria to arboviruses in Africa. Lancet Planet. Health. 2020;4:e416–e423. doi: 10.1016/S2542-5196(20)30178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simo FBN, et al. Dengue virus infection in people residing in Africa: A systematic review and meta-analysis of prevalence studies. Sci. Rep. 2019;9:13626. doi: 10.1038/s41598-019-50135-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchwald AG, Hayden MH, Dadzie SK, Paull SH, Carlton EJ. Aedes-borne disease outbreaks in West Africa: A call for enhanced surveillance. Acta Trop. 2020;209:105468. doi: 10.1016/j.actatropica.2020.105468. [DOI] [PubMed] [Google Scholar]
- 7.Ushijima Y, et al. Surveillance of the major pathogenic arboviruses of public health concern in Gabon, Central Africa: increased risk of West Nile virus and dengue virus infections. BMC Infect. Dis. 2021;21:265. doi: 10.1186/s12879-021-05960-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowman L, McCall PJ. Evaluating the evidence for effectiveness of vector control of dengue outbreaks by systematic review and meta-analysis. Am. J. Trop. Med. Hyg. 2014;91:231. [Google Scholar]
- 9.Programme, W. H. O. G. M. Vector alert: Anopheles stephensi invasion and spread. (Geneva, 2019).
- 10.Faulde MK, Rueda LM, Khaireh BA. First record of the Asian malaria vector Anopheles stephensi and its possible role in the resurgence of malaria in Djibouti. Horn of Africa. Acta Tropica. 2014;139:39–43. doi: 10.1016/j.actatropica.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Carter TE, et al. First detection of Anopheles stephensi Liston, 1901 (Diptera: culicidae) in Ethiopia using molecular and morphological approaches. Acta Trop. 2018;188:180–186. doi: 10.1016/j.actatropica.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Tadesse FG, et al. Anopheles stephensi Mosquitoes as Vectors of Plasmodium vivax and falciparum, Horn of Africa, 2019. Emerg. Infect. Dis. 2021;27:603–607. doi: 10.3201/eid2702.200019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed A, et al. Invasive Malaria Vector Anopheles stephensi Mosquitoes in Sudan, 2016–2018. Emerg. Infect. Dis. 2021;27:2952–2954. doi: 10.3201/eid2711.210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed A, Khogali R, Elnour MB, Nakao R, Salim B. Emergence of the invasive malaria vector Anopheles stephensi in Khartoum State Central Sudan. Parasites Vectors. 2021;14:511. doi: 10.1186/s13071-021-05026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Organization, W. H. WHO malaria threats map, https://apps.who.int/malaria/maps/threats/.
- 16.Seyfarth M, Khaireh BA, Abdi AA, Bouh SM, Faulde MK. Five years following first detection of Anopheles stephensi (Diptera: Culicidae) in Djibouti, Horn of Africa: Populations established-malaria emerging. Parasitol. Res. 2019;118:725–732. doi: 10.1007/s00436-019-06213-0. [DOI] [PubMed] [Google Scholar]
- 17.Gutu MA, et al. Another dengue fever outbreak in Eastern Ethiopia—An emerging public health threat. PLoS Negl. Trop. Dis. 2021;15:e0008992. doi: 10.1371/journal.pntd.0008992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulchandani R, et al. A community-level investigation following a yellow fever virus outbreak in South Omo Zone South-West Ethiopia. PeerJ. 2019;7:e6466. doi: 10.7717/peerj.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balkew M, et al. Geographical distribution of Anopheles stephensi in eastern Ethiopia. Parasit. Vectors. 2020;13:35. doi: 10.1186/s13071-020-3904-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinka ME, et al. A new malaria vector in Africa: predicting the expansion range of Anopheles stephensi and identifying the urban populations at risk. Proc. Natl. Acad. Sci. U.S.A. 2020;117:24900–24908. doi: 10.1073/pnas.2003976117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamlet A, et al. The potential impact of Anopheles stephensi establishment on the transmission of Plasmodium falciparum in Ethiopia and prospective control measures. BMC Med. 2022;20:135. doi: 10.1186/s12916-022-02324-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Organization, W. H. Vector alert: Anopheles stephensi invasion and spread (World Health Organization, 2019).
- 23.Thomsen PF, Willerslev E. Environmental DNA—An emerging tool in conservation for monitoring past and present biodiversity. Biol. Cons. 2015;183:4–18. doi: 10.1016/j.biocon.2014.11.019. [DOI] [Google Scholar]
- 24.Ficetola GF, Miaud C, Pompanon F, Taberlet P. Species detection using environmental DNA from water samples. Biol. Let. 2008;4:423–425. doi: 10.1098/rsbl.2008.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison JB, Sunday JM, Rogers SM. Predicting the fate of eDNA in the environment and implications for studying biodiveristy. Proc. R. Soc. B. 2019;286:20191409. doi: 10.1098/rspb.2019.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odero J, Gomes B, Fillinger U, Weetman D. Detection and quantification of Anopheles gambiae sensu lato mosquito larvae in experimental aquatic habitats using environmental DNA (eDNA) Wellcome Open Res. 2018;3:26. doi: 10.12688/wellcomeopenres.14193.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider J, et al. Detection of invasive mosquito vectors using environmental DNA (eDNA) from water samples. PLoS ONE. 2016;11:e0162493. doi: 10.1371/journal.pone.0162493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acford-Palmer, H. et al. Identification of two insecticide resistance markers in Ethiopian Anopheles stephensi mosquitoes using a multiplex amplicon sequencing assay targeting vgsc, rdl, gste2 and ace1 loci. Res. Square (2023). 10.21203/rs.3.rs-2416521/v1. [DOI] [PMC free article] [PubMed]
- 29.Collins EL, et al. A next generation targeted amplicon sequencing method to screen for insecticide resistance mutations in Aedes aegypti populations reveals a rdl mutation in mosquitoes from Cabo Verde. PLoS Negl. Trop. Dis. 2022;16:e0010935. doi: 10.1371/journal.pntd.0010935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zogo, B. et al. Identification and characterization of Anopheles spp. breeding habitats in the Korhogo area in northern Côte d'Ivoire: A study prior to a Bti-based larviciding intervention. Parasites Vectors12, 146 (2019). [DOI] [PMC free article] [PubMed]
- 31.Hamza AM, Rayah El, el, A. A qualitative evidence of the breeding sites of Anopheles arabiensis Patton (Diptera: Culicidae) in and Around Kassala Town, Eastern Sudan. Int. J. Insect Sci. 2016;8:65–70. doi: 10.4137/IJIS.S40071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ndiaye A, et al. Mapping the breeding sites of Anopheles gambiae s.l. in areas of residual malaria transmission in central western Senegal. PLoS ONE. 2020;15:e0236607. doi: 10.1371/journal.pone.0236607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattah PA, et al. Diversity in breeding sites and distribution of Anopheles mosquitoes in selected urban areas of southern Ghana. Parasit. Vectors. 2017;10:25. doi: 10.1186/s13071-016-1941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fillinger U, Sonye G, Killeen GF, Knols BG, Becker N. The practical importance of permanent and semipermanent habitats for controlling aquatic stages of Anopheles gambiae sensu lato mosquitoes: Operational observations from a rural town in western Kenya. Tropical Med. Int. Health. 2004;12:1274–1289. doi: 10.1111/j.1365-3156.2004.01335.x. [DOI] [PubMed] [Google Scholar]
- 35.Walker K, Lynch M. Contributions of Anopheles larval control to malaria suppression in tropical Africa: Review of achievements and potential. Med. Vet. Entomol. 2007;21:2–21. doi: 10.1111/j.1365-2915.2007.00674.x. [DOI] [PubMed] [Google Scholar]
- 36.Nambunga IH, et al. Aquatic habitats of the malaria vector Anopheles funestus in rural south-eastern Tanzania. Malar. J. 2020;19:219. doi: 10.1186/s12936-020-03295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Organization, W. H. Manual for monitoring insecticide resistance in mosquito vectors and selecting appropriate interventions (Geneva, 2022).
- 38.Nkya TE, et al. Impact of agriculture on the selection of insecticide resistance in the malaria vector Anopheles gambiae: A multigenerational study in controlled conditions. Parasit. Vectors. 2014;7:480. doi: 10.1186/s13071-014-0480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hien AS, et al. Evidence that agricultural use of pesticides selects pyrethroid resistance within Anopheles gambiae s.l. populations from cotton growing areas in Burkina Faso West Africa. PLoS ONE. 2017;12:e0173098. doi: 10.1371/journal.pone.0173098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahn, J. et al. Modeling marine cargo traffic to identify countries in Africa with greatest risk of invasion by Anopheles stephensi. Sci. Rep.13, 876 (2023). 10.1038/s41598-023-27439-0. [DOI] [PMC free article] [PubMed]
- 41.Feldmann AM, Ponnudurai T. Selection of Anopheles stephensi for refractoriness and susceptibility to Plasmodium falciparum. Med. Vet. Entomol. 1989;3:41–52. doi: 10.1111/j.1365-2915.1989.tb00473.x. [DOI] [PubMed] [Google Scholar]
- 42.Merkes C, et al. Reporting the limits of detection (LOD) and quantification (LOQ) for environmental DNA assays. Environ. DNA. 2020;2:271–282. doi: 10.5066/P9AKHU1R. [DOI] [Google Scholar]
- 43.RStudio: Integrated Development for R. (Bostom, MA, 2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The qPCR datasets generated during this study are contained within Supplementary File S1. Sequence data generated by this study will be available at Sequence Read Archive (SRA) BioProject PRJEB56471 (accession numbers: ERS13525860-ERS13525863).