

Abstract
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract characterized in many patients by extraintestinal manifestations. One of the most common comorbidities seen in IBD patients is a significant reduction in their bone mass. The pathogenesis of IBD is mainly attributed to the disrupted immune responses in the gastrointestinal mucosa and putative disruptions in the gut microbiomes. The excessive inflammation of the gastrointestinal tract activates different systems, such as the RANKL/RANK/OPG and the Wnt pathways linked with bone alterations in IBD patients, thereby suggesting a multifactorial etiology. The mechanism responsible for the reduced bone mineral density in IBD patients is thought to be multifactorial, and, so far, the principal pathophysiological pathway has not been well established. However, in recent years, many investigations have increased our understanding of the effect of gut inflammation on the systemic immune response and bone metabolism. Here, we review the main signaling pathways associated with altered bone metabolism in IBD.
Keywords: Inflammatory bowel disease, bone loss, RANKL/RANK/OPG pathway, Wnt pathways
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract. It encompasses 2 conditions, Crohn’s disease (CD) and ulcerative colitis (UC), that can deteriorate the quality of life and lead to life-threatening complications [1]. Recent epidemiological data highlight the differences in the epidemiology of IBD. Although it was considered a disease of the West, accumulating data from the East reveals that the incidence of IBD has risen [2]. IBD has been associated with osteoporosis and bone loss and IBD patients usually display low bone mineral density (BMD) [3]. Although our understanding of the exact mechanisms associated with bone loss remains unclear [4], it has been established that excessive inflammation in IBD reduces BMD and progressively increases the risk for osteopenia and osteoporosis [3]. More specifically, osteopenia was found in 22-55% of CD patients and 32-67% of UC patients, and osteoporosis in 3-6% and 4-50%, respectively [5]. Studies have shown that the risk of a spontaneous fracture in IBD patients is 40% higher than in the general population [6]. Various risk factors have been associated with the reduction in BMD of IBD patients, including age >65 years [7,8], menopause, smoking, poor diet, lack of physical activity, glucocorticoid use, as well as vitamin D deficiency [9]. Vitamin D deficiency can cause insufficient calcium absorption and secondary hyperparathyroidism, a condition that stimulates osteoclastic activity and eventually results in a substantial increase in osteoporosis [10].
Bone homeostasis may also be affected by the gut microbiome, known to play an essential role in the regulation of the host’s health and physiology. Research data indicate that gut microbiota influence the immune and endocrine systems and the gut-brain axis [11], and may be associated with the pathogenesis of osteoporosis by inducing the production of circulating cytokines, such as interleukin (IL)-17, tumor necrosis factor (TNF)-α, and receptor activator of nuclear factor (NF)-κΒ ligand (RANKL) [12]. In IBD patients in particular, a reduction in microbiome diversity has been reported to involve an increase in the abundance of Bacteroidetes and Proteobacteria and a decrease in Firmicutes compared to control individuals. How dysbiosis affects the multiple biological systems involved in IBD development, including bone homeostasis, has not been sufficiently determined [13].
In addition, the signaling networks involved in bone loss in IBD patients are under investigation. Therefore, this review aims to provide an overview of the interaction between IBD and metabolic bone alterations through the regulation of the osteoprotegerin receptor (OPG)/RANKL ratio and the proteins dickkopf-1 (DKK-1), sclerostin (SOST), and periostin (POSTN), which regulate the Wnt signaling pathway in osteocytes.
Inflammation and immune response
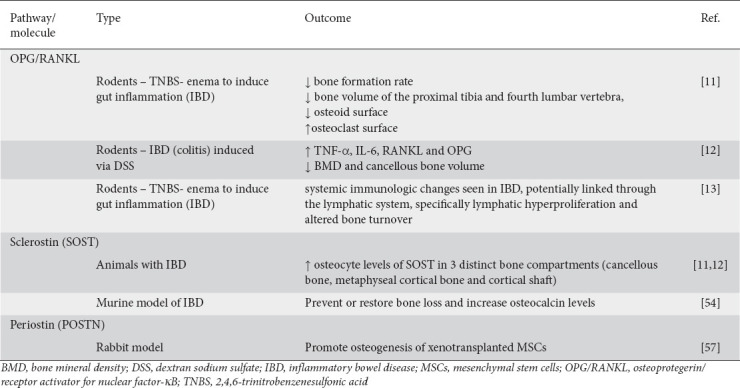
Researchers have not yet succeeded in completely decoding the main mechanisms responsible for the reduced BMD in IBD patients. Most data on the potential mechanistic link between IBD and how it affects bone metabolism are derived from rodent studies. In 2016 a study examined the influence of IBD on osteocyte proteins using 2,4,6-trinitrobenzenesulfonic acid (TNBS) enema to induce gut inflammation. Four weeks later immunohistochemical analysis of the distal femur revealed that %TNF-α+, %IL-6+, %RANKL+, and %OPG+ osteocytes were increased in the cancellous bone of the experimental group. These observations were also accompanied by a lower bone formation rate and a lower bone volume of the proximal tibia and fourth lumbar vertebra, a lower osteoid surface and a higher osteoclast surface. The inflammatory state of the experimental model potentially affected bone resorption and bone formation by modifying the regulatory protein profile in the osteocytes, thus resulting in IBD-induced bone loss [14]. In 2019 the same authors examined the therapeutic potential of irisin as an anti-inflammatory treatment in a mild IBD rodent model. IBD (colitis) was induced via dextran sodium sulfate (DSS). The authors verified their previous findings: specifically, DSS animals had elevated levels of TNF-α, IL-6, RANKL, and OPG, with lower BMD and cancellous bone volume [15]. Using the TNBS rodent model of IBD, another study proposed that damage in the gut triggers the systemic immunologic changes seen in IBD, potentially linked through the lymphatic system: specifically, the lymphatic hyperproliferation and the altered bone turnover induced by TNF-α/RANKL-driven pathology [16].
The immune response in IBD is dysregulated and is characterized not only by the increased presence of B-cells and antibody production, together with excessive activation of effector T cells, but also by increased production of proinflammatory factors that stimulate osteoclast formation [6,9,17]. The RANKL/OPG ratio can be increased during inflammatory conditions possibly caused by the elevated levels of cytokines (Fig. 1). For example, cytokines such as TNF-α and IL-1 promote inflammation and bone resorption [18]. Anti-TNF-α therapy has been shown to decrease OPG, a member of the TNF-α receptor family, in IBD patients and potentially improve markers of bone metabolism such as procollagen type I N-terminal propeptide, osteocalcin, and alkaline phosphatase [15,19-22]. Early studies showed that IL-6 plays a well-established role in the pathology of osteoporosis, as well as in other diseases such as rheumatoid arthritis [23]. Genetic differences in IL-6 and IL-1 receptor antagonist genes were also associated with the clinical course of IBD and the extent of bone loss [24].
Figure 1.
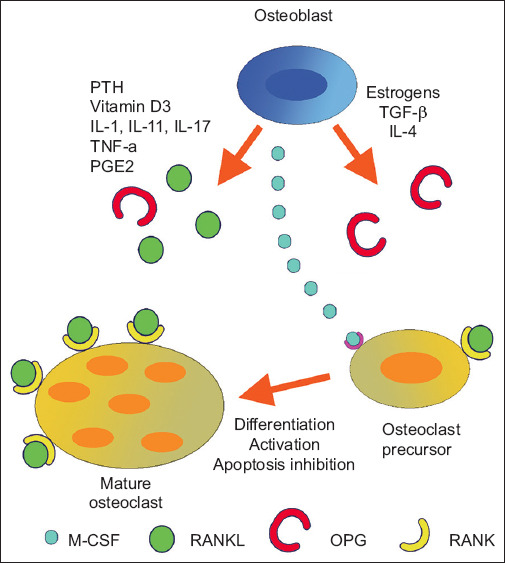
The RANK-RANKL-OPG system in bone remodeling. Factors secreted by the inflamed mucosa, such as IL-1, IL-6 and TNF-α, stimulate the expression of RANKL, thereby inducing osteoclast formation indirectly [25]
PTH, parathormone; IL, interleukin; TNF, tumor necrosis factor; PGE2, prostaglandin E2; TGF, transforming growth factor; M-CSF, macrophage colony stimulating factor; OPG, osteoprotegerin; RANK, receptor activator of nuclear factor (NF)-κB; RANKL, receptor activator of NF-kB ligand
It is well documented that IBD patients present elevated levels of proinflammatory cytokines such as IL-6, TNF-α, and IL-1β [26]. These proinflammatory cytokines may affect bone metabolism by inducing RANKL expression. RANKL is expressed on the surface of osteoclasts and binds either to an osteoclast precursor expressing the RANKL receptor, or to RANK (receptor activator of NF-κB) receptors, or the OPG receptor. If RANKL binds to the RANK receptors, then the osteoclasts differentiate and mature, resulting in increased bone loss. OPG inhibits this interaction by binding to RANKL, thus protecting the skeleton from excessive bone resorption. The RANK-RANKL-OPG system seems to be the most common pathway by which various molecules regulate bone homeostasis (Fig. 2).
Figure 2.

The RANKL/OPG ratio is an important determinant of bone mass and skeletal integrity. A high OPG/RANKL ratio prevents osteoclast differentiation, whereas a low OPG/RANKL ratio favors osteoclast differentiation leading to bone formation and bone resorption, respectively [25]
M-CSF, macrophage colony stimulating factor; OPG, osteoprotegerin; RANK, receptor activator of nuclear factor (NF)-κB; RANKL, receptor activator of NF-kB ligand
A low OPG/RANKL ratio could be responsible for bone loss in patients with IBD. Early studies showed that the RANKL/OPG ratio is related to bone mass and IBD treatment [27]. A study of 180 patients with an established diagnosis of IBD (111 CD and 69 UC) examined the activation of the RANKL/OPG pathway and how it affects BMD. A control group of healthy individuals matched for age and sex was also included in the study. An analysis of OPG and soluble RANKL (sRANKL) levels was performed in plasma (112 patients and 45 healthy controls) and supernatant of colonic explant cultures (CEC) (68 patients, 28 with UC and 40 with CD). OPG plasma levels were significantly higher in IBD patients (especially CD patients). On the other hand, sRANKL plasma levels (free unbound and not complexed with OPG) were similar among control and IBD patients. OPG plasma levels were negatively correlated with the femoral neck (r=0.408) and lumbar spine BMD (r=0.376), with osteoporotic patients exhibiting significantly higher OPG and significantly lower sRANKL plasma levels than patients with osteopenia or with normal BMD. CEC analysis showed that the intestine was a potential source of OPG in CD and UC patients (with increases of 3.4-fold and 3.8-fold, respectively). IBD patients treated with corticosteroids, or combined azathioprine and corticosteroids, presented significantly lower sRANKL levels compared with patients who received no specific treatment. This study demonstrated that the alterations in RANKL/OPG are associated with IBD. Moreover, the authors suggested that their observations (OPG and sRANKL regulation), together with those from murine models of IBD and bone loss, indicate that RANKL/OPG may play a part in the bone loss seen in IBD patients [28]. However, in another study, IBD patients (37 CD patients and 37 UC patients) had lower OPG serum levels than healthy volunteers (n=37) [29,30]. Soluble RANKL levels were not significantly different between IBD patients and the control group. However, IL-6 levels were 4.8-fold and 2.5-fold higher in CD and UC patients, respectively, and IL-6 was assumed to exert a pivotal role in modulating BMD, potentially through the OPG/sRANKL system. Thus, patients with CD were at higher risk of developing a condition with lower bone density (osteopenia and osteoporosis). In the former study [28], the mean disease duration was 3.12 and 4.69 years for CD and UC, respectively, whereas in the latter studies [29,30] the respective durations were 8.03 and 8.05 years. Thus, the initial increase in OPG levels may be a potential mechanism for controlling homeostasis, which aims to inhibit RANKL or TNF-α-induced osteoclastogenesis and maintain normal bone density.
Wnt signaling pathway
The identification of the role of the Wnt signaling pathway (wingless-type) in the regulation of osteoblast function highlighted its crucial contribution to the regulation of bone mass [31]. Although its exact mechanism of action remains unclear, the β-catenin signaling pathway interacts with bone morphogenic protein 2 to induce bone mass formation [32]. The Wnt signaling pathway is a signaling system found in different types of cells, where it regulates various biological functions (such as tissue regeneration, cell differentiation, bone remodeling, etc.) and it is a combination of 3 pathways: the canonical Wnt; the plane cell polar; and the Wnt-Ca(2+) signal transduction pathways. The regulatory role of the Wnt/β-catenin pathway in the immune response has been highlighted in recent years, especially in autoimmune diseases and immune-related inflammations [33]. Regarding IBD, the Wnt/β-catenin pathway was found to exert anti-inflammatory effects and its role is now under investigation [33]. In the canonical Wnt-pathway, β-catenin is activated through the binding of extracellular Wnt ligands to the frizzled and lipoprotein receptor-related protein (LRP) 5/6 coreceptors at the cell surface [34]. In this way, the Wnt pathway controls the production of the β-catenin transcription factor. On the other hand, non-canonical Wnt signaling is independent of β-catenin transcriptional function and is triggered by Wnt or frizzled signaling [34]. The absence of Wnt ligands leads to the degradation of cytoplasmic β-catenin through the ubiquitin-proteasome system. In the presence of Wnt-ligand, intracellular β-catenin accumulates in cells and translocates to the cell nucleus, where it binds to transcription factors that modulate genes related to bone formation [34] (Fig. 3).
Figure 3.
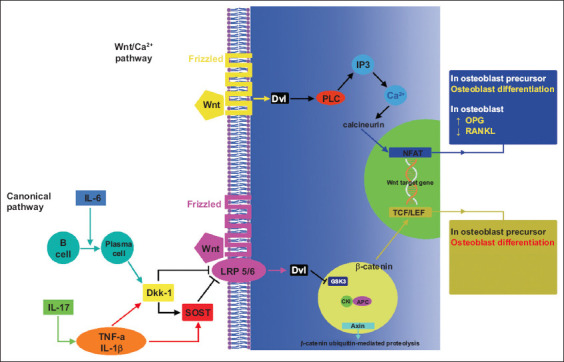
Wnt signaling in bone formation. The lower half of the figure depicts the canonical pathway and the upper half the Wnt/Ca2+ pathway [35]
IL, interleukin; TNF, tumor necrosis factor; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor-κB ligand; Dkk-1, dickkopf-1; SOST, sclerostin; LRP, lipoprotein receptor-related protein; PLC, phospholipase C; NFAT, nuclear factor of activated T-cells; TCF, T cell factor; LEF, lymphoid enhancer factor; GSK3, glycogen synthase kinase-3β; CKI, casein kinase I; APC, adenomatous polyposis coli; Dvl, dishevelled; IP3, inositol 1,4,5-triphosphate-3
The Wnt signaling pathway is regulated by a variety of factors including secreted frizzled-related proteins, Cerberus, Wnt inhibitory factor-1, and DKK-1 [36]. Tissue analysis of normal colonic and small intestinal mucosal samples from CD and UC patients revealed that the expression of several of the above-mentioned Wnt signaling pathway components is altered [37]. Some of them are upregulated while others are downregulated. Wnt signaling has an essential role in maintaining Paneth cell differentiation [38], and impaired Wnt signaling (T cell factor [TCF]-4 and LRP6 dysfunction) has been shown to decrease Paneth cell-produced α-defensins [39]. Moreover, the increased number of M2 macrophages commonly found in the mucosa of UC patients was related to the activation of Wnt signaling [40]. Recently animal studies showed that the Wnt5a-Ror2 axis promotes interferon-g signaling, leading to enhanced IL-12 expression [41].
The interactions of the proteins DKK-1, SOST, and POSTN with the Wnt signaling pathway in IBD are described briefly in the following paragraphs.
DKK-1
DKK-1, a member of the Dickkopf family, is a protein that consists of 2 cysteine-rich domains as well as a signal sequence. DKK-1 is an endogenous inhibitor of the Wnt/β-catenin pathway, competing with Wnt ligands for LRP5/6 receptors [42,43]. High circulating levels of DKK-1 can affect bone metabolism, as animal studies have shown that inhibition of DKK-1 by monoclonal antibodies stimulates bone formation [44]. DKK-1 regulates bone metabolism via the Wnt signaling pathway and increases gene expression of vascular endothelial growth factor, Runt-related transcription factor 2, and several other genes associated with cell growth and osteogenesis [45]. A role for DKK-1 in the etiology and progression of various autoimmune diseases (such as osteoarthritis, ankylosing spondylitis (AS), and rheumatoid arthritis) has also been proposed [46]. Previous studies suggested that glucocorticoids could increase DKK-1 expression by inhibiting the β-catenin pathway [47].
A recent study among 21 pediatric patients diagnosed with CD found that these patients have higher DKK-1 serum levels than healthy control subjects (n=142). DKK-1 levels in CD patients were also correlated with levels of erythrocyte sedimentation rate, C-reactive protein, and albumin. Moreover, mRNA colonic mucosa expression of DKK-1 was elevated in these patients, whereas β-catenin was decreased [48]. The authors concluded that DKK-1 could play a significant role in the pathogenesis of CD, and that high levels of serum DKK-1 can be utilized as a marker of inflammation.
SOST
SOST, a glycoprotein almost exclusively produced by osteocytes, is integral in the maintenance of normal bone mass and strength. Once secreted, it binds to the low-density LRP5/6 Wnt-coreceptors of the bone lining cells (inactive osteoblasts) and inhibits the Wnt/β-catenin signaling pathway [49,50], resulting in reduced osteoblastogenesis and bone formation. Through this mechanism, SOST regulates OPG in a dose-dependent manner, causing the RANK/mRNA ratio to increase, and exerting a catabolic action by promoting osteoclast formation and osteocyte activity [51]. Recent studies in IBD rodent models have shown elevated osteocyte levels of SOST in 3 distinct bone compartments (cancellous bone, metaphyseal cortical bone and cortical shaft) of animals with IBD [14,15]. On the other hand, low levels of SOST are associated with activation of the Wnt/β-catenin signaling pathway and hence enhancement of osteoblast activity [52]. Sclerostin production is also induced by TNF-α, released by activated T lymphocytes seen in inflammatory conditions such as IBD [53].
In a recent prospective cohort of patients with IBD screened for the presence of articular symptoms, lower levels of serum SOST were strongly correlated with the presence of IBD-associated spondyloarthritis (SpA/IBD). These patients also exhibited higher serum levels of anti-SOST immunoglobulin G (IgG), considered a putative marker of disease activity. The decreased levels of SOST serum levels, probably driven by the presence and concentration of the anti-SOST IgG, could lead to the development of axial joint inflammation. Thus, the early detection of anti-SOST-IgG might indicate the risk for axial SpA [54]. In SpA-IBD patients, SOST and anti-SOST-IgG serum levels were significantly correlated with lipopolysaccharide and soluble CD14 serum levels [55]. These 2 biomarkers are indicators of microbial translocation and innate immunity activation, respectively. These findings support a potential connection between dysbiosis and epithelial intestinal impairment with SOST levels in such patients [55]. Previous experiments in a murine model of IBD have shown that treatment with SOST antibody could prevent or restore bone loss and increase osteocalcin levels, considered a marker of bone formation [56].
POSTN
POSTN, a protein encoded by the Postn gene, interacts with several cell surface integrin molecules [57]. It was recently shown that POSTN interacts with integrin-αv and promotes allergic inflammation through the activation of NF-κB signaling in keratinocytes [58]. Recently it was shown in a rabbit model that POSTN interacts with the Wnt/β-catenin pathway to promote osteogenesis of xenotransplanted mesenchymal stem cells in response to cytotoxic T lymphocyte antigen 4, a molecule that regulates T-cell anergy and apoptosis but also increases immune tolerance [59].
The role of POSTN in AS and its relationship with biomarkers of the Wnt signaling pathway has also been studied. One study included 98 consecutive AS patients and 49 healthy controls. It was found that serum POSTN and DKK-1 levels in AS patients were significantly lower than in healthy controls, and especially in patients with active disease. Interestingly, serum POSTN levels could be independently predicted from DKK-1 and IL-8 levels, indicating that POSTN might play a role in new bone formation through the Wnt signaling pathway [60]. The downregulation of POSTN in AS patients with highly active disease and its potential contribution to disease pathogenesis through an interaction with the Wnt signaling pathway was also verified in a later study with an almost identical study population size (97 consecutive AS patients and 48 healthy controls) [61].
The relationship of POSTN with IBD was also investigated in a recent study of 144 pediatric patients. There were no significant differences in plasma POSTN levels between pediatric patients with CD or UC and control subjects; however, the authors reported that POSTN levels were significantly elevated during remission compared to active CD [62]. Patients with UC exhibit higher expression of POSTN in the lamina propria [63]. Moreover, POSTN could be considered a potential target for IBD treatment, given the ability of POSTN to mediate intestinal inflammation through the activation of NF-κB [64]. It seems that POSTN might have a role in inflammation and mucosal repair; nonetheless, more studies are required to understand POSTN’s role in the inflammatory process in IBD [62].
Dysbiosis-associated inflammation and bone metabolism
Dysbiosis has been implicated in the pathophysiology of many inflammatory diseases [65-67], such as several rheumatic diseases, rheumatoid arthritis, systemic lupus erythematosus, fibromyalgia, systemic sclerosis, AS, and Sjögren’s syndrome, and IBD [65]. Biological mechanisms linking gut dysbiosis to systemic inflammation have been postulated. In particular, the connection between the gut microbiome and bone health in IBD is not yet fully understood. Some researchers suggest that the NOD1 and NOD2 signaling pathways might be responsible for the effects of the gut microbiota on bone metabolism [68]. The bone function could be affected by activated immune cells released from the gut, or by metabolites produced by specific species of intestinal microbes. Ongoing research is focused on determining the impact of specific cytokines and changes in the microbiome on bone metabolism during IBD, and specifically on the impact of dysbiosis and the IL-12/23 signaling axis on IBD-associated bone loss; however, hard data are still lacking [69].
Dysbiosis-related osteoporosis studied in animal models of postmenopausal osteoporosis (mice with ovariectomy [OVX]) [12] has shown that probiotic treatment with Lactobacillus spp. prevents OVX-induced bone loss initiated by the increase of inflammatory markers such as IL-17a, TNF and RANKL [70], or reduces TNF-α and IL-1β levels and increases OPG in the mouse cortical bone [71]. Studies in germ-free mice also revealed that alterations in gut microbiota are correlated with bone mass metabolism [70,72] potentially through the production of pro-inflammatory and pro-osteoclastogenic cytokines and the enhanced production of TNF-α from T cells [73]. Recent placebo-controlled trials of probiotic supplementation in early postmenopausal women [74-76] and women with low bone density [77] also confirm the findings in animal studies. However, data on dysbiosis and IBD-related osteoporosis is currently scarce.
Concluding remarks
IBD has been associated with altered bone metabolism. However, the signaling pathways originating from the gut, which alter the systemic immune response and generate signals that in turn affect bone metabolism, are not yet fully understood. Data from IBD-related bone loss studies are scarce. In particular, there is a lack of clinical studies with a substantial sample size investigating several potential markers and signaling pathways suspected of affecting bone metabolism in patients with IBD (Table 1). There are studies in rodent models that provide us with useful data to explore in IBD patients (Table 2). These studies pinpoint the changes in specific markers, such as DKK-1 and sclerostin as, well as the Wnt-catenin pathway. Therefore, controlled studies in IBD patients, including patients with new-onset IBD not receiving treatment (to exclude any confounding effects from therapy) and patients with diagnosed IBD under treatment, are necessary to establish the signaling pathways and the potential time frames to initiate targeted treatment aimed at specific molecules.
Table 1.
Main outcomes of the human studies included in the review
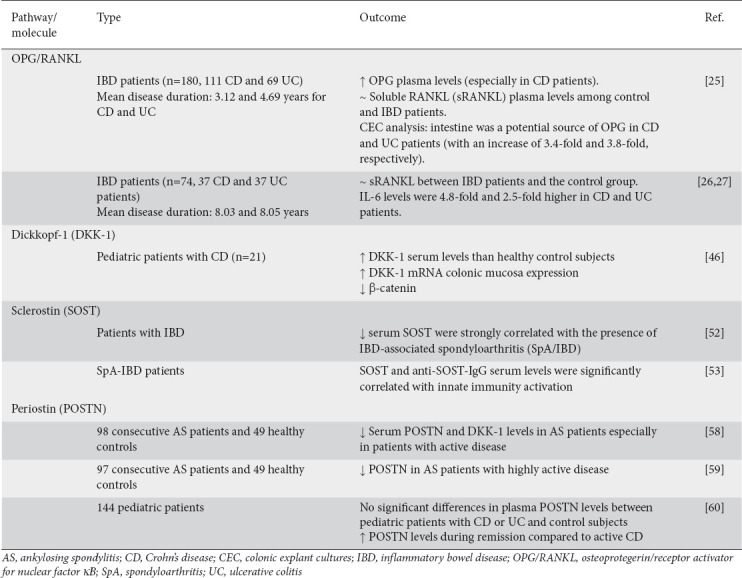
Table 2.
Main outcomes of the animal studies included in the review
Biography
“Agios Panteleimon” General Hospital of Nikaia-Piraeus; “Agia Varvara” General Hospital of Western Attica, Nikaia; Aretaieion Hospital, Athens; “P. & A. Kyriakou” Children’s Hospital, Athens; Aretaieion University Hospital, Medical School of Athens, Greece
Footnotes
Conflict of Interest: None
References
- 1.Baumgart DC, Carding SR. Inflammatory bowel disease:cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2021;18:56–66. doi: 10.1038/s41575-020-00360-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lima CA, Lyra AC, Rocha R, Santana GO. Risk factors for osteoporosis in inflammatory bowel disease patients. World J Gastrointest Pathophysiol. 2015;6:210–218. doi: 10.4291/wjgp.v6.i4.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lima CA, Lyra AC, Mendes CMC, et al. Bone mineral density and inflammatory bowel disease severity. Braz J Med Biol Res. 2017;50:e6474. doi: 10.1590/1414-431X20176374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katz S, Weinerman S. Osteoporosis and gastrointestinal disease. Gastroenterol Hepatol (N Y) 2010;6:506–517. [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein CN, Blanchard JF, Leslie W, Wajda A, Yu BN. The incidence of fracture among patients with inflammatory bowel disease. A population-based cohort study. Ann Intern Med. 2000;133:795–799. doi: 10.7326/0003-4819-133-10-200011210-00012. [DOI] [PubMed] [Google Scholar]
- 7.Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis:pathophysiology and therapy. Osteoporos Int. 2007;18:1319–1328. doi: 10.1007/s00198-007-0394-0. [DOI] [PubMed] [Google Scholar]
- 8.Mitra R. Adverse effects of corticosteroids on bone metabolism:a review. PM R. 2011;3:466–471. doi: 10.1016/j.pmrj.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein CN, Leslie WD. The pathophysiology of bone disease in gastrointestinal disease. Eur J Gastroenterol Hepatol. 2003;15:857–864. doi: 10.1097/00042737-200308000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Mouli VP, Ananthakrishnan AN. Review article:vitamin D and inflammatory bowel diseases. Aliment Pharmacol Ther. 2014;39:125–136. doi: 10.1111/apt.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behera J, Ison J, Tyagi SC, Tyagi N. The role of gut microbiota in bone homeostasis. Bone. 2020;135:115317. doi: 10.1016/j.bone.2020.115317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Wang X, Zhang C, Liu Z, Li C, Ren Z. Gut microbiota and bone diseases:a growing partnership. Front Microbiol. 2022;13:877776. doi: 10.3389/fmicb.2022.877776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santana PT, Rosas SLB, Ribeiro BE, Marinho Y, de Souza HSP. Dysbiosis in inflammatory bowel disease:pathogenic role and potential therapeutic targets. Int J Mol Sci. 2022;23 doi: 10.3390/ijms23073464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metzger CE, Narayanan A, Zawieja DC, Bloomfield SA. Inflammatory bowel disease in a rodent model alters osteocyte protein levels controlling bone turnover. J Bone Miner Res. 2017;32:802–813. doi: 10.1002/jbmr.3027. [DOI] [PubMed] [Google Scholar]
- 15.Metzger CE, Narayanan SA, Elizondo JP, et al. DSS-induced colitis produces inflammation-induced bone loss while irisin treatment mitigates the inflammatory state in both gut and bone. Sci Rep. 2019;9:15144. doi: 10.1038/s41598-019-51550-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narayanan SA, Metzger CE, Bloomfield SA, Zawieja DC. Inflammation-induced lymphatic architecture and bone turnover changes are ameliorated by irisin treatment in chronic inflammatory bowel disease. FASEB J. 2018;32:4848–4861. doi: 10.1096/fj.201800178R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Redlich K, Hayer S, Ricci R, et al. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J Clin Invest. 2002;110:1419–1427. doi: 10.1172/JCI15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone. 1999;25:255–259. doi: 10.1016/s8756-3282(99)00162-3. [DOI] [PubMed] [Google Scholar]
- 19.Franchimont N, Putzeys V, Collette J, et al. Rapid improvement of bone metabolism after infliximab treatment in Crohn's disease. Aliment Pharmacol Ther. 2004;20:607–614. doi: 10.1111/j.1365-2036.2004.02152.x. [DOI] [PubMed] [Google Scholar]
- 20.Miheller P, Muzes G, Rácz K, et al. Changes of OPG and RANKL concentrations in Crohn's disease after infliximab therapy. Inflamm Bowel Dis. 2007;13:1379–1384. doi: 10.1002/ibd.20234. [DOI] [PubMed] [Google Scholar]
- 21.Pazianas M, Rhim AD, Weinberg AM, Su C, Lichtenstein GR. The effect of anti-TNF-alpha therapy on spinal bone mineral density in patients with Crohn's disease. Ann N Y Acad Sci. 2006;1068:543–556. doi: 10.1196/annals.1346.055. [DOI] [PubMed] [Google Scholar]
- 22.Veerappan SG, O'Morain CA, Daly JS, Ryan BM. Review article:the effects of antitumour necrosis factor-a on bone metabolism in inflammatory bowel disease. Aliment Pharmacol Ther. 2011;33:1261–1272. doi: 10.1111/j.1365-2036.2011.04667.x. [DOI] [PubMed] [Google Scholar]
- 23.Manolagas SC. The role of IL-6 type cytokines and their receptors in bone. Ann N Y Acad Sci. 1998;840:194–204. doi: 10.1111/j.1749-6632.1998.tb09563.x. [DOI] [PubMed] [Google Scholar]
- 24.Schulte CM, Dignass AU, Goebell H, Röher HD, Schulte KM. Genetic factors determine extent of bone loss in inflammatory bowel disease. Gastroenterology. 2000;119:909–920. doi: 10.1053/gast.2000.18158. [DOI] [PubMed] [Google Scholar]
- 25.Castrogiovanni P, Trovato FM, Szychlinska MA, Nsir H, Imbesi R, Musumeci G. The importance of physical activity in osteoporosis. From the molecular pathways to the clinical evidence. Histol Histopathol. 2016;31:1183–1194. doi: 10.14670/HH-11-793. [DOI] [PubMed] [Google Scholar]
- 26.Redlich K, Smolen JS. Inflammatory bone loss:pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012;11:234–250. doi: 10.1038/nrd3669. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein CN, Sargent M, Leslie WD. Serum osteoprotegerin is increased in Crohn's disease:a population-based case control study. Inflamm Bowel Dis. 2005;11:325–330. doi: 10.1097/01.mib.0000164015.60795.ca. [DOI] [PubMed] [Google Scholar]
- 28.Moschen AR, Kaser A, Enrich B, et al. The RANKL/OPG system is activated in inflammatory bowel disease and relates to the state of bone loss. Gut. 2005;54:479–487. doi: 10.1136/gut.2004.044370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krela-Kaźmierczak I, Wysocka E, Szymczak A, et al. Osteoprotegerin, s-RANKL, and selected interleukins in the pathology of bone metabolism in patients with Crohn's disease. Prz Gastroenterol. 2016;11:30–34. doi: 10.5114/pg.2015.52589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krela-Kaźmierczak I, Szymczak-Tomczak A, Łykowska-Szuber L, et al. Interleukin 6, osteoprotegerin, sRANKL and bone metabolism in inflammatory bowel diseases. Adv Clin Exp Med. 2018;27:449–453. doi: 10.17219/acem/75675. [DOI] [PubMed] [Google Scholar]
- 31.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1:a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 32.Mbalaviele G, Sheikh S, Stains JP, et al. Beta-catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J Cell Biochem. 2005;94:403–418. doi: 10.1002/jcb.20253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jridi I, Canté-Barrett K, Pike-Overzet K, Staal FJT. Inflammation and Wnt signaling:target for immunomodulatory therapy? Front Cell Dev Biol. 2021;8:615131. doi: 10.3389/fcell.2020.615131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rochefort GY. The osteocyte as a therapeutic target in the treatment of osteoporosis. Ther Adv Musculoskelet Dis. 2014;6:79–91. doi: 10.1177/1759720X14523500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cici D, Corrado A, Rotondo C, Cantatore FP. Wnt signaling and biological therapy in rheumatoid arthritis and spondyloarthritis. Int J Mol Sci. 2019;20:5552. doi: 10.3390/ijms20225552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- 37.Hughes KR, Sablitzky F, Mahida YR. Expression profiling of Wnt family of genes in normal and inflammatory bowel disease primary human intestinal myofibroblasts and normal human colonic crypt epithelial cells. Inflamm Bowel Dis. 2011;17:213–220. doi: 10.1002/ibd.21353. [DOI] [PubMed] [Google Scholar]
- 38.Gersemann M, Stange EF, Wehkamp J. From intestinal stem cells to inflammatory bowel diseases. World J Gastroenterol. 2011;17:3198–3203. doi: 10.3748/wjg.v17.i27.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koslowski MJ, Kübler I, Chamaillard M, et al. Genetic variants of Wnt transcription factor TCF-4 (TCF7L2) putative promoter region are associated with small intestinal Crohn's disease. PLoS One. 2009;4:e4496. doi: 10.1371/journal.pone.0004496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cosín-Roger J, Ortiz-MasiáD , Calatayud S, et al. M2 macrophages activate WNT signaling pathway in epithelial cells:relevance in ulcerative colitis. PLoS One. 2013;8:e78128. doi: 10.1371/journal.pone.0078128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato A, Kayama H, Shojima K, et al. The Wnt5a-Ror2 axis promotes the signaling circuit between interleukin-12 and interferon-g in colitis. Sci Rep. 2015;5:10536. doi: 10.1038/srep10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mao B, Wu W, Davidson G, et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002;417:664–667. doi: 10.1038/nature756. [DOI] [PubMed] [Google Scholar]
- 43.Semënov MV, Tamai K, Brott BK, Kühl M, Sokol S, He X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol. 2001;11:951–961. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 44.Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. 2012;33:747–783. doi: 10.1210/er.2011-1060. [DOI] [PubMed] [Google Scholar]
- 45.Pinzone JJ, Hall BM, Thudi NK, et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood. 2009;113:517–525. doi: 10.1182/blood-2008-03-145169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma Y, Zhang X, Wang M, et al. The serum level of Dickkopf-1 in patients with rheumatoid arthritis:A systematic review and meta-analysis. Int Immunopharmacol. 2018;59:227–232. doi: 10.1016/j.intimp.2018.04.019. [DOI] [PubMed] [Google Scholar]
- 47.De Keyser F, Baeten D, Van den Bosch F, et al. Structure-modifying capacity of anti-tumour necrosis factor-alpha therapy in ankylosing spondylitis. Drugs. 2004;64:2793–2811. doi: 10.2165/00003495-200464240-00005. [DOI] [PubMed] [Google Scholar]
- 48.Kim MJ, Choe YH. Correlation of Dickkopf-1 with Inflammation in Crohn disease. Indian Pediatr. 2019;56:929–932. [PubMed] [Google Scholar]
- 49.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 51.Appel H, Ruiz-Heiland G, Listing J, et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2009;60:3257–3262. doi: 10.1002/art.24888. [DOI] [PubMed] [Google Scholar]
- 52.Sgambato D, Gimigliano F, De Musis C, et al. Bone alterations in inflammatory bowel diseases. World J Clin Cases. 2019;7:1908–1925. doi: 10.12998/wjcc.v7.i15.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Briot K, Geusens P, Em Bultink I, Lems WF, Roux C. Inflammatory diseases and bone fragility. Osteoporos Int. 2017;28:3301–3314. doi: 10.1007/s00198-017-4189-7. [DOI] [PubMed] [Google Scholar]
- 54.Luchetti MM, Ciccia F, Avellini C, et al. Sclerostin and antisclerostin antibody serum levels predict the presence of axial spondyloarthritis in patients with inflammatory bowel disease. J Rheumatol. 2018;45:630–637. doi: 10.3899/jrheum.170833. [DOI] [PubMed] [Google Scholar]
- 55.Luchetti MM, Ciccia F, Avellini C, et al. Gut epithelial impairment, microbial translocation and immune system activation in inflammatory bowel disease-associated spondyloarthritis. Rheumatol (Oxford) 2021;60:92–102. doi: 10.1093/rheumatology/keaa164. [DOI] [PubMed] [Google Scholar]
- 56.Eddleston A, Marenzana M, Moore AR, et al. A short treatment with an antibody to sclerostin can inhibit bone loss in an ongoing model of colitis. J Bone Miner Res. 2009;24:1662–1671. doi: 10.1359/jbmr.090403. [DOI] [PubMed] [Google Scholar]
- 57.Hamilton DW. Functional role of periostin in development and wound repair:implications for connective tissue disease. J Cell Commun Signal. 2008;2:9–17. doi: 10.1007/s12079-008-0023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masuoka M, Shiraishi H, Ohta S, et al. Periostin promotes chronic allergic inflammation in response to Th2 cytokines. J Clin Invest. 2012;122:2590–2600. doi: 10.1172/JCI58978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F, Luo K, Rong Z, et al. Periostin upregulates Wnt/β-catenin signaling to promote the osteogenesis of CTLA4-modified human bone marrow-mesenchymal stem cells. Sci Rep. 2017;7:41634. doi: 10.1038/srep41634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akar S, Uslu S, Kozaci LD, et al. 603 Periostin may have a role in ankylosing spondylitis and it is associated with Wnt signalling pathway regulators. Arthritis Rheumatol. 2014;10(Suppl):S265. [Google Scholar]
- 61.Solmaz D, Uslu S, Kozacı D, et al. Evaluation of periostin and factors associated with new bone formation in ankylosing spondylitis:periostin may be associated with the Wnt pathway. Int J Rheum Dis. 2018;21:502–509. doi: 10.1111/1756-185X.13186. [DOI] [PubMed] [Google Scholar]
- 62.Coelho T, Sonnenberg-Riethmacher E, Gao Y, et al. Expression profile of the matricellular protein periostin in paediatric inflammatory bowel disease. Sci Rep. 2021;11:6194. doi: 10.1038/s41598-021-85096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kikuchi Y, Kashima TG, Nishiyama T, et al. Periostin is expressed in pericryptal fibroblasts and cancer-associated fibroblasts in the colon. J Histochem Cytochem. 2008;56:753–764. doi: 10.1369/jhc.2008.951061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koh SJ, Choi Y, Kim BG, et al. Matricellular protein periostin mediates intestinal inflammation through the activation of nuclear factor kB signaling. PLoS One. 2016;11:e0149652. doi: 10.1371/journal.pone.0149652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Wei J, Zhang W, et al. Gut dysbiosis in rheumatic diseases:a systematic review and meta-analysis of 92 observational studies. EBioMedicine. 2022;80:104055. doi: 10.1016/j.ebiom.2022.104055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeGruttola AK, Low D, Mizoguchi A, Mizoguchi E. Current understanding of dysbiosis in disease in human and animal models. Inflamm Bowel Dis. 2016;22:1137–1150. doi: 10.1097/MIB.0000000000000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elias-Oliveira J, Leite JA, Pereira IS, et al. NLR and intestinal dysbiosis-associated inflammatory illness:drivers or dampers? Front Immunol. 2020;11:1810. doi: 10.3389/fimmu.2020.01810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ohlsson C, Nigro G, Boneca IG, Bäckhed F, Sansonetti P, Sjögren K. Regulation of bone mass by the gut microbiota is dependent on NOD1 and NOD2 signaling. Cell Immunol. 2017;317:55–58. doi: 10.1016/j.cellimm.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 69.Peek CT. The impact of dysbiosis and the IL-12/23 signaling axis on IBD-associated bone loss. Grantome. 2022. [Accessed 27 January 2023]. Available from https://grantome.com/grant/NIH/F30-DK120114-02 .
- 70.Li JY, Chassaing B, Tyagi AM, et al. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J Clin Invest. 2016;126:2049–2063. doi: 10.1172/JCI86062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ohlsson C, Engdahl C, Fåk F, et al. Probiotics protect mice from ovariectomy-induced cortical bone loss. PLoS One. 2014;9:e92368. doi: 10.1371/journal.pone.0092368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan J, Herzog JW, Tsang K, et al. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc Natl Acad Sci U S A. 2016;113:E7554–E7563. doi: 10.1073/pnas.1607235113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.D'Amelio P, Grimaldi A, Di Bella S, et al. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity:a key mechanism in osteoporosis. Bone. 2008;43:92–100. doi: 10.1016/j.bone.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 74.Jansson PA, Curiac D, Lazou A I, et al. Probiotic treatment using a mix of three Lactobacillus strains for lumbar spine bone loss in postmenopausal women:a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Rheumatol. 2019;1:e154–e162. doi: 10.1016/S2665-9913(19)30068-2. [DOI] [PubMed] [Google Scholar]
- 75.Takimoto T, Hatanaka M, Hoshino T, et al. Effect of Bacillus subtilis C-3102 on bone mineral density in healthy postmenopausal Japanese women:a randomized, placebo-controlled, double-blind clinical trial. Biosci Microbiota, Food Health. 2018;37:87–96. doi: 10.12938/bmfh.18-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jafarnejad S, Djafarian K, Fazeli MR, Yekaninejad MS, Rostamian A, Keshavarz SA. Effects of a multispecies probiotic supplement on bone health in osteopenic postmenopausal women:a randomized, double-blind, controlled trial. J Am Coll Nutr. 2017;36:497–506. doi: 10.1080/07315724.2017.1318724. [DOI] [PubMed] [Google Scholar]
- 77.Nilsson AG, Sundh D, Bäckhed F, Lorentzon M. Lactobacillus reuteri reduces bone loss in older women with low bone mineral density:a randomized, placebo-controlled, double-blind, clinical trial. J Intern Med. 2018;284:307–317. doi: 10.1111/joim.12805. [DOI] [PubMed] [Google Scholar]