

Abstract

First-tier MS-based newborn screening by flow injection analysis can have high presumptive positive rates, often due to isomeric/isobaric compounds or poor biomarker specificity. These presumptive positive samples can be analyzed by second-tier screening assays employing separations such as liquid chromatography–mass spectrometry (LC–MS/MS), which increases test specificity and drastically reduces false positive referrals. The ability to screen for multiple disorders in a single multiplexed test simplifies workflows and maximizes public health laboratories’ resources. In this study, we developed and validated a highly multiplexed second-tier method for dried blood spots using a hydrophilic interaction liquid chromatography (HILIC) column coupled to an MS/MS system. The LC–MS/MS method was capable of simultaneously detecting second-tier biomarkers for maple syrup urine disease, homocystinuria, methylmalonic acidemia, propionic acidemia, glutaric acidemia type 1, glutaric acidemia type 2, guanidinoacetate methyltransferase deficiency, short-chain acyl-CoA dehydrogenase deficiency, adrenoleukodystrophy, and Pompe disease.
Newborn screening (NBS) is a public health program where newborns are screened for various disorders at birth. NBS is performed using a variety of methods including physical exams, pulse oximetry, immunoassays, molecular assays, and mass spectrometry.1−3 Dried blood spots (DBS) are collected from newborns and are analyzed to identify metabolic defects. Because mass spectrometry can measure metabolic phenotypes directly, it has been widely adopted for the screening of metabolic disorders in newborns.1,3 Over time, tandem mass spectrometry (MS/MS) has enabled the quick measurement of dozens of biomarkers that serve as indicators for metabolic diseases and facilitated the rapid expansion of disorders screened universally hours after birth.4−8
In some cases, the most specific and/or selective biomarkers for a disease need to be separated from interfering components using liquid chromatography coupled to MS/MS (LC–MS/MS) and/or complex sample preparation.9 To maximize throughput, NBS laboratories analyze specimens using flow injection analysis MS/MS (FIA–MS/MS) as a first-tier screen which often involves the assay of less specific and/or sensitive biomarkers, with cutoff values set to avoid false negatives.9 Laboratories then either refer the baby for diagnostic screening, which may lead to a lot of false positive referrals or pass presumptive positive results from this low specificity screening assay to a second-tier screening assay that uses the more specific and/or sensitive biomarkers to reduce false positive referrals.
The Secretary of the Department of Health and Human Services recommends which diseases should be adopted by state programs by providing the recommended uniform screening panel (RUSP).10 The specificity and/or sensitivity of the following metabolic diseases included in the RUSP core conditions can be further improved using LC–MS/MS-based second-tier screening:11−19 maple syrup urine disease (MSUD), homocystinuria (HCU), methylmalonic acidemia, propionic acidemia (PA), isovaleric acidemia (IVA), glutaric acidemia type 1 (GA-1), adrenoleukodystrophy (ALD), congenital adrenal hyperplasia (CAH), and Pompe disease. In addition, malonic acidemia (MAL), glutaric acidemia type 2 (GA 2), and short-chain acyl-CoA dehydrogenase deficiency (SCAD) are secondary conditions on RUSP that use LC–MS/MS in their second-tier screening assay.10 Furthermore, guanidinoacetate methyltransferase deficiency (GAMT), Ornithine transcarbamylase deficiency (OTC), and Krabbe disease can also benefit from LC–MS/MS-based second-tier screening.3,20,21 For a breakdown of these diseases and previously documented corresponding second-tier biomarkers of interest, see Supporting Information Table S1.
Some of the current challenges associated with the adoption of second-tier screening are that (i) they only screen for one or very few disorders requiring laboratories to maintain several assays at the same time and (ii) the reflex rate to second-tier screening for some disorders is low. These challenges often lead laboratories to not adopt second-tier screening for these disorders, batch enough specimens before performing the assay or outsource the second-tier screening to reference labs. In either of the latter cases, several day delays are introduced to the reporting of results. There are several second-tier screening LC–MS/MS assays that only screen for one disorder by screening one or a few biomarkers.11,16,22,23 The highest number of RUSP disorders multiplexed into a second-tier screening assay to date was described in 2010 by Turgeon et al.,13 where three biomarkers were analyzed by LC–MS/MS to provide second-tier screening for five RUSP disorders.
ALD, introduced to the RUSP in 2016, is the only MS/MS-screened disorder for which second-tier screening is a requirement due to endogenous interferences driving high false positive referrals during FIA–MS/MS-based screening. Currently, NBS laboratories reflex up to 3% of daily ALD specimens to second-tier screening, but the assay only screens for ALD.16 There is a need to multiplex ALD biomarkers with second-tier biomarkers for other disorders to allow daily, comprehensive, in-house second-tier screening for multiple disorders regardless of their reflex rate.
Hydrophilic interaction liquid chromatography (HILIC) is a mode of chromatography that effectively separates hydrophilic metabolites. This mode of chromatography has the potential to enable the separation of many different second-tier biomarkers, many of which are hydrophilic, without derivatization.24 Because of the potential for a simpler, more multiplexed method, there are several studies that have examined the use of HILIC on biomarkers of interest.25 In the DBS matrix, HILIC has been used in NBS as an HCU second-tier assay analyzing the biomarker total-homocysteine (t-Hcy).12 The OTC biomarker orotic acid (OA) has also been measured in DBS and urine with a 5 min HILIC method.26 As a last example, a HILIC column similar to the one validated in this study has been used to develop a multiplexed method with the MSUD biomarker, alloisoleucine (aIle), and the argininosuccinate acid lyase deficiency biomarker, argininosuccinate.27 However, multiplexing second-tier methods into one or a few highly multiplexed NBS methods remains understudied.
The aim of this study was to develop and validate a universal second-tier NBS assay using LC–MS/MS. A diverse set of 19 second-tier biomarkers for the screening of aminoacidopathies, organic acid disorders, fatty acid oxidation disorders, Pompe disease, and ALD were successfully multiplexed. An additional 9 biomarkers were evaluated for inclusion in the method, including biomarkers for IVA, CAH, and Krabbe. The biomarkers investigated span a wide range of chemical classes (amino acids, organic acids, acylcarnitines, steroids, and lipids) and physicochemical properties, making them difficult to extract and analyze with a single assay. Priority was given to the multiplexing of second-tier screening biomarkers from disorders with the highest false-positive referral rates such as ALD, HCU, methylmalonic acidemia, and PA.
Experimental Section
Materials
Information on chemicals used in this study is presented in detail in the supplementary materials (Supporting Information Table S2).
DBS Manufacturing
DBS for method validation were created in the following manner. O+ packed red blood cells stored in CPDA-1 (Tennessee Blood Services) were washed with saline three times, hematocrit adjusted to 50% using hormone-depleted serum (SeraCare, catalog number 1800-0006), and freeze-lysed for >1 week at −20 °C. Unlabeled biomarkers dissolved in the solvents noted in Supporting Information Table S3 were added to the blood to make an enriched pool that was 92.5% blood and 7.5% biomarker stock solutions. An unenriched base pool was made with hematocrit adjusted to match the enriched pool by the addition of saline to 7.5% of the total unenriched base pool. The enriched pool was mixed with the unenriched pool to create a series of 10 enriched pools and an unenriched base pool. There was a fold change in the enrichment concentration of 1.7 between levels; level 1 was unenriched and level 11 was the most enriched (Supporting Information Table S3). These were then spotted with 100 μL of blood/spot onto filter paper cards (Eastern Business Forms, 903 no circle) using a Titertek syringe pump pipetting robot model # 43020 and allowed to dry overnight before being stored at −20 °C with desiccant (Multisorb, catalog number 02-00039AG105) until needed.
LC–MS/MS Setup
The chromatographic separation was performed on an Imtakt Intrada amino acid column 150 × 2 mm, 3 μm, with a Waters Acquity UPLC BEH C18 2.1 × 5 mm, 1.7 μm VanGuard Pre-Column. The chromatographic column was heated at 30 °C. Gradient elution was used, with 0.2% formic acid (FA) in acetonitrile (ACN) as mobile phase A (MBA) and an aqueous solution containing 40 mM ammonium formate (AmFA) and 666 μM oxalic acid as mobile phase B. The gradient elution program was as follows: 0 min 90% MPA 0.35 mL/min, 1 min 85% MPA 0.35 mL/min, 7 min 85% MPA 0.35 mL/min, 9.5 min 0% MPA 0.35 mL/min, 11 min 0% MPA 0.35 mL/min, 11.3 min 90% 0.6 mL/min, 12 min 90% 1 mL/min, 13.9 min 90% 1 mL/min, and 14 min 90% MPA 0.35 mL/min. The LC–MS/MS platform was a Waters TQ-XS mass spectrometer coupled to a Waters Acquity I-class plus binary UPLC system. The MRMs used in these studies are detailed in Supporting Information Tables S4 and S5. Differences from this chromatographic method during early method development are noted in supplemental figure legends. Timed SRMs were used to minimize the number of transitions followed at one time and maximize sensitivity. For the application of the method section, an Agilent Ultivo 6465B with a 1290 Infinity II LC system was used with the same chromatographic column and mobile phases described above.
Extraction Optimization
Extraction optimization experiments were performed using DBS-based quality control (QC) materials from the Centers of Disease Control and Prevention (CDC) Newborn Screening Quality Assurance Program (NSQAP). The QC material lot# D1814 used had the following relevant biomarkers enriched: 50 μM methylmalonic acid (MMA), 50 μM malonic acid (MA), 50 μM ethylmalonic acid (EMA), 50 μM homocysteine dimer (Hcy), and 25 μM methylcitric acid (MCA). The QC material was analyzed in triplicate for each condition. Aside from noted changes, the same extraction protocol was used as was applied for validation.
The initial extraction optimization examined the effect of changing the extraction solution composition with differing percentages of methanol (MeOH)/water and ACN/water including 100/0, 90/10, 80/20, and 70/30 with or without 0.1% FA. In addition, a three-way split of MeOH, ACN, and water (33/33/33) was tried with and without 0.1% FA. The end concentrations of the internal standards (IS) in the extraction solutions were 1 μM for creatine-D3 (CRE-D3), guanidinoacetic acid-13C2,15N (GUAC-13C2,15N), creatinine-D3 (CRN-D3), OA-15N2, 3-hygroxyglutaric acid-D5 (3-HGA–D5), and Hcy-D8; 0.4 μM for LPC 26:0 -D4; 2 μM for MMA–D3, EMA–D3, MA-13C3, and MCA–D3; and 2.5 μM for aIle-D10, isoleucine-D10,15N (Ile-D10,15N), leucine-D3 (Leu-D3), and valine-D8 (Val-D8).
A follow-up extraction optimization tested several aspects of sample preparation, namely, the use of additives, reducing agent, and extraction temperature. At this point, 1 μM 2-hydroxyglutaric acid-13C5 (2-HGA-13C5) was added to the standard IS mix for better evaluation of 2-HGA extraction. All tests used 80% ACN and 20% water, but several solvent variations were tested, namely, different percentages of FA (0, 0.05, 0.1, and 0.2%) and different concentrations of oxalic acid (1, 3, and 9 mM) with a set FA concentration (0.1%). Also, 80% ACN and 20% water with 0% FA and 3 mM oxalic acid were tested. All solvents were examined with the following conditions: room temperature (RT) extraction with dithiothreitol (DTT) reduction, 45 °C extraction with DTT reduction, RT extraction followed by tris(2-carboxyethyl)phosphine (TCEP) reduction, and 45 °C extraction followed by TCEP reduction. For the TCEP reduction, 12 μL 15 mM TCEP was added to water after extraction and shaken for 5 min at RT before drying down.
Analysis of both extraction experiments looked for high-fold changes relative to the mean biomarker value over all treatments. The calculated concentration, IS peak area, and unlabeled biomarker peak area fold changes were all considered when evaluating the different conditions. A high peak area, indicated by a high-fold change, for the endogenous biomarker and IS was considered of particular importance because a low biomarker area could mean low extraction and a low IS area could mean precipitation of IS. A low signal in either could mean a higher limit of quantification (LOQ) value than may otherwise be possible. The calculated concentration was considered of secondary importance and was monitored for abrupt changes in the calculated analyte concentration.
Validation
The following optimized conditions were used for method validation. First, 100 μL of the extraction solution containing 80% ACN, 20% water, 0.1% FA, 1 mM oxalic acid, and IS were added to a 1/8″ hole punch in a 96-well plate, followed by the addition of 12 μL of 8 mg/mL DTT in water. This mix was sealed and shaken for 45 min at RT. Then, the extract was transferred to fresh wells and dried under nitrogen for 30 min. The dried extracts were then resuspended while shaking in 100 μL of 90% ACN, 10% water, 0.18% FA, 4 mM AmFA, and 66.6 μM oxalic acid for 5 min. IS concentrations in the extraction solution were 1 μM for CRE-D3, GUAC-13C2,15N, CRN-D3, OA-15N2, 3-HGA–D5, 2-HGA-13C5, MMA–D3, MCA–D3, AIlle-D10, Ile-D10,15N, Leu-D3, Val-D8, and Hcy-D8; 0.01 μM for LPC 20:0-D4, LPC 22:0-D4, and LPC 24:0-D4; and 0.2 μM for LPC 26:0-D4; and 0.5 μM for MA-13C3 and EMA–D3.
Accuracy
For testing the accuracy of the LC–MS/MS method, stocks of unlabeled biomarkers were spiked into aliquots of the extraction solution for spiked concentrations adjusted for direct comparison to a 1/8″ (3.1 μL) DBS sample; see Supporting Information Table S6. The dilutions were done in triplicate with 3 levels of spiking and an unenriched level. Each dilution was used to extract duplicate samples on two different days with two unenriched matrixes (level 1 DBS and NSQAP QC material lot# A2013). Accuracy was calculated as percent spike recovery for each target analyte 100*([mean concentration for each condition] – [mean concentration unenriched DBS])/(theoretical spiked concentration). The goal was to have accuracy values between 85 and 115%.
Imprecision
To test the imprecision of the method, 3 of the enriched levels were chosen to measure in duplicate over 20 runs with 1 run per day and 2 different analysts. The 3 enrichment levels were chosen so biomarker imprecision was evaluated below and above typical clinical cutoffs. The resulting data were used to calculate the relative standard deviation (RSD) as 100*(total standard deviation)/mean. The goal was to have an RSD of ≤ 15% for each analyte.
Reproducibility
Data from imprecision analyses were used to calculate reproducibility. The mean result for each analyte from each analyst was calculated and reproducibility for each analyte was calculated as |100*[mean analyst 1]/[mean analyst 2] – 100|. The goal was to have reproducibility values ≤ 15%.
LOQ
The LOQ was determined with the Taylor method28 using all 11 levels of DBS materials created during this study. These materials were measured in singlets over 11 days. Measurements that were >3× the standard deviation (SD) for the enriched level in question were removed from the analysis as outliers. Also, samples with an IS peak area of <1/10× the average IS peak area were removed as failed injections. For the Taylor estimation, each biomarker had 3 enrichment levels selected for analysis with the conditions that the highest enrichment selected must have a measured concentration at least 2-fold the unenriched concentration and the lowest possible high enrichment must be selected that satisfies the first condition. Then, the SD was plotted against the measured concentration and the intercept of the resulting linear fit was the estimate of SD at 0 (S0).28 The recovery of the assay was accounted for by taking the LOQ data and plotting the measured concentration of all levels versus enriched concentration. The slope of this line is the fraction of material put into DBS for this method that was recovered (recovery). The LOQ was calculated as 10*S0/recovery. In cases where the S0 was negative, the SD at the lowest enrichment DBS level was used as S0.
Recovery
The recovery and accuracy comparison uses percent recovery from DBS determined by taking the recovery described in LOQ and multiplying by 100. Percent recovery from DBS values is compared to accuracy values from the highest spike concentration to isolate effects from DBS creation and extraction from effects of the LC–MS/MS method.
Linearity
The linearity analysis used all 10 enriched DBS levels, which were analyzed in quadruplicate on a single day. These data were used to calculate the R2 value for each analyte. The goal was to have R2 values of ≥ 0.98.
Carryover
For carryover analysis, filter paper-only samples were evaluated in triplicate after other filter paper-only samples (L0 to L0) as a baseline or after high enrichment L11 DBS samples (L11 to L0) to establish carryover. The final blanked carryover was calculated by taking the percent ratio of the peak areas, 100*(L11 to L0)/L11, as the unblanked carryover for each L11 to L0 pair; averaging the triplicate results of the unblanked carryover; calculating the percent ratio of the peak areas, 100*[L0 to L0]/L11, as a baseline of the assay; averaging the triplicate baseline values; and subtracting the baseline from the unblanked carryover to get the final blanked carryover. Ideal carryover values were <1%.
Stability
For stability tests, level 4 and level 10 of the DBS materials were used in triplicate for the following tests: DBS freeze-thawed three times, DBS stored at RT 24 h, extract stored at 4 °C 24 h, and extract stored at RT 24 h. Results were considered acceptable if they fell inside a 3xSD window around the imprecision mean established in the imprecision experiment.
Ruggedness
For ruggedness tests, the same levels, replication, and criteria were used as in stability for the following tests: 0.2 ± 0.02% FA in mobile phase A, 40 ± 4 mM AmFA in mobile phase B, 30 ± 3 °C column temperature, 30 °C extraction temperature, and 8 ± 0.8 mg/mL DTT added prior to extraction.
Specificity
For specificity, a mix of known interferents of biomarkers, namely, hydroxyproline enriched to 260 μM for aIle, succinic acid enriched to 1.0 mM for MMA, and methylsuccinic acid (MSA) and glutaric acid (GA) both enriched to 39 μM for EMA were spiked into the extracts of unenriched DBS to establish if these interfering substances change the calculated concentration of their respective biomarkers. Reported spiked interferent concentrations were adjusted to reflect concentrations that would have been in the 1/8″ (3.1 μL) DBS sample if they had originated from the DBS. Acceptable results had a biomarker concentration change <20% when calculating the change in the biomarker concentration, using the formula |100*([spiked]-[not spiked])/[not spiked]|.
Results and Discussion
LC Optimization
Experiments initially focused on optimizing the resolution of the isobars Leu, aIle, and Ile. First, the percentage of ACN varied with differing concentrations of AmFA (Supporting Information Figure S1). Then, the optimal column temperature and flow rate were determined (Supporting Information Figures S2 and S3). For these experiments, optimum conditions for separating Leu, Ile, and aIle are 15/85 water/ACN, 5 mM AmFA, and 0.2% FA with a column temperature of 30 °C and a flow rate of ∼0.35 mL/min. An evaluation of variable FA percentage in the mobile phase shows 0.2% FA increases the signal for several organic acids compared to 0.05% FA while avoiding signal losses seen in some amino acids at 1% FA (Supporting Information Figure S4A). Under optimal FA conditions (0.2% FA), the addition of oxalic acid to the mobile phase boosts the sensitivity of several compounds, especially MCA, with optimal results at 100 μM (Supporting Information Figure S4B). After oxalic acid addition, AmFA was adjusted to 6 mM to compensate for retention and resolution shifts introduced by oxalic acid, therefore maintaining the aforementioned optimal resolution of Leu, aIle, and Ile. To facilitate the separation of additional biomarkers using a gradient, the solvent components were split into mobile phases that approximate the optimized isocratic conditions described above when mixed as 85% mobile phase A (0.2% FA in ACN) and 15% mobile phase B (40 mM AmFA and 666 μM oxalic acid in water). To provide adequate retention of organic acids while maintaining the resolution of Leu, aIle, and Ile and keeping the total LC–MS/MS method time <15 min, the optimized gradient elution used has several steps, as described in the Experimental Section. These include a starting mix of 90/10 organic/aqueous with a 1 min linear gradient to conditions adequate for the resolution of Leu, aIle, and Ile that is maintained until the elution of Leu, aIle, and Ile, followed by a linear gradient to the 100% aqueous mobile phase to elute the rest of the hydrophilic compounds. The method ends with a high flow rate re-equilibration to the initial conditions, the total method time is 14 min, and the maximum back pressure is 6700 psi (Figure 1).
Figure 1.

HILIC method LC–MS/MS profile for labeled and unlabeled second-tier analytes of interest in high enrichment QC materials. Unlabeled analytes have gray traces and labeled analytes have black traces. Abbreviations: Percent mobile phase B (%B).
The present method prioritized the detection of organic acids and resolution of Leu, aIle, and Ile, leading to compromises that did not work for some biomarkers of interest (Supporting Information Table S7). Initially, we tried to add steroids typically used in CAH second-tier assays, namely, 17-α-hydroxyprogesterone (17-OHP), 21-deoxycortisol, 11-deoxycortisol, cortisol, and androstenedione. However, all analytes elute early in the chromatography; 21-deoxycortisol does not resolve from 11-deoxycortisol or 17-deoxycortisol and 17-OHP coelutes with the known interferent deoxycorticosterone. 21-Deoxycortisol and 11-deoxycortisol were shown to separate with 5% aqueous starting mobile phase, but this percentage organic negatively affected the chromatography of 3-HGA and 2-HGA. We also needed to remove the Krabbe biomarker psychosine from the panel due to its high retention under these chromatographic conditions. The IVA biomarker, isovalerylcarnitine, needed to be dropped from the panel as well because it coelutes with the known interferents 2-methylbutyrylcarnitine and pivaloylcarnitine under these chromatographic conditions. The GA I biomarker GA was dropped because it coelutes with MSA. Some methods that lack oxalic acid were able to separate GA from MSA, but the addition of oxalic acid to the mobile phase was too important for organic acid sensitivity to drop. However, GA I is retained in this method because 3-HGA is the pathognomonic marker for GA I and GA is a less specific but potentially useful biomarker. For a complete list of investigated biomarkers that need to be separated before detection by mass spectrometry, see Supporting Information Table S7.
Extraction Optimization
For the initial extraction optimization, LPC 26:0, t-Hcy, and MCA were shown to be affected the most by changes in the extraction solvent and proved to be the most problematic for reaching the sensitivity required (Figure 2). LPC 26:0 declined with higher proportions of water, particularly in mixtures with MeOH. The t-Hcy peak area declines with increasing organic and has a higher peak area in ACN when FA is added. The MCA peak area is highest in conditions with MeOH and high-water content with no FA. To see parallel effects observed in IS fold changes and resulting effects on concentration, see Supporting Information Figures S5 and S6.
Figure 2.
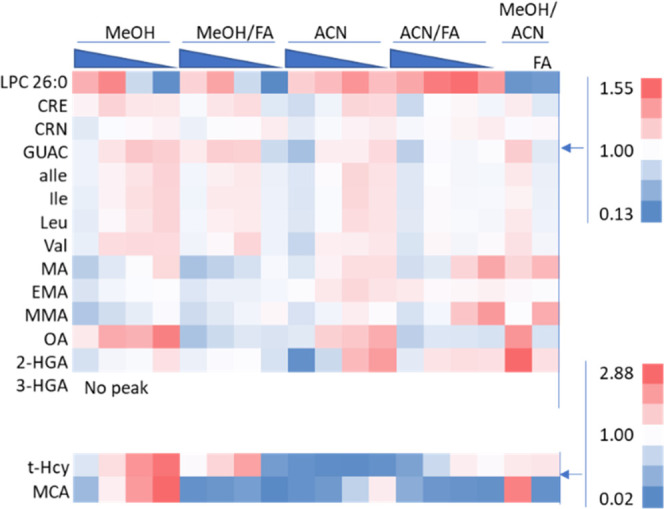
Heat map showing the effect of the extraction solvent composition on the extracted biomarker peak area fold change relative to the mean peak area of each analyte. Solvents tested include either equal percentages of MeOH/ACN/water (MeOH/ACN) or variable percentages of MeOH/water (MeOH) and ACN/water (ACN), 100/0, 90/10, 80/20, and 70/30, with or without 0.1% FA. Above 1 (mean peak area) is red, below 1 is blue, and 1 is white. The material extracted was the NSQAP QC material lot# D1814.
Further optimization focused on additives, extraction temperature, and reducing agents to maximize the peak area of MCA, t-Hcy, and LPC 26:0. It was decided that the baseline extraction solvent for this would be a compromise between the optimal LPC 26:0 and t-Hcy solvent conditions in the prior experiment: 80% ACN, 20% water, and 0.1% FA. This follow-up extraction optimization has the most dramatic fold changes in t-Hcy and MCA, with LPC 26:0 closely monitored due to its unique chemistry and high variability in the prior experiment (Figure 3). The MCA unlabeled biomarker area increases by 82× when 9 mM oxalic acid is added to the extraction solution with RT extraction and DDT reduction. However, this concentration of oxalic acid had a negative effect on the t-Hcy peak area. The oxalic acid concentration that is considered a good compromise between MCA and the t-Hcy peak area is 1 mM oxalic acid when using DTT as the reducing agent. When using TCEP as the reducing agent, the negative effect on t-Hcy is not noticeable and the area for t-Hcy peaked at 9 mM oxalic acid. However, the use of TCEP causes a problematic decline in the t-Hcy peak area compared to DTT. LPC 26:0 yielded smaller peak areas when using 45 °C and/or TCEP. Tests on different FA concentrations (0, 0.05, and 0.2%) show no improvement compared to 0.1% FA. To see the parallel changes observed in the IS area and concentration, see Supporting Information Figures S7 and S8. For these reasons, the optimal conditions for extraction are RT extraction with DTT reduction and an extraction solvent consisting of 80% ACN, 20% water, 0.1% FA, and 1 mM oxalic acid. The addition of oxalic acid to improve the MCA extraction/peak area from DBS is a novel result that could be of great importance to the development of other methods if underivatized MCA is not reaching the required sensitivity.
Figure 3.

Heat map showing the effects of varying extraction conditions on the extracted biomarker peak area fold change relative to the mean peak area of each analyte. Conditions tested include DTT or TCEP reduction, room temperature or 45 °C extraction, variable FA at (0, 0.05, 0.1, or 0.2%), or variable oxalic acid (Oxalic acid at 1, 3, or 9 mM) using 0.1% FA. In addition, 3 mM oxalic acid with no FA was tested (*). All conditions use 80% ACN with 20% water. Above 1 is red, below 1 is blue, and 1 is white. The material extracted was the NSQAP QC material lot# D1814.
Method Validation
Overall, the method was performed within desired parameters for most tested conditions. The following discussion addresses metrics that were outside of the ideal results but are ultimately considered acceptable. When observing the recovery of biomarkers from DBS, it is clear that many analytes are not close to 100% recovery (Supporting Information Table S8). However, this calculation is only based on unlabeled biomarker/IS ratios, as is the standard practice in virtually all NBS laboratories. The use of calibrant DBS materials in the future could be used to correct recovery and prevent inaccuracy. Most NBS assays are not accurate for many analytes, but they are still considered to be fit-for-purpose as they are precise, and cutoffs can be set adequately to account for any inaccuracies (i.e., due to decreased recoveries). The only analyte with an average imprecision and reproducibility above 15% is t-Hcy, with 20 and ∼18%, respectively (Table 1 and Supporting Information Table S9). Those imprecision results, although slightly outside our initial goal, are still acceptable/fit-for-purpose due to the significant difference in DBS t-Hcy concentrations between healthy babies and babies diagnosed with homocystinuria. t-Hcy is the analyte with the most complicated extraction, with a reduction step required before detection. Likewise, when looking at the analytical accuracy of the mass spectrometer by spiking analytes into the extraction solvent, only 3-HGA shows a value of 80%, consistently below the goal of 85–115% (Supporting Information Table S8). It is possible this is due to a deuterium effect originating from the deuterated IS, but this could also be corrected using calibrators or 13C-labeled IS in the assay.
Table 1. HILIC Method Validation Results for Imprecision, LOQ, Linearity, and Carryover.
| analyte | average RSD (% CV) | LOQ (μM) | linearity (R2) | carryover (%) |
|---|---|---|---|---|
| CRN | 8.9 | 11.2 | 0.996 | 0.0014 |
| Val | 15 | 115 | 0.993 | 0.059 |
| t-Hcy | 20 | 6.85 | 0.985 | 0.025 |
| GUAC | 6.1 | 0.456 | 0.996 | –0.056 |
| Leu | 5.5 | 24.2 | 0.997 | 0.061 |
| CRE | 6.1 | 115 | 0.989 | 0.014 |
| MA | 7.2 | 4.29 | 0.997 | 0.16 |
| MMA | 8.6 | 1.51 | 0.997 | 0.47 |
| EMA | 6.1 | 0.867 | 0.997 | 0.032 |
| 3-HGA | 10 | 0.753 | 0.998 | 0.50 |
| 2-HGA | 5.4 | 9.65 | 0.993 | 0.12 |
| OA | 5.6 | 0.297 | 0.998 | 0.026 |
| MCA | 5.7 | 0.305 | 0.998 | 0.029 |
| LPC 20:0 | 8.2 | 0.0809 | 0.998 | 0.032 |
| LPC 22:0 | 7.5 | 0.0159 | 0.998 | 0.00018 |
| LPC 24:0 | 9.3 | 0.0294 | 0.991 | 0.0051 |
| LPC 26:0 | 14 | 0.0264 | 0.992 | 0.010 |
| Ile | 11 | 9.85 | 0.994 | 0.050 |
| aIle | 11 | 0.347 | 0.992 | 0.065 |
The ruggedness and stability tests highlighted where special attention needs to be given during the execution of this method. Ruggedness testing highlights the importance of proper solvent preparation with MMA, aIle, and Ile outside 3×SD of the imprecision mean when FA is increased or AmFA is decreased (Supporting Information Table S10). Stability tests showed that extracts should not be stored at RT for 24 h with CRN and LPC 24:0 outside 3xSD of the imprecision mean under this condition (Supporting Information Table S11).
The LOQ, specificity, carryover, and linearity components of the validation were performed as desired for all analytes. LOQ has several analytes with high concentrations in the unenriched material, and as a result, when determining LOQ, the analysis did not have measurements anywhere near 0 to use when estimating S0, as is recommended for the Taylor method. As a result, some analytes with higher endogenous concentrations such as CRN, Val, Leu, CRE, Ile, and 2-HGA may have lower LOQs than reported in this study (Table 1). For specificity, the addition of interferents caused a minimal change in MMA (8%), aIle (17%), and EMA (10%) concentrations. All carryover and linearity results are satisfactory (Table 1).
Application of the Method
While residual newborn specimens were unable to be obtained due to their rarity, DBS specimens that were produced in-house were evaluated as unknowns to support the clinical validity of the assay. These blinded specimens included a proficiency testing (PT) specimen that was produced for the NSQAP ALD PT program. This specimen was enriched with ALD biomarkers LPC 24:0 and LPC 26:0. A specimen that was produced as a high-concentration MSUD QC was also analyzed. This specimen was enriched with MSUD biomarker aIle. In a normal, unenriched DBS, alloisoleucine, being the pathognomonic marker for MSUD, is not observed, while there is an approximately 9-fold difference for LPC 26:0 between the enriched and unenriched specimen (Figure 4A–C), providing clear distinction of normal specimens as compared to specimens outside of normal limits.
Figure 4.
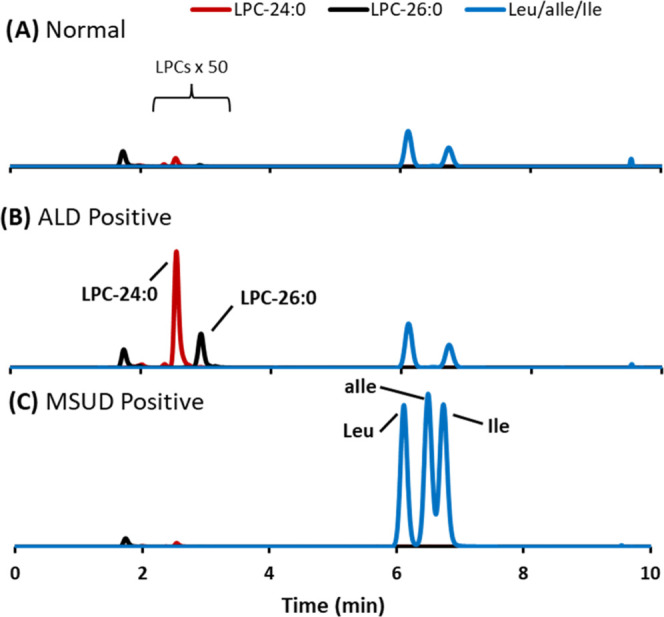
Extracted ion chromatograms of biomarkers for adrenoleukodystrophy (ALD) and maple syrup urine disease (MSUD) in a (A) normal specimen, (B) ALD-positive DBS enriched with LPC 24:0 and LPC 26:0 produced for ALD proficiency testing, and (C) MSUD-positive DBS enriched with Leu, aIle, and Ile produced for MSUD quality control.
Conclusions
A novel HILIC-MS/MS second-tier NBS assay was developed, optimized, and analytically validated for the quantification of a diverse set of 19 metabolites that serve as biomarkers for 11 NBS disorders. Analytical validation of this highly multiplexed method demonstrated its suitability for NBS by evaluating accuracy, imprecision, reproducibility, LOQ, recovery, linearity, carryover, stability, ruggedness, and specificity. Careful optimization of biomarker extraction and chromatographic conditions allowed for adequate limits of quantitation to be achieved for challenging analytes such as organic acids and separation of isomers such as Leu, Ile, and aIle without previous derivatization. To the best of our knowledge, this is the highest multiplexed second-tier NBS assay to date in terms of both metabolites and disorders screened.
One of the strengths of this novel method stems from the ability to multiplex ALD biomarkers, mainly LPC C26:0, which, under first-tier NBS, produces a lot of analytical false positives (requiring second-tier screening), with second-tier biomarkers for multiple disorders with much lower reflex rates to second-tier screening. The approach overcomes current challenges faced by NBS laboratories where they either must maintain several assays for a very small number of presumptive positives requiring second-tier screening for most disorders, or they must outsource second-tier screening to a reference laboratory causing reporting delays for time-critical disorders.
We estimate that up to 4% of daily newborn specimens could be reflexed to this second-tier screening assay based on cutoffs and presumptive positives rates published in the literature for the screening of MSUD, HCU, methylmalonic acidemia, PA, MAL, GA-1, GAMT, ALD, Pompe disease, and SCAD. This assay will allow NBS laboratories to perform second-tier screening daily, eliminating previously encountered delays associated with low presumptive positive specimen quantities that needed to be analyzed with assays that were performed only once or twice per week.
Acknowledgments
The findings and conclusions in this study are those of the authors and do not necessarily represent the views of the U.S. Department of Health and Human Services or the U.S. Centers for Disease Control and Prevention. Use of trade names and commercial sources is for identification only and does not constitute an endorsement by the U.S. Department of Health and Human Services or the U.S. Centers for Disease Control and Prevention. This project was supported in part by appointment to the Science Education and Workforce Development Programs at Oak Ridge National Laboratory, administered by ORISE through the U.S. Department of Energy Oak Ridge Institute for Science and Education. Dr. Dimitrios Platis’ contribution to this work was performed at the Centers for Disease Control and Prevention during a 3-month sabbatical.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c03098.
Details of experimental results expressed as figures and tables and method details expressed as tables (PDF)
Author Contributions
§ M.B.K. and D.P. contributed equally to this work.
Author Contributions
D.P., T.L., and M.B.K. executed experiments; M.B.K., S.I., A.P., D.P., and K.P. designed experiments; and M.B.K., C.C., S.I., A.P., D.P., and K.P. contributed to the writing of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Chace D. H.; De Jesús V. R.; Haynes C. A.. Analytical Perspectives on the Use of Dried Blood Spots and Mass Spectrometry in Newborn Screening. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; Wiley, 2015; pp 1–26. [Google Scholar]
- Torresani T.; Grüters A.; Scherz R.; Burckhardt J. J.; Harras A.; Zachmann M. Improving the efficacy of newborn screening for congenital adrenal hyperplasia by adjusting the cut-off level of 17α-hydroxyprogesterone to gestational age. Screening 1994, 3, 77–84. 10.1016/0925-6164(94)90003-5. [DOI] [Google Scholar]
- Sinclair G. B.; van Karnebeek C. D. M.; Ester M.; Boyd F.; Nelson T.; Stockler-Ipsiroglu S.; Vallance H. A three-tier algorithm for guanidinoacetate methyltransferase (GAMT) deficiency newborn screening. Mol. Genet. Metab. 2016, 118, 173–177. 10.1016/j.ymgme.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Millington D. S.; Kodo N.; Terada N.; Roe D.; Chace D. H. The analysis of diagnostic markers of genetic disorders in human blood and urine using tandem mass spectrometry with liquid secondary ion mass spectrometry. Int. J. Mass Spectrom. Ion Processes 1991, 111, 211–228. 10.1016/0168-1176(91)85056-R. [DOI] [Google Scholar]
- Chace D. H.; Sherwin J. E.; Hillman S. L.; Lorey F.; Cunningham G. C. Use of phenylalanine-to-tyrosine ratio determined by tandem mass spectrometry to improve newborn screening for phenylketonuria of early discharge specimens collected in the first 24 hours. Clin. Chem. 1998, 44, 2405–2409. 10.1093/clinchem/44.12.2405. [DOI] [PubMed] [Google Scholar]
- Chace D. H.; Hillman S. L.; Millington D. S.; Kahler S. G.; Roe C. R.; Naylor E. W. Rapid diagnosis of maple syrup urine disease in blood spots from newborns by tandem mass spectrometry. Clin. Chem. 1995, 41, 62–68. 10.1093/clinchem/41.1.62. [DOI] [PubMed] [Google Scholar]
- Chace D. H.; Hillman S. L.; Van Hove J. L.; Naylor E. W. Rapid diagnosis of MCAD deficiency: quantitative analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin. Chem. 1997, 43, 2106–2113. 10.1093/clinchem/43.11.2106. [DOI] [PubMed] [Google Scholar]
- Turgeon C.; Magera M. J.; Allard P.; Tortorelli S.; Gavrilov D.; Oglesbee D.; Raymond K.; Rinaldo P.; Matern D. Combined Newborn Screening for Succinylacetone, Amino Acids, and Acylcarnitines in Dried Blood Spots. Clin. Chem. 2008, 54, 657–664. 10.1373/clinchem.2007.101949. [DOI] [PubMed] [Google Scholar]
- Chace D. H.; Hannon W. H. Impact of Second-Tier Testing on the Effectiveness of Newborn Screening. Clin. Chem. 2010, 56, 1653–1655. 10.1373/clinchem.2010.153494. [DOI] [PubMed] [Google Scholar]
- US Department of Health and Human Services Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children. Recommended Uniform Screening Panel, 2020. http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/recommendedpanel/index.html. [DOI] [PubMed]
- Oglesbee D.; Sanders K. A.; Lacey J. M.; Magera M. J.; Casetta B.; Strauss K. A.; Tortorelli S.; Rinaldo P.; Matern D. Second-tier test for quantification of alloisoleucine and branched-chain amino acids in dried blood spots to improve newborn screening for maple syrup urine disease (MSUD). Clin. Chem. 2008, 54, 542–549. 10.1373/clinchem.2007.098434. [DOI] [PubMed] [Google Scholar]
- Okun J. G.; Gan-Schreier H.; Ben-Omran T.; Schmidt K. V.; Fang-Hoffmann J.; Gramer G.; Abdoh G.; Shahbeck N.; Al Rifai H.; Al Khal A. L.; Haege G.; Chiang C.-C.; Kasper D. C.; Wilcken B.; Burgard P.; Hoffmann G. F. Newborn Screening for Vitamin B(6) Non-responsive Classical Homocystinuria: Systematical Evaluation of a Two-Tier Strategy. JIMD Rep. 2017, 32, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon C. T.; Magera M. J.; Cuthbert C. D.; Loken P. R.; Gavrilov D. K.; Tortorelli S.; Raymond K. M.; Oglesbee D.; Rinaldo P.; Matern D. Determination of Total Homocysteine, Methylmalonic Acid, and 2-Methylcitric Acid in Dried Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 2010, 56, 1686–1695. 10.1373/clinchem.2010.148957. [DOI] [PubMed] [Google Scholar]
- Janzen N.; Steuerwald U.; Sander S.; Terhardt M.; Peter M.; Sander J. UPLC-MS/MS analysis of C5-acylcarnitines in dried blood spots. Clin. Chim. Acta 2013, 421, 41–45. 10.1016/j.cca.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Al-Dirbashi O. Y.; Kölker S.; Ng D.; Fisher L.; Rupar T.; Lepage N.; Rashed M. S.; Santa T.; Goodman S. I.; Geraghty M. T.; Zschocke J.; Christensen E.; Hoffmann G. F.; Chakraborty P. Diagnosis of glutaric aciduria type 1 by measuring 3-hydroxyglutaric acid in dried urine spots by liquid chromatography tandem mass spectrometry. J. Inherited Metab. Dis. 2011, 34, 173–180. 10.1007/s10545-010-9223-2. [DOI] [PubMed] [Google Scholar]
- Haynes C. A.; De Jesús V. R. Improved analysis of C26:0-lysophosphatidylcholine in dried-blood spots via negative ion mode HPLC-ESI-MS/MS for X-linked adrenoleukodystrophy newborn screening. Clin. Chim. Acta 2012, 413, 1217–1221. 10.1016/j.cca.2012.03.026. [DOI] [PubMed] [Google Scholar]
- Janzen N.; Peter M.; Sander S.; Steuerwald U.; Terhardt M.; Holtkamp U.; Sander J. Newborn Screening for Congenital Adrenal Hyperplasia: Additional Steroid Profile using Liquid Chromatography-Tandem Mass Spectrometry. J. Clin. Endocrinol. Metab. 2007, 92, 2581–2589. 10.1210/jc.2006-2890. [DOI] [PubMed] [Google Scholar]
- Tortorelli S.; Eckerman J. S.; Orsini J. J.; Stevens C.; Hart J.; Hall P. L.; Alexander J. J.; Gavrilov D.; Oglesbee D.; Raymond K.; Matern D.; Rinaldo P. Moonlighting newborn screening markers: the incidental discovery of a second-tier test for Pompe disease. Genet. Med. 2018, 20, 840–846. 10.1038/gim.2017.190. [DOI] [PubMed] [Google Scholar]
- Matern D.; Tortorelli S.; Oglesbee D.; Gavrilov D.; Rinaldo P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: the Mayo Clinic experience (2004-2007). J. Inherited Metab. Dis. 2007, 30, 585–592. 10.1007/s10545-007-0691-y. [DOI] [PubMed] [Google Scholar]
- D’Apolito O.; Garofalo D.; la Marca G.; Russo A. D.; Corso G. Reference intervals for orotic acid in urine, plasma and dried blood spot using hydrophilic interaction liquid chromatography–tandem mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 883–884, 155–160. 10.1016/j.jchromb.2011.09.054. [DOI] [PubMed] [Google Scholar]
- Chuang W. L.; Pacheco J.; Zhang X. K.; Martin M. M.; Biski C. K.; Keutzer J. M.; Wenger D. A.; Caggana M.; Orsini J. J. Jr. Determination of psychosine concentration in dried blood spots from newborns that were identified via newborn screening to be at risk for Krabbe disease. Clin. Chim. Acta 2013, 419, 73–76. 10.1016/j.cca.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Turgeon C. T.; Orsini J. J.; Sanders K. A.; Magera M. J.; Langan T. J.; Escolar M. L.; Duffner P.; Oglesbee D.; Gavrilov D.; Tortorelli S.; Rinaldo P.; Raymond K.; Matern D. Measurement of psychosine in dried blood spots—a possible improvement to newborn screening programs for Krabbe disease. J. Inherited Metab. Dis. 2015, 38, 923–929. 10.1007/s10545-015-9822-z. [DOI] [PubMed] [Google Scholar]
- Maase R.; Skrinska V.; Younes N.; Hassan L.; Mitri R.; Matern D.; Rinaldo P.; Turgeon C. A Rapid Screening Method for the Measurement of Neonatal Total Homocysteine in Dried Blood Spots by Liquid Chromatography-Tandem Mass Spectrometry. Int. J. Neonatal Screen. 2017, 3, 32. 10.3390/ijns3040032. [DOI] [Google Scholar]
- Buszewski B.; Noga S. Hydrophilic interaction liquid chromatography (HILIC)--a powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. 10.1007/s00216-011-5308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew E. M.; Lewis L.; Rao P.; Nalini K.; Kamath A.; Moorkoth S. Novel HILIC-ESI-MS method for urinary profiling of MSUD and methylmalonic aciduria biomarkers. J. Chromatogr. Sci. 2019, 57, 715–723. 10.1093/chromsci/bmz045. [DOI] [PubMed] [Google Scholar]
- D’Apolito O.; Garofalo D.; Paglia G.; Zuppaldi A.; Corso G. Orotic acid quantification in dried blood spots and biological fluids by hydrophilic interaction liquid chromatography tandem mass spectrometry. J. Sep. Sci. 2010, 33, 966–973. 10.1002/jssc.200900758. [DOI] [PubMed] [Google Scholar]
- Griffin C.; Ammous Z.; Vance G. H.; Graham B. H.; Miller M. J. Rapid quantification of underivatized alloisoleucine and argininosuccinate using mixed-mode chromatography with tandem mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2019, 1128, 121786 10.1016/j.jchromb.2019.121786. [DOI] [PubMed] [Google Scholar]
- Taylor J. K.Quality Assurance of Chemical Measurements, 1st ed; Routledge: New York, 1987. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.