

Abstract
Molecular docking plays a major role in academic and industrial drug screening and discovery processes. Despite the availability of numerous docking software packages, there is a lot of scope for improvement for the docking algorithms in terms of becoming more reliable to replicate the experimental binding results. Here, we propose a combinatorial or consensus docking approach where complementary powers of the existing methods are captured. We created a meta-docking protocol by combining the results of AutoDock4.2, LeDock, and rDOCK programs as these are freely available, easy to use, and suitable for large-scale analysis and produced better performance on benchmarking studies. Rigorous benchmarking analyses were undertaken to evaluate the scoring, posing, and screening capability of our approach. Further, the performance measures were compared against one standard state-of-the-art commercial docking software, GOLD, and one freely available software, PLANTS. Performances of MetaDOCK for scoring, posing, and screening the protein–ligand complexes were found to be quite superior compared to the reference programs. Exhaustive molecular dynamics simulation and molecular mechanics Poisson–Boltzmann and surface area-based free energy estimation also suggest better energetic stability of the docking solutions produced by our meta-approach. We believe that the MetaDOCK approach is a useful packaging of the freely available software and provides a better alternative to the scientific community who are unable to afford costly commercial packages.
Introduction
Molecular docking has become an integral tool and technique and plays a major role in the efficacy of the high-throughput virtual screening protocols implemented in academic and industrial drug screening and discovery processes. In recent years, significant improvement in high-performance computing, optimized software and environmental platforms, and enriched publicly accessible compound libraries have aided the computational screening methods to become more accurate, effective, and useful.
Over the last few decades, numerous docking software packages have been developed for both academic and commercial purposes. Similarly, a number of publications in the last 20–25 years involving “docking” have also increased exponentially. A large number of excellent review articles and comparative studies have enriched the theoretical knowledge and practicability of the available programs.1−7 Among the many freely available docking programs, DOCK,8 AutoDOCK,9 AutoDock Vina,10 rDOCK,11 OEDOCKING,12,13 SWISSDOCK,14 UCSF DOCK,15 iGEMDOCK,16 HADDOCK,17 LeDock,18−20 and PLANTS21 are quite popular, user-friendly, and successful. Similarly, many commercial software like GOLD,22,23 Glide,24−26 LigandFit,27 molecular operating environment (MOE) Dock,28,29 Surflex-Dock,30 FlexX,31,32 and ICM33 are used quite regularly by both academic and industrial communities.
Most of the programs incorporated two important aspects of the docking protocol: an algorithm to search/sample different conformations and an objective scoring function to rank the probable docking solutions. Various sampling algorithms like matching algorithm,34−36 incremental construction,37−39 multiple copy simultaneous search,40−42 Monte Carlo (MC) methods,43,44 genetic algorithms (GAs),45−47 and swarm optimization methods48−51 have been developed to implement three different approaches like shape matching, systematic search, and stochastic search. On the other hand, scoring functions involve estimating the binding affinity between the protein and ligand. Scoring functions can be divided into force-field-based, empirical-, and knowledge-based assumptions and approximations.52−62 However, another important factor that differentiates various docking programs is the flexibility of the ligand and protein. Initial docking programs, like DOCK15 and FTDOCK,63 considered both the ligand and the receptor as rigid entities. However, most of the recently developed and/or updated programs implement flexible ligand and rigid receptor body docking approach, while a handful of programs GOLD,22,23 AutoDock,9 FlexX,31,32 ICM,33 and so forth managed to adopt flexibility for both ligand and protein receptor.
Hence, it is essential to understand the advantages and limitations of the available programs in order to be able to utilize the most compatible programs for a specific class of ligands and/or receptors. Rigorous and objective benchmarking studies enable us to assess the specific strength and limitation of the docking methodologies5−7,64−68 and subsequently can provide guidelines to improve the overall docking accuracy via complementary combinatorial approaches. Some of the recent and most exhaustive benchmarking studies5,6,69 showed that no single docking program has dominative advantages with regard to sampling and scoring power than other programs and individual programs have a lot of scope for improvement to become ideal choice for general docking experiments. As most of the current docking programs implement different search algorithms and scoring functions, their output usually turns out to be quite different. Thus, some of the docking programs can accurately predict the high-affinity compounds for a given receptor or ligand, while other programs may fail to perform at the same level. Hence, it is logical to believe that effective combination of different docking tools with varying sampling algorithms and scoring functions into a single platform may be a practical approach to achieve better predictions for docking-based virtual screening.
Combinatorial or consensus docking approaches have been used previously by some research groups which advocated reasonable success in limited dataset as well for specific targets.66,70−76 All these studies generally showed that consensus docking/scoring via a meta-docking approach was more effective than single docking programs or scoring function. Similarly, the results also suggested that combination of particular types of programs and/or scoring functions was less crucial as most of the double or triple combinations yielded similar performance results.
Commercial docking programs are very efficient and have served the drug industry and academic community very well. However, their prices are becoming quite steep day by day, whereas their accuracies are not getting significantly better compared to freely available programs. Hence, we thought of undertaking a consensus docking approach where we can utilize freely available software and combine their output in an efficient way so that the final predicted solution becomes much better compared to the individual programs and commercial programs. Here, we have used three popular freely available docking programs, AutoDock4.2,77 LeDock,18−20 and rDOCK,11 to create a meta-docking approach. The selection of the programs was done based on their ease of use, suitability for large-scale analysis, and performance on previous benchmarking studies. However, more programs can easily be added to the framework in order to complement the existing ones. Top ranked solutions from each program are pooled together and rescored before implementing a clustering filter based on root-mean-square deviation (rmsd) less than or equal to 2.5 Å between any two docking poses to be clustered together. Highest scored (rescore) solution from the largest cluster was selected as the most likely docking solution. Comprehensive and curated datasets (“refined” and “core” sets, respectively) from the PDBbind database78,79 were used following the standard docking benchmarking procedures. Scoring, posing, and screening capability of our approach, named MetaDOCK, were verified using rigorous benchmarking, and subsequently the performance measures were compared against one standard state-of-the-art commercial docking software, GOLD.22,23 Performances of MetaDOCK for scoring, posing, and screening the protein–ligand complexes were found to be quite superior compared to the GOLD program. Further, exhaustive molecular dynamics (MD) simulation followed by free energy estimation of 130 docking complexes using molecular mechanics Poisson–Boltzmann and surface area (MM-PBSA) method also showed much more stabilized fraction of the complexes derived by MetaDOCK than that produced by GOLD. We also present a simple, user-friendly web-based platform available at http://www.hpppi.iicb.res.in/metadock/index.html for the users to execute the MetaDOCK package and receive probable docking solutions. We believe that the MetaDOCK approach is a useful packaging of the freely available software and its benchmarking performance supports its usage for the scientific community who are unable to afford costly commercial alternatives.
Materials and Methods
Dataset
Docking Dataset
The PDBbind database78,79 provides a comprehensive collection of experimental binding affinity data for the biomolecular complexes deposited in the Protein Data Bank (PDB).80 PDBbind provides experimentally measured binding affinity data (i.e., Kd, Ki, or IC50 values) along with processed structural files of the protein and ligand molecules of each protein–ligand complex. In this study, we have used both the “refined set” and “core set” data from PDBbind v.2019 consisting of 4854 and 285 complexes, respectively.
Docking Decoy
Directory of Useful Decoys-Enhanced (DUD-E) database81,82 was used to generate ligand sets containing active and decoy ligands for 10 proteins, which are also part of the PDBbind dataset. For 10 protein targets, 10 active and 500 decoy ligands were collected from the DUD-E database. Overall, a total of 510 ligands were screened by the docking programs.
Meta-docking Approach
We implemented a meta-docking approach where three freely available programs, AutoDock4.2,77 LeDock,18−20 and rDOCK,11 were used to create the “MetaDOCK” package. The five best docking solutions from each of the three programs were pooled together and were rescored using AutoDock Vina10 program. Rescored solutions were clustered based on the similarity of the docking poses where any two docked complexes having rmsd less than or equal to 2.5 Å were clustered together. Highest scored (rescore) solution from the largest cluster was selected as the most likely docking solution. Only those complexes were considered for further analysis where at least 5 docking solutions each were produced by AutoDock4.2, LeDock, and rDOCK. Thus, out of the 4854 and 285 complexes from the “refined” and “core” datasets, MetaDOCK solutions were obtained for 3661 and 165 complexes, respectively.
To compare the results of MetaDOCK, we have also generated docking solutions using a well-known commercially available program called GOLD.22,23 GOLDscore was utilized to rank the docked solutions, and the top/highest rank solution was chosen for further comparison (GOLD_TS approach). Similarly, GOLD solutions were clustered based on the similarity of the docking poses, and top solution from the largest cluster was selected as the most likely solution (GOLD_LCTS approach). An rmsd cutoff of 2.5 Å was also implemented to cluster the docked solutions. Similarly, another non-commercial docking program, PLANTS,83 was also used to compare the performance of MetaDOCK. 3661 and 165 complexes from the “refined” and “core” sets were found to be common among the MetaDOCK, GOLD, and PLANTS methods.
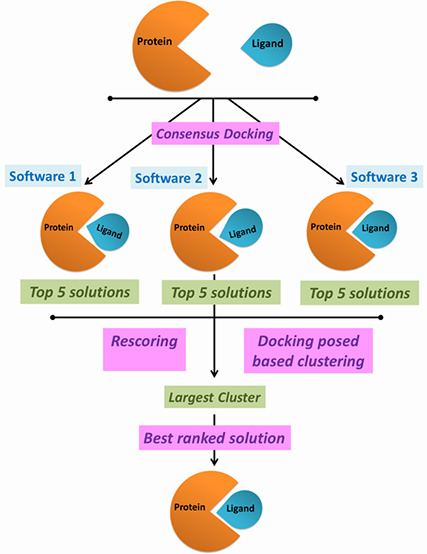
Figure 1 provides a flowchart and pictorial overview of the MetaDOCK and reference approaches.
Figure 1.

Schematic overview of the MetaDOCK and the reference docking protocols employed in this study.
Molecular Docking Programs
Autodock v4.2,77 which employs a Lamarckian GA, was used to score protein–ligand complexes. Population size and the number of energy assessments were set to 150 and 2 500 000, respectively. All other default parameters were used to calculate the docking scores.
LeDock18−20 is based on simulated annealing and evolutionary optimization of the ligand poses (position and orientation). Default options were used to generate docking solutions.
rDOCK11 implements GA-based search techniques followed by MC and simplex minimization (MIN) stages to generate low energy ligand poses. Default parameters were used to generate docking poses.
The GOLD software22,23 also utilizes GA for optimizing the fitness score of possible docking. The following parameters were used in the docking cycles: population size (100), selection pressure (1.100000), number of operations (100,000), number of islands (5), niche size (2), crossover weight (95), mutate weight (95), and migrate weight (10). The docking poses were ranked based on the GOLDscore of GOLD software.
PLANTS83 was also used as a reference docking program. PLANTS is based on a class of stochastic optimization algorithms called ant colony optimization. The default settings were used while docking the proteins and ligands from the PDBbind datasets.
Performance Analysis
The performance of the MetaDOCK approach was evaluated using three different criteria: scoring, posing, and screening power analysis.
Scoring Power Analysis
Scoring power of the docking approaches was evaluated by calculating correlation coefficient between the docking score of the best solution with respect to the experimentally derived binding affinity of the original complex (log Kd) extracted from the PDBbind dataset (refined and core sets). Correlation coefficients were calculated for the docking scores of the most likely solutions from MetaDOCK, GOLD, and PLANTS approaches.
Posing Power Analysis
Posing power of the docking algorithms was testified by calculating the rmsd between the poses of the docked and experimentally derived crystal complexes. Docking result was considered to be a success if the resultant rmsd between the corresponding docked and experimentally solved complexes becomes equal or less than 2 Å. Cumulative frequencies of the docking solutions below a specific rmsd value were calculated using in-house perl codes.
Screening Power Analysis
The capability of distinguishing the docking of true or active ligands from false or decoy ligands for a given receptor protein was testified via screening analysis. As mentioned before, for 10 protein targets, 10 active/true and 500 decoy/false ligands were collected from the DUD-E database. Overall, a total of 510 ligands were screened by the docking programs. For each of the 10 proteins, active and decoy ligands were docked via MetaDOCK and GOLD protocols and the docking scores for each selected complex were ranked and further utilized to generate the receiver operating characteristic (ROC) curve and area under the curve (AUC) statistics.
Ligand and Binding Pocket-Specific Performance Estimation
Performance and success of the docking protocols were evaluated based on the ligand and binding pocket-specific properties. For example, ligands were categorized based on the number of atoms and rotatable bonds. Similarly, ligand binding pockets of the receptor proteins were also categorized based on their surface area. The success rate of the docking protocols was evaluated for each category.
Molecular Dynamics-Based Energy Estimation
Molecular Dynamics Analyses
The three-dimensional (3D) structures of docked complexes derived from MetaDOCK and GOLD_LCTS protocols were subjected to MD simulation using the GROMACSv4.5.3 simulation package.84 Coordinates and topology files of the receptor and ligand molecules were generated with Amberff99sb85 and ACPYPE (AnteChamber PYthon Parser interface),86 respectively. Steepest-descent87 and conjugate-gradient88 MIN algorithms were used for complexes embedded within TIP3P water89-filled simulation box. Equilibration was done under NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature) conditions. Trajectories were saved at the interval of 0.02 ps, and a total of 500 000 snapshots were recorded from 10 ns simulation at a temperature of 300 K and a pressure of 1 atm. In total, 2600 ns or 2.6 μs of MD simulations was run for 260 (MetaDOCK: 130, GOLD_LCTS: 130) protein–ligand complexes.
Estimation of the Free Energy of Binding of Small Molecules
Estimation of the free energy of the docking complexes can be performed using end-point binding free energy calculation approaches such as MM-PBSA and molecular mechanics generalized Born surface area (MM-GBSA) methods.90 In this study, MM-PBSA was used considering the force field (Amberff99sb) compatibility and duration of the simulation.91 The gmx_MMPBSA tool was used to estimate the binding affinity of the receptor–ligand complexes derived from MD simulation-based ensemble structures.92 Binding free energies were calculated from 50 snapshots extracted at the interval of 200 ps from the 10 ns simulation trajectory. Thus, in total, 13 000 intermediate snapshot complex structures were used to calculate binding free energies of the complexes derived from MetaDOCK and GOLD_LCTS docking protocols. Interaction entropy (IE) module was used for the calculation of entropic component of the binding free energy.93
Construction of the MetaDOCK Server
A web-based server platform is created (URL: http://www.hpppi.iicb.res.in/metadock/index.html) where the MetaDOCK approach is implemented into a PHP-based front end and a python-based back end wrapper package.
Server Input Option
Users need to provide their receptor protein in PDB format, while the ligand 3D conformation needs to be submitted in MOL2 format. Additionally, the binding pocket information needs to be submitted in the prescribe format. Example input files are provided at the MetaDOCK server site for reference.
Server Output Option
The most likely solution of the given docked complex is represented in a 3D graphical view window. Additionally, coordinate files of the top three ranked docked complexes are provided for download option. Along with this, a dendrogram plot generated using the rmsd matrix for the largest cluster solutions is also displayed on the result page.
Results
Assessment of Scoring
Assessment of scoring power of the docking approaches was evaluated via drawing correlation between docking scores of most likely solutions and experimentally derived binding affinity values represented as logarithm of dissociation constant (log Kd). Figure S1 shows the correlation between the docking scores of complexes derived from MetaDOCK component programs (AutoDock4.2, LeDock, and rDOCK) and experimentally derived binding affinity of the original complex. Similarly, Figure 2 provides scatter plots of docking score versus log Kd value for each docking complex resulted by MetaDOCK, GOLD, and PLANTS. Interestingly, MetaDOCK-derived docking scores correlate much better (R2 = 0.37 and 0.05) with experimental affinities than that derived by the GOLD- and PLANTS-based approaches.
Figure 2.

Scoring power analysis and comparison. Scatter plots of the docking scores and the experimental binding free energies (log Kd) for the core (A–D) and refined sets (E–H), respectively. Trend line is colored in black, while the correlation coefficients between docking scores and experimental binding free energies are represented by R2 values. A–E, B–F, C–G, and D–H show data for MetaDOCK, GOLD_LCTS, GOLD_TS, and PLANTS, respectively. LCTS: largest cluster top score; TS: top score.
Assessment of Posing
In order to test the efficacy of the docking solutions predicted by the MetaDOCK, GOLD, and PLANTS approaches, posing similarity between the docked and native crystal ligand was evaluated via calculating the rmsd values. Lower rmsd values indicate better similarity and reproduction of the original crystal posing by the docking approaches. Posing efficacies of the individual programs were also tested and compared. Overall, LeDock performs slightly better than other two programs. However, the combination of the three programs in the form of MetaDOCK results similar or better posing efficacy (Figure S2). Figure 3 plots cumulative fractions of the docked complexes with respect to the rmsd values between docked and native complexes. It is evident that MetaDOCK outperforms the GOLD and PLANTS approaches for both refined and core dataset (Figure 3A,B), respectively. Almost 72% of MetaDOCK complexes from the core dataset (165 complexes) fall on or below 2.0 Å cutoff, which determines the success of the docking protocol, whereas only 36, 42, and 49% GOLD (derived from largest cluster and higher scoring approaches) and PLANTS docking solutions are found to be successful. For the refined set (3661 complexes), even higher success (73%) is achieved by MetaDOCK compared to GOLD- and PLANTS-based approaches (60, 63, and 45%, respectively) (Figure 3B). Figure 3C,D also shows that MetaDOCK-derived solutions possess overall lower rmsd with respect to the native complex than that achieved by the GOLD and PLANTS approaches for core (165 complexes) and refined datasets (3661complexes), respectively.
Figure 3.

Posing power analysis and comparison. Cumulative fractions of the docking complexes at each rmsd threshold are plotted in (A,B) for the core and refined datasets, respectively. Accuracy of the poses on or below 2 Å rmsd is marked by black dotted lines. Average rmsd of the docked complexes with respect to their corresponding original complexes is shown in (C,D) for the core and refined datasets, respectively. Error bars indicate standard error. *** = p-value ≤ 0.001.
Assessment of Screening
Screening capability of the MetaDOCK approach was evaluated by utilizing a dataset that contains active or native ligand for a receptor protein along with several inactive or decoy ligands that possess similar two-dimensional or 3D properties with respect to the native ligand. For 10 randomly selected protein targets, 10 active/true and 500 decoy/false ligands were collected from the DUD-E database. Overall, a total of 510 ligands were screened by the docking programs (Table S1). Docking scores were ranked, and ROC curve and AUC statistics were generated. Figure 4 shows the AUC values extracted from the corresponding ROC plots. Overall, for these 10 cases, MetaDOCK approach-based screening between true and false ligands achieved better performance as compared to the GOLD approach (Figure 4).
Figure 4.
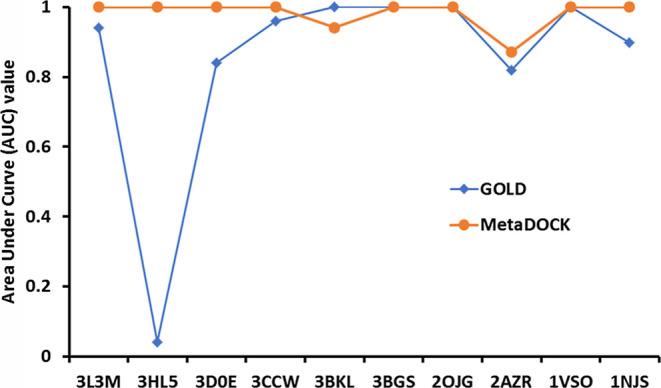
Screening power analysis and comparison. AUC values extracted from the ROC plots generated by calculating the true positive rate (TPR) and false positive rate (FPR) are plotted. TPR and FPR were calculated based on the docking scores of active and decoy ligands for each of the 10 randomly selected target proteins provided in the DUD-E database. PDB codes for the target proteins are marked in the X-axis.
Ligand and Binding Pocket-Specific Performance Estimation
In order to check if the performance of the MetaDOCK is biased for specific types of ligands, we further compared the success of docking for ligands categorized into groups based on their size and rotatable bonds. Ligands were categorized into four groups based on the number of atoms they possess. Categories of ligand size (LS) with 18 atoms or less (LS ≤ 18), 19 to 25 (18 < LS≥25), 26 to 35 (25 < LS ≥ 35), and greater than 35 (LS ≥ 35) contained similar number complexes. Figure 5A shows a higher number of MetaDOCK-derived complexes having lower rmsd (≤2 Å) compared to that achieved by the GOLD docking complexes for all the size categories. However, complexes with more than 2 Å rmsd are found to be consistently higher in GOLD results compared to the MetaDOCK. Figure 5B–E suggests much lower average rmsd for MetaDOCK-derived complexes categorized into various size bins of the ligands.
Figure 5.

Ligand size (LS)-based docking performance analysis and comparison. (A) Plot of the number of successful docking complexes (rmsd ≤ 2 Å) and complexes with rmsd > 2 Å for four categories of ligand size, LS ≤ 18, 18 < LS ≤ 25, 25 < LS < 35, LS ≥ 35. (B–E) Plot of average rmsd of the docked complexes with respect to their corresponding original complexes for various ligand size categories. Ligand size is categorized based on the number of atoms. *** indicates p-value ≤ 0.001.
We have also investigated whether MetaDOCK performs specifically better for ligands with higher or lower rotatable bonds. The numbers of MetaDOCK complexes having lower rmsd (≤2 Å) compared to that achieved by the GOLD approaches are found to be markedly higher (Figure 6A), while complexes with ≥2 Å rmsd are found to be higher in GOLD docking approaches. Average rmsd for MetaDOCK docking complexes is found to be consistently lower for all the ligands having lesser or higher number of rotatable bonds (Figure 6B–E).
Figure 6.

Rotatable bond count (RBC)-based docking performance analysis and comparison. (A) Plot of the number of successful docking complexes (rmsd ≤2 Å) and complexes with rmsd > 2 Å for four categories of RBC of the ligands, RBC < 4, 4 ≤ RBC < 6, 6 ≤ RBC < 10, RBC ≥ 10. (B–E) Plot of average rmsd of the docked complexes with respect to their corresponding original complexes for various RBC categories. RBC is categorized based on the number of rotatable bonds. *** indicates p-value ≤ 0.001.
A similar trend is also observed when the performance of the docking protocols was examined for smaller (<4000 Å2) and larger (≥4000 Å2) binding pockets of the receptor proteins (Figure S3). In both categories, much higher numbers of MetaDOCK complexes having better rmsd values were obtained compared to that achieved by the GOLD docking protocols.
Estimation of Energy of the Docking-Derived Complexes Via MM-PBSA
Stability and the binding energies of the docked complexes were further estimated via utilization of the MD-based ensemble conformer generation followed by the IE module of MM-PBSA analysis. 130 MetaDOCK and 130 GOLD-derived receptor–ligand docked complexes from the core dataset were successfully undergone MD simulation, and subsequent ensembles of intermediate complexes were utilized to estimate the binding energy of the initial complex. The gmx_MMPBSA package was used to calculate the binding free energy (see Methods). Table S2 provides the binding free energy (ΔG) and its enthalpy (ΔH) and entropy (TΔS) components of the 260 docked complexes for which the MD-based MM-PBSA analysis results could be obtained. Figure 7A plots the gain/loss of binding free energies (ΔΔG; ΔG of MetaDOCK complex minus ΔG of GOLD complex) of the docked complexes derived by MetaDOCK with respect to complexes obtained via GOLD docking. 66% of the complexes that underwent MD-based binding energy estimation show binding free energy gain by the MetaDOCK approach (Figure 7A) compared to 34% obtained by the GOLD approach. Figure 7B plots the distribution of MD-derived binding free energies of the docked complexes with respect to their rmsd values when compared to the native complex. Interestingly, 78% MetaDOCK complexes obtained negative (favorable) binding free energies (ΔGs) compared to 72% of the GOLD docking complexes. However, 63% of the MetaDOCK complexes with negative ΔGs were found to be successful (rmsd ≤ 2 Å) compared to only 40% of the GOLD-derived complexes with negative ΔGs after MD simulations.
Figure 7.

Molecular dynamics (MD) simulation and MM-PBSA analysis-based energy estimation of the docked complexes. 130 + 130 receptor–ligand docked complexes produced by MetaDOCK and GOLD_LCTS, respectively, were subjected to MD simulations, and ensembles of intermediate complexes were utilized to estimate the binding energy of the initial complex using the MM-PBSA analysis. (A) Plot of the gain/loss of binding free energies (ΔΔG; ΔG of MetaDOCK complex minus ΔG of GOLD complex) of the docked complexes derived by MetaDOCK with respect to complexes obtained via GOLD docking. Gain of binding free energies by MetaDOCK is colored by green, while loss is marked in red color. (B) Plot of the fraction of docking complexes with negative/positive binding free energies (ΔG or DG) along with docking success cutoff represented by rmsd ≤2 Å.
MetaDOCK Web Server
MetaDOCK server provides a simple front end to upload structures of input protein and ligand along with the binding pocket information. The back end of the server runs the programs in the background and displays the most likely complex solution in an interactive 3D graphical window. The coordinate file of the docked complex is given for download. Figure S4 provides a snapshot of the MetaDOCK server input and output options.
Discussion
Contribution of molecular docking to structure-based drug design, virtual screening, and optimization of lead compounds has become even more prominent during the recent COVID-19 pandemic time where the modeling community has been employed to study a wide range of potential pharmaceutical molecules and/or repurposed existing drugs against the SARS-CoV2 proteins using molecular modeling and docking approaches.
However, still, there is a lot of scope of improvement for the docking algorithms to successfully simulate and replicate the experimental binding results. In addition to mechanistic and algorithmic improvements, several additive meta-approaches have been associated with molecular docking protocols to improve the overall accuracy.94−101 Here, we propose a combinatorial or consensus docking approach where complementary power of the existing methods is captured and most likely docking solution was extracted utilizing posing similarity-based statistics. We created a meta-docking protocol by combining the results of AutoDock4.2, LeDock, and rDOCK programs as these are freely available, easy to use, and suitable for large-scale analysis and produced better performance on benchmarking studies. Docking solutions from each program are pooled together and rescored before implementing a clustering-based posing similarity filter. PDBbind database was used to evaluate the scoring, posing, and screening capability of our approach. Further, the performance measures were compared against one standard state-of-the-art commercial docking software, GOLD, and one freely available program, PLANTS. We also implemented an exhaustive MD simulation and MM-PBSA-based free energy estimation to investigate the energetic stability of the docking solution produced by our meta-approach. This MetaDOCK approach is made freely available at http://www.hpppi.iicb.res.in/metadock/index.html for the users to perform docking experiments.
Rigorous comparison and benchmarking analyses have been performed to check the efficacy of the MetaDOCK approach. A well curated and reliable dataset is very important to add the reliability of the benchmarking process. Hence, we have used the curated protein–ligand docking complexes from the PDBbind database where a reasonably large number of dataset (refined set) as well as highly curated core set are also available for testing molecular docking efficiencies. Protein and ligands 3661 and 165 complexes from the “refined” and “core” set were successfully re-docked by the MetaDOCK component docking software, namely, AutoDock4.2, LeDock, and rDOCK. We employed three pronged evaluation criteria using scoring, posing, and screening power analysis to test the performance of the MetaDOCK approach. Scoring power analysis showed much better correlation between docking scores derived from the MetaDOCK method with experimental affinities compared to that derived by the GOLD- and PLANTS-based approaches. More importantly, posing power comparison clearly showed much better performance of the MetaDOCK approach in capturing the original/native pose. This finding is definitely exciting and advocates the utilization of combinatorial or consensus docking approaches over state-of-the-art, expensive, and expertise-dependent software packages. It must be noted that no special criteria was considered in the selection of the individual component algorithms in the MetaDOCK approach. However, it was noticed that LeDock generally performs best among the three followed by rDOCK and AutoDock4.2. Combinations with other freely available algorithms may yield better or worse performance although previous benchmarking studies have suggested that most of the double or triple combinations yielded similar performance results.1 Similarly, there was no specific reason to choose GOLD as a reference commercial program. We selected GOLD based on its popularity, credibility, and availability with us as a standalone version. Unfortunately, we could not procure other commercial software like Glide,24−26 MOE,28,29 ICM,33 and so forth to include them in the comparison study.
Capability of distinguishing an active ligand from inactive (decoy) ligands by the docking programs was testified using screening power analysis. DUD-E database is an extremely useful resource, which is designed to help benchmarking of molecular docking programs by providing challenging decoys. For 10 protein targets, 10 active and 500 decoy ligands were screened by the docking programs. AUC values derived from the ROC statistics suggest better performance of MetaDOCK compared to GOLD approaches.
Finally, the stability of the docked complexes was evaluated by estimating the binding free energies of the ensemble complex structures derived from MD simulation runs followed by MM-PBSA analysis. An exhaustive MD protocol was implemented where 130 docked complexes each from MetaDOCK and GOLD were subjected to MD run followed by energy estimation. In total, 13 000 intermediate snapshot complex (50 from each MD run) structures were extracted from the MD simulation runs with a combined simulation time of 2600 ns. More than 66% of the docked complexes derived by MetaDOCK approach showed a gain in stability represented by gain of binding free energies. This result further ascertains better efficacy of the MetaDOCK approach. However, comparison of the binding free energy with respect to the original, experimentally solved complex is warranted. Hence, a subset consisting randomly selected 25 complexes from the 130 complexes was further utilized to check whether MetaDOCK-derived docked complexes gaining similar or better binding energies with respect to the original complex. Figure S5 indeed shows that more MetaDOCK complexes possess better binding energy profile with respect to the original complex than that achieved by the GOLD docking complexes.
The MetaDOCK webserver platform provides an opportunity for the users to run their docking experiments on the fly and receive a reasonably reliable docking solution for further analysis and verifications. Although it does not include any novel docking algorithm, careful combination of existing programs and statistical analysis enables MetaDOCK to be a useful software for the scientific community who are unable to afford costly commercial alternatives.
Acknowledgments
S.C. acknowledges CSIR-IICB for infrastructural and financial support. IMK acknowledges DBT, Government of India, for his Ph.D. fellowship (DBT/2019/IICB/1213).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c07619.
Scatter plots of the docking scores and the experimental binding free energies (log Kd) for the core and refined sets for the individual programs used in MetaDOCK approach, difference of rmsd of the complexes generated by AutoDock4, LeDock, and rDOCK, respectively, with respect to the most likely complex derived from the MetaDOCK approach, binding pocket surface area (BPSA)-based docking performance analysis and comparison, screenshots of the MetaDOCK web server input and output options, molecular dynamics (MD) simulation and MM-PBSA analysis-based energy estimation of the docked complexes, list of decoy (ZINC database ID) and active (SMILE) ligands, and binding free energy (ΔG) and its enthalpy (ΔH) and entropy (TΔS) components of the docked complexes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Prieto-Martínez F. D.; Arciniega M.; Medina-Franco J. L. Molecular docking: current advances and challenges. TIP, Rev. Espec. Cienc. Quim.-Biol. 2018, 21, 65–87. 10.22201/fesz.23958723e.2018.0.143. [DOI] [Google Scholar]
- Pinzi L.; Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. 10.3390/ijms20184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagadala N. S.; Syed K.; Tuszynski J. Software for molecular docking: a review. Biophys. Rev. 2017, 9, 91–102. 10.1007/s12551-016-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X. Y.; Zhang H. X.; Mezei M.; Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Sun H.; Yao X.; Li D.; Xu L.; Li Y.; Tian S.; Hou T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. 10.1039/c6cp01555g. [DOI] [PubMed] [Google Scholar]
- Çınaroğlu S. S.; Timuçin E. Comparative Assessment of Seven Docking Programs on a Nonredundant Metalloprotein Subset of the PDBbind Refined. J. Chem. Inf. Model. 2019, 59, 3846–3859. 10.1021/acs.jcim.9b00346. [DOI] [PubMed] [Google Scholar]
- Zev S.; Raz K.; Schwartz R.; Tarabeh R.; Gupta P. K.; Major D. T. Benchmarking the Ability of Common Docking Programs to Correctly Reproduce and Score Binding Modes in SARS-CoV-2 Protease Mpro. J. Chem. Inf. Model. 2021, 61, 2957–2966. 10.1021/acs.jcim.1c00263. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D.; Blaney J. M.; Oatley S. J.; Langridge R.; Ferrin T. E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- Österberg F.; Morris G. M.; Sanner M. F.; Olson A. J.; Goodsell D. S. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins 2002, 46, 34–40. 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. A.D. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Carmona S.; Alvarez-Garcia D.; Foloppe N.; Garmendia-Doval A. B.; Juhos S.; Schmidtke P.; Barril X.; Hubbard R. E.; Morley S. D. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- Kelley B. P.; Brown S. P.; Warren G. L.; Muchmore S. W. POSIT: Flexible Shape-Guided Docking For Pose Prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. 10.1021/acs.jcim.5b00142. [DOI] [PubMed] [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen W. J.; Balius T. E.; Mukherjee S.; Brozell S. R.; Moustakas D. T.; Lang P. T.; Case D. A.; Kuntz I. D.; Rizzo R. C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. M.; Chen C. C. GEMDOCK: a generic evolutionary method for molecular docking. Proteins: Struct., Funct., Genet. 2004, 55, 288–304. 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. HADDOCK: A Protein–Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Caflisch A. Discovery of ZAP70 inhibitors by high-throughput docking into a conformation of its kinase domain generated by molecular dynamics. Bioorg. Med. Chem. Lett. 2013, 23, 5721–5726. 10.1016/j.bmcl.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Unzue A.; Zhao H.; Lolli G.; Dong J.; Zhu J.; Zechner M.; Dolbois A.; Caflisch A.; Nevado C. The ″Gatekeeper″ Residue Influences the Mode of Binding of Acetyl Indoles to Bromodomains. J. Med. Chem. 2016, 59, 3087–3097. 10.1021/acs.jmedchem.5b01757. [DOI] [PubMed] [Google Scholar]
- Liu N.; Xu Z. Using LeDock as a docking tool for computational drug design. IOP Conf. Ser. Earth Environ. Sci. 2019, 218, 012143. 10.1088/1755-1315/218/1/012143. [DOI] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. Empirical Scoring Functions for Advanced Protein–Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Venkatachalam C. M.; Jiang X.; Oldfield T.; Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graphics Modell. 2003, 21, 289–307. 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
- Vilar S.; Cozza G.; Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- Corbeil C. R.; Williams C. I.; Labute P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. 2012, 26, 775–786. 10.1007/s10822-012-9570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer R.; Jain A. N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput.-Aided Mol. Des. 2012, 26, 687–699. 10.1007/s10822-011-9533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rarey M.; Kramer B.; Lengauer T.; Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- Kramer B.; Rarey M.; Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 1999, 37, 228–241. . [DOI] [PubMed] [Google Scholar]
- Neves M. A.; Totrov M.; Abagyan R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J. Comput.-Aided Mol. Des. 2012, 26, 675–686. 10.1007/s10822-012-9547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brint A. T.; Willett P. Algorithms for the Identification of Three-Dimensional Maximal Common Substructures. J. Chem. Inf. Comput. Sci. 1987, 27, 152–158. 10.1021/ci00056a002. [DOI] [Google Scholar]
- Fischer D.; Norel R.; Wolfson H.; Nussinov R. Surface motifs by a computer vision technique: searches, detection, and implications for protein-ligand recognition. Proteins 1993, 16, 278–292. 10.1002/prot.340160306. [DOI] [PubMed] [Google Scholar]
- Norel R.; Fischer D.; Wolfson H. J.; Nussinov R. Molecular surface recognition by a computer vision-based technique. Protein Eng. 1994, 7, 39–46. 10.1093/protein/7.1.39. [DOI] [PubMed] [Google Scholar]
- Rarey M.; Kramer B.; Lengauer T.; Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- DesJarlais R. L.; Sheridan R. P.; Dixon J. S.; Kuntz I. D.; Venkataraghavan R. Docking flexible ligands to macromolecular receptors by molecular shape. J. Med. Chem. 1986, 29, 2149–2153. 10.1021/jm00161a004. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Kuntz I. D. Conformational analysis of flexible ligands in macromolecular receptor sites. J. Comput. Chem. 1992, 13, 730–748. 10.1002/jcc.540130608. [DOI] [Google Scholar]
- Miranker A.; Karplus M. Functionality maps of binding sites: a multiple copy simultaneous search method. Proteins 1991, 11, 29–34. 10.1002/prot.340110104. [DOI] [PubMed] [Google Scholar]
- Eisen M. B.; Wiley D. C.; Karplus M.; Hubbard R. E. HOOK: a program for finding novel molecular architectures that satisfy the chemical and steric requirements of a macromolecule binding site. Proteins 1994, 19, 199–221. 10.1002/prot.340190305. [DOI] [PubMed] [Google Scholar]
- Böhm H. J. LUDI rule-based automatic design of new substituents for enzyme inhibitor leads. J. Comput.-Aided Mol. Des. 1992, 6, 593–606. 10.1007/BF00126217. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S.; Lauble H.; Stout C. D.; Olson A. J. Automated docking in crystallography: analysis of the substrates of aconitase. Proteins 1993, 17, 1–10. 10.1002/prot.340170104. [DOI] [PubMed] [Google Scholar]
- Hart T. N.; Read R. J. A multiple-start Monte Carlo docking method. Proteins 1992, 13, 206–222. 10.1002/prot.340130304. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. . [DOI] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Oshiro C. M.; Kuntz I. D.; Dixon J. S. Flexible ligand docking using a genetic algorithm. J. Comput.-Aided Mol. Des. 1995, 9, 113–130. 10.1007/bf00124402. [DOI] [PubMed] [Google Scholar]
- Kennedy J.; Eberhart R. C.. Particle swarm optimization. Proceedings of the IEEE International Conference on Neural Networks, 1995;Vol. 4, pp 1942–1948.
- Chen H. M.; Liu B. F.; Huang H. L.; Hwang S. F.; Ho S. Y. SODOCK: swarm optimization for highly flexible protein-ligand docking. J. Comput. Chem. 2007, 28, 612–623. 10.1002/jcc.20542. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. A.D. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zhao L.; Li W.; Zhao D.; Song M.; Yang Y. FIPSDock: a new molecular docking technique driven by fully informed swarm optimization algorithm. J. Comput. Chem. 2013, 34, 67–75. 10.1002/jcc.23108. [DOI] [PubMed] [Google Scholar]
- Kollman P. A. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. 10.1021/cr00023a004. [DOI] [Google Scholar]
- Åqvist J.; Luzhkov V. B.; Brandsdal B. O. Ligand binding affinities from MD simulations. Acc. Chem. Res. 2002, 35, 358–365. 10.1021/ar010014p. [DOI] [PubMed] [Google Scholar]
- Böhm H. J. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Comput.-Aided Mol. Des. 1998, 12, 309–323. 10.1023/a:1007999920146. [DOI] [PubMed] [Google Scholar]
- Gehlhaar D. K.; Verkhivker G. M.; Rejto P. A.; Sherman C. J.; Fogel D. B.; Fogel L. J.; Freer S. T. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming. Chem. Biol. 1995, 2, 317–324. 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Verkhivker G. M.; Bouzida D.; Gehlhaar D. K.; Rejto P. A.; Arthurs S.; Colson A. B.; Freer S. T.; Larson V.; Luty B. A.; Marrone T.; Rose P. W. Deciphering common failures in molecular docking of ligand-protein complexes. J. Comput.-Aided Mol. Des. 2000, 14, 731–751. 10.1023/a:1008158231558. [DOI] [PubMed] [Google Scholar]
- Jain A. N. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J. Comput.-Aided Mol. Des. 1996, 10, 427–440. 10.1007/bf00124474. [DOI] [PubMed] [Google Scholar]
- Eldridge M. D.; Murray C. W.; Auton T. R.; Paolini G. V.; Mee R. P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- Muegge I.; Martin Y. C. A General and Fast Scoring Function for Protein–Ligand Interactions: A Simplified Potential Approach. J. Med. Chem. 1999, 42, 791–804. 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- Mitchell J. B. O.; Laskowski R. A.; Alex A.; Thornton J. M. BLEEP?potential of mean force describing protein-ligand interactions: I. Generating potential. J. Comput. Chem. 1999, 20, 1165–1176. . [DOI] [Google Scholar]
- Ishchenko A. V.; Shakhnovich E. I. SMall Molecule Growth 2001 (SMoG2001): An Improved Knowledge-Based Scoring Function for Protein–Ligand Interactions. J. Med. Chem. 2002, 45, 2770–2780. 10.1021/jm0105833. [DOI] [PubMed] [Google Scholar]
- Feher M.; Deretey E.; Roy S. BHB: a simple knowledge-based scoring function to improve the efficiency of database screening. J. Chem. Inf. Comput. Sci. 2003, 43, 1316–1327. 10.1021/ci030006i. [DOI] [PubMed] [Google Scholar]
- Gabb H. A.; Jackson R. M. M. J.; Sternberg M. J. E. Modelling protein docking using shape complementarity, electrostatics and biochemical information 1 1Edited by J. Thornton. J. Mol. Biol. 1997, 272, 106–120. 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- Hartshorn J. M.; Mortenson P. N. Diverse, high-quality test set for the validation of protein-ligand native and cross-docking performance. J. Med. Chem. 2007, 50, 726–741. 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- Plewczynski D.; Łaźniewski M.; Augustyniak R.; Ginalski K. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem. 2011, 32, 742–755. 10.1002/jcc.21643. [DOI] [PubMed] [Google Scholar]
- Grinter S. Z.; Yan C.; Huang S. Y.; Jiang L.; Zou X. Automated Large-Scale File Preparation, Docking, and Scoring: Evaluation of ITScore and STScore Using the 2012 Community Structure-Activity Resource Benchmark. J. Chem. Inf. Model. 2013, 53, 1905–1914. 10.1021/ci400045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm-Ganamet K. L.; Smith R. D.; Dunbar J. B. Jr.; Stuckey J. A.; Carlson H. A. CSAR benchmark exercise 2011-2012: evaluation of results from docking and relative ranking of blinded congeneric series. J. Chem. Inf. Model. 2013, 53, 1853–1870. 10.1021/ci400025f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. C. Beware of docking!. Trends Pharmacol. Sci. 2015, 36, 78–95. 10.1016/j.tips.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Tuccinardi T.; Poli G.; Romboli V.; Giordano A.; Martinelli A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. 10.1021/ci500424n. [DOI] [PubMed] [Google Scholar]
- Charifson P. S.; Corkery J. J.; Murcko M. A.; Walters W. P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- Wang R.; Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 2001, 41, 1422–1426. 10.1021/ci010025x. [DOI] [PubMed] [Google Scholar]
- Wang R.; Lu Y.; Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- Chaput L.; Mouawad L. Efficient conformational sampling and weak scoring in docking programs? Strategy of the wisdom of crowds. J. Cheminf. 2017, 9, 37. 10.1186/s13321-017-0227-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta P. N.; Das K. K. Inhibition activities of catechol diether based non-nucleoside inhibitors against the HIV reverse transcriptase variants: Insights from molecular docking and ONIOM calculations. J. Mol. Graphics Modell. 2017, 75, 294–305. 10.1016/j.jmgm.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Fani N.; Sattarinezhad E.; Bordbar A. K. Identification of new 2,5-diketopiperazine derivatives as simultaneous effective inhibitors of αβ-tubulin and BCRP proteins: Molecular docking, Structure–Activity Relationships and virtual consensus docking studies. J. Mol. Struct. 2017, 1137, 362–372. 10.1016/j.molstruc.2017.02.049. [DOI] [Google Scholar]
- Onawole A. T.; Sulaiman K. O.; Adegoke R. O.; Kolapo T. U. Identification of potential inhibitors against the Zika virus using consensus scoring. J. Mol. Graphics Modell. 2017, 73, 54–61. 10.1016/j.jmgm.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Fang X.; Lu Y.; Wang S. The PDBbind Database: Collection of Binding Affinities for Protein–Ligand Complexes with Known Three-Dimensional Structures. J. Med. Chem. 2004, 47, 2977–2980. 10.1021/jm030580l. [DOI] [PubMed] [Google Scholar]
- Wang R.; Fang X.; Lu Y.; Yang C. Y.; Wang S. The PDBbind Database: Methodologies and updates. J. Med. Chem. 2005, 48, 4111–4119. 10.1021/jm048957q. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K.; Irwin J. J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. Empirical Scoring Functions for Advanced Protein–Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Case D. A.; Cheatham T. E. 3rd.; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa da Silva A. W.; Vranken W. F. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocedal J.; Wright S. J.. Numerical Optimization. Numerical Optimization; Glynn P., Robinson S. M., Eds.; Springer Nature, 2006; pp 1–664. [Google Scholar]
- Straeter T. A.On the Extension of the Davidon-Broyden Class of Rank One, Quasi-Newton Minimization Methods to an Infinite Dimensional Hilbert Space with Applications to Optimal Control Problems; North Carolina State Univ.: Raleigh, NC, United States, 1971. [Google Scholar]
- Mark P.; Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. 10.1021/jp003020w. [DOI] [Google Scholar]
- Genheden S.; Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discovery 2015, 10, 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.; Sun H.; Wang J.; Wang Z.; Liu H.; Zhang J. Z. H.; Hou T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- Valdés-Tresanco M. S.; Valdés-Tresanco M. E.; Valiente P. A.; Moreno E. gmx_MMPBSA A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- Duan L.; Liu X.; Zhang J. Z. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722. 10.1021/jacs.6b02682. [DOI] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- Weiner S. J.; Kollman P. A.; Case D. A.; Singh U. C.; Ghio C.; Alagona G.; Profeta S. Jr.; Weiner P. A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins. J. Am. Chem. Soc. 1984, 106, 765–784. 10.1021/ja00315a051. [DOI] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. 10.1002/jcc.540040211. [DOI] [Google Scholar]
- Gervasio F. L.; Laio A.; Parrinello M. Flexible docking in solution using metadynamics. J. Am. Chem. Soc. 2005, 127, 2600–2607. 10.1021/ja0445950. [DOI] [PubMed] [Google Scholar]
- Barducci A.; Bonomi M.; Parrinello M. Metadynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 826–843. 10.1002/wcms.31. [DOI] [Google Scholar]
- Clark A. J.; Tiwary P.; Borrelli K.; Feng S.; Miller E. B.; Abel R.; Friesner R. A.; Berne B. J. Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J. Chem. Theory Comput. 2016, 12, 2990–2998. 10.1021/acs.jctc.6b00201. [DOI] [PubMed] [Google Scholar]
- Lemkul J. A.; Bevan D. R. Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. J. Phys. Chem. B 2010, 114, 1652–1660. 10.1021/jp9110794. [DOI] [PubMed] [Google Scholar]
- Zeller F.; Zacharias M. Efficient calculation of relative binding free energies by umbrella sampling perturbation. J. Comput. Chem. 2014, 35, 2256–2262. 10.1002/jcc.23744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.