

Abstract

Quantum chemistry is a promising application for noisy intermediate-scale quantum (NISQ) devices. However, quantum computers have thus far not succeeded in providing solutions to problems of real scientific significance, with algorithmic advances being necessary to fully utilize even the modest NISQ machines available today. We discuss a method of ground state energy estimation predicated on a partitioning of the molecular Hamiltonian into two parts: one that is noncontextual and can be solved classically, supplemented by a contextual component that yields quantum corrections obtained via a Variational Quantum Eigensolver (VQE) routine. This approach has been termed Contextual Subspace VQE (CS-VQE); however, there are obstacles to overcome before it can be deployed on NISQ devices. The problem we address here is that of the ansatz, a parametrized quantum state over which we optimize during VQE; it is not initially clear how a splitting of the Hamiltonian should be reflected in the CS-VQE ansätze. We propose a “noncontextual projection” approach that is illuminated by a reformulation of CS-VQE in the stabilizer formalism. This defines an ansatz restriction from the full electronic structure problem to the contextual subspace and facilitates an implementation of CS-VQE that may be deployed on NISQ devices. We validate the noncontextual projection ansatz using a quantum simulator and demonstrate chemically precise ground state energy calculations for a suite of small molecules at a significant reduction in the required qubit count and circuit depth.
1. Introduction
Quantum computers promise to yield solutions to complex problems that have previously been unattainable by classical means, yet experimental demonstration remains challenging. To date, some of the largest molecules simulated on noisy intermediate-scale quantum (NISQ) hardware are H12–albeit only a Hartree–Fock calculation–conducted by Google using just 12 of the 53 qubits available on their superconducting quantum processor Sycamore,1 and H2O performed independently by IonQ using 3 qubits of an unspecified proprietary trapped ion device2 and by IBM using 5 of the 27 qubits on the now-decommissioned ibmq_dublin superconducting device.3
Due to the limitations of short coherence times, restrictive qubit connectivity and high noise floors that characterize the NISQ era, we are not able to harness the full state-space afforded to these machines. To circumvent the above issues, we turn to the class of variational quantum algorithms, of which the Variational Quantum Eigensolver (VQE)4 is most widely studied. In contrast with eigenvalue-finding algorithms requiring fault-tolerant machines such as Quantum Phase Estimation (QPE),5 which necessitates state evolution over an extended period of coherence, VQE executes a large ensemble of comparatively shallow parametrized circuits to estimate energy expectation values, informing a classical optimizer that updates the parameter settings before reinitialization of the quantum circuit. Its success is predicated on the variational principle, meaning the ground state energy of the system bounds expectation values from below.6
However, VQE is not without its challenges. First of all, the parametrized quantum state mentioned above, known as an ansatz, needs to be constructed carefully: It must be sufficiently expressible so the subspace of quantum states it spans contains the true ground state. On the other hand, if the ansatz is too expressible, we run into the problem of barren plateaus7 where we observe vanishing gradients. This is more often a symptom of “hardware efficient” ansätze,1,8−12 which aim to access the largest possible region of Hilbert space for the fewest number of native quantum gates.
To avoid barren plateaus, one must take into account some of the underlying problem structure to define ansatz circuits whose images are confined to a smaller but more targeted region of Hilbert space. Within this category are “chemically inspired” ansätze that represent sequences of electronic excitation operators in circuit; unitary coupled cluster (UCC)13,14 is widely acknowledged as the gold standard for electronic structure simulations, albeit computationally very expensive in practice.
More recently, we have seen the development of hybrid ansätze that bridge the gap between hardware efficiency and chemical motivation. For example, Gard et al.15 designed a compact circuit designed to conserve molecule symmetries such as particle number and spin, while Adaptive Derivative-Assembled Pseudo-Trotter (ADAPT) VQE16−19 describes a more complete approach to scalable quantum chemistry simulations by defining selection criteria of ansatz terms from a pool of excitation operators.
Second, the energy estimation procedure in VQE invokes the measurement problem; in order to mitigate statistical error, many prepare-and-measure cycles are necessary to achieve sufficient precision in the estimate. The advances made in recent years toward measurement reduction techniques are expansive20−29 and range from classical pre/postprocessing of the measurement information, such as in classical shadow tomography,30,31 to Hamiltonian term-grouping schemes and reductions in the number of Hamiltonian terms at a cost of coherent resource, such as in unitary partitioning.32−36 Combined with techniques of error mitigation,37−44 one can optimize VQE with the objective of maximal NISQ resource utilization.
In this work, we are concerned with Contextual Subspace VQE (CS-VQE),45 which describes a method of partitioning the molecular Hamiltonian into disjoint parts so that an electronic structure problem may be simulated to some degree on the available quantum device, even when the dimension of the full problem is too great to be encoded on the number of qubits available. This is supplemented by some classical overhead, but this often permits one to achieve chemical precision (to within 1.6 mHa ≈ 4 kJ/mol of the full configuration interaction (FCI) energy) at a saving of qubits, as indicated by Kirby et al.45 We highlight the relevance of chemical precision over accuracy here; since a minimal basis set (STO-3G) will be used throughout our benchmark, one should not expect agreement with experimentally obtained molecular energies and thus chemical accuracy is not an appropriate phrase. A finer basis set such as cc-pVxZ where x = D, T, Q, etc. should be used if one wishes to assess accuracy; however, this comes at the cost of increased qubits.
There has since
been further research into the use of classical
estimates of the electronic structure problem to reduce the resource
requirements on quantum hardware. In particular, Classically Boosted
VQE (CB-VQE)46 identifies classically tractable
states and excludes them from the quantum simulation, alleviating
some measurement and fidelity requirements of the VQE routine. CS-VQE
also bears a resemblance to the qubit reduction technique of qubit
tapering,47,48 which exploits 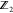 symmetries of the Hamiltonian; the differences
and similarities are highlighted herein and by Kirby et al.45
symmetries of the Hamiltonian; the differences
and similarities are highlighted herein and by Kirby et al.45
There are still a number of problems to address before CS-VQE may be successfully deployed on real quantum hardware, most notably with regard to the ansatz, which is the principal focus of this work. To aid this objective, we place the method on a strong theoretical footing of stabilizer subspaces and projections therein; this reformulation is better suited to efficient implementation, which is being addressed through the Symmer project.49 This rephrasing of CS-VQE illuminates the matter of constructing ansätze for the contextual subspace and renders this method compatible with contemporary approaches to ansatz construction such as ADAPT-VQE.
2. Preliminaries
The notation used throughout shall be to write operators in standard capital font (A, B, C, etc.), with the exception of single-qubit Pauli operators being written in the form
| 1 |
for p ∈ {0, 1, 2,
3}. Sets are denoted by script letters 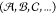 and vector spaces by bold script typeface
and vector spaces by bold script typeface 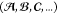 . The state space of N qubits
may be identified with the 2N dimensional
Hilbert space
. The state space of N qubits
may be identified with the 2N dimensional
Hilbert space  , with the space of (bounded) linear operators
acting upon
, with the space of (bounded) linear operators
acting upon 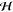 denoted
denoted 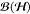 .
.
We introduce the Pauli group, 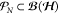 , consisting of operators
, consisting of operators 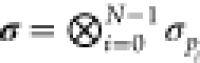 for pi ∈ {0, 1, 2, 3}, up to multiplication by ±1, ±i. Note the distinction between the bold font σ denoting tensor products and σp a single-qubit Pauli operator; we will sometimes write
for pi ∈ {0, 1, 2, 3}, up to multiplication by ±1, ±i. Note the distinction between the bold font σ denoting tensor products and σp a single-qubit Pauli operator; we will sometimes write 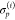 to index explicitly the qubit position
to index explicitly the qubit position 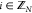 on which it acts. We shall also make use
of the commutator [A, B] ≔ AB – BA and anticommutator {A, B}
≔ AB + BA, defined for operators
on which it acts. We shall also make use
of the commutator [A, B] ≔ AB – BA and anticommutator {A, B}
≔ AB + BA, defined for operators  , which are zero when A and B commute/anticommute, respectively.
, which are zero when A and B commute/anticommute, respectively.
An N-qubit Hamiltonian can be written in the form
| 2 |
for a set of Pauli operators  ; specifying real coefficients ensures that
; specifying real coefficients ensures that 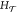 is Hermitian. The objective
of quantum chemistry simulations is to estimate the ground state energy
is Hermitian. The objective
of quantum chemistry simulations is to estimate the ground state energy
| 3 |
where 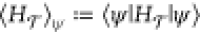 is the expectation value of
is the expectation value of 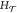 with respect to some quantum state
with respect to some quantum state 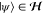 . Many physical properties of the target
system are determined by the ground state, motivating this goal.
. Many physical properties of the target
system are determined by the ground state, motivating this goal.
The Variational Quantum Eigensolver (VQE) quantum–classical
hybrid algorithm4 is the most widely studied
means of achieving this on NISQ hardware. VQE requires a parametrized
ansatz state, 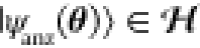 , whose parameters θ are
manipulated within a classical optimization scheme that aims to minimize
the energy expectation value
, whose parameters θ are
manipulated within a classical optimization scheme that aims to minimize
the energy expectation value
| 4 |
evaluated via many prepare-and-measure cycles. The choice of ansatz restricts us to a subspace of quantum states and therefore must be carefully designed to be sufficiently expressible so as to capture the true ground state of the system.
A common form of ansatz state, particularly in relation to the electronic structure problem, is
| 5 |
where  is some fixed reference state in which
the quantum circuit is initialized and
is some fixed reference state in which
the quantum circuit is initialized and 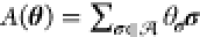 for parameters
for parameters 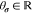 and Pauli operators
and Pauli operators  ; the unitary eiA(θ) effects excitations above the reference state.
Such ansätze as unitary coupled cluster (UCC)13,14 may by expressed by our choice of A (taking as
reference the Hartree–Fock state), in addition to any others
based on the theory of excitation operators such as ADAPT-VQE.16−19 The quantum advantage in VQE stems from the ability to prepare classically
intractable states from our parametrized ansatz circuits.
; the unitary eiA(θ) effects excitations above the reference state.
Such ansätze as unitary coupled cluster (UCC)13,14 may by expressed by our choice of A (taking as
reference the Hartree–Fock state), in addition to any others
based on the theory of excitation operators such as ADAPT-VQE.16−19 The quantum advantage in VQE stems from the ability to prepare classically
intractable states from our parametrized ansatz circuits.
3. Projections onto Stabilizer Subspaces
Given an operator 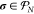 , the space of quantum states
, the space of quantum states 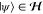 that it stabilizes are those satisfying σ|ψ⟩ = |ψ⟩, the +1-eigenspace
of σ. Extending this notion to an Abelian subgroup
of Pauli operators
that it stabilizes are those satisfying σ|ψ⟩ = |ψ⟩, the +1-eigenspace
of σ. Extending this notion to an Abelian subgroup
of Pauli operators 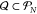 , there is an induced vector space
, there is an induced vector space 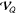 of states stabilized by the elements of
of states stabilized by the elements of  .
.
A particularly useful definition
is that of a Hamiltonian symmetry, taken here to
mean a set 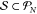 of Pauli operators such that
of Pauli operators such that
| 6 |
In other words, a symmetry of 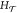 is any set of Pauli operators that commute
universally among
is any set of Pauli operators that commute
universally among  , which we may extend to an Abelian group
, which we may extend to an Abelian group 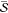 , the closure of
, the closure of  under operator multiplication, which we
shall call a symmetry group.
under operator multiplication, which we
shall call a symmetry group.
Note the setting
in which we present symmetries here is stricter
than the conventional definition, which considers any operator S that commutes with the Hamiltonian, i.e., 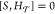 , to be a symmetry. Such an operator need
not commute with the individual terms as we require here. For example,
in the Fermionic picture, the number operator
, to be a symmetry. Such an operator need
not commute with the individual terms as we require here. For example,
in the Fermionic picture, the number operator 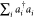 (where a is the Fermionic
annihilation operator and its Hermitian conjugate a† represents the creation operator) commutes with
the full second-quantized molecular Hamiltonian, but not with an arbitrary
excitation term.
(where a is the Fermionic
annihilation operator and its Hermitian conjugate a† represents the creation operator) commutes with
the full second-quantized molecular Hamiltonian, but not with an arbitrary
excitation term.
The operators of  will in general be algebraically dependent,
but the theory of stabilizers50 ensures
the existence of a set of independent generators
will in general be algebraically dependent,
but the theory of stabilizers50 ensures
the existence of a set of independent generators  such that
such that  . Now, recall that the Clifford group consists
of unitary operators
. Now, recall that the Clifford group consists
of unitary operators  (meaning
(meaning 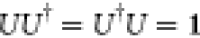 ) with the property
) with the property  , i.e., U normalizes the
Pauli group. We may construct a Clifford operation U mapping each symmetry generator to distinct single-qubit Pauli operators
σp, where we are free to choose p ∈ {1, 2, 3}. More precisely, there exists a subset
of qubit positions
, i.e., U normalizes the
Pauli group. We may construct a Clifford operation U mapping each symmetry generator to distinct single-qubit Pauli operators
σp, where we are free to choose p ∈ {1, 2, 3}. More precisely, there exists a subset
of qubit positions 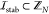 satisfying
satisfying 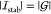 and a bijective map
and a bijective map 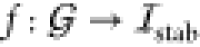 such that
such that
| 7 |
This is a powerful concept that provides a mechanism for reducing the number of qubits in the Hamiltonian while preserving its energy spectrum. This is at the core of qubit tapering,47,48 in which it is observed that
| 8 |
implying the rotated Hamiltonian 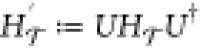 consists solely of identity or Pauli σp operators in the qubit positions indexed
by
consists solely of identity or Pauli σp operators in the qubit positions indexed
by 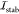 . Taking expectation values, one may replace
the qubits
. Taking expectation values, one may replace
the qubits 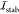 by their eigenvalues νi = ±1; each assignment
by their eigenvalues νi = ±1; each assignment
| 9 |
defines a symmetry sector and at least one such sector will contain the true solution to the eigenvalue problem. Note the other sectors still have physical significance and may, for example, relate to solutions with different particle numbers or to excited states. Ancillary data files are provided in which we report the symmetry generators and corresponding sector for the Hamiltonians representing the molecular systems listed in Table 1.
Table 1. Systems Investigated to Benchmark the Noncontextual Projection Ansatza.
| molecular
systems |
number
of qubits |
||||
|---|---|---|---|---|---|
| name | charge | mult. | full | taper | CS-VQEb |
| Be | 0 | 1 | 10 | 5 | 3 |
| B | 0 | 2 | 10 | 5 | 3 |
| LiH | 0 | 1 | 12 | 8 | 4 |
| BeH | +1 | 1 | 12 | 8 | 6 |
| HF | 0 | 1 | 12 | 8 | 4 |
| BeH2 | 0 | 1 | 14 | 9 | 7 |
| H2O | 0 | 1 | 14 | 10 | 7 |
| F2 | 0 | 1 | 20 | 16 | 10 |
| HCl | 0 | 1 | 20 | 17 | 4 |
All in the STO-3G basis.
Indicates the fewest number of qubits required to achieve chemical precision.
A quantum state consistent with any such sector must
be stabilized
by the operators 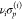 , and we may define a projection onto the
corresponding stabilizer subspace. In general, a projection is defined
to be an idempotent operator
, and we may define a projection onto the
corresponding stabilizer subspace. In general, a projection is defined
to be an idempotent operator 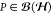 , i.e., P2 = P; the projection onto the ±1-eigenspace of a single-qubit
Pauli operator σp for p ∈ {1, 2, 3} may be written
, i.e., P2 = P; the projection onto the ±1-eigenspace of a single-qubit
Pauli operator σp for p ∈ {1, 2, 3} may be written
| 10 |
States with no component inside the chosen eigenspace are mapped to zero and observe that
| 11 |
for q ∈ {1, 2, 3}.
Let 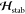 be the reduced Hilbert space supported
by the stabilized qubits
be the reduced Hilbert space supported
by the stabilized qubits 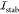 and
and 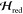 its complement such that
its complement such that 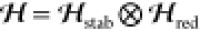 . Given an assignment of eigenvalues
. Given an assignment of eigenvalues  , we may project onto the corresponding
sector via
, we may project onto the corresponding
sector via
 |
12 |
and subsequently perform a partial
trace over the stabilized qubits  . This is effected by the unique linear
map
. This is effected by the unique linear
map 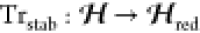 satisfying the property
satisfying the property 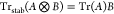 for all
for all 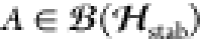 and
and 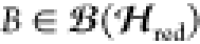 .
.
Finally, we may define the full stabilizer subspace projection map
| 13 |
which, using the linearity of Trstab, yields a reduced Hamiltonian
 |
14 |
where 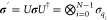 and we have written
and we have written 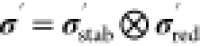 . The new coefficients
. The new coefficients 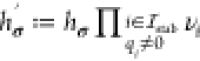 differ from hσ by a sign dependent on the chosen symmetry sector.
differ from hσ by a sign dependent on the chosen symmetry sector.
In qubit tapering, U is taken as eq 7 with the corresponding basis  a generating set for a full Hamiltonian
symmetry.47,48 Assuming identification of the correct sector,
the ground state energy of the
a generating set for a full Hamiltonian
symmetry.47,48 Assuming identification of the correct sector,
the ground state energy of the  -qubit reduced Hamiltonian
-qubit reduced Hamiltonian 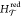 will coincide with the true value of the
full system
will coincide with the true value of the
full system 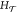 .
.
This stabilizer projection procedure
is straightforward with respect
to the Hamiltonian, since the stabilized qubits contain only operators
with nonzero image under conjugation with Pν. However, suppose we were to take another observable 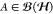 and wish to determine a reduced form on
and wish to determine a reduced form on 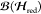 that is consistent with the reduced Hamiltonian
that is consistent with the reduced Hamiltonian  . This may be achieved by following precisely
the same process that was applied to
. This may be achieved by following precisely
the same process that was applied to 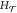 , but the symmetry
, but the symmetry 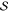 will not in general be a symmetry of A and therefore the “symmetry-breaking” terms
(those which anticommute with the generators
will not in general be a symmetry of A and therefore the “symmetry-breaking” terms
(those which anticommute with the generators  ) will vanish under projection onto the
stabilizer subspace, as per eq 11. Letting
) will vanish under projection onto the
stabilizer subspace, as per eq 11. Letting 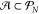 be the set of terms in the Pauli-basis
expansion of A, observe that
be the set of terms in the Pauli-basis
expansion of A, observe that
 |
15 |
recalling that qi indicates the type of single-qubit Pauli acting
on qubit position 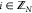 in some tensor product σ, defined in Section 2.
in some tensor product σ, defined in Section 2.
The resulting form is identical to eq 14,
except we are explicit that the terms surviving projection are only
those whose qubit positions indexed by 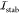 consist exclusively of identity and Pauli
σp operators; this is trivially
true for the Hamiltonian by construction. Most importantly, this extends
the stabilizer subspace projection to ansätze defined on the
full system for use in variational algorithms. It should be noted
that the above operations are classically tractable and can be implemented
efficiently in the symplectic representation of Pauli operators.51,52
consist exclusively of identity and Pauli
σp operators; this is trivially
true for the Hamiltonian by construction. Most importantly, this extends
the stabilizer subspace projection to ansätze defined on the
full system for use in variational algorithms. It should be noted
that the above operations are classically tractable and can be implemented
efficiently in the symplectic representation of Pauli operators.51,52
It would be remiss of us not to draw attention to the likeness
of eq 13 to Positive Operator-Valued Measures
(POVMs);53 indeed, the projectors (eq 12) define a complete set of Kraus operators.54 The stabilizer subspace
projection procedure is reduced to a matter of enforcing a partial
measurement over some subsystem of the full problem, for which the
relevant outcomes have been determined via an auxiliary method. For
example, this could involve identifying a quantum state with a known
nonzero overlap with the true ground state; measuring the symmetry
generators 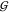 in this state will yield the correct sector.
in this state will yield the correct sector.
Hartree–Fock often provides such a state for electronic structure problems, although it is not immune to failure; this is particularly true in the strongly correlated regime. In these cases, we should defer to more effective reference states such as those obtained from Møller–Plesset perturbation theory (MP), coupled-cluster (CC) methods, and so on. One can imagine a hierarchy of increasingly precise ground state approximations, for which we should hope to obtain at some point a nonzero overlap with the true ground state.
4. CS-VQE in the Stabilizer Formalism
We now describe the Contextual Subspace VQE (CS-VQE) method in the stabilizer setting introduced in Section 3. CS-VQE partitions the Hamiltonian (2) into two disjoint components, one that is noncontextual and another that is contextual, which provides quantum corrections to the former via VQE.45 Explicitly, this allows us to write
| 16 |
where 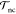 is a noncontextual set of Pauli operators
and
is a noncontextual set of Pauli operators
and 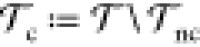 is what remains, which will in general
be contextual.
is what remains, which will in general
be contextual.
CS-VQE differs from qubit tapering (described
in Section 3) in the
following way: the
latter exploits existing (i.e., physical) symmetries of the Hamiltonian,
whereas in CS-VQE, we impose additional “pseudosymmetries”
derived from the noncontextual Hamiltonian. This results in a loss
of information, since any terms of 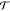 not commuting with the symmetry generators
will vanish under projection.
not commuting with the symmetry generators
will vanish under projection.
4.1. The Noncontextual Problem
The notion
of contextuality goes back to the Bell–Kochen–Specker
theorem.55−57 Here we use an explicit condition for the noncontextuality
of a set of Pauli operators, developed by Kirby and Love58 and independently by Raussendorf et al.59 Strictly speaking, this condition tests for
strong measurement contextuality. In this setting, a set 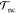 is understood to be noncontextual if and
only if commutation forms an equivalence relation on
is understood to be noncontextual if and
only if commutation forms an equivalence relation on  , where we have defined the sub-Hamiltonian
symmetry
, where we have defined the sub-Hamiltonian
symmetry 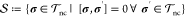 . There is an implied structure
. There is an implied structure
| 17 |
where the  are equivalence classes with respect to
commutation–in other words, elements of the same class commute
and across classes they anticommute. Conversely, such a set of Pauli
operators is contextual if and only if commutation fails to be transitive
on
are equivalence classes with respect to
commutation–in other words, elements of the same class commute
and across classes they anticommute. Conversely, such a set of Pauli
operators is contextual if and only if commutation fails to be transitive
on 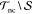 .
.
The symmetry 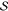 can be expanded by taking pairwise products
within equivalence classes, since {Ci, Cj} = 0 for
can be expanded by taking pairwise products
within equivalence classes, since {Ci, Cj} = 0 for 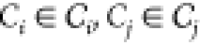 with i ≠ j, it is the case that
with i ≠ j, it is the case that  and we may define
and we may define 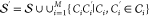 . As before, in Section 3,
. As before, in Section 3, 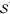 induces a symmetry group for which one
may define independent generators
induces a symmetry group for which one
may define independent generators  and a Clifford operation
and a Clifford operation 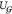 mapping the generators to single-qubit
Pauli operators; the expectation value over these qubits will again
be determined by an assignment
mapping the generators to single-qubit
Pauli operators; the expectation value over these qubits will again
be determined by an assignment  of eigenvalues, analogous to the selection
of a symmetry sector in qubit tapering.
of eigenvalues, analogous to the selection
of a symmetry sector in qubit tapering.
From each equivalence
class 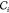 , we select a representative Ci and construct an observable
, we select a representative Ci and construct an observable 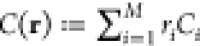 where
where 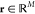 and |r| = 1. Kirby and Love60 found that quantum states
and |r| = 1. Kirby and Love60 found that quantum states  stabilized by the operators
stabilized by the operators 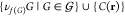 are consistent with a classical objective
function η(ν, r) (derived in
the Supporting Information), in the sense
that η(ν, r) coincides with
the noncontextual energy expectation value
are consistent with a classical objective
function η(ν, r) (derived in
the Supporting Information), in the sense
that η(ν, r) coincides with
the noncontextual energy expectation value 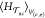 for all parametrizations (ν, r). This is a consequence of the joint probability
distribution chosen over the phase-space points of their (epistricted)
model.60,61
for all parametrizations (ν, r). This is a consequence of the joint probability
distribution chosen over the phase-space points of their (epistricted)
model.60,61
The noncontextual energy spectrum is therefore parametrized by two vectors: the ±1 eigenvalue assignments ν, determining the contribution of the universally commuting terms, and r, encapsulating the remaining pairwise anticommuting classes. In this sense, we may refer to (ν, r) as a state of the noncontextual Hamiltonian itself, abstracted from quantum states of the corresponding stabilizer subspace. Optimizing over these parameters, we obtain the noncontextual ground state energy
| 18 |
and call an element (ν, r) of the preimage 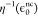 a noncontextual ground state of
a noncontextual ground state of 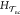 . Let us denote by
. Let us denote by 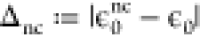 the absolute error with respect to the
true ground state energy.
the absolute error with respect to the
true ground state energy.
As a classical estimate to the ground
state energy of the full
Hamiltonian 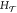 , in Section 5, we found the difference between the noncontextual
ground state and Hartree–Fock energy to be negligible for each
of the molecules simulated, since the heuristic used to choose
, in Section 5, we found the difference between the noncontextual
ground state and Hartree–Fock energy to be negligible for each
of the molecules simulated, since the heuristic used to choose 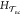 prioritizes diagonal Hamiltonian terms.
In principle, it may be an improvement upon Hartree–Fock as
the noncontextual set can also take into account an off-diagonal contribution
within the anticommuting classes. This is highly dependent on the
chosen form of noncontextual set; a reformulation in terms of graphs,
e.g., representing Pauli operators as nodes with (non)adjacency indicating
(anti)commutation, will allow one to identify what the equivalent
problem(s) are in computer science and therefore draw upon the vast
body of existing research and select the best algorithms designed
to solve such computational problems of graph theory. It should be
noted that the “optimal” noncontextual subset will not
necessarily be that which minimizes the noncontextual ground state
energy and some consideration of the resulting quantum corrections
must inform this choice, which remains an open question.
prioritizes diagonal Hamiltonian terms.
In principle, it may be an improvement upon Hartree–Fock as
the noncontextual set can also take into account an off-diagonal contribution
within the anticommuting classes. This is highly dependent on the
chosen form of noncontextual set; a reformulation in terms of graphs,
e.g., representing Pauli operators as nodes with (non)adjacency indicating
(anti)commutation, will allow one to identify what the equivalent
problem(s) are in computer science and therefore draw upon the vast
body of existing research and select the best algorithms designed
to solve such computational problems of graph theory. It should be
noted that the “optimal” noncontextual subset will not
necessarily be that which minimizes the noncontextual ground state
energy and some consideration of the resulting quantum corrections
must inform this choice, which remains an open question.
4.2. Quantum Corrections
Our simulation
approach has thus far been strictly classical–now we arrive
at the quantum element of CS-VQE. We have derived a classical estimate
of the ground state energy from the noncontextual part of the Hamiltonian 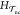 ; however, the contextual component
; however, the contextual component 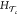 has so far been neglected.
has so far been neglected.
While C(r) is not a stabilizer in the strict sense
(it is not an element of the Pauli group), it is unitarily equivalent
to one as a linear combination of anticommuting Pauli elements. Similar
to the symmetry generators 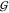 , it is possible to define a unitary operation UC mapping C(r) onto a single-qubit Pauli operator, following the
approach of unitary partitioning.32−36 However, unlike the
, it is possible to define a unitary operation UC mapping C(r) onto a single-qubit Pauli operator, following the
approach of unitary partitioning.32−36 However, unlike the 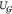 rotation,
rotation,  is not Clifford as it collapses M terms onto a single Pauli operator and can therefore introduce
additional terms to the Hamiltonian. Kirby et al.45 cautioned that, in principle, this increase in Hamiltonian
complexity could be exponential in the number of equivalence classes M, namely, a scaling of
is not Clifford as it collapses M terms onto a single Pauli operator and can therefore introduce
additional terms to the Hamiltonian. Kirby et al.45 cautioned that, in principle, this increase in Hamiltonian
complexity could be exponential in the number of equivalence classes M, namely, a scaling of 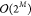 . However, Ralli et al.36 demonstrated that the general scaling for this sequence
of rotations (SeqRot) method is
. However, Ralli et al.36 demonstrated that the general scaling for this sequence
of rotations (SeqRot) method is  where x ∈ [1, 2];
that is, still exponential, yet the necessary conditions to obtain
the worst-case x = 2 are contrived and have not been
observed for any molecular Hamiltonians investigated to date. Regardless,
one may circumvent this potentially adverse scaling entirely by implementing
the linear combination of unitaries (LCU) approach to unitary partitioning,33,35 which is only quadratic in the number of equivalence classes
where x ∈ [1, 2];
that is, still exponential, yet the necessary conditions to obtain
the worst-case x = 2 are contrived and have not been
observed for any molecular Hamiltonians investigated to date. Regardless,
one may circumvent this potentially adverse scaling entirely by implementing
the linear combination of unitaries (LCU) approach to unitary partitioning,33,35 which is only quadratic in the number of equivalence classes  .36
.36
Appending C(r) to our set of generators 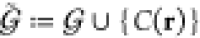 and defining
and defining  , there exists a subset of qubit indices
, there exists a subset of qubit indices 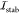 satisfying
satisfying  and a bijective map
and a bijective map 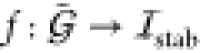 such that
such that  for each
for each  . We reiterate that p ∈
{1, 2, 3} may be chosen at will; the approach taken by Kirby et al.45 is to select p = 3 to enforce
diagonal generators.
. We reiterate that p ∈
{1, 2, 3} may be chosen at will; the approach taken by Kirby et al.45 is to select p = 3 to enforce
diagonal generators.
Suppose we have a quantum state |ψ(ν,r)⟩ that is consistent
with 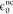 ; since the rotated state
; since the rotated state  must be stabilized by
must be stabilized by 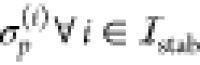 , the qubit positions
, the qubit positions 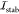 must be fixed. This implies a decomposition
must be fixed. This implies a decomposition
| 19 |
where |b(ν,r)⟩ represents a single basis state of 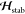 and
and 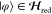 is independent of the parameters (ν, r). The expectation value of the full
Hamiltonian may be expressed as
is independent of the parameters (ν, r). The expectation value of the full
Hamiltonian may be expressed as
| 20 |
where  contains only the terms of the contextual
Hamiltonian that commute with all the noncontextual generators, just
as in eq 15. It was observed by Kirby et al.45 that any term which anticommutes with at least
one noncontextual generator must have zero expectation value, and
our stabilizer subspace projection captures this fact.
contains only the terms of the contextual
Hamiltonian that commute with all the noncontextual generators, just
as in eq 15. It was observed by Kirby et al.45 that any term which anticommutes with at least
one noncontextual generator must have zero expectation value, and
our stabilizer subspace projection captures this fact.
Inspecting
eq 20, we may optimize freely
over quantum states φ, i.e., we are not constrained by the noncontextual
ground state within 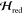 . In fact, we may absorb the noncontextual
ground state energy into the reduced contextual Hamiltonian
. In fact, we may absorb the noncontextual
ground state energy into the reduced contextual Hamiltonian
| 21 |
defining the contextual subspace Hamiltonian; this form is obtained naturally when applying the stabilizer subspace projection to the full Hamiltonian, which automatically includes the noncontextual energy by fixing the corresponding eigenvalue assignments.
Now, we may perform unconstrained VQE to obtain a quantum-corrected estimate
| 22 |
of the true ground state energy with absolute
error 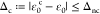 . We have equality when the stabilizers
span every qubit position, which is the case when
. We have equality when the stabilizers
span every qubit position, which is the case when 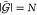 since the generators must be algebraically
independent: this means the initial quantum correction is trivial
as the noncontextual part determines the entire system.
since the generators must be algebraically
independent: this means the initial quantum correction is trivial
as the noncontextual part determines the entire system.
For
instances of the electronic structure problem, there is no
guarantee that 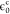 will achieve chemical precision (Δc < 1.6 mHa ≈ 4 kJ/mol) and, indeed, it might not
improve upon the noncontextual estimate (although it will never be
worse, due to the variational principle applying in this case). However,
one can easily define a subset of
will achieve chemical precision (Δc < 1.6 mHa ≈ 4 kJ/mol) and, indeed, it might not
improve upon the noncontextual estimate (although it will never be
worse, due to the variational principle applying in this case). However,
one can easily define a subset of 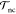 that is again noncontextual; this is achieved
by discarding one of the noncontextual generators
that is again noncontextual; this is achieved
by discarding one of the noncontextual generators  , along with the operators that it generates.
We now append the discarded operators to the contextual Hamiltonian,
relaxing the stabilizer constraint on the qubit position f(G) and permitting a search over its Hilbert space.
This process may be iterated until the noncontextual set is exhausted
and we recover full VQE. This means that, unless the ground state
energy of
, along with the operators that it generates.
We now append the discarded operators to the contextual Hamiltonian,
relaxing the stabilizer constraint on the qubit position f(G) and permitting a search over its Hilbert space.
This process may be iterated until the noncontextual set is exhausted
and we recover full VQE. This means that, unless the ground state
energy of 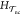 and H coincides, CS-VQE
will improve upon the noncontextual energy using less quantum resources
than full VQE; this is more rigorously defined in the next section.
and H coincides, CS-VQE
will improve upon the noncontextual energy using less quantum resources
than full VQE; this is more rigorously defined in the next section.
In summary, what we have described here is a technique of scaling the relative sizes of the noncontextual (read classical) and contextual (read quantum) simulations in a reciprocal manner. We can therefore trade-off quantum and classical workloads in CS-VQE.
4.3. Expanding the Contextual Subspace
Now we describe the process of growing the contextual subspace more
rigorously. We select a subset of noncontextual generators 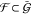 whose stabilizer constraints we mean to
enforce and construct a new noncontextual set
whose stabilizer constraints we mean to
enforce and construct a new noncontextual set  ; the contextual set is expanded accordingly
by appending the terms not generated by
; the contextual set is expanded accordingly
by appending the terms not generated by 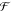 , i.e.,
, i.e.,  . As before, there exists a unitary operation
. As before, there exists a unitary operation 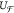 , a subset of qubit indices
, a subset of qubit indices 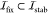 , and a bijective map
, and a bijective map 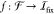 satisfying
satisfying 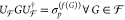 (the rotation
(the rotation 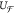 may or may not be Clifford depending on
whether C(r) is among the stabilizers
we wish to fix).
may or may not be Clifford depending on
whether C(r) is among the stabilizers
we wish to fix).
Denote by 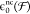 the ground state energy of the new noncontextual
Hamiltonian
the ground state energy of the new noncontextual
Hamiltonian  with absolute error
with absolute error  . While this is weaker as an estimate of
the true ground state energy of the full system, at the very least
we are guaranteed to recover the initial noncontextual ground state
energy from performing a simulation of the expanded contextual subspace,45 which we describe below.
. While this is weaker as an estimate of
the true ground state energy of the full system, at the very least
we are guaranteed to recover the initial noncontextual ground state
energy from performing a simulation of the expanded contextual subspace,45 which we describe below.
The stabilizer
constraints of 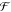 are enforced over the Hilbert space
are enforced over the Hilbert space 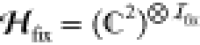 of qubits indexed by
of qubits indexed by 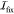 , whereas we may perform a VQE simulation
over
, whereas we may perform a VQE simulation
over 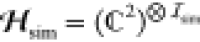 , the Hilbert space of the remaining
, the Hilbert space of the remaining 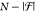 qubits indexed by
qubits indexed by 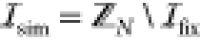 . Invoking the stabilizer subspace projection
map
. Invoking the stabilizer subspace projection
map  with the eigenvalue assignments
with the eigenvalue assignments  yields an expanded contextual subspace
Hamiltonian
yields an expanded contextual subspace
Hamiltonian
| 23 |
Performing an 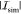 -qubit VQE simulation over the contextual
subspace, we obtain a new quantum-corrected estimate
-qubit VQE simulation over the contextual
subspace, we obtain a new quantum-corrected estimate
| 24 |
with an error satisfying  . Recall that
. Recall that 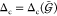 corresponds with the contextual error when
we enforce the full set of noncontextual stabilizers.
corresponds with the contextual error when
we enforce the full set of noncontextual stabilizers.
Observe
that, when 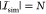 , we are simply performing full VQE over
the entire system; this occurs when we do not enforce the stabilizer
constraint for any of the noncontextual generators, i.e.,
, we are simply performing full VQE over
the entire system; this occurs when we do not enforce the stabilizer
constraint for any of the noncontextual generators, i.e., 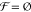 . Therefore, it must be the case that
. Therefore, it must be the case that
| 25 |
Furthermore, given a nested sequence of generator
subsets 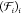 with
with 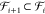 , then
, then 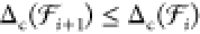 and the convergence is monotonic. In this
way, CS-VQE describes an interpolation between a purely classical
estimate of the ground state energy and a full VQE simulation of the
Hamiltonian. In the context of electronic structure calculations,
this often permits one to achieve chemical precision at a saving of
qubit resources, as indicated by Kirby et al.45 for a suite of tapered test molecules of up to 18 qubits. We note
in eq 25 that the quality of the chosen ansatz
and optimization procedure will limit the actual error one may achieve
in practice. This statement instead indicates that, for an appropriate
level of contextual subspace approximation, it is possible to construct
a reduced Hamiltonian whose exact ground state lies within some error
threshold of the true value.
and the convergence is monotonic. In this
way, CS-VQE describes an interpolation between a purely classical
estimate of the ground state energy and a full VQE simulation of the
Hamiltonian. In the context of electronic structure calculations,
this often permits one to achieve chemical precision at a saving of
qubit resources, as indicated by Kirby et al.45 for a suite of tapered test molecules of up to 18 qubits. We note
in eq 25 that the quality of the chosen ansatz
and optimization procedure will limit the actual error one may achieve
in practice. This statement instead indicates that, for an appropriate
level of contextual subspace approximation, it is possible to construct
a reduced Hamiltonian whose exact ground state lies within some error
threshold of the true value.
Suppose we wish to find the optimal
contextual subspace Hamiltonian
of size N′ < N. The problem
reduces to minimizing the error 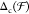 over the
over the  generator subsets
generator subsets 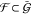 satisfying
satisfying 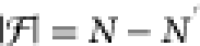 . CS-VQE is highly sensitive to this choice
and remains a vital open question for the continued success of the
technique. For chemistry applications, we grow the contextual subspace
until the CS-VQE error attains chemical precision, which means finding
the minimal
. CS-VQE is highly sensitive to this choice
and remains a vital open question for the continued success of the
technique. For chemistry applications, we grow the contextual subspace
until the CS-VQE error attains chemical precision, which means finding
the minimal 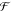 such that
such that 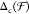 < 1.6 mHa. In general, we will not have
access to a target energy and so will not necessarily know when the
desired precision is achieved; instead, we might choose the largest
contextual subspace accommodated by the available quantum resource
or iterate until the VQE convergence is within some fixed bound.
< 1.6 mHa. In general, we will not have
access to a target energy and so will not necessarily know when the
desired precision is achieved; instead, we might choose the largest
contextual subspace accommodated by the available quantum resource
or iterate until the VQE convergence is within some fixed bound.
Greedily selecting combinations of d ≤ N generators that yield the greatest reduction in error
is an effective stabilizer relaxation ordering heuristic, where iterating k < N/d involves a
search of depth d over N–dk elements, thus necessitating  CS-VQE simulations. Taking d = 2 produces a good balance between efficiency and efficacy,45 but there is room for more targeted approaches
that exploit some structure of the underlying problem. For example,
in quantum chemistry problems, it could be that one should relax the
stabilizers that have nontrivial action near the Fermi level, between
the highest occupied molecular orbital (HOMO) and lowest unoccupied
molecular orbital (LUMO). Excitations clustered around this gap are
more likely to appear in the true ground state and should therefore
not be assigned definite values under the noncontextual projection.
This idea comes from the theory of pseudopotential approximations,62 in which it is observed that chemically relevant
electrons are predominantly those of the valence space, whereas the
core may be “frozen”, thus reducing the electronic complexity.
CS-VQE simulations. Taking d = 2 produces a good balance between efficiency and efficacy,45 but there is room for more targeted approaches
that exploit some structure of the underlying problem. For example,
in quantum chemistry problems, it could be that one should relax the
stabilizers that have nontrivial action near the Fermi level, between
the highest occupied molecular orbital (HOMO) and lowest unoccupied
molecular orbital (LUMO). Excitations clustered around this gap are
more likely to appear in the true ground state and should therefore
not be assigned definite values under the noncontextual projection.
This idea comes from the theory of pseudopotential approximations,62 in which it is observed that chemically relevant
electrons are predominantly those of the valence space, whereas the
core may be “frozen”, thus reducing the electronic complexity.
Alternatively, one might define a Hamiltonian term-importance metric that considers coefficient magnitudes63 or second-order response with respect to a perturbation of the Hartree–Fock state.64 In relation to this, it is also not clear which features of a molecular system mean that it might be more or less amenable to CS-VQE; additional insight here would allow one to predict how many qubits will be required to simulate a given problem to chemical precision.
It is not fully understood how CS-VQE relates to active space techniques more generally, but this would be an interesting pursuit for future work. For example, the downfolding technique of subsystem embedding subalgebra coupled cluster (SES-CC)65 presents a compelling approach that iteratively decouples excitations σ = σint + σext into an “internal” part that belongs to a chosen excitation subalgebra and its “external” complement that may additionally be combined with the double unitary coupled cluster (DUCC) ansatz.66 This yields an effective Hamiltonian Hexteff(DUCC) = (P + Qint) e–σextH eσext(P + Qint) where P projects onto the reference state and Qint onto the subspace of excitations generated by σint. This has a similar form to our stabilizer subspace projection (eq 13); indeed, it might be possible to reproduce SES-CC under a qubit mapping within the contextual subspace framework by identifying an appropriate noncontextual sub-Hamiltonian and stabilizer subspace.
A benchmark of this and other dimensionality reduction methodologies such as projection-based embedding (PBE)67 would be valuable. Furthermore, CS-VQE can be layered on top of these techniques to yield hybrid methods that might outperform any of them on their own; this is a consideration that we plan to take forward into further work, with the goal of deployment on larger molecular systems and basis sets.
4.4. The Noncontextual Projection Ansatz
CS-VQE has thus far not been applied to systems exceeding 18 qubits, and the resulting reduced Hamiltonians (eq b23) have been solved by direct diagonalization;45 clearly, this will not scale to larger systems, with the required classical memory increasing exponentially. Instead, they must be simulated by performing VQE routines, but defining an ansatz for the contextual subspace provided an obstacle to achieving this in practice.
However, having now placed the problem within the stabilizer formalism described in Section 3, we have already introduced (in Sections 4.1–4.3) the tools necessary to restrict an ansatz of the form in eq 5–defined over the full system–to the contextual subspace (eq 23). The approach adopted here is equivalent to that which we defined for qubit tapering in eq 15. To restrict a parametrized ansatz operator
| 26 |
in line with the stabilizer constraints 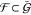 , we may simply call upon the stabilizer
subspace projection map
, we may simply call upon the stabilizer
subspace projection map  once more, which yields a restricted ansatz
state
once more, which yields a restricted ansatz
state
| 27 |
where
| 28 |
Any rotated ansatz term 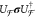 that is not identity or a Pauli σp on some subset of the qubit positions indexed
by
that is not identity or a Pauli σp on some subset of the qubit positions indexed
by 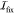 will vanish.
will vanish.
The restricted reference
state 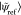 is obtained from an effective partial projective
measurement of
is obtained from an effective partial projective
measurement of 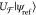 (see the discussion on POVMs in Section 3) with outcomes
defined by ν′, which yields a product state
(see the discussion on POVMs in Section 3) with outcomes
defined by ν′, which yields a product state
| 29 |
where we have explicitly demarcated the separability
across  and
and 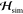 . The postmeasurement state
. The postmeasurement state 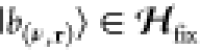 on the noncontextual subspace represents
a single basis vector and can therefore be disregarded, leaving just
the state of the contextual subspace; this we take as reference for
our restricted ansatz. If the unitary partitioning rotations are not to be applied, then the
on the noncontextual subspace represents
a single basis vector and can therefore be disregarded, leaving just
the state of the contextual subspace; this we take as reference for
our restricted ansatz. If the unitary partitioning rotations are not to be applied, then the 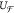 rotation is trivial over
rotation is trivial over  and we incur no expense in coherent resource.
However, if one does enforce the operator C(r) over the contextual subspace, there might be some nontrivial
component of the rotation that must be applied in-circuit to ensure
that the ansatz lies within the correct subspace; referring to Section 4.2, for the SeqRot
approach this will consist of at most
and we incur no expense in coherent resource.
However, if one does enforce the operator C(r) over the contextual subspace, there might be some nontrivial
component of the rotation that must be applied in-circuit to ensure
that the ansatz lies within the correct subspace; referring to Section 4.2, for the SeqRot
approach this will consist of at most  CNOT operations in-circuit, whereas LCU
is probabilistic due to the nature of block-encoding.35 Given a hardware-efficient ansatz, one may neglect this
since the optimizer should compensate the parameters accordingly.
CNOT operations in-circuit, whereas LCU
is probabilistic due to the nature of block-encoding.35 Given a hardware-efficient ansatz, one may neglect this
since the optimizer should compensate the parameters accordingly.
We may now define the contextual subspace energy expectation function
| 30 |
with 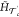 as in eq 23, at which
point we have reduced the problem to standard VQE, performed over
a subspace of the full problem.
as in eq 23, at which
point we have reduced the problem to standard VQE, performed over
a subspace of the full problem.
5. Simulation Results
The molecular systems that were simulated to benchmark the noncontextual projection ansatz for CS-VQE are given in Table 1. The molecule geometries were obtained from the Computational Chemistry Comparison and Benchmark Database (CCCBDB)68 and their Hamiltonians were constructed using IBM’s Qiskit Nature69 with PySCF as the underlying quantum chemistry package.70
Before we evaluate the efficacy of our noncontextual projection
ansatz, there are a few features of eq 27 that
should be highlighted. First of all, from the discussion following
eq 29, we potentially apply some component of
the operation 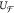 in-circuit, introducing further gates that
will contribute additional noise. However, when the reference state
is taken to be that of Hartree–Fock, we observed Uψref to coincide with the noncontextual ground state. This
is an artifact of the noncontextual set construction heuristic prioritizing
diagonal entries, used within both this work and that of Kirby et
al.45 This need not always be the case,
but for the molecular systems investigated, this allows us to avoid
performing
in-circuit, introducing further gates that
will contribute additional noise. However, when the reference state
is taken to be that of Hartree–Fock, we observed Uψref to coincide with the noncontextual ground state. This
is an artifact of the noncontextual set construction heuristic prioritizing
diagonal entries, used within both this work and that of Kirby et
al.45 This need not always be the case,
but for the molecular systems investigated, this allows us to avoid
performing 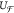 in-circuit and instead take the noncontextual
ground state as our reference. Since we choose to rotate the noncontextual
symmetry generators onto Pauli σ3 operators here,
this may be prepared by applying a Pauli σ1 in each
of the qubit positions
in-circuit and instead take the noncontextual
ground state as our reference. Since we choose to rotate the noncontextual
symmetry generators onto Pauli σ3 operators here,
this may be prepared by applying a Pauli σ1 in each
of the qubit positions 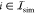 such that νi = −1 so that the corresponding reference state is stabilized
by the relevant operators νiσ3(i). This is visible in Figure 2, in which the VQE routine is initiated with the optimization
parameters zeroed, i.e., θ = 0, and
since eiÃ(0) = 1, optimization begins at the noncontextual ground state energy.
such that νi = −1 so that the corresponding reference state is stabilized
by the relevant operators νiσ3(i). This is visible in Figure 2, in which the VQE routine is initiated with the optimization
parameters zeroed, i.e., θ = 0, and
since eiÃ(0) = 1, optimization begins at the noncontextual ground state energy.
Figure 2.

Validation of the noncontextual projection approach to ansatz construction for CS-VQE (eq 27), used here in conjunction with ADAPT-VQE.16−19 We plot (on a log10 scale) the absolute error of wave function simulations conducted for the suite of trial molecules outlined in Table 1, each shown to achieve chemical precision; the horizontal axis indicates the algorithm step counter with each shaded region a separate ADAPT-VQE cycle. Adaptive moment estimation (Adam)73 is the classical optimizer taken in the VQE routine performed over the contextual subspace for each ADAPT-VQE cycle, and the settings used are as follows: tolerance = 10–4, learning rate = 10–2, β1 = 0.4, β2 = 0.999, ϵ = 10–8. The parameter gradients ∂Ẽ(θ)/∂θi, required for both operator pool term selection and VQE, were computed using the parameter shift rule.74
Second, application of the unitary partitioning rotations UC to the ansatz operator A(θ) may introduce additional terms by
a worst-case scaling factor of 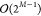 where M is the number
of equivalence classes in eq 17, although the
true scaling is unlikely to be this severe as discussed in Section 4.2. We obtained
M = 2 for all of the molecules tested, in which case SeqRot is identical
to the asymptotically favorable LCU method. In fact, for small M ≪ N, SeqRot may generate fewer
terms than LCU (Ralli et al. presented a toy problem with M = 3 in which this was the case36) and therefore our choice of SeqRot here is valid given that the
noncontextual set
where M is the number
of equivalence classes in eq 17, although the
true scaling is unlikely to be this severe as discussed in Section 4.2. We obtained
M = 2 for all of the molecules tested, in which case SeqRot is identical
to the asymptotically favorable LCU method. In fact, for small M ≪ N, SeqRot may generate fewer
terms than LCU (Ralli et al. presented a toy problem with M = 3 in which this was the case36) and therefore our choice of SeqRot here is valid given that the
noncontextual set 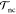 construction heuristic prioritizes the
universally commuting terms
construction heuristic prioritizes the
universally commuting terms 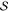 in eq 17. Different
heuristics may lead to larger values for M, in which
case, we recommend an adoption of LCU for implementations of CS-VQE.
in eq 17. Different
heuristics may lead to larger values for M, in which
case, we recommend an adoption of LCU for implementations of CS-VQE.
Despite this, upon the subsequent projection of A(θ), it is possible that a significant number
of terms will vanish. This is highly dependent on the quality of the
initial ansatz and how heavily it is supported on the stabilized qubit
positions 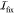 . Figure 1 presents circuit depths of the noncontextual projection
ansatz as a proportion of the base ansatz from which it is derived,
in this case the unitary coupled-cluster singles and doubles (UCCSD)
operator. A net reduction in circuit depth is observed, which is quite
dramatic up to the point of reaching chemical precision in the CS-VQE
routine; in Table 2, we give the specific number of ansatz terms before and after application
of the noncontextual projection to UCCSD and UCCSDT for the fewest
number of qubits permitting chemical precision.
. Figure 1 presents circuit depths of the noncontextual projection
ansatz as a proportion of the base ansatz from which it is derived,
in this case the unitary coupled-cluster singles and doubles (UCCSD)
operator. A net reduction in circuit depth is observed, which is quite
dramatic up to the point of reaching chemical precision in the CS-VQE
routine; in Table 2, we give the specific number of ansatz terms before and after application
of the noncontextual projection to UCCSD and UCCSDT for the fewest
number of qubits permitting chemical precision.
Figure 1.

Ideal CS-VQE errors (left-hand axis) and corresponding noncontextual projection ansatz circuit depths as a proportion of the full UCCSD operator from which it is derived (right-hand axis) against the number of qubits simulated.
Table 2. Number of Pauli Terms 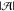 for a Selection of (Tapered) Ansätzea.
for a Selection of (Tapered) Ansätzea.
| number
of terms in ansatz operator |
||||
|---|---|---|---|---|
| molecule | 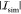 |
UCCSDT (full/proj)b | UCCSD (full/proj)b | ADAPT-VQEc |
| Be | 3 | (48/6) | (48/6) | 5 |
| B | 3 | (48/12) | (32/4) | 3 |
| LiH | 4 | (704/53) | (192/53) | 5 |
| BeH+ | 6 | (646/191) | (166/79) | 11 |
| HF | 4 | (92/57) | (92/57) | 4 |
| BeH2 | 7 | (1312/352) | (224/96) | 10 |
| H2O | 7 | (1892/942) | (324/238) | 21 |
| F2 | 10 | (176/114) | (176/114) | 12 |
| HCl | 4 | (348/40) | (348/40) | 4 |
The number of qubits in the contextual subspace over which the ansatz is projected; each tuple (full/proj) gives the number of terms pre- and postprojection.
The number of ADAPT-VQE cycles required to achieve chemical precision, with the operator pool consisting of the projected UCCSD terms.
In order to identify a compact ansatz that closely captures the underlying chemistry with minimal redundancy, we employ the ADAPT-VQE methodology.16−19 The algorithm centers around an operator pool from which terms are selected in line with a gradient-based argument and appended to a dynamically expanding ansatz whose parameters are optimized at each cycle via VQE. The particular approach we implement here is that of qubit-ADAPT-VQE,17 following on from iterative qubit coupled cluster,71 which searches at the level of Jordan–Wigner encoded Pauli operators; the seminal ADAPT-VQE paper16 instead defines its operator pool over Fermionic excitations.
The Jordan–Wigner transformation72 maps a single Fermionic annihilation operator onto two Pauli operators
 |
31 |
with the creation operator given by its Hermitian
conjugate ai†. Therefore, an excitation
on 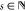 spin orbitals of the form
spin orbitals of the form
| 32 |
is represented by 22s Pauli operators under this encoding. In the unitary coupled cluster theory, we are interested rather in the operator a–a† to ensure unitarity upon exponentiation; this may be expressed by 22s–1 Pauli terms.
As such, after a mapping onto qubits via the Jordan–Wigner transformation, single, double, and triple excitations account for 2, 8, and 32 Pauli operator terms, respectively; while these are required to enforce various electronic symmetries in the ansatz state, not all are necessary to reach chemical precision. This idea lies behind qubit-ADAPT-VQE, which will select only the necessary Pauli terms and therefore yields considerably reduced circuit depths.17
To leverage ADAPT-VQE in the context of
CS-VQE, we define an operator
pool 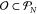 and apply to it the stabilizer subspace
projection (eq 13) to define a reduced pool
and apply to it the stabilizer subspace
projection (eq 13) to define a reduced pool  for the corresponding contextual subspace.
Projecting the full pool in this way will ensure that any symmetries S present will be preserved, since
for the corresponding contextual subspace.
Projecting the full pool in this way will ensure that any symmetries S present will be preserved, since 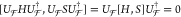 , allowing us to incorporate some chemical
intuition into the contextual subspace despite an abstraction from
the original problem; one could define a reduced pool directly, but
care should be taken to avoid the inclusion of symmetry-breaking terms
that may needlessly increase the complexity of the ADAPT-VQE procedure.
The algorithm is then executed as normal, only terminating once the
ADAPT-VQE energy is chemically precise with respect to the FCI energy;
for scalability, one should terminate computation when the largest
gradient in magnitude falls below some predefined threshold, since
the true ground state energy will not in general be known. In the Supporting Information, we provide a detailed
description of the specific ADAPT-VQE implementation used within this
work.
, allowing us to incorporate some chemical
intuition into the contextual subspace despite an abstraction from
the original problem; one could define a reduced pool directly, but
care should be taken to avoid the inclusion of symmetry-breaking terms
that may needlessly increase the complexity of the ADAPT-VQE procedure.
The algorithm is then executed as normal, only terminating once the
ADAPT-VQE energy is chemically precise with respect to the FCI energy;
for scalability, one should terminate computation when the largest
gradient in magnitude falls below some predefined threshold, since
the true ground state energy will not in general be known. In the Supporting Information, we provide a detailed
description of the specific ADAPT-VQE implementation used within this
work.
For the following, we take our pool 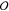 to be the terms of the UCCSD operator for
each of the molecules in Table 1 before tapering and projecting into the relevant contextual
subspace. In Figure 2, we present the ADAPT-VQE convergence data
with expectation values obtained via exact wave function (statevector)
calculations (i.e., no statistical/hardware noise); chemical precision
is achieved in each instance. We used the adaptive moment estimation
(Adam)73 classical optimizer and computed
parameter gradients as per the parameter shift rule.74 Adam has been adopted for previous research in VQE for
its resilience to noise, although it exhibits relatively slow convergence
compared with other optimizers75,76 such as Broyden–Fletcher–Goldfarb–Shanno
(BFGS)77 and quantum natural gradient (NatGrad);78 the latter might be preferable for future work.
to be the terms of the UCCSD operator for
each of the molecules in Table 1 before tapering and projecting into the relevant contextual
subspace. In Figure 2, we present the ADAPT-VQE convergence data
with expectation values obtained via exact wave function (statevector)
calculations (i.e., no statistical/hardware noise); chemical precision
is achieved in each instance. We used the adaptive moment estimation
(Adam)73 classical optimizer and computed
parameter gradients as per the parameter shift rule.74 Adam has been adopted for previous research in VQE for
its resilience to noise, although it exhibits relatively slow convergence
compared with other optimizers75,76 such as Broyden–Fletcher–Goldfarb–Shanno
(BFGS)77 and quantum natural gradient (NatGrad);78 the latter might be preferable for future work.
The number of ADAPT-VQE cycles (and therefore the
number of terms
in the resulting ansatz operator) are presented in Table 2, alongside the size of the
projected UCCSD operator pool used; one observes a significant reduction
in the number of terms. The optimized ADAPT-VQE ansatz operators are
reported in ancillary data files, along with a description of the
smallest CS-VQE problem permitting chemical precision. This includes
the optimal noncontextual generator subset 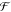 , the resulting noncontextual projection
ansatz (eq 27), the restricted reference state
, the resulting noncontextual projection
ansatz (eq 27), the restricted reference state 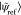 (eq 29), the target
error
(eq 29), the target
error  (eq 25), and that
which was actually achieved in our VQE simulations (Figure 2). We also include the corresponding
contextual subspace Hamiltonians for reproducibility.
(eq 25), and that
which was actually achieved in our VQE simulations (Figure 2). We also include the corresponding
contextual subspace Hamiltonians for reproducibility.
Extracting
the optimal parameter configuration θmin from the wave function simulations in Figure 2, we subsequently assess the
effect of sampling noise on the simulation error with our ansatz circuit
preparing the optimal quantum state 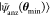 . Note that, for each of the molecular systems
in Table 1, θmin is given explicitly in the ancillary data files.
. Note that, for each of the molecular systems
in Table 1, θmin is given explicitly in the ancillary data files.
To achieve an absolute error of Δ > 0, one should expect
to perform  shots (for each term of the Hamiltonian).4 Conversely, suppose we are allocated a quantity
shots (for each term of the Hamiltonian).4 Conversely, suppose we are allocated a quantity  of shots; the obtained error should be
of the order
of shots; the obtained error should be
of the order 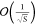 . In order to increase estimate accuracy,
we collected the Pauli terms into qubit-wise commuting (QWC) groups25 using the graph-coloring functionality of NetworkX;79 such groups may be measured simultaneously.
. In order to increase estimate accuracy,
we collected the Pauli terms into qubit-wise commuting (QWC) groups25 using the graph-coloring functionality of NetworkX;79 such groups may be measured simultaneously.
In Figure 3, the number of shots S = 2n for n = 0, ..., 20 carried out per QWC group is varied, and we observe the root mean-square error (RMSE) over 20 realizations of the ground state energy estimate, plotted on a log–log scale. For clarity, note the only source of noise here is that which arises from statistical variation of the quantum circuit sampling; we have not introduced hardware noise in the form of imperfect quantum gates or decoherence.
Figure 3.

Each of the plots in
panels a–i correspond with Figure 2a–i and illustrate
the statistical effect of sampling noise at the optimal parametrization θmin determined from the ADAPT-VQE statevector
simulations in Figure 2. We plot the root-mean-square error (RMSE) for 20 “realizations”
of the ground state energy estimate with S ≤
220 shots executed via IBM’s QASM simulator; determining
the line of best fit m·log10(S) + c with respect to the log–log
data indicates a decay in error of  .
.
Two error regimes are observed, one of which is
quite trivial:
at high shot counts, we see a plateau resulting from the optimal error 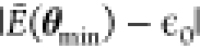 being recovered. To assess the convergence
properties outside of this limiting region, we plot a line of best
fit m·log10(S) + c among the data not exhibiting such behavior; since the
data is represented on a log–log scale, this corresponds with
a decay in error of
being recovered. To assess the convergence
properties outside of this limiting region, we plot a line of best
fit m·log10(S) + c among the data not exhibiting such behavior; since the
data is represented on a log–log scale, this corresponds with
a decay in error of 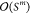 . In each plot of Figure 3, we obtain m ≈ −0.5,
meaning the RMSE follows the predicted decay of
. In each plot of Figure 3, we obtain m ≈ −0.5,
meaning the RMSE follows the predicted decay of 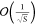 .
.
In every simulation bar F2, chemical precision was achieved within S = 220 ≈ 106 shots per QWC group. However, our shot budget could be reduced by implementing more advanced allocation strategies, for example, according to the magnitude of Hamiltonian term coefficients80 or a classical shadow tomography approach.30,31
6. Conclusions
We have placed CS-VQE on the theoretical footing of stabilizer subspace projections, which allows one to compare it against other qubit reduction techniques such as qubit tapering.47,48 Tapering defines a projection dependent on a symmetry of the full Hamiltonian and preserves the ground state energy exactly, whereas CS-VQE is approximate and projects onto a contextual subspace consistent with the symmetry of the noncontextual sub-Hamiltonian, augmented by an anticommuting contribution. In combination, the two techniques can effect a significant reduction in quantum resource requirements, as illustrated by Kirby et al.45 and in Figure 1.
Previously, the only obstacle to building a CS-VQE framework that would be faithful to deployment on quantum devices was that of the ansatz, which has been addressed within this work. Furthermore, we demonstrated how CS-VQE may be combined with the ADAPT-VQE16−19 ansatz construction framework by applying our noncontextual projection to the operator pool; validation was presented in Figure 2 in which we achieved chemical precision for the suite of small molecules outlined in Table 1. This combination provides considerable flexibility in both qubit count and circuit depth, allowing one to identify a reduced problem that may be simulated on the available quantum resource.
A number of research questions concerning
the scalability of CS-VQE
remain; we recapitulate these here. First, the success of CS-VQE is
sensitive to the generator subset 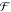 one chooses to constrain in the stabilizer
subspace projection. To date, the most effective method for choosing
this subset has been a greedy-search heuristic necessitating
one chooses to constrain in the stabilizer
subspace projection. To date, the most effective method for choosing
this subset has been a greedy-search heuristic necessitating 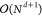 VQE simulations where d ≤ N is the search depth; this is expensive
for NISQ hardware, and there is room for more targeted heuristics.
For example, we may draw on chemical intuition to inform the selection
of a contextual subspace that captures information about the underlying
electronic structure problem. The second obstacle lies in the approach
taken to construct the noncontextual sub-Hamiltonian. There is currently
no intuition as to what constitutes an effective choice here, although
it should be noted that the “optimal” noncontextual
subset will not necessarily be that which minimizes the noncontextual
ground state energy; some consideration of the resulting contextual
subspaces must come into the construction of the noncontextual problem.
We leave these issues for future work.
VQE simulations where d ≤ N is the search depth; this is expensive
for NISQ hardware, and there is room for more targeted heuristics.
For example, we may draw on chemical intuition to inform the selection
of a contextual subspace that captures information about the underlying
electronic structure problem. The second obstacle lies in the approach
taken to construct the noncontextual sub-Hamiltonian. There is currently
no intuition as to what constitutes an effective choice here, although
it should be noted that the “optimal” noncontextual
subset will not necessarily be that which minimizes the noncontextual
ground state energy; some consideration of the resulting contextual
subspaces must come into the construction of the noncontextual problem.
We leave these issues for future work.
The natural next step is to execute this method on a NISQ computer, challenging the current best-in-class electronic structure simulations from Google, IonQ, and IBM.1−3 To achieve this goal, CS-VQE could be combined with techniques of measurement reduction20−35 and error mitigation.37−44
Finally, we have written an open-source Python package that facilitates the stabilizer subspace projection techniques of this work, with in-built tapering and CS-VQE functionality. We welcome the reader to make use of our code,49 which is freely available on GitHub.
Acknowledgments
T.W. and A.R. acknowledge support from the Unitary Fund and the Engineering and Physical Sciences Research Council (EP/S021582/1 and EP/L015242/1, respectively). T.W. also acknowledges support from CBKSciCon Ltd., Atos, Intel and Zapata. W.K. and P.J.L. acknowledge support by the NSF STAQ project (PHY-1818914). W. K. acknowledges support from the National Science Foundation, Grant No. DGE-1842474. P.V.C. is grateful for funding from the European Commission for VECMA (800925) and EPSRC for SEAVEA (EP/W007711/1). We would like to thank both Atos and the Leibniz Supercomputing Centre (LRZ), who each provided access to separate Atos Quantum Learning Machine (QLM) simulators that aided with the computational workload.
Data Availability Statement
In the interest of reproducibility, we supply in the Supporting Information the tapering parameters, CS-VQE model data, and noncontextual projection ansätze which permit chemical precision for the fewest number of qubits with respect to the molecular systems listed in Table 1; the raw data for these results are supplied in ancillary files hosted at https://arxiv.org/abs/2204.02150 along with an explanatory notebook. This will provide sufficient information for the reader to reproduce Figures 1, 2 and 3.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.2c00910.
Further information concerning circuit construction, deriving the noncontextual objective function one must solve to perform CS-VQE and a closer look at the ADAPT-VQE algorithm (PDF)
Tapering parameters, CS-VQE model data and noncontextual projection ansätze which permit chemical precision for the fewest number of qubits with respect to the molecular systems listed in Table 1—also includes the raw data required to reproduce Figures 1, 2 and 3, with an accompanying notebook (ZIP)
The authors declare no competing financial interest.
Supplementary Material
References
- Arute F.; Arya K.; Babbush R.; Bacon D.; Bardin J. C.; Barends R.; Boixo S.; Broughton M.; Buckley B. B.; Buell D. A.; et al. Hartree-Fock on a superconducting qubit quantum computer. Science 2020, 369, 1084–1089. 10.1126/science.abb9811. [DOI] [PubMed] [Google Scholar]
- Nam Y.; Chen J.-S.; Pisenti N. C.; Wright K.; Delaney C.; Maslov D.; Brown K. R.; Allen S.; Amini J. M.; Apisdorf J.; et al. Ground-state energy estimation of the water molecule on a trapped-ion quantum computer. npj Quantum Information 2020, 6, 33. 10.1038/s41534-020-0259-3. [DOI] [Google Scholar]
- Eddins A.; Motta M.; Gujarati T. P.; Bravyi S.; Mezzacapo A.; Hadfield C.; Sheldon S.. Doubling the size of quantum simulators by entanglement forging. arXiv preprint 2021, https://arxiv.org/abs/2104.10220.
- Peruzzo A.; McClean J.; Shadbolt P.; Yung M.-H.; Zhou X.-Q.; Love P. J.; Aspuru-Guzik A.; O’Brien J. L. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 2014, 5, 4213. 10.1038/ncomms5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaev A. Y.Quantum measurements and the Abelian stabilizer problem. arXiv preprint 1995, https://arxiv.org/abs/quant-ph/9511026.
- Griffiths D. J.; Schroeter D. F.. Introduction to quantum mechanics; Cambridge University Press, 2018. [Google Scholar]
- McClean J. R.; Boixo S.; Smelyanskiy V. N.; Babbush R.; Neven H. Barren plateaus in quantum neural network training landscapes. Nat. Commun. 2018, 9, 4812. 10.1038/s41467-018-07090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wecker D.; Hastings M. B.; Troyer M. Progress towards practical quantum variational algorithms. Phys. Rev. A 2015, 92, 042303. 10.1103/PhysRevA.92.042303. [DOI] [Google Scholar]
- Kandala A.; Mezzacapo A.; Temme K.; Takita M.; Brink M.; Chow J. M.; Gambetta J. M. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets. Nature 2017, 549, 242–246. 10.1038/nature23879. [DOI] [PubMed] [Google Scholar]
- Rattew A. G.; Hu S.; Pistoia M.; Chen R.; Wood S.. A domain-agnostic, noise-resistant, hardware-efficient evolutionary variational quantum eigensolver. arXiv preprint 2019, https://arxiv.org/abs/1910.09694.
- Wiersema R.; Zhou C.; de Sereville Y.; Carrasquilla J. F.; Kim Y. B.; Yuen H. Exploring entanglement and optimization within the hamiltonian variational ansatz. PRX Quantum 2020, 1, 020319. 10.1103/PRXQuantum.1.020319. [DOI] [Google Scholar]
- Harrigan M. P.; Sung K. J.; Neeley M.; Satzinger K. J.; Arute F.; Arya K.; Atalaya J.; Bardin J. C.; Barends R.; Boixo S.; et al. Quantum approximate optimization of non-planar graph problems on a planar superconducting processor. Nat. Phys. 2021, 17, 332–336. 10.1038/s41567-020-01105-y. [DOI] [Google Scholar]
- Taube A. G.; Bartlett R. J. New perspectives on unitary coupled-cluster theory. Int. J. Quantum Chem. 2006, 106, 3393–3401. 10.1002/qua.21198. [DOI] [Google Scholar]
- Romero J.; Babbush R.; McClean J. R.; Hempel C.; Love P. J.; Aspuru-Guzik A. Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz. Quantum Science and Technology 2019, 4, 014008. 10.1088/2058-9565/aad3e4. [DOI] [Google Scholar]
- Gard B. T.; Zhu L.; Barron G. S.; Mayhall N. J.; Economou S. E.; Barnes E. Efficient symmetry-preserving state preparation circuits for the variational quantum eigensolver algorithm. npj Quantum Information 2020, 6, 10. 10.1038/s41534-019-0240-1. [DOI] [Google Scholar]
- Grimsley H. R.; Economou S. E.; Barnes E.; Mayhall N. J. An adaptive variational algorithm for exact molecular simulations on a quantum computer. Nat. Commun. 2019, 10, 3007. 10.1038/s41467-019-10988-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. L.; Shkolnikov V.; Barron G. S.; Grimsley H. R.; Mayhall N. J.; Barnes E.; Economou S. E. qubit-adapt-vqe: An adaptive algorithm for constructing hardware-efficient ansätze on a quantum processor. PRX Quantum 2021, 2, 020310. 10.1103/PRXQuantum.2.020310. [DOI] [Google Scholar]
- Shkolnikov V.; Mayhall N. J.; Economou S. E.; Barnes E.. Avoiding symmetry roadblocks and minimizing the measurement overhead of adaptive variational quantum eigensolvers. arXiv preprint 2021, https://arxiv.org/abs/2109.05340.
- Fedorov D. A.; Alexeev Y.; Gray S. K.; Otten M.. Unitary Selective Coupled-Cluster Method. arXiv preprint 2021, https://arxiv.org/abs/2109.12652.
- Babbush R.; Wiebe N.; McClean J.; McClain J.; Neven H.; Chan G. K.-L. Low-depth quantum simulation of materials. Physical Review X 2018, 8, 011044. 10.1103/PhysRevX.8.011044. [DOI] [PubMed] [Google Scholar]
- Wang D.; Higgott O.; Brierley S. Accelerated variational quantum eigensolver. Phys. Rev. Lett. 2019, 122, 140504. 10.1103/PhysRevLett.122.140504. [DOI] [PubMed] [Google Scholar]
- Verteletskyi V.; Yen T.-C.; Izmaylov A. F. Measurement optimization in the variational quantum eigensolver using a minimum clique cover. J. Chem. Phys. 2020, 152, 124114. 10.1063/1.5141458. [DOI] [PubMed] [Google Scholar]
- Jena A.; Genin S.; Mosca M.. Pauli partitioning with respect to gate sets. arXiv preprint 2019, https://arxiv.org/abs/1907.07859.
- Gokhale P.; Angiuli O.; Ding Y.; Gui K.; Tomesh T.; Suchara M.; Martonosi M.; Chong F. T.. Minimizing state preparations in variational quantum eigensolver by partitioning into commuting families. arXiv preprint 2019, https://arxiv.org/abs/1907.13623.
- Yen T.-C.; Verteletskyi V.; Izmaylov A. F. Measuring all compatible operators in one series of single-qubit measurements using unitary transformations. J. Chem. Theory Comput. 2020, 16, 2400–2409. 10.1021/acs.jctc.0c00008. [DOI] [PubMed] [Google Scholar]
- Huggins W. J.; McClean J. R.; Rubin N. C.; Jiang Z.; Wiebe N.; Whaley K. B.; Babbush R. Efficient and noise resilient measurements for quantum chemistry on near-term quantum computers. npj Quantum Information 2021, 7, 23. 10.1038/s41534-020-00341-7. [DOI] [Google Scholar]
- Gokhale P.; Angiuli O.; Ding Y.; Gui K.; Tomesh T.; Suchara M.; Martonosi M.; Chong F. T. O(N3) measurement Cost for Variational Quantum Eigensolver on Molecular Hamiltonians. IEEE Transactions on Quantum Engineering 2020, 1, 1–24. 10.1109/TQE.2020.3035814. [DOI] [Google Scholar]
- Torlai G.; Mazzola G.; Carleo G.; Mezzacapo A. Precise measurement of quantum observables with neural-network estimators. Physical Review Research 2020, 2, 022060. 10.1103/PhysRevResearch.2.022060. [DOI] [Google Scholar]
- Crawford O.; van Straaten B.; Wang D.; Parks T.; Campbell E.; Brierley S. Efficient quantum measurement of Pauli operators in the presence of finite sampling error. Quantum 2021, 5, 385. 10.22331/q-2021-01-20-385. [DOI] [Google Scholar]
- Huang H.-Y.; Kueng R.; Preskill J. Predicting many properties of a quantum system from very few measurements. Nat. Phys. 2020, 16, 1050–1057. 10.1038/s41567-020-0932-7. [DOI] [Google Scholar]
- Hadfield C.; Bravyi S.; Raymond R.; Mezzacapo A.. Measurements of quantum Hamiltonians with locally-biased classical shadows. arXiv preprint 2020, https://arxiv.org/abs/2006.15788.
- Izmaylov A. F.; Yen T.-C.; Lang R. A.; Verteletskyi V. Unitary partitioning approach to the measurement problem in the variational quantum eigensolver method. J. Chem. Theory Comput. 2020, 16, 190–195. 10.1021/acs.jctc.9b00791. [DOI] [PubMed] [Google Scholar]
- Zhao A.; Tranter A.; Kirby W. M.; Ung S. F.; Miyake A.; Love P. J. Measurement reduction in variational quantum algorithms. Phys. Rev. A 2020, 101, 062322. 10.1103/PhysRevA.101.062322. [DOI] [Google Scholar]
- Bonet-Monroig X.; Babbush R.; O’Brien T. E. Nearly optimal measurement scheduling for partial tomography of quantum states. Physical Review X 2020, 10, 031064. 10.1103/PhysRevX.10.031064. [DOI] [Google Scholar]
- Ralli A.; Love P. J.; Tranter A.; Coveney P. V. Implementation of measurement reduction for the variational quantum eigensolver. Physical Review Research 2021, 3, 033195. 10.1103/PhysRevResearch.3.033195. [DOI] [Google Scholar]
- Ralli A.; Weaving T.; Tranter A.; Kirby W. M.; Love P. J.; Coveney P. V.. Unitary Partitioning and the Contextual Subspace Variational Quantum Eigensolver. arXiv preprint 2022, https://arxiv.org/abs/2207.03451. [DOI] [PMC free article] [PubMed]
- Temme K.; Bravyi S.; Gambetta J. M. Error mitigation for short-depth quantum circuits. Phys. Rev. Lett. 2017, 119, 180509. 10.1103/PhysRevLett.119.180509. [DOI] [PubMed] [Google Scholar]
- Li Y.; Benjamin S. C. Efficient variational quantum simulator incorporating active error minimization. Physical Review X 2017, 7, 021050. 10.1103/PhysRevX.7.021050. [DOI] [Google Scholar]
- Endo S.; Benjamin S. C.; Li Y. Practical quantum error mitigation for near-future applications. Physical Review X 2018, 8, 031027. 10.1103/PhysRevX.8.031027. [DOI] [Google Scholar]
- Kandala A.; Temme K.; Córcoles A. D.; Mezzacapo A.; Chow J. M.; Gambetta J. M. Error mitigation extends the computational reach of a noisy quantum processor. Nature 2019, 567, 491–495. 10.1038/s41586-019-1040-7. [DOI] [PubMed] [Google Scholar]
- Giurgica-Tiron T.; Hindy Y.; LaRose R.; Mari A.; Zeng W. J.. Digital zero noise extrapolation for quantum error mitigation. 2020 IEEE International Conference on Quantum Computing and Engineering (QCE); IEEE: 2020; pp 306–316. [Google Scholar]
- He A.; Nachman B.; de Jong W. A.; Bauer C. W. Zero-noise extrapolation for quantum-gate error mitigation with identity insertions. Phys. Rev. A 2020, 102, 012426. 10.1103/PhysRevA.102.012426. [DOI] [Google Scholar]
- Endo S.; Cai Z.; Benjamin S. C.; Yuan X. Hybrid quantum-classical algorithms and quantum error mitigation. J. Phys. Soc. Jpn. 2021, 90, 032001. 10.7566/JPSJ.90.032001. [DOI] [Google Scholar]
- Huggins W. J.; McArdle S.; O’Brien T. E.; Lee J.; Rubin N. C.; Boixo S.; Whaley K. B.; Babbush R.; McClean J. R. Virtual distillation for quantum error mitigation. Physical Review X 2021, 11, 041036. 10.1103/PhysRevX.11.041036. [DOI] [Google Scholar]
- Kirby W. M.; Tranter A.; Love P. J. Contextual Subspace Variational Quantum Eigensolver. Quantum 2021, 5, 456. 10.22331/q-2021-05-14-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radin M. D.; Johnson P.. Classically-Boosted Variational Quantum Eigensolver. arXiv preprint 2021, https://arxiv.org/abs/2106.04755.
- Bravyi S.; Gambetta J. M.; Mezzacapo A.; Temme K.. Tapering off qubits to simulate fermionic Hamiltonians. arXiv preprint 2017, https://arxiv.org/abs/1701.08213.
- Setia K.; Chen R.; Rice J. E.; Mezzacapo A.; Pistoia M.; Whitfield J. D. Reducing qubit requirements for quantum simulations using molecular point group symmetries. J. Chem. Theory Comput. 2020, 16, 6091–6097. 10.1021/acs.jctc.0c00113. [DOI] [PubMed] [Google Scholar]
- Ralli A.; Weaving T.. symmer. https://github.com/UCL-CCS/symmer, 2022.
- Gottesman D.Stabilizer codes and quantum error correction. Ph.D. Thesis, California Institute of Technology, 1997. [Google Scholar]
- Gottesman D.An introduction to quantum error correction and fault-tolerant quantum computation. Quantum information science and its contributions to mathematics; Proceedings of Symposia in Applied Mathematics; American Mathematical Society: 2010; pp 13–58.
- Dehaene J.; De Moor B. Clifford group, stabilizer states, and linear and quadratic operations over GF (2). Phys. Rev. A 2003, 68, 042318. 10.1103/PhysRevA.68.042318. [DOI] [Google Scholar]
- Nielsen M. A.; Chuang I. L.. Quantum Computation and Quantum Information, 10th Anniversary edition; Cambridge University Press, 2010. [Google Scholar]
- Kraus K.; Böhm A.; Dollard J. D.; Wootters W.. States, effects, and operations: fundamental notions of quantum theory. Lectures in Mathematical Physics at the University of Texas at Austin; Springer-Verlag: 1983, Vol. 190. [Google Scholar]
- Bell J. S. On the Einstein Podolsky Rosen paradox. Physics Physique Fizika 1964, 1, 195. 10.1103/PhysicsPhysiqueFizika.1.195. [DOI] [Google Scholar]
- Bell J. S. On the problem of hidden variables in quantum mechanics. Rev. Mod. Phys. 1966, 38, 447. 10.1103/RevModPhys.38.447. [DOI] [Google Scholar]
- Kochen S.; Specker E. P.. The logico-algebraic approach to quantum mechanics; Springer: 1975; pp 293–328. [Google Scholar]
- Kirby W. M.; Love P. J. Contextuality test of the nonclassicality of variational quantum eigensolvers. Phys. Rev. Lett. 2019, 123, 200501. 10.1103/PhysRevLett.123.200501. [DOI] [PubMed] [Google Scholar]
- Raussendorf R.; Bermejo-Vega J.; Tyhurst E.; Okay C.; Zurel M. Phase-space-simulation method for quantum computation with magic states on qubits. Phys. Rev. A 2020, 101, 012350. 10.1103/PhysRevA.101.012350. [DOI] [PubMed] [Google Scholar]
- Kirby W. M.; Love P. J. Classical simulation of noncontextual Pauli Hamiltonians. Phys. Rev. A 2020, 102, 032418. 10.1103/PhysRevA.102.032418. [DOI] [Google Scholar]
- Spekkens R. W.Quantum Theory: Informational Foundations and Foils; Springer Netherlands: Dordrecht, 2016; pp 83–135. [Google Scholar]
- Schwerdtfeger P. The pseudopotential approximation in electronic structure theory. ChemPhysChem 2011, 12, 3143–3155. 10.1002/cphc.201100387. [DOI] [PubMed] [Google Scholar]
- Wecker D.; Bauer B.; Clark B. K.; Hastings M. B.; Troyer M. Gate-count estimates for performing quantum chemistry on small quantum computers. Phys. Rev. A 2014, 90, 022305. 10.1103/PhysRevA.90.022305. [DOI] [Google Scholar]
- Poulin D.; Hastings M. B.; Wecker D.; Wiebe N.; Doherty A. C.; Troyer M.. The Trotter step size required for accurate quantum simulation of quantum chemistry. arXiv preprint 2014, https://arxiv.org/abs/1406.4920.
- Kowalski K. Properties of coupled-cluster equations originating in excitation sub-algebras. J. Chem. Phys. 2018, 148, 094104. 10.1063/1.5010693. [DOI] [Google Scholar]
- Bauman N. P.; Bylaska E. J.; Krishnamoorthy S.; Low G. H.; Wiebe N.; Granade C. E.; Roetteler M.; Troyer M.; Kowalski K. Downfolding of many-body Hamiltonians using active-space models: Extension of the sub-system embedding sub-algebras approach to unitary coupled cluster formalisms. J. Chem. Phys. 2019, 151, 014107. 10.1063/1.5094643. [DOI] [PubMed] [Google Scholar]
- Ralli A.; Williams M. I.; Coveney P. V.. A Scalable Approach to Quantum Simulation via Projection-based Embedding. arXiv preprint, arXiv:2203.01135, 2022, https://arxiv.org/abs/2203.01135.
- Johnson R. D., IIINIST Computational Chemistry Comparison and Benchmark Database. NIST Standard Reference Database Number 101, 2020, https://cccbdb.nist.gov/.
- Anis M. S.; Mitchell A.; Abraham H.; Agarwal R.; Agliardi G.; Aharoni M.; Ajith V.; Akhalwaya I. Y.; Aleksandrowicz G.; Alexander T.; et al. Qiskit: An Open-source Framework for Quantum Computing, 2021, 10.5281/zenodo.2573505. [DOI]
- Sun Q.; Berkelbach T. C.; Blunt N. S.; Booth G. H.; Guo S.; Li Z.; Liu J.; McClain J. D.; Sayfutyarova E. R.; Sharma S.; et al. PySCF: the Python-based simulations of chemistry framework. Wiley Interdisciplinary Reviews: Computational Molecular Science 2018, 8, e1340 10.1002/wcms.1340. [DOI] [Google Scholar]
- Ryabinkin I. G.; Lang R. A.; Genin S. N.; Izmaylov A. F. Iterative qubit coupled cluster approach with efficient screening of generators. J. Chem. Theory Comput. 2020, 16, 1055–1063. 10.1021/acs.jctc.9b01084. [DOI] [PubMed] [Google Scholar]
- Jordan P.; Wigner E. P.. The Collected Works of Eugene Paul Wigner; Springer, 1993; pp 109–129. [Google Scholar]
- Kingma D. P.; Ba J.. Adam: A method for stochastic optimization. arXiv preprint 2014, https://arxiv.org/abs/1412.6980.
- Parrish R. M.; Hohenstein E. G.; McMahon P. L.; Martinez T. J.. Hybrid quantum/classical derivative theory: Analytical gradients and excited-state dynamics for the multistate contracted variational quantum eigensolver. arXiv preprint 2019, https://arxiv.org/abs/1906.08728.
- Wierichs D.; Gogolin C.; Kastoryano M. Avoiding local minima in variational quantum eigensolvers with the natural gradient optimizer. Physical Review Research 2020, 2, 043246. 10.1103/PhysRevResearch.2.043246. [DOI] [Google Scholar]
- Pellow-Jarman A.; Sinayskiy I.; Pillay A.; Petruccione F. A comparison of various classical optimizers for a variational quantum linear solver. Quantum Information Processing 2021, 20, 202. 10.1007/s11128-021-03140-x. [DOI] [Google Scholar]
- Fletcher R.Practical methods of optimization; John Wiley & Sons: 2013. [Google Scholar]
- Stokes J.; Izaac J.; Killoran N.; Carleo G. Quantum natural gradient. Quantum 2020, 4, 269. 10.22331/q-2020-05-25-269. [DOI] [Google Scholar]
- Hagberg A.; Swart P.; S Chult D.. Exploring network structure, dynamics, and function using NetworkX. Proceedings of the 7th Python in Science Conference; 2008. [Google Scholar]
- Arrasmith A.; Cincio L.; Somma R. D.; Coles P. J.. Operator sampling for shot-frugal optimization in variational algorithms. arXiv preprint 2020, https://arxiv.org/abs/2004.06252.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In the interest of reproducibility, we supply in the Supporting Information the tapering parameters, CS-VQE model data, and noncontextual projection ansätze which permit chemical precision for the fewest number of qubits with respect to the molecular systems listed in Table 1; the raw data for these results are supplied in ancillary files hosted at https://arxiv.org/abs/2204.02150 along with an explanatory notebook. This will provide sufficient information for the reader to reproduce Figures 1, 2 and 3.