

Abstract

Retinoic acid receptor-related orphan receptor γ-t (RORγt) and GPBAR1, a transmembrane G-protein-coupled receptor for bile acids, are attractive drug targets to develop clinically relevant small modulators as potent therapeutics for autoimmune diseases. Herein, we designed, synthesized, and evaluated several new bile acid-derived ligands with potent dual activity. Furthermore, we performed molecular docking and MD calculations of the best dual modulators in the two targets to identify the binding modes as well as to better understand the molecular basis of the inverse agonism of RORγt by bile acid derivatives. Among these compounds, 7 was identified as a GPBAR1 agonist (EC50 5.9 μM) and RORγt inverse agonist (IC50 0.107 μM), with excellent pharmacokinetic properties. Finally, the most promising ligand displayed robust anti-inflammatory activity in vitro and in vivo in a mouse model of 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis.
Introduction
Immune cells in the gastrointestinal tract and liver are continuously exposed to microbial antigens generated by the intestinal microbiota. However, because the maintenance of immune homeostasis in enterohepatic tissues is essential to prevent the onset of inflammation, mechanisms that regulate the development of a tolerogenic phenotype by immune cells in the gastrointestinal tract and liver are only partially known.
Bile acids (BAs)1 are steroidal molecules that represent the end product of cholesterol metabolism, exerting their regulatory function by activating a family of receptors known as the bile acid-activated receptors (BARs). Among these, the farnesoid X receptor (FXR) and the G-protein-coupled bile acid receptor (GPBAR1, also commonly named M-BAR or Takeda G-protein-coupled receptor 5, TGR5) have proven to be druggable, and several selective or dual ligands have been developed.2−5 Both FXR and GPBAR1 are expressed by cells of innate immunity, and their activation reverses intestinal and systemic inflammation in a variety of preclinical models.
However, the lack of expression on cells of adaptive immunity limits the development of FXR and GPBAR1 agonists for therapeutic purposes in those settings where T cells play a mechanistic role.6 The retinoic acid receptor-related orphan receptors (RORs) represent key regulators of many physiological and pathological processes. Among ROR isoforms, RORγ is highly expressed in lymphoid organs, skeletal muscles, adipose tissues, kidneys, and liver, and RORγt plays a role in regulating TH17 cells, a subset of T helper lymphocytes whose activation drives intestinal inflammation in IBD patients.4,7,8 Additionally, the overexpression of RORγt in naïve CD4+ T cells leads to the induction and development of TH17 cells, whereas its inhibition reduces the autoimmune response4,9 and holds promise in the treatment of colitis, autoimmune diseases, and metabolic disorders.8,10 Several cholesterol intermediates and metabolites exhibit high affinity toward RORγt and act as agonists or inverse agonists.11,12 The mechanism of action of RORγt agonists is explained through the stabilization of the H-bond between His479 and Tyr502 on Helix 11 (H11) and Helix 12 (H12), respectively, allowing the recruitment of transcriptional coactivators on target genes. On the other hand, inverse agonists of RORγt disrupt the H-bond between H11 and H12 through a mechanism of interaction that includes Leu324, Trp317, His479, and Tyr502, preventing the recruitment of transcriptional coactivators and inhibiting TH17 cell differentiation both in vitro and in vivo.13−18
The specific structural motifs of RORγt inverse agonists rely on the presence of a hydrogen bond acceptor at one end of the molecule and a lipophilic and rigid motif on the other side, appropriately spaced.17,19 RORγt cocrystallized structures demonstrated that the AF-2 domain at the C-terminus of the receptor, together with helices H3, H4, and H5, forms a charge clamp pocket that facilitates the binding of coactivators. Mutagenesis studies of this region confirmed that such a charged clamp pocket is required for hydroxycholesterols to affect RORγt activity.14
While RORγt expression is limited to TH17 cells and innate lymphoid 3 cells (ILC3s), GPBAR1 is expressed by the cells of innate immunity, therefore developing dual RORγt antagonists/GPBAR1 agonists might provide a unique opportunity to simultaneously target the innate immunity along with effector mechanisms in the TH17 pathway, tightening the spectrum of activity and limiting the potential side effects linked to the nonselective inhibition of the whole immune system.5,20−25
Building on this background,23 we have designed a new series of bile acid-derived ligands with potential multitarget activity (compounds 1–21, Figure 1). Synthesis, pharmacological profiling, and computational studies resulted in the discovery of 3 and 7 as GPBAR1 agonists and RORγt inverse agonists. Further, the excellent pharmacokinetic profile of 7 placed this novel hybrid ligand in a privileged position for preclinical studies, and deep in vivo pharmacological evaluation demonstrated its therapeutical potential in the treatment of colitis. Therefore, here, we present, in a proof-of-concept study, the first evidence that dual modulation of RORγt/GPBAR1 represents a new armamentarium in IBD pharmacological treatment.
Figure 1.
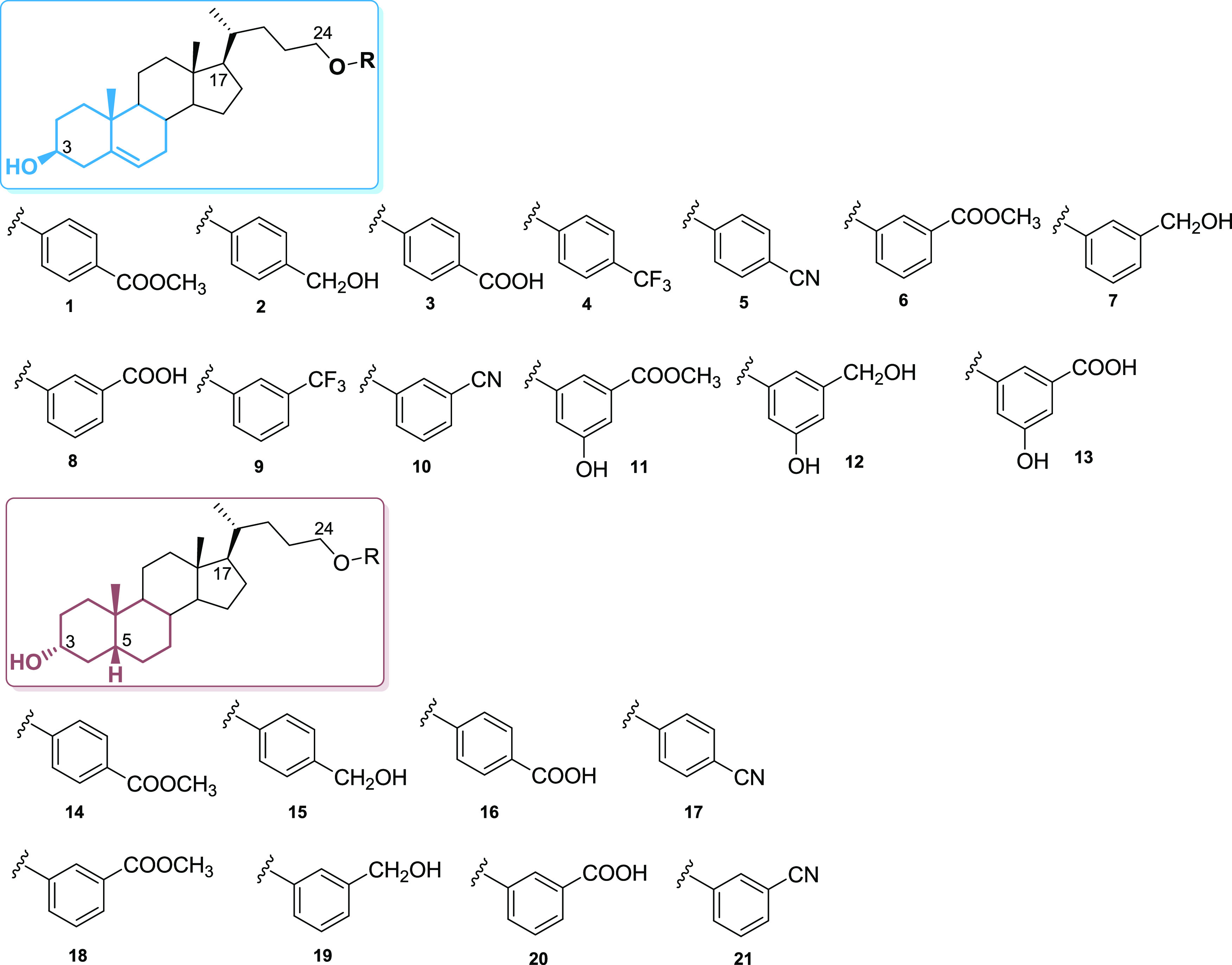
Chemical structure of 1–21.
Results and Discussion
Starting from hyodeoxycholic acid and lithocholic acid, two different subsets of compounds, focusing on the ring A and B junction and modifying the side chain in length and in the nature of the end-group, have been prepared (Figure 1).
The preparation of subset 1 (compounds 1–13, Scheme 1) was obtained from compound 23(26) by alcohol protection at C3 and reduction at the side-chain methyl ester to give 24, which was used in multiple Mitsunobu reactions with various phenols (Scheme 1) to produce, after deprotection at C3, derivatives 4, 5, 9, and 10 and the expected methyl esters (1, 6, and 11).27 Finally, LiBH4 reduction and basic hydrolysis afforded the corresponding alcohols (compounds 2, 7, and 12) and carboxylic acids (compounds 3, 8, and 13), respectively.
Scheme 1. Synthesis of Analogues 1–21.
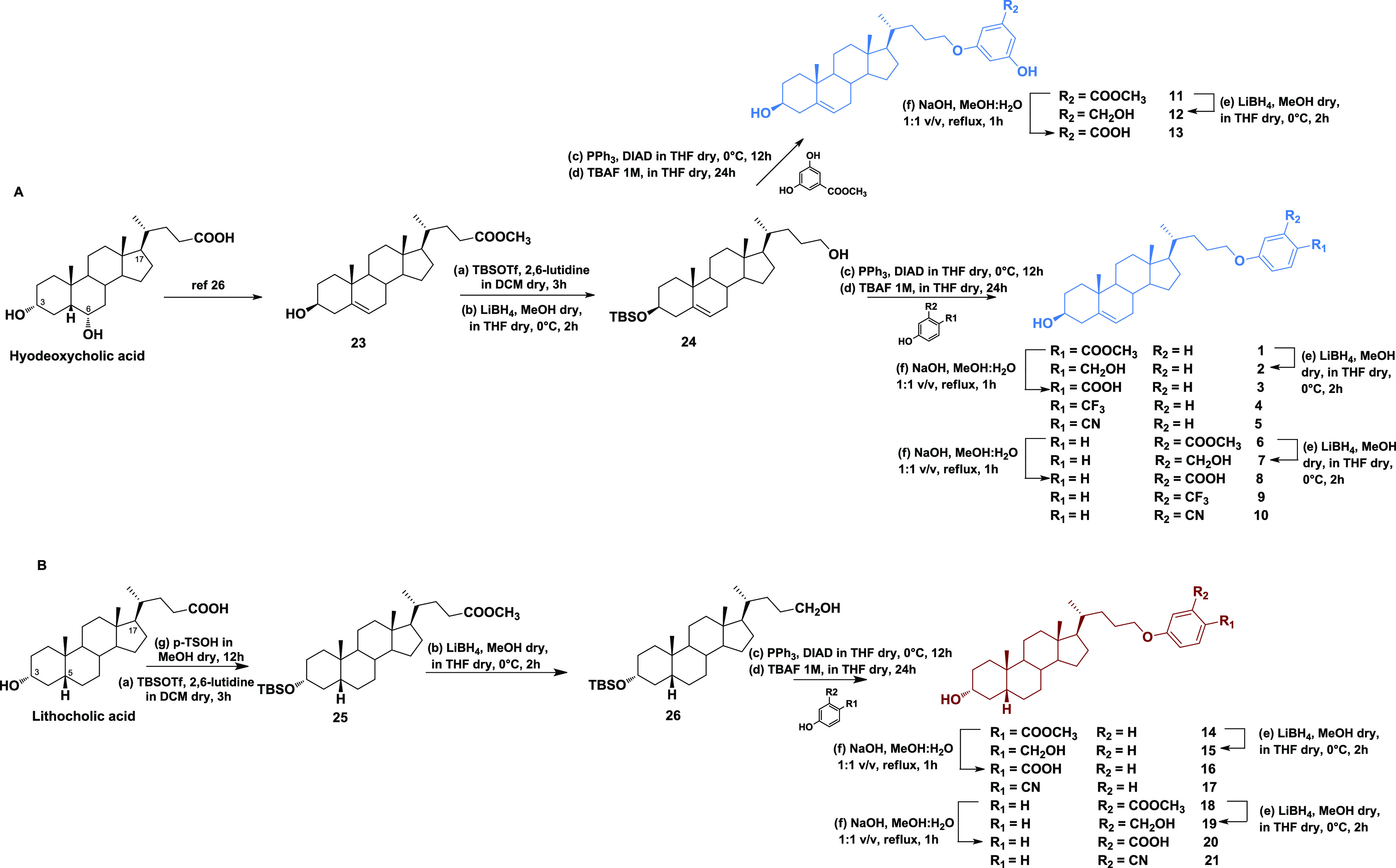
For the synthesis of the second subset of compounds (14–21), lithocholic acid was used as a starting material. To enhance the selectivity of the Mitsunobu coupling at C24,28 we protected the C3 alcoholic function before LiBH4 reduction of ester in the side chain. Intermediate 26 was coupled with four different phenols to produce 14, 17, 18, and 21. LiBH4 reduction and basic hydrolysis on methyl esters 14 and 18 furnished the alcohols 15 and 19 and the carboxylic acids 16 and 20, respectively.
A selection of new derivatives (compounds 2–5, 7–10, 12, 13, 15–17, and 19–21) was first tested in the luciferase reporter assay on HEK-293T cells transfected with GPBAR1. Particularly, we decided to exclude from our assays the ester derivatives (compounds 1, 6, 11, 14, and 18) because of their tendency to be hydrolyzed in vivo. Results of these assays, reported in Table 1, revealed that several compounds exhibited good efficacy as GPBAR1 agonists and, notably, 7, 15, and 19 showed similar activity to taurolithocholic acid (TLCA), the most potent endogenous GPBAR1 agonist.
Table 1. Efficacy on GPBAR1 and RORγtabc.
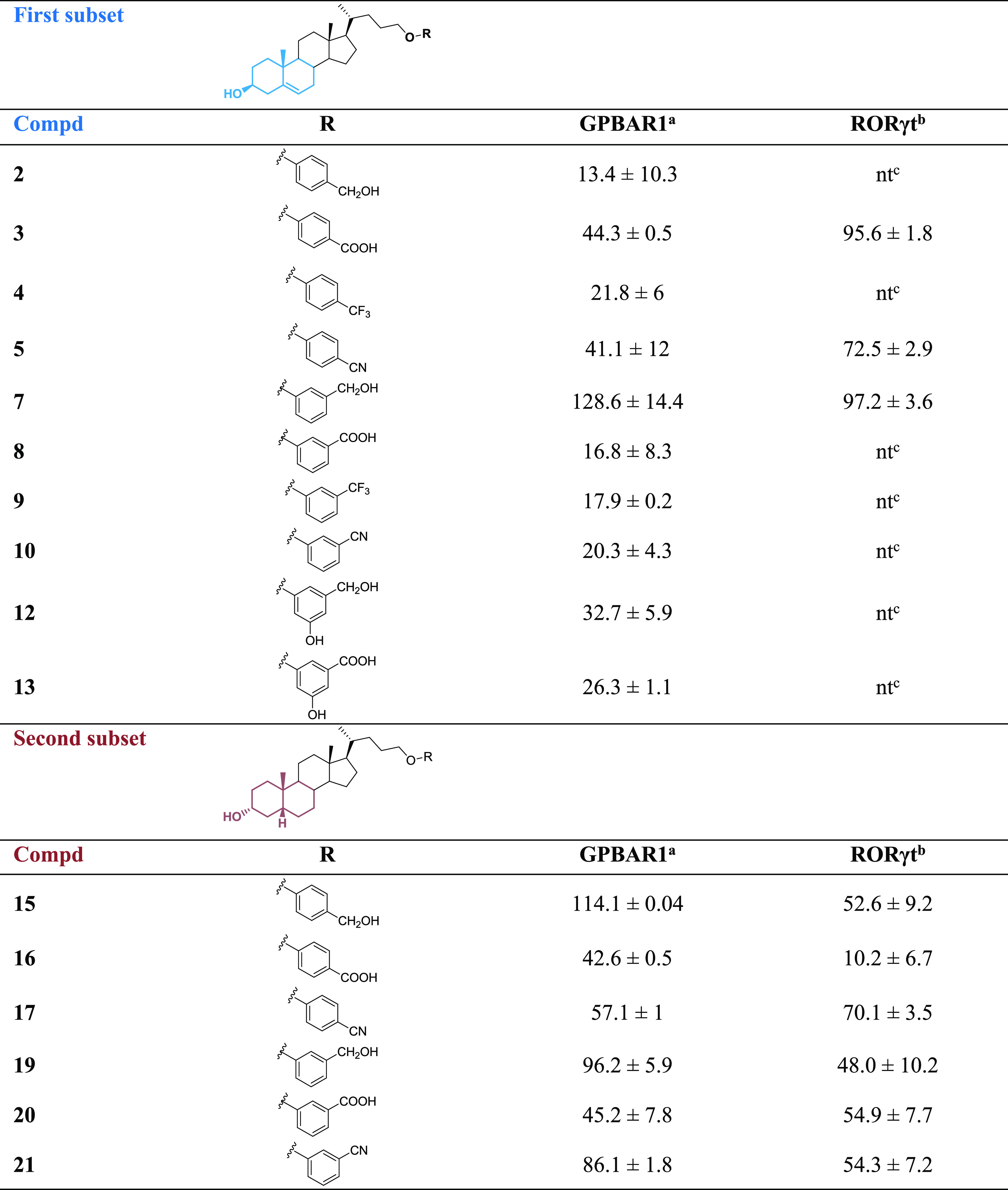
Eff. (%) is the maximum efficacy of the compound (10 μM) relative to TLCA (10 μM) as 100 in transactivation of a cAMP-responsive element (CRE) on HEK-293T cells; results are the mean of two experiments ± SD.
Eff. is the efficacy of the compound (10 μM) as % of inhibition calculated from the residual activity of RORγt, defining the wells containing only vehicle (DMSO) as 100% control and the wells without the protein as 0% control.
nt means not tested.
All of the compounds showing an efficacy greater than 40% were further analyzed on RORγt to identify new tools for the treatment of IBD.
To evaluate inverse-agonist activities against human RORγt of the selected compounds, the human RORγ reporter assay system was used. Moreover, the ability of these compounds to interfere with the RORγt-SRC-1 interaction was also evaluated in a cell-free α-screen assay. The efficacies of all tested compounds (3, 5, 7, 15–17, and 19–21) are reported in Table 1. Notably, apart from 17, all compounds of the second subset, endowed with the bile acid shape (cis A/B ring junction), showed lower efficacy against RORγt than the corresponding derivatives belonging to the first subset, with 3, 5, and 7 showing the best efficacy on RORγt. Compounds 5 and 17 resulted in being less soluble in aqueous buffers (10 and 15 μM, respectively), thus hampering further pharmacological evaluations and prompting us to investigate the most promising compounds, 3 and 7.
As reported in Figure 2, 3 and 7 exhibited good activity against both receptors, representing the best match in terms of efficacy and potency. These compounds exerted a concentration-dependent effect on the activation of CREB in HEK-293T cells transfected with GPBAR1 with an EC50 of 0.22 and 5.9 μM, respectively. Despite the low value of EC50 for 3, it should be considered that the above potency is related to an efficacy of about 44% when compared to TLCA (Table 1), whereas the efficacy of 7 was about 129%. Moreover, 3 and 7 inhibited the binding of RORγt-LBD to coactivator peptide SRC-1 with an IC50 of 1.19 and 0.107 μM, respectively. The potency of 7 was also confirmed in the human RORγt reporter assay, revealing an IC50 of 0.193 μM. Ursolic acid and digoxin were used as the assay control (IC50 values of 0.116 and 0.237 μM, respectively). Moreover, both compounds exhibited excellent selectivity with no off-target activity toward metabolic nuclear receptors, such as FXR, LXRα, and LXR β (Table S1 in the SI).
Figure 2.
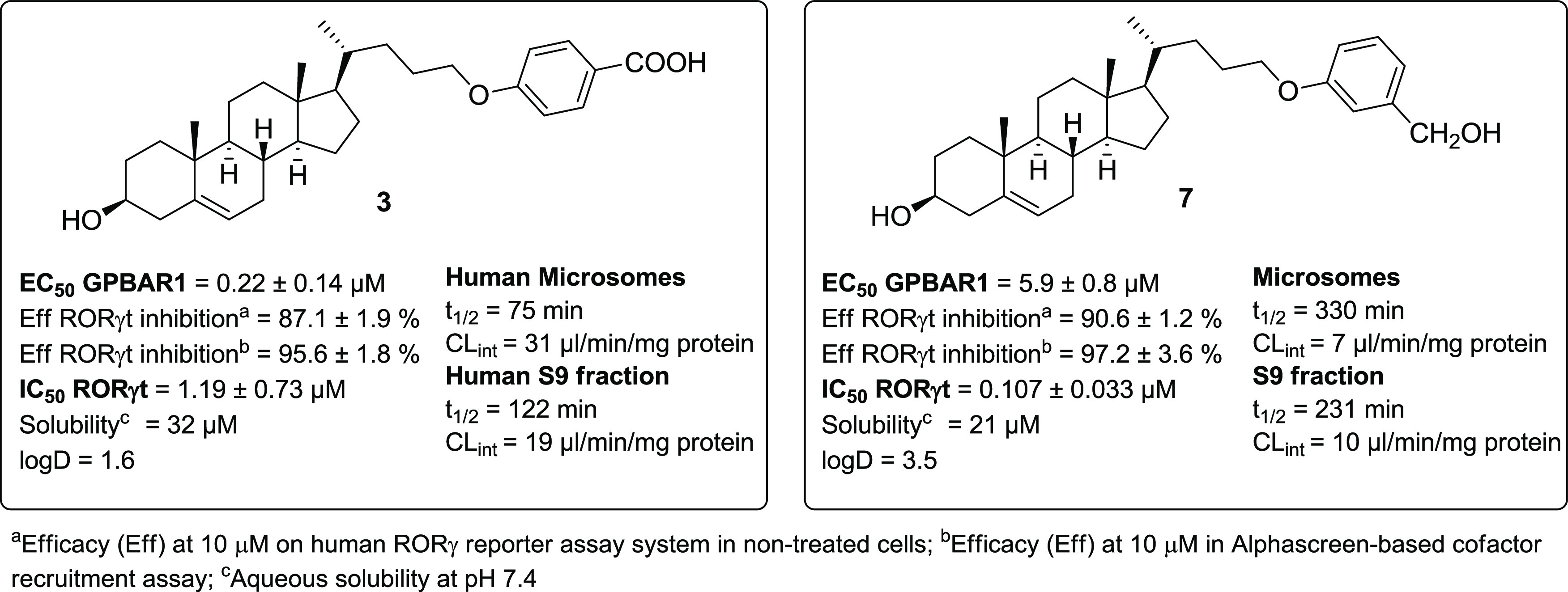
In vitro assays. An overview of the efficacy, potency, and preliminary PK parameters of compounds 3 and 7.
Compounds 3 and 7 were further investigated to assess their aqueous solubility, LogD, and the liability to hepatic metabolizing enzymes (Figure 2). Both the liver microsomal fraction and S9 fraction were employed. The compounds resulted in being sufficiently soluble in an aqueous buffer at pH 7.4 and were endowed with proper LogD values. Moreover, in vitro treatment with human liver microsomes and human liver S9 fractions demonstrated a very promising t1/2 value of 330 min (CLint = 7 μL/min/mg protein) for compound 7. All of these data highlight the pharmacological potential of 7 in associating potency and efficacy in the dual modulation of RORγt and GPBAR1 with excellent metabolic stability.
Computational Studies
To elucidate the binding mode of the most promising dual-activity derivatives, 3 and 7, toward RORγt and GPBAR1, molecular modeling studies based on docking and MD calculations, which are widely employed techniques to study ligand/protein binding interactions,29−31 have been carried out. First, we performed molecular docking calculations using the Glide software package (SI).32 As the starting structure, the human crystallographic structure of the active conformation of the RORγt-LBD domain with PDB ID 3l0j(33) was employed. For the GPBAR1 receptor, we used the 3D model developed in 2014.34 The docking calculations identified binding modes of 3 and 7 to RORγt and GPBAR1 (SI Figures S1 and S3) in agreement with those previously reported for structurally similar ligands.33 To assess the docking results in a more realistic condition, where protein flexibility and the water effect are taken into account, the best docking pose for both 3 and 7 underwent a 1 μs MD simulation in RORγt in an explicit solvent. The results showed that, after a few ns of simulation, 3 underwent a conformational rearrangement with respect to the initial docking pose (Figure 3, panels A and C), leading to an extended conformation of the flexible chain at C-17, while the hydroxyl group at C-3 kept a stable H-bond with the Gln286 side chain on H2 as found by docking. The steroidal scaffold was placed in the amphipathic pocket between H4 and H5, where it was further stabilized by a set of hydrophobic interactions established with the side chains of Leu324, Val361, Val376, and Phe378. The aromatic ring was located between H4 and H11, where it took part in aromatic interactions with Trp317 and His479. The carboxyl group on the ligand side chain moved during the MD simulation to form an H-bond with Arg482 located on H11 (Figure 3, panel A). Finally, the above conformational change and the stacking interaction between the aromatic ring in the ligand’s side chain and the side chain of His479 caused the destruction of the H-bond between His479 and Tyr502 and a slight shift of H12, altering the binding site of the coactivator (SI Figure S4).
Figure 3.

Representation of the MD binding mode of (A) 3 and (B) 7 in RORγ and (E) 3 and (F) 7 in the GPBAR1 homology model. The ligands are represented as pink and gold sticks, respectively, and the interacting residues of the receptor are shown in gray and labeled, with oxygen atoms in red and nitrogens in blue. The receptor is represented as ribbons with its helix labeled. Hydrogens are omitted for the sake of clarity and H-bonds are displayed as dashed lines. Plots of average RMSD calculated on the heavy atoms of the steroidal scaffold of (C) and (G) 3 and (D) and (H) 7.
At the end of the production run of 7, the ligand’s binding mode was stabilized by different interactions (Figure 3, panels B and D). The hydroxyl group at C-3 H-bonded Arg264, whereas the steroidal scaffold was stabilized by hydrophobic interactions with Leu324, Val361, Val376, and Phe388. The methylene linker on the side chain at C-17 interacted with Leu391, Ile397, and Ile400, whereas the ethereal oxygen H-bonded Cys393. Finally, the aromatic ring was stabilized between H7 and H11 by aromatic interactions with Trp317, His479, and Phe486. A direct interaction of the 7 side chain with Tyr502 caused a more marked weakening of the H-bond network between His479 and Tyr502 (SI Figure S4), stabilizing the open form of H12, which hampered the recruitment of the coactivator. Our results agree with the available literature data,11 showing that the mechanism of the action of inverse agonists relies on the disruption of the H-bond between His479 and Tyr502. This event destabilizes the active form of the receptor and hampers the recruitment of the transcriptional coactivator SRC.
During the MD simulations, both the ligands showed the so-called “push–pull” mechanism of action, i.e., pushing Trp317 and pulling H479 or Y502, already proposed for other inverse agonists.17,18 In more detail, the ligands induced a flip of W317 into a position that did not allow the H-bond interaction between His479 and Thr502, while interacting with one of such residues. As the final effect, the tertiary structure of H11–12 is perturbed, thus affecting the recruitment of the coactivator and target gene expression (SI Figure S4). Overall, our MD simulations indicated that 3 and 7 work as RORγt inverse agonists.
To further evaluate the capability of 3 and 7 to act as inverse agonists, we also performed 500 ns of MD simulations of the cocrystallized agonist ligand, 22R-hydroxycholesterol, in the active form of RORγt (PDB ID 3l0j).33 At variance with compounds 3 and 7, the agonist stabilizes the active conformation of RORγt (see Figures S4 and S5 and the Supporting Information for details).
In addition, 1 μs MD calculations were performed on the docking binding modes of 3 and 7 to GPBAR1. After 150 ns, 3 underwent a change in its binding mode, which was then maintained until the end of the simulation (Figure 3, panel G). The hydroxyl group at C-3 was still forming an H-bond with the Glu169 backbone, and the steroidal scaffold was located between transmembrane helices (TM) 3 and 5, formed by Tyr89, Asn93, Phe96, Leu97, Leu174, Leu263, Leu266, and Trp267 and stabilized by hydrophobic interactions. The aromatic ring on the side chain was also involved in hydrophobic interactions with Leu18, Leu68, and Leu71 and in a t-shaped π-stacking with Trp63 (Figure 3, panel E).
Compound 7 binding mode changed after 100 ns of MD (Figure 3, panel H) to achieve a more stable conformation, like the one described for 3. The steroidal scaffold was in the same pocket of 3, and it established hydrophobic interactions with the side chain of several residues, such as Leu14, Trp63, Leu68, Leu71, Leu97, Leu174, Leu263, Leu266, and Trp267, and H-bonds between the meta-CH2OH group and Ser267 (Figure 3, panel F), the hydroxyl group at C-3 and the backbone of Glu169, and the ethereal oxygen and the side chain of Ser21 that further contributed to stabilize compound 3 in the binding pocket.
The higher affinity of 3 than 7 (Figure 2) can be explained through the electrostatic potential surface calculated on GPBAR1 (Figure 4), where the charge distributions of the binding pocket showed the preference to accommodate 3, which is negatively charged.
Figure 4.
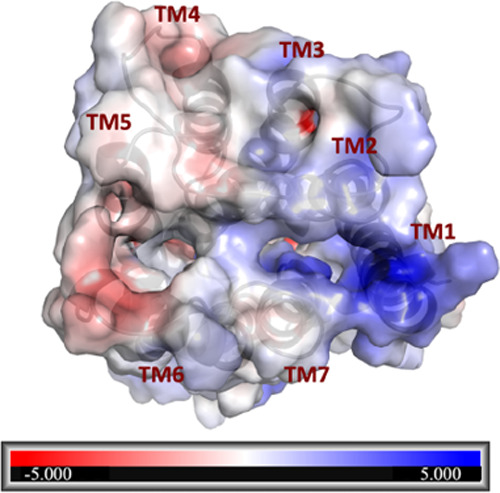
Electrostatic potential map calculated on GPBAR1. The surface is colored according to the electrostatic potential, where the red color (negative potential) indicates an excess of negative charges and the blue color (positive potential) is an excess of positive charges. The white regions correspond to fairly neutral potentials. The values are expressed in atomic units (a.u).
In Vitro Pharmacological Evaluation
We have first investigated whether 3 and 7 modulate the inflammatory response of U937 cells, a human monocytic cell, to lipopolysaccharide (LPS) and TNFα. Cells were coincubated with or without compounds at 0.1, 1, 10, 100, and 1000 nM and dexamethasone (Dex) at 5 μM was used as a control (Figure 5).
Figure 5.

U937 cells activated with LPS (100 ng/mL) plus TNFα (100 ng/mL) for 24 h, alone or in combination with 3 or 7 at 0.1, 1, 10, 100, and 1000 nM. Dexamethasone (Dex) was used at 5 μM. Quantitative real-time PCR analysis of the expression of proinflammatory genes IL-1β (A and E), TNFα (B and F), IL-6 (C and G), and M1 marker CD11c (D and H). These data are normalized to TBP mRNA expression. Data are derived from five replicates. Results represent the mean ± SEM. #p < 0.05 vs NT group and *p < 0.05 vs LPS plus TNFα group. Analysis of variance (ANOVA) was used for statistical comparisons.
Exposure of U937 to LPS plus TNFα induced an increase in the production of proinflammatory cytokines IL-1β, TNFα, and IL-6, also increasing the expression of CD11c, a specific marker of the proinflammatory M1 subtype of macrophages (Figure 5). Compound 3 reduced the expression of proinflammatory cytokines and CD11c. At the highest concentration of 1 μM, a reduction in the expression of all measured genes was observed by 30–50% compared to cells exposed only to LPS plus TNFα (Figure 5). Also, 7 (Figure 5) reverted the inflammatory state induced in U937 cells by LPS plus TNFα by reducing the expression of all proinflammatory markers measured with a concentration-dependent effect (Figure 5).
Therefore, 3 and 7 showed comparable immunomodulatory activity in vitro, but the data shown in Figure 2 indicated a much higher metabolic stability for 7 compared to 3 with a very high t1/2 value. For these reasons, we focused our attention on 7 in the subsequent study in vivo.
The effect exerted by 7 was tested in a mouse model of TNBS-induced colitis. The severity of colitis was relieved by 7 in a dose-dependent manner, as demonstrated by the trend in the percentage of body weight and the colitis disease activity index (CDAI) (Figure 6, panels A, B). All three doses of 7 decreased the damage to the colon as demonstrated by both the macroscopic analysis (Figure 6, panels C, D) and the histological analysis using H&E staining (Figure 6, panel E).
Figure 6.

Mice treated with TNBS were administered with the vehicle or 7 at doses of 10, 20, or 30 mg/kg/day by gavage from day 0 to day 4. (A) Change in body weight, (B) CDAI score, (C) colon length, (D) ratio between colon weight and colon length, (E) H&E staining on colon sections of the control, TNBS-treated, and TNBS plus various concentrations of 7 treated mice. Results are the mean ± SEM of 5–10 mice per group. *p < 0.05 vs TNBS group and #p < 0.05 vs NT group.
To gain deeper insight into the immunomodulatory effect exerted by 7, we have investigated the expression of various cytokines and immune cell markers in the colon of mice rendered colitic by TNBS administration (Figure 7). The development of colon inflammation in this model is associated with a robust induction in the expression of the proinflammatory cytokines, IL-1β, IL-6, and TNFα (Figure 7, panel A), along with a downregulation of the anti-inflammatory cytokines IL-10 and TGFβ (Figure 7, panel B). Furthermore, TNBS colitis results in robust recruitment in the colon lamina propria of various leukocyte subsets including macrophages and TH17 lymphocytes.
Figure 7.
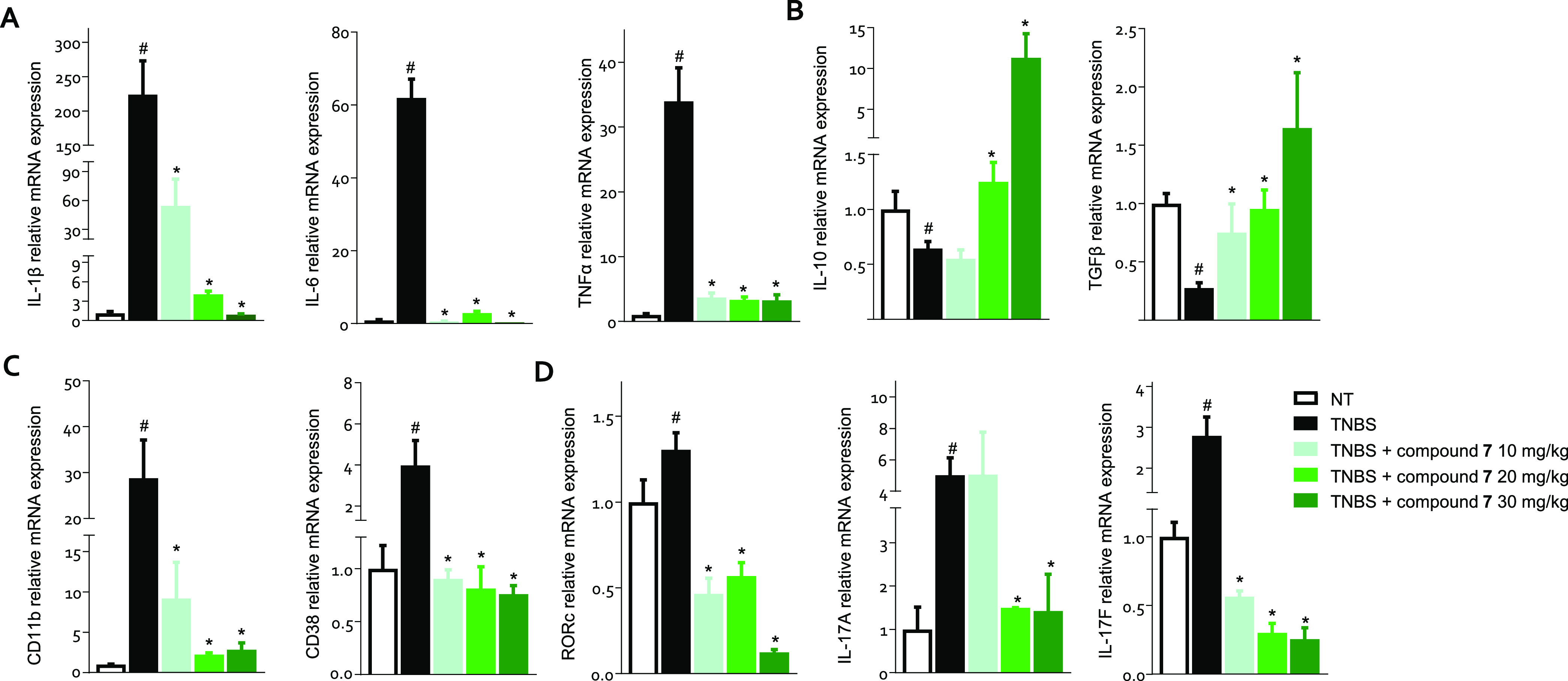
C57 male mice treated with TNBS were administered with the vehicle or 7 at doses of 10, 20, or 30 mg/kg/day by gavage from day 0 to day 4. The relative mRNA expression of (A) proinflammatory (IL-1β, IL-6, and TNFα) and (B) anti-inflammatory cytokines (IL-10 and TGFβ), (C) macrophage markers, and (D) TH17 cell markers in the colon assayed by RTPCR. Data are normalized to the expression of Gapdh, Tbp, and Actb mRNA. Results are the mean ± SEM of 5–10 mice per group. *p < 0.05 vs TNBS group and #p < 0.05 vs NT group.
Treating mice with TNBS induced a strong increase in the colonic expression of CD11b, a macrophage marker (Figure 7, panel C), suggesting an influx of monocytes into the inflamed tissue. These cells showed a proinflammatory signature, an M1 phenotype, as evidenced by the upregulation of the CD38 gene (Figure 7, panel C). The analysis of the expression of Rorc, the gene encoding RORγt, and of IL-17A and F demonstrated a robust inflow of TH17 RORγt+ cells in the lamina propria of colitis mice (Figure 7, panel D). Compound 7 reverted this inflammatory pattern in a dose-dependent manner and also promoted a strong increase in the expression of IL-10, a potent anti-inflammatory mediator, which represents one of the mechanisms underlying the anti-inflammatory effect of GPBAR1 activation (Figure 7, panels A, B).35 The administration of 7 blunted the inflow of macrophages into the lamina propria of the colon, as demonstrated by the downregulation of CD11b, and reversed the polarization toward the proinflammatory M1 phenotype, as assessed by the decreased expression of CD38 (Figure 7, panel C). On adaptive immunity, the administration of 7 decreased the expression of Rorc, IL-17A, and F, negatively modulating the immune response of TH17 lymphocytes (Figure 7, panel D).
Together, these data support the notion that 7 might represent a promising candidate for the drug therapy of IBD thanks to its ability to modulate both the innate immune response by acting on macrophages and adaptive immunity by inhibiting the polarization/activation of TH17 lymphocytes.33
To further investigate the role of simultaneous modulation of RORγt and GPBAR1 in protecting against the development of colitis, TNBS-treated mice were force-fed with 7, BAR501, a selective GPBAR1 agonist,36 or ML209, a RORγt inverse agonist (Figure 8).37 All three compounds attenuated the development of colitis, highlighting the role of both receptors in the model. Accordingly, compound 7 exerted a greater protective effect than the two selective agents, as demonstrated by the attenuation of the CDAI score, measurement of colon length, and histological damage (Figure 8, panels A–D).
Figure 8.
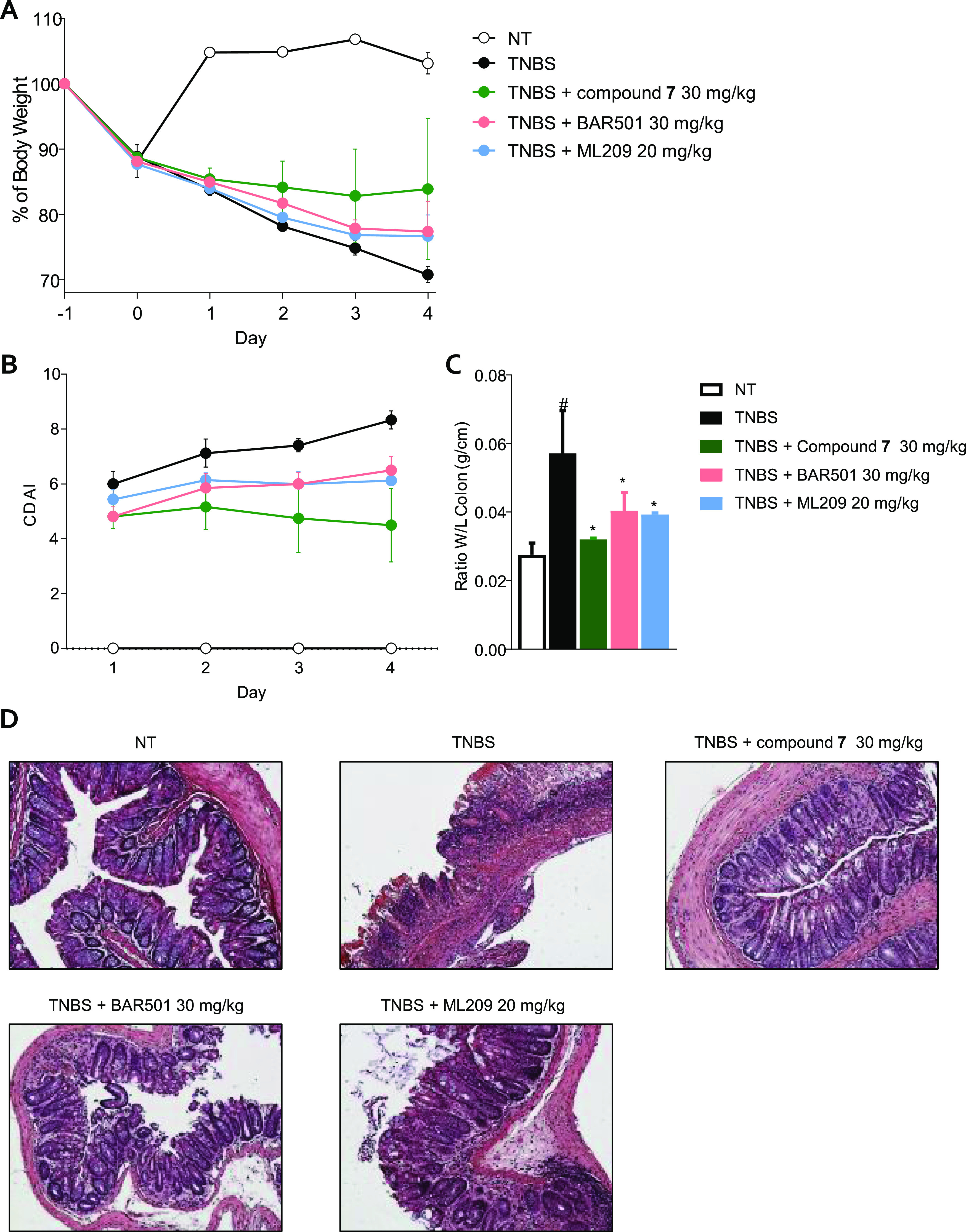
Mice treated with TNBS were administered with the vehicle or 7 30 mg/kg/day, BAR501 30 mg/kg/day, and ML209 20 mg/kg/day by gavage from day 0 to day 4. (A) Change in body weight, (B) CDAI score, (C) ratio between colon weight and colon length, and (D) H&E staining on colon sections of each experimental group. Results are the mean ± SEM of 5–8 mice per group. *p < 0.05 vs TNBS group and #p < 0.05 vs NT group.
Conclusions
The present study reports the discovery of a family of new steroidal derivatives acting as GPBAR1 agonists and RORγt inverse agonists that might be suitable for the treatment of autoimmune disorders. Our results highlighted a different profile activity of the two series described in the study, driven by the shape of the steroidal scaffold. The cholesterol derivatives (first subset) resulted in being more prone to bind RORγt, while on the contrary, the steroidal scaffold of the second subset (bent shape-BAs-like) exhibited better activity toward GPBAR1. Docking studies suggested that the nonplanar structure of the steroidal scaffold of compounds belonging to the second subset could bind the RORγt binding pocket after a rotation of the steroidal core, determining the weakening of the network of hydrophobic interactions, the loss of the H-bond of the 3-hydroxyl group to Gln286, and forcing the side chain in a more hindered and apolar region. As regards the second subset, it should be noted that all compounds belong to the LCA scaffold with modification in the carboxylic acid on the side chain, and the results on compound 17, discarded for its low solubility, are consistent with the inhibition of RORγt transcriptional activity previously reported for gut microbiota-LCA metabolites (mainly 3-oxo-LCA),38,39 suggesting that it is also possible to exploit the bile acid scaffold to design dual GPBAR1 agonists and RORγt inverse agonists. Moreover, molecular dynamics simulations showed that the flexibility of the side chain at C17 played a key role in assuming the right conformation to bind both GPBAR1 and RORγt binding pockets, allowing the interaction of the aromatic ring of compounds 3 and 7 to the key residues involved in the receptor activation, Glu169 and Ser20 in GPBAR1, or deactivation, Trp379, His479 and Tyr502 in RORγt.
Results from pharmacokinetics analysis revealed that 7 was endowed with excellent metabolic stability, thus deploying the newly hybrid ligand designed for simultaneous modulation of GPBAR1 and RORγt in a privileged position to enter preclinical studies.
Data on in vitro experiments showed that the activation of GPBAR1 by 7 modulates the polarization of monocytes by blocking the differentiation toward the proinflammatory M1 phenotype and the production of proinflammatory cytokines, counteracting the proinflammatory effect exerted by LPS plus TNFα.
Of interest is the effect of 7 in a mouse model of colitis induced by TNBS, showing that 7 relieved signs and symptoms of colitis in a dose-dependent manner, protecting, at 30 mg/kg dose, from weight loss and reduced colon inflammation. Further analysis of colonic cytokines and immune cell markers showed that the beneficial effect of 7 was due to a profound modulation of both innate and adaptive immune responses. In fact, treating mice with 7 reduced the expression of CD38, a specific marker for M1 macrophages, a proinflammatory subset of intestinal macrophages, along with Rorc, a marker for TH17 lineage, indicating a reduction of these two cell populations in the colon’s lamina propria. The beneficial effect exerted on macrophages and TH17 cells led to a consequent reduction in the expression of the proinflammatory cytokines produced by these cells resulting in reduced inflammation in the colon.
In summary, in the present study, we describe the synthesis, the in vitro and in vivo pharmacological characterization, and the structural requisites of the ligand–receptor interaction of a novel family of cholesterol derivatives including potent dual modulators of RORγt/GPBAR1. This study resulted in the identification of compound 7, the first example of a potent GPBAR1 agonist and RORγt inverse agonist, that effectively reverts colitis in a validated mouse model of IBD.
Acknowledgments
This work was partially supported by a grant from the University of Naples Federico II “Finanziamento della Ricerca in Ateneo,” Linea B-Project MoDiGa and by a grant from the Italian MIUR/PRIN 2017 (2017FJZZRC). V.L. acknowledges the support from the European Research Council (“CoMMBi” ERC grant agreement No. 101001784) and the Swiss National Supercomputing Centre (CSCS) [project ID s1150].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c07907.
Docking calculations, Figures S1–S5, experimental procedures, 1H NMR spectra of compounds 1–21 (Figures S6–S26), and HPLC traces of compounds 3 and 7 (Figures S27 and S28) (PDF)
Author Contributions
⊥ B.F., R.R., and C.F. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): S. F. and A. Z. have filed a patent application in the name of PRECISION BIO-THERAPEUTICS S.R.L., a spinoff of the University of Perugia, on the same compounds described in this paper.
Supplementary Material
References
- Ridlon J. M.; Kang D.-J.; Hylemon P. B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- Fiorucci S.; Carino A.; Baldoni M.; Santucci L.; Costanzi E.; Graziosi L.; Distrutti E.; Biagioli M. Bile Acid Signaling in Inflammatory Bowel Diseases. Dig. Dis. Sci. 2021, 66, 674–693. 10.1007/s10620-020-06715-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang J. Y. L. Bile Acids: Regulation of Synthesis. J. Lipid Res. 2009, 50, 1955–1966. 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M.; Okamoto A. Y.; Repa J. J.; Tu H.; Learned R. M.; Luk A.; Hull M. V.; Lustig K. D.; Mangelsdorf D. J.; Shan B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Biagioli M.; Carino A.; Fiorucci C.; Marchianò S.; di Giorgio C.; Roselli R.; Magro M.; Distrutti E.; Bereshchenko O.; Scarpelli P.; Zampella A.; Fiorucci S. GPBAR1 Functions as Gatekeeper for Liver NKT Cells and Provides Counterregulatory Signals in Mouse Models of Immune-Mediated Hepatitis. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 447–473. 10.1016/j.jcmgh.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicky P.; Unger S.; Becher B. Targeting Interleukin-17 in Chronic Inflammatory Disease: A Clinical Perspective. J. Exp. Med. 2020, 217, e20191123. 10.1084/jem.20191123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten A. M. Retinoid-Related Orphan Receptors (RORs): Critical Roles in Development, Immunity, Circadian Rhythm, and Cellular Metabolism. Nucl. Recept. Signal. 2009, 7, nrs.07003 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; Unutmaz D.; Zou Y.-R.; Sunshine M. J.; Pierani A.; Brenner-Morton S.; Mebius R. E.; Littman D. R. Requirement for RORγ in Thymocyte Survival and Lymphoid Organ Development. Science 2000, 288, 2369–2373. 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- Ivanov I. I.; McKenzie B. S.; Zhou L.; Tadokoro C. E.; Lepelley A.; Lafaille J. J.; Cua D. J.; Littman D. R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Burris T. P.; Busby S. A.; Griffin P. R. Targeting Orphan Nuclear Receptors for Treatment of Metabolic Diseases and Autoimmunity. Chem. Biol. 2012, 19, 51–59. 10.1016/j.chembiol.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Luo X.; Wu D.; Xu Y. ROR Nuclear Receptors: Structures, Related Diseases, and Drug Discovery. Acta Pharmacol. Sin. 2015, 36, 71–87. 10.1038/aps.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten A. M.; Takeda Y.; Slominski A.; Kang H. S. Retinoic Acid-Related Orphan Receptor γ (RORγ): Connecting Sterol Metabolism to Regulation of the Immune System and Autoimmune Disease. Curr. Opin. Toxicol. 2018, 8, 66–80. 10.1016/j.cotox.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt L. A.; Kumar N.; Nuhant P.; Wang Y.; Lauer J. L.; Liu J.; Istrate M. A.; Kamenecka T. M.; Roush W. R.; Vidović D.; Schürer S. C.; Xu J.; Wagoner G.; Drew P. D.; Griffin P. R.; Burris T. P. Suppression of TH17 Differentiation and Autoimmunity by a Synthetic ROR Ligand. Nature 2011, 472, 491–494. 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Kumar N.; Crumbley C.; Griffin P. R.; Burris T. P. A Second Class of Nuclear Receptors for Oxysterols: Regulation of RORα and RORγ Activity by 24S-Hydroxycholesterol (Cerebrosterol). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 917–923. 10.1016/j.bbalip.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh J. R.; Leung M. W. L.; Huang P.; Ryan D. A.; Krout M. R.; Malapaka R. R. v.; Chow J.; Manel N.; Ciofani M.; Kim Sv.; Cuesta A.; Santori F. R.; Lafaille J. J.; Xu H. E.; Gin D. Y.; Rastinejad F.; Littman D. R. Digoxin and Its Derivatives Suppress TH17 Cell Differentiation by Antagonizing RORγt Activity. Nature 2011, 472, 486–490. 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita-Sato S.; Ito S.; Isobe T.; Ohyama T.; Wakabayashi K.; Morishita K.; Ando O.; Isono F. Structural Basis of Digoxin That Antagonizes RORγt Receptor Activity and Suppresses Th17 Cell Differentiation and Interleukin (IL)-17 Production. J. Biol. Chem. 2011, 286, 31409–31417. 10.1074/jbc.M111.254003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallen J.; Izaac A.; Be C.; Arista L.; Orain D.; Kaupmann K.; Guntermann C.; Hoegenauer K.; Hintermann S. Structural States of RORγt: X-Ray Elucidation of Molecular Mechanisms and Binding Interactions for Natural and Synthetic Compounds. ChemMedChem 2017, 12, 1014–1021. 10.1002/cmdc.201700278. [DOI] [PubMed] [Google Scholar]
- Noguchi M.; Nomura A.; Doi S.; Yamaguchi K.; Hirata K.; Shiozaki M.; Maeda K.; Hirashima S.; Kotoku M.; Yamaguchi T.; Katsuda Y.; Crowe P.; Tao H.; Thacher S.; Adachi T. Ternary Crystal Structure of Human RORγ Ligand-Binding-Domain, an Inhibitor and Corepressor Peptide Provides a New Insight into Corepressor Interaction. Sci. Rep. 2018, 8, 17374 10.1038/s41598-018-35783-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; Magnuson S. Modulators of the Nuclear Receptor Retinoic Acid Receptor-Related Orphan Receptor-γ (RORγ or RORc). J. Med. Chem. 2014, 57, 5871–5892. 10.1021/jm401901d. [DOI] [PubMed] [Google Scholar]
- Keitel V.; Donner M.; Winandy S.; Kubitz R.; Häussinger D. Expression and Function of the Bile Acid Receptor TGR5 in Kupffer Cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- Klepsch V.; Moschen A. R.; Tilg H.; Baier G.; Hermann-Kleiter N. Nuclear Receptors Regulate Intestinal Inflammation in the Context of IBD. Front. Immunol. 2019, 10, 1070. 10.3389/fimmu.2019.01070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickael M. E.; Bhaumik S.; Basu R. Retinoid-Related Orphan Receptor RORγt in CD4+ T-Cell–Mediated Intestinal Homeostasis and Inflammation. Am. J. Pathol. 2020, 190, 1984–1999. 10.1016/j.ajpath.2020.07.010. [DOI] [PubMed] [Google Scholar]
- Sun N.; Yuan C.; Ma X.; Wang Y.; Gu X.; Fu W. Molecular Mechanism of Action of RORγt Agonists and Inverse Agonists: Insights from Molecular Dynamics Simulation. Molecules 2018, 23, 3181. 10.3390/molecules23123181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagioli M.; Marchianò S.; Carino A.; di Giorgio C.; Santucci L.; Distrutti E.; Fiorucci S. Bile Acids Activated Receptors in Inflammatory Bowel Disease. Cells 2021, 10, 1281. 10.3390/cells10061281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gege C. Retinoic Acid-Related Orphan Receptor Gamma t (RORγt) Inverse Agonists/Antagonists for the Treatment of Inflammatory Diseases – Where Are We Presently?. Expert Opin. Drug. Discovery 2021, 16, 1517–1535. 10.1080/17460441.2021.1948833. [DOI] [PubMed] [Google Scholar]
- Sepe V.; Ummarino R.; D’Auria M. V.; Mencarelli A.; D’Amore C.; Renga B.; Zampella A.; Fiorucci S. Total Synthesis and Pharmacological Characterization of Solomonsterol A, a Potent Marine Pregnane-X-Receptor Agonist Endowed with Anti-Inflammatory Activity. J. Med. Chem. 2011, 54, 4590–4599. 10.1021/jm200241s. [DOI] [PubMed] [Google Scholar]
- Terasawa T.; Ikekawa N.; Morisaki M. Photochemical Reaction of Cholesterol Analogs with a Carbene-Generating Substituent on the Side Chain. Chem. Pharm. Bull. 1986, 34, 935–936. 10.1248/cpb.34.935. [DOI] [Google Scholar]
- Averin A. D.; Ranyuk E. R.; Lukashev N. V.; Beletskaya I. P. Synthesis of Nitrogen- and Oxygen-Containing Macrocycles—Derivatives of Lithocholic Acid. Chem. Eur. J. 2005, 11, 7030–7039. 10.1002/chem.200500784. [DOI] [PubMed] [Google Scholar]
- Anzini M.; Braile C.; Valenti S.; Cappelli A.; Vomero S.; Marinelli L.; Limongelli V.; Novellino E.; Betti L.; Giannaccini G.; Lucacchini A.; Ghelardini C.; Norcini M.; Makovec F.; Giorgi G.; Ian Fryer R. Ethyl 8-Fluoro-6-(3-Nitrophenyl)-4 H -Imidazo[1,5- a][1,4]Benzodiazepine-3-Carboxylate as Novel, Highly Potent, and Safe Antianxiety Agent. J. Med. Chem. 2008, 51, 4730–4743. 10.1021/jm8002944. [DOI] [PubMed] [Google Scholar]
- Anzini M.; Valenti S.; Braile C.; Cappelli A.; Vomero S.; Alcaro S.; Ortuso F.; Marinelli L.; Limongelli V.; Novellino E.; Betti L.; Giannaccini G.; Lucacchini A.; Daniele S.; Martini C.; Ghelardini C.; di Cesare Mannelli L.; Giorgi G.; Mascia M. P.; Biggio G. New Insight into the Central Benzodiazepine Receptor–Ligand Interactions: Design, Synthesis, Biological Evaluation, and Molecular Modeling of 3-Substituted 6-Phenyl-4 H -Imidazo[1,5- a][1,4]Benzodiazepines and Related Compounds. J. Med. Chem. 2011, 54, 5694–5711. 10.1021/jm2001597. [DOI] [PubMed] [Google Scholar]
- Deplano A.; Morgillo C. M.; Demurtas M.; Björklund E.; Cipriano M.; Svensson M.; Hashemian S.; Smaldone G.; Pedone E.; Luque F. J.; Cabiddu M. G.; Novellino E.; Fowler C. J.; Catalanotti B.; Onnis V. Novel Propanamides as Fatty Acid Amide Hydrolase Inhibitors. Eur. J. Med. Chem. 2017, 136, 523–542. 10.1016/j.ejmech.2017.05.033. [DOI] [PubMed] [Google Scholar]
- Glide, Version 7.1; Schrödinger, LLC: New York, NY, 2019.
- Jin L.; Martynowski D.; Zheng S.; Wada T.; Xie W.; Li Y. Structural Basis for Hydroxycholesterols as Natural Ligands of Orphan Nuclear Receptor RORγ. Mol. Endocrinol. 2010, 24, 923–929. 10.1210/me.2009-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amore C.; di Leva F. S.; Sepe V.; Renga B.; del Gaudio C.; D’Auria M. V.; Zampella A.; Fiorucci S.; Limongelli V. Design, Synthesis, and Biological Evaluation of Potent Dual Agonists of Nuclear and Membrane Bile Acid Receptors. J. Med. Chem. 2014, 57, 937–954. 10.1021/jm401873d. [DOI] [PubMed] [Google Scholar]
- Biagioli M.; Carino A.; Cipriani S.; Francisci D.; Marchianò S.; Scarpelli P.; Sorcini D.; Zampella A.; Fiorucci S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. 10.4049/jimmunol.1700183. [DOI] [PubMed] [Google Scholar]
- Festa C.; Renga B.; D’Amore C.; Sepe V.; Finamore C.; de Marino S.; Carino A.; Cipriani S.; Monti M. C.; Zampella A.; Fiorucci S. Exploitation of Cholane Scaffold for the Discovery of Potent and Selective Farnesoid X Receptor (FXR) and G-Protein Coupled Bile Acid Receptor 1 (GP-BAR1) Ligands. J. Med. Chem. 2014, 57, 8477–8495. 10.1021/jm501273r. [DOI] [PubMed] [Google Scholar]
- Huh J. R.; Englund E. E.; Wang H.; Huang R.; Huang P.; Rastinejad F.; Inglese J.; Austin C. P.; Johnson R. L.; Huang W.; Littman D. R. Identification of Potent and Selective Diphenylpropanamide RORγ Inhibitors. ACS Med Chem Lett. 2013, 4, 79–84. 10.1021/ml300286h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibaut M. M.; Bindels L. B. Crosstalk between Bile Acid-Activated Receptors and Microbiome in Entero-Hepatic Inflammation. Trends Mol. Med. 2022, 28, 223–236. 10.1016/j.molmed.2021.12.006. [DOI] [PubMed] [Google Scholar]
- Song X.; Sun X.; Oh S. F.; Wu M.; Zhang Y.; Zheng W.; Geva-Zatorsky N.; Jupp R.; Mathis D.; Benoist C.; Kasper D. L. Microbial Bile Acid Metabolites Modulate Gut RORγ+ Regulatory T Cell Homeostasis. Nature 2020, 577, 410–415. 10.1038/s41586-019-1865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.