

Abstract

In recent years, molecular hybridization strategies have developed into a potent strategy for drug discovery. A series of novel thiopyrano[2,3-d]thiazoles linked to the pyrazole moiety was designed and developed as anticancer agents by a molecular hybridization. Target compounds were synthesized and characterized by spectroscopic tools as well as X-ray crystallography analysis as in the case of thiopyrano[2,3-d]thiazole derivative 5a. The MTT assay was used to demonstrate the in vitro efficacy of compounds 5a–g and 7a–j on MCF-7 and HePG-2. The results showed that some cycloadducts such as bromophenyl-4-thioxo-2-thiazolidinone 3e, 4-methylphenyl derivative of thiopyrano[2,3-d]thiazole 5d, and 6-substituted-thiopyrano[2,3-d]thiazoles 7e–j displayed good to excellent IC50 in the range of 10.08 ± 1.5 to 25.95 ± 2.8 μg/mL against the MCF-7 cell line and from 7.83 ±2.1 to 13.37 ± 1.2 μg/mL against the HePG-2 cell line. To explore the enzymatic tests for isozymes hCAIX and hCAXII, the most promising eight compounds 3e, 5d, and 7e–j with IC50 ranging from 7.83 ± 2.1 to 25.95 ± 2.8 μM were chosen. Compound 7e exhibited an IC50 (0.067 ± 0.003 μM) similar to that of the standard drug AZA against CAIX (0.059 ± 0.003 μM)). For CAXII, the compound 7i had an IC50 equal to 0.123 ± 0.007 μM compared to that of AZA (0.083 ± 0.005 μM). In addition, using flow cytometry, cell cycle analysis and apoptosis studies in HePG-2 were performed for the two potent anticancer and selective carbonic anhydrase agents (7e and 7i). An enzymatic assay of these two compounds against caspase-9 was also examined. Interestingly, the molecular docking studies revealed that compounds 7e and 7i successfully embedded themselves in the active pockets of the CAIX and CAXII enzymes through different interactions. Overall, the novel thiopyrano[2,3-d]thiazole-pyrazole hybrids (7e and 7i) were suggested to be potent and selective inhibitors of CAIX and CAXII.
1. Introduction
Cancer cells have many characteristics that distinguish them from normal cells, such as a more acidic extracellular pH, a slightly alkaline cytoplasmic pH, and a lower than normal oxygen (hypoxia). Hence, the reduced oxygen level, perturbed pH balance, upregulated glucose metabolism, and other aspects of the tumor microenvironment have been viewed as an opportunities to develop antitumor drugs. These drugs specifically target tumor cells without affecting normal cells.1,2 Studies demonstrated that CA IX and CA XII are biomarkers with therapeutic component for many cancer types.3−8
It has been established that carbonic anhydrases (CAs) are crucial for clinical medicine and drug development. Zinc ion, which present in the active site of carbonic anhydrase (CA, EC 4.2.1.1), interacts with water molecules and histidine residues via its tetrahedral structure. Through the hydration reaction, the zinc ion binds to the water molecule and catalyzes the transformation of carbon dioxide into bicarbonate ions and protons. This process occurs through a nucleophilic mechanism of metal hydroxide.
CA is crucial to the body’s natural defense mechanism.9−12 Mammals have different 16-CA isozymes, with CA I, CA II, CA III, CA VII, and CA XIII being the first five cytosolic forms. CA IV, CA IX, CA XII, CA XIV, and CA XV are the second five membrane-bound isozymes, along with two mitochondrial forms (CA VA, CA VB), and a secreted CA isozyme (CA VI).13 The extracellular domain of the transmembrane proteins known as CA IX and CA XII isoform enzymes contains catalytic activity and is important in the control of the tumor microenvironment. Due to its modest expression in normal cells but high expression in solid tumors, CA IX is of particular relevance.14−16 However, declines in CAIX or CAXII activity seem to have an impact on the pH of the tumor microenvironment, which inhibits the survival and growth of tumor cells.17,18
This feature makes CA IX/XII appealing targets for anti-cancer agents when taken as a whole. This is because of both CAIX and CAXII have well-established roles in the process of tumorigenesis. In preclinical research, a variety of classes of inhibitors and biologics that target CAIX/CAXII have been examined in the processes of cancer signaling, tumor development, acidification, and metastasis. These findings offer encouraging evidence that inhibiting CAIX/CAXII catalytic activity lowers the potential for growth, proliferation, and metastasis of a number of aggressive malignancies both in vitro and in vivo.19−22 Classical carbonic anhydrase inhibition occurred by compounds concerning sulfonamide-based (SO2NH2) zinc-binding groups (ZBG). These compounds interact with deprotonated catalytic zinc in a tetrahedral shape, which inhibits CA activity by displacing the water/hydroxide ions bound to the zinc (Figure 1). Some sulfonamide drugs are currently being used clinical setting as CA inhibitors, although 3 to 6% of the general population are allergic to them because of untreatable sulfur allergies.
Figure 1.

Some known sulfonamide drugs as CA inhibitors.
Moreover, nonsulfonamide CA inhibitors like phenols, polyamines, carboxylic acids, coumarins, thiocoumarins, and their derivatives exhibit a binding mechanism that is a typical of conventional sulfonamide CA inhibitors, as they are anchored to zinc-bound water/hydroxide ions or bind outside the active site to inhibit substrate entry (Figure 2). Many hCAIX inhibitors of such concept had synthesized, including aromatic sulfonamides, open saccharins and isatin derivatives. For example, currently, in clinical trials, 4-[(4-fluorophenyl)carbamoyl]aminobenzene sulfonamide (SLC-0111) seems to be a potent CA inhibitor.23−25 It is also conceivable for other inhibitor binding mechanisms to attach to the allosteric or activating region without interacting directly with zinc ions.
Figure 2.

Some nonsulfonamide CA inhibitors.
Important heterocyclic rings with many beneficial biological properties include thiazolidinone core, which have anti-inflammatory, antibacterial, antifungal, antiviral, anticancer, anticonvulsant, and antitubercular properties.26−28 Furthermore, glitazone and their analogues were also recognized as CAII inhibitors such as nonsulfonamide 5-phenyl-2,4-thiazolidinedione (Figure 3). The findings suggested a connection between the zinc cation at the active site and the acidic nitrogen ring (Figure 4).29 In addition, other glitazone derivatives like pioglitazone, rosiglitazone, and troglitazone demonstrated considerable binding and inhibition of the CAII isozyme through interaction between the acidic imide NH and the active site zinc.30 As an example, the interaction between rosiglitazone and active site of CAII has an ionic bond of 2.0 Å between the imide nitrogen and the active site Zn cation and only one hydrogen bond between the imide and Thr199 of 3.1 Å.
Figure 3.

Glitazone derivatives as CAII inhibitors.
Figure 4.

Interaction of glitazone, rosiglitazone, and RhA-1 with the active site of CAII.
Other 4-thiazolidinones interact with the zinc active site through a carbonyl group on a ring that is not the acidic imide NH ring. As an illustration, 3-[(4-hydroxy-3-methoxybenzylidene)amino]-2-thioxothiazolidin-4-one (RhA-1) forms hydrogen bonds with Asn 62 and Thr 200, and the benzyl ring forms an aromatic hydrogen bond with His 94. The 2-thioxothiazolidine scaffold that formed an aromatic bond interacted with Zn 265 of hCA II (Figure 4).31 Hence, thiazolidinedione derivatives may represent exciting opportunities for the development of novel CA inhibitors as future drugs (Figure 4).
The molecular hybridization strategy is a novel approach to develop hybrid multifunctional molecules by combining two or more pharmacophores in a single scaffold. Another instance is the formation of the innovative anti-tuberculosis (TB) chemotype benzo[d]thiazol-2-yl(piperazin-1-yl)-meth-anone through hybridization of the piperazine moiety with benzo[d]thiazole-2-carboxamide.32 Furthermore, hybridization of ethyl 1-phenyl-1H-benzo[d]imidazole-2-carboxylate with alicyclic amines yielded benzo[d]imidazole-2-carboxamides as anti-TB agents33 and others.34−37 The presence of two or more biologically active pharmacophores within a single unit not only synergizes the biological activity but also enhances the ability to interact with more than one biological target.38 Moreover, the pyrazole ring system also has a wide range of biological activities, including antifungal,39 antimicrobial,40 anticancer,41−43 anti-AIDS,44 and antidepressants.45 The pyrazole ring system is an important bioactive ingredient in over-the-counter drugs such as pyrazomycin (anticancer drug), floxan (anti-inflammatory drug), and difenamizole (nonsteroidal anti-inflammatory drug) (Figure 5).
Figure 5.

Marked drugs of pyrazole derivatives.
Considering the foregoing and continuing to synthesize diverse ring systems with potential biological applications,46−58 molecular hybridization approaches were applied to synthesize two series of compounds by combining the above nuclei. The first series contained of pyrazoles connected to thiopyrano[2,3-d]thiazole hybrids (5a–g) via N-aryl maleimides with 3a–d, and the second series consisted of pyrazole-linked thiopyrano[2,3-d]thiazoles hybrid (7a–j) with a cyano or ester group (Figure 6).
Figure 6.

Design strategy for the synthesis of thiopyrano[2.3-d]thiazoles (color codes indicate general chemical structures).
2. Results and Discussion
2.1. Chemistry
A Knoevenagel condensation was carried out between 4-thioxo-2-thiazolidinone 1 and 1-phenyl-3-aryl-1H-pyrazole-4-carbaldehydes 2a–e, providing 5-pyrazolyl-methylene-4-thioxo-2-thiazolidinones 3a–e according to Scheme 1. The chemical structures of 3a–e were proven by spectroscopic tools. As an example, compound 3a revealed two singlet signals at δ 8.70 and 13.75 ppm representing the pyrazole-H-5 and NH protons, respectively, along with an aryl multiplet in the range of δ 7.29 to 8.0 ppm in the 1H NMR chart. 13C NMR revealed significant signals at δ 153.12, 170.25, and 194.79 ppm corresponding to pyrazole-C-3, carbonyl, and thiocarbonyl carbons, respectively, and along with it are signals corresponding to aromatic carbons, thiazolidine-C-5 and vinyl-C. The IR spectrum of 3a showed characteristic absorption bands at 3434 and 1736 cm–1 due to the NH and C=O groups, respectively. Mass spectrometry showed an ion peak at m/z = 363 (M+). The proposed mechanism is that a base deprotonates the alpha-carbon of 4-thioxo-2-thiazolidinone 1, to give an anion. This anion attacks the carbonyl function in aldehydes 2 to produce an aldol-like intermediate, which undergoes dehydration to produce the final products as shown in Scheme 1.59
Scheme 1. Synthesis of 5-[(3-Aryl-1-phenyl-1H-pyrazol-4-yl)methylene]-4-thioxo-2-thiazolidinones 3a–e.

Additionally, compounds 3a–e were reacted with N-aryl maleimides 4a–c in refluxing acetic acid to yield 1:1 adducts 5a–g (Scheme 2). The assigned structures for the products 5a–g were established on spectroscopic tools. In compound 5e, the IR spectrum represent absorption bands at 3445, 1716 and 1674 cm–1 corresponding to N–H and two C=O groups, respectively. The 1H NMR chart contains a singlet at δ 3.81 ppm to the OCH3 group, a doublet of doublets at δ 4.20 ppm with J values of 3.0 and 9.0 Hz corresponding to H-6, a doublet at δ 4.92 ppm with a J value of 3.0 Hz due to H-5, and a doublet signal at δ 4.94 ppm with a J value of 9.0 Hz attributed to H-7 in addition to the multiplet at δ 7.07–7.96 ppm corresponding to the aryl protons and two singlets at δ 8.56 and 11.75 ppm attributed to pyrazole-H-5 and NH protons, respectively. Its mass spectrum exhibited, among other the fragments, ion peaks at m/z = 566 (M+). The formation of compounds 5a–g proceeds via a concerted mechanism based on the hetero-Diels–Alder reaction, using compounds 3a–g as heterodienes and N-aryl maleimides 4a–c as dienophiles, as shown in Scheme 2.60
Scheme 2. Synthesis of 8-(3-Aryl-1-phenyl-1H-pyrazol-4-yl)-6-arylpyrrolo[3′,4′:5,6]thiopyrano[2,3-d]thiazole-2,5,7-triones 5a–g.

Derived from proton nuclear magnetic resonance data, 5a–g have only one stereoisomer, (Z-isomer) arising from the deshielding of vinyl proton, which is effected by the C=S moiety of the thiazole ring, leading to a lower magnetic field (δ ≈ 7.88–7.94 ppm) than the E-isomer appearing at higher magnetic fields (δ ≈ 7.30–7.60 ppm).61 This consumption was also proven by X-ray crystallographic analysis of compound 5a. We noticed from determination of X-ray data that the most important aspect of this structure is the relative stereochemistry of the H atoms at the stereoisomeric C-25, C-27, and C-10 centers. The C-25 and C-27 hydrogen atoms have a cis-axial–axial orientation, while the C-10 hydrogen atom occupies an axial position relative to the dihydrothiopyran ring (Figure 7).
Figure 7.

X-ray crystallography of cycloadduct 5a.
Under the same reaction conditions, 3a–e reacted with dienophiles 6a,b to yield the 1:1 cycloadducts, which was formulated as 7-(3-aryl-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano[2,3-d]thiazole derivatives 7a–j (Scheme 3). From the spectral data, structures 7a–j were assigned to these adducts, so the 7d’s IR spectrum showed absorption bands at 3442, 2245, and 1644 cm–1 corresponding to the N–H, CN, and CO groups, respectively. The 1H NMR chart appeared a multiplet signal at δ 3.44–3.64 ppm assigned to H-5, a multiplet signal at δ 3.99 ppm to H-6, and a doublet signal at δ 4.53 ppm with a J value of 5.1 Hz to H-7. Also clarified were multiplet signals ranging from δ 7.33–7.92 ppm assigned to aryl protons and two singlets at δ 8.61 and 11.49 ppm corresponding to pyrazole-H-5 and NH protons (D2O exchangeable), respectively. The 13C NMR spectrum observed significant signals at 27.03, 31.54, and 33.13 ppm for C-5, C-7, and C-6, respectively. Mass spectrometry showed ion peaks at m/z = 452 (M++2), 451 (M++1), and 450 (M+), consistent with the molecular formula C22H15ClN4OS2.
Scheme 3. Synthetic Route of 7-(3-Aryl-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano[2,3-d]thiazole Derivatives 7a–j.

3. Biological Activity
3.1. Anticancer Activity
The cytotoxicity of the tested compounds 3a–e, 5a, 5d–g, and 7a–j assessed human breast cancer (MCF-7) and hepatocellular carcinoma (HePG-2) by the MTT assay and utilizing doxorubicin as a standard drug. The data on in vitro cytotoxicity were expressed as IC50, which is the compound concentration (in μg/mL) that decreased cell viability by 50% (Tables 1–3 and Figures 8–10). The MCF-7 and HePG-2 cell lines were shown to be sensitive to the tested compounds based on how the synthetic compounds affected the proliferation of the two cancer cell lines.
Table 1. IC50 Values of Compounds 3a–e against MCF-7 and HePG-2 Cell Lines.
| compound no. | R | IC50 in μg/mL MCF-7 | IC50 in μg/mL HePG-2 |
|---|---|---|---|
| doxorubicin | 4.17 ± 0.2 | 5.57 ± 0.4 | |
| 3a | H | 52.11 ± 3.1 | 65.75 ± 3.3 |
| 3b | Me | 56.27 ± 3.4 | 62.16 ± 3.2 |
| 3c | OMe | 37.59 ± 3.4 | 31.22 ± 2.6 |
| 3d | Cl | 30.84 ± 2.2 | 38.20 ± 2.7 |
| 3e | Br | 11.16 ± 1.5 | 10.66 ± 1.0 |
Table 3. IC50 Values of Compounds 7a–j against MCF-7 and HePG-2 Cell Lines.
| compound no. | R | X | IC50 in μg/mL MCF-7 | IC50 in μg/mL HePG-2 |
|---|---|---|---|---|
| doxorubicin | 4.17 ± 0.2 | 5.57 ± 0.4 | ||
| 7a | H | CN | 23.76 ± 1.9 | 26.84 ± 3.3 |
| 7b | Me | CN | 35.03 ± 2.3 | 30.42 ± 1.1 |
| 7c | OMe | CN | 29.43 ± 1.1 | 24.12 ± 2.9 |
| 7d | Cl | CN | 67.32 ± 3.7 | 73.05 ± 4.3 |
| 7e | Br | CN | 10.10 ± 2.1 | 9.11 ± 3.2 |
| 7f | H | COOEt | 15.90 ± 2.3 | 13.37 ± 1.2 |
| 7g | Me | COOEt | 12.30 ± 2.4 | 11.87 ± 2.7 |
| 7h | OMe | COOEt | 11.36 ± 1.5 | 12.18 ± 1.1 |
| 7i | Cl | COOEt | 10.08 ± 1.5 | 7.83 ± 2.1 |
| 7j | Br | COOEt | 11.80 ± 4.0 | 10.26 ± 4.8 |
Figure 8.

Graph representing the IC50 values of compounds 3a–e against MCF-7 and HePG-2 cell lines, indicating that 3e is the most potent cytotoxic agent in this series using doxorubicin as standard drug.
Figure 10.

Graph representing the IC50 values of compounds 7a–j against MC-7 and HePG-2 cell lines, indicating that compounds 7e–j have good anticancer activity against MCF-7 and HePG-2.
Preliminary in vitro anticancer screening results indicate that most of the compounds tested exhibited IC50 ranging from 10.08 ± 1.5 to 67.32 ± 3.7 μg/mL against the MCF-7 cell line. In the first series of 3a–e, compounds 3a and 3b showed weak activity against the two cell lines tested, while compounds 3c and 3d showed moderate activity. Moreover, compound 3e exhibits a strong anticancer activity against MCF-7 cell line with IC50 = 11.16 ± 1.5 μg/mL. On the other hand, the sensitivity of compounds 3a–e to the HePG-2 cell line showed an IC50 ranged from 7.83 ± 1.5 to 73.05 ± 4.3 μg/mL. Compound 3e showed strong cytotoxic activity with IC50 = 7.83 ± 1.5 μg/mL among the other prepared series 3a–e (Table 1 and Figure 8). In a second series of thiopyrano[2,3-d]thiazoles 5a–g, the most active compound 5d exhibited IC50 = 25.40 ± 2.1 and 21.31 ± 1.8 μg/mL, against MCF-7 and HePG-2 cells, respectively, while compound 5g was weakly active toward the two cell lines in this series (Table 2 and Figure 9). The last series, cycloadducts 7a–j revealed moderate to strong anticancer activity against MCF-7 and HePG-2 cell lines. Thus, 7e and 7i were the most potent anticancer activity of all the tested compounds in this series, with IC50 = 10.10 ± 2.1 and 10.08 ± 1.5 μg/mL, respectively, for MCF-7, while HePG-2 have IC50 = 9.11 ± 3.2 and 7.83 ± 2.1 μg/mL, respectively. Other compounds 7g, 7h, and 7j appeared strong cytotoxicity against MCF-7 and HePG-2, while compounds 7a–c and 7f showed moderate anticancer activity, whereas compound 7d showed weak activity as shown in Table 3 and Figure 10
Table 2. IC50 Values of Compounds 5a and 5d–g against MCF-7 and HePG-2 Cell Lines.
| compound no. | R | X | IC50 in μg/mL MCF-7 | IC50 in μg/mL HePG-2 |
|---|---|---|---|---|
| doxorubicin | 4.17 ± 0.2 | 5.57 ± 0.4 | ||
| 5a | H | H | 30.30 ± 0.4 | 38.42 ± 2.1 |
| 5d | Me | H | 25.95 ± 2.8 | 21.31 ± 1.8 |
| 5e | OMe | H | 35.72 ± 72 | 30.70 ± 0.3 |
| 5f | Cl | H | 25.40 ± 2.1 | 29.0 ± 2.1 |
| 5g | Br | H | 42.91 ± 2.7 | 44.96 ± 3.0 |
Figure 9.

Graph showing the IC50 values of compounds 5a and 5d–g against MCF-7 and HePG-2 cell lines, indicating that 5d in this series has a good cytotoxic effect using the MTT colorimetric method and doxorubicin as a reference.
3.2. Structure–Activity Relationship (SAR)
Based on the data revealed, it is possible to conclude structure–activity relationships for the synthesized target compounds, as shown in Figure 11.
Figure 11.

Structure–activity relationship (SAR) of the targets 3, 5a–g, 7e, and 7i.
(1) As demonstrated in Figure 11, the substituents on the benzene ring and thiopyrano moiety have a significant impact on the activity and broadness, which can be summed up as follows.
(2) The presence of a bromine atom on a phenyl ring incorporated to a pyrazole moiety (electron-withdrawing group, −ve inductive effect) in the starting 3a–e is required to show the broad spectrum cytotoxic activity against MCF-7 and HePG-2, which represents 3e.
(3) On the other hand, the presence of a methyl group in the phenyl ring linked to the pyrazole moiety reduces the activity against MC-F7 and HePG-2 as in 3b.
(4) In series a of cycloadducts 5a and 5d–g, introduction of a bromine atom into the phenyl ring of the pyrazole moiety reduced activity against MCF-7 and HePG-2 as in 5g.
(5) The results from the previous part of this series show that the presence of an electron donating group, such as a methoxy group, enhanced the cytotoxic activity toward the two cell lines (compound 5e).
(6) Compound 5f showed almost moderate activity against MCF-7 and HePG-2 due to the presence of a chlorine atom as an electron withdrawing group.
(7) The results for the final cycloadducts 7a–j show that the presence of an electron withdrawing group (Br) at the 4-position of the phenyl ring of the pyrazole moiety increases the negative inductive effect on the reactive center of the molecule, resulting in increased cytotoxic activity for compounds 7e, 7i, and 7j, expect compound 7d.
(8) Compounds with a methyl or methoxy group as a donor group at the 4-position of the phenyl ring and cyano group as an electron-withdrawing group at the thiopyran moiety showed moderate cytotoxicity activity (compounds 7b and 7c).
(9) Compounds with a methyl or methoxy group at the 4-position of the phenyl ring and an ester group on the thiopyran moiety showed the similar effects and increased the cytotoxic activity (compounds 7g and 7h).
(10) Compounds 5a, 7a, and 7f, with no substituents on the phenyl ring, showed moderate cytotoxic activity. Therefore, compounds 3e, 5d, and 7e–j were chosen for enzyme inhibition investigation.
3.3. In Vitro Cellular Viability
Data obtained from cell viability in WI38 (human lung fibroblast cell line) are listed in Table 4 and Figure 12 for compounds 3a–e, 5a, 5d–g, and 7a–j. The results reveal that compounds 3a, 3b, and 5g with moderate anticancer activity in the MCF-7 and HePG-2 revealed strong cell viability at IC50 = 28.20 ± 2.3, 25.27 ± 2.1, and 24.86 ± 2.0 μg, respectively, on WI38. Compounds 7e, 7h, and 7i, with strong anticancer activity with IC50 = 27.05 ± 3.3, 25.34 ± 2.2, and 19.49 ± 1.6 μg, respectively, show high cell survival values at WL38.
Table 4. IC50 of the Target Compounds against Human Lung Fibroblast Cell Line WI38.
| compound no. | IC50 in μg/mL WI38 |
|---|---|
| DOX | 8.87 ± 0.6 |
| 3a | 28.20 ± 2.3 |
| 3b | 25.27 ± 2.1 |
| 3c | 64.53 ± 3.8 |
| 3d | 71.21 ± 4.2 |
| 3e | 41.48 ± 2.9 |
| 5a | 65.68 ± 1.1 |
| 5d | 49.86 ± 3.4 |
| 5e | 51.67 ± 3.7 |
| 5f | 43.02 ± 3.1 |
| 5g | 24.86 ± 2.0 |
| 7a | 54.64 ± 3.5 |
| 7b | 41.35 ± 1.7 |
| 7c | 51.91 ± 3.4 |
| 7d | 39.49 ± 1.7 |
| 7e | 27.05 ± 3.3 |
| 7f | 44.50 ± 4.5 |
| 7g | 30.31 ± 1.7 |
| 7h | 25.34 ± 2.2 |
| 7i | 19.49 ± 1.6 |
| 7j | 33.76 ± 2.5 |
Figure 12.

Cell viability in the human lung fibroblast cell line WI38 for all synthetic compounds.
3.4. Enzyme Inhibition
3.4.1. In Vitro hCA-IX and hCA-XII Inhibition
The most potent anticancer compounds 3e, 5d, 7e, and 7e–j were evaluated for their potential as inhibitors of human carbonic anhydrase IX and XII isoforms using acetazolamide (AZA) as a standard. All the tested compounds significantly inhibited CAIX activity, with IC50 values ranging from 0.067 ± 0.003 to 0.928 ± 0.045 μM. As demonstrated in Table 5 and Figure 13, the IC50 values for CAXII ranged from 0.123 ± 0.007 to 1.047 ± 0.057 μM.
Table 5. Carbonic Anhydrase Inhibition Data of the Tested Compounds 3e, 5d, 7e–i, and Standard Drug AZA.
| compound | CAIX IC50 (μM) | CA XII IC50 (μM) |
|---|---|---|
| 3e | 0.349 ± 0.011 | 0.556 ± 0.016 |
| 5d | 0.336 ± 0.016 | 0.604 ± 0.033 |
| 7e | 0.067 ± 0.003 | 0.202 ± 0.011 |
| 7f | 0.928 ± 0.045 | 1.047 ± 0.057 |
| 7g | 0.390 ± 0.019 | 0.566 ± 0.031 |
| 7h | 0.133 ± 0.006 | 0.409 ± 0.022 |
| 7i | 0.287 ± 0.014 | 0.123 ± 0.007 |
| 7j | 0.239 ± 0.03 | 0.589 ± 0.03 |
| AZA | 0.059 ± 0.003 | 0.083 ± 0.005 |
Figure 13.

Graph comparing the carbonic anhydrase inhibition data for the target compounds 3e, 5d, and 7e–j with the standard drug acetazolamide (AZA), showing that compound 7e is a potent and selective CAIX inhibitor and compound 7i is a potent and selective CAXII inhibitor compared to the reference drug.
Data listed in Table 5 showed that compound 7e was more potent (IC50 = 0.067 ± 0.003 μM) against the CAIX isoform like AZA (IC50 = 0.059 ± 0.003 μM). Compound 7h has lower activity against hCAIX than that of compound 7e, with an IC50 value of 0.133 ± 0.006. Compounds 3e, 5d, and 7g showed nearly identical IC50 values 0.349 ± 0.011, 0.336 ± 0.016, and 0.390 ± 0.019 μM, respectively. Compounds 7i and 7j also have IC50 in the same range equal to 0.287 ± 0.014 and 0.239 ± 0.03 μM, respectively. Compound 7f was found to be the weakest active hit in the series with an IC50 = 0.928 ± 0.045 μM (Table 5 and Figure 13). In the CAXII assay studies, compound 7i was the most potent hCAXII inhibitor in the series, with IC50 = 0.123 ± 0.007 μM, compared to the standard drug. Compound 7e was found to have a lower IC50 than 7i (0.202 ± 0.011 μM). On the other hand, compounds 3e, 5d, 7g, 7h, and 7j have nearly identical activity against CAXII, with IC50 values of 0.556 ± 0.016, 0.604 ± 0.033, 0.566 ± 0.031, 0.049 ± 0.022 and 0.589 ± 0.03 μM, respectively, but compound 7f still showed the weakest activity against CAXII with an IC50 = 1.047 ± 0.057 μM, compared to the other compounds tested in this series as shown in Table 5 and Figure 13. Thus, it is noteworthy that compounds 7e (contains 4-bromo) and 7i (containing 4-Cl atom) showed excellent inhibition against CAIX and CAXII isoforms because of their electron-withdrawing groups. Compound 7e was a more potent and selective CAIX inhibitor than the other prepared series and compatible with the standard drug AZA, while compound 7i was potent and selective to CAXII inhibitor and its IC50 is close to AZA. Based on the above information, compounds 7e and 7i were selected for further biological investigation.
3.4.2. Cell Apoptosis and Necrosis
Based on the data obtained from the cytotoxic activity and carbonic anhydrase inhibitory effect, compounds 7e and 7i appear to be the most potent and selective cytotoxic effect agents in the HePG-2 cell line, exhibiting significant inhibitory potency against the two carbonic anhydrases, CAIX and CAXII. To demonstrate the mechanistic effects on the cell cycle of compounds 7e and 7i (at concentrations of IC50 values), with the lowest IC50 values in the HePG-2 cell line, we selected them for further investigation. Flow cytometry was performed on MCF-7 cell lines using propidium iodide (PI) and annexin V-FITC to determine the cell number at each mitosis. After treatment of thiopyrano[2,3-d]thiazoles 7e and 7i with HePG-2 for 24 h, the corresponding red (PI) and green (FITC) fluorescence was detected by flow cytometry. It was found that treatment of HePG-2 cells with cycloadducts 7e and 7i significantly increased the percentage of annexin V-FITC-positive apoptotic cells, containing the early from 0.55 to 11.91 (lower right cytogram quadrant) for compound 7e and from 0.55 to 28.71 for compound 7i compared to control. However, in the late apoptosis, the results showed that the proportion of annexin V-FITC-positive apoptotic cells for compound 7e was 21.24 (upper right cytogram quadrant). Late apoptosis of compound 7i is 9.51. In conclusion, compounds 7e and 7i increase the early apoptotic ratios by 21-fold and 52-fold compared to control cells, whereas the late apoptotic % increases approximately 101-fold and 45-fold, respectively. Moreover, compounds 7e and 7i exhibited nearly the same necrosis results of 3-fold compared to the control cell HePG-2 (1.39). These results indicated that compound 7i, which was investigated, had an apoptosis inducing effect on HepG-2 cells at an early stage, while compound 7e had a stronger apoptotic inducing effect at a late stage, and both compounds 7e and 7i induced the apoptosis rather than the necrotic pathway through the programmed cell death pathways (Table 6 and Figures 14–17).
Table 6. Cell Content % in Initial and Late Apoptosis on HePG-2 Cells Treated with Compounds 7e and 7i besides the Percent of Necrosis.
| apoptosis |
||||
|---|---|---|---|---|
| compound | total | early | late | necrosis |
| 7e/HePG2 | 36.28 | 11.91 | 21.24 | 3.13 |
| 7i/HePG2 | 41.13 | 28.71 | 9.51 | 2.91 |
| Cont.HePG2 | 2.15 | 0.55 | 0.21 | 1.39 |
Figure 14.

Chart showing the percent of cells in apoptosis and necrosis on HePG-2 cells treated with 7e and 7i.
Figure 17.
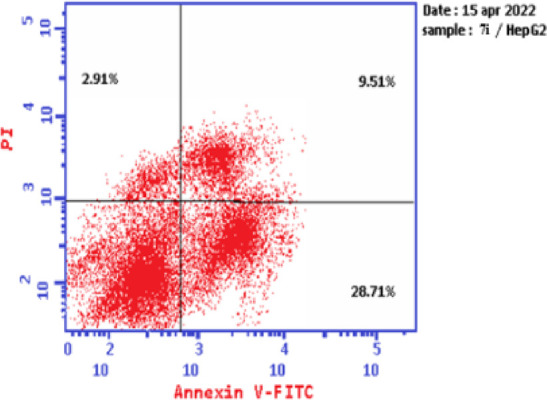
Graph showing the cell apoptosis and necrosis in HePG-2 cells treated with 7i.
Figure 15.
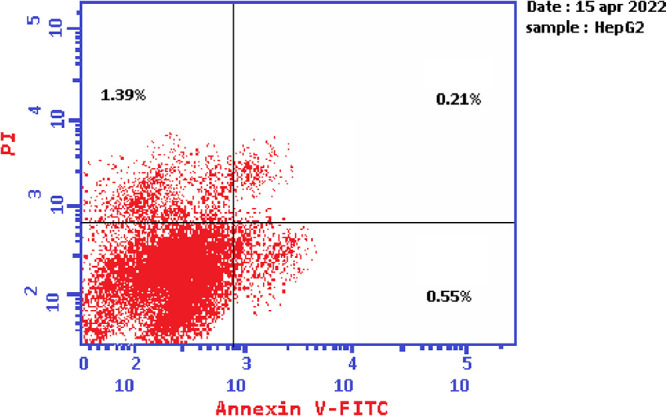
Graph showing the cell population in apoptosis and necrosis in HePG-2.
Figure 16.
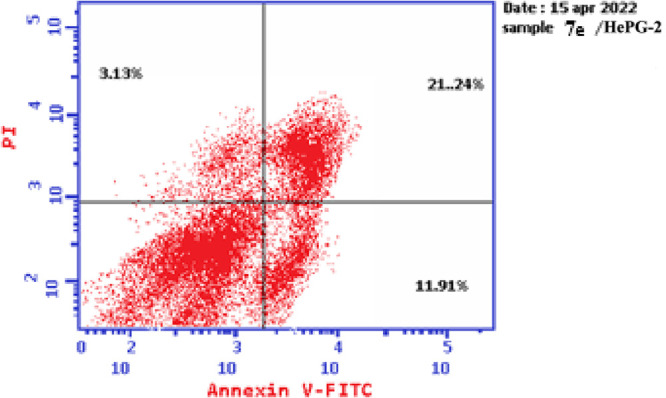
Graph representing the cell apoptosis and necrosis in HePG-2 cells treated with 7e.
3.4.3. Cell Cycle Detection
The effects of thiopyrano[2,3-d]thiazoles 7e and 7i on cell cycle progression were performed on HePG-2 cancer cell lines, through a DNA flow cytometry. HePG-2 cells were treated with cycloadducts 7e and 7i at IC50 concentrations (9.11 and 7.83, respectively) for 24 h. The results of this assay demonstrated that exposure of HePG-2 cells to compounds 7e and 7i led to the percentage of cells in the S phase and G2/M was significantly increased by 7e and 7i. Compared to the control, compound 7e increased the cell ratio by 1.2-fold in phase S, while compound 7i increased the cell ratio by 1.6-fold in phase G2/M. HepG-2 cells. Thus, the HepG2 was arrested in phase S in the cell cycle by compound 7e, whereas cells were arrested in phase G2/M by compound 7i (Table 7 and Figures 18–21). Therefore, compounds 7e and 7i bind to the molecules involved in S and G2/M phases, respectively.
Table 7. Cell Cycle Analysis of Compounds 7e and 7i.
| code | %G0–G1 | %S | %G2/M | comment |
|---|---|---|---|---|
| 7e/HePG2 | 41.39 | 44.25 | 14.36 | cell growth arrest@S |
| 7i/HePG2 | 39.56 | 32.51 | 27.93 | cell growth arrest@G2/M |
| Cont.HePG2 | 46.32 | 35.99 | 17.69 |
Figure 18.

Cell cycle graph of the most potent compounds 7e and 7i.
Figure 21.
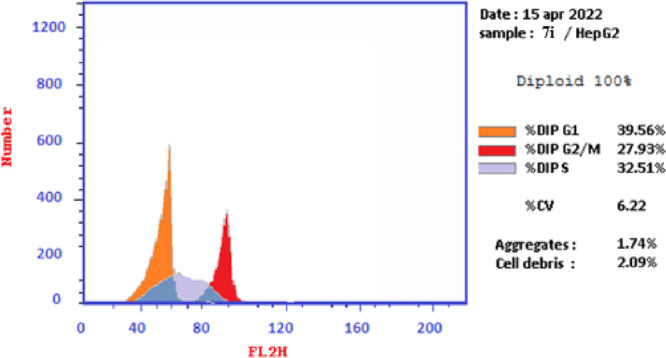
Graph showing the DNA content % in the HePG-2 treated with 7ivia division phases expressing the arrest of the cell cycle in the G2/M phase.
Figure 19.

Graph showing the DNA content % in the HePG-2.
Figure 20.

Graph showing the DNA content % in the HePG-2 treated with 7evia division phases expressing the arrest of cell cycle in the S phase.
3.4.4. Cell Cycle Analysis
3.5. Caspase 9 Assay
Apoptosis is a tightly controlled form of cell death that gets rid of unhealthy or dysfunctional cells. A range of internal or environmental factors can cause apoptosis. One of two signaling routes can be activated by a stimulus, depending on its source (internal or external). Both processes require caspases, which can be divided into initiator caspases and effector caspases. Caspases are aspartate-specific cysteine proteases. Caspases play a vital role in initiating the apoptotic process. Caspase activation ensures a controlled method of degradation of cellular components with minimal impact on surrounding tissue. Caspase-9 is a key initiator caspase in the apoptotic pathway and is activated during endogenous trigger apoptosis.62 When HePG-2 cells were treated with 7e and 7i, an increase in caspase-9 concentration was observed, as shown in Table 8. Compared to control, compound 7e showed a significant increase (almost 4-fold) in the level of active caspase-9. Compound 7i also showed significant increase by 3-fold compared to the control as shown in Table 8 and Figures 22 and 23.
Table 8. Results of Caspase 9.
| Casp9 | ||
|---|---|---|
| compound | OD | β-actine |
| 7e/HepG2 | 0.813 | + |
| 7i/HepG2 | 0.609 | + |
| Cont.HepG2 | 0.186 | + |
Figure 22.

Impact of 7e and 7i on caspase 9, indicating that compounds 7e and 7i have an effect on it.
Figure 23.

Effect of the targets 7e and 7i on Caspase 9 using Western Blot method.
Image J analysis was also performed to estimate the protein levels in caspase-9, as shown in Figure 26. Analysis revealed that the level in caspase cells, treated with compound 7i, was 2.98-fold compared to the untreated cell, whereas compound 7e resulted in greater levels of caspase-9 than in untreated cells by 7.143-fold (the values calculated after normalization to β-actin). These results are consistent with those obtained from measuring caspase 9 activity utilizing Western Blot assay.
Figure 26.

2D and 3D ligand interaction of 7ea and 7eb with the amino acids of the 5U0F active site.
3.6. Molecular Docking Study
Molecular Operating Environment (MOE 2015. 10) software was used to explore the potential binding modes for the most active compounds, 7e, 7i, and 7h, as well as their isomers, with the active sites of CAIX and CAXII [(R)-5-(2,4-dimethoxybenzyl)-2-thioxothiazolidin-4-one)] (5U0F in PDB) in order to explain the observed carbonic anhydrase inhibitory activity and to understand the potential mechanism (Figure 24).
Figure 24.

The most active compounds 7e, 7i, and 7h against CAIX and CAXII with (R)-5-(2,4-dimethoxybenzyl)-2-thioxothiazolidin-4-one) [PDB code: 5U0F].
The standard drug AZA binds to the active site and provides four interactions. This included the interaction of one hydrogen bond of the sulfoxide function group (S=O) with Thr A199 with a bond length of 2.79 Å, and the ionic interaction of the zinc ion (Zn 301) with another sulfoxide group. The binding energy of interactions is −2.2591 kcal/mol and rmsd-fine 1.4460 as depicted in Table 9 and Figure 25. The most active 7e, which is found in the two isomers 7ea and 7eb, presented the interactions for adopted by the co-crystallized ligand binding to the active site amino acids including His A94, which interacts with the phenyl group in the thiazole ring by an arene-H bond as well as an ionic bond between the carbonyl of the thiazole and zinc ion 301 for 7ea (Figure 26), whereas the second isomer, 7eb, exhibited an ionic interaction between Zn 301 and the cyano group, in addition to the interaction between the sulfur atom of the thiazole ring and Thr 200 with a bond length of 3.75 Å (Figure 26). The interaction binding energy of isomer 7ea (−2.2591 kcal/mol) is smaller than that of 7eb (−4.6934 K cal/mol), so the isomer 7ea is favored in the interaction. Compound 7i is also found in two isomers, 7ia and 7ib. Isomer 7ia binds to the active pocket of 5U0F by zinc ion 301 with a thiazole ring carbonyl group (Figure 27). On the other hand, carbonyl group of 7ib, with rmsd_refine 0.9538 at S = −5.6139 binds to Asn 62 together with arene-H with Leu 128 (Figure 27). The isomer 7ia is therefore more acceptable due to the ionic interaction of the Zn ion. Similarly, compound 7h also interacts with the pock of the active site of 5U0F by an ionic bond with zinc ion 301 as demonstrated in Figure 28 for 7ha, whereas 7hb has one interaction between the carbonyl group and His 64 of S = −5.1544 kcal/mol (Figure 28). From the interaction patterns of compounds 7e, 7i, and 7h, we concluded that the presence of the carbonyl group on thiazole ring is important for the interaction with the active site pocket of 5U0F (see table 3).
Table 9. Interactions of Compounds 7e, 7i, 7h, and AZA with 5U0F.
| compound no. | energy S kcal/mol | rmsd–refine | amino acids contributed in the interaction | length of bonds Å |
|---|---|---|---|---|
| AZA | –2.2591 | 1.4460 | Thr 199 | H-bond 2.97 Å (S=O) |
| Zn 301 | ionic bond (S=O) | |||
| 7ea | –6.5441 | 1.3601 | His 94 | arene-H of phenyl ring |
| Zn 301 | C=O thiazole ring | |||
| 7eb | –4.6934 | 0.9475 | Zn 301 | CN group |
| Thr 200 | arene-H of S-thiazole | |||
| 7ia | –6.7493 | 2.0325 | Zn 301 | C=O of thiazole ring |
| 7ib | –5.6139 | 0.9538 | Asn 62 | C=O thiazole ring |
| Leu 128 | arene-H of phenyl ring | |||
| 7ha | –6.9086 | 1.8485 | Zn 301 | C=O of thiazole ring |
| 7hb | –6.1544 | 1.7606 | His 64 | C=O of thiazole ring |
Figure 25.

2D and 3D of AZA with an active pocket of 5U0F.
Figure 27.

2D and 3D ligand interaction of 7ia and 7ib with the amino acids of the 5U0F active site.
Figure 28.

2D and 3D ligand interaction of 7ha and 7 hb with the amino acids of the 5U0F active site.
4. Conclusions
A new class of thiopyrano[2,3-d]thiazoles were synthesized and assessed for their anticancer activity. Cycloadducts 7e and 7i displayed potent anticancer activity at low concentrations. The antiproliferative activity of cycloadducts 7e and 7i appeared to correlate well with their ability to inhibit both CAXI and CAXII isoforms at submicromolar levels. Cycloadduct 7e is emerged as the most potent hCAIX inhibitor with an IC50 value of 0.067 ± 0.003 μM, while compound 7i is also a more potent and selective CAXII inhibitor (IC50 = 0.123 ± 0.007 μM) compared to the standard drug acetazolamide AZA. Therefore, we screened cycloadducts 7e and 7i for cell cycle disruption of HePG-2 and found that the % of annexinV-FITC positive apoptotic cells increased from 11.91 to 21.24% for compound 7e and decreased from 28.71 to 9.51% for compound 7i in cells. This investigated the mechanistic pathways of compounds 7e and 7i in cells and confirmed that they significantly increased the levels of active caspase-9. Finally, to rationalize the obtained results, we performed molecular docking studies of thiopyranothiazoles 7e and 7i within CAIX and XII active sites (PDB; 5U0F). Thiopyrano[2,3-d]thiazoles 7e and 7i are privileged multitargeted scaffolds for the development of novel anticancer drugs as well as CAIX and CAXII inhibitors.
5. Experimental Section
5.1. Chemistry
The uncorrected melting points for all materials were calculated using an Electrothermal (9100) device, and the IR spectra were captured using KBr pellets on a Perkin Elmer 1430 spectrophotometer. The NMR spectra were recorded on a Varian Mercury VXR-300 NMR spectrometer and recorded at 300 and 75 MHz (1H and 13C NMR spectra, respectively) using DMSO-d6 as solvent, with results are expressed as δ values. A Shimadzu GCMS-QP 1000 Ex mass spectrometer was used to measure the mass spectra at a voltage 70 eV. At Cairo University’s Microanalyses Center, an elemental analysis was carried out using a Vario EL III Elemental CHNS analyzer. The University of Zürich’s Institute of Organic Chemistry conducted the X-ray crystallographic analysis. The pharmacy faculty at Egypt’s Mansoura University engaged in anticancer activity. At VACSERA (holding company for biological products and vaccines Cairo, Egypt), the enzyme inhibition, cell cycle, apoptosis, and caspase-9 were measured. Beginning materials 1, 2a–e, and 4a–c were acquired using the procedures previously described.63−65
5.1.1. Synthesis of 5-[(3-Aryl-1-phenyl-1H-pyrazol-4-yl)methylene]-4-thioxo-2-thiazolidinone Derivatives 3a–e
5.2. General Procedure
3-Aryl-1-phenyl-1H-pyrazole-4-carbaldehydes 2a–e, 4-thioxo-2-thiazolidinone 1, and fused sodium acetate were all refluxed in 20 mL of glacial acetic acid for 2 h before being left to stand at room temperature overnight. The precipitate that resulted was filtered, washed with water, dried, and then recrystallized from the mixture of ethanol and dioxane.
5.2.1. 5-[(1,3-Diphenyl-1H-pyrazol-4-yl)methylene]-4-thioxo-2-thiazolidinone (3a)
Red crystals; 56%; mp 271 °C; IR (KBr, cm–1) 3434 (NH), 1736 (CO); 1H NMR (DMSO-d6): δ = 7.29–8.0 (m, 11H, 10 Ar-H and olefinic-H), 8.70 (s, 1H, pyrazole-H), 13.75 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 116.40, 119.41, 125.70, 127.55, 127.98, 128.42, 129.50, 129.62, 130.34, 133.98, 138.50, 139.16, 153.12, 170.25, 194.79; m/z (%) = 364 (6.6, M++1), 363 (22.1, M+), 286 (4.6), 232 (5.7), 209 (1.7), 144 (3.7), 77 (100.0). Anal. calcd for C19H13N3OS2 (363.46): C, 62.79; H, 3.61; N, 11.56; S, 17.64. Found: C, 62.52; H, 3.88; N, 11.74; S, 17.41%.
5.2.2. 5-[(3-(4-Methylphenyl)-1-phenyl-1H-pyrazol-4-yl)methylene]-4-thioxo-2-thiazolidinone (3b)
Orange crystals; 40%; mp 293 °C; IR (KBr, cm–1) 3427 (NH), 1729 (CO); 1H NMR (DMSO-d6): δ = 2.41 (s, 3H, CH3), 7.37–8.03 (m, 10H, 9 Ar-H and vinylic-H), 8.75 (s, 1H, pyrazole-H), 13.70 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 20.87, 116.37, 119.48, 126.59, 127.63, 128.02, 128.29, 128.72, 128.55, 129.63, 138.65, 138.81, 154.67, 170.31, 194.65; m/z (%) = 378 (18.1, M++1), 377 (76.2, M+), 301 (8.20), 284 (58.8), 208 (17.9), 91 (19.9), 77 (100.0). Anal. calcd for C20H15N3OS2 (377.48): C, 63.64; H, 4.01; N, 11.13; S, 16.99. Found: C, 63.50; H, 4.20; N, 11.32; S, 16.84%.
5.2.3. 5-[(3-(4-Methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)methylene]-4-thioxo-2-thiazolidinone (3c)
Red crystals; 42%; mp 306 °C; IR (KBr, cm–1) 3429 (NH), 1728 (CO); 1H NMR (DMSO-d6): δ = 3.85 (s, 3H, OCH3), 7.08–8.37 (m, 10H, 9 Ar-H and vinylic-H), 8.74 (s, 1H, pyrazole-H), 13.73 (s, 1H, NH, D2O exchangeable); m/z (%) = 394 (26.5, M++1), 393 (100.0, M+), 300 (60.5), 224 (14.6), 104 (19.3), 77 (94.8). Anal. calcd for C20H15N3O2S2 (393.48): C, 61.05; H, 3.84; N, 10.68; S, 16.30. Found: C, 61.21; H, 4.03; N, 10.41; S, 16.07%.
5.2.4. 5-[(3-(4-Chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene]-4-thioxo-2-thiazolidinone (3d)
Red crystals; 55%; mp 300 °C; IR (KBr, cm–1) 3433 (NH), 1735 (CO); 1H NMR (DMSO-d6): δ = 7.29–8.0 (m, 10H, 9 Ar-H and olefinic-H), 8.70 (s, 1H, pyrazole-H), 13.75 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 116.40, 119.39, 125.71, 127.59, 127.99, 128.46, 129.26, 129.62, 130.02, 130.34, 133.98, 138.50, 139.1, 153.11, 170.20, 194.74; m/z (%) = 399 (31.8, M++2), 398 (20.3, M++1), 397 (69.1, M+), 304 (46.9), 275 (12.5), 104 (17.6), 77 (100.0). Anal. calcd for C19H12ClN3OS2 (397.90): C, 57.35; H, 3.04; Cl, 8.91; N, 10.56; S, 16.12. Found: C, 57.14; H, 3.22; Cl, 9.05 N, 10.38; S, 15.96%.
5.2.5. 5-[(3-(4-Bromophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene]-4-thioxo-2-thiazolidinone (3e)
Red crystals; 67%; mp >300 °C; IR (KBr, cm–1) 3431 (NH), 1730 (CO); 1H NMR (DMSO-d6): δ = 7.26–8.01 (m, 10H, 9 Ar-H and olefinic-H), 8.71 (s, 1H, pyrazole-H), 13.73 (s, 1H, NH, D2O exchangeable); m/z (%) = 443 (42.8, M++2), 442 (15.6, M++1), 441 (43.4, M+), 350 (23.1), 348 (23.7), 275 (17.1), 231 (13.8), 104 (17.1), 77 (100.0). Anal. calcd for C19H12BrN3OS2 (442.35): C, 51.59; H, 2.73; Br, 18.06; N, 9.50; S, 14.50. Found: C, 51.33; H, 2.95; Br, 18.22; N, 9.22; S, 14.72%.
4.1.3. Synthesis of 8-(3-Aryl-1-phenyl-1H-pyrazol-4-yl)-6-arylpyrrolo[3′,4′:5,6]-thiopyrano[2,3-d]thiazole-2,5,7-trione 5a–g
5.3. General Procedure
N-Aryl maleimides 4a–c were added to the suspension of the 5-arylmethylene-4-thioxo-2-thiazolidinone derivatives 3a–e (0.01 mol) in glacial acetic acid (20 mL) (0.01 mol). The mixture was refluxed until decolorization occurred and was then left at room temperature for the night. The resulting solid product was filtered out and recrystallized from an ethanol and dioxane combination.
5.3.1. 8-(1,3-Diphenyl-1H-pyrazol-4-yl)-6-phenyl-7a,8-dihydropyrrolo[3′,4′:5,6]-thiopyrano[2,3-d]thiazole-2,5,7(3H,4aH,6H)-trione (5a)
Yellow crystals; 53%; mp 230 °C; IR (KBr, cm–1) 3440 (NH), 1714, 1677 (CO); 1H NMR (DMSO-d6): δ = 4.34 (dd, 1H, J = 3.6 Hz, 8.7 Hz, H-6), 4.41 (d, 1H, J = 3.9 Hz, H-5), 4.98 (d, 1H, J = 8.4 Hz, H-7), 7.02–7.84 (m, 15H, Ar-H), 8.71 (s, 1H, pyrazole-H), 11.83 (s, 1H, NH, D2O exchangeable). Anal. calcd for C29H20N4O3S2 (536.62): C, 64.91; H, 3.76; N, 10.44; S, 11.95. Found: C, 64.72; H, 3.95; N, 10.76; S, 11.78%.
5.3.2. 8-(1,3-Diphenyl-1H-pyrazol-4-yl)-6-(4-methoxyphenyl)-7a,8-dihydropyrrolo[3′,4′:5,6]-thiopyrano[2,3-d]thiazole-2,5,7(3H,4aH,6H)-trione (5b)
White crystals; 89%; mp 232 °C; IR (KBr, cm–1) 3440 (NH), 1714, 1677 (CO); 1H NMR (DMSO-d6): δ = 3.77 (s, 3H, OCH3), 4.13 (dd, 1H, J = 3.0 Hz, 9 Hz, H-6), 4.90 (d, 1H, J = 2.7 Hz, H-5), 4.93 (d, 1H, J = 9 Hz, H-7), 7.03–7.98 (m, 14H, Ar-H), 8.60 (s, 1H, pyrazole-H), 11.73 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 31.37, 40.06, 48.17, 55.36, 110.12, 114.34, 118.29, 118.66, 119.77, 124.34, 126.29, 127.81, 128.00, 128.30, 128.72, 129.44, 132.85, 139.36, 150.29, 159.19, 170.50, 174.70, 174.94. m/z (%) = 566 (0.32 M+), 363 (100), 330 (2.29), 319 (5.38), 270 (0.43), 104 (5.05), 77 (27.82). Anal. calcd for C30H22N4O4S2 (566.65): C, 63.59; H, 3.91; N, 9.89; S, 11.32. Found: C, 63.74; H, 3.76; N, 10.12; S, 11.09%.
5.3.3. 6-(4-Chlorophenyl)-8-(1,3-diphenyl-1H-pyrazol-4-yl)-7a,8-dihydropyrrolo-[3′,4′:5,6]-thiopyrano[2,3-d]thiazole-2,5,7(3H,4aH,6H)-trione (5c)
White crystals; 85%; mp 252 °C; IR (KBr, cm–1) 3250 (NH), 1712, 1679, 1685 (CO); 1H NMR (DMSO-d6): δ = 4.11 (dd, 1H, J = 3.3 Hz, 9 Hz, H-6), 4.91–4.94 (m, 2H, H-5 and H-6), 7.17–7.96 (m, 14H, Ar-H), 8.57 (s, 1H, pyrazole-H), 11.70 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6): δ = 31.23, 42.92, 48.15, 109.93, 118.21, 118.46, 119.74, 126.25, 127.92, 128.33, 128.70, 129.18, 129.42, 130.54, 132.77, 133.20, 139.28, 143.92, 150.16, 170.47, 174.36, 174.57. m/z (%) = 572 (1.03, M+ +1), 363 (97.74), 330 (2.52), 319 (5.87), 302 (2.06), 270 (1.20), 104 (4.81), 90 (8.04), 77 (100). Anal. calcd for C29H19ClN4O3S2 (571.07): C, 60.99; H, 3.35; Cl, 6.21; N, 9.81; S, 11.23. Found: C, 60.68; H, 3.54; Cl, 6.40; N, 10.0; S, 11.02%.
5.3.4. 8-(3-(4-Methylphenyl)-1-phenyl-1H-pyrazol-4-yl)-6-phenyl-7a,8-dihydro-pyrrolo[3′,4′:5,6]-thiopyrano[2,3-d]thiazole-2,5,7(3H,4aH,6H)-trione (5d)
White crystals; 56%; mp 210 °C; IR (KBr, cm–1) 3440 (NH), 1714, 1682, 16,677 (CO); 1H NMR (DMSO-d6): δ = 2.36 (s, 3H, CH3), 4.35 (dd, 1H, J = 3.6 Hz, 8.4 Hz, H-6), 4.43 (d, 1H, J = 3.6 Hz, H-5), 4.98 (d, 1H, J = 8.4 Hz, H-7), 7.06–7.84 (m, 14H, Ar-H), 8.74 (s, 1H, pyrazole-H), 11.89 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 20.77, 34.05, 46.31, 49.48, 66.33, 114.95, 116.92, 118.42, 119.55, 126.55, 126.60, 128.00, 128.65, 128.80, 129.12, 129.27, 129.47, 129.53, 129.63, 131.72, 137.71, 139.28, 151.71, 170.44, 174.00, 174.10. Anal. calcd for C30H22N4O3S2 (550.65): C, 65.44; H, 4.03; N, 10.17; S, 11.65. Found: C, 65.27; H, 3.90; N, 10.39; S, 11.83%.
5.3.5. 8-(3-(4-Methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)-6-phenyl-7a,8-dihydro-pyrrolo-[3′,4′:5,6]thiopyrano[2,3-d]thiazole-2,5,7(3H,4aH,6H)-trione (5e)
Pale yellow crystals; 87%; mp 182 °C; IR (KBr, cm–1) 3440 (NH), 1714, 1682 (CO); 1H NMR (DMSO-d6): δ = 3.81 (s, 3H, OCH3), 4.20 (dd, 1H, J = 3.0 Hz, 9.0 Hz, H-6), 4.92 (d, 1H, J = 3.0 Hz, H-5), 4.94 (d, 1H, J = 9.0 Hz, H-7), 7.07–7.96 (m, 14H, Ar-H), 8.56 (s, 1H, pyrazole-H), 11.75 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 31.42, 43.06, 48.15, 55.14, 110.16, 114.15, 116.80, 119.55, 119.71, 124.73, 125.20, 126.14, 128.14, 129.10, 129.41, 131.83, 139.40, 150.13, 159.32, 170.53, 174.52, 174.80; m/z (%) = 567 (4.8, M++1), 566 (12.4, M+), 522 (11.1), 435 (7.0), 393 (23.3), 173 (27.6), 104 (21.8), 77 (100.0). Anal. calcd for C30H22N4O4S2 (566.65): C, 63.59; H, 3.91; N, 9.89; S, 11.32. Found: C, 63.41; H, 3.69; N, 10.20; S, 11.58%.
5.3.6. 8-(3-(4-Chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)-6-phenyl-7a,8-dihydro-pyrrolo[3′,4′:5,6]-thiopyrano[2,3-d]thiazole-2,5,7(3H,4aH,6H)-trione (5f)
Yellow crystals; 85%; mp 251 °C; IR (KBr, cm–1) 3250 (NH), 1695 (CO); 1H NMR (DMSO-d6): δ = 4.07 (dd, 1H, J = 3.3 Hz, 8.7 Hz, H-6), 4.84 (d, 1H, J = 8.7 Hz, H-7), 4.94 (d, 1H, J = 3.0 Hz, H-5), 7.11–7.58 (m, 12H, Ar-H), 7.79 (d, 1H, J = 8.4 Hz, Ar), 7.92 (d, 1H, J = 8.4 Hz, Ar), 8.55 (s, 1H, pyrazole-H), 11.70 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6): δ = 31.36, 43.12, 48.17, 109.92, 118.36, 118.64, 120.13, 126.47, 126.65, 128.68, 128.82, 129.18, 129.56, 129.78, 131.77, 131.84, 133.09, 139.28, 149.07, 170.61, 174.65, 174.83; m/z (%) = 572 (0.51, M++1), 571 (0.47, M+), 521 (0.17), 425 (0.11), 397 (100), 355 (1.94), 304 (3.38), 91 (6.62), 77 (85.09). Anal. calcd for C29H19ClN4O3S2 (571.07): C, 60.99; H, 3.35; Cl, 6.21; N, 9.81; S, 11.23. Found: C, 61.22; H, 3.59; Cl, 6.02; N, 9.65; S, 11.08%.
5.3.7. 8-(3-(4-Bromophenyl)-1-phenyl-1H-pyrazol-4-yl)-6-phenyl-7a,8-dihydro-pyrrolo[3′,4′:5,6]-thiopyrano[2,3-d]thiazole-2,5,7(3H,4aH,6H)-trione (5g)
White crystals; 85%; mp 245 °C; IR (KBr, cm–1) 3442 (NH), 1716, 1679 (CO); 1H NMR (DMSO-d6): δ = 4.05 (dd, 1H, J = 3.0 Hz, 8.7 Hz, H-6), 4.83 (d, 1H, J = 9 Hz, H-7), 4.42 (d, 1H, J = 2.7 Hz, H-5), 7.10–8.0 (m, 14H, Ar-H), 8.54 (s, 1H, pyrazole-H), 11.70 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6): δ = 31.7, 43.12, 48.16, 109.92, 118.36, 118.64, 120.15, 121.75, 126.65, 128.72, 128.82, 129.18, 129.54, 130.05, 129.78, 131.73, 131.84, 132.12, 139.27, 143.54, 144.41, 149.09, 170.62, 174.64, 174.84; m/z (%) = 616 (0.18, M++1), 615 (0.14, M+), 441 (4.81), 399 (1.66), 350 (3.68), 173 (3.03), 129 (2.10), 104 (4.53), 77 (100.0). Anal. calcd for C29H19BrN4O3S2 (615.52): C, 56.59; H, 3.11; Br, 12.98; N, 9.10; S, 10.42. Found: C, 56.41; H, 3.00; Br, 13.22; N, 9.34; S, 10.18%.
5.3.8. Synthesis of 7-(3-Aryl-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano[2,3-d]thiazole Derivatives 7a–j
5.4. General Procedure
Either acrylonitrile (6a) or ethyl acrylate (6b) was added to 3a–e (0.01 mol), suspended in glacial acetic acid (20 mL) (0.01 mol). The mixture was refluxed until decolorization occurred followed by an overnight resting period at room temperature. The resulting solid product was filtered out and recrystallized from an ethanol and dioxane mixture.
5.4.1. 7-(1,3-Diphenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano-[2,3-d]thiazole-6-carbonitrile (7a)
White crystals; 66%; mp 264 °C; IR (KBr, cm–1) 3126 (NH), 2245 (CN), 1639 (CO); 1H NMR (DMSO-d6): δ = 3.45–3.66 (m, 2H, H-5), 3.98–4.03 (m, 1H, H-6), 4.50 (d, 1H, J = 5.4 Hz, H-7), 7.31–7.66 (m, 8H, Ar-H), 7.92 (d, 1H, J = 7.8 Hz, Ar-H), 8.60 (s, 1H, pyrazole-H), 11.49 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 27.57, 32.08, 33.66, 105.63, 118.82, 119.24, 119.58, 120.49, 127.03, 128.80, 129.16, 129.99, 132.61, 139.59, 144.80, 144.89, 151.68, 170.55. Anal. calcd for C22H16N4OS2 (416.52): m/z (%) = 416 (29.02, M+), 379 (2.14, M+), 363 (100), 330 (7.04.92), 319 (12.08), 287 (6.49), 270 (36.84), 172 (2–08), 104 (14.26), 77 (65.16). C, 63.44; H, 3.87; N, 13.45; S, 15.40. Found: C, 63.70; H, 3.98; N, 13.26; S, 15.18%.
5.4.2. 7-(3-(4-Methylphenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetra-hydro-2H-thiopyrano-[2,3-d]thiazole-6-carbonitrile (7b)
White crystals; 41%; mp 252 °C; IR (KBr, cm–1) 3435 (NH), 2246 (CN), 1637 (CO); 1H NMR (DMSO-d6): δ = 2.37 (s, 3H, CH3), 3.44–3.64 (m, 2H, H-5), 3.95–3.99 (m, 1H, H-6), 4.48 (d, 1H, J = 5.1 Hz, H-7), 7.29–7.35 (m, 3H, Ar-H), 7.48–7.53 (m, 4H, Ar-H), 7.90 (d, 2H, J = 7.4 Hz, Ar-H), 8.57 (s, 1H, pyrazole-H), 11.48 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 20.90, 27.19, 31.71, 33.27, 105.29, 118.38, 118.87, 119.14, 120.08, 126.57, 128.30, 128.70, 129.36, 129.60, 137.78, 139.23, 144.70, 151.30, 170.18. Anal. calcd for C23H18N4OS2 (430.55): m/z (%) = 430 (3.65, M+), 377 (11.33), 334 (1.70), 313 (1.67), 284 (2.23), 245 (4.11), 173 (1.39), 104 (3.27), 77 (100.0). C, 64.16; H, 4.21; N, 13.01; S, 14.90. Found: C, 63.96; H, 3.99; N, 13.22; S, 15.12%.
5.4.3. 7-(3-(4-Methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetra-hydro-2H-thiopyrano[2,3-d]thiazole-6-carbonitrile (7c)
White crystals; 46%; mp 274 °C; IR (KBr, cm–1) 3440 (NH), 2243 (CN), 1639 (CO); 1H NMR (DMSO-d6): δ = 3.81 (s, 3H, OCH3), 3.42–3.61 (m, 2H, H-5), 3.96–4.01 (m, 1H, H-6), 4.50 (d, 1H, J = 5.1 Hz, H-7), 7.32–7.94 (m, 9H, Ar-H), 8.64 (s, 1H, pyrazole-H), 11.47 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 27.03, 31.55, 33.12, 55.1, 104.96, 118.44, 118.81, 119.33, 120.21, 126.83, 128.80, 129.22, 129.64, 130.02, 131.15, 133.24, 139.17, 150.23, 170.12. Anal. calcd for C23H18N4O2S2 (446.54): C, 61.86; H, 4.06; N, 12.55; S, 14.36. Found: C, 61.99; H, 3.85; N, 13.78; S, 14.19%.
5.4.4. 7-(3-(4-Chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetra-hydro-2H-thiopyrano[2,3-d]thiazole-6-carbonitrile (7d)
White crystals; 44%; mp 300 °C; IR (KBr, cm–1) 3442 (NH), 2245 (CN), 1644 (CO); 1H NMR (DMSO-d6): δ = 3.44–3.64 (m, 2H, H-5), 3.99–4.04 (m, 1H, H-6), 4.53 (d, 1H, J = 5.1 Hz, H-7), 7.32–7.72 (m, 2H, Ar-H), 7.49–7.52 (m, 1H, Ar-H), 7.53 (d, 2H, J = 7.5 Hz, Ar-H), 7.66 (d, 2H, J = 7.8 Hz, Ar-H), 7.92 (d, 2H, J = 7.2 Hz, Ar-H) 8.61 (s, 1H, pyrazole-H), 11.49 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 27.02, 31.54, 33.13, 104.92, 118.43, 118.70, 119.21, 120.11, 126.66, 128.69, 128.99, 129.51, 130.07, 131.04, 133.09, 139.05, 149.99, 170.05; m/z (%) = 452 (4.2, M++2), 451 (2.7, M++1), 450 (9.8, M+), 397 (70.5), 304 (43.5), 104 (18.2), 77 (100.0). Anal. calcd for C22H15ClN4OS2 (450.96): C, 58.59; H, 3.35; Cl, 7.86; N, 12.42; S, 14.22. Found: C, 58.36; H, 3.18; Cl, 7.99; N, 12.65; S, 14.41%.
5.4.5. 7-(3-(4-Bromophenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano-[2,3-d]thiazole-6-carbonitrile (7e)
White crystals; 42%; mp 299 °C; IR (KBr, cm–1) 3441 (NH), 2246 (CN), 1647 (CO); 1H NMR (DMSO-d6): δ = 3.43–3.50 (m, 1H, H-5), 3.59–3.64 (m, 1H, H-5), 3.96–3.99 (m, 1H, H-6), 4.51 (d, 1H, J = 5.4 Hz, H-7), 7.32–8.02 (m, 9H, Ar-H), 8.76 (s, 1H, pyrazole-H), 11.49 (s, 1H, NH, D2O exchangeable). m/z (%) = 496 (3.21, M++1), 495 (1.38, M+), 459 (1.19), 443 (53.64), 382 (3.61), 350 (26.66), 319 (10.09), 275 (14.67), 231 (8.98), 181 (5.22), 172 (4.38), 129 (6.47), 104 (15.09), 77 (100.0). Anal. calcd for C22H15BrN4OS2 (495.41): C, 53.34; H, 3.05; Br, 16.13; N, 11.31; S, 12.94. Found: C, 53.50; H, 3.20; Br, 15.96; N, 11.21; S, 12.75%.
5.4.6. Ethyl 7-(1,3-Diphenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano[2,3-d]thiazole-6-carboxylate (7f)
White crystals; 71%; mp 242 °C; IR (KBr, cm–1) 3433 (NH), 1725, 1649 (CO); 1H NMR (DMSO-d6): δ = 0.69 (t, 3H, J = 7.2 Hz, CH2CH3), 3.16–3.29 (m, 2H, H-5), 3.46–3.50 (m, 1H, H-6), 3.76 (q, 2H, J = 7.2 Hz, CH2CH3), 4.56 (d, 1H, J = 5.4 Hz, H-7), 7.31–7.60 (m, 8H, Ar-H), 7.92–7.95 (m, 2H, Ar-H), 8.58 (s, 1H, pyrazole-H), 11.38 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 13.33, 23.54, 30.51, 45.01, 60.30, 106.38, 118.17, 119.48, 120.05, 126.37, 128.11, 128.31, 128.52, 128.66, 129.45, 132.83, 139.19, 144.12, 144.22, 150.99, 170.61. m/z (%) = 463 (6.49, M+), 363 (27.97), 319 (7.41), 302 (8.63), 270 (15.69), 171 (1.91), 104 (13.62), 77 (100.0). Anal. calcd for C24H21N3O3S2 (463.10): C, 62.18; H, 4.57; N, 9.06; S, 13.83. Found: C, 61.98; H, 4.25; N, 9.32; S, 14.02%.
5.4.7. Ethyl 7-(3-(4-Methylphenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano[2,3-d]thiazole-6-carboxylate (7g)
White crystals; 45%; mp 252 °C; IR (KBr, cm–1) 3447 (NH), 1731, 1678 (CO); 1H NMR (DMSO-d6): δ = 0.741 (t, 3H, J = 6.9 Hz, CH2CH3), 2.36 (s, 3H, CH3), 3.12–3.33 (m, 2H, H-5), 3.47–3.52 (m, 1H, H-6), 3.75 (q, 2H, J = 6.9 Hz, CH2CH3), 4.55 (d, 1H, J = 5.1 Hz, H-7), 7.29 (d, 2H, J = 7.5 Hz, Ar-H), 7.31–7.50 (m, 5H, Ar-H), 7.91 (d, 2H, J = 7.8 Hz, Ar-H), 8.56 (s, 1H, pyrazole-H), 11.39 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 13.22, 20.75, 23.62, 30.52, 44.98, 60.25, 106.42, 118.08, 119.33, 119.92, 126.16, 128.14, 128.26, 129.07, 129.32, 129.93, 137.35, 139.20, 150.99, 170.47. Anal. calcd for C25H23N3O3S2 (477.12): C, 62.87; H, 4.85; N, 8.80; S, 13.43. Found: C, 62.74; H, 4.99; N, 9.00; S, 13.21%.
5.4.8. Ethyl 7-(3-(4-Methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetrahydro-2H-thiopyrano[2,3-d]thiazole-6-carboxylate (7h)
White crystals; 84%; mp 250 °C; IR (KBr, cm–1) 3444 (NH), 1736, 1640 (CO); 1H NMR (DMSO-d6): δ = 0.73 (t, 3H, J = 6.9 Hz, CH2CH3), 3.13–3.35 (m, 2H, H-5), 3.49–3.53 (m, 1H, H-6), 3.77–3.83 (m, 5H, OCH3 and CH2CH3), 4.55 (d, 1H, J = 5.1 Hz, H-7), 7.05 (d, 2H, J = 8.7 Hz, Ar-H), 7.26–7.54 (m, 5H, Ar-H), 7.91 (d, 2H, J = 8.1 Hz, Ar-H), 8.54 (s, 1H, pyrazole-H), 11.39 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 13.31, 23.68, 30.59, 45.02, 55.14, 60.29, 106.51, 114.01, 118.07, 119.32, 119.83, 125.19, 126.12, 128.17, 129.33, 129.53, 139.25, 150.86, 159.18, 170.53; m/z (%) = 494 (5.7, M++1), 493 (17.5, M+), 393 (100.0), 300 (54.3), 224 (13.4), 104 (17.7) 77 (73.7). Anal. calcd for C25H23N3O4S2 (493.11): C, 60.83; H, 4.70; N, 8.51; S, 12.99. Found: C, 60.99; H, 4.63; N, 8.74; S, 12.78%.
5.4.9. Ethyl 7-(3-(4-Chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetra-hydro-2H-thiopyrano[2,3-d]thiazole-6-carboxylate (7i)
White crystals; 41%; mp 254 °C; IR (KBr, cm–1) 3117 (NH), 1735, 1640 (CO); 1H NMR (DMSO-d6): δ = 0.73 (t, 3H, J = 6.9 Hz, CH2CH3), 3.12–3.38 (m, 2H, H-5), 3.46–3.55 (m, 1H, H-6), 3.75 (q, 2H, J = 6.9 Hz, CH2CH3), 4.52 (d, 1H, J = 5.4 Hz, H-7), 7.05 (d, 2H, J = 9 Hz, Ar-H), 7.27–7.32 (m, 1H, Ar-H), 7.45–7.53 (m, 4H, Ar-H), 7.90 (d, 2H, J = 7.8 Hz, Ar-H), 8.54 (s, 1H, pyrazole-H), 11.37 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): δ = 13.76, 24.04, 30.96, 45.41, 60.73, 106.91, 114.47, 118.48, 119.77, 120.26, 125.57, 126.60, 128.65, 129.82, 129.96, 139.65, 144.16, 151.25, 159.59, 170.07. Anal. calcd for C24H20ClN3O3S2 (497.99): C, 57.88; H, 4.05; Cl, 7.12; N, 8.44; S, 12.88. Found: C, 57.56; H, 3.89; Cl, 7.36; N, 8.78; S, 12.62%.
5.4.10. Ethyl 7-(3-(4-Bromophenyl)-1-phenyl-1H-pyrazol-4-yl)-2-oxo-3,5,6,7-tetra-hydro-2H-thiopyrano[2,3-d]thiazole-6-carboxylate (7j)
White crystals; 41%; mp 256 °C; IR (KBr, cm–1) 3447 (NH), 1732, 1679 (CO); 1H NMR (DMSO-d6): δ = 0.73 (t, 3H, J = 6.9 Hz, CH2CH3), 3.12–3.29 (m, 2H, H-5), 3.43–3.47 (m, 1H, H-6), 3.75 (q, 2H, J = 6.9 Hz, CH2CH3), 4.54 (d, 1H, J = 5.4 Hz, H-7), 7.29–7.99 (m, 9H, Ar-H), 8.65 (s, 1H, pyrazole-H), 11.39 (s, 1H, NH, D2O exchangeable). m/z (%) = 543 (0.93, M++1), 542 (0.41, M+), 441 (3.40), 348 (2.16), 319 (0.85), 275 (1.25), 231 (1.27), 155 (1.24), 104 (5.33), 77 (100.0). Anal. calcd for C24H20BrN3O3S2 (542.47): C, 53.14; H, 3.72; Br, 14.73; N, 7.75; S, 11.82. Found: C, 53.32; H, 3.95; Br, 14.63; N, 7.56; S, 11.99%.
5.5. Crystallographic Analysis
To determine the three dimensional structure of the titled structure, a study was conducted. Below is a table of geometries. MaXus was used to complete computations and draw various diagrams (Bruker Nonius, Delft and MacScience, Japan).
Crystal data for 5a: C29H20N4O3S2, = Mr = 536.632, triclinic, a = 8.4142(3), b = 10.5253(4), c = 14.5668(6) Å, α = 90.911 (2)°, β = 100.943(2)°, γ = 95.418(3)°, V = 1260.19(8) Å3, Z = 2, Dx = 1.414 Mg/m–3, Mo Kα radiation, λ = 0.71073 Å, Cell parameters from 2935, T = 298 K, θ = 2.910–27.485°, θmax = 27.57°, Rint = 0.030. Refinement on F2; R(all) = 0.133, R(gt) = 0.039, wR(ref) = 0.069, wR(all) = 0.098, wR(gt) = 0.069. S(ref) = 1.224, S(all) = 1.198, S(gt) = 1.228, 2557 reflections and 343 parameters, μ = 0.25 mm–1. Deposition number (CCDC) 1544355. The Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 (0) 1223 336,033; email: deposit@ccdc.cam.ac.uk, is where you can get these data for free (see Supporting Information).
5.6. Biological Activity Methods
6. Materials and Methods
Holding company for biological products and vaccines (VACSERA), Cairo, Egypt, received the human normal lung fibroblast (WI38), hepatocellular carcinoma (HePG-2), and mammary gland breast cancer (MCF-7) cell lines from ATCC and employed doxorubicin as a standard drug.
6.1. MTT Assay
The MTT test was performed to assess the inhibitory effect of the substances on cell growth in the aforementioned cell lines. This colorimetric technique relies on mitochondrial succinate dehydrogenase, which transforms the yellow tetrazolium bromide (MTT) into a purple formazan derivative in living cells. A 10% fetal bovine serum was added to RPMI-1640 medium in which cell lines were grown.
The cells were grown in a 5% CO2 incubator at 37 °C with addition of antibiotics (100 units/mL penicillin and 100 g/mL streptomycin). Under 96-well plates, cell lines were seeded at a density of 1.0 × 104 cells/well for 48 h. Then, the cells were exposed to various concentrations of compounds and incubated for 24 h. For 4 h, 20 μL of a 5 mg/mL MTT solution was incubated and then underwent drug treatment for 24 h. Each well received 100 μ of DMSO to dissolve the purple formazan. A plate reader was used to measure and record the absorbance at 570 nm (EXL 800, USA). The relative cell viability percentage was determined as (A570 of the treated sample/A570 of the untreated sample) times 100.66,67
6.2. Enzyme Inhibition
hCAIX and hCAXII enzyme inhibition assay was performed: human carbonic anhydrase IX (hCAIX), containing a C-terminal His-tag, MW = 40 kDa, produced in an Sf9 cell expression system infected with a baculovirus 1400 pmol/min/g of specific activity; recombinant human carbonic anhydrase XII (hCAXII) source from the mouse myeloma cell line, NS0-derived Ala25Gln291, with a C-terminal 10His tag N-terminal Sequence Analysis Ala25 (see Supporting Information).
6.3. Method
p-Nitrophenyl acetate is hydrolyzed through CA to produce p-nitrophenol. The intensity of the yellow color of p-nitrophenol is proportional to the amount of the substrate, p-nitrophenyl acetate buffer 0.1 M (pH 7.4). After incubation at room temperature for 3 min, the absorbance of the sample at 348 nm was measured using a spectrophotometer. A standard curve was derived from a series of p-nitrophenol solutions ranging from 0 to 100 nmol/mL68,69 (see Supporting Information).
6.4. In Vitro Cell Cycle Analysis
In vitro cell cycle analysis: HePG-2 cells are precultured in a 25 cm2 cell culture flask. The investigated chemicals 7e and 7i were utilized in the cell treatment at their IC50 values after being individually dissolved in the necessary medium, which was RPMI-1640. Cells were extracted after an overnight treatment, carefully fixed with 70% (v/v) ethanol in PBS and then suspended in PBS containing 40 g/mL propidium iodide (PI), 0.1 mg/mL RNase, and 0.1% (v/v) Triton X-100 in a darkened room. A flow cytometer fitted with an argon-ion laser at a wavelength of 488 nm was used to analyze the cell cycle after 30 min at 37 °C and quantify the proportion of cells in G1, S, and G2/M.
6.5. Annexin V-FITC Apoptosis Assay
HePG-2 cells were collected and treated with 7e and 7i individually for 48 h in the annexin V FITC apoptosis test. The cells were then collected, repeatedly rinsed with PBS twice, and centrifuged. After that, using the apoptosis detection kit (BD Biosciences, San Jose, CA) in accordance with the manufacturer’s instructions, the cells were treated with annexin V-FITC and propidium iodide (PI). A flow cytometer was used to examine PI binding and annexin V-FITC binding.
6.6. Western Blot Analysis
According to the abovementioned information of the most potent inhibitors 7e and 7i against CAIX and CAXII, a further study was carried out as Western Blot analysis in gel electrophoresis and immunoblot analysis of proteins.70,71
6.7. Method
To separate proteins according to their sizes, sodium dodecyl sulfate polyacrylamide gel electrophoresis is employed. It is generally used in conjunction with western blotting (immunoblotting) to establish the presence and/or relative abundance of a target protein in a sample containing a complex protein combination. In this approach, the total protein in each sample is loaded, and then using an electric current to transport the proteins through the gel matrix, the samples are electrophoretically separated. The proteins must first come into touch with a detergent as SDS, which denatures and negatively charges the proteins. A molecular weight marker that generates bands of known size is used to help identify the protein of interest. Once the proteins separated, an electric current is used to transfer the gel to a polyvinylidene fluoride membrane, allowing the proteins to migrate from the gel onto the membrane. A secondary antibody that binds to the complex via its antibody side is added in order to detect a specific protein on the membrane after a primary antibody to that protein has been added to form a protein–antibody complex. Most frequently, the secondary antibody is joined to an enzyme that reacts with the substrate to produce light. The Biorad Imager can record this luminescence, which is proportional to the volume of the protein that interacted with the antibody. The chemical technique is in the Supporting Information.
6.8. Molecular Docking
Molecular Operating Environment (MOE) software version 2015.1072 was used for the molecular docking study. The original ligand is present in the CA IX and CA XII X-ray crystal structures (PDB: 5U0F) [(5R)-5-[(2,4-dimethoxyphenyl)methyl]-2-sulfanylidene-1,3-thiazolidin-4].73 Protein was prepared via 3D protonation; the water molecules and ligand that were not implicated in the active site were removed; and the default protocol was then used to build the active site. Then, hydrogen atoms were added to the protein and reduced until the RMS gradient and RMS distance were both 0.01 kcal mol–l and 0.1, respectively. London dG was chosen as the docking score energy and triangular matcher as the placement technique. The original ligand, AZA, self-docked in the active site to start the docking process. The compounds 7e and 7i were generated from Chemdraw 14.0, then subjected to 3D protonation with minimized energy, and saved as an mdb file as a ligand atom that was then used in the docking protocol and then replaced the co-crystallized AZA under the same methods.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06954.
All spectral data such as IR, 1H NMR, 13C NMR, and IR for newly synthesized compounds and raw data for the biological part (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Pouyssegur J.; Dayan F.; Mazure N. M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- Neri D.; Supuran C. T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discovery 2011, 10, 767–777. 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Chiche J.; Ilc K.; Laferriére J.; Trottier E.; Dayan F.; Mazure N. M.; Horn M. C. B.; Pouyssegur J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- Kallio H.; Rodriguez A. M.; Hilvo M.; Hyrskyluoto A.; Parkkila S. Cancer-associated carbonic anhydrases IX and XII: effect of growth factors on gene expression in human cancer cell lines. J. Cancer Mol. 2010, 5, 73–78. [Google Scholar]
- Ondriskova E.; Debreova M.; Pastorekova S.. Tumor-associated carbonic anhydrases IX and XII. In: Supuran CT; De Simone G, editors. Carbonic anhydrases as biocatalysts from theory to medical and industrial applications; Elsevier: Amsterdam: 2015; pp. 169–205. [Google Scholar]
- Kuijk S. J. V.; Yaromina A.; Houben R.; Niemans R.; Lambin P.; Dubois L. J. Prognostic significance of carbonic anhydrase IX expression in cancer patients: a meta-analysis. Front. Oncol. 2016, 6, 69. 10.3389/fonc.2016.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kciuk M.; Gielecinska A.; Mujwar S.; Mojzych M.; Marciniak B.; Drozda R.; Kontek R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J. Enzyme Inhib. Med. Chem. 2022, 37, 1278–1298. 10.1080/14756366.2022.2052868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. 10.1042/BCJ20160115. [DOI] [PubMed] [Google Scholar]
- Boone C. D.; Pinard M.; McKenna R.; Silverman D. Catalytic mechanism of α-class carbonic anhydrases: CO2 hydration and proton transfer. Subcell. Biochem. 2014, 75, 31–52. 10.1007/978-94-007-7359-2_3. [DOI] [PubMed] [Google Scholar]
- McKenna R.; Supuran C. T. Carbonic anhydrase inhibitors drug design. Subcell. Biochem. 2014, 75, 291–323. 10.1007/978-94-007-7359-2_15. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2012, 27, 759–772. 10.3109/14756366.2012.672983. [DOI] [PubMed] [Google Scholar]
- Tonissen K. F.; Poulsen S. A. Carbonic anhydrase XII inhibition overcomes P-glyco-protein-mediated drug resistance: a potential new combination therapy in cancer. Cancer Drug Resist. 2021, 4, 343–355. 10.20517/cdr.2020.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost S. C.; McKenna R.. Carbonic anhydrase: Mechanism, regulation, links to disease, and industrial applications. Subcellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Pinard M. A.; Mahon B.; McKenna R. Probing the surface of human carbonic anhydrase for clues towards the design of isoform specific inhibitors. BioMed Res. Int. 2015, 2015, 1–15. 10.1155/2015/453543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mboge M. Y.; McKenna R.; Frost S. C.. Advances in anti-cancer drug development targeting carbonic anhydrase IX and XII. InTopics in Anti-Cancer Research; Bentham Science Publishers: Sharjah, United Arab Emirates. 2016, 5, 3–42, 10.2174/9781681083339116050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Tu C.; Wang H.; Silverman D. N.; Frost S. C. Catalysis and pH control by membrane-associated carbonic anhydrase IX in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2011, 286, 15789–15796. 10.1074/jbc.M110.188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon B. P.; Bhatt A.; Socorro L.; Driscoll J. M.; Okoh C.; Lomelino C. L.; Mboge M. Y.; Kurian J. J.; Tu C.; Agbandje-McKenna M.; Frost S. C.; McKenna R. The structure of carbonic anhydrase IX is adapted for low-pH catalysis. Biochemistry 2016, 55, 4642–4653. 10.1021/acs.biochem.6b00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lounnas N.; Rosilio C.; Nebout M.; Mary D.; Griessinger E.; Neffati Z.; Chiche J.; Spits H.; Hagenbeek T. J.; Asnafi V.; Poulsen S. A.; Supuran C. T.; Peyron J. F.; Imbert V. Pharmacological inhibition of carbonic anhydrase XII interferes with cell proliferation and induces cell apoptosis in T-cell lymphomas. Cancer Lett. 2013, 333, 76–88. 10.1016/j.canlet.2013.01.020. [DOI] [PubMed] [Google Scholar]
- Yang J. S.; Lin C. W.; Chuang C. Y.; Su S. C.; Lin S. H.; Yang S. F. Carbonic anhydrase IX overexpression regulates the migration and progression in oral squamous cell carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 9517–9524. 10.1007/s13277-015-3692-8. [DOI] [PubMed] [Google Scholar]
- Hsieh M. J.; Chen K. S.; Chiou H. L.; Hsieh Y. S. Carbonic anhydrase XII promotes invasion and migration ability of MDA-MB-231 breast cancer cells through the p38 MAPK signaling pathway. Eur. J. Cell Biol. 2010, 89, 598–606. 10.1016/j.ejcb.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Pacchiano F.; Carta F.; McDonald P. C.; Lou Y.; Vullo D.; Scozzafava A.; Dedhar S.; Supuran C. T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. 10.1021/jm101541x. [DOI] [PubMed] [Google Scholar]
- Sugrue M. F. Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog. Retin. Eye Res. 2000, 19, 87–112. 10.1016/S1350-9462(99)00006-3. [DOI] [PubMed] [Google Scholar]
- Asiedu M.; Ossipov M. H.; Kaila K.; Price T. J. Acetazolamide and midazolam act synergistically to inhibit neuropathic pain. Pain. 2010, 148, 302–308. 10.1016/j.pain.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta F.; Cesare L. M. D.; Pinard M.; Ghelardini C.; Scozzafava A.; McKenna R.; Supuran C. T. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg. Med. Chem. 2015, 23, 1828–1840. 10.1016/j.bmc.2015.02.027. [DOI] [PubMed] [Google Scholar]
- Lesyk R. Drug design: 4-thiazolidinones applications. Part 2. Pharmacological profiles. J Med. Sci. 2020, 89, e407 10.20883/medical.e407. [DOI] [Google Scholar]
- Lesyk R. B.; Zimenkovsky B. S.; Kaminskyy D. V.; Kryshchyshyn A. P.; Havrylyuk D. Y.; Atamanyuk D. V.; Subtelna I. Y.; Khylyuk D. V. Thiazolidinone motif in anticancer drug discovery. Experience of DH LNMU medicinal chemistry scientific group. Biopolym. Cell 2011, 27, 107–117. 10.7124/bc.000089. [DOI] [Google Scholar]
- Agrawal N. Synthetic and therapeutic potential of 4-thiazolidinone and its analogs. Curr. Chem. Lett. 2021, 10, 119–138. [Google Scholar]
- Mueller S. L.; Chrysanthopoulos P. K.; Halili M. A.; Hepburn C.; Nebl T.; Supuran C. T.; Nocentini A.; Peat T. S.; Poulsed S.-A. The Glitazone class of drugs as carbonic anhydrase. Molecules 2021, 26, 3010. 10.3390/molecules26103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palla R.; Distratis C.; Cominotto R.; Panichi V.; Pozzetti G.; Bionda A.; Neri M.; Frattarelli L.; Renal effects of etozolin in man. In diuretics: Basic, pharmacological, and clinical Aspects. Developments in Nephrology; Andreucci V. E.; Dal Canton A., Eds.; Springer: Boston, MA, USA, 1987; 18, 553–555, ISBN 978–1–4612-9227-2. [Google Scholar]
- Bayindir S.; Caglayan C.; Karaman M.; Gülcin İ. The green synthesis and molecular docking of novel N-substituted rhodanines as effective inhibitors for carbonic anhydrase and acetylcholinesterase enzymes. Bioorg. Chem. 2019, 90, 103096 10.1016/j.bioorg.2019.103096. [DOI] [PubMed] [Google Scholar]
- Pancholia S.; Dhameliya T. M.; Shah P.; Jadhavar P. S.; Sridevi J. P.; Yogeshwari P.; Sriram D.; Chakraborti A. K. 3-Benzo[d]thiazol-2-yl(piperazin-1-yl)methanones as new anti-mycobacterial chemotypes: Design, synthesis, biological evaluation and 3D-QSAR studies. Eur. J. Med. Chem. 2016, 116, 187–199. 10.1016/j.ejmech.2016.03.060. [DOI] [PubMed] [Google Scholar]
- Dhameliya T. M.; Patel K. I.; Tiwari R.; Vagolu S. K.; Panda D.; Sriram D.; Chakraborti A. K. Design, synthesis, and biological evaluation of benzo[d]imidazole-2-carboxamides as new anti-TB agents. Bioorg. Chem. 2021, 107, 104538 10.1016/j.bioorg.2020.104538. [DOI] [PubMed] [Google Scholar]
- Kapileswar S.; Garg S. K.; Raj R.; Priyank P.; Meena V. S.; Rohit G.; Banerjee U. C.; Chakraborti A. K. 2-(2-Arylphenyl)benzoxazole As a Novel Anti-Inflammatory Scaffold: Synthesis and Biological Evaluation. ACS Med. Chem. Lett. 2014, 5, 512–516. 10.1021/ml400500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P.; Dhameliva T. M.; Bansal R.; Nautival M.; Kommi D. N.; Jadhavar P. S.; Sridevi J. P.; Yogeeswari P.; Sriram D.; Charkraborti A. K. 2-N-Arylalkylbenzo-[d]thiazole-2-carboxamides as anti-mycobacterial agents: design, new methods of synthesis and biological evaluation. Med. Chem. Commun. 2014, 5, 1489–1495. 10.1039/C4MD00224E. [DOI] [Google Scholar]
- Dhameliva T. M.; Tiwari R.; Banerjee A.; Pancholia S.; Sriram D.; Panda D.; Chakraborti A. K. 4-Benzo[d]thiazole-2-carbanilides as new anti-TB chemotypes: Design, synthesis, biological evaluation, and structure-activity relationship. Eur. J. Med. Chem. 2018, 155, 364–380. 10.1016/j.ejmech.2018.05.049. [DOI] [PubMed] [Google Scholar]
- Bhagat S.; Supriya M.; Pathak S.; Sriram D.; Chakraborti A. K. α-Sulfonamidophosphonates as new anti-mycobacterial chemotypes: Design, development of synthetic methodology, and biological evaluation. Bioorg. Chem. 2019, 82, 246–252. 10.1016/j.bioorg.2018.09.023. [DOI] [PubMed] [Google Scholar]
- Jadhavara P. S.; Patela K. I.; Dhameliyaa T. M.; Sahaa M.; Vajaa M. D.; Krishnab V. S.; Sriramb D.; Chakrabortia A. K. Benzimidazoquinazolines as new potent anti-TB chemotypes: Design, synthesis, and biological evaluation. Bioorg. Chem. 2020, 99, 103774 10.1016/j.bioorg.2020.103774. [DOI] [PubMed] [Google Scholar]
- Kerru N.; Singh P.; Koorbanally N.; Jaj R.; Kumar V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2015, 142, 179–212. 10.1016/j.ejmech.2017.07.033. [DOI] [PubMed] [Google Scholar]
- Kryshchyshyn A.; Roman O.; Lozynskyi A.; Lesyk R. Thiopyrano[2,3-d]thiazoles as new efficient scaffolds in medicinal chemistry. Sci. Pharm. 2018, 86, 26. 10.3390/scipharm86020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardiansah B. Recent reports on pyrazole-based bioactive compounds as candidate for anticancer agents. Asian J. Pharm. Clin. Res. 2017, 12, 45. 10.22159/ajpcr.2017.v10i12.22065. [DOI] [Google Scholar]
- Kumar R. S.; Arif A. I.; Ahmed A.; Idhayadhulla A. Antiinflammatory and antimicrobial activities of novel pyrazole analogues. Saudi J. Biol. Sci. 2016, 23, 614. 10.1016/j.sjbs.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennani F. E.; Doudach L.; Cherrah Y.; Ramli Y.; Karrouch K.; Amsar M.; Faouz M. E. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 2020, 97, 103470 10.1016/j.bioorg.2019.103470. [DOI] [PubMed] [Google Scholar]
- Kuthyala S.; Sheikh S.; Prabhu A.; Rekha P. D.; Karikannar N. G.; Shankar M. K. Synthesis, Characterization, and Anticancer Studies of Some Pyrazole-Based Hybrid Heteroatomics. ChemistrySelect 2020, 5, 10827–10834. 10.1002/slct.202002483. [DOI] [Google Scholar]
- Maciejewska N.; Olszewski M.; Jurasz J.; Serocki M.; Dzierzynska M.; Cekala K.; ieczerzak E.; Baginski M. Novel chalcone-derived pyrazoles as potential therapeutic agents for the treatment of non-small cell lung cancer. Sci. Rep. 2021, 12, 3703. 10.1038/s41598-022-07691-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir A.; Altintop M. D.; Kaplancikli Z. A.; Can O. D.; Ozkay U. D.; Zitouni G. T. Synthesis and evalution of new 1,5-diaryl-3-[4(methyl-sulfonyl)phenyl]-4,5-dihydro-1H-pyrazole derivatives as potential antidepressant agents. Molecules 2015, 20, 2668–2684. 10.3390/molecules20022668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metwally N. H.; Badawy M. A.; Okpy D. S. Synthesis and anticancer activity of some new thiopyrano[2,3-d]thiazoles incorporating pyrazole moiety. Chem. Pharm. Bull. 2015, 63, 495–503. 10.1248/cpb.c14-00885. [DOI] [PubMed] [Google Scholar]
- Metwally N. H.; Abdelrazek F. M.; Eldaly S. M. Synthesis and anticancer activity of some new heterocyclic compounds based on 1-cyanoacetyl-3,5-dimethylpyrazole. Res. Chem. Intermed. 2016, 42, 1071–1089. 10.1007/s11164-015-2074-6. [DOI] [Google Scholar]
- Metwally N. H.; Abdelrazek F. M.; Eldaly S. M. Synthesis, molecular docking, and biological evaluation of some novel bis-heterocyclic compounds based N,N′-([1,1′-biphenyl]-4,4′-diyl)bis(2-cyanoacetamide) as potential anticancer agents. J. Heterocycl. Chem. 2018, 55, 2668–2682. 10.1002/jhet.3290. [DOI] [Google Scholar]
- Metwally N. H.; Deeb E. A. Synthesis, anticancer assessment on human breast, liver and colon carcinoma cell lines and molecular modeling study using novel pyrazolo[4,3-c]pyridine derivatives. Bioorg. Chem. 2018, 77, 203–214. 10.1016/j.bioorg.2017.12.032. [DOI] [PubMed] [Google Scholar]
- Metwally N. H.; Badawy M. A.; Okpy S. M. Green synthesis of some new thiopyrano[2,3-d][thiazolo][1,3]thiazoles using lemon juice and their antibacterial activity. Synth. Commun. 2018, 48, 2496–2509. 10.1080/00397911.2018.1495234. [DOI] [Google Scholar]
- Metwally N. H.; Mohamed M. S.; Ragb E. A. Design, synthesis, anticancer evaluation, mol-ecular docking and cell cycle analysis of 3-methyl-4,7-dihydropyrazolo[1,5-a]pyrimidine derivatives as potent histone lysine demethylases (KDM) inhibitors and apoptosis inducers. Bioorg. Chem. 2019, 88, 102929 10.1016/j.bioorg.2019.102929. [DOI] [PubMed] [Google Scholar]
- Metwally N. H.; Saad G. R.; Abdwahab E. A. Grafting of multiwalled carbon nanotubes with pyrazole derivatives: Characterization, antimicrobial activity and molecular docking study. Int. J. Nanomed. 2019, 20, 6645–6659. 10.2147/IJN.S182699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metwally N. H.; Mohamed M. S. New imidazolone derivatives comprising a benzoate or sulfonamide moiety as anti-inflammatory and antibacterial inhibitors: Design, synthesis, selective COX-2, DHFR and molecular-modeling study. Bioorg. Chem. 2020, 99, 103438 10.1016/j.bioorg.2019.103438. [DOI] [PubMed] [Google Scholar]
- Metwally N. H.; Abdallah S. O.; Mohsen M. M. A. Design, green one-pot synthesis and molecular docking study of novel N,N-bis(cyanoacetyl) hydrazines and bis-coumarins as effective inhibitors of DNA gyrase and topoisomerase IV. Bioorg. Chem. 2020, 97, 103672 10.1016/j.bioorg.2020.103672. [DOI] [PubMed] [Google Scholar]
- Metwally N. H.; Mohamed M. S.; Deeb E. A. Synthesis, anticancer evaluation, CDK2 inhibition, and apoptotic activity assessment with molecular docking modeling of new class of pyrazolo[1,5-a]pyrimidines. Res. Chem. Intermed. 2021, 47, 5027–5060. 10.1007/s11164-021-04564-x. [DOI] [Google Scholar]
- Metwally N. H.; Abd-Elmoety A. S. Novel fluorinated pyrazolo[1,5-a]pyrimidines: In a way from synthesis and docking studies to biological evaluation. J. Mol. Struct. 2022, 1257, 132590 10.1016/j.molstruc.2022.132590. [DOI] [Google Scholar]
- Metwally N. H.; Badawy M. A.; Okpy D. S. Synthesis, biological evaluation of novel thiopyrano[2,3-d]thiazoles incorporating arylsulfonate moiety as potential inhibitors of tubulin polymerization, and molecular modeling studies. J. Mol. Struct. 2022, 1258, 132848. 10.1016/j.molstruc.2022.132648. [DOI] [Google Scholar]
- Mase N.; Horibe T. Organocatalytic Knoevenagel condensations by means of carbamic acid ammonium salts. Org. Lett. 2013, 15, 1854–1857. 10.1021/ol400462d. [DOI] [PubMed] [Google Scholar]
- Kryshchyshyn A.; Roman O.; Lozynskyi A.; Lesyk R. Thiopyrano[2,3-d]thiazoles as new efficient scaffolds in medicinal chemistry. Sci. Pharm. 2018, 86, 26. 10.3390/scipharm86020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozynskyi A.; Matiychuk V.; Karpenko O.; Gzella A. K.; Lesyk R. Tandem hetero-Diels–Alder-hemiacetal reaction in the synthesis of new chromeno[4′,3′:4,5] thiopyrano[2,3-d]thiazoles. Heterocycl. Commun. 2017, 23, 1–5. 10.1515/hc-2016-0176. [DOI] [Google Scholar]
- Li P.; Zhou L.; Zhao T.; Liu X.; Zhang P.; Liu Y.; Zheng X.; Li O. Caspase-9: structure, mechanisms and clinical application. Oncotarget. 2017, 8, 23996–24008. 10.18632/oncotarget.15098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaritsa I. D.; Baranov S. N.; Grischuk A. P. 4-Thiazolidines, derivatives and analogs. Chem. Heterocycl. Compd. 1967, 3, 533–534. 10.1007/BF00481594. [DOI] [Google Scholar]
- Kiro M. A.; Abdel-Rahman M. O.; Gadalla K. Z. The vilsmeier-haack reaction-III-cyclization of hydrazones to pyrazoles. Tetrahedron Lett. 1696, 2, 109–110. [Google Scholar]
- Cava M. P.; Deana A. A.; Muth K.; Mitchell M. N-Phenylmaleimides. Org. Synth. 1961, 41, 93–95. [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Denizot F.; Lang R. Rapid colorimetric assay for cell growth and survival. J. Immunol. Methods 1986, 89, 271–277. 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Ozdemir H. O.; Cetinkaya R. Effects of glycation on erythrocyteanhydrase-I and II patients with diabetes mellitus. Turk. J. Med. Sci. 2000, 30, 135–141. [Google Scholar]
- Hilvo M.; Baranauskiene L.; Salzano A. M.; Scaloni A.; Matulis D.; Innocenti A.; Scozzafava A.; Kulomaa S. M.; Nordlund H. R.; Supuran C. T.; Parkkila S. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J. Biol. Chem. 2008, 283, 27799–27809. 10.1074/jbc.M800938200. [DOI] [PubMed] [Google Scholar]
- Burnett W. N. Western blotting: electrophoretic transfer of protein from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 1981, 112, 195–203. 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Sambrook J.; Fritsch E. F.; Maniatis T.. Molecular cloning: A laboratory manual; Cold Spring Harbor Laboratory: New York. 1989. [Google Scholar]
- C. C. Group , Chemical Computing Group and Molecular Networks Announce the Integration of CORINA into MOE; C. C. Group; 2007. [Google Scholar]
- Chrysanthopoulos P. K.; Mujumdar P.; Woods L. A.; Dolezal O.; Ren B.; Peat T. S.; Poulsen S. A. Identification of a New Zinc Binding Chemotype by Fragment Screening. J. Med. Chem. 2017, 60, 7333–7349. 10.1021/acs.jmedchem.7b00606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.