

ABSTRACT
Mycobacterium tuberculosis (Mtb) is a bacterium that exclusively resides in human hosts and remains a dominant cause of morbidity and mortality among infectious diseases worldwide. Host protection against Mtb infection is dependent on the function of immunity-related GTPase clade M (IRGM) proteins. Polymorphisms in human IRGM associate with altered susceptibility to mycobacterial disease, and human IRGM promotes the delivery of Mtb into degradative autolysosomes. Among the three murine IRGM orthologs, Irgm1 has been singled out as essential for host protection during Mtb infections in cultured macrophages and in vivo. However, whether the paralogous murine Irgm genes, Irgm2 and Irgm3, play roles in host defense against Mtb or exhibit functional relationships with Irgm1 during Mtb infection remains undetermined. Here, we report that Irgm1−/− mice are indeed acutely susceptible to aerosol infection with Mtb, yet the additional deletion of the paralogous Irgm3 gene restores protective immunity to Mtb infections in Irgm1-deficient animals. Mice lacking all three Irgm genes (panIrgm−/−) are characterized by shifted lung cytokine profiles at 5 and 24 weeks postinfection, but control disease until the very late stages of the infection, when panIrgm−/− mice display increased mortality compared to wild-type mice. Collectively, our data demonstrate that disruptions in the balance between Irgm isoforms is more detrimental to the Mtb-infected host than total loss of Irgm-mediated host defense, a concept that also needs to be considered in the context of human Mtb susceptibility linked to IRGM polymorphisms.
KEYWORDS: IRGM, host genetics, inflammation, tuberculosis
INTRODUCTION
Interest in cell-autonomous immune mechanisms that act downstream of interferon (IFN) signaling led to the discovery of four IFN-responsive families of dynamin-like GTPase proteins (1). Among these, the immunity-related GTPases (IRGs) have been implicated in host resistance to many intracellular pathogens, including Toxoplasma gondii, Listeria monocytogenes, Mycobacterium tuberculosis (Mtb), Salmonella typhimurium, and Chlamydia trachomatis in mouse models of infection (2). C57BL/6 mice possess 21 IRG genes, which are divided into two subclasses (“GMS” or “GKS”) based on the amino acid sequence encoded in the G1 motif of their N-terminal GTP-binding domains (3). The genome of C57BL/6 mice contains three GMS genes (Irgm1, Irgm2, and Irgm3). In contrast, through a series of fascinating evolutionary events, IRG genes have mostly been lost from the human genome, leaving IRGM as the only known homologue of the murine GMS genes. It is expressed as five different splice variants (IRGMa–e) (3). Despite its high degree of similarity, IRGM was initially presumed to be a pseudogene due to its truncated GTP-binding domain and lack of IFN-dependent expression (4). However, subsequent studies have identified protective functions for IRGM in autoimmunity or immune responses to infection via its intersection with the autophagy pathway (5 to 9). What factors have driven the differential expansion and deletion of IRG genes between mice and humans, and what the relative fitness costs or benefits of retaining or losing the IRG system are, remain intriguing questions (10). Studies that expand our understanding of how the IRG system functions in mice during infection with diverse pathogens simultaneously offer useful points of comparison for examining the function of IRGM in humans.
Mtb is a facultative intracellular bacterium that causes the death of ~1.4 million people worldwide annually (11). Mtb has coevolved with humans for thousands of years and is adept at manipulating the immune responses of macrophages, its primary host cell niche (12, 13). Multiple studies have proposed that Irgm1 is important for host protection during mycobacterial infection. It was previously shown that mice lacking Irgm1 exhibit extensive lung damage associated with large lesions, are unable to control Mtb burden, and rapidly succumb to aerosol infection (14). Similarly, Irgm1−/− mice intravenously infected with either Mtb or Mycobacterium avium survive through the acute stage of infection but cannot successfully control bacterial growth and die by ~8 to 16 weeks postinfection (14, 15). The human ortholog, IRGM, has also been linked to control of mycobacterial infection. Various polymorphisms in IRGM are associated with increased or decreased risk of active pulmonary TB; however, these associations may be host population and bacterial strain dependent (16 to 21). Interestingly, in a cohort of the Han population of Hubei Province, China, there was a direct relationship between a variant haplotype (−1208A/−1161C/−947T) that decreased transcriptional activity of the IRGM promoter, reduced IRGM expression in patient peripheral blood mononuclear cells (PBMCs), and increased risk of pulmonary TB disease (19). Conversely, a haplotype (−1208A/−1161C/−947C) that increases IRGM transcription was associated with reduced TB disease risk in two Chinese cohorts (17, 19). Proposed explanations for the requirement of Irgm1/IRGM during mycobacterial infection include promotion of optimal macrophage phagolysosome function via autophagy, or prevention of IFN-γ-dependent death of T cells that results in severe lymphopenia (5, 14, 15, 22).
Observations across studies that examined mice with additional Irgm deficiencies indicate that the three genes (Irgm1, Irgm2, and Irgm3) have nonredundant functions and complex interregulatory relationships, as evidenced by mice displaying differential susceptibilities to infection with various intracellular pathogens depending on which Irgm genes are inactivated (1). For example, Irgm1−/− mice exhibit dysregulated host protection during infection with S. typhimurium, but this phenotype can be partially or entirely countered when mice are deficient in both Irgm1 and Irgm3 (Irgm1/3−/−) (23). Mice infected with T. gondii or C. trachomatis on the other hand require Irgm3 expression for host protection (24). Although Irgm-deficient mice are universally susceptible to T. gondii, Irgm2 and Irgm1/3 have differential roles in the cell-autonomous response to infection, regulating the recruitment of distinct effectors to the parasitophorous vacuole (2, 25). However, whether Irgm2 and Irgm3 exhibit functional interactions with Irgm1 that significantly influence the host response during mycobacterial infection is not yet established. In this work, we investigated if mice deficient in both Irgm1 and Irgm3, or the full repertoire of IRGM proteins, exhibit differences in disease progression during Mtb infection. We show that mice deficient in both Irgm1 and Irgm3 are not susceptible to Mtb, exhibiting a rescue phenotype compared to Irgm1-deficient mice. We also demonstrate that despite significant changes in the levels of certain disease-associated cytokines in their lungs, mice deficient in all three IRGM proteins show the same level of host protection as wild-type mice until almost 1 year following infection. Therefore, disruption of the interregulatory relationships between functionally divergent Irgm isoforms is a key driver of susceptibility to Mtb infection in mice.
RESULTS
Irgm1−/− mice are acutely susceptible to infection with Mtb.
To investigate the significance of IRGM proteins in host protection during Mtb infection, we first sought to recapitulate the established observation that Irgm1−/− mice are highly susceptible to Mtb (14, 15). Wild-type (WT) C57BL/6 and Irgm1−/− mice were infected with a low dose of Mtb strain H37Rv by the aerosol route. IFN-γ receptor knockout (IFNγR−/−) mice were included as a control to represent the complete loss of downstream IFN-γ signaling. Consistent with the results of previously published studies, the bacterial burden in Irgm1−/− lungs was significantly higher (~1.2 log10 CFU, P < 0.01) than WT at 5 weeks postinfection (Fig. 1A). Similarly, the bacterial burden in the spleens of Irgm1−/− mice was increased (~1 log10 CFU, P < 0.01) relative to WT (Fig. 1B). In a separate survival experiment, mice were infected with a low dose of Mtb by the aerosol route. All Irgm1−/− mice and IFNγR−/− mice succumbed to Mtb infection by 6 weeks postinfection, while WT mice survived beyond 150 days postinfection (Fig. 1C). Taken together, these data corroborate previously published results and indicate that mice lacking Irgm1 are acutely susceptible to Mtb, exhibiting uncontrolled bacterial burden and early death (14, 15).
FIG 1.
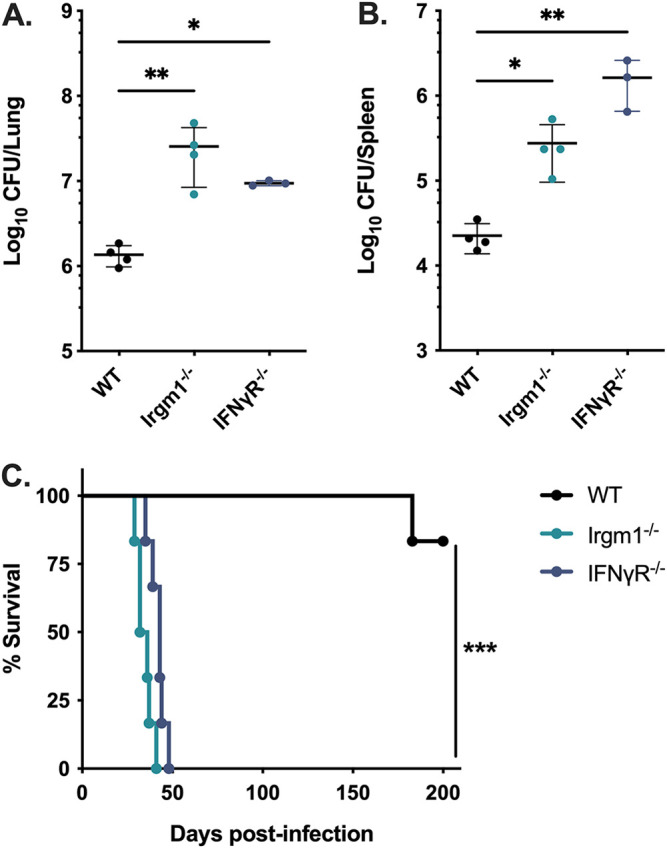
Mice lacking Irgm1 rapidly succumb to pulmonary Mtb infection. WT, Irgm1−/−, and IFNγR−/− mice were infected with Mtb H37Rv by the aerosol route in a single batch (day 1, 50 to 200 CFU). Lungs and spleens were collected at 5 weeks postinfection and used to quantify bacterial CFU. (A) Bacterial burden in the lungs and (B) spleens of mice. Each point represents a single mouse; data are from one experiment, with 4 male mice per group, and are representative of two similar experiments. Statistics were determined via Kruskal-Wallis test by ranks and Dunn’s multiple-comparison test (*, P < 0.05; **, P < 0.01). (C) WT, Irgm1−/−, and IFNγR−/− mice were infected with Mtb H37Rv by the aerosol route in a single batch (day 1, 50 to 150 CFU), and their relative survival was quantified (Mantel-Cox test; ***, P < 0.001). Data are from one experiment with 6 male mice per group and are representative of two similar experiments.
Host protection against Mtb is restored in mice deficient in both Irgm1 and Irgm3.
While Irgm1-deficient mice are highly susceptible to Mtb infection, the role of the two remaining IRGM proteins (IRGM2 and IRGM3) in host protection remains unclear. Because murine IRGM proteins exhibit nonredundant functions in host protection during other infections, we tested whether knocking out Irgm3 in an Irgm1−/− background (Irgm1/3−/−) alters host susceptibility to Mtb. In contrast with Irgm1−/− hosts, mice deficient in both Irgm1 and Irgm3 controlled Mtb burden in the lungs (Fig. 2A) and spleens (Fig. 2B), similar to WT mice at 4 weeks postinfection. Moreover, Irgm1/3−/− mice maintained control of Mtb burden up to 100 days postinfection (Fig. S1A and B in the supplemental material). Additionally, Irgm1/3−/− mice exhibited no survival defect relative to WT mice up to 200 days postinfection with Mtb (Fig. 2C). These results demonstrate that knocking out Irgm3 in Irgm1-deficient mice rescues control of Mtb burden and restores long-term survival.
FIG 2.
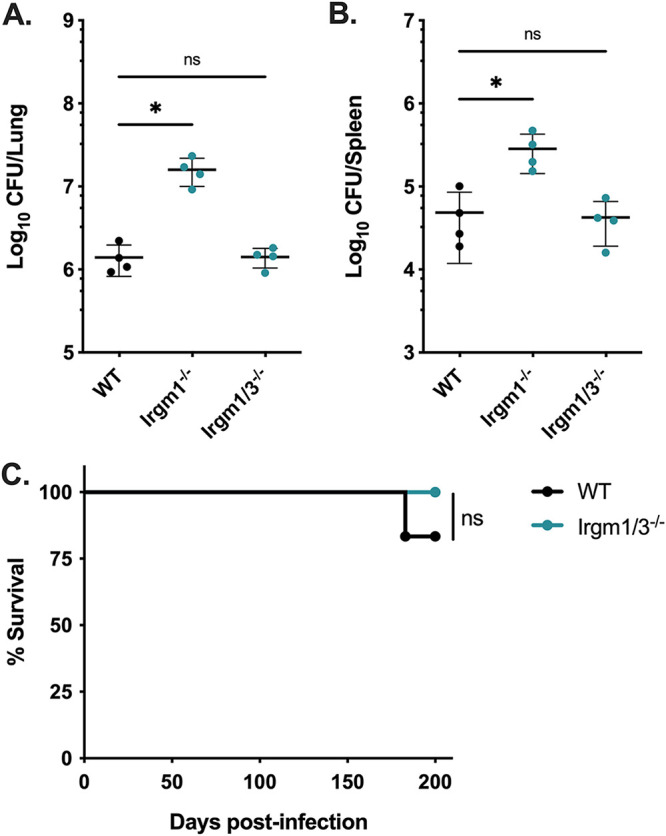
Mice deficient in both Irgm1 and Irgm3 are not susceptible to Mtb infection. WT, Irgm1−/−, and Irgm1/3−/− mice were infected with Mtb H37Rv by the aerosol route in a single batch (day 1, 50 to 150 CFU). Lungs and spleens were collected at 4 weeks postinfection and used to quantify bacterial CFU. (A) Bacterial burden in the lungs and (B) spleens of mice. Each point represents a single mouse; data are from one experiment, with 4 female mice per group, and are representative of 4 similar experiments. Statistics were determined via Kruskal-Wallis test by ranks and Dunn’s multiple-comparison test (*, P < 0.05; ns, not significant). (C) WT or Irgm1/3−/− mice were infected with Mtb H37Rv by the aerosol route in the same batch as Fig. 1C (day 1, 50 to 150 CFU), and their relative survival was quantified (Mantel-Cox test; ns, not significant). Data are from one experiment with 6 male mice per group and are representative of 3 similar experiments.
panIrgm−/− mice maintain control at 1 month postinfection with Mtb.
Although Irgm1/3−/− mice controlled Mtb burden and did not have a survival defect, it remained possible that Irgm2 is required to maintain control in the context of Irgm1/3 deficiency. To determine whether deficiency in the entire Irgm locus increases susceptibility to Mtb after the onset of Th1 immunity, we infected panIrgm−/− (Irgm1−/− Irgm2−/− Irgm3−/−) mice with Mtb H37Rv by the aerosol route and compared their susceptibility to WT mice. At 5 weeks postinfection, the bacterial loads in the lungs and spleens of panIrgm−/− mice were not significantly different from WT mice (Fig. 3A and B). Lung sections from each group of mice were stained with hematoxylin and eosin (H&E) and used to estimate the degree of tissue damage (Fig. S2A). Consistent with the CFU data, the relative area of lung damage in WT and panIrgm−/− mice was comparable (Fig. 3C and D). These results contrast sharply with the phenotype of Irgm1−/− mice following Mtb infection reported in the literature and in our experiments, where lung CFU are between 1 and 2-log10 higher than WT (Fig. 1) and more than 70% of the air space is obstructed by lesions at 4 to 5 weeks postinfection (14). Altogether, our results suggest that the full repertoire of mouse IRGM proteins is dispensable for host resistance at 1-month postinfection.
FIG 3.

Mtb disease phenotypes in panIrgm−/− mice at 5 weeks postinfection. WT or panIrgm−/− mice were infected with H37Rv Mtb by the aerosol route in a single batch (day 1, 200 to 250 CFU). At 5 weeks postinfection, samples were collected from the lungs and spleens for phenotyping disease susceptibility and cytokines. (A) Bacterial burden in the lungs and (B) spleens of mice. (C) Relative area of damaged tissue and (D) representative images from formalin-fixed paraffin embedded lung sections that were H&E stained and used for damage quantification. (E) Concentration of cytokines (M-CSF, CXCL2, TNFα, or CCL5) in lung homogenates from infected mice. Each point represents a single mouse; data are from one experiment, with 4 to 6 male mice per group. Statistics were determined via Mann-Whitney test (*, P < 0.05; ns, not significant).
Next, we characterized the cytokine profile in the lungs of panIrgm−/− mice at 5 weeks postinfection relative to WT mice. Analysis of a 32-plex cytokine array performed on lung homogenates revealed that panIrgm−/− lungs contained significantly less M-CSF, CXCL2, TNFα, and CCL5 (P < 0.05) than WT mice (Fig. 3E). Importantly, the levels of IFN-γ were not significantly different between WT and panIrgm−/− lungs (Fig. S2B). Taken together, we conclude that the full repertoire of IRGM proteins in mice significantly influences a suite of cytokine responses in the lung early after the onset of adaptive immunity, but without substantially altering disease susceptibility at this stage of infection.
panIrgm−/− mice exhibit higher bacterial burden and altered cytokines during late-stage Mtb infection.
To investigate disease progression in the panIrgm−/− mice, we measured disease metrics at 24 weeks postinfection. We found a slight but significant (~0.4 Log10; P < 0.05) increase in lung CFU of panIrgm−/− mice relative to WT (Fig. 4A). There was no significant difference in the bacterial burden in the spleens (Fig. 4B). The relative area of damaged lung tissue was estimated from H&E-stained tissue sections, and there was no statistically significant difference between WT and panIrgm−/− mice (Fig. 4C and D; Fig. S3B). However, we noted that despite little variability in lung CFU numbers among the panIrgm−/− mice, the degree of lung tissue damage was more variable among the panIrgm−/− mice. Examples of both highly damaged and minimally damaged lungs relative to WT were present among panIrgm−/− sections (Fig. 4D).
FIG 4.

panIrgm−/− mice bacterial burden and cytokine response at 24 weeks postinfection. WT or panIrgm−/− mice were infected with H37Rv Mtb by the aerosol route in the same batch as Fig. 3 (day 1, 200 to 250 CFU). At 24 weeks postinfection, samples were collected from the lungs and spleens for phenotyping disease susceptibility. (A) Bacterial burden in the lungs and (B) spleens of mice. (C) Relative area of damaged tissue and (D) representative images from formalin-fixed paraffin-embedded lung sections that were H&E stained and used for damage quantification. Examples of relatively highly damaged (“panIrgm−/− high”) and minimally damaged (“panIrgm−/− low”) lungs are shown. (E) Cytokines were quantified by multiplex ELISA. Shown are concentrations of select cytokines in lung homogenates from infected mice. Each point represents a single mouse; data are from one experiment, with 4 or 5 female mice per group. Statistics were determined via Mann-Whitney test (*, P < 0.05, or exact P value shown for trends above significance threshold; ns, not significant).
Because panIrgm−/− mice demonstrated altered bacterial burden in the lungs at 24 weeks, we additionally investigated the cytokine response at this time point. Here, we observed a significant decrease in CCL5 (P < 0.05) and a trend toward decreased CXCL2 (P = 0.0635) in panIrgm−/− lungs (Fig. 4E). IFN-γ levels were unaffected by the loss of all three IRGM proteins (Fig. S3A). When we analyzed the remaining cytokines in the 32-plex panel, we identified several that were associated with the loss of IRGM proteins at the 24-week time point. Specifically, IL-10 was significantly (P < 0.05) decreased in panIrgm−/− mice relative to WT mice, while CXCL1 was significantly (P < 0.05) increased (Fig. 4E). Overall, our results indicate that the loss of all Irgm genes shifts the abundance of select cytokines in the lungs of mice during chronic disease.
panIrgm−/− mice show impaired survival following Mtb infection.
Considering panIrgm−/− mice showed elevated lung bacterial burden and altered cytokines at 24 weeks postinfection, we investigated the overall survival of WT and panIrgm−/− mice over the course of infection. panIrgm−/− mice began to die earlier (P < 0.01), with less than a 40% chance of survival by 52 weeks postinfection compared to ~90% survival in WT mice (Fig. 5). Thus, the loss of all IRGM proteins impacts overall disease susceptibility to Mtb but does so very late in the course of disease in mice.
FIG 5.
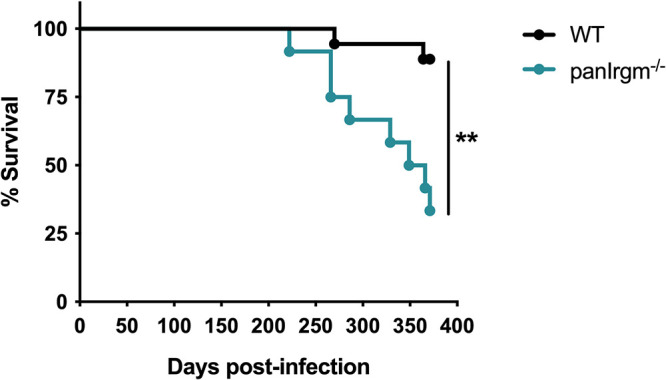
panIrgm−/− mice survival phenotype after long-term Mtb infection. WT or panIrgm−/− mice were infected with H37Rv Mtb by the aerosol route in the same batch as Fig. 3 and 4 (day 1, 200 to 250 CFU). Relative survival of WT and panIrgm−/− mice following Mtb infection by the aerosol route (Mantel-Cox test; **, P < 0.01). Data are from one experiment with 12 to 18 male mice per group.
DISCUSSION
Cell-intrinsic immunity is a fundamental mechanism through which hosts defend against a variety of pathogens, including viruses, bacteria, and parasites. IFN-γ is vital in coordinating cell-autonomous immune responses, yet the context-dependent mechanisms by which downstream IRGs facilitate host protection are incompletely understood. Comparing the functions of human and murine IRGM proteins in the context of different pathogens and during autoimmune disease continues to provide new insights into the ways these proteins regulate IFN-γ-dependent immune responses (26). In this work, we examined whether functional relationships among Irgm1, Irgm2, and Irgm3 genes significantly influence host protection in the context of early (4 to 5 weeks) and late (24 weeks) stages of Mtb infection in mice. Our results suggest that a balance between IRGM proteins is required to effectively protect against TB.
Similar to previous studies, we found that Irgm1−/− mice succumb to Mtb infection rapidly with uncontrolled bacterial replication (14, 15). Various interrelated explanations for the extreme susceptibility of Irgm1−/− mice to Mtb have been proposed, including defective phagosome maturation, impaired autophagosome biogenesis and/or delivery of mycobacteria to late endosome/lysosome compartments, and dysregulated T-cell survival (5, 8, 14, 15). However, the contribution of each of these mechanisms to the loss of host protection remains a matter of debate and is dependent on the pathogen. For example, Irgm1−/− mice are also susceptible to S. typhimurium, L. monocytogenes, C. trachomatis, and T. gondii infection (26). During T. gondii infection, Irgm1 contributes to cell-autonomous control of the pathogen by regulating activation of GKS IRG proteins and their accumulation on the parasitophorous vacuole membrane, ultimately coordinating destruction of the pathogen (27). In the context of mycobacterial and listerial infections, it was initially proposed that IRGM1 is critical for phagocytosis, directly localizing to the phagosome membrane to promote maturation (14, 28, 29). However, experiments using improved antibodies and more extensive controls to dissect IRGM1 localization during mycobacterial and listerial infection in vitro could not confirm that IRGM1 directly associates with the phagosomal membrane (30).
In the context of both IRGM1 and IRGM3, studies examining the simultaneous loss of both proteins discovered that the balance of different IRGM proteins modulates disease outcome in a pathogen-dependent manner. While the susceptibility of Irgm1−/− mice to S. typhimurium is reversed in Irgm1/3−/− mice, the susceptibility remains during infection with C. trachomatis and T. gondii, suggesting distinct mechanisms of protection that are dependent on the pathogen (23, 24). We observed that at the onset of adaptive immunity, Mtb infection resembles S. typhimurium, with the rapid disease progression of Irgm1−/− mice being entirely reversed in Irgm1/3−/− mice that survive far into the chronic phase of disease with no changes in bacterial control. Mtb infection perturbs mitochondrial functions in macrophages, and even in uninfected Irgm1-deficient mice macrophages exhibit defective mitochondrial quality control (31, 32). Taken together, this suggests that rather than directly controlling Mtb replication in macrophages, a balance between IRGM1 and IRGM3 may be required to maintain disease tolerance by regulating mitochondrial functions, and that loss of this balance is the source of susceptibility to Mtb in Irgm1−/− mice.
How IRGM1 and IRGM3 together regulate the overall host response during Mtb infection is not entirely clear and may be multifactorial. Taken together, past studies indicate that IRGM functions overlap both autophagy and type I IFN pathways; importantly, both of these are previously proposed correlates of TB disease progression (32 to 35). Specifically, previous studies indicate that the loss of murine Irgm1 or human IRGM is consistently associated with defects in autophagy during infection. This autophagy dysfunction drives a shift toward proinflammatory metabolism, increased inflammasome activation, and death of proliferating T cells that results in lymphopenia (36, 37). As mentioned above, the loss of Irgm1 also affects mitochondrial quality control, which leads to exacerbated type I IFN responses in macrophages (32). Our results highlight the importance of Irgm balance in controlling protective responses, independent of bacterial replication, but the underlying mechanisms driving unbalanced Irgm responses remain to be understood. Given the complex roles of type I IFNs in TB disease (6, 34), it will be important to confirm how the balance of Irgm1 and Irgm3 in mice affects type I IFN responses during Mtb infection, and whether this is related to mistargeting of GKS proteins in immune cells (32). Although detailed mechanistic analysis suggests that IRGM1 does not directly localize to the mycobacterial-containing vacuole and is not likely a direct effector at the phagosome, it remains possible that the loss of Irgm1 leads to increased IRGM3 activity and dysfunction in maintaining stability of intracellular membrane compartments (30).
Given the clear genetic interactions between Irgm1 and Irgm3 during Mtb infection, it was also important to consider the role Irgm2 plays in TB susceptibility using a mouse lacking all three IRGM proteins (panIrgm−/−). We observed that panIrgm−/− mice showed no changes to bacterial burden or lung damage early after the initiation of adaptive immunity. However, there were some significant differences in cytokine abundance in the lungs. At a much later stage of disease, panIrgm−/− mice displayed marginally increased bacterial burden in the lungs and more variable tissue damage, and ultimately died earlier than wild-type animals. The late presentation of a survival defect in panIrgm−/− mice, following over 300 days of infection, further suggests that IRGM proteins do not strongly contribute to direct control of Mtb replication, as proposed previously. Instead, it is more likely that the IRGM proteins are involved in controlling the inflammatory environment and tissue damage in the lungs, which appears to be critically important during very late stages of infection and/or advanced host age. Of the cytokines examined, we consistently observed changes in chemokines that drive immune cell recruitment into the infected lung environment, including CXCL1, which is a known correlate of TB disease severity in diverse mice and humans (38 to 43). Fully understanding how IRGM proteins balance the function of each other will require further investigation into mice lacking either IRGM2 or IRGM3 alone or in combination with each other. It is possible that loss of these regulators will modulate inflammation during Mtb infection. For example, loss of IRGM2 has been associated with disrupted inflammatory networks in models of systemic sepsis (44, 45).
The late emergence of a survival defect in panIrgm−/− mice during Mtb infection contrasts sharply with T. gondii infection, where panIrgm−/− mice survive poorly, at a frequency only slightly higher than Irgm1−/− or Irgm1/3−/− mice (23, 25). Our data clearly indicate a need for IRGM proteins very late during infection. Age is a factor known to contribute to TB disease susceptibility, and changes during aging, including lower nutrition and immunosuppression or immune dysregulation, function via pleotropic mechanisms (46, 47). Exemplifying the phenomenon of “inflammaging,” IL-12, TNFα, and IL-1β increase in the lungs as the host ages, yet cause and effect mechanisms for this are difficult to isolate. Whether IRGM proteins are required to maintain lung protection, perhaps contributing to the regulation of inflammatory responses, as the host ages is an important hypothesis to consider. How our results relate to human IRGM functions also remains to be determined. For example, it is unclear whether IRGM polymorphisms that have been identified as correlates of TB control or progression alter cell-autonomous control of Mtb in human macrophages, and/or have pleiotropic effects on the immune response to Mtb via autophagy and metabolism.
In conclusion, in this work we have begun to address the need for a balance of IRGM proteins during Mtb infection in mice for long-term protection. Irgm1 is necessary for early control of Mtb infection in mice when it is lost individually, but the imbalance created by the loss of Irgm1 can be repaired by eliminating Irgm3 or all IRGM proteins. However, as the infection progresses to a very late stage in an aging host, the loss of all IRGM proteins becomes detrimental to host survival and is associated with early death.
MATERIALS AND METHODS
Ethics statement.
Mouse studies were performed in strict accordance using the recommendations from the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the Office of Laboratory Animal Welfare. Mouse studies were performed using protocols approved by the Institutional Animal Care and Use Committee (IACUC) for each institution, in a manner designed to minimize pain and suffering in Mtb-infected animals. IACUC numbers for each institution include the University of Massachusetts Medical School (A3306-01); Duke University (A221-20-11); and Michigan State (PROTO202200127). Any animal that exhibited severe disease signs was immediately euthanized in accordance with IACUC-approved endpoints.
Mouse strains and infection with Mtb.
WT C57BL/6J mice were purchased from The Jackson Laboratory (number 000664). panIrgm−/− mice were generated by the lab of Jörn Coers at Duke University as described previously (44). All mice were housed in a specific pathogen-free facility under standard conditions (12 h light/dark, food and water ad libitum). Mice were infected with Mtb between 8 and 12 weeks of age with the H37Rv strain of Mtb (PDIM positive). For aerosol infections, Mtb was cultured in 7H9 media supplemented with oleic acid-albumin-dextrose-catalase OADC enrichment (Middlebrook) and 0.05% Tween 80 (Fisher). Prior to all in vivo infections, Mtb cultures were washed, resuspended in phosphate-buffered saline (PBS) containing 0.05% Tween 80, and sonicated or filtered through a 40-μM filter to generate a single-cell suspension. For infections of WT, Irgm1−/−, and Irgm1/3−/− mice, an inoculum between 50 and 200 CFU was delivered by an aerosol-generating Glas-Col chamber. For WT versus panIrgm−/− infections, an inoculum of ~200 to 250 CFU was delivered by an aerosol-generating Madison chamber (University of Wisconsin at Madison) to the groups of mice as indicated. To determine the inoculation dose, 5 mice were euthanized at 1 day postinfection and CFU were enumerated from lung homogenates, as described below.
Bacterial burden quantification.
At 1 day, 4 or 5 weeks, or 24 weeks postinfection, mice were euthanized by overdose with isoflurane (Covetrus), and the spleens and lungs were removed aseptically. For enumeration of viable bacteria, organs were individually homogenized in PBS-Tween 80 (0.05%) by bead beating (MP Biomedical), and 10-fold dilutions were plated on 7H10 agar (Middlebrook) plates containing OADC enrichment (Middlebrook) and 50 μg/mL Carbenicillin, 10 μg/mL Amphotericin B, 25 μg/mL Polymyxin B, and 20 μg/mL Trimethoprim (Sigma). Plates were incubated at 37°C for 3 to 4 weeks, and individual colonies were enumerated to calculate CFU. The mice euthanized at 1 day postinfection were used to determine the infection dose for each experiment, reported as the range of CFU among 5 individual day-1 mice.
Lung pathology and estimation of damage.
In parallel with bacterial burden quantification, one lung lobe from each mouse was reserved for histology and fixed in 10% neutral buffered formalin. Lungs were submitted to the Duke University Pathology core facility where they were paraffin embedded, sectioned at 5 μM, and stained with hematoxylin and eosin. Lung sections were imaged at ×10 magnification and stitched into whole-lung images for each mouse (Keyence). The relative area of damaged tissue was estimated using QuPath v0.3.2 (48). For this, a pixel classifier was trained to identify damaged versus undamaged areas of H&E-stained mouse lung sections, based on 132 damaged annotations and 96 undamaged annotations across 11 training images, which were imported to train the ANN_MLP classifier at moderate resolution. The quality of the classifier was visually inspected across diverse samples by overlaying live annotation predictions with each H&E image. The resulting pixel classifier was loaded into QuPath and used to estimate the damaged area of each lung sample, relative to the total lung area (Fig. S1 and S2).
Quantification of cytokines in tissue homogenates.
Murine lung homogenates were centrifuged to remove cellular debris, and the supernatants were filtered through 0.2-μM filters. Thirty-two cytokines/chemokines were quantified via a Discovery Assay (Eve Technologies; MD31). In this assay, Fluorescence Intensity values were measured for all samples and a standard curve with expected concentrations for each cytokine. Observed Concentration values (pg/mL) were then interpolated for each sample and cytokine based on their respective standard curves and Fluorescence Intensity values. Observed Concentration values provided in the Eve Technologies results were used for all downstream analyses. From this cytokine panel, IL-3, IL-4, and IL-12p70 fell below the detectable limit at both time points and were excluded from further analysis.
Statistical analysis.
Statistical analyses were performed using Prism 9 (Graph Pad) software. Bacterial burden, lung damage, and cytokine differences between two groups were analyzed using the Mann-Whitney test. When more than two groups were compared, Kruskal-Wallis test by ranks and Dunn’s multiple-comparison test were selected. Differences in survival were graphed using Kaplan-Meier curves, and statistical significance was assigned via Mantel-Cox testing. Throughout, P value thresholds are noted as ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ACKNOWLEDGMENTS
We thank Greg Taylor for insightful manuscript comments, Summer Harris and Emily Hunt for technical assistance, and members of the Olive, Smith, Sassetti, and Coers labs for helpful feedback. This work was funded by National Institutes of Health grants AI148243 and AI103197 to J.C.; AI132130 to C. M. Sassetti; AI165618 to A.J.O.; and a Whitehead Scholar Award and an NIH Director’s New Innovator Award (GM146458) to C. M. Smith. Biocontainment work was partially performed in the Duke Regional Biocontainment Laboratory, which received partial support for construction from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (UC6-AI058607; G20-AI167200).
Footnotes
Supplemental material is available online only.
Contributor Information
Andrew J. Olive, Email: oliveand@msu.edu.
Clare M. Smith, Email: clare.m.smith@duke.edu.
Sabine Ehrt, Weill Cornell Medicine.
REFERENCES
- 1.Pilla-Moffett D, Barber MF, Taylor GA, Coers J. 2016. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol 428:3495–3513. 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor GA, Feng CG, Sher A. 2007. Control of IFN-gamma-mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases). Microbes Infect 9:1644–1651. 10.1016/j.micinf.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Bekpen C, Xavier RJ, Eichler EE. 2010. Human IRGM gene “to be or not to be.” Semin Immunopathol 32:437–444. 10.1007/s00281-010-0224-x. [DOI] [PubMed] [Google Scholar]
- 4.Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, Glowalla E, Leptin M, Howard JC. 2005. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol 6:R92. 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh SB, Davis AS, Taylor GA, Deretic V. 2006. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313:1438–1441. 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 6.Jena KK, Mehto S, Nath P, Chauhan NR, Sahu R, Dhar K, Das SK, Kolapalli SP, Murmu KC, Jain A, Krishna S, Sahoo BS, Chattopadhyay S, Rusten TE, Prasad P, Chauhan S, Chauhan S. 2020. Autoimmunity gene IRGM suppresses cGAS-STING and RIG-I-MAVS signaling to control interferon response. EMBO Rep 21:e50051. 10.15252/embr.202050051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehto S, Jena KK, Nath P, Chauhan S, Kolapalli SP, Das SK, Sahoo PK, Jain A, Taylor GA, Chauhan S. 2019. The Crohn's Disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol Cell 73:429–445.e7. 10.1016/j.molcel.2018.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Jain A, Choi SW, da Silva GPD, Allers L, Mudd MH, Peters RS, Anonsen JH, Rusten TE, Lazarou M, Deretic V. 2020. Mammalian Atg8 proteins and the autophagy factor IRGM control mTOR and TFEB at a regulatory node critical for responses to pathogens. Nat Cell Biol 22:973–985. 10.1038/s41556-020-0549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petkova DS, Viret C, Faure M. 2012. IRGM in autophagy and viral infections. Front Immunol 3:426. 10.3389/fimmu.2012.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bekpen C, Marques-Bonet T, Alkan C, Antonacci F, Leogrande MB, Ventura M, Kidd JM, Siswara P, Howard JC, Eichler EE. 2009. Death and resurrection of the human IRGM gene. PLoS Genet 5:e1000403. 10.1371/journal.pgen.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.WHO. 2019. Global tuberculosis report 2019. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 12.Liu CH, Liu H, Ge B. 2017. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol 14:963–975. 10.1038/cmi.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brites D, Gagneux S. 2015. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev 264:6–24. 10.1111/imr.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacMicking JD, Taylor GA, McKinney JD. 2003. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science 302:654–659. 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 15.Feng CG, Collazo-Custodio CM, Eckhaus M, Hieny S, Belkaid Y, Elkins K, Jankovic D, Taylor GA, Sher A. 2004. Mice deficient in LRG-47 display increased susceptibility to mycobacterial infection associated with the induction of lymphopenia. J Immunol 172:1163–1168. 10.4049/jimmunol.172.2.1163. [DOI] [PubMed] [Google Scholar]
- 16.Intemann CD, Thye T, Niemann S, Browne EN, Amanua Chinbuah M, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, Helm S, Rusch-Gerdes S, Horstmann RD, Meyer CG. 2009. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5:e1000577. 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Che N, Li S, Gao T, Zhang Z, Han Y, Zhang X, Sun Y, Liu Y, Sun Z, Zhang J, Ren W, Tian M, Li Y, Li W, Cheng J, Li C. 2010. Identification of a novel IRGM promoter single nucleotide polymorphism associated with tuberculosis. Clin Chim Acta 411:1645–1649. 10.1016/j.cca.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Bahari G, Hashemi M, Taheri M, Naderi M, Eskandari-Nasab E, Atabaki M. 2012. Association of IRGM polymorphisms and susceptibility to pulmonary tuberculosis in Zahedan, Southeast Iran. Scientific World Journal 2012:950801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan L, Ke Z, Ma J, Guo Y, Li Y. 2016. IRGM gene polymorphisms and haplotypes associate with susceptibility of pulmonary tuberculosis in Chinese Hubei Han population. Tuberculosis (Edinb) 96:58–64. 10.1016/j.tube.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Xie H, Li C, Zhang M, Zhong N, Chen L. 2017. Association between IRGM polymorphisms and tuberculosis risk: a meta-analysis. Medicine (Baltimore) 96:e8189. 10.1097/MD.0000000000008189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang D, Chen J, Shi C, Jing Z, Song N. 2014. Autophagy gene polymorphism is associated with susceptibility to leprosy by affecting inflammatory cytokines. Inflammation 37:593–598. 10.1007/s10753-013-9773-1. [DOI] [PubMed] [Google Scholar]
- 22.Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, Komatsu M, Deretic V. 2010. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 12:1154–1165. 10.1038/ncb2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry SC, Daniell XG, Burroughs AR, Indaram M, Howell DN, Coers J, Starnbach MN, Hunn JP, Howard JC, Feng CG, Sher A, Taylor GA. 2009. Balance of Irgm protein activities determines IFN-γ-induced host defense. J Leukoc Biol 85:877–885. 10.1189/jlb.1008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coers J, Gondek DC, Olive AJ, Rohlfing A, Taylor GA, Starnbach MN. 2011. Compensatory T cell responses in IRG-deficient mice prevent sustained Chlamydia trachomatis infections. PLoS Pathog 7:e1001346. 10.1371/journal.ppat.1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dockterman J, Fee BE, Taylor GA, Coers J. 2021. Murine Irgm paralogs regulate nonredundant functions to execute host defense to Toxoplasma gondii. Infect Immun 89:e0020221. 10.1128/IAI.00202-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunn JP, Feng CG, Sher A, Howard JC. 2011. The immunity-related GTPases in mammals: a fast-evolving cell-autonomous resistance system against intracellular pathogens. Mamm Genome 22:43–54. 10.1007/s00335-010-9293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, Frickel EM, Valdivia RH, Coers J. 2013. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog 9:e1003414. 10.1371/journal.ppat.1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tiwari S, Choi HP, Matsuzawa T, Pypaert M, MacMicking JD. 2009. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P2 and PtdIns(3,4,5)P3 promotes immunity to mycobacteria. Nat Immunol 10:907–917. 10.1038/ni.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shenoy AR, Kim BH, Choi HP, Matsuzawa T, Tiwari S, MacMicking JD. 2007. Emerging themes in IFN-γ-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology 212:771–784. 10.1016/j.imbio.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Springer HM, Schramm M, Taylor GA, Howard JC. 2013. Irgm1 (LRG-47), a regulator of cell-autonomous immunity, does not localize to mycobacterial or listerial phagosomes in IFN-γ-induced mouse cells. J Immunol 191:1765–1774. 10.4049/jimmunol.1300641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patrick KL, Watson RO. 2021. Mitochondria: powering the innate immune response to Mycobacterium tuberculosis infection. Infect Immun 89:e00687-20. 10.1128/IAI.00687-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rai P, Janardhan KS, Meacham J, Madenspacher JH, Lin WC, Karmaus PWF, Martinez J, Li QZ, Yan M, Zeng J, Grinstaff MW, Shirihai OS, Taylor GA, Fessler MB. 2021. IRGM1 links mitochondrial quality control to autoimmunity. Nat Immunol 22:312–321. 10.1038/s41590-020-00859-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paik S, Kim JK, Chung C, Jo EK. 2019. Autophagy: a new strategy for host-directed therapy of tuberculosis. Virulence 10:448–459. 10.1080/21505594.2018.1536598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moreira-Teixeira L, Mayer-Barber K, Sher A, O'Garra A. 2018. Type I interferons in tuberculosis: foe and occasionally friend. J Exp Med 215:1273–1285. 10.1084/jem.20180325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. 2015. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528:565–569. 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coers J, Brown HM, Hwang S, Taylor GA. 2018. Partners in anti-crime: how interferon-inducible GTPases and autophagy proteins team up in cell-intrinsic host defense. Curr Opin Immunol 54:93–101. 10.1016/j.coi.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alwarawrah Y, Danzaki K, Nichols AG, Fee BE, Bock C, Kucera G, Hale LP, Taylor GA, MacIver NJ. 2022. Irgm1 regulates metabolism and function in T cell subsets. Sci Rep 12:850. 10.1038/s41598-021-04442-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmed M, Thirunavukkarasu S, Rosa BA, Thomas KA, Das S, Rangel-Moreno J, Lu L, Mehra S, Mbandi SK, Thackray LB, Diamond MS, Murphy KM, Means T, Martin J, Kaushal D, Scriba TJ, Mitreva M, Khader SA. 2020. Immune correlates of tuberculosis disease and risk translate across species. Sci Transl Med 12:eaay0233. 10.1126/scitranslmed.aay0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, Gatti DM, Kramnik I, Yener B, Gurcan M, Beamer G. 2015. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech 8:1141–1153. 10.1242/dmm.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, Fallert Junecko BA, Reinhart TA, Kolls J, Baez-Saldana R, Cruz-Lagunas A, Rodriguez-Reyna TS, Kumar NP, Tessier P, Roth J, Selman M, Becerril-Villanueva E, Baquera-Heredia J, Cumming B, Kasprowicz VO, Steyn AJ, Babu S, Kaushal D, Zuniga J, Vogl T, Rangel-Moreno J, Khader SA. 2013. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med 188:1137–1146. 10.1164/rccm.201304-0803OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, Cho SN, Via LE, Barry CE, III, 2010. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137:122–128. 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lovewell RR, Baer CE, Mishra BB, Smith CM, Sassetti CM. 2021. Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol 14:229–241. 10.1038/s41385-020-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, Smith CM, Phuah JY, Long JE, Dubuke ML, Palace SG, Goguen JD, Baker RE, Nambi S, Mishra R, Booty MG, Baer CE, Shaffer SA, Dartois V, McCormick BA, Chen X, Sassetti CM. 2017. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol 2:17072. 10.1038/nmicrobiol.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finethy R, Dockterman J, Kutsch M, Orench-Rivera N, Wallace GD, Piro AS, Luoma S, Haldar AK, Hwang S, Martinez J, Kuehn MJ, Taylor GA, Coers J. 2020. Dynamin-related Irgm proteins modulate LPS-induced caspase-11 activation and septic shock. EMBO Rep 21:e50830. 10.15252/embr.202050830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eren E, Planes R, Bagayoko S, Bordignon PJ, Chaoui K, Hessel A, Santoni K, Pinilla M, Lagrange B, Burlet-Schiltz O, Howard JC, Henry T, Yamamoto M, Meunier E. 2020. Irgm2 and Gate-16 cooperatively dampen Gram-negative bacteria-induced caspase-11 response. EMBO Rep 21:e50829. 10.15252/embr.202050829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piergallini TJ, Turner J. 2018. Tuberculosis in the elderly: why inflammation matters. Exp Gerontol 105:32–39. 10.1016/j.exger.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajagopalan S. 2001. Tuberculosis and aging: a global health problem. Clin Infect Dis 33:1034–1039. 10.1086/322671. [DOI] [PubMed] [Google Scholar]
- 48.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW. 2017. QuPath: open source software for digital pathology image analysis. Sci Rep 7:16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download iai.00510-22-s0001.xlsx, XLSX file, 0.03 MB (34.5KB, xlsx)
Fig. S1 to S3. Download iai.00510-22-s0002.pdf, PDF file, 2.3 MB (2.3MB, pdf)