

Abstract
Background
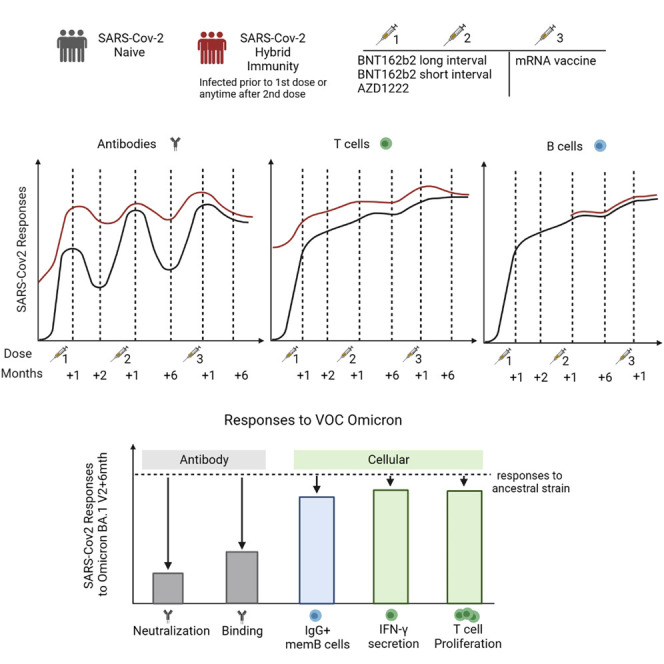
Both infection and vaccination, alone or in combination, generate antibody and T cell responses against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). However, the maintenance of such responses—and hence protection from disease—requires careful characterization. In a large prospective study of UK healthcare workers (HCWs) (Protective Immunity from T Cells in Healthcare Workers [PITCH], within the larger SARS-CoV-2 Immunity and Reinfection Evaluation [SIREN] study), we previously observed that prior infection strongly affected subsequent cellular and humoral immunity induced after long and short dosing intervals of BNT162b2 (Pfizer/BioNTech) vaccination.
Methods
Here, we report longer follow-up of 684 HCWs in this cohort over 6–9 months following two doses of BNT162b2 or AZD1222 (Oxford/AstraZeneca) vaccination and up to 6 months following a subsequent mRNA booster vaccination.
Findings
We make three observations: first, the dynamics of humoral and cellular responses differ; binding and neutralizing antibodies declined, whereas T and memory B cell responses were maintained after the second vaccine dose. Second, vaccine boosting restored immunoglobulin (Ig) G levels; broadened neutralizing activity against variants of concern, including Omicron BA.1, BA.2, and BA.5; and boosted T cell responses above the 6-month level after dose 2. Third, prior infection maintained its impact driving larger and broader T cell responses compared with never-infected people, a feature maintained until 6 months after the third dose.
Conclusions
Broadly cross-reactive T cell responses are well maintained over time—especially in those with combined vaccine and infection-induced immunity (“hybrid” immunity)—and may contribute to continued protection against severe disease.
Funding
Department for Health and Social Care, Medical Research Council.
Keywords: SARS-CoV-2, COVID-19, COVID vaccine, T cells, antibody, immunity
Graphical abstract
Context and significance
There is public concern about waning immunity to SARS-CoV-2. In addition, booster vaccines might be less effective as the virus changes. As such, how measures of immunity to SARS-CoV-2 relate to protection is a key question. Researchers from the PITCH consortium in the UK observed in healthcare workers that antibodies drop after SARS-CoV-2 vaccination; however, T cell responses do not. After a third vaccine dose, the immune response lasted longer and recognized different variants. It also made much less difference which vaccine had been used for the first immunizations, while before the third dose, the response to mRNA vaccines was the strongest. As time goes on, the differences in immune response to the vaccine or the virus even out.
Moore et al. studied antibody and cellular responses to COVID-19 vaccines before and after dose 3. Antibody responses waned, but T cell responses were well maintained. T cells recognized Omicron variants better and for longer than antibodies. Differences due to vaccine regimen and previous infection evened out over time.
Introduction
As vaccines have been deployed to tackle the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, crucial questions have emerged regarding long-term maintenance of protective immunity against disease. The appearance of viral variants leading to successive waves of infection has clearly shown the limits of vaccine protection against infection.1 Despite this, vaccine protection against severe disease has been well maintained across the recent Delta2 and Omicron BA.13 waves. To understand the underlying immune responses that determine these population-level observations, large-scale studies of individuals with high exposure to SARS-CoV-2, such as healthcare workers (HCWs), can provide valuable insights, as has been demonstrated by the SARS-CoV-2 Immunity and Reinfection Evaluation (SIREN) study in the UK.4 , 5 , 6 Protective Immunity from T Cells in Healthcare Workers (PITCH), a study aligned closely with SIREN, is focused on the longitudinal analysis of antiviral T and B cell responses after infection and/or vaccination with BNT162b2 (Pfizer/BioNTech) or AZD1222 (Oxford/AstraZeneca). PITCH has already provided data indicating that the extended interval vaccine regimen for BNT162b2 mRNA vaccine deployed in the UK was associated with enhanced antibody and CD4+ T cell helper responses.7 All immune responses were strongly enhanced by prior SARS-CoV-2 infection.
The long-term impacts of prior exposure, vaccine regimen, and vaccine type have not been fully defined, especially at the level of T cell responses. Characterizing the response to vaccines and infections in healthy people is essential to determine future vaccination policies, while identification of vulnerable non-responders can inform additional interventions, such as extra booster doses of vaccine and/or monoclonal antibody therapies. Correlations with protection from infection at a population level have been observed for binding8 , 9 and neutralizing antibodies.9 , 10 , 11 , 12 , 13 The role of other, non-neutralizing antibody functions, such as antibody-dependent natural killer (NK) cell activity, antibody-dependent phagocytosis, or complement deposition, requires further investigation.14 , 15 , 16 However, monitoring of SARS-CoV-2-specific T cell immunity is also essential, as T cell defense is potentially a key explanation for lower case hospitalization and mortality for the Omicron variant compared with earlier variants,17 despite Omicron’s high mortality in unvaccinated populations.18 T cells are a cornerstone of antiviral defense, orchestrating the immune response, including cytotoxic activity against virally infected cells and optimizing production of antibodies from B cells.19 Macaque20 and human21 , 22 , 23 studies support this key role for T cells in protection against the severe effects of SARS-CoV-2 infection, potentially alongside functional antibody properties beyond neutralization.24 , 25 In some cases, cross-reactive T cells are associated with protection against infection in exposed seronegative groups.26 There is also evidence of SARS-CoV-2-specific cell responses in highly exposed HCWs without seroconversion,27 and expansion of pre-existing RNA-polymerase-specific T cells in seronegative SARS-CoV-2 infection.28
There is a body of emerging data on the waning of antibody responses, especially after the shorter dose interval regimen for BNT162b2.29 , 30 Waning of antibody is associated with loss of protection against infection,6 , 31 whereas protection against severe disease is relatively well maintained.1 , 2 , 3 , 32 , 33 , 34 T cell responses to spike protein post vaccination do not correlate strongly with binding or neutralizing antibody responses.7 Importantly, while antibodies generated in response to vaccination neutralize Omicron much less well than the ancestral strain,35 , 36 the T cell response to SARS-CoV-2 is minimally affected by mutations in the Alpha, Beta, Gamma, and Delta variants of concern,7 , 37 and 75%–85% preserved against the Omicron BA.1 variant.38 , 39 , 40 , 41 , 42 , 43 , 44 Given that, at this point in the pandemic, public health decisions are increasingly being made around limiting severe disease rather than preventing milder infections in the community, having robust data at scale that indicates the trajectory of the T cell responses after different vaccine regimens is of increasing value. The impact of subsequent vaccine dosing on T and B cell responses is additionally a key focus in such decision making.
We previously observed higher anti-spike binding, higher neutralizing antibody responses, and lower spike-specific T cell magnitude but increased interleukin (IL)-2 production 1 month after second dose when BNT162b2 was delivered with a longer dosing interval (median 10 weeks) compared with the licensed shorter (3–4 weeks) interval.7 This pattern was reproduced in an elderly population,45 and the antibody findings have been confirmed in the larger SIREN cohort.46 Evidence of improved vaccine effectiveness with a longer dose interval was reported in a study of two Canadian provinces.47
In the study presented here, our objective was to explore the characteristics of adaptive and humoral immunity following two and three vaccine doses, to consider the longer-term impacts of regimen variation, vaccine type (including the Oxford-AZ ChadOx1-based vaccine), and infection over time. We observed the long term impact of prior infection even after two doses of vaccine, which is consistent with protection documented in SIREN.6 We saw no decline in T cell responses over time regardless of vaccine regimen; this contrasts with waning of both binding and neutralizing antibody (NAb) titers, which remained strongest and broadest in the long-interval BNT162b2 group. The third dose of vaccine boosted binding antibody responses such that differences seen between vaccine regimens after only two doses were reduced, as were differences associated with prior infection. Overall, the data indicate a stable pool of T cell memory is induced and maintained across vaccine types/regimens, consistent with the sustained impact of vaccination with or without prior infection in protection against severe disease.
Results
Participants vaccinated with a primary course and a booster dose of COVID vaccine
We studied 684 participants who had been vaccinated with a primary course of COVID-19 vaccine between 9th December 2020 and 23rd May 2021 (Table 1 and Figure 1 A). In total, 592 participants received a primary course of BNT162b2 vaccine (Pfizer), of whom 84 participants received the second dose of BNT162b2 vaccine after a short (3–5 weeks, median 24 days) interval, and 508 participants received the second dose of BNT162b2 vaccine after an extended (6–17 weeks, median 71 days) interval.7 Ninety-two participants received a primary course of AZD1222 vaccine administered with an interval of 7–23 weeks (median 74 days). The median age of all participants was 43 years (range 22–77 years), and 73.8% of participants were female, reflecting the demographic of HCWs in the UK and consistent with our previous reports and the wider SIREN cohort.
Table 1.
Demographic characteristics of participants in the study
| Total N | All |
AZ |
Pfizer short |
Pfizer long |
pa value |
|---|---|---|---|---|---|
| 684 | 92 | 84 | 508 | NT | |
| Dosing intervals | |||||
| Median days | 71 | 74 | 24 | 71 | NT |
| Median weeks | 10 | 11 | 3 | 10 | NT |
| IQR (days) | 63–77 | 64.75–78 | 21–27 | 66–78 | NT |
| Maximum days | 158 | 158 | 38 | 120 | NT |
| Minimum days | 14 | 53 | 0 | 0 | NT |
| Range (days) | 14–158 | 53:158 | 0:38 | 0:120 | NT |
| Infection status | |||||
| Naive, N (%) | 342 (50.0) | 45 (51.1) | 49 (41.7) | 248 (52.4) | NT |
| Total previous SARS-CoV-2, N (%) | 342 (50.0) | 47 (48.9) | 35 (58.3) | 266 (48.8) | 0.22 |
| Previous infection at baseline, Nb | 269 | 39 | 30 | 200 | NT |
| PCR + breakthrough infections, Nc | 33 | 5 | 4 | 24 | NT |
| Seroconverted during study, Nd | 49 | 6 | 1 | 42 | NT |
| Age | |||||
| Maximum age (years) | 77 | 77 | 71 | 71 | NT |
| Minimum age (years) | 22 | 22 | 22 | 22 | NT |
| Age range (years) | 22–77 | 22–77 | 22–71 | 22–71 | NT |
| Median age (years) | 43 | 43 | 45 | 43 | NT |
| Interquartile age range (years) | 33–52.3 | 27–56 | 37–55 | 33–51.25 | NT |
| Sex | |||||
| Female, N (%) | 505 (73.8) | 68 (73.9) | 50 (59.5) | 387 (76.2) | NT |
| Male, N (%) | 179 (26.2) | 24 (26.1) | 34 (40.5) | 121 (23.8) | 0.006 |
| Ethnicity | |||||
| White, N (%)e | 464 (83.8) | 71 (79.8) | 56 (84.8) | 337 (84.5) | NT |
| Asian, N (%)e | 56 (10.1) | 12 (13.5) | 5 (7.6) | 39 (9.8) | NT |
| Black, N (%)e | 7 (1.3) | 0 (0.0) | 1 (1.5) | 6 (1.5) | NT |
| Other, N (%)e | 27 (4.9) | 6 (6.7) | 4 (6.1) | 17 (4.3) | NT |
| Unreported, N | 130 | 3 | 18 | 109 | NT |
NT, not tested.
Differences between the groups were assessed using the chi-square test.
Previous infection at baseline (time of first vaccination) = previous PCR + SARS-CoV-2 +/− anti-nucleocapsid IgG positive at baseline.
PCR + breakthrough infections include nine re-infections that were in the “previous infection at baseline” group.
Seroconverted during study = no documented PCR+, lateral flow test, or suspected SARS-CoV-2 infection but asymptomatic rise in anti-nucleocapsid IgG (MSD) above assay positivity threshold and >2× baseline.
Percentage using total reported ethnicities as the denominator.
Figure 1.

Study design and T cell and IgG responses 6 months after vaccine dose 2, time course of T cell binding IgG and B cell responses for all participants, and cross section of responses 1 month post dose 3 after two doses of BNT162b2 (short or long interval) or AZD1222 vaccine
(A) Schematic representation of vaccination and phlebotomy time points. Figure created using BioRender.
(B) Association of membrane (M) and nucleocapsid (N) protein-specific T cell and SARS-CoV-2 N-specific IgG responses in participants 6 months after second dose and 28 days after third dose (hence participants can have >1 value) by infection status.
(C) Comparison of IFNγ ELISpot responses to spike (S, ancestral strain) from cryopreserved peripheral blood mononuclear cells (PBMCs) in naive (gray circles) participants 6 months after two doses of BNT162b2 (Pfizer-BioNTech) delivered with a short dosing interval short, 3–5 weeks, n = 33), or a long dosing interval (long, 6–17 weeks, n = 116), or 6 months after two doses of AZD1222 (AstraZeneca) vaccine (AZ, n = 29), or previously infected (closed red circles infected at baseline, open red circles infected during study) BNT162b2 short (n = 13), previously infected BNT162b2 long (n = 94), and AZ (n = 16)-vaccinated individuals.
(D) Effect of vaccine regimen and infection status on SARS-CoV-2 S-specific IgG responses in naive short (n = 38), long (n = 170), and AZ (n = 39) and previously infected short (n = 18), long (n = 99), and AZ (n = 28)-vaccinated individuals.
(E) Effect of vaccine regime and infection status on SARS-CoV-2 RBD-specific IgG responses in naive short (n = 38), long (n = 169), and AZ (n = 37) and previously infected short (n = 18), long (n = 99), and AZ (n = 28)-vaccinated individuals.
(F) Time course comparison of T cell responses to SARS-CoV-2 spike by IFNγ ELISpot assay for all vaccine regimens up to 6 months post third dose (n = 613).
(G) Time course comparison of IgG antibody response to SARS-CoV-2 spike by MSD assay for all vaccine regimens up to 6 months post third dose (n = 680).
(H) Time course comparison of B cell responses to SARS-CoV-2 spike by B cell ELISpot assay for all vaccine regimens up to 1 month post third dose.
(I) Comparison of T cell responses 1 month after the third booster dose by primary vaccine regimen (BNT162b2 short, long, or AZD1222).
(J) Comparison of IgG antibody responses 1 month after the third booster dose by primary vaccine regimen.
(K) Comparison of B cell responses 1 month after the third booster dose by primary vaccine regimen. Gray circles, naive individuals; red circles, hybrid immunity. ELISpot values are expressed as spot-forming units per million (SFU/106) PBMCs. Data displayed are responses to peptide pools representing the sum of S1 and S2 units of S (ancestral strain). IgG responses were measured in serum 6 months after the second dose using multiplexed MSD immunoassays and are shown in arbitrary units (AU)/mL. Bars represent the median. Comparisons within groups were compared with Kruskal-Wallis and Dunn’s multiple comparisons test (C–K) and Spearman’s tests (B), with two-tailed p values shown above linking lines for significant differences with p ≤ 0.05. Where p values are absent, comparison was not statistically significant (p > 0.05). Dashed lines in (B) represent thresholds for a positive response: SARS-CoV-2 N IgG based on the mean concentrations measured in 103 pre-pandemic sera +3 SDs (3,874 AU/mL); SARS-CoV-2 M and N IFNγ ELISpot assay, mean +2 SDs of the DMSO wells across all experiments in the study (33 SFU/106). Unpaired comparisons between naive and hybrid immune time points were tested with the Mann-Whitney test.
Symptomatic infection and asymptomatic anti-nucleocapsid seroconversion were common during the study period
During follow-up of this cohort (May 2021 to March 2022), some participants became infected during the SARS-CoV-2 waves of Delta and Omicron BA.1 or BA.2 (Table 1). Thirty-three participants developed symptomatic COVID-19 confirmed by positive SARS-CoV-2 PCR assay. A further 49 participants had evidence of asymptomatic infection between 1 and 6 months after the second vaccine, reflected by SARS-CoV-2 nucleocapsid (N) antibody seroconversion detected in the 6-month samples. After accounting for those infections, half the cohort (342 participants) met the definition of infection naive at the time of the third vaccination. In addition, 11 participants of 21 followed up to 6 months post third dose became infected with Omicron variants.
We measured T cell responses 6 months after the second vaccine dose and found that participants infected between 1 and 6 months after the second dose had similar T cell responses to those infected prior to their first vaccine dose (Figure S1). Spike immunoglobulin (Ig) G, measured by MesoScale Discovery (MSD), was lower in those infected during the study compared with those infected before vaccination, but was higher than infection-naive participants. Therefore, in this report, participants with natural infection at any point were analyzed together as a “hybrid immunity” group, regardless of when the infection occurred in relation to vaccine doses.
6 months post second vaccination, T cell IFNγ ELISpot responses are greatest following BNT162b2 short dose interval at 6 months and are augmented in participants with hybrid immunity
In infection-naive participants, at 6 months post vaccine dose 2, there was no significant difference in the T cell response by interferon gamma (IFNγ) ELISpot assay between the three primary vaccine groups, although there was a trend toward higher T cell responses in those who received BNT162b2 vaccine with a short interval (median 3 weeks) than those groups who were vaccinated with a BNT162b2 long interval (median 10 weeks), or the group vaccinated with AZD1222 (Figure 1C). This difference was significant for the BNT162b2 short- and long-interval groups 1 month after the second dose.7 Spike-specific T cell responses 6 months after the second vaccination were considerably greater in all groups (105 spot-forming units [SFU]/106 peripheral blood mononuclear cells [PBMCs], interquartile range [IQR] 48–240) than the historical median responses we observed using the same assay in this cohort pre-vaccination in 202015 6 months after wave 1 infection (44 SFU/106 PBMC, IQR 1–107).
For anti-spike binding antibody responses, levels were higher for BNT162b2 recipients than AZD1222 recipients irrespective of the dosing interval (Figure 1D). A similar pattern was apparent for receptor-binding domain (RBD) antibody (Figure 1E). As was observed at 1 month post second dose, T cell and antibody responses were greater in magnitude in those who were previously infected at any point before the 6-month post-second-dose sample was collected (Figures 1C–1E). T cell responses against M and N were, as expected, higher in those with hybrid immunity, and correlated with N antibody levels (Figure 1B).
After a booster (third) vaccine, IFNγ ELISpot T cell responses were equivalent in all groups irrespective of primary vaccine regimen.
Over the 6-month period following the second vaccine dose, T cell IFNγ responses were well maintained, with a modest fall that did not reach statistical significance, and, overall, were boosted significantly after the third dose in both naive and hybrid immune participants (Figure 1F). This apparent boost was accounted for by the largest group, the BNT162b2 long-interval group (Figure S2D), while the other, smaller groups did not achieve statistical significance (Figure S2A and S2G). These responses were well maintained for 6 months following the third dose, with no significant change in T cell response between 1 and 6 months post third dose, although fewer hybrid immune participants were tested at this time point (Figure 1F). One month after the third vaccine dose, participants receiving all three vaccine regimens had equivalent T cell IFNγ responses (Figure 1I). The post-dose-3 boosting effect did not generate T cell responses any higher than those measured 28 days post dose 2, but responses were higher than those measured 6 months post dose 2 value. Thus, all groups derived a detectable benefit on the T cell response from the third vaccine dose.
Infection leads to boosting of IFNγ ELISpot T cell responses following all vaccine regimens
Spike-specific T cell responses were higher in those with hybrid immunity compared with infection naive. This was the case for both BNT162b2-vaccinated groups (Figures S2A and S2D) but was not the case in the AZD1222 group (Figure S2G). T cell responses were still higher 1 month post dose 3 in those who had been previously infected with SARS-CoV-2 (Figure 1F). However, by 6 months post dose 3, the spike-specific T cell response in naive and hybrid immune participants was equivalent (Figure 1F). M and N responses were higher in BNT162b2-vaccinated participants with hybrid immunity (Figure S3A and S3D), but this difference was not seen in the AZD1222 group (Figure S3G). Between 1 month post dose 2 and 1 month post dose 3, even in the group who did not seroconvert to N, we detected a rise in the T cell response to M and N in the BNT162b2 long-interval naive group (the largest group), which became significant 1 month after the third dose (Figure S3D), and appeared to increase still further at the 6-month time point (although this was not significant). Given that T cell responses are more sustained than antibody responses, this presumably reflects people who became asymptomatically infected but whose subsequent samples were taken after waning of the N antibody response.
Humoral responses wane quickly but are boosted by third dose vaccination
After the second vaccine dose, binding antibody responses decreased sharply. The median SARS-CoV-2 spike IgG titer (MSD) decreased 5.6-fold in naive vaccine recipients and 3.3-fold in the hybrid immunity group by 6 months (Figure 1G). Participants who received the different vaccine regimens followed similar patterns (Figures S2B, S2E, and S2H). Naive participants who received AZD1222 had lower spike antibody titers post second dose than those receiving BNT162b2 regimens, but these titers were then boosted 25-fold by the third (BNT162b2 mRNA) vaccine dose (Figure S2H). One month after the third dose, spike antibody IgG binding levels increased back to similar levels to those measured 1 month post dose 2 (Figure 1G). By 6 months after the third dose, the rate of waning was less than after the second dose and was less in the naive group than in the hybrid immune group. The naive group waned by 1.4-fold between 1 and 6 months after the third dose, which was not significant, compared with 5.8-fold after the second dose. The hybrid immune group waned 1.9-fold between 1 and 6 months post dose 3, compared with 3.2-fold in the equivalent period after the second dose. The reduction was significant for the hybrid group, and brought it down to a level equivalent to that of the naive group by 6 months post dose 3.
After the third dose, there was no significant difference in the magnitude of the spike binding IgG response between vaccine regimens (Figure 1J). Overall, a subtle (1.4-fold) but significant increase in spike IgG remained between previously infected and naive participants 1 month after the third dose (Figure 1G). The IgG levels measured post dose 3 were significantly greater than those measured post dose 2 in naive participants, but those with hybrid immunity did not derive additional benefit from the levels 1 month post dose 2, although there was a substantial boost over the 6 months post dose 2 level in this group. The RBD binding response followed the same pattern as the total spike response (Figure S3B, S3E, and S3H) and N antibody titers were unchanged by vaccination in the hybrid immune group (Figure S3C, S3F, and S3I). However, some of the naive participants did show rises in N antibody between 1 and 6 months post third dose. This time period corresponded to the very large wave of Omicron BA.1 in the UK, and likely represents subclinical infection in some of our participants.
Memory B cell responses were measured by IgG ELISpot in a subset of 106 participants (Figures 1H, S2C, S2F, and S2I). Six months after the second dose, memory B cell frequencies were similar between naive and hybrid immunity group, and these responses were preserved, with no statistically significant difference from 1 month post second dose. In the whole dataset, memory B cell responses were not affected by the third vaccine dose, although there was a significant increase in the BNT162b2 long-interval and AZD1222 groups in both naive participants (Figures S2F and S2I) and in the BNT162b2 long-interval participants with hybrid immunity (Figure S2F). Unlike the T cell IFNγ response, where there was still an advantage in those previously infected, there was no increase in the memory B cell response in those previously infected in any group (Figures 1H and 1K).
These data indicate that, although antibody levels decline between the second and third vaccine doses, T and B cell responses are well maintained across this period. Hybrid immunity conferred an advantage on the magnitude of the T cell and antibody response at all time points, including after the third vaccine dose, but did not for the B cell response. The third vaccine dose boosted immunity back to previous levels, or greater, with a tendency to even out any earlier differences between two-dose vaccine regimens.
Our cohort was mostly female, but the BNT162b2 short-interval group contained significantly more male participants (30 of 84, 40.5%, p = 0.006, chi-square test; Table 1). We did not detect any significant differences in responses between the three vaccine regimens, but, to ensure that there was no potential for the imbalance in male participants to influence this, we ran regression models to investigate the influence of age, sex, previous SARS-CoV-2 infection, and vaccine regimen on log10 transformed spike-specific IgG and T cell responses (see Table S1 [antibody] and Table S2 [T cell] regression analysis). Multivariable models indicated that previous infection was independently associated with both IgG responses (Table S1) and T cell responses, but that male sex was inversely associated with T cell responses (Table S2). Multivariable models were used to explore the effect of sex within each vaccine regimen group for IgG and T cell responses. Sex had no effect on IgG responses in all three vaccine groups (Table S1). Responses were negatively associated with age and associated with previous infection in the BNT162b2 long-interval group. For T cell responses, previous infection was associated with T cell responses in the BNT162b2 groups, and male sex was negatively associated with T cell responses only in the AZD1222 group (Table S2). Therefore, the male imbalance did not affect the measurement of responses in the BNT162b2 short-interval group. Although we found evidence that T cell responses to a booster mRNA vaccine are weaker in men who have received a primary course of AZD1222, this must be viewed with caution as it is based on only 24 participants.
Polyfunctional T cell responses are detectable 6 months after vaccination, with enhancement in individuals with hybrid immunity
T cell responses measured by intracellular cytokine staining (ICS) were lower at 6 months post second dose in AZD1222-vaccinated participants compared with BNT162b2 recipients (Figure 2 A), in line with the ELISpot findings. T cell function was similar between the two BNT162b2 groups, and there was less IL-2 and tumor necrosis factor (TNF) made by the AZD1222 group (Figure 2A). These differences evened out in the hybrid immunity group. CD8+ T cells made a substantial fraction of the IFNγ, at least half on average (Figure 2B), with a trend to more in the AZD1222 group, as known for chimpanzee adenovirus vectored vaccines.48 Very little IL-2 was made by CD8+ T cells; the overwhelming majority of the IL-2 response came from CD4+ T cells on a per-individual basis, irrespective of vaccination regimen (Figure 2C). All groups of participants made polyfunctional T cell responses, which we defined as IFNγ/IL-2/TNF triple-positive cells (Figure 2D). There were no differences between vaccine regimens in those with hybrid immunity, who uniformly had polyfunctional responses detectable.
Figure 2.

Analysis of spike-specific T cell responses by flow cytometry
(A and B) Cryopreserved PBMCs from a subset of 95 participants who received BNT162b2 (Pfizer/BioNTech) with a short or long dosing interval or AZD1222 (AstraZeneca) 1 month after the second dose were analyzed by ICS and flow cytometry. The individual cytokine expression levels of total IFNγ, IL-2, or TNF are shown as a percentage of (A) the CD4+ T cell population (top) or (B) the CD8+ T cell population (bottom). Populations were analyzed by gating on single, live, CD3+ cells (Figure S4). Short, BNT162b2 short interval; long, BNT162b2 long interval; AZ, AZD1222. Naive participants are shown as gray circles and hybrid immunity group are shown as red circles. Horizontal bars represent the median.
(C) The T cell populations responsible for IFNγ or IL-2 expression were assessed as the proportion of IFNγ or IL-2 expressed by CD4+ T cells, calculated by dividing the cytokine production in CD4+ T cells by the total cytokine production in response to spike in both CD4+ and CD8+ T cells. Horizontal bars represent the median.
(D) Polyfunctionality was evaluated by combined expression of IFNγ, IL-2, and TNF in CD4+ and CD8+ T cells, showing the percentage of cells making all three cytokines. Naive short, n = 20; naive long, n = 15; naive AZ, n = 14; hybrid immunity short, n = 13; hybrid immunity long, n = 17; hybrid immunity AZ, n = 16. Unpaired comparisons across two groups were performed using the Mann-Whitney test with two-tailed p values shown above linking lines when two-tailed p ≤ 0.05. Horizontal bars represent the median.
CD4+ and CD8+ proliferation responses to SARS-CoV-2 spike are higher in previously infected participants
We also assessed cellular responses to SARS-CoV-2 using T cell proliferation, a measure more biased toward central memory responses than IFNγ assays. T cell proliferation to spike S1 and S2 peptide pools was higher in previously infected AZD1222-vaccinated and the short-interval BNT162b2 group compared with naive individuals with a 3- to 8-fold increase in the median responses of CD4+ and CD8+ T cells respectively (Figures 3A, 3B, and 3D), thus confirming the enduring increase in cellular memory conferred by infection combined with vaccination. As expected, responses to M and N were absent in the majority of naive individuals (Figures 3C and 3E), with only one sample per vaccination regimen showing slightly elevated CD4+ T cell proliferation (3%–11%), which was not explained by N seroconversion (Figure 3C). Differences between vaccination regimens were only apparent in the BNT162b2-vaccinated hybrid immunity groups with significantly increased CD8 responses to S1, S2, and M in the short compared with the long interval.
Figure 3.

T cell proliferation to SARS-CoV-2 at 6 months after the primary vaccine course of two doses of BNT162b2 or AZD1222
T cell proliferation to SARS-CoV-2 peptide pools was assessed by flow cytometry in PBMCs from 73 participants who had received either BNT162b2 with a short or long vaccine dosing interval or AZD1222 vaccine and were either naive or were previously infected (either at baseline or during the course of the study).
(A) Relative frequency of CD4+ and CD8+ T cells proliferating to individual peptide pools spike S1, spike S2, membrane (M), and N protein in naive (n = 39) and hybrid immunity (n = 34) individuals. Gray color, missing value.
(B–E) (B and D) Proliferation to S1 and S2 and (C and E) M and N protein in CD4+ (B and C) and CD8+ (D and E) T cells are shown across the three vaccine regimens separated by exposure status (naive versus hybrid immunity). Individual data points and median with IQR are displayed for naive short, n = 16; naive long, n = 15; naive AZ, n = 8; hybrid immunity short, n = 11; hybrid immunity long, n = 12; hybrid immunity AZ, n = 11. Comparisons between naive and hybrid immunity within each vaccine regimen were performed using the Mann-Whitney test, and comparisons between the three vaccine regimens within the naive and previously infected groups was performed using the Kruskal-Wallis test and Dunn’s multiple comparisons correction. Two-tailed p values are shown only for statistically significant comparisons (p ≤ 0.05). Fold change between medians of two groups are shown in brackets next to or under p value. (Fold change is not shown for those comparisons where there was no proliferation detected in one of the groups.) Gray circles, naive individuals; red circles, participants with hybrid immunity. Central horizontal bars represent the median, and error bars represent the IQR.
The antibody response to SARS-CoV-2 broadens after the third vaccine dose, including enhanced neutralization activity against Omicron BA.1
Despite the differences between the naive vaccine groups in binding antibody 6 months after the second dose (Figure 1D), there was no significant difference in neutralization capacity of sera from these participants against the ancestral Victoria strain (Figure 4 A). Neutralization titers were lower against Delta and lower still against Omicron BA.1 compared with Victoria, as previously described.35 , 36 The BNT162b2 long-interval group had higher neutralizing titers against Delta than the short-interval group, as they did 28 days after the second dose.7 Using a surrogate neutralization assay on the MSD platform, which measures inhibition of spike-ACE2 binding, we measured neutralization of a wider range of variants. We also observed differences with the BNT162b2 long-interval group having higher antibody titers than the other groups (Figure 4B). Although there was a trend for higher titers in the BNT162b2 short group compared with the AZD1222 group, this did not reach significance. The surrogate neutralization assay showed a good correlation with the live virus focus reduction neutralization assay for Victoria, Delta, and Omicron variants (Figure S5).
Figure 4.

Neutralizing antibody and ACE2 inhibition titer profiles against SARS-CoV-2 variants of concern 6 months after two doses of BNT162b2 or AZD1222 and 1 month after a third vaccine with BNT162b2
(A and C) Focus reduction neutralization assay 50% (FRNT50) antibody titers against the Victoria isolate (orange), Delta (B.1.617.2, purple), and Omicron BA.1 (B.1.1.529 BA.1, blue) taken from infection-naive participants. FRNT50 is the reciprocal dilution of the concentration of serum required to produce a 50% reduction in infectious focus-forming units of virus in Vero cells (ATCC CCL-81). Participants either received two doses of BNT162b2 (Pfizer-BioNTech) vaccine delivered in a short (3–5 weeks, n = 20) or long (6–17 weeks, n = 20) dosing interval or two doses of AZD1222 (AstraZeneca) vaccine (AZ, n = 16). Neutralizing antibody titers are shown in (A) 6 months after the second dose and, for the same individuals, (C) 1 month after a third booster dose of mRNA vaccine for all participants. Geometric mean neutralizing titers with 95% confidence intervals are shown.
(E) Comparison of the data from (A) and (C), plotted as means with error bars by vaccine regimen 6 months after the second vaccine (V2 + 6 months), 1 month after the third booster mRNA vaccine (V3 + 1 month). The range of fold change (median) between V2 + 6 months and V3 + 1 month for the three vaccine regimens (short, dashed line; long, solid line; and AZ, dotted line) is shown in brackets for each variant. Points represent the median, and error bars represent the IQR. Data in (A), (C), and (E) from the short group (n = 20) have been previously published.35
(B and D) Impact of short or long BNT162b2 vaccine dosing interval and AZ on the ability of sera to inhibit ACE2 binding to SARS-CoV-2 spike (Victoria isolate, Delta (B.1.617.2), Omicron BA.1 (B.1.1.529 BA.1), Alpha (B.1.1.7), Beta (B.1.351), and Gamma (P.1)) (B) 6 months after the second dose and (D) 1 month after a third booster dose with mRNA vaccine. ACE2 inhibition was analyzed using a multiplexed MSD assay and performed at a serum dilution of 1:10 at V2 + 6 months and 1:100 at V3 + 1 month. Data are shown as percentage of inhibition. Bars represent the median with 95% confidence intervals. Naive, short, n = 20; naive, long, n = 20; naive, AZ, n = 16 for V2 + 6 months; naive, short, n = 19; naive, long, n = 20; naive, AZ, n = 10 for V3 + 1 month. Vaccine regimens were compared with the Kruskal-Wallis nonparametric test and Dunn’s multiple comparisons correction, with two-tailed p values shown above linking lines when two-tailed p ≤ 0.05, and fold changes are shown between the columns.
After the third dose of vaccine, neutralization capacity against both the Delta and Omicron BA.1 variants increased. Our previous report in this cohort demonstrated that the neutralization of Omicron BA.1 was significantly higher 28 days after three doses of BNT162b2 compared with 28 days after two doses.35 No differences were observed between vaccine groups after the third dose (Figure 4C). These differences also evened out in the ACE2 inhibition assay, although there was some saturation of the assay (Figure 4D). Therefore, although the overall level of binding antibody increased minimally (only in the naive group) between 28 days after the second and 28 days after the third dose (Figure 1G), the neutralization capacity of the antibody response broadened and the gap between groups closed (Figure 4E). Thus, we observed a higher quality of response after the third dose, paralleling what has been seen for clinical effectiveness of a booster dose against Omicron.
In a smaller subset of naive participants, we extended these analyses to BA.2 and BA.3 (for MSD binding and ACE2 inhibition), 6 months post dose 2 and 1 month post dose 3. In order to determine the lasting effects of the booster dose on Omicron variants post dose 3, we studied a further 115 participants for IgG binding to Omicron variants, and 45 participants for live virus neutralization to Omicron BA.1, BA.2, and BA.5 6 months post dose 3. These assays showed that IgG binding to Omicron BA.1, 2, and 3 spike was lower than that for the ancestral strain but persisted well 6 months after the third dose (Figures 5A–5C), including binding to BA.4/5, which we measured at this time point. ACE2 inhibition by antibody was reduced for Omicron BA.1–3, and ancestral and Omicron responses waned (Figures 5D–5F). However, the spread of responses at 6 months post dose 3 was wide, and by this point 11 of the 21 participants had contracted Omicron infections (Figure 5F). Virus neutralization for BA.1, BA.2, and BA.5 showed similar levels of neutralization for BA.1 and BA.2, and a slight drop for BA.5 (Figure 5G). These responses waned significantly by 6 months, but, in the subgroup of 11 people who became infected with between 1 and 6 months post dose 3, responses were significantly higher to Omicron variants but not to the ancestral virus (Figure 5G). Neutralization responses correlated with ACE2 inhibition for most participants (Figures S5D–S5I), with some evidence of saturation of the ACE2 assay. Importantly, overall, we detected less waning 6 months after the third dose than at the same time point after the second dose.
Figure 5.

Antibody responses to Omicron subvariants up to 6 months after dose 3
(A–C) IgG binding measured on the MSD platform to spike from ancestral SARS-CoV-2 (orange) and the BA.1 (blue), BA.2 (gray), BA.3 (brown), BA.3 (maroon), and BA.5 (green) Omicron variants at 6 months after two doses of BNT162b2 (A) and 1 month after dose 3 of BNT162b2 vaccine in infection-naive participants (n = 21) (B) and 6 months post dose 3 of BNT162b2 vaccine (C) in both infection-naive participants (n = 60) and in participants who became infected with an Omicron variant between 1 and 6 months after dose 3 (n = 55). Central horizontal bars represent the median, and error bars represent the IQR.
(D–F) ACE2 inhibition by plasma from the same donors in (A)–(C) at 6 months post dose 2 (D), 1 month post dose 3 (E), and 6 months post dose 3 (F). ACE2 inhibition was performed at a serum dilution of 1 in 100 to account for saturation of the assay, as seen in Figure 6. Comparisons between responses to ancestral and Omicron variants were made using Friedman’s test, with two-tailed p values of significant differences (p ≤ 0.05) shown above linking line. Central horizontal bars represent the median, and error bars represent the 95% confidence interval.
(G) Neutralizing antibody was measured at 1 month post dose 3 and 6 months post dose 3 by FRNT50 for Victoria strain (orange), BA.1 (blue), BA.2 (gray), and BA.5 (green) Omicron variants in participants who remained infection naive (n = 33) and those who became infected in between 1 and 6 months after dose 3 (n = 11). Filled circles indicate participants who remain infection naive, and participants who became infected with an Omicron variant between 1 month and 6 months after the third vaccine dose are indicated in unfilled circles. Paired comparisons between 1 and 6 months after vaccine were tested using the Wilcoxon signed-rank test, and comparisons between groups were tested using the Mann-Whitney test.
Cross-reactive T and B cell responses to the Omicron variant are preserved compared with the ancestral strain (Victoria) after second and third vaccine doses
We investigated the effect of the third vaccine dose on T cell and B cell responses to Omicron variants, in recognition of reduced vaccine effectiveness against infection with Omicron but preservation of protection against severe disease. First, we tested responses to Omicron BA.1 at 6 months post dose 2, similar to the situation for many people when Omicron first appeared in the UK in November 2021. Unlike neutralizing antibody responses, which were much lower for Omicron BA.1 6 months after the second dose (Figure 4A), and lower but with the gap narrowed after the third dose (Figure 4C), T cell and B cell ELISpot responses were much less affected. Using flow cytometry in the same participants in whom we studied multiple cytokine responses to spike, we did not detect any differences in the functionality of CD4 or CD8 T cell responses to Omicron BA.1 at 6 months post dose 2 (Figures 6A and 6B), although the total proportion of the IFNγ response in CD4+ cells dropped slightly (Figure S6A).
Figure 6.

Comparison of cytokine response at 6 months after dose 2 against ancestral strain and Omicron BA.1 variant according to infection status
Longitudinal comparison of T cell and B cell responses against ancestral strain and Omicron BA.1 variant according to vaccine regimen and infection status.
(A) Comparison of percentage IFNγ, TNF, and IL-2-positive CD4 T cells against ancestral strain and Omicron BA.1 variant by ICS of cryopreserved PBMCs in either infection-naive participants or participants with hybrid immunity. Box plots represent the median and IQR and whiskers represent 1.5× the IQR.
(B) Comparison of percentage IFNγ, TNF, and IL-2-positive CD8 T cells against ancestral strain and Omicron BA.1 variant by ICS of PBMCs in either infection-naive participants or participants with hybrid immunity. Box plots represent the median and IQR and whiskers represent 1.5× the IQR.
(C and D) Pairwise comparison of T cell responses to spike from ancestral strain and Omicron BA.1 variant from PBMCs by IFNγ ELISpot assay (C) in participants 6 months after primary vaccine course (two doses of BNT162b2 or AstraZeneca), n = 215, and (D) 1 month after third BNT162b2 vaccine dose, n = 175. Displayed are responses to peptide pools representing the sum of S1 and S2 units of S from ancestral strain and Omicron variant.
(E) Pairwise comparison of IFNγ ELISpot responses in a subset of participants (n = 36) to only the 51 out of 178 peptides spanning spike that have mutations in Omicron BA.1 compared with the ancestral strain.
(F) T cell responses to spike from ancestral strain and Omicron BA.1, BA.2, and BA.4/5 variants in PBMCs from naive (n = 28) and hybrid immune (n = 46) donors by IFNγ ELISpot assay. Horizontal lines represent the median.
(G–I) (G) Pairwise comparison of B cell responses to S in ancestral strain and Omicron BA.1 variant from PBMCs in participants 1 month after vaccine dose 2 (n = 12); (H) 6 months post second vaccine dose (n = 43); (I) 1 month after third vaccine dose (n = 80). Orange circles, responses against Victoria variant; blue circles, responses against Omicron BA.1 variant. Displayed are responses to peptide pools representing S1 and S2 units of S from ancestral and Omicron variants. ELISpot values are expressed as antibody SFU/106 PBMCs. Horizontal lines represent median values. Comparisons between responses to ancestral and Omicron variants were made using Wilcoxon matched-pairs signed-rank test, with two-tailed p values of significant differences (p ≤ 0.05) shown above linking line.
Using the more sensitive IFNγ ELISpot assay, the proportion of ancestral SARS-CoV-2 T cell responses that were relatively preserved for Omicron BA.1 on a per-individual basis was very high 6 months after the second dose (median 94%, IQR 75–110), and 1 month after a third dose, (median 90%, IQR 70–104), although the difference between ancestral strain and Omicron was significant by Wilcoxon matched-pairs signed-rank test (Figures 6C and 6D). Analysis of T cell ELISpot responses comparing only the peptides affected by mutations did reveal a drop (Figure 6E; median 53%, IQR 22–75), but this was not enough to have an impact on the T cell response for all of spike. We extended this analysis at 6 months post dose 3 for 46 hybrid immune and 28 naive participants. We tested ancestral SARS-CoV-2 spike peptides alongside those from Omicron BA.1, BA.2, and BA.4/5 (Figure 6F). At this point, there was no difference detected between the T cell response to any Omicron variant in either group by 6 months post third vaccine dose.
For B cells, responses to Omicron BA.1 were lower compared with the ancestral Victoria strain 1 month after the second dose (median 59% Omicron relative to ancestral SARS-CoV-2, IQR 56–67, p = 0.0005) (Figure 6G), 6 months after the second dose (median 57%, IQR 45–64, p < 0.0001) (Figure 6H), and 1 month after a third dose, (median 69%, IQR 58–78, p < 0.0001) (Figure 6I). This still represents a relative preservation of B cell immunity, compared with the absolute loss of neutralizing antibodies to Omicron after two vaccines (Figures 4A and 4C).
We also measured the effect of Omicron on proliferative responses of T cells in some participants. No changes were observed for CD4+ and CD8+ T cell proliferation in the naive group, although numbers of naive participants were limited (Figures S6B and S6D). In the hybrid immunity group, we observed a significant but modest drop in the proliferative response of CD4+ and CD8+ T cells to Omicron BA.1 spike S2 (Figure S6C, p = 0.0115) and S1 pool (Figure S6E, p = 0.034) respectively compared with ancestral spike. Overall, T and B cell responses to the Omicron BA.1 variant were well preserved compared with antibody responses.
Discussion
Our study reports robust immunity to SARS-CoV-2 spike, including to Omicron subvariants for all three primary vaccine regimens—BNT162b2 with a short (3–4 weeks) dosing interval, BNT162b2 with a long (6–17 weeks) dosing interval, and AZD1222—following boosting with an mRNA vaccine. Over the course of the COVID-19 pandemic, vaccines have significantly reduced the link between the number of infections with SARS-CoV-2 and the numbers of hospital admissions and deaths due to COVID-19. Although there has been continual evolution of viral variants, which have evaded the antibody response to varying degrees,49 vaccines have retained more effectiveness against severe disease than against overall infection.1 , 3 , 50 Emerging evidence implicates T cells as one potential mechanism for this protection, perhaps in addition to non-neutralizing antibody functions.23 , 24 , 25 , 51 The presence of both T cell and antibody responses gives the greatest protection from infection23 and from death in severe disease,21 an observation that is also supported by studies in a macaque model.20
Here, in a cohort of participants that overlaps with the SIREN study, in which vaccine effectiveness has been shown,6 we have observed that responses after a third dose of COVID vaccine have different dynamics: binding and neutralizing antibodies wane over the 6 months following the second dose, whereas B and T cell ELISpot responses wane much less over that interval. At 6 months post second dose, T cells secrete multiple cytokines and proliferate, indicating a broad range of memory function is retained by these cells. In addition, T cell responses are higher 6 months after vaccination in uninfected participants than they were in unvaccinated HCW 6 months after wave 1 infection in 2020, in a previous study of this cohort.15 Our findings are similar to those of Maringer et al., who also found that T cell responses were preserved more than antibody responses between the primary course and booster vaccination,52 although we also found a benefit with the third dose, likely due to increased power from a much larger sample size.
The third vaccine dose boosted all responses from their nadir post dose 2. The relative magnitude of the T cell boost was smaller compared with the antibody boost, but T cell responses had not waned to the same degree prior to the third dose. The third vaccine dose led to peak T cell levels that were higher than their previous peak 1 month post second dose. In contrast, the boost to binding antibody response achieved by the third dose did not exceed the previous peak achieved post dose 2. Interestingly, although a third dose of vaccine did not achieve higher peak binding antibody levels, the neutralizing capacity of the antibody response was much greater post dose 3 compared with post dose 2, replicating earlier observations.35 We observed that the B cell response also declined less in the 6 months after second vaccination than did the neutralizing antibody response, and this implies many of these cells make antibody that binds, but does not neutralize, the virus.
With each successive vaccine dose, and up to 1 month after the third vaccine dose, participants who had been naturally infected with SARS-CoV-2 had their antibody and T cell responses boosted and the absolute values achieved were consistently higher than those who had not been naturally infected. These observations are particularly important when evaluating the relative benefit of a third vaccine dose, which we demonstrate achieved statistically significant boosting effects even in the presence of hybrid immunity. These differences finally evened out by 6 months after the third dose. The ex vivo immunogenicity benefits of hybrid immunity demonstrated here align with evidence of the enhanced clinical effectiveness of vaccination in the presence of hybrid immunity.6 Superior vaccine effectiveness has also been observed against Omicron BA.1, BA.2, and BA.4/5 infections in those with hybrid immunity, compared with vaccination or infection alone.53 , 54 A recent systematic review comparing a range of estimates of protection from previous infection, vaccination, and hybrid immunity has also found that hybrid immunity provides the greatest and most sustained protection.55
We could also still detect an influence of the dose interval of BNT162b2 vaccine at 6 months after second vaccination. However, after the third vaccine dose, these differences had largely evened out and were no longer significant between the groups. T cell and antibody responses to spike were lower 6 months after primary vaccination course for AZD1222 compared with either BNT162b2 dosing regimen. These findings are compatible with previous reports for antibodies56 , 57 and lower vaccine effectiveness against infection,1 although vaccine effectiveness against hospitalization has been well preserved. After the AZD1222-primed recipients received a heterologous boost with mRNA vaccine, robust and similar cellular and antibody immunity including against Omicron BA.1 variant was seen for all three regimens studied. We detected a possible influence of male sex on reducing T cell responses to a third dose of mRNA vaccine in people who had received a primary course of AZD1222 vaccine. However, this finding was based on a small number of participants so must be viewed with caution. The larger parent SIREN study would have greater potential to answer this question definitively, although the public health relevance of this observation is diminishing over time.
The third dose gave a broad immune response that could recognize all the variants tested. This included neutralization of the Omicron BA.1, BA.2, and BA.5 lineages. The few participants who were followed out to 6 months post dose 3 and had an Omicron infection (11 participants) increased their neutralizing antibody responses to Omicron and not to Victoria, providing no evidence of immune imprinting (or antigenic sin) as has been recently suggested to occur with Omicron.58 More recent population-level evidence from Denmark and the UK suggests that Omicron BA.1 or BA.2 infection in combination with vaccination is more protective against Omicron BA.5 than Alpha or Delta infection.54 , 59 This may be due to waning immunity, antigenic difference, or both, rather than imprinting. We have not tested the effect of hybrid immunity on subsequent responses to Omicron; such work is ongoing. However, we found no evidence of antigenic sin for responses after Omicron infections, which were larger than the corresponding increase in antibody to the ancestral vaccine virus. T cell responses were less affected by viral variants that antibodies, likely due to the wider range of epitopes available to T cells compared with antibodies, where protective responses are more focused. Our findings are in line with those of others, who have also observed that antibodies decline more rapidly than T cell responses.60 We found that T cell responses after the third dose were durable out to 6 months post dose, and that at this point, overall, ancestral, and Omicron strains were recognized equally well.
Despite public concern about loss of immunity over time post infection and/or vaccines, we find ample evidence of strong and durable immunity and memory responses that are likely to sustain protection against severe COVID-19 long term. Further booster vaccinations are likely to be most beneficial for preventing severe disease in the clinically vulnerable and may lead to a reduction in hospitalization rates. People with immune compromise are now receiving fourth or even fifth vaccine doses in the UK and other countries, and parallel studies of durability of immunity in such populations are needed. The role of further booster vaccines for HCWs requires onward longitudinal follow-up of this cohort and others, but prevention of infection in HCWs continues to be desirable to minimize infection-related absence, nosocomial transmission, and risks of long COVID.61
Limitations of the study
Our study has a number of limitations. (1) As with other HCW studies, our cohort has a female majority and is predominantly in people reporting white ethnicity; it may not therefore fully represent other populations. We have not observed any impact of sex or ethnicity in this study or our previous reports.7 , 62 (2) Our longitudinal cohort does not include never-vaccinated participants, because all the HCWs engaged with our studies across six sites took up vaccination. However, we have been able to compare responses 6 months after vaccination (in 2021) with historical data using the same assay in a subset of the same cohort in 2020, 6 months after wave 1 (ancestral strain) infection before vaccine were available,15 and demonstrate that vaccine-induced responses in infection-naive HCWs are higher than infection-induced responses. (3) We were not able to perform all assays on all participants at all time points, due to lack of sample availability, missed follow-up visits, and/or laboratory capacity. This means that not all our data are longitudinal, although many are. To account for this, we have used unpaired testing in all our comparisons. (4) We only performed neutralizing antibody measurements on naive participants due to the labor intensity and interpretation requiring matching with infecting variant strain, and this information was limited. (5) We defined hybrid immunity in participants as previously testing PCR positive for SARS-CoV-2, or seroconversion to anti-N positivity during the study. However, some of the group labeled naive could have been exposed to SARS-CoV-2, because up to 60% of vaccinated people may not develop anti-N antibody, and the N sequence differs between variants.63 , 64 As time went on, the N antibody levels rose in our naive participants, even though many remained below the assay threshold for a positive N response. As hybrid immunity evolves in the population, it will become increasingly difficult to define the shrinking group of people who have never been infected with SARS-CoV-2. (6) For people with vaccine breakthrough infections since the second vaccine dose, infecting sequence data were not always available. However, we know that the majority of this report covers a period in time when Delta was the predominant variant, with 68% and 88% of the sampling complete for this study by 1st December 2021 and 1st January 2022 respectively. (7) Finally, we have not addressed mucosal immunity in this report, and this is the subject of ongoing work. Antibody can be readily detected in the mucosa post infection with SARS-CoV-2.65 Cellular and antibody responses have been also detected in the mucosa after COVID vaccination,66 , 67 but at low levels, and their role in protection remains unclear.
In summary, we have observed that SARS-CoV-2-specific cellular immune responses are better maintained compared with antibodies in the 6 months following the second dose of COVID-19 vaccine. The third dose of vaccine confers a measurable benefit to these responses irrespective of the primary course, including in people who have previously been infected (hybrid immunity), who therefore may also stand to benefit from a third dose. The third dose also induces better antibody recognition of SARS-CoV-2 variants, including Omicron BA.1. Our findings allow establishment of the dynamics of the immune response post infection and vaccination in a healthy population of working age, which can then be used as a benchmark for evaluating immunity in vulnerable groups, and provides the first glimpse of evolving hybrid immunity driven by ongoing viral exposure in vaccinated populations.
Consortia
The members of the PITCH Consortium are Hibatullah Abuelgasim, Emily Adland, Zahra Ahmed, Hossain Delowar Akther, Ahmed Alhussni, Ali Amini, M. Azim Ansari, Rachel Anslow, Carolina V. Arancibia-Cárcamo, Ana Atti, James Austin, Angela Bailey, Martin Bayley, Alice Bridges-Webb, Helen Brown, Holly Caborn, Jeremy Chalk, Meera Chand, Anu Chawla, Senthil Chinnakannan, Elizabeth Clutterbuck, Debbie Cross, Joseph Cutteridge, Sophie Davies, Catherine de Lara, Lucy Denly, Ben Diffey, Stavros Dimitriadis, Timothy Donnison, Thomas M Drake, Maeva Dupont, Elena Efstathiou, David Eyre, Alex Fairman, Sarah Foulkes, John Frater, Siobhan Gardiner, Javier Gilbert-Jarmillo, Philip Goulder, Jessica Gregory, Carl-Philipp Hackstein, Sophie Hambleton, Muzlifah Haniffa, Helen Hanson, Kate Harrington, Jenny Haworth, Carole Hays, Jennifer Holmes, Fatima Mariam Ilyas, Jasmin Islam, Anni Jämsén, Chris Jones, Geraldine Jones, Mwila Kasanyinga, Sinead Kelly, Maqsood Khan, Jon Kilby, Rosemary Kirk, Michael L. Knight, Allan Lawrie, Lian Lee, Lauren Lett, Katy Lillie, Nicholas Lim, Alison Lye, Spyridoula Marinou, Chloe Matthewman, Jessica McNeill, Gracie Mead, Hema Mehta, Haniffa Muzlifah, Christopher Norman, Denise O'Donnell, Ane Ogbe, Juyeon Park, Brendan A.I. Payne, Gareth Platt, Sonia Poolan, Nicholas Provine, Narayan Ramamurthy, Nichola Robinson, Leigh Romaniuk, Patpong Rongkard, Ayoub Saei, Oliver L. Sampson, Donal Skelly, Jarmila S. Spegarova, Gareth Stephens, Emily Stephenson, Rachel Stimpson, Krishanthi Subramaniam, Chloe Tanner, Lydia J. Taylor, Chitra Tejpal, Sarah Thomas, Neal Townsend, Simon Travis, Nicola Trewick, Stephanie Tucker, Helena Turton, Zara Valiji, Adam Watson, Lisa Watson, Esme Weeks, Rachel Whitham, Jayne Willson, Barbara Wilson, Robert Wilson, Steven Wood, Huiyuan Xiao, and Amira A.T. Zawia.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PerCP mouse anti-human CD3 (Clone: UCHT1, IgG1, κ) | Biolegend, UK | Cat#300428; RRID: AB_893298 |
| FITC mouse anti-human CD3 (Clone: UCHT1, IgG1, κ) | Biolegend, UK | Cat#300440; RRID: AB_2562046 |
| APC mouse anti-human CD4 (Clone: RPA-T4, IgG1, κ) | Biolegend, UK | Cat#300514; RRID: AB_314082 |
| PE-Cy7 mouse anti-human CD8 (Clone: RPA-T8, IgG1, κ) | Biolegend, UK | Cat#301012; RRID: AB_314130 |
| BV510 mouse anti-human CD8 (Clone: RPA-T8, IgG1, κ) | Biolegend, UK | Cat#301048; RRID: AB_2561942 |
| APC-Fire750 mouse anti-human CD14 (Clone: M5E2, IgG2a, κ) | Biolegend, UK | Cat#301854; RRID: AB_2632660 |
| PE mouse anti-human IFNγ Clone: 4S.B3, IgG1, κ) | Biolegend, UK | Cat#502508; RRID: AB_315233 |
| FITC mouse anti-human TNFα (Clone: MAb11, IgG1, κ) | Biolegend, UK | Cat#502906; RRID: AB_315258 |
| PE-Cy7 rat anti-human IL-2 (Clone: MQ1-17H12, IgG2a, κ) | Thermo Fisher (eBioscience) | Cat#25-7029-42; RRID: AB_2573517 |
| Purified NA/LE mouse anti-human CD28 (Clone: CD28.2) | BD Biosciences, UK | Cat#555725; RRID:AB_396068 |
| Purified NA/LE mouse anti-human CD49d (Clone: 9F10) | BD Biosciences, UK | Cat#555501; RRID:AB_2130052 |
| SARS-CoV-2 (B.1.617.2; AY.1, AY.2, AY.3) Stabilized Spike Glycoprotein, His-Strep-Tag (HEK293) | The Native Antigen Company | Cat#REC31975-100 |
| Human anti-NP (mAb206) | Dejnirattisai et al. 2021 | N/A |
| Anti-Human IgG (Fc specific)-Peroxidase | Sigma | Cat#A0170; RRID:AB_257868 |
| Bacterial and Virus Strains | ||
| SARS-CoV-2 (Australia/VIC01/2020) | Caly et al., 2020 | N/A |
| SARS-CoV-2/B.1.617.2 (delta) | Wendy Barclay and Thushan De Silva | N/A |
| SARS-CoV-2/B.1.1.529 BA.1 (omicron) | John Radcliffe Hospital, Oxford | N/A |
| SARS-CoV-2/B.1.1.529 BA.2 (omicron) | John Radcliffe Hospital, Oxford | N/A |
| SARS-CoV-2/ B.1.1.529 BA.5 (omicron) | John Radcliffe Hospital, Oxford | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TrueBlue Peroxidase Substrate | Insight Biotechnology | Cat#5510-0030 |
| Dulbecco’s Modified Eagle Medium, high glucose | Sigma-Aldrich | Cat#D5796 |
| Custom synthesized peptides (18-mers) | Mimotopes | See supplementary file Data S1.xlsx for sequences of the peptide library. http://www.mimotopes.com |
| Dimethyl Sulfoxide | Sigma | Cat#D2650-100ML |
| RPMI-1640 Medium with Sodium bicarbonate, no L-Glutamine | Sigma | Cat#R0883 |
| L-Glutamine | Sigma | Cat#G7513 |
| Penicillin/Streptomycin | Sigma | Cat#P0781 |
| Fetal Bovine Serum | Sigma | Cat#F9665-500ML |
| Lymphoprep | StemCell Technology | Cat#07861 |
| L-Glutamine–Penicillin–Streptomycin solution | Sigma | Cat#G1146 |
| GlutaMAX™ Supplement | Gibco | Cat#35050061 |
| Phosphate buffered saline tablets | Thermo Fisher Scientific | Cat#12821680 |
| Fetal Bovine Serum | Gibco | Cat#12676029 |
| Carbonate/bicarbonate capsules | Sigma Aldrich | Cat#C3041-100CAP |
| ProMix CEF peptide pool | Proimmune, Oxford | Cat#PX-CEF |
| Phytohemagglutinin-L | Sigma Aldrich | Cat#11249738001 |
| Concanavalin A | Merck | Cat# C5275-5MG |
| Carboxymethyl cellulose | Sigma | Cat#C4888 |
| Tween 20 | Sigma Aldrich | Cat#P2287-500ml |
| 1-Step™ NBT/BCIP Substrate Solution | Life Technologies | Cat#34042 |
| LIVE/DEAD fixable near-IR dead cell stain kit | Thermo Fisher Scientific | Cat#L34975 |
| CellTrace™ Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat#C34557 |
| Fixation/Permeabilization Solution Kit | BD Biosciences, UK | Cat#554714 |
| Cell staining buffer | Biolegend, UK | Cat#420201 |
| Brefeldin A 1000x | Biolegend, UK | Cat#420601 |
| Cell activation cocktail w/o BFA (contains PMA/Ionomycin) 500x | Biolegend, UK | Cat#423301 |
| Anti-Mouse Ig, κ/Negative Control Compensation Particles Set | BD Biosciences, UK | Cat#552843 |
| ArC Amine Reactive Compensation Bead Kit | Thermo Fisher Scientific | Cat#A10346 |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | Cat#14190144 |
| 37% Formaldehyde solution | Merck, UK | Cat#F8775 |
| Gibco™ Fetal Bovine Serum, qualified, heat inactivated | Thermo Fisher Scientific, UK | Cat#10500064 |
| RPMI-1640 Medium with sodium bicarbonate but without L-Glutamine | Merck, UK | Cat#R0883 |
| Bovine Serum Albumin (BSA) | Merck, UK | Cat#A9418 |
| Critical Commercial Assays | ||
| V-PLEX COVID-19 Coronavirus Panel 3 Kit | Meso Scale Discovery, Rockville, MD USA | Cat#K15399U-2 |
| V-PLEX SARS-CoV-2 Panel 23 Kit | Meso Scale Discovery, Rockville, MD, USA | cat#15570U |
| V-PLEX SARS-CoV-2 Panel 25 Kit | Meso Scale Discovery, Rockville, MD, USA | cat#15586U |
| V-PLEX SARS-CoV-2 Panel 27 Kit | Meso Scale Discovery, Rockville, MD, USA | cat#K15609U |
| Human IgA/IgG FluoroSpotFLEX kit | Mabtech | Cat#X-06G05R-10 |
| Human memory B-cell stimpack | Mabtech | Cat#3660-1 |
| Human IFNγ ELISpot Basic kit | Mabtech | Cat#3420-2A |
| Deposited Data | ||
| Evolution of long-term vaccine induced and hybrid immunity in healthcare workers after different COVID19 vaccine regimens: a longitudinal cohort study. Moore et al. | Mendeley Data | https://doi.org/10.17632/fyp26zjgmj.1 |
| Experimental Models: Cell Lines | ||
| Vero cells | ATCC | Cat#CCL-81 |
| Software and Algorithms | ||
| Discovery Bench 4.0 | Meso Scale Discovery, Rockville, MD, USA | Immunoassay Analysis Software | Meso Scale Discovery |
| Prism 8.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| IBM SPSS Software 26 | IBM | https://www.ibm.com |
| AID ELISpot software 8.0 | Autoimmun Diagnostika | http://www.elispot.com/products/software |
| Flowjo 10.7.1 | BD Biosciences | https://www.flowjo.com/ |
| R version 4.0.4 (2021-02-15) -- "Lost Library Book" | Web-based open source software | https://www.r-project.org |
| R studio version 1.1.463 | Web-based open source software | https://www.rstudio.com |
| Other | ||
| MACS Quant flow cytometer | Miltenyi Biotec, Germany | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lance Turtle (lance.turtle@liverpool.ac.uk).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Study design and sample collection
In this prospective, observational, cohort study, participants were recruited into the PITCH study from across six centres (Birmingham, Cambridge, Liverpool, Newcastle, Oxford and Sheffield). Individuals consenting to participate were recruited by word of mouth, hospital e-mail communications and from hospital-based staff screening programmes for SARS-CoV-2, including HCWs enrolled in the national SIREN study at three sites (Liverpool, Newcastle and Sheffield). Eligible participants were adults aged 18 or over, and currently working as an HCW, including allied support and laboratory staff, or were volunteers linked to the hospital. The majority of participants were sampled for previous reports in this PITCH cohort.7 , 27 , 37 , 62 Participants were sampled for the current study between 4 January 2021 and 15 February 2022, with the majority of the sampling complete before the omicron BA.1 variant emerged in the UK (68% of sampling was prior to December 2021 and 88% was prior to January 2022).
Participants had received one of three vaccine regimens: “Short” - two doses of BNT162b2 (Pfizer/BioNTech) administered with the manufacturer’s licenced dosing interval (median 24 days, IQR 21-27); “Long” - two doses of BNT162b2 (Pfizer/BioNTech) administered with an extended dosing interval (median 71 days, IQR 66-78); and “AZ” - two doses of AZD1222 (Oxford/AstraZeneca), administered a median 74 days (IQR 65-78) apart. All participants then received a third “booster” dose of BNT162b2, a median of 207 days, (IQR 191-233) days after the second dose, regardless of primary vaccine regimen. Participants underwent phlebotomy for assessment of immune responses one (median 28 days, IQR 26-32) and six (median 185 days, IQR 173-200) months after the second dose of vaccine, and one month after the third dose of vaccine (median 31 days, IQR 28-37). Clinical information including BNT162b2 and AZD1222 vaccination dates, date of any SARS-CoV-2 infection (either prior to vaccination or during the study) defined by a positive PCR test and/or detection of antibodies to spike or nucleocapsid protein, presence or absence of symptoms, time between symptom onset and sampling, age, sex and ethnicity of participant was recorded. Key information on demographics and vaccine dose intervals is shown in Table 1. Participants’ information on sex, age, and race was self-reported. Information on gender and socioeconomic status was not collected.
Participants were considered to be SARS-CoV-2 exposed if they had ever been PCR or lateral flow device positive for SARS-CoV-2, irrespective of symptoms. In addition, participants were considered exposed to SARS-CoV-2 if they seroconverted with N antibody on the mesoscale discovery (MSD) assay. N seroconversion was defined as an N antibody level over the cut-off threshold of 3874 previously defined using pre-pandemic samples,7 and at least a 2-fold increase over the baseline value. Participants who did not meet any of these criteria were considered to be infection-naïve.
PITCH is a sub-study of the SIREN study, which was approved by the Berkshire Research Ethics Committee, Health Research 250 Authority (IRAS ID 284460, REC reference 20/SC/0230), with PITCH recognised as a sub-study on 2 December 2020. SIREN is registered with ISRCTN (Trial ID:252 ISRCTN11041050). Some participants were recruited under aligned study protocols. In Birmingham participants were recruited under the Determining the immune response to SARS-CoV-2 infection in convalescent health care workers (COCO) study (IRAS ID: 282525). In Liverpool some participants were recruited under the “Human immune responses to acute virus infections” Study (16/NW/0170), approved by North West - Liverpool Central Research Ethics Committee on 8 March 2016, and amended on 14th September 2020 and 4th May 2021. In Oxford, participants were recruited under the GI Biobank Study 16/YH/0247, approved by the research ethics committee (REC) at Yorkshire & The Humber - Sheffield Research Ethics Committee on 29 July 2016, which has been amended for this purpose on 8 June 2020. In Sheffield, participants were recruited under the Observational Biobanking study STHObs (18/YH/0441), which was amended for this study on 10 September 2020. We also included some participants from Cambridge from a study approved by the National Research Ethics Committee and Health Research Authority (East of England – Cambridge Research Ethics Committee (SCORPIO study, SARS-CoV-2 vaccination response in obesity amendment of ‘‘NIHR BioResource’’ 17/EE/0025).The study was conducted in compliance with all relevant ethical regulations for work with human participants, and according to the principles of the Declaration of Helsinki (2008) and the International Conference on Harmonization (ICH) Good Clinical Practice (GCP) guidelines. Written informed consent was obtained for all participants enrolled in the study.
Peripheral blood mononuclear cells (PBMCs), plasma and serum were separated and cryopreserved. Some of the immune response data from one month after the second dose has been previously reported,7 as has some of the neutralising antibody data for HCWs receiving a short dosing interval for BNT162b2.35 The study size was selected because this number was feasible for the six clinical and laboratory sites to study, and consistent with our track record of significant findings at this scale.
Method details
Primary cell cultures
Vero cells (ATCC CCL-81) were cultured in Dulbecco’s Modified Eagle Medium, high glucose (Sigma-Aldrich), supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco), 100 units/mL penicillin, 100 μg/mL streptomycin, 2mM L-Glutamine (Sigma) and 2mM GlutaMax (Gibco).
Viral stocks
SARS-CoV-2/human/AUS/VIC01/202068 was provided by Public Health England. Delta (B.1.617.2) virus was kindly provided Wendy Barclay and Thushan de Silva. Omicron (B.1.1.529) BA.1, BA.2 and BA.5 were isolated from swabs obtained from the John Radcliffe Hospital, Oxford by culture in Vero E6-TMPRSS2 cells. For assays, Viruses were grown in Vero (ATCC CCL-81) cells. Cells were infected with the SARS-CoV-2 virus using an MOI of 0.0001. Virus containing supernatant was harvested at 80% cytopathic effect, and spun at 3000 rpm at 4 °C before storage at -80 °C. Viral titers were determined by a focus-forming assay on Vero cells. Viral stocks were sequenced to verify that they contained the expected spike protein sequence and no changes to the furin cleavage sites.
Meso Scale Discovery (MSD) IgG binding assay
IgG responses to SARS-CoV-2, SARS-CoV-1, MERS-CoV, seasonal coronaviruses or SARS-CoV-2 variants were measured using multiplexed MSD immunoassays: The V-PLEX COVID-19 Coronavirus Panel 3 (IgG) (cat# K15399U) from Meso Scale Discovery, Rockville, MD USA. A MULTI-SPOT® 96-well, 10 spot plate was coated with three SARS CoV-2 antigens (Spike (S), Receptor-Binding Domain (RBD), Nucleoprotein (N)), SARS-CoV-1 and MERS-CoV spike trimers, spike proteins from seasonal human coronaviruses, HCoV-OC43, HCoV-HKU1, HCoV-229E and HCoV-NL63, and bovine serum albumin (negative control). The V-PLEX COVID-19 Coronavirus Panel 25 (cat# 15583U) and Panel 27 (cat# 15606U) Kits were coated with SARS-CoV-2 variant spike antigens including B (Victoria), B.1.1.529 BA.1/omicron BA.1; BA.2/omicron BA.2; BA.3/omicron BA.3; BA.4/omicron BA.4 and/or BA.5/omicron BA.5. Multiplex MSD assays were performed as per the manufacturer’s instructions. To measure IgG antibodies, 96-well plates were blocked with MSD Blocker A for 30 minutes. Following washing with washing buffer, samples diluted 1:1,000-30,000 in diluent buffer, MSD standard and undiluted internal MSD controls, were added to the wells. After 2-hour incubation and a washing step, detection antibody (MSD SULFO-TAG™ anti-human IgG antibody, 1/200) was added. Following washing, MSD GOLD™ read buffer B was added and plates were read using a MESO® SECTOR S 600 reader. The standard curve was established by fitting the signals from the standard using a 4-parameter logistic model. Concentrations of samples were determined from the electrochemiluminescence signals by back-fitting to the standard curve and multiplying by the dilution factor. Concentrations are expressed in Arbitrary Units/ml (AU/ml). Cut-offs were determined for each SARS-CoV-2 antigen (S, RBD and N) based on the mean concentrations measured in 103 pre-pandemic sera + 3 Standard Deviations. Cut-offs were: S, 1160 AU/ml; RBD, 1169 AU/ml; and N, 3874 AU/ml.
MSD ACE2 inhibition assay
The V-PLEX SARS-CoV-2 Panel 23 (ACE2) (cat#15570U), Panel 25 (ACE2) (cat#15586U) and Panel 27 (cat# K15609U) Kits, from MSD, Rockville, MD, multiplexed MSD immunoassays, were also used to measure the ability of human sera to inhibit ACE2 binding to SARS-CoV-2 spike antigens including B (Victoria), B.1.1.7/alpha, B.1.351/beta P.1/gamma, B.1.617.2/delta, B.1.1.529 BA.1/omicron BA.1; BA.2/omicron BA.2; BA.3/omicron BA.3; BA.4/omicron BA.4; BA.5/omicron BA.5). A MULTI-SPOT 96-well, 10 spot plate was coated with SARS-CoV-2 spike antigens including these ones above-mentioned. Multiplex MSD Assays were performed as per manufacturer’s instructions. To measure ACE2 inhibition, 96-well plates were blocked with MSD Blocker for 30 minutes. Plates were then washed in MSD washing buffer, and samples were diluted 1:10 – 1:100 in diluent buffer. Neutralizing activity was determined by measuring the presence of antibodies able to block the binding of ACE2 to SARS-CoV-2 spike proteins from Victoria spike, B.1.1.7/alpha, B.1.617.2/delta, B.1.351/beta, P.1/gamma and B.1.1.529 BA.1/omicron BA.1; BA.2/omicron BA.2; BA.3/omicron BA.3; BA.4/omicron BA.4; BA.5/omicron BA.5 and was expressed as percentage of ACE2 inhibition in comparison to the blanks on the same plate. Furthermore, internal controls and the WHO SARS-CoV-2 Immunoglobulin international standard (NIBSC 20/136) were added to each plate. After a 1-hour incubation, recombinant human ACE2-SULFO TAG was added to all wells. After a further 1-hour, plates were washed and MSDGOLD Read Buffer B was added, plates were then immediately read using a MESO SECTOR S 600 Reader.
Focus reduction neutralisation assay (FRNT)
The neutralisation potential of antibodies (Ab) was measured using a Focus Reduction Neutralisation Test (FRNT), where the reduction in the number of the infected foci is compared to a negative control well without antibody. Briefly, serially diluted Ab or plasma was mixed with SARS-CoV-2 strain Victoria or P.1 and incubated for 1 hr at 37C. The mixtures were then transferred to 96-well, cell culture-treated, flat-bottom microplates containing confluent Vero cell monolayers in duplicate and incubated for a further 2 hr followed by the addition of 1.5% semi-solid carboxymethyl cellulose (Sigma) overlay medium to each well to limit virus diffusion. A focus forming assay was then performed by staining Vero cells with human anti-nucleocapsid monoclonal Ab (mAb206) followed by peroxidase-conjugated goat anti-human IgG (A0170; Sigma). Finally, the foci (infected cells) approximately 100 per well in the absence of antibodies, were visualized by adding TrueBlue Peroxidase Substrate (Insight Biotechnology). Virus-infected cell foci were counted on the classic AID ELISpot reader using AID ELISpot software. The percentage of focus reduction was calculated and IC50 was determined using the probit program from the SPSS package. In order to reduce confounding arising from exposure to different SARS-CoV-2 variants, these experiments were conducted only on participants who were naive at the time of sampling 6-months post second vaccine dose, as defined by no history of positive PCR or lateral flow test for SARS-CoV-2, and no anti-N IgG seroconversion during the study.
T cell interferon-gamma (IFNγ) ELISpot Assay
The PITCH ELISpot Standard Operating Procedure has been published previously.62 Interferon-gamma (IFNγ) ELISpot assays were set up from cryopreserved PBMCs using the Human IFNγ ELISpot Basic kit (Mabtech 3420-2A). A single protocol was agreed across the centres and is available on the PITCH website (http://www.pitch-study.org/). PBMCs were thawed and rested for 3-6 hours in R10 media: RPMI 1640 (Sigma) supplemented with 10% (v/v) Fetal Bovine Serum (Sigma), 2mM L-Glutamine (Sigma) and 1mM Penicillin/Streptomycin (Sigma) in a humidified incubator at 37∘C, 5% CO2, prior to stimulation with peptides. PBMCs were then plated in duplicate or triplicate at 200,000 cells/well in a MultiScreen-IP filter plate (Millipore, MAIPS4510) previously coated with capture antibody (clone 1-D1K) and blocked with R10. PBMCs were then stimulated with overlapping peptide pools (18-mers with 10 amino acid overlap, Mimotopes, see supplementary Data S1.xlsx, peptide library, and STAR Methods) representing the spike (S), Membrane (M) or nucleocapsid (N) SARS-CoV-2 proteins at a final concentration of 2 μg/ml for 16 to18 hours in a humidified incubator at 37∘C, 5% CO2. For selected individuals, pools representing spike protein of the Omicron (BA.1) variant were included. Pools consisting of CMV, EBV and influenza peptides at a final concentration of 2μg/ml (CEF; Proimmune) and concanavalin A or phytohemagglutinin L (PHA-L, Sigma) were used as positive controls. DMSO was used as the negative control at an equivalent concentration to the peptides. After the incubation period as well as all subsequent steps wells were washed with PBS/0.05% (v/v) Tween20 (Sigma). Wells were incubated with biotinylated detection antibody (clone 7-B6-1) followed by incubation with the ELISpot Basic kit streptavidin-ALP. Finally colour development was carried out using the 1-step NBT/BCIP substrate solution (Thermo Scientific) for 5 minutes at RT. Colour development was stopped by washing the wells with tap water. Air dried plates were scanned and analysed with either the AID Classic ELISpot reader (software version 8.0, Autoimmune Diagnostika GmbH, Germany) or the ImmunoSpot® S6 Alfa Analyser (Cellular Technology Limited LLC, Germany). Antigen-specific responses were quantified by subtracting the mean spots of the negative control wells from the test wells and the results were expressed as spot-forming units (SFU)/106 PBMCs. Samples with a mean spot value greater than 50 spots in the negative control wells were excluded from the analysis.
For comparison of responses to omicron BA.1 we firstly compared responses to 178 peptides spanning all of spike (S1 and S2) for the ancestral (wild type) and the omicron BA.1 variant, then secondly, we compared responses to the 51 peptides representing the regions of spike with mutations in omicron BA.1, again comparing ancestral and omicron BA.1. To reduce the disproportionate impact of background noise, samples with a total response to ancestral spike of <33 SFU/106 PBMCs were excluded from analysis, with this cut off threshold calculated as the mean + 2 standard deviations of the DMSO wells across all experiments in the study. The % of the T cell response to ancestral strain that was preserved against omicron BA.1 was calculated for each paired sample then expressed as the median and IQR for the group.
Memory B cell fluorospot assay
Cryopreserved PBMCs were thawed and cultured for 72 hours with polyclonal stimulation containing 1 μg/ml R848 and 10 ng/ml IL-2 from the Human memory B cell stimpack (Mabtech). Using the Human IgA/IgG FluoroSpotFLEX kit (Mabtech), stimulated PBMCs were then added at 2x105 cells/well to fluorospot plates coated with 10 μg/ml Sars-CoV-2 spike glycoprotein diluted in PBS. Plates were incubated for 16 hours in a humidified incubator at 37∘C, 5% CO2 and developed according to the manufacturer’s instructions (Mabtech). Analysis was carried out with AID ELISpot software 8.0 (Autoimmun Diagnostika). All samples were tested in triplicates and response was measured as spike- specific spots per million PBMCs with PBS background subtracted.
Intracellular cytokine stimulation assay
In a subset of donors (n=95), selected at random from all three vaccine regimens and previous SARS-CoV-2 infection, T cell responses were characterised further using intracellular cytokine staining (ICS) after stimulation with overlapping SARS-CoV2 peptide pools. In brief, cryopreserved PBMCs were thawed, rested for 4-5 hours in R10 media and then plated at 1x106 cells/well in a 96 well U-bottom plate together with co-stimulatory molecules anti-CD28 and anti-CD49d (both BD). Peptide pools (spanning ancestral (B.1) spike, omicron BA.1 spike, ancestral membrane (M) and nucleocapsid (N) proteins) were added at 2 μg/ml final concentration for each peptide. DMSO (Sigma) was used as the negative control at the equivalent concentration to the peptides. As a positive control, cells were stimulated with 1x cell activation cocktail containing phorbol-12-myristate 13-acetate (PMA) at 81μM and ionomycin at 1.3μM final concentration (Biolegend). The cells were then incubated in a humidified incubator at 37°C, 5% CO2 for 1 hour before incubating for a further 15 hours in the presence of 5μg/ml Brefeldin A (Biolegend). Flow cytometry staining was performed as described below.
Proliferation assay
T cell proliferation assessed the magnitude of memory responses to SARS-CoV2 spike, M and N protein in the CD4+ and CD8+ T cell pool in 73 individuals selected for the ICS assay, with 27 participants from the BNT162b2 short interval group (16 naïve and 11 with hybrid immunity), 27 participants from the BNT162b2 long interval group (15 naïve and 12 with hybrid immunity) and 19 participants from the AZD1222 group (8 naïve and 11 with hybrid immunity). CellTraceTM Violet (CTV, Invitrogen) labelling and stimulation with SARS-CoV-2 peptide pools spanning ancestral spike (divided into two pools, S1 and S2), omicron (BA.1) spike (S1 and S2), ancestral M and N protein, as well as a control peptide mix, CEF (1μg/ml per peptide) was carried as in previously published work.27 Cells were incubated in RPMI 1640 (Sigma) supplemented with 10% human AB serum (Sigma), 2mM L-glutamine (Sigma) and 1 mM Penicillin/Streptomycin (Sigma) in a 96 well U-bottom plate at 250,000 cells per well in single or duplicate depending on cell availability. DMSO added at the same concentration to SARS-CoV-2 peptides served as negative control and 2ug/ml PHA-L as positive control. Cells were placed in a humidified incubator at 37∘C, 5% CO2. Half a media change was performed on day 4 and cells were harvested for flow cytometry staining on day 7 as described below. Data were expressed as relative frequency of proliferating cells within single, live CD4+ T cells and CD8+ T cells respectively. Background was subtracted from stimulated samples and samples were excluded due to high background (DMSO control >2% proliferation in any T cell subset,) or less than 1000 events in the single, live CD3+ gate (10 samples in total were excluded). Responses to individual peptide pools and summed responses to total spike (S1+S2) and M+NP were reported.
Flow cytometry straining and analysis
Details for antibodies are listed in Table S2. All washes and extracellular staining steps for PBMC were carried out in cell staining buffer (Biolegend) for ICS samples and PBS for proliferation samples. At the end of the culture period, PBMCs were washed once and subsequently stained with near-infrared fixable live/dead stain (Invitrogen) together with a cocktail of fluorochrome-conjugated primary human-specific antibodies against CD4, CD8, CD14 (all Biolegend) as well as human Fc blocking reagent (Miltenyi Biotec) for ICS and CD3, CD4 and CD8 (all Biolegend) for proliferation samples. Cells were stained at 4°C in the dark for 20 minutes, followed by one wash. Proliferation samples were then fixed with a 4% formaldehyde solution (Sigma) for 10min at 4°C, washed and stored in PBS in the fridge for up to one day. ICS samples were fixed and permeabilized in Cytofix/Cytoperm buffer (BD) for 20 min at 4°C, washed with 1x Perm buffer (BD) once followed by staining with the following primary human-specific antibodies diluted in Perm buffer: CD3, IFN-γ, TNF (all Biolegend), IL-2 (eBioscience) for 20 min at 4°C followed by one wash in 1x Perm buffer. Cells were stored in cell staining buffer in the fridge for up to one day. Samples were acquired on a MACSQuant analyser 10 and X (Miltenyi Biotec) and analysis was performed using FlowJo software version 10.8.1 (BD Biosciences). Example gating strategies are shown in Figure S4.
Quantification and statistical analysis
Continuous variables are displayed with median and interquartile range (IQR). Unpaired comparisons across two groups were performed using the Mann-Whitney test, and across three groups using the Kruskal-Wallis test with Dunn’s multiple comparisons test. Paired comparisons were performed using the Wilcoxon matched pairs signed rank test. Two-tailed P values are displayed. Statistical analyses were done using R version 4.0.2 (R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/) using the tidyverse packages69 and GraphPad Prism 9.3.1.
Statistical regression models
Multivariate regression models were created to estimate the associations between variables in the study cohort and antibody and T cell immune response. Variables included age, sex, ethnicity, previous infection, time point and vaccine dosing interval. Interactions and co-linearity between variables were explored and variables analysed in separate models where necessary. Generalized linear models were created to estimate associations between the variables sex (discrete), age (continuous), Ethnicity (discrete), previous infection (discrete), and vaccine interval regimen (discrete) on spike ELISpot response (spike B SFU/106; log transformed) or spike IgG response (SARS-Cov-2 S AU/ml; log transformed).
Linear models were created to estimate associations between variables sex (discrete), age (continuous), previous infection (discrete), vaccine regimen (discrete) and Ethnicity (discrete), on spike ELISpot response (IFNγ SFU/106 PBMC; log transformed) or spike IgG response (SARS-Cov-2 S AU/ml; log transformed). GLM models were performed in R /R studio separately for antibody and T cell responses. Models were constructed for all participants and by vaccine regimen to investigate for a differential effect of sex within the BNT162b2 short interval group. Summary tables were reported. To check assumptions were met, residuals vs fitted and residual and normal Q-Q diagnostic plots were created.
Examples of R code used to construct models are:
model_MSD <- glm(MSD_Spike_log ∼ Group + Sex + Age + Exposure, data = Data_antibody)
model_ELISpot <- glm(MSD_Spike_log ∼ Group + Sex + Age + Exposure, data = Data_ ELISpot)
The data objects (Data_antibody and Data_ELISpot) are replaced by the subset data for each vaccine regimen respectively for the models constructed by vaccine regimen.
Acknowledgments
We are grateful to all our healthcare worker colleagues who participated in the study. For the Birmingham participants, the study was carried out at the National Institute for Health Research (NIHR)/Wellcome Trust Birmingham Clinical Research Facility. Laboratory studies were undertaken by the Clinical Immunology Service, University of Birmingham. This work was funded by the UK Department of Health and Social Care as part of the PITCH Consortium, UKRI as part of Investigation of Proven Vaccine Breakthrough by SARS-CoV-2 Variants in Established UK Healthcare Worker cohorts: SIREN consortium & PITCH Plus Pathway, MR/W02067X/1, with contributions from UKRI/NIHR through the UK Coronavirus Immunology Consortium (UK-CIC), the Huo Family Foundation, and the National Institute for Health Research (UKRIDHSC COVID-19 Rapid Response Rolling Call, grant reference number COV19-RECPLAS). E.B. and P.K. are NIHR Senior Investigators, and P.K. is funded by WT109965MA. S.J.D. is funded by an NIHR Global Research Professorship (NIHR300791). T.d.S. is funded by a Wellcome Trust Intermediate Clinical Fellowship (110058/Z/15/Z). R.P.P. is funded by a Career Re-entry Fellowship (204721/Z/16/Z). C.J.A.D. is funded by a Wellcome Clinical Research Career Development Fellowship (211153/Z/18/Z). J.M. and G.S. are funded by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS), China (grant number 2018-I2M-2-002), Schmidt Futures, the Red Avenue Foundation, and the Oak Foundation. The Wellcome Center for Human Genetics is supported by the Wellcome Trust (grant 090532/Z/09/Z). P.C.M. is funded by Wellcome (110,110z/15/Z), the Francis Crick Institute, and the University College London Hospital NIHR Biomedical Research Center. J.E.D.T. is supported by the Medical Research Council (MR/W020564/1) and (MC_UU_0025/12). L.T. is supported by the Wellcome Trust (grant number 205228/Z/16/Z), the National Institute for Health Research Health Protection Research Unit (NIHR HPRU) in Emerging and Zoonotic Infections (EZI) (NIHR200907), and the Centre of Excellence in Infectious Diseases Research (CEIDR) and the Alder Hey Charity. The HPRU-EZI at the University of Liverpool is in partnership with UK Health Security Agency (UKHSA), in collaboration with Liverpool School of Tropical Medicine and the University of Oxford. D.G.W. is supported by an NIHR Advanced Fellowship in Liverpool. M.C., S.L., L.T., and T.T. are supported by the US Food and Drug Administration Medical Countermeasures Initiative contract 75F40120C00085. The Sheffield Teaching Hospitals Observational Study of Patients with Pulmonary Hypertension, Cardiovascular and other Respiratory Diseases (STH-ObS) was supported by the British Heart Foundation (PG/11/116/29,288). The STH-ObS Chief Investigator Allan Laurie is supported by a British Heart Foundation Senior Basic Science Research fellowship (FS/18/52/33808). We gratefully acknowledge financial support from the UK Department of Health and Social Care via the Sheffield NIHR Clinical Research Facility award to the Sheffield Teaching Hospitals Foundation NHS Trust. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health and Social Care, Public Health England, or the US Food and Drug Administration.
Author contributions
Conceptualization, L.T., S.J.D., P.K., T.d.S., S.H., V.H., C.J.A.D., R.P.P., A.R., M.C., and G.S.; methodology, S.J.D., P.K., L.T., S.C.M., B.K., S.L., T.d.S., C.J.A.D., A.R., M.C., G.S., C.D., N.G., S.H., and V.H.; formal analysis, B.K., S.C.M., S.J.D., L.T., T.d.S., C.D., S.L., D.T.S., W.D., A.S.D., S.A., and J.D.W.; investigation, B.K., R.P.P., S.L., C.L., W.D., S.A., N.M., S.F., S.A.-T., S.C.M., T.T., L.M.H., A.A., R.B., A.R.N., S.L.D., E.C.H., L.H.B., P.S., A.C., A.B.-W., L.S.R., A.L., J.K.T., H.H., I.G., M.P., P.Z., T.A.H.N., J.M.N., P.A., E.P., T.M., I.N., A.H., A. Shields, L.S., D.G.W., A.B., and M.Z.; resources, A.B., L.T., and E.B.; data curation, S.C.M. and A.D.; writing – original draft, L.T., S.J.D., and P.K.; writing – review & editing, B.K., S.C.M., S.L., T.d.S., S.L.D., S.J., D.G.W., C.P.C., K.J., P.C.M., A.J.P., J.M., E.B., A.R., M.C., and G.S.; visualization, S.C.M., S.L., B.K., S.A., J.D.W., L.T., and S.J.D.; supervision, B.K., C.P.C., K.J., J.F., A.J.P., S.L.R.-J., J.E.D.T., R.P.P., J.M., E.B., S.H., V.H., C.D., C.J.A.D., A.R., M.C., G.S., T.d.S., L.T., P.K., and S.J.D.; project administration, A.S.D.; funding acquisition, P.K., S.J.D., L.T., T.d.S., C.J.A.D., A.R., S.H., and V.H.; statistical analyses, S.C.M., B.K., S.L., and L.T., overseen by L.T., S.J.D., T.d.S., and P.K. The following authors had unrestricted access to all the data: S.C.M., L.T., S.J.D., and P.K. All authors approved the final draft of the manuscript and take responsibility for its content, including the accuracy of the data.
Declaration of interests
S.J.D. is a Scientific Advisor to the Scottish Parliament on COVID-19, for which she receives a fee. A.J.P. is Chair of UK Department of Health and Social Care’s (DHSC) Joint Committee on Vaccination and Immunisation (JCVI) but does not participate in policy decisions on COVID-19 vaccines. He was previously a member of the WHO’s SAGE. The views expressed in this article do not necessarily represent the views of DHSC, JCVI, or WHO. A.J.P. is chief investigator on clinical trials of Oxford University’s COVID-19 vaccine funded by NIHR. Oxford University has entered a joint COVID-19 vaccine development partnership with AstraZeneca. G.S. sits on the GSK Vaccines Scientific Advisory Board and is a founder member of RQ Biotechnology.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: February 16, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2023.02.004.
Supplemental information
Data and code availability
-
•
IFNγ ELISpot data, MSD data, angiotensin-converting enzyme (ACE)2 inhibition and neutralisation data derived from human samples have been deposited at: https://data.mendeley.com/datasets/fyp26zjgmj/1 and are publicly available as of the date of publication. De-identified participant metadata is available in the same location.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- 1.UK Health Security Agency . UK Health Security Agency; 2022. COVID-19 Vaccine Surveillance Report: 12 May 2022 (Week 19) [Google Scholar]
- 2.Tartof S.Y., Slezak J.M., Fischer H., Hong V., Ackerson B.K., Ranasinghe O.N., Frankland T.B., Ogun O.A., Zamparo J.M., Gray S., et al. Effectiveness of mRNA BNT162b2 COVID-19 vaccine up to 6 months in a large integrated health system in the USA: a retrospective cohort study. Lancet. 2021;398:1407–1416. doi: 10.1016/S0140-6736(21)02183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews N., Stowe J., Kirsebom F., Toffa S., Rickeard T., Gallagher E., Gower C., Kall M., Groves N., O’Connell A.-M., et al. Covid-19 vaccine effectiveness against the omicron (B.1.1.529) variant. N. Engl. J. Med. 2022;386:1532–1546. doi: 10.1056/NEJMoa2119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall V.J., Foulkes S., Charlett A., Atti A., Monk E.J.M., Simmons R., Wellington E., Cole M.J., Saei A., Oguti B., et al. SARS-CoV-2 infection rates of antibody-positive compared with antibody-negative health-care workers in England: a large, multicentre, prospective cohort study (SIREN) Lancet. 2021;397:1459–1469. doi: 10.1016/S0140-6736(21)00675-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall V.J., Foulkes S., Saei A., Andrews N., Oguti B., Charlett A., Wellington E., Stowe J., Gillson N., Atti A., et al. COVID-19 vaccine coverage in health-care workers in England and effectiveness of BNT162b2 mRNA vaccine against infection (SIREN): a prospective, multicentre, cohort study. Lancet. 2021;397:1725–1735. doi: 10.1016/S0140-6736(21)00790-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall V., Foulkes S., Insalata F., Kirwan P., Saei A., Atti A., Wellington E., Khawam J., Munro K., Cole M., et al. Protection against SARS-CoV-2 after Covid-19 vaccination and previous infection. N. Engl. J. Med. 2022;386:1207–1220. doi: 10.1056/NEJMoa2118691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payne R.P., Longet S., Austin J.A., Skelly D.T., Dejnirattisai W., Adele S., Meardon N., Faustini S., Al-Taei S., Moore S.C., et al. Immunogenicity of standard and extended dosing intervals of BNT162b2 mRNA vaccine. Cell. 2021;184:5699–5714.e11. doi: 10.1016/j.cell.2021.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Earle K.A., Ambrosino D.M., Fiore-Gartland A., Goldblatt D., Gilbert P.B., Siber G.R., Dull P., Plotkin S.A. Evidence for antibody as a protective correlate for COVID-19 vaccines. Vaccine. 2021;39:4423–4428. doi: 10.1016/j.vaccine.2021.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert P.B., Montefiori D.C., McDermott A.B., Fong Y., Benkeser D., Deng W., Zhou H., Houchens C.R., Martins K., Jayashankar L., et al. Immune correlates analysis of the mRNA-1273 COVID-19 vaccine efficacy clinical trial. Science. 2022;375:43–50. doi: 10.1126/science.abm3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng S., Phillips D.J., White T., Sayal H., Aley P.K., Bibi S., Dold C., Fuskova M., Gilbert S.C., Hirsch I., et al. Correlates of protection against symptomatic and asymptomatic SARS-CoV-2 infection. Nat. Med. 2021;27:2032–2040. doi: 10.1038/s41591-021-01540-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khoury D.S., Cromer D., Reynaldi A., Schlub T.E., Wheatley A.K., Juno J.A., Subbarao K., Kent S.J., Triccas J.A., Davenport M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021;27:1205–1211. doi: 10.1038/s41591-021-01377-8. [DOI] [PubMed] [Google Scholar]
- 12.Addetia A., Crawford Katharine H.D., Dingens A., Zhu H., Roychoudhury P., Huang M.-L., Jerome Keith R., Bloom Jesse D., Greninger Alexander L. Neutralizing antibodies correlate with protection from SARS-CoV-2 in humans during a fishery vessel outbreak with a high attack rate. J. Clin. Microbiol. 2020;58:e02107–e02120. doi: 10.1128/JCM.02107-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore P.L., Moyo-Gwete T., Hermanus T., Kgagudi P., Ayres F., Makhado Z., Sadoff J., Le Gars M., van Roey G., Crowther C., et al. Neutralizing antibodies elicited by the Ad26.COV2.S COVID-19 vaccine show reduced activity against 501Y.V2 (B.1.351), despite protection against severe disease by this variant. bioRxiv. 2021 doi: 10.1101/2021.06.09.447722. Preprint at. [DOI] [Google Scholar]
- 14.Ewer K.J., Barrett J.R., Belij-Rammerstorfer S., Sharpe H., Makinson R., Morter R., Flaxman A., Wright D., Bellamy D., Bittaye M., et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 2021;27:270–278. doi: 10.1038/s41591-020-01194-5. [DOI] [PubMed] [Google Scholar]
- 15.Tomic A., Skelly D.T., Ogbe A., O'Connor D., Pace M., Adland E., Alexander F., Ali M., Allott K., Azim Ansari M., et al. Divergent trajectories of antiviral memory after SARS-CoV-2 infection. Nat. Commun. 2022;13:1251. doi: 10.1038/s41467-022-28898-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplonek P., Cizmeci D., Fischinger S., Collier A-r, Suscovich T., Linde C., Broge T., Mann C., Amanat F., Dayal D., et al. mRNA-1273 and BNT162b2 COVID-19 vaccines elicit antibodies with differences in Fc-mediated effector functions. Sci. Transl. Med. 2022;14:eabm2311. doi: 10.1126/scitranslmed.abm2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nyberg T., Ferguson N.M., Nash S.G., Webster H.H., Flaxman S., Andrews N., Hinsley W., Bernal J.L., Kall M., Bhatt S., et al. Comparative analysis of the risks of hospitalisation and death associated with SARS-CoV-2 omicron (B.1.1.529) and delta (B.1.617.2) variants in England: a cohort study. Lancet. 2022;399:1303–1312. doi: 10.1016/S0140-6736(22)00462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mefsin Y.M., Chen D., Bond H.S., Lin Y., Cheung J.K., Wong J.Y., Ali S.T., Lau E.H.Y., Wu P., Leung G.M., et al. Epidemiology of infections with SARS-CoV-2 omicron BA.2 variant, Hong Kong, January-March 2022. Emerg. Infect. Dis. 2022;28:1856–1858. doi: 10.3201/eid2809.220613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sette A., Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell. 2021;184:861–880. doi: 10.1016/j.cell.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMahan K., Yu J., Mercado N.B., Loos C., Tostanoski L.H., Chandrashekar A., Liu J., Peter L., Atyeo C., Zhu A., et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature. 2021;590:630–634. doi: 10.1038/s41586-020-03041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kedzierska K., Thomas P.G. Count on us: T cells in SARS-CoV-2 infection and vaccination. Cell Rep. Med. 2022;3:100562. doi: 10.1016/j.xcrm.2022.100562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molodtsov I.A., Kegeles E., Mitin A.N., Mityaeva O., Musatova O.E., Panova A.E., Pashenkov M.V., Peshkova I.O., Alsalloum A., Asaad W., et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)–specific T cells and antibodies in coronavirus disease 2019 (COVID-19) protection: a prospective study. Clin. Infect. Dis. 2022;75:e1–e9. doi: 10.1093/cid/ciac278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplonek P., Fischinger S., Cizmeci D., Bartsch Y.C., Kang J., Burke J.S., Shin S.A., Dayal D., Martin P., Mann C., et al. mRNA-1273 vaccine-induced antibodies maintain Fc effector functions across SARS-CoV-2 variants of concern. Immunity. 2022;55:355–365.e4. doi: 10.1016/j.immuni.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartsch Y.C., Tong X., Kang J., Avendaño M.J., Serrano E.F., García-Salum T., Pardo-Roa C., Riquelme A., Cai Y., Renzi I., et al. Omicron variant Spike-specific antibody binding and Fc activity are preserved in recipients of mRNA or inactivated COVID-19 vaccines. Sci. Transl. Med. 2022;14:eabn9243. doi: 10.1126/scitranslmed.abn9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kundu R., Narean J.S., Wang L., Fenn J., Pillay T., Fernandez N.D., Conibear E., Koycheva A., Davies M., Tolosa-Wright M., et al. Cross-reactive memory T cells associate with protection against SARS-CoV-2 infection in COVID-19 contacts. Nat. Commun. 2022;13:80. doi: 10.1038/s41467-021-27674-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogbe A., Kronsteiner B., Skelly D.T., Pace M., Brown A., Adland E., Adair K., Akhter H.D., Ali M., Ali S.-E., et al. T cell assays differentiate clinical and subclinical SARS-CoV-2 infections from cross-reactive antiviral responses. Nat. Commun. 2021;12:2055. doi: 10.1038/s41467-021-21856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swadling L., Diniz M.O., Schmidt N.M., Amin O.E., Chandran A., Shaw E., Pade C., Gibbons J.M., Le Bert N., Tan A.T., et al. Pre-existing polymerase-specific T cells expand in abortive seronegative SARS-CoV-2. Nature. 2022;601:110–117. doi: 10.1038/s41586-021-04186-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldberg Y., Mandel M., Bar-On Y.M., Bodenheimer O., Freedman L., Haas E.J., Milo R., Alroy-Preis S., Ash N., Huppert A. Waning immunity after the BNT162b2 vaccine in Israel. N. Engl. J. Med. 2021;385:e85. doi: 10.1056/NEJMoa2114228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naaber P., Tserel L., Kangro K., Sepp E., Jürjenson V., Adamson A., Haljasmägi L., Rumm A.P., Maruste R., Kärner J., et al. Dynamics of antibody response to BNT162b2 vaccine after six months: a longitudinal prospective study. Lancet Reg. Health. Eur. 2021;10:100208. doi: 10.1016/j.lanepe.2021.100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei J., Pouwels K.B., Stoesser N., Matthews P.C., Diamond I., Studley R., Rourke E., Cook D., Bell J.I., Newton J.N., et al. Antibody responses and correlates of protection in the general population after two doses of the ChAdOx1 or BNT162b2 vaccines. Nat. Med. 2022;28:1072–1082. doi: 10.1038/s41591-022-01721-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg E.S., Dorabawila V., Easton D., Bauer U.E., Kumar J., Hoen R., Hoefer D., Wu M., Lutterloh E., Conroy M.B., et al. Covid-19 vaccine effectiveness in New York state. N. Engl. J. Med. 2022;386:116–127. doi: 10.1056/NEJMoa2116063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carazo S., Skowronski D.M., Brisson M., Barkati S., Sauvageau C., Brousseau N., Gilca R., Fafard J., Talbot D., Ouakki M., et al. Protection against omicron (B.1.1.529) BA.2 reinfection conferred by primary omicron BA.1 or pre-omicron SARS-CoV-2 infection among health-care workers with and without mRNA vaccination: a test-negative case-control study. Lancet Infect. Dis. 2023;23:45–55. doi: 10.1016/S1473-3099(22)00578-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin D.-Y., Gu Y., Wheeler B., Young H., Holloway S., Sunny S.-K., Moore Z., Zeng D. Effectiveness of Covid-19 vaccines over a 9-month period in North Carolina. N. Engl. J. Med. 2022;386:933–941. doi: 10.1056/NEJMoa2117128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dejnirattisai W., Huo J., Zhou D., Zahradník J., Supasa P., Liu C., Duyvesteyn H.M.E., Ginn H.M., Mentzer A.J., Tuekprakhon A., et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell. 2022;185:467–484.e15. doi: 10.1016/j.cell.2021.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt F., Muecksch F., Weisblum Y., Da Silva J., Bednarski E., Cho A., Wang Z., Gaebler C., Caskey M., Nussenzweig M.C., et al. Plasma neutralization of the SARS-CoV-2 omicron variant. N. Engl. J. Med. 2022;386:599–601. doi: 10.1056/NEJMc2119641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skelly D.T., Harding A.C., Gilbert-Jaramillo J., Knight M.L., Longet S., Brown A., Adele S., Adland E., Brown H., et al. Medawar Laboratory Team Two doses of SARS-CoV-2 vaccination induce robust immune responses to emerging SARS-CoV-2 variants of concern. Nat. Commun. 2021;12:5061. doi: 10.1038/s41467-021-25167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keeton R., Tincho M.B., Ngomti A., Baguma R., Benede N., Suzuki A., Khan K., Cele S., Bernstein M., Karim F., et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature. 2022;603:488–492. doi: 10.1038/s41586-022-04460-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarke A., Coelho C.H., Zhang Z., Dan J.M., Yu E.D., Methot N., Bloom N.I., Goodwin B., Phillips E., Mallal S., et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell. 2022;185:847–859.e11. doi: 10.1016/j.cell.2022.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Y., Cai C., Grifoni A., Müller T.R., Niessl J., Olofsson A., Humbert M., Hansson L., Österborg A., Bergman P., et al. Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat. Med. 2022;28:472–476. doi: 10.1038/s41591-022-01700-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GeurtsvanKessel C.H., Geers D., Schmitz K.S., Mykytyn A.Z., Lamers M.M., Bogers S., Scherbeijn S., Gommers L., Sablerolles R.S.G., Nieuwkoop N.N., et al. Divergent SARS-CoV-2 Omicron–reactive T and B cell responses in COVID-19 vaccine recipients. Sci. Immunol. 2022;7:eabo2202. doi: 10.1126/sciimmunol.abo2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madelon N., Heikkilä N., Sabater Royo I., Fontannaz P., Breville G., Lauper K., Goldstein R., Grifoni A., Sette A., Siegrist C.A., et al. Omicron-specific cytotoxic T-cell responses after a third dose of mRNA COVID-19 vaccine among patients with multiple sclerosis treated with ocrelizumab. JAMA Neurol. 2022;79:399–404. doi: 10.1001/jamaneurol.2022.0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J., Chandrashekar A., Sellers D., Barrett J., Jacob-Dolan C., Lifton M., McMahan K., Sciacca M., VanWyk H., Wu C., et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature. 2022;603:493–496. doi: 10.1038/s41586-022-04465-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Marco L., D’Orso S., Pirronello M., Verdiani A., Termine A., Fabrizio C., Capone A., Sabatini A., Guerrera G., Placido R., et al. Assessment of T-cell reactivity to the SARS-CoV-2 omicron variant by immunized individuals. JAMA Netw. Open. 2022;5:e2210871. doi: 10.1001/jamanetworkopen.2022.10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parry H., Bruton R., Stephens C., Bentley C., Brown K., Amirthalingam G., Hallis B., Otter A., Zuo J., Moss P. Extended interval BNT162b2 vaccination enhances peak antibody generation. NPJ Vaccines. 2022;7:14. doi: 10.1038/s41541-022-00432-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otter A.D., D’Arcangelo S., Whitaker H., Hewson J., Foulkes S., Atti A., Cole M., Linley E., Tonge S., Hettiarachchi N., et al. Determinants of SARS-CoV-2 anti-spike antibody levels following BNT162b2 vaccination: cross-sectional analysis of 6,000 SIREN study participants. medRxiv. 2022 doi: 10.1101/2022.04.21.22274025. Preprint at. [DOI] [Google Scholar]
- 47.Skowronski D.M., Febriani Y., Ouakki M., Setayeshgar S., El Adam S., Zou M., Talbot D., Prystajecky N., Tyson J.R., Gilca R., et al. Two-dose severe acute respiratory syndrome coronavirus 2 vaccine effectiveness with mixed schedules and extended dosing intervals: test-negative design studies from British Columbia and Quebec, Canada. Clin. Infect. Dis. 2022;75:1980–1992. doi: 10.1093/cid/ciac290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barnes E., Folgori A., Capone S., Swadling L., Aston S., Kurioka A., Meyer J., Huddart R., Smith K., Townsend R., et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Transl. Med. 2012;4:115ra1. doi: 10.1126/scitranslmed.3003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., et al. COVID-19 Genomics UK COG-UK Consortium SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tartof S.Y., Slezak J.M., Puzniak L., Hong V., Xie F., Ackerson B.K., Valluri S.R., Jodar L., McLaughlin J.M. Durability of BNT162b2 vaccine against hospital and emergency department admissions due to the omicron and delta variants in a large health system in the USA: a test-negative case control study. Lancet Respir. Med. 2022;10:689–699. doi: 10.1016/S2213-2600(22)00101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scurr M.J., Lippiatt G., Capitani L., Bentley K., Lauder S.N., Smart K., Somerville M.S., Rees T., Stanton R.J., Gallimore A., et al. Magnitude of venous or capillary blood-derived SARS-CoV-2-specific T cell response determines COVID-19 immunity. Nat. Commun. 2022;13:5422. doi: 10.1038/s41467-022-32985-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maringer Y., Nelde A., Schroeder S.M., Schuhmacher J., Hörber S., Peter A., Karbach J., Jäger E., Walz J.S. Durable spike-specific T-cell responses after different COVID-19 vaccination regimens are not further enhanced by booster vaccination. Sci. Immunol. 2022;7:eadd3899. doi: 10.1126/sciimmunol.add3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Altarawneh H.N., Chemaitelly H., Ayoub H.H., Tang P., Hasan M.R., Yassine H.M., Al-Khatib H.A., Smatti M.K., Coyle P., Al-Kanaani Z., et al. Effects of previous infection and vaccination on symptomatic omicron infections. N. Engl. J. Med. 2022;387:21–34. doi: 10.1056/NEJMoa2203965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei J., Matthews P.C., Stoesser N., Newton J.N., Diamond I., Studley R., Taylor N., Bell J.I., Farrar J., Kolenchery J., et al. Correlates of protection against SARS-CoV-2 Omicron variant and anti-spike antibody responses after a third/booster vaccination or breakthrough infection in the UK general population. medRxiv. 2022 doi: 10.1101/2022.11.29.22282916. Preprint at. [DOI] [Google Scholar]
- 55.Bobrovitz N., Ware H., Ma X., Li Z., Hosseini R., Cao C., Selemon A., Whelan M., Premji Z., Issa H., et al. Protective effectiveness of previous SARS-CoV-2 infection and hybrid immunity against the omicron variant and severe disease: a systematic review and meta-regression. Lancet Infect. Dis. 2023;S1473-3099:00801-5. doi: 10.1016/S1473-3099(22)00801-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ward H., Whitaker M., Flower B., Tang S.N., Atchison C., Darzi A., Donnelly C.A., Cann A., Diggle P.J., Ashby D., et al. Population antibody responses following COVID-19 vaccination in 212,102 individuals. Nat. Commun. 2022;13:907. doi: 10.1038/s41467-022-28527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wall E.C., Wu M., Harvey R., Kelly G., Warchal S., Sawyer C., Daniels R., Adams L., Hobson P., Hatipoglu E., et al. AZD1222-induced neutralising antibody activity against SARS-CoV-2 Delta VOC. Lancet. 2021;398:207–209. doi: 10.1016/S0140-6736(21)01462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reynolds C.J., Pade C., Gibbons J.M., Otter A.D., Lin K.M., Muñoz Sandoval D., Pieper F.P., Butler D.K., Liu S., Joy G., et al. Immune boosting by B.1.1.529 (Omicron) depends on previous SARS-CoV-2 exposure. Science. 2022;377:eabq1841. doi: 10.1126/science.abq1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansen C.H., Friis N.U., Bager P., Stegger M., Fonager J., Fomsgaard A., Gram M.A., Christiansen L.E., Ethelberg S., Legarth R., et al. Risk of reinfection, vaccine protection, and severity of infection with the BA.5 omicron subvariant: a nation-wide population-based study in Denmark. Lancet Infect. Dis. 2023;23:167–176. doi: 10.1016/S1473-3099(22)00595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Z., Mateus J., Coelho C.H., Dan J.M., Moderbacher C.R., Gálvez R.I., Cortes F.H., Grifoni A., Tarke A., Chang J., et al. Humoral and cellular immune memory to four COVID-19 vaccines. Cell. 2022;185:2434–2451. doi: 10.1016/j.cell.2022.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Antonelli M., Penfold R.S., Merino J., Sudre C.H., Molteni E., Berry S., Canas L.S., Graham M.S., Klaser K., Modat M., et al. Risk factors and disease profile of post-vaccination SARS-CoV-2 infection in UK users of the COVID Symptom Study app: a prospective, community-based, nested, case-control study. Lancet Infect. Dis. 2022;22:43–55. doi: 10.1016/S1473-3099(21)00460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Angyal A., Longet S., Moore S.C., Payne R.P., Harding A., Tipton T., Rongkard P., Ali M., Hering L.M., Meardon N., et al. T-cell and antibody responses to first BNT162b2 vaccine dose in previously infected and SARS-CoV-2-naive UK health-care workers: a multicentre prospective cohort study. Lancet. Microbe. 2022;3:e21–e31. doi: 10.1016/S2666-5247(21)00275-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whitaker H.J., Gower C., Otter A.D., Simmons R., Kirsebom F., Letley L., Quinot C., Ireland G., Linley E., Ribeiro S., et al. Nucleocapsid antibody positivity as a marker of past SARS-CoV-2 infection in population serosurveillance studies: impact of variant, vaccination, and choice of assay cut-off. medRxiv. 2021 doi: 10.1101/2021.10.25.21264964. Preprint at. [DOI] [Google Scholar]
- 64.Follmann D., Janes H.E., Buhule O.D., Zhou H., Girard B., Marks K., Kotloff K., Desjardins M., Corey L., Neuzil K.M., et al. Antinucleocapsid antibodies after SARS-CoV-2 infection in the blinded phase of the randomized, placebo-controlled mRNA-1273 COVID-19 vaccine efficacy clinical trial. Ann. Intern. Med. 2022;175:1258–1265. doi: 10.7326/M22-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fröberg J., Gillard J., Philipsen R., Lanke K., Rust J., Van Tuijl D., Teelen K., Bousema T., Simonetti E., Van Der Gaast-De Jongh C.E., et al. SARS-CoV-2 mucosal antibody development and persistence and their relation to viral load and COVID-19 symptoms. Nat. Commun. 2021;12:5621. doi: 10.1038/s41467-021-25949-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ssemaganda A., Nguyen H.M., Nuhu F., Jahan N., Card C.M., Kiazyk S., Severini G., Keynan Y., Su R.-C., Ji H., et al. Expansion of cytotoxic tissue-resident CD8+ T cells and CCR6+CD161+ CD4+ T cells in the nasal mucosa following mRNA COVID-19 vaccination. Nat. Commun. 2022;13:3357. doi: 10.1038/s41467-022-30913-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sano K., Bhavsar D., Singh G., Floda D., Srivastava K., Gleason C., PARIS Study Group. Carreño J.M., Simon V., Krammer F., et al. SARS-CoV-2 vaccination induces mucosal antibody responses in previously infected individuals. Nat. Commun. 2022;13:5135. doi: 10.1038/s41467-022-32389-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caly L., Druce J., Roberts J., Bond K., Tran T., Kostecki R., Yoga Y., Naughton W., Taiaroa G., Seemann T., et al. Isolation and rapid sharing of the 2019 novel coronavirus (SARS-CoV-2) from the first patient diagnosed with COVID-19 in Australia. Med. J. Aust. 2020;212:459–462. doi: 10.5694/mja2.50569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the tidyverse. J. Open Source Softw. 2019;4:1686. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
IFNγ ELISpot data, MSD data, angiotensin-converting enzyme (ACE)2 inhibition and neutralisation data derived from human samples have been deposited at: https://data.mendeley.com/datasets/fyp26zjgmj/1 and are publicly available as of the date of publication. De-identified participant metadata is available in the same location.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.