

Abstract
Ongoing research has highlighted the significance of the cross-play of macrophages and mesenchymal stem cells (MSCs). Lysine-specific demethylase 6B (KDM6B) has been shown to control osteogenic differentiation of MSCs by depleting trimethylated histone 3 lysine 27 (H3K27me3). However, to date, the role of KDM6B in bone marrow-derived macrophages (BMDMs) remains controversial. Here, a chromatin immunoprecipitation assay (ChIP) proved that KDM6B derived from osteogenic-induced BMSCs could bind to the promoter region of BMDMs’ brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein-1 (BMAL1) gene in a coculture system and activate BMAL1. Transcriptome sequencing and experiments in vitro showed that the overexpression of BMAL1 in BMDM could inhibit the TLR2/NF-κB signaling pathway, reduce pyroptosis, and decrease the M1/M2 ratio, thereby promoting osteogenic differentiation of BMSCs. Furthermore, bone and macrophage dual-targeted GSK-J4 (KDM6B inhibitor)-loaded nanodiscs were synthesized via binding SDSSD-apoA-1 peptide analogs (APA) peptide, which indirectly proved the critical role of KDM6B in osteogenesis in vivo. Overall, we demonstrated that KDM6B serves as a positive circulation trigger during osteogenic differentiation by decreasing the ratio of M1/M2 both in vitro and in vivo. Collectively, these results provide insight into basic research in the field of osteoporosis and bone repair.
Keywords: KDM6B; osteogenesis; macrophage; circadian rhythm; H3K27me3, BMAL1
Introduction
Osteoporosis, the most prevalent bone disorder worldwide, is defined as a systemic bone disease characterized by low bone mass and deterioration of bone tissue microstructure. Ultimately, osteoporosis leads to an increased susceptibility to bone fractures.1,2 The worldwide prevalence of osteoporosis is nearly 20%, with a higher incidence in the female population.3 More recently, epigenetic regulation is emerging as a key factor in cell differentiation, thus offering a potential avenue in disease treatment.4 More specifically, epigenetic modification determines gene expression and responds to environmental cues to guide cell lineage specification. Trimethylated histone 3 lysine 27 (H3K27me3), a key marker of gene suppression, is known as one of the most dominant epigenetic signatures.5,6 The suppression of H3K27me3 resulted in a downregulation of mesenchymal stem cell (MSC) lineage specification to adipocytes and was also found to orchestrate osteoblast activity and bone formation in mice.7,8
MSCs have been extensively studied for their applications in stem-cell-based regeneration. MSCs are precursor cells of bone and cartilage tissue, which have been shown to regenerate ectopic bone tissue in vivo. It has been reported that lysine-specific demethylase 4B (KDM4B) and lysine-specific demethylase 6B (KDM6B) are most strongly induced by bone morphogenetic protein (BMP)-4/7 and promote osteogenic differentiation of human MSCs.9 KDM6B is recruited to the BMP2 and HOXC6 promoters, resulting in the removal of H3K27me3 markers and activation of the transcription of BMP2 and HOXC6.10 Jmjd3 (KDM6B)-mediated H3K27 demethylation is considered a crucial factor for regulating M2 macrophage development, leading to anti-helminth host responses.11 In addition, macrophages are well known to play a pivotal role in the induction of bone mesenchymal stem cells toward osteoblastic fate.12,13 Emerging evidence has demonstrated that the cross-talk between monocyte-macrophage-osteoclasts and bone marrow stromal cells (BMSCs)-osteoblasts is a pathological cornerstone in osteoporosis.14 Macrophages are critically important for the recruitment and regulation of MSC differentiation during bone regeneration. In fact, animal studies have demonstrated that macrophages are essential for fracture healing.15 The bone remodeling cycle is regulated by local and systemic factors. Activated macrophages can drive bone formation and are also involved in tissue repair.16
Circadian rhythmicity is maintained by a well-documented molecular clock involving the transcription of circadian-related genes such as period (PER1, PER2, PER3) and cryptochrome (CRY1, CRY2), which are activated by the dimerization of brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein-1 (BMAL1) and circadian locomotor output cycles kaput (CLOCK).17 Such circadian rhythm genes have been identified in the hypothalamic suprachiasmatic nucleus (SCN) and in peripheral cells, including osteoclasts,18 BMSCs,19 and immune cells.20 Thus, it is essential to understand the underlying mechanisms between osteoporosis and circadian rhythmicity. Over the past decades, a mounting body of research has revealed the link between BMAL1 and skeletal development. Holding a strong oscillatory profile, BMAL1 has been found to be vital for embryonic development, intramembranous ossification, osteoblast differentiation, and bone formation.21,22 However, in this context, the evidence on circadian rhythm and macrophage polarization is scarce. While regulating rhythmic gene expression in macrophages, BMAL1 also interacts with STAT6 and other factors to maintain homeostasis of macrophage function.23 With BMAL1 serving as a key metabolic sensor in macrophages, its deficiency results in a metabolic shift of increased glycolysis and mitochondrial respiration, leading to an enhanced pro-inflammatory state, energy stress, and Hif-1α-dependent metabolic reprogramming.24,25 Therefore, it is crucial to decipher the relationship between the rhythmicity of macrophages and BMSCs in the process of osteoblastic differentiation.
Our results demonstrated that the increased KDM6B can bind the promoter region of BMAL1 in bone marrow-derived macrophages (BMDMs) and demethylate H3K27m3 during BMSC-induced osteogenesis. After treatment with GSK-J4 (inhibitor of KDM6B), the expression of rhythm-related proteins in BMDM was significantly reduced. In addition, cell-level experiments revealed that KDM6B promoted the differentiation of BMDM into M2. Endogenous and synthetic lipoproteins, such as high-density lipoprotein (HDL) nanodiscs, have emerged as a promising drug delivery system (DDS) for small molecules, peptides, and nucleic acids due to their inherent long half-life in vivo and passive targeting characteristics.26,27 Based on our recently reported work on reprogrammable peptide analogs of apolipoprotein A-I (APA) for tunable nanodisc engineering,28 we propose a GSK-J4-loaded nanodisc (ND-GSK-J4) targeting macrophages in bone by introducing a bone formation surface-targeting peptide-modified membrane scaffold (SDSSD-APA) and administered it to mice. After administration, it was noted that macrophages in the bone marrow of mice tended to exhibit the M1 type, and bone loss increased. Finally, we developed a lentivirus-mediated BMAL1 overexpression (OE) model and performed transcriptome sequencing of BMAL1-OE and BMAL1-negative control (NC) BMDMs. Thus, we demonstrated that KDM6B released during osteogenic differentiation of BMSCs can promote the overexpression of BMAL1 in BMDM and decrease the ratio of M1/M2 by inhibiting the TLR2/NF-κB pathway, ultimately reducing macrophage pyroptosis and promoting osteogenesis. A comprehensive understanding of this cellular cross-play in the osteogenic microenvironment may help develop effective treatment modalities.
Results and Discussion
Bone and Macrophage Dual-Targeted Nanodiscs Were Designed as the Carrier for GSK-J4 to Study the Function of KDM6B in Vivo
Recent research efforts have studied the mechanism of interaction between macrophages and MSCs in vitro. In infarcted myocardium injected with MSCs, macrophages adjacent to MSCs showed strong expression of Arg-1.29 However, to date, the role of KDM6B in BMDM polarization is still controversially discussed.30
To elucidate the association between the differentiation ability of BMSCs and BMDMs in bone marrow in vivo, we synthesized a bone and macrophage dual-target-GSK-J4 (KDM6B inhibitor)-delivering nanodisc (DN-GSK-J4). The schematic diagram of this nanodisc synthesis is illustrated in Figure 1a. Crude SDSSD-APA peptide was purified and analyzed by high-performance liquid chromatography (HPLC) and mass spectrometry (MS, Supplementary Figure S1). Characterizations of ND and ND-GSK-J4 were performed by dynamic light scattering (DLS) and transmission electron microscopy (TEM). As shown, the diameter of the ND ranged from 8 to 15 nm, with the diameter increasing to 15–30 nm when loaded with GSK-J4 (Figure 1b). ND and ND-GSK-J4 were measured again after 4 weeks at 4 °C via DLS, and the results indicated that the diameter distribution range was not significantly different from that when it was first synthesized, demonstrating its good colloidal stability (Figure 1b). The TEM results also reflected the differences in size and shape between ND and ND-GSK-J4 (Figure 1c). Nanodiscs were associated with Dio and showed green fluorescence. To explore the targeting of the nanodiscs at the cellular level, we co-incubated Dio-ND-GSK-J4 with BMSCs or BMDMs. The results showed that—at the same dosage—the green fluorescence of the BMDM group was significantly stronger, indicating macrophage targeting of ND-GSK-J4 (Figure 1d). We then injected mice with 200 μL of Dio-ND-GSK-J4 and Dio-unloaded/untargeted-ND (Dio-NC-ND, without SDSSD peptide) through the tail vein. After 12 h, in vivo imaging revealed that both Dio-ND-GSK-J4 and Dio-NC-ND are mainly metabolized by the liver and Dio-ND-GSK-J4 clearly targeted the bone structure (compared to Dio-NC-ND, Figure 1e).
Figure 1.

Characterization of ND and ND-GSK-J4. (a) Preparation of GSK-J4-loaded nanodiscs (ND-GSK-J4). (b, c) Characterization of ND and ND-GSK-J4 via DLS and TEM (scale bar: 200/50 nm, 100/25 nm). (d) Dio-ND-GSK-J4 were co-incubated with BMSCs or BMDMs. The macrophage target effect in vitro was confirmed using a cellular uptake analysis (scale bar: 10 μm). (e) The bone target effect was confirmed using a Vivo FX PRO imaging system.
KDM6B Derived from BMSC Osteogenic Differentiation Process Accumulated at the BMAL1 E-box of BMDMs
KDM6B was identified as the most upregulated among 28 histone demethylases in BMP-4/7-induced human MSCs.9 We, therefore, tried to induce osteogenic differentiation with conventional differentiation culture medium (“osteo+”, containing dexamethasone, ascorbic acid, and β-glycerophosphate).31 Quantitative polymerase chain reaction (PCR) revealed that the expression level of KDM6B was significantly higher than in the noninduced state (Figure 2a). Macrophages and MSCs are closely associated in the immune microenvironment of the bone marrow. Thus, we speculated whether the significantly increased KDM6B production by MSCs during the process of osteogenic differentiation might be related to macrophages. Over 500 transcription factors were found to be responsible for the polarization of macrophages that are directly regulated by KDM6B-mediated demethylation via chromatin immunoprecipitation-sequencing (ChIP-Seq) analysis.11 BMAL1, a key circadian rhythm gene, determines macrophage fate through epigenetic events. We found that the binding level of H3K27me3 in the promoter region of BMAL1 of BMDM was significantly different in WT and KDM6B (Jmjd3)–/– mice (analyzed using Cistrome Data Browser and UCSC Genome Browser on Mouse, Supplementary Figure S2).11 In order to substantiate the results, we extracted BMDMs from WT mice and found that H3K27me3 could bind to the promoter region of BMAL1 and the binding level is significantly increased after GSK-J4 treatment via the ChIP assay and DNA agarose gel electrophoresis (Figure 2b). The binding of H3K27me3 in the promoter region of BMAL1 then showed a circadian rhythm oscillation (Figure 2c). To investigate whether the demethylation of the BMAL1 promoter region of BMDMs is due to KDM6B produced by BMSCs during the osteogenic differentiation process, we seeded BMDMs in lower chambers of a 24-well Transwell plate and BMSCs in the upper chambers. The results showed that the rhythm oscillation of H3K27me3 in the promoter region of BMAL1 of BMDMs was significantly impaired when cocultured with induced BMSCs (Figure 2c). We also detected the mRNA expression of BMAL1 and found that, in the induced BMSC group, the expression of BMAL1 in BMDM was also significantly increased, which might be related to the impaired inhibitory effect of H3K27me3 (Figure 2d). In BMDMs’ chromatin, KDM6B was enriched at the BMAL1 E-box and showed a circadian rhythm (Figure 2e,f). After adding the KDM6B inhibitor GSK-J4, oscillation of KDM6B at the promoter region of BMAL1 was significantly impaired (compared to the control group via the ChIP assay; Figure 2f). We extracted mouse liver tissue and bone marrow cells at different time points and performed PCR assays. BMAL1 mRNA expression was found to be subject to rhythmic oscillation, while the KDM6B level remained relatively stable. The aforementioned results demonstrated that the KDM6B level itself had no circadian rhythm, while the protein level of KDM6B recruited and bound by the BMAL1 promoter region was subject to circadian fluctuations (Figure 2g). However, the reason for this phenomenon remains unclear, and it may be that there is a complex of rhythm-regulating molecules and KDM6B to regulate the BMAL1 gene,32 which we will further explore in the future.
Figure 2.

KDM6B accumulated at the promoter region of BMAL1 in BMDMs during BMSC osteogenic differentiation. (a) BMSCs and BMDMs were extracted and seeded in the upper or lower chamber of Transwell plates. BMSC osteogenic differentiation culture medium was added (osteo+), and KDM6B mRNA expression levels were detected in all lower chambers via RT-qPCR. (b) The ChIP assay and DNA agarose gel electrophoresis were performed to test the binding level of H3K27me3 in the promoter region of BMAL1. (c) The ChIP assay was performed with synchronized mature BMDMs at specific intervals to show the circadian rhythm oscillation of H3K27me3 binding to the promoter region of BMAL1. (d) BMAL1 mRNA expression levels were also detected in all lower chambers via RT-qPCR. (e, f) The level of KDM6B binding to the promoter region of BMAL1 in BMDMs was increased when BMSCs in the upper chamber were osteogenically differentiated, whereas the addition of GSK-J4 led to the suppression of binding. (g) RT-qPCR was performed to investigate the circadian rhythm oscillation level of BMAL1 and KDM6B in the murine liver and bone marrow (circadian time (CT) 0–12: yellow background; CT12–24: gray background).
KDM6B Was Required for Normal Circadian Function of BMDMs
PCR testing was performed to investigate whether KDM6B is required for the expression of circadian rhythm-related genes in BMDMs. The assays revealed that the level of BMAL1 in BMDMs was significantly suppressed after stimulation with GSK-J4 (Figure 3a). To explore the effect of BMAL1 gene inhibition on macrophages, we introduced BMAL1 conditional knockout (BMAL1–/–) mice. The results of the mouse tail genotype identification are illustrated in Figure 3b. Next, we harvested bone marrow from WT and BMAL1–/– mice. Flow cytometry revealed that there was a significant difference in the proportion of BMDMs (F4/80+CD11b+) in the bone marrow cells of the two mouse types, which suggested a certain degree of damage to the monocyte–macrophage system caused by BMAL1 knockout (Figure 3c,d). To further investigate the effect of KDM6B on the expression levels of circadian rhythm-related genes, we synchronized mature BMDMs with dexamethasone and then stimulated them with GSK-J4. Cells were collected for RNA extraction at CT0, 3, 6, 9, 12, 15, 18, or 21 and for protein extraction at CT0, 4, 8, 12, 16, or 20. PCR results demonstrated that the peaks of mRNA expression of BMAL1, CLOCK, and Cry 1 appeared in CT9, while the trough of mRNA expression of Per1, Per2, and NR1D1 appeared at CT15, CT18, and CT12, respectively. After GSK-J4 stimulation, the expression levels of circadian rhythm genes in BMDMs were significantly decreased, and no obvious circadian rhythm was observed in the expression levels of BMAL1 (Figure 3e,f). Western blotting (WB) results also confirmed the impairment of BMDM circadian rhythms by GSK-J4 (Figure 3g).
Figure 3.

KDM6B played an integral role in the circadian rhythm of BMDMs. (a) GSK-J4 suppressed the BMAL1 mRNA level in BMDMs, as indicated by RT-qPCR testing. (b) DNA gel electrophoresis results of mouse tail identification. (c, d) Identification of the proportion of macrophages in the bone marrow of wild-type and BMAL-KO mice using flow cytometry and the corresponding statistical analysis. (e, f) Synchronized mature macrophages were stimulated by GSK-J4, and the mRNA and protein expressions of circadian rhythm-related genes were detected by RT-qPCR and WB at specific intervals (circadian time (CT) 0–12: yellow background; CT12–24: gray background).
KDM6B Served as a Positive Circulation Trigger for the Osteogenic Differentiation of BMSCs by Decreasing the Proportion of M1/M2 in Vitro
Based on the observation that BMAL1 knockout affected the overall proportion of macrophages, we investigated whether changes in KDM6B levels from osteogenic differentiated BMSCs will modify the phenotype of macrophages. First, we noted that the transcription and translation levels of BMAL1 in BMDMs were significantly increased after recombinant human KDM6B protein stimulation (Figure 4a). Flow cytometry results then showed that M2 (F4/80+CD11b+CD206+) levels were significantly elevated, while the level of M1 (F4/80+CD11b+CD86+) was markedly decreased after stimulation by KDM6B (Figure 4b,c). In addition, immunofluorescence revealed that the expression of Arg1 (a marker of M2) was upregulated by KDM6B and downregulated by GSK-J4, with opposite trends for the M1 marker INOS (Figure 4d). We also simulated the osteogenic effect of MSCs in the macrophage-immune environment of bone marrow by a Transwell assay. It became evident that the positive rate of ALP and Alizarin Red was higher in the BMDM and BMSCs coculture group than in the group of BMSCs alone, which may be caused by phenotype-altered macrophages. Therefore, BMSCs were simultaneously stimulated with KDM6B-pretreated or GSK-J4-pretreated BMDMs. We found that the inhibitory effect of GSK-J4 on osteogenic differentiation was also pronounced in the coculture system. We hypothesized that this was related to the changes caused by the increased M1 polarization in macrophages affected by GSK-J4 (Figure 4e, statistical results shown in Supplementary Figure S3).
Figure 4.

KDM6B decreased the M1/M2 ratio and promoted BMSC osteogenic differentiation in the BMSCs–BMDMs coculture system. (a) RT-qPCR and WB analyses were performed to analyze the effect of KDM6B and its inhibitor GSK-J4 on the expression of BMAL1 in BMDMs. (b) Flow cytometry was used to detect the effects of KDM6B and its inhibitor GSK-J4 on macrophage polarization. (c) Statistical results of flow cytometry. (d) Immunofluorescence of macrophage polarization and statistical analysis (scale bar: 100 μm). (e) BMDMs or BMSCs were added to the upper or lower chambers of the Transwell plates, and the BMSCs in the lower chamber were osteogenically induced. The osteogenic differentiation of BMSCs in the lower chamber was evaluated via ALP staining or Alizarin Red S staining (scale bar: 100 μm).
Bone and Macrophage Dual-Targeted Nanodiscs ND-GSK-J4 Induced Osteoporosis in Vivo via Knockdown of KDM6B
To investigate the association between the differentiation ability of BMSCs and BMDMs in vivo, we introduced an ovariectomy (OVX) murine model, with the femur and its blood supply being intact (when compared to the femoral fracture model). Accordingly, ND-GSK-J4 could effectively pass the capillaries of the intact femur and act on its bone marrow, further affecting the entire femur. Therefore, we divided the mice (female, 6 weeks) into three groups: namely, (i) sham surgery group (removal of part of the adipose tissue near the ovary), (ii) bilateral OVX group, and (iii) ND-GSK-J4 tail vein injection group. All mice were sacrificed after 10 weeks, with the femurs being harvested for further analyses. The results of the micro-CT analysis showed that—in comparison to the sham surgery cohort—both the OVX group and the ND-GSK-J4 group were associated with different degrees of decrease in bone mineral density (BMD), trabecular bone volume/trabecula volume (Tb. BV/TV), trabecular number (Tb. N), and trabecular separation/spacing (Tb. Sp) (Figure 5a,b). Hematoxylin–-eosin (HE) staining validated this result (Figure 5c). Bone dynamic fluorescence staining of calcein and alizarin-3-methylimnodiacetic acid indicated that the bone formation was significantly decreased in the OVX group and ND-GSK-J4 group (Figure 5d,f). Further, an increased proportion of osteoclasts in the OVX group and ND-GSK-J4 group was detected using TRAP staining (Figure 5e). To investigate whether these results were associated with the macrophage phenotypic changes induced by macrophage-targeted GSK-J4, we simultaneously performed immunofluorescence (IF) staining for INOS and Arg1 in the femurs of these mice: while the M2 marker Arg1 was significantly downregulated in the bone tissue of the OVX group and ND-GSK-J4 group, the M1 marker INOS was significantly increased (Figure 5g,h). The results of flow cytometry of the femoral bone marrow (harvested presacrifice) were consistent with the IF staining findings (Figure 5i,j). In conclusion, bone and macrophage dual-targeted GSK-J4 significantly increased the M1/M2 ratio and suppressed femoral osteogenesis and bone formation.
Figure 5.

Inhibition of KDM6B in bone macrophages led to osteoporosis in vivo. (a, b) Six-week-old female C57BL/6J mice were divided into three groups, which underwent sham surgery, OVX surgery, or ND-GSK-J4 tail vein injection (once a week). Micro-CT analysis after 10 weeks and its statistical results of BMD, Tb. BV/TV, Tb. N and Tb. Sp. (c) HE staining of femurs from the three groups (scale bar: 500 μm). (d) Mineral apposition rate (MAR) analysis was performed in all three groups before the mice were sacrificed (scale bar: 50 μm). (e) TRAP staining and its statistical results of mouse femurs from sham, OVX, and ND-GSK-J4 groups. (f) Quantitation of MAR. (g) Immunofluorescence was used to evaluate the polarization of macrophages in the bone marrow of the three murine groups (scale bar: 50/500 μm, N = 5). (h) Quantitation of INOS-positive and Arg1-positive cells in the immunofluorescence assay. (i, j) Flow cytometry assay and quantitation of CD86-positive and CD206-positive cells in F4/80+CD11b+ cells.
KDM6B Regulated the Link between the Polarization of BMDMs and the Osteogenic Differentiation of BMSCs by Promoting BMAL1 Expression
Aiming to decipher the role of KDM6B in the progression of osteoporosis at the cellular level, we isolated BMDMs from mice of the sham surgery and OVX cohorts. Subsequent PCR results indicated that the mRNA expression levels of KDM6B and BMAL1 were significantly lower in BMDMs derived from OVX mice (compared to those harvested from the sham surgery cohort). In contrast to GSK-J4, which significantly downregulated the expression of BMAL1 in BMDMs isolated from the sham surgery cohort, the addition of KDM6B restored the expression level of BMAL1 in OVX-derived BMDMs (Figure 6a). This result was also confirmed using the WB assay (Figure 6b). After demonstrating that KDM6B and its inhibitor GSK-J4 could modify the polarization of BMDMs and thus affect the osteogenic differentiation of BMSCs in vitro and in vivo, we prepared BMAL1-overexpressing lentiviruses to infect BMDMs (MOI: 50, Supplementary Figure S4). Simultaneously, we isolated BMDMs from BMAL1–/– mice to obtain a BMAL1-KO BMDM system. BMDMs of the two systems (BMAL1-OE and BMAL1-KO) were used to confirm that KDM6B or GSK-J4 mainly modulates the polarization of BMDMs by affecting BMAL1. The results of PCR and WB showed that the mRNA and protein levels of BMAL1 were significantly reduced in the BMAL-KO group, whereas the expression of BMAL1 in the BMAL1-OE group was upregulated, indicating that the BMAL1-OE and BMAL1-KO BMDM systems were successfully established (Figure 6c,d). Using flow cytometry and immunofluorescence, we demonstrated that BMDMs in the BMAL1-KO group tended to polarize toward M1, while BMDMs in the BMAL1-OE group preferentially polarized toward M2 (Figure 6e–g). Subsequently, we cocultured the BMDMs and BMSCs of the three groups, with the BMSCs in the lower layer being subjected to osteogenic differentiation. The ALP and Alizarin Red levels of BMSCs in the BMAL1-KO group were decreased. Conversely, the ALP and Alizarin Red levels of BMSCs in the BMAL1-OE group were increased (Figure 6h). Furthermore, we observed that the protein expression levels of bone-related genes such as collagen I, ALP, RUNX2, and OCN were significantly higher in BMSCs from the BMAL1-OE group (Figure 6i).
Figure 6.

KDM6B orchestrated the polarization of BMDMs and the osteogenic differentiation of BMSCs by regulating BMAL1 expression. (a, b) BMDMs were isolated from the sham surgery and OVX cohorts, and RT-qPCR and WB assay were performed to detect the effect of KDM6B and GSK-J4. (c, d) BMAL1-KO BMDMs and BMAL1-OE BMDMs were isolated from BMAL1-KO mice and lentivirus-infected BMDMs, respectively. RT-qPCR and the WB assay were performed for subsequent analyses. (e–g) Flow cytometry and immunofluorescence were used to evaluate the results of macrophage polarization in BMAL1 knockout and overexpression systems (scale bar: 100 μm). (h) BMSCs were seeded in the lower chamber of the Transwell plate, while BMDMs with different treatments were added to the upper chamber. Analysis of osteogenic differentiation of BMSCs in the lower chamber was completed by ALP staining or Alizarin Red S staining (scale bar: 100 μm). (i) The WB results show the expression of osteogenesis-related genes.
BMAL1 Decreased the Ratio of M1/M2 via the TLR2/NF-κB Signaling Pathway
After we confirmed that osteogenic induced BMSCs mainly affect the expression of BMAL1 in BMDMs through KDM6B and thus change the phenotype of BMDMs, we collected BMDMs and BMAL1-OE BMDMs to perform transcriptome sequencing and to further explore the mechanism through which BMAL1 orchestrates phenotypic shifts in BMDMs. We could show that the transcript levels of 896 genes were upregulated (p < 0.05, log2FC > 1), and 755 genes were downregulated (p < 0.05, log2FC < 1) in the BMAL1-OE group (Figure 7a). Heatmaps of differential genes, GO analysis results of upregulated genes, KEGG analysis results of upregulated genes, and GO analysis results of downregulated genes are shown in Supplementary Figure S5. The results of the KEGG analysis of downregulated genes are shown in Figure 7b. We could provide evidence that distinct hyper-inflammatory or immune-related disease pathways such as those commonly observed in rheumatoid arthritis or inflammatory bowel disease were downregulated. Among the downregulated genes, we noticed that some genes typically related to M1 macrophage polarization or function were inhibited, including CD86, IL-6ra, IL-1b, and TLR2 (Figure 7c). Our analysis further demonstrated that TLR2 was associated with inflammatory pathways characteristic of autoimmunopathies such as rheumatoid arthritis or inflammatory bowel disease (Figure 7b). Further, the TLR2/NF-κB signaling pathway has been implicated in the differentiation and function of macrophages. Therefore, we examined the expression levels of TLR2, P-P65, and IκB-α in controls, BMAL1-KO, and BMAL1-OE BMDMs. The NF-κB protein normally forms a homo/heterodimer with p65 and p50. In the cellular cytoplasm, it is commonly bound to the inhibitory protein IκB to form a trimeric inactive complex.33 We found that the TLR2 protein level of BMDMs in the BMAL-OE group was significantly inhibited, with the downstream p-p65 level being also decreased, while the expression level of its inhibitory protein IκB-α was increased (Figure 7d). The results in the BMAL1-KO group were the opposite. When the cells of these three groups were stained with propidium iodide (PI)/Hoechst for detection of pyroptosis, we found that the positive rate of PI in the BMAL1-KO group was significantly increased, while the positive rate of PI in the BMAL-OE group was significantly decreased (Figure 7e). We speculated that the expression of BMAL1 was related to pyroptosis and, therefore, performed further WB experiments. The results suggested that NLRP3 was significantly induced in BMAL1-KO BMDMs. Moreover, protein expressions of pro-IL-1β and IL-1β in BMAL1-KO BMDMs were significantly elevated (Figure 7f). The NLRP3 signaling pathway in the BMAL1-OE group was inhibited and found to be protective against macrophage pyroptosis. In order to investigate whether the inhibition of TLR2 can directly affect BMDMs, we divided BMDMs into three groups (PBS, si-NC, or si-TLR2 transfection). Flow cytometry results showed that si-TLR2 indeed reduced the M1/M2 ratio of BMDMs (Figure 7g). However, following the coculture of the three BMDMs groups with BMSCs, the positive rate of ALP staining and alizarin red staining was significantly increased in the si-TLR2 group (Figure 7h). We collected the BMSCs and found that the protein expression levels of bone-related genes such as ALP, RUNX2, and OCN were also significantly higher in the si-TLR2 group (Figure 7i). In summary, we demonstrated at the cellular level that KDM6B produced during the osteogenic differentiation of BMSCs can promote the expression of BMAL1 by binding to the promoter region of the BMAL1 gene in BMDMs to demethylate H3K27me3. This process seemed to protect macrophages from pyroptosis and reduce the M1/M2 ratio by inhibiting the TLR2/NF-kB pathway.
Figure 7.

BMAL1 regulated macrophage polarization via the TLR2/NF-κB signaling pathway. (a) BMAL1-NC and BMAL1-OE BMDMs were collected, and transcriptome sequencing was performed. A volcanic map showing differentially expressed genes (DEGs). (b) KEGG analysis of downregulated genes. (c) Heat map of portions of DEGs. (d) Genes related to the TLR2/NF-κB signaling pathway were analyzed in the control, BMAL1-KO, and BMAL1-OE groups via WB assay. (e, f) Pyroptosis of BMDMs was analyzed via PI/Hoechst staining and WB assay of NLRP3 inflammasome-related genes (scale bar: 100 μm). (g) BMDMs were treated with PBS or transfected with si-NC or si-TLR2. Flow cytometry was used to visualize the polarization of BMDM. (h) Treated BMDMs were cocultured with BMSCs using Transwell plates. Analysis of the osteogenic differentiation of BMSCs in the lower chamber was performed using ALP staining or Alizarin Red S staining (scale bar: 100 μm). (i) WB results showing the expression of osteogenesis-related genes in BMSCs.
Knockdown of BMAL1 in Vivo Increased the Level of TLR2
In vivo, we performed immunofluorescence colocalization of macrophage markers CD68 and BMAL1. Therefore, we collected femurs of the sham, OVX, and ND-GSK-J4 groups. We found that the proportion of BMAL1 positive in CD68-positive macrophages in the ND-GSK-J4 group was significantly decreased, indicating that GSK-J4 can affect the expression of BMAL1 by inhibiting KDM6B (Figure 8a). The immunofluorescence colocalization results of the macrophage markers CD68 and TLR2 in the ND-GSK-J4 group and the OVX group showed that the proportion of double-positive cells was significantly higher compared to the sham group, pointing toward an activation of the TLR2 signaling pathway (Figure 8b). In combination with the increase of the M1/M2 ratio in the ND-GSK-J4 group, we could confirm that GSK-J4 targeting bone-marrow-derived macrophages can suppress the expression of BMAL1 by inhibiting KDM6B, activate the TLR2 signal pathway, and promote the increase of the M1/M2 ratio to hinder osteogenesis in vivo. Schematic illustration of cellular experiments was concluded in Figure 9.
Figure 8.

Inhibition of KDM6B in bone macrophages resulted in suppression of BMAL1 and activation of TLR2 in vivo. (a, b) Femurs from sham surgery, OVX, and ND-GSK-J4 groups were analyzed for CD68-positive macrophages and BMAL1 or TLR2 colocalization using immunofluorescence.
Figure 9.
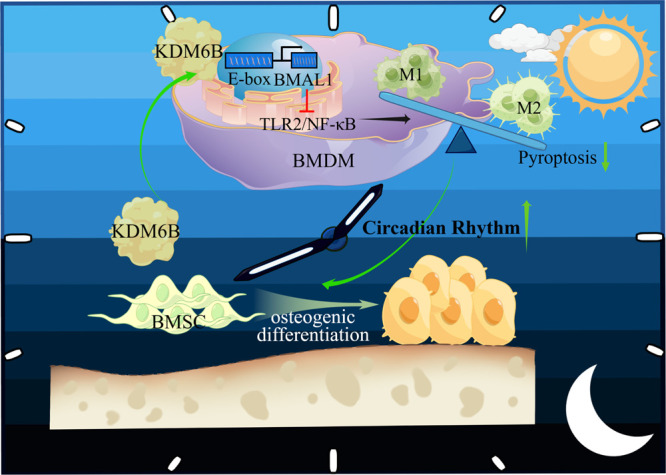
Schematic illustration of cellular experiments. KDM6B served as a positive circulation trigger for osteogenic differentiation of BMSCs by decreasing the proportion of M1/M2 ratio. Mechanistically, KDM6B derived from osteogenic-induced BMSCs can bind to the promoter region of BMDMs’ BMAL1 gene and activate it in a coculture system. Furthermore, elevated BMAL1 in BMDMs could inhibit the TLR2/NF-κB signaling pathway, reduce pyroptosis, and decrease the M1/M2 ratio.
This study is not without limitations. In adult bone marrow, osteoblasts and adipocytes share a common precursor referred to as MSC. The bone marrow microenvironment is a dynamic system, which interacts via autocrine, paracrine, and endocrine signaling.34 Bone marrow provides a suitable microenvironment for osteogenesis and maintaining bone homeostasis under physiological conditions. However, the bone marrow microenvironment gradually changes under pathological conditions such as osteoporosis. This change is characterized by increased oxidative stress, chronic inflammation, inhibition of osteoblastic signal, and an increase of osteoblastic inhibitors. This article mainly focuses on the relationship between BMSCs and BMDMs in the bone marrow microenvironment. However, further research is needed to ascertain the complex lipogenic function of MSCs.35 At the same time, bone remodeling involves the cross-talk of osteoclasts and osteoblasts. Macrophages play a major role in the activation and formation of osteoclasts and differentiate from monocytes under the influence of M-CSF.36 The effects of macrophages on the adipogenic differentiation of MSCs and osteoclasts will be discussed in future articles. Protein aggregation, inactivation, and a hyperactive immune response still represent challenging problems for sufficient protein delivery. In our in vivo experiments investigating the function of KDM6B, we adopted an indirect method to target bone macrophages and ensure the local delivery of their inhibitor GSK-J4. Due to the high molecular weight of KDM6B protein, it is difficult to design a nano delivery system for it that simultaneously targets bone and macrophages. However, given the development of nano drug delivery systems in recent years, promising potential solutions such as polymer nanoparticles, lipid nanocarriers, protein metal–organic framework (MOF) complexes, and inorganic nanoparticles (silica, gold, magnetic framework, and carbon framework) are under investigation to overcome this problem. Although the nanoparticles presented in this study improved the mechanistic functionality of KDM6B as a bridge between MSCs and BMDMs, they cannot be used to treat osteoporosis. In the near future, we will refine the design method and develop more targeted drugs that can promote osteogenesis and improve the bone microenvironment in vivo.
Our experimental results on the TLR2/NF-κB pathway align with previous research work, as we found that even OVX-derived BMSCs secreted a certain amount of KDM6B during osteogenesis induction, although this amount was significantly reduced compared to control conditions.33 Based on our finding that KDM6B can promote polarization of macrophages toward M2, we hypothesized that MSCs deploy other pathways to counteract bone loss even if the body’s overall osteogenic capacity is reduced.
Conclusion
In this study, we showed that KDM6B secreted during the osteogenic differentiation of MSCs can further promote its differentiation by regulating the epigenetics and the phenotype of BMDMs, representing a positive feedback regulation process. We also investigated the underlying mechanism of how KDM6B can reduce the M1/M2 ratio by conducting in vitro functional experiments and mRNA-sequencing technology. The results of the ChIP assay experiments suggested that KDM6B can demethylate H3K27me3 in the BMAL1 promoter region of BMDM, thus activating the BMAL1 gene. Additional pathway and functional tests indicated that BMAL1 overexpression in BMDMs reduces the production of inflammasomes in macrophages and pyroptosis and decreases the proportion of M1 via the TLR2/NF-kb pathway. Bone and macrophage dual-targeted GSK-J4 (KDM6B inhibitor)-loaded nanodiscs indirectly proved the critical role of KDM6B in osteogenesis in vivo. Collectively, KDM6B served as a positive circulation trigger for the osteogenic differentiation of BMSCs by decreasing the proportion of M1/M2 both in vivo and in vitro.
Materials and Methods
Cell Culture, Viral Infection, and Transfection
BMSCs isolated from wild-type C57BL/6J mice (female, 6 weeks old) were cultured in RPMI 1640 (#11875093, Gibco, Grand Island, NY, USA) containing 10% FBS (#10099141, Gibco) and 1% penicillin/streptomycin (#SV30010, Hyclone, USA). To obtain BMDMs, 50 ng/mL of macrophage colony-stimulating factor (M-CSF, #216-MC, R&D System, Minneapolis, MN, USA) was added for 7 days, as previously described by our group.37 Viral packaging systems were purchased from Genechem (GV640, Shanghai Genechem Co., Ltd., Shanghai, China). For viral inoculation, BMDMs were incubated with the viral supernatants (MOI = 50) in a complete medium for 24 h. The medium was switched to standard culture media 24 h postinfection. Si-TLR2 and siRNA-NC were purchased from Tsingke Biotechnology Co., Ltd. (Wuhan, China). The siRNA transfection was conducted with Lipofectamine 3000 reagent (Invitrogen, L3000015, Carlsbad, CA, USA) according to the manufacturer’s manuals.
ChIP Assays
ChIP-PCR was performed according to the manufacturer’s instructions (Beyotime, P2078). Mature BMDMs were cultured in cell culture dishes (100 mm; Corning, cat. no. 430167). For protein–DNA cross-linking, 270 μL of formaldehyde was added, and the cells were incubated at 37 °C for 10 min. The reaction was quenched with 1.1 mL of glycine solution (10×) for 5 min at room temperature. Cells were washed twice with ice-cold PBS containing 1 mM PMSF. Cells were collected in the 2.0 mL EP tube with 106 cells per tube. A 0.2 mL amount of SDS lysis buffer containing 1 mM PMSF was used per tube for cell lysis. The cross-linked DNA was sonicated with an ultrasonic cell disruptor at 60% power for five pulses of 10 s each. A 1.8 mL amount of ChIP dilution buffer containing 1 mM PMSF was added to each EP tube. A 20 μL sample was saved as the input sample. Immunoprecipitation was performed overnight using 5 μg of trimethyl-histone H3-K27 rabbit pAb (1:100, #A2363, Abclonal, China), anti-KDM6B antibody (1:100, ab38113, Abcam) or a negative control antibody. Immunoprecipitates were bound to Protein A+G agarose/salmon sperm DNA and then washed once with a low salt immune complex wash buffer, once with a high salt immune complex wash buffer, once with a LiCl immune complex wash buffer, and twice with a TE buffer. Elution was performed in an elution buffer (1% SDS, 0.1 M NaHCO3). DNA–protein cross-linking was reversed with a 5 M NaCl solution. DNA purification was performed as previously described.38 Purified DNA was further used for qPCR and PCR. Targeted DNA amplification was conducted via PCR with a 2× rapid taq master mix (Vazyme, P222-AA). Standard agarose gel electrophoresis was performed using a 1% gel.
Real-Time Reverse Transcriptase–Polymerase Chain Reaction (Real-Time RT-PCR) and Western Blotting
The total protein concentration was determined using a BCA protein assay kit (#P0012S, Beyotime, China), and protein samples were separated on a SDS-PAGE gel, transferred to Immobilon-PSQ PVDF membranes (#ISEQ00010, Millipore, MA, USA), and probed with primary antibodies against BMAL1 (1:1000, #A4714, Abclonal), CLOCK (1:1000, #A5633, Abclonal), CRY1 (1:1000, #A6890, Abclonal), PER1 (1:1000, #A8449, Abclonal), PER2 (1:1000, #A13168, Abclonal), NR1D1 (1:1000, #A18602, Abclonal), KDM6B (1:1000, #A17382, Abclonal), ALP (1:1000, #ab229126, Abcam), OCN (1:1000, #ab93876, Abcam), collagen I (1:1000, #ab34710, Abcam), RUNX2 (1:1000, #A2851, Abclonal), TLR2 (1:500, #A19125, Abclonal), p-p65 (1:500, #AP0475, Abclonal), IκBα (1:1000, #A11168, Abclonal), NLRP3 (1:1000, #A5652, Abclonal), cleaved caspase-1 (1:1000, #A21296, Abclonal), and IL1β (1:1000, #A16288, ABclonal). After incubation with a secondary antibody for 1 h at room temperature, the blots were imaged with an ECL kit (#BL520A, Biosharp, China) and ChemiDoc MP imaging system (Biorad, USA). After treatment, the total RNA was extracted using Trizol reagent (Invitrogen, USA) and quantified with a NanoDrop 2000 spectrophotometer (Thermo Scientific). The total RNA in cells was isolated and purified by the RNAiso reagent (Takara,9109, Japan). HiScript III-RT SuperMix for qPCR (Vazyme, R323-01, Nanjing, China) was used to obtain cDNA, and Nanodrop 2000 was used for quantitative detection of the mRNA levels. AceQ qPCR SYBR Green master mix (Vazyme, Q111-C1, Nanjing, China) and a Biorad CFX96 system were used to perform RT-PCR according to the manufacturer’s instructions. Specific primers are presented in Table S1 (Supporting Information).
Animals, BMAL1-Deficient Mice, Ovariectomized Mice Model, and Micro-CT
Six-week-old female C57BL/6J mice were purchased from The Center of Experimental Animals, Tongji Medical College, Huazhong University of Science and Technology (HUST), and housed under specific-pathogen-free (SPF) conditions. All animal experiments received consent from The Institutional Animal Care and Use Committee at Tongji Medical College, HUST. For BMAL1-deficient mice (Bmal1–/–, B6.129-Arntltm1Bra/J mice), homozygous mice were produced by breeding heterozygous BMAL1-deficient mating pairs (Bmal1+/–) that were kindly provided by Prof. Lili Chen (Wuhan Union Hospital, China).39 Mouse tail genotyping was performed with the quick genotyping assay kit for mouse tail (Beyotime, D7283s) according to the manufacturer’s instructions. DNA was isolated from the mouse tails. Targeted DNA amplification was conducted via PCR with a 2× rapid taq master mix (Vazyme, P222-AA). Standard agarose gel electrophoresis was performed using a 1% gel. Fat tissue excision near the ovaries was performed in the sham group, and bilateral ovariectomy surgery was performed in the OVX group mice. The sham group and OVX group received 200 μL of PBS, and the ND-GSK-J4 group received 200 μL of ND-GSK-J4 by tail vein injection weekly. Mice were sacrificed after 10 weeks, and the femurs were collected for micro-CT analysis (n = 5 per group). The femurs were scanned by the Bruker SkyScan 1176 scanner mCT system (2400 frames, 5 frames/frame, 37 kV, 121 mA) and fixed overnight using paraformaldehyde. BMD, Tb. BV/TV, Tb. N, and Tb. Sp were analyzed in volume of interest with the CTan software (Bruker).
Circadian Rhythm Induction
C57BL/6J mice were purchased and housed under SPF conditions. Mice from all groups were routinely housed under 12:12 light/dark cycle conditions (LD12:12), with the light being switched on from 8:00 a.m. CST. As for circadian rhythm induction of BMDMs, the medium of BMDMs from six-well plates or 24-well plates was removed and replaced with fresh complete medium containing 1 μmol/L dexamethasone (Sigma) for 1 h. Then the culture medium was replaced with fresh complete medium again, and this timing was set as circadian time 0 (CT0). No further medium changes from this point were performed before harvest. Individual plates were harvested for total RNA and protein at preset time intervals.
Flow Cytometric Analysis
To determine the proportion of M1 and M2, bone marrow was extracted and mixed with red blood cell lysis buffer (Solarbio, Beijing, Chian) and further stained with 1 μL of BV421 Zombie Violet fixable viability kit (#423113, Biolegend, USA) in the dark at room temperature for live/dead analysis. As described previously,37 1 μL of FITC anti-mouse F4/80 (#157309, Biolegend), 1 μL of APC anti-mouse/human CD11b (#101211, Biolegend), 1 μL of APC/cyanine7 anti-mouse CD86 (#105029, Biolegend), 1 μL of PE/cyanine7 anti-mouse CD206 (#141719, Biolegend), and intracellular fixation and permeabilization buffer set (#88-8824-00, eBioscience, USA) were used to stain and differentiate M1 and M2 cells. Cells were analyzed with flow cytometry (BD, LSRFortessa X-20, USA) and FlowJo_V10 software.
Histological Analysis, Immunofluorescence, and Mineral Apposition Rate (MAR) Measurement
Femurs were collected after micro-CT scanning, decalcified using 10% EDTA solution for 15 days, and then embedded in paraffin. Samples were sectioned at 5–7 μm and stained with HE or TRAP (#G1050-50T, Servicebio) according to the manufacturer’s instructions. The primary antibodies anti-INOS (1:100, #A14031, ABclonal), anti-Arg1(1:100, #A4923, ABclonal), anti-CD68 (1:100, #A6554, ABclonal), anti-TLR2 (1:50, #A11225, ABclonal), and anti-BMAL1 (1:200, # ab3350, Abcam) were used for an immunofluorescence assay. For MAR measurement, 25 mg/kg calcein (Aladdin, Shanghai, China) and 50 mg/kg alizarin-3-methyliminodiacetic acid (A3882, Sigma) dissolved in 2% sodium bicarbonate solution (Aladdin) were intraperitoneally injected into mice 7 and 2 days prior to sacrificing the mice, respectively. After overnight fixation with paraformaldehyde, dehydrated and undecalcified femurs were embedded in methyl methacrylate and then underwent hard tissue slicing (HistoCore AUTOCUT, Leica, Germany). Histology was visualized using an automatic slice scanning system (SV120, Olympus, Japan).
ALP Activity Assay and Alizarin Red S Staining
To induce osteogenic differentiation, 1 × 105 BMSCs were cultured in 0.1% gelatin-coated 24-well plates with osteogenic-inducing media (#HUXMX-90021, Cyagen, USA) for 1 or 3 weeks. We then used the ALP assay kit (Beyotime, Shanghai, China) and Alizarin Red S staining solution (Cyagen, USA) according to the manufacturer’s instructions.
Synthesis of Peptide
To target bone,40 SDSSD peptide was fused to the N-terminal of apoA-1 peptide analogs (APA) through an SSG linker. The SDSSD-APA peptide (SDSSDSSGLEELREKLRPLLEELREKLRPLLEELREKLRPLLEELREKLRPL) was synthesized via solid-phase peptide synthesis (SPPS) using 9-fluorenylmethyloxycarbonyl (Fmoc) as protecting group. The protective group was removed by trifluoroacetic acid, and then the crude peptide was purified and analyzed by HPLC (Waters2695-2487) and MS (Waters ZQ2000). The purified SDSSD-APA peptide was lyophilized and stored at −20 °C.
Preparation of Empty Nanodiscs and GSK-J4-Loaded Nanodiscs (ND-GSK-J4)26,41,42
The ability of ND to target macrophages was accomplished by DPPS,43 and the ND assemblies were prepared according to a previously reported method with some modifications.28 Briefly, 1,2-dipalmitoyl-sn-glycerol-3-phosphocholine (DPPC) and 1,2-dipalmitoyl-sn-glycero-3-phospho-l-serine (DPPS) were dissolved in chloroform in a glass tube at a 9:1 molar ratio. Chloroform was dried using a gentle argon gas stream, and the tube was placed in a vacuum desiccator overnight to remove residual solvent. MSP buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 0.5 mM EDTA) containing sodium cholate (NaCh) was added to hydrate the dried lipid film to obtain a lipid stock. MSP buffer, lipid stock, and APA or SDSSD-APA peptide solution were successively added to prepare negative control nanodiscs (NC-ND, without bone-targeting ability) or empty nanodiscs (ND, with bone-targeting ability). The assembly mixture was incubated for 2 h at 41 °C, and then the mixture was shaken for 1 h at 41 °C, 120 rpm. The assembly was analyzed and further purified by size exclusion chromatography (SEC) using a Superdex 200 Increase 10/300 GL column (GE Healthcare) at a flow rate of 0.5 mL/min. Prior to incubation and shaking, a GSK-J4 solution or fluorescent dye (DiO) was additionally added to prepare ND-GSK-J4, Dio-NC-ND, or Dio-ND-GSK-J4 in a similar process.
Dynamic Light Scattering and Transmission Electron Microscopy44,45
The particle sizes of purified samples were measured using DLS with a Nano ZS90 Zetasizer (Malvern Instruments, UK). Every sample was measured in quadruplicates (9 runs) at 25 °C. A small droplet of each sample was pipetted onto a carbon-coated copper grid until natural drying and then negatively stained with 2% NaPT (w/v). All images were recorded using a Talos F200X transmission electron microscope (ThermoFisher, USA) operating at an accelerating voltage of 200 kV.
Cellular Uptake in Vitro and Targeting of Nanodiscs in Vivo
BMDMs were cultured in confocal dishes (⦶ 35 mm) with 1.5 mL of complete medium containing 50 μL of ND-GSK-J4 or ND. BMDMs were fixed and stained with TRITC-conjugated phalloidin (#FAK100, Sigma) and DAPI when cell fusion reached 30%. Twelve hours after the tail vein injection of 200 μL of ND-GSK-J4 or ND, mice were sacrificed and the heart, liver, spleen, lung, kidney, and femur were harvested. The distribution of ND-GSK-J4 or ND was observed with the Vivo FX PRO imaging system (Bruker, Karlsruhe, Germany).
RNA Sequencing
The total RNA extracted from mature BMDMs and BMAL1-OE BMDMs was prepared before the establishment of the sequencing library for each RNA sample using the NEBNext Ultra RNA library prep kit (Illumina). Sequencing was performed on an Illumina Hiseq platform completed by Wuhan SeqHealth Tech Co., Ltd. (project number: KC2022-G0183). Fastp (version 0.23.0) was selected to filter raw reads containing adapter, low quality, or poly-N. Paired-end clean reads were then aligned to the reference genome to obtain transcript information. The RPKM (reads per kilobase per million reads) of each gene was calculated based on the mapped reads (millions) and exon length (KB). Then, the screening of differentially expressed genes (DEGs) in two groups (three biological replicates per group) was completed by the DESeq2 R package (1.16.1). Genes with a p-value < 0.05 and absolute value of log fold-change > 1 were assigned as DEGs. The ClusterProfiler R package was used to obtain a gene ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis of DEGs.
Statistical Analysis
Data are expressed as mean ± standard deviation (SD) with biological replicates. Statistical analysis was performed using Student’s t test (two groups). Significance was set at p < 0.05. Figures were created using Origin 2021 (OriginLab, MA, USA).
Acknowledgments
This work was supported by the Department of Science and Technology of Hubei Province (No. 2021CFB425, No. 2020BCB004) and National Science Foundation of China (No. 82072444, No. 82272491, No. 82002313, No. 82001887). Abstract graphic and Figure 9 were created by Figdraw. We thank the support provided by The Center of Experimental Animals, Huazhong University of Science and Technology.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c12440.
Table of primers; figures of purified SDSSD-APA peptide measured using HPLC and MS, ChIP-seq analysis results of H3K27me3 levels of WT BMDMs and KDM6B (Jmjd3) – /– BMDMs,11 statistical results of ALP staining and Alizarin Red staining in Figure 4e, MOI of BMAL1-overexpression lentivirus infection in BMDMs, heatmap of differentially expressed genes, GO analysis and KEGG analysis results of upregulated genes, GO analysis results of downregulated genes (PDF)
Author Contributions
L.C., C.Y., W.X., and Y.X. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Song S.; Guo Y.; Yang Y.; Fu D. Advances in Pathogenesis and Therapeutic Strategies for Osteoporosis. Pharmacol Ther 2022, 237, 108168. 10.1016/j.pharmthera.2022.108168. [DOI] [PubMed] [Google Scholar]
- Compston J. E.; McClung M. R.; Leslie W. D. Osteoporosis. Lancet 2019, 393 (10169), 364–376. 10.1016/S0140-6736(18)32112-3. [DOI] [PubMed] [Google Scholar]
- Salari N.; Ghasemi H.; Mohammadi L.; Behzadi M. H.; Rabieenia E.; Shohaimi S.; Mohammadi M. The Global Prevalence of Osteoporosis in the World: A Comprehensive Systematic Review and Meta-Analysis. J. Orthop Surg Res. 2021, 16 (1), 609. 10.1186/s13018-021-02772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W.; Liu Y.; Chen T.; Zhang X.; Lv L.; Jin C.; Jiang Y.; Shi L.; Zhou Y. The Epigenetic Promotion of Osteogenic Differentiation of Human Adipose-Derived Stem Cells by the Genetic and Chemical Blockade of Histone Demethylase LSD1. Biomaterials 2014, 35 (23), 6015–6025. 10.1016/j.biomaterials.2014.04.055. [DOI] [PubMed] [Google Scholar]
- Shi Y. Histone Lysine Demethylases: Emerging Roles in Development, Physiology and Disease. Nat. Rev. Genet 2007, 8 (11), 829–833. 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- Crea F.; Sun L.; Mai A.; Chiang Y. T.; Farrar W. L.; Danesi R.; Helgason C. D. The Emerging Role of Histone Lysine Demethylases in Prostate Cancer. Mol. Cancer 2012, 11, 52. 10.1186/1476-4598-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H.; Liao L.; An Y.; Su X.; Liu S.; Shuai Y.; Zhang X.; Jin Y. Suppression of EZH2 Prevents the Shift of Osteoporotic MSC Fate to Adipocyte and Enhances Bone Formation During Osteoporosis. Mol. Ther 2016, 24 (2), 217–229. 10.1038/mt.2015.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Cai M.; Zhong J.; Yang L.; Xiao J.; Jin F.; Xue H.; Liu X.; Liu H.; Zhang Y.; Jiang D.; Hong A.; Ji X.; Wang Z.; Zhang G.; Wang X. The Long Noncoding RNA Lnc-Ob1 Facilitates Bone Formation by Upregulating Osterix in Osteoblasts. Nat. Metab 2019, 1 (4), 485–496. 10.1038/s42255-019-0053-8. [DOI] [PubMed] [Google Scholar]
- Ye L.; Fan Z.; Yu B.; Chang J.; al Hezaimi K.; Zhou X.; Park N.-H.; Wang C.-Y. Histone Demethylases KDM4B and KDM6B Promotes Osteogenic Differentiation of Human MSCs. Cell Stem Cell 2012, 11 (1), 50–61. 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Lee H. L.; Suh J. S.; Deng P.; Lee C. R.; Bezouglaia O.; Mirnia M.; Chen V.; Zhou M.; Cui Z. K.; Kim R. H.; Lee M.; Aghaloo T.; Hong C.; Wang C. Y.. The ERα/KDM6B Regulatory Axis Modulates Osteogenic Differentiation in Human Mesenchymal Stem Cells. Bone Res. 2022, 10 ( (1), ), 10.1038/s41413-021-00171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T.; Takeuchi O.; Vandenbon A.; Yasuda K.; Tanaka Y.; Kumagai Y.; Miyake T.; Matsushita K.; Okazaki T.; Saitoh T.; Honma K.; Matsuyama T.; Yui K.; Tsujimura T.; Standley D. M.; Nakanishi K.; Nakai K.; Akira S. The Jmjd3-Irf4 Axis Regulates M2Macrophage Polarization and Host Responses against Helminth Infection. Nat. Immunol 2010, 11 (10), 936–944. 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- Xiong Y.; Chen L.; Yan C.; Zhou W.; Yu T.; Sun Y.; Cao F.; Xue H.; Hu Y.; Chen D.; Mi B.; Liu G. M2Macrophagy-Derived Exosomal MiRNA-5106 Induces Bone Mesenchymal Stem Cells towards Osteoblastic Fate by Targeting Salt-Inducible Kinase 2 and 3. J. Nanobiotechnology 2020, 18 (1), 66. 10.1186/s12951-020-00622-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mahon O. R.; Browe D. C.; Gonzalez-Fernandez T.; Pitacco P.; Whelan I. T.; von Euw S.; Hobbs C.; Nicolosi V.; Cunningham K. T.; Mills K. H. G.; Kelly D. J.; Dunne A. Nano-Particle Mediated M2Macrophage Polarization Enhances Bone Formation and MSC Osteogenesis in an IL-10 Dependent Manner. Biomaterials 2020, 239, 119833. 10.1016/j.biomaterials.2020.119833. [DOI] [PubMed] [Google Scholar]
- Xu R.; Shen X.; Si Y.; Fu Y.; Zhu W.; Xiao T.; Fu Z.; Zhang P.; Cheng J.; Jiang H. MicroRNA-31a-5p from Aging BMSCs Links Bone Formation and Resorption in the Aged Bone Marrow Microenvironment. Aging Cell 2018, 17 (4), e12794. 10.1111/acel.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajarinen J.; Lin T.; Gibon E.; Kohno Y.; Maruyama M.; Nathan K.; Lu L.; Yao Z.; Goodman S. B. Mesenchymal Stem Cell-Macrophage Crosstalk and Bone Healing. Biomaterials 2019, 196, 80–89. 10.1016/j.biomaterials.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.-H.; Yang M.-Y.. The Role of Macrophage in the Pathogenesis of Osteoporosis. Int. J. Mol. Sci. 2019, 20 ( (9), ), 2093. 10.3390/ijms20092093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson C. M.; Kohrt W. M.; Buxton O. M.; Everson C. A.; Wright K. P.; Orwoll E. S.; Shea S. A. The Importance of the Circadian System & Sleep for Bone Health. Metabolism 2018, 84, 28–43. 10.1016/j.metabol.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L.; Patel M. S.; Bradley A.; Wagner E. F.; Karsenty G. The Molecular Clock Mediates Leptin-Regulated Bone Formation. Cell 2005, 122 (5), 803–815. 10.1016/j.cell.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Cha S.; Wang J.; Lee S. M.; Tan Z.; Zhao Q.; Bai D.. Clock-Modified Mesenchymal Stromal Cells Therapy Rescues Molecular Circadian Oscillation and Age-Related Bone Loss via MiR142–3p/Bmal1/YAP Signaling Axis. Cell Death Discov 2022, 8 ( (1), ), 10.1038/s41420-022-00908-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque N.; Cermakian N. Circadian Clocks in the Immune System. J. Biol. Rhythms 2015, 30 (4), 277–290. 10.1177/0748730415577723. [DOI] [PubMed] [Google Scholar]
- Chen G.; Tang Q.; Yu S.; Xie Y.; Sun J.; Li S.; Chen L.. The Biological Function of BMAL1 in Skeleton Development and Disorders. Life Sciences; Elsevier Inc., 2020, 10.1016/j.lfs.2020.117636. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Xu T.; Li Y.; Fei W.; Yang G.; Hong Y. Inhibition of CRY2 by STAT3/MiRNA-7–5p Promotes Osteoblast Differentiation through Upregulation of CLOCK/BMAL1/P300 Expression. Mol. Ther Nucleic Acids 2020, 19, 865–876. 10.1016/j.omtn.2019.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo M.; Cao X.; Zhang H.; Lau C. W.; Hong H.; Chen F. M.; Huang Y.; Chawla A.; Tian X. Y. Loss of Myeloid Bmal1 Exacerbates Hypertensive Vascular Remodelling through Interaction with STAT6 in Mice. Cardiovasc. Res. 2022, 118, 2859. 10.1093/cvr/cvab336. [DOI] [PubMed] [Google Scholar]
- Timmons G. A.; Carroll R. G.; O’Siorain J. R.; Cervantes-Silva M. P.; Fagan L. E.; Cox S. L.; Palsson-McDermott E.; Finlay D. K.; Vincent E. E.; Jones N.; Curtis A. M.. The Circadian Clock Protein BMAL1 Acts as a Metabolic Sensor In Macrophages to Control the Production of Pro IL-1β. Front Immunol. 2021, 12, 10.3389/fimmu.2021.700431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander R. K.; Liou Y.-H.; Knudsen N. H.; Starost K. A.; Xu C.; Hyde A. L.; Liu S.; Jacobi D.; Liao N.-S.; Lee C.-H.. Bmal1 Integrates Mitochondrial Metabolism and Macrophage Activation. Elife 2020, 9, 10.7554/eLife.54090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Chen X.; Chen Z.; Lv Y.; Li Y.; Li S.; Xu W.; Mo Y.; Wang X.; Chen Z.; Chen T.; Wang T.; Wang Z.; Wu M.; Wang J. An in Silico Approach to Reveal the Nanodisc Formulation of Doxorubicin. Front Bioeng Biotechnol 2022, 10, 859255. 10.3389/fbioe.2022.859255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariwal J.; Ma H.; Altenberg G. A.; Liang H. Nanodiscs: A Versatile Nanocarrier Platform for Cancer Diagnosis and Treatment. Chem. Soc. Rev. 2022, 51 (5), 1702–1728. 10.1039/D1CS01074C. [DOI] [PubMed] [Google Scholar]
- Xu D.; Chen X.; Li Y.; Chen Z.; Xu W.; Wang X.; Lv Y.; Wang Z.; Wu M.; Liu G.; Wang J. Reconfigurable Peptide Analogs of Apolipoprotein A-I Reveal Tunable Features of Nanodisc Assembly. Langmuir 2023, 10.1021/acs.langmuir.2c03082. [DOI] [PubMed] [Google Scholar]
- Cho D.-I.; Kim M. R.; Jeong H.; Jeong H. C.; Jeong M. H.; Yoon S. H.; Kim Y. S.; Ahn Y. Mesenchymal Stem Cells Reciprocally Regulate the M1/M2 Balance in Mouse Bone Marrow-Derived Macrophages. Exp Mol. Med. 2014, 46 (1), e70. 10.1038/emm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bossche J.; Neele A. E.; Hoeksema M. A.; de Winther M. P. J. Macrophage Polarization: The Epigenetic Point of View. Curr. Opin Lipidol 2014, 25 (5), 367–373. 10.1097/MOL.0000000000000109. [DOI] [PubMed] [Google Scholar]
- Langenbach F.; Handschel J. Effects of Dexamethasone, Ascorbic Acid and β-Glycerophosphate on the Osteogenic Differentiation of Stem Cells in Vitro. Stem Cell Res. Ther 2013, 4 (5), 117. 10.1186/scrt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTacchio L.; Le H. D.; Vollmers C.; Hatori M.; Witcher M.; Secombe J.; Panda S. Histone Lysine Demethylase JARID1a Activates CLOCK-BMAL1 and Influences the Circadian Clock. Science 2011, 333 (6051), 1881–1885. 10.1126/science.1206022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X.; Bao X.; Weng X.; Bai X.; Feng Y.; Huang J.; Liu S.; Jia H.; Yu B. The Protective Effect of Quercetin on Macrophage Pyroptosis via TLR2/Myd88/NF-KB and ROS/AMPK Pathway. Life Sci. 2022, 291, 120064. 10.1016/j.lfs.2021.120064. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Cai D.; Bai X. Macrophages Regulate the Progression of Osteoarthritis. Osteoarthritis Cartilage 2020, 28 (5), 555–561. 10.1016/j.joca.2020.01.007. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu X.; Zuo B.; Zhang L. The Role of Bone Marrow Microenvironment in Governing the Balance between Osteoblastogenesis and Adipogenesis. Aging Dis 2016, 7 (4), 514–525. 10.14336/AD.2015.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.; Wan Y. Molecular Determinants for the Polarization of Macrophage and Osteoclast. Semin Immunopathol 2019, 41 (5), 551–563. 10.1007/s00281-019-00754-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi B.; Chen L.; Xiong Y.; Yang Y.; Panayi A. C.; Xue H.; Hu Y.; Yan C.; Hu L.; Xie X.; Lin Z.; Zhou W.; Cao F.; Xiao X.; Feng Q.; Liu G. Osteoblast/Osteoclast and Immune Cocktail Therapy of an Exosome/Drug Delivery Multifunctional Hydrogel Accelerates Fracture Repair. ACS Nano 2022, 16, 771. 10.1021/acsnano.1c08284. [DOI] [PubMed] [Google Scholar]
- Bowler C.; Benvenuto G.; Laflamme P.; Molino D.; Probst A. v.; Tariq M.; Paszkowski J. Chromatin Techniques for Plant Cells. Plant Journal 2004, 39 (5), 776–789. 10.1111/j.1365-313X.2004.02169.x. [DOI] [PubMed] [Google Scholar]
- Yu S.; Tang Q.; Chen G.; Lu X.; Yin Y.; Xie M.; Long Y.; Zheng W.; Guo F.; Shao L.; Shi A.; Chen L. Circadian Rhythm Modulates Endochondral Bone Formation via MTR1/AMPKβ1/BMAL1 Signaling Axis. Cell Death Differ. 2022, 29 (4), 874–887. 10.1038/s41418-021-00919-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Ye X.; Cai M.; Liu X.; Xiao J.; Zhang C.; Wang Y.; Yang L.; Liu J.; Li S.; Kang C.; Zhang B.; Zhang Q.; Wang Z.; Hong A.; Wang X. Osteoblast-Targeting-Peptide Modified Nanoparticle for SiRNA/MicroRNA Delivery. ACS Nano 2016, 10 (6), 5759–5768. 10.1021/acsnano.5b07828. [DOI] [PubMed] [Google Scholar]
- Hagn F.; Nasr M. L.; Wagner G. Assembly of Phospholipid Nanodiscs of Controlled Size for Structural Studies of Membrane Proteins by NMR. Nat. Protoc 2018, 13 (1), 79–98. 10.1038/nprot.2017.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveetil R.; Nguyen K.; Vinogradova O. Nanodiscs and Solution NMR: Preparation, Application and Challenges. Nanotechnol Rev. 2017, 6 (1), 111–126. 10.1515/ntrev-2016-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros N. B.; Migliaccio V.; Facundo V. A.; Ciancaglini P.; Stábeli R. G.; Nicolete R.; Silva-Jardim I. Liposomal-Lupane System as Alternative Chemotherapy against Cutaneous Leishmaniasis: Macrophage as Target Cell. Exp Parasitol 2013, 135 (2), 337–343. 10.1016/j.exppara.2013.07.022. [DOI] [PubMed] [Google Scholar]
- Eichmann C.; Campioni S.; Kowal J.; Maslennikov I.; Gerez J.; Liu X.; Verasdonck J.; Nespovitaya N.; Choe S.; Meier B. H.; Picotti P.; Rizo J.; Stahlberg H.; Riek R. Preparation and Characterization of Stable α-Synuclein Lipoprotein Particles. J. Biol. Chem. 2016, 291 (16), 8516–8527. 10.1074/jbc.M115.707968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chromy B. A.; Arroyo E.; Blanchette C. D.; Bench G.; Benner H.; Cappuccio J. A.; Coleman M. A.; Henderson P. T.; Hinz A. K.; Kuhn E. A.; Pesavento J. B.; Segelke B. W.; Sulchek T. A.; Tarasow T.; Walsworth V. L.; Hoeprich P. D. Different Apolipoproteins Impact Nanolipoprotein Particle Formation. J. Am. Chem. Soc. 2007, 129 (46), 14348–14354. 10.1021/ja074753y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.