

Abstract
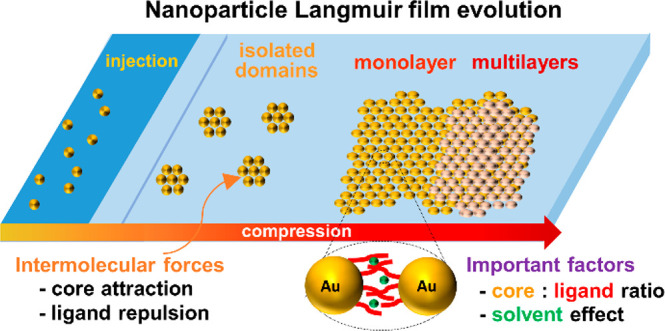
Recently the focus of the Langmuir–Blodgett technique as a method of choice to transfer monolayers from the air/water interface onto solid substrates in a controllable fashion has been shifting toward purely hydrophobic gold and silver nanoparticles. The fundamental interactions between particles that become relevant in the absence of polar groups range from dispersive attractions from the metal cores and repulsions between ligand shells to weaker entropic factors. The layer evolution is explored, starting with interfacial self-assembly upon solution spreading and domain and circular island formation, which subsequently merge into a complete monolayer and finally form multilayers or macroscopic wrinkles. Moreover, structural properties such as the core:ligand size ratio are investigated in the context of dispersive forces, whereby the nanoparticles with small cores and long ligands tend not to aggregate sufficiently to produce continuous films, those with large cores and short ligands were found to aggregate irreversibly, and those in between the two extremes were concluded to be able to form highly organized crystalline films. Similarly, the characteristics of the spreading solution such as the concentration and the solvent type crucially influence the film crystallinity, with the deciding factor being the degree of affinity between the capping ligand and the solvent used for spreading. Finally, the most common strategies employed to enhance the mechanical stability of the metal nanoparticle films along with the recent attempts to functionalize the particles in attempts to improve their applicability in the industry are summarized and evaluated in relation to their future prospects. One of the objectives of this feature article is to elucidate the differences between hydrophobic metal nanoparticles and typical amphiphilic molecules that the majority of the literature in the field describes and to familiarize the reader with the knowledge required to design Langmuir–Blodgett nanoparticle systems as well as the strategies to improve existing ones.
Introduction
The systematic study of Langmuir monolayers has its origins as far back as the turn of the 19th and 20th centuries when Agnes Pockels and Irving Langmuir performed their pioneering work on oil molecules on the surface of water. Their combined efforts demonstrated that insoluble monolayers floating on top of the aqueous subphase can be compressed using movable barriers and that this process results in a detectable change in surface tension. Further work initiated by Langmuir and refined by Katherine Blodgett concluded that multilayers composed of such films can be deposited on vertically placed substrates through successive immersion and extraction across the interface in what today is called the Langmuir–Blodgett (LB) technique. Another variation of this deposition method was explored by Vincent Schaefer, who demonstrated that lowering a horizontally oriented substrate until it makes contact with the air/water interface covered with molecules can lead to the transfer of a monolayer onto the substrate in what is referred to as the Langmuir–Schaefer (LS) technique.1 Both techniques are depicted in Figure 1(a).
Figure 1.

(a) Schematic showing Langmuir compression and merging of NP domains and successive transfer onto a solid substrate using LB and LS techniques. (b) Geometries of various X-ray surface-specific techniques. GIWAXS and GISAXS provide information about lateral and normal ordering of the film at atomic/molecular and mesoscopic length scales, respectively. XRR is used to determine the thickness, density, and roughness of the film by probing electron density perpendicular to the surface. Adapted with permission from ref (22). Copyright 2020 Springer Nature.
Since the early days of the technique, multiple new methods to analyze both the films on the water surface and various substrates after the deposition have been developed and optimized, as evidenced by thousands of publications and multiple reviews and book chapters released in the field. However, the vast majority of this work focuses on amphiphilic molecules such as fatty acids and phospholipids, which are known to form very stable Langmuir layers.1−3
Recently, however, the focus of LB research has been gradually shifting to the study of nonamphiphilic molecules, which can range from macrocyclic molecules4 and ionic liquids5 to carbon-based nanomaterials6 and metal nanoparticles (NPs).7 The absence of hydrogen bonds that stabilize the Langmuir layer introduces a new challenge for successfully ordering and transferring nonamphiphilic films. However, the newly sparked interest in the field is driven by a vast array of potential applications due to their optical and electrical properties as well as facile functionalization coupled with improved mechanical characteristics of recent nonamphiphiles that cannot be matched by their typical long-chain ancestors.1
Although a number of excellent reviews describing new methodology and the future of the LB technique in surface science have been published recently, we feel that inadequate attention has been attributed to fully hydrophobic molecules and, in particular, metal NPs.1,7,8 In this feature article, we focus on thiol-monolayer-protected gold NPs and nanoclusters (or clusters), which are small (<2 nm), atomically precise nanosystems.9 To familiarize the reader with the field, we begin by providing a brief introduction of Langmuir and LB films as well as a description of analytical techniques, although the technical details are outside of the scope of this paper and can be found in the cited literature.1,2,7,8 We then proceed to fundamental differences in intramolecular interactions at the air/water interface between typical amphiphilic LB molecules and metal NPs. The subsequent sections of this feature article are devoted to the evolution of a metal NP film throughout a typical Langmuir experiment and the impact of the structural properties of NPs and the spreading solution on the ordering in the film. We address this issue by focusing on individual factors such as NP size, ligand length/type, or the solvent used for dispersion. Finally, we consider recently discovered gold nanoclusters and speculate about the future of gold NP film deposition through the use of the LB technique.
Typical Langmuir Films and Analytical Techniques
A typical Langmuir monolayer is prepared by spreading an amphiphilic molecule dissolved in a volatile solvent on top of a water surface. While the hydrophobic tail endows the molecule with the required insolubility in water, the hydrophilic head anchors the amphiphile to the water molecules in the subphase via hydrogen bonds. Another crucial parameter for the solvent is the spreading coefficient Sc calculated using the following equation
where γ refers to the interfacial tension between two media and the indices w, a, and s correspond to water, air, and solvent, respectively. A positive Sc will result in lowering of the interfacial tension upon introducing the organic solvent, meaning that the spreading is spontaneous.1 This is fundamentally different from casting a drop of the solution directly on a solid substrate, where in most experiments with NPs the droplet does not wet the support (depending on the substrate’s hydrophilicity) and the interactions between molecules are governed by their diffusive properties and the evaporating speed of the solvent.10−12 This often results in nonequilibrium structures and multiple phases being present. On the other hand, the surface of ultrapure water used in LB experiments has a considerably higher surface energy than a solid substrate, and provided that adhesive forces dominate cohesive forces, the molecules will occupy the entire area available at the air/water interface.1 Following a rapid evaporation of a thinly spread solvent, a film is formed upon compression under equilibrium conditions.13 Consequently, the entire film will be composed of the same uniform phase, depending on the given surface pressure.14
The spreading of molecules in 2D can be likened to gas molecules filling the entire volume available to them in 3D and introduces us to the concept of surface manometry.1 Surface pressure (π) is defined as the degree of reduction of the surface tension of water by the presence of a monolayer at the air/water interface and can be recorded by means of a Wilhelmy plate method as the area available to the molecules is decreased by compressing the movable barriers at a constant temperature. Plotting the change in surface pressure against the area per molecule (Am) gives rise to π–Am isotherms, which indicate the formation of quasi-2D analogs corresponding to gaseous, liquid expanded, liquid condensed, and solid phases. Upon reduction of the area, the isotherm can show multiple steep transitions as the molecules exert more force on each other, which is accompanied by an increase in positional and orientational order.2 The extrapolation of a specific region of the isotherm to the zero surface pressure on the Am axis provides a straightforward way to estimate the area occupied by each molecule in the given phase.15
Apart from standard optical microscopy, a prominent analytical technique of the Langmuir monolayers is Brewster angle microscopy (BAM). With the incident polarized laser beam falling on the water surface at the Brewster angle (∼53°), it is ensured that the reflected intensity is minimal for water and that any changes in the refractive index of the interface imposed by the monolayer domains are easily picked up by the camera. The reflected light intensity provides information about the thickness of the film, the surface density of the molecules, and the film morphology on the mesoscopic scale. Moreover, in some cases the optical anisotropy can be used to determine the collective tilt of the molecules.15,16 The orientation of the molecules on the water surface can be further studied with vibrational spectroscopies including sum-frequency generation spectroscopy (SFG) and a range of infrared-based techniques.1,17,18 These techniques, however, have a limited use for metal NPs, since the contribution of organic groups covering the spherical core is weak.
A high-electron-density metal core, however, is an advantage when it comes to tapping into the structural details on the nanoscopic scale of NP films using X-ray scattering. Grazing incidence X-ray diffraction (GIXD) is a surface-specific adaption of the well-known X-ray diffraction in 3D.13 The technique is sensitive enough to study the molecular organization of a Langmuir monolayer that fulfills the Bragg condition in 2D. High sensitivity is obtained by producing an evanescent wave propagating along the air/water interface containing the monolayer.19 This is achieved by keeping the incident X-ray below the critical angle for total reflection. Changes in the setup geometry (typically adjustments of the vertical and horizontal detection angles, see Figure 1(b)) lead to different properties being probed in a range of surface-specific adaptations of the well-known X-ray techniques such as small-angle or wide-angle X-ray scattering (GISAXS and GIWAXS respectively, where GI stands for grazing incidence) or X-ray reflectivity (XRR).20−22
XRR probes the electron density only in the direction perpendicular to the surface, whereby the interference of all the reflected waves (in and out of phase) produces a reflectivity curve. Model fitting allows the elucidation of individual properties of each layer, including its thickness, roughness, and electron density, typically presented in an intuitive scattering-length density (SLD) plot.23 Combining XRR with an X-ray technique with an xy component (such as GIXD) provides extremely useful microscopic information about ligand interdigitation, ordering of particles, and vertical organization of LB films of metal NPs.21
The range of analytical techniques that can be applied following the deposition onto solid substrates gets largely expanded to any surface-specific technique that is sensitive enough to detect the given monolayer. Therefore, the spectroscopic and X-ray-/neutron-scattering techniques discussed above are not excluded from that list. An important expansion used ubiquitously for transferred films is scanning probe microscopy (SPM). Since the early days of SPM, numerous alterations have been conceived, which, on top of determining surface topography, allowed the study of electrical, dielectric, magnetic, and mechanical properties of the film.24
For example, in atomic force microscopy (AFM) the topographic images are recorded by measuring the atomic force between an ultrasmall tip and the surface. In its most common configuration, the tapping mode, the tip is oscillated above the sample close to the resonant frequency of the cantilever. The change in amplitude is used to obtain a topographic image of the sample while the change in phase of oscillation provides information about the tip–sample interactions including the mechanical properties.21,25 The latter can be further elucidated from a range of nanomechanical AFM modes such as bimodal AFM26−28 or force–distance curve measurements (nanoindentation).29−31
It should be noted that the substrate requirements for a given technique can be a limiting factor as different hydrophilicities and roughnesses of the surface will affect the transfer efficiency. Moreover, it has been shown that the mode of transfer (LB vs LS) can influence the hydrophobicity of the fabricated surface.32
All of the information about Langmuir films and analytical techniques so far included in this feature article apply both to typical amphiphilic molecules and their hydrophobic counterparts. The next section, however, will discuss the interactions of nonamphiphilic molecules on the water surface with a particular focus devoted to Au and Ag spherical NPs. These interactions are drastically different from those of typical amphiphilic molecules discussed in most Langmuir/LB reviews to date.
Interactions of Nonamphiphilic Molecules
Amphiphilic Langmuir monolayers exhibit a strong stabilizing effect primarily driven by enthalpic interaction between the polar heads of the molecules and the water subphase as new hydrogen bonds are formed upon spreading of the film. In the absence of polar groups, multiple factors contribute to the stability of a monolayer. In the case of NPs, van der Waals (vdW) attractions and steric repulsions are the dominant interactions between the particles.13 The attractive dispersion forces are expected to scale geometrically with NP size and to increase considerably with a high degree of monodispersity, which allows the contacts between particles to be maximized.14,33 On the other hand, the interdigitated ligands (typically thiols) are a source of steric repulsion between the NPs,34 the exact nature of which depends on many factors including the length and chemical nature of the capping ligand,17 their density on the metal core,13,35 and the solvent.36 It has even been suggested that NPs’ cores might act only as anchors for the thiols, with themselves not directly influencing the mechanical properties of the Langmuir film. This claim is justified by the argument that the metal cores are typically separated by 1 to 2 nm, which considerably exceeds the range of chemical and vdW interaction potentials.37 A comprehensive study of LB films based on a range of core:ligand size ratios showed that this statement might indeed be true for small particles capped with long ligands. However, in the case of LB films based on larger NPs with shorter ligands, the dispersion effects dominate.14
It has been seen that large dodecanethiol-capped Au NPs (7.5 nm core) can irreversibly aggregate into nonequilibrium structures predicted by the diffusion-limited-aggregation (DLA) model, which demonstrates the importance of strong attractive dispersion forces.14 In an extreme case, it has been shown that at very high surface pressures the alkanethiols capping the surface of Au NPs can be removed, leading to the formation of strong metallic bonds and subsequent fusion of spherical NPs into nanowires.36 Regardless, the ligand interactions preclude the treatment of NPs as hard spheres, which eliminates some of the interactions that might be typically important for films based on polystyrene or silica spheres, such as capillary forces.38
Given the complexity of the system, the direct effect of the core is extremely difficult to isolate as changes to its type or size will often affect other factors that crucially influence the intermolecular forces. Indeed, comparisons by Kim et al. of dodecanethiol-capped metal NPs with an average diameter of 6 nm have shown that films of Au NPs exhibited a clear 2D-hexagonal packing while those based on Ag NPs appeared to be amorphous. However, in spite of evident differences in packing as well as the mechanical properties of the two films, it could not be concluded that the type of metal core had an effect, since the polydispersity of the two types of NPs was significantly different (with the Ag NPs being more polydisperse).37
The relative polydispersity also tends to increase with the decreasing NPs size.37 This means that in the case of smaller NPs, the contribution of attractive dispersion forces decreases primarily due to weaker vdW forces but second due to decreased contacts resulting from higher polydispersity. Gupta and Manjuladevi explore various examples of LB films based on nonamphiphilic molecules and suggest that, in the absence of enthalpic interactions in the form of hydrogen bonds between molecules of study and the water subphase, entropic factors may contribute to the layer stability.15
For instance, an increase in entropy can originate from the rearrangement of the interfacial water molecules and the disorder introduced by hydrogen-bonds breaking as the hydrophobic molecules approach each other through hydrophobic interactions.39,40 Alternatively, NPs that exhibit a degree of polydispersity as well as hydrophobic molecules that can attain multiple orientations on the water surface can lead to the frustration in the orientation of the water molecules, facilitating the monolayer formation by increasing the entropy of the system.41,42
In spite of the interplay between entropic gains resulting from high polydispersity and efficient molecular ordering due to high monodispersity, in the case of most of the LB films of noble metal NPs that have been described in the literature, the latter effect seems to dominate. Higher-stability films are obtained when the polydispersity is low and the contacts between NPs are maximized. For instance, Kim et al., when comparing Ag NPs with Au NPs, attribute the decrease in mechanical stability of the Ag NP film to the higher polydispersity of the constituting particles.37 Similarly, when comparing films of more-monodisperse Ag NPs and less-monodisperse Au films, Heath et al. obtained better ordered films with the former, demonstrating that the degree of polydispersity might indeed be more important than the metal type.14
Film Evolution
Domain Formation
Following the injection of the solution onto the water surface and the rapid evaporation of the volatile solvent, small crystalline domains of NPs are formed.13 Multiple studies employing GIXD or similar surface-sensitive X-ray techniques20,21,27,33 have shown that the same correlation peaks are often observed irrespective of the surface pressure, suggesting that the interparticle separation distance is established in the initial stages of the Langmuir experiment. Nevertheless, the crystallinity of the small domains that are formed is highly dependent on the polydispersity of the NP solution,33 and the interparticle separation distance depends on the thiol ligand length and its concentration on the surface of the metal core.13,35
A mechanism of particle assembly following the injection has been proposed by Huang et al., as shown in Figure 2(a). As the solvent evaporates, the convective flows (js in Figure 2(a)(i)) carry the particles toward the surface of the solvent (jp), where they assemble (convective assembly). After further evaporation of the solvent until it reaches a thickness of one NP (Figure 2(a)(ii)), the surface tension significantly increases the attractive interactions between NPs, leading to the formation of closely packed arrays (Figure 2(a)(iii)).43 Furthermore, the evaporation of the solvent increases the vdW attractions between NPs due to the diminishing effect of the dielectric shielding of the solvent molecules.44
Figure 2.

(a) Proposed mechanism of the self-assembly of NPs after the injection: (i) convective assembly of NPs at the solvent/air interface; (ii) increase of the dispersive interaction between NPs upon solvent thinning; and (iii) formation of closely packed arrays. Reprinted with permission from ref (43). Copyright 2001 American Vacuum Society. (b) Optical microscopy images of Ag NP films at different stages of compression: (i) monolayer, (ii) hash phase, (iii) wrinkling, and (iv) giant V-folds. Reprinted with permission from ref (37). Copyright 2011 AIP Publishing.
The events occurring
before the solvent thinning
to one NP thickness
can be related to the kinetic models proposed by Bigioni et al. for
drop casting experiments. The diffusion of an NP in the bulk of the
drop can be estimated from the Stokes–Einstein equation D = kBT/6πηr, where D is the diffusion
constant, kB is Boltzmann’s constant, T is the temperature, η is the solvent viscosity,
and r is the hydrodynamic radius of the NP. As the
drop evaporates, the interface descends toward the substrate with
a velocity v, which is determined primarily by the
evaporation speed and is typically much higher than the diffusion
speed of the NP, and therefore is impinged upon by NPs with flux f = −cv, where c is the concentration of the NPs. If the NP impinges on the interface
within the interfacial diffusion length l of an existing
domain, then it can be incorporated or it can desorb back to the bulk
drop. This length is defined by the equation 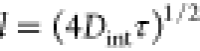 , where Dint is the diffusion constant for the air–liquid interface and
τ is the time the NP moves along the interface. Interestingly, l has been found to increase with the free thiol concentration
in the solution. NPs can also impinge directly on a domain, in which
case they can move along the domain and be incorporated at its perimeter
or at a defect.11
, where Dint is the diffusion constant for the air–liquid interface and
τ is the time the NP moves along the interface. Interestingly, l has been found to increase with the free thiol concentration
in the solution. NPs can also impinge directly on a domain, in which
case they can move along the domain and be incorporated at its perimeter
or at a defect.11
This model was proposed and tested experimentally for drop casting experiments, where the thickness of the droplet considerably exceeds the thickness of the solvent used for spreading Langmuir films where the solvent completely wets a fluid substrate (water). Nevertheless, the mechanism relies on the movement of the NPs in the bulk solvent and the evaporation of the air–liquid interface and therefore can be used to justify the differences in film morphology obtained within the LB framework when different solvents or NPs are used in the spreading solution. Furthermore, it explains why drop casting experiments lead to the initial formation of a small number of islands, which grow to relatively large sizes following a constant flux of NPs as the solvent evaporates over a prolonged amount of time.13 It also rationalizes why many nonequilibrium structures are formed in drop casting experiments. On the other hand, in LB the evaporation of the solvent is comparably very rapid, which leads to the formation of many small separated islands distributed over a large available area, which upon compression are merged together under equilibrium conditions.
Given how rapid the evaporation events are after the spreading of the NP solution, the current techniques cannot establish whether the interparticle distance is established during or following the solvent evaporation. Indeed, when comparing drop-cast and LB films of the same NPs, the latter typically has a smaller interparticle separation.13
This is consistent with an initial interfacial self-assembly as described for drop casting by Bigioni et al.11 and subsequent reduction of the interparticle separation as suggested by Huang et al. and described above.43 This reduction occurs due to the surface tension and the diminishing dielectric shielding effect of the solvent once it thins enough. Although the details of this mechanism are currently unresolved, it is clear that the processes heavily depend on the evaporation speed of the solvent, the concentration of NPs in the solution, and the size of the NP.
Coalescence into a Monolayer
The subsequent evolution of noble metal NP films upon compression of the barriers can be followed by the coloring caused by the surface plasmon resonance (SPR) as seen in the optical microscopy images shown in Figure 2(b) in the case of a film based on 6 nm dodecanethiol-capped Ag NPs. Although the wavelength of the SPR is size-dependent, in this size regime most Ag and Au NP films will exhibit yellowish and purplish coloring, respectively.37,45 However, compared to their frequency of SPR in a solution, the films of NPs that are in close proximity in the 2D lattice tend to exhibit modifications in their dielectric environment due to neighboring particles, resulting in shifts of the SPR to lower energy.46
At early stages of the Langmuir experiment, the aforementioned crystalline domains formed upon solvent evaporation merge together to form mesoscopic 2D islands. With subsequent compression, these islands rotate and rearrange with respect to each other to eventually form a complete monolayer characterized by the lack of distinct features (Figure 2(b)(i)).
The merging of domains upon compression is also well demonstrated by scanning electron microscope (SEM) images of a dodecanethiol-ligated Au NP film (8.5 nm diameter), transferred onto a hydrophobized silicon substrate by a horizontal lift (LS) as shown in Figure 3(a).47 It should be noted that with the reduction of the area, it is the interdomain distance that gets reduced while the spacing between individual NPs within the domains remains constant, as known from GIXD.13 Our recent fast AFM measurements on LB films of Au38(SC2H4Ph)24 cluster films transferred onto Si demonstrate that crystalline domains extending over hundreds of nanometers of particles as small as 1.6 nm diameter can be imaged (Figure 3(c)).21 Another example of a surface-pressure-dependent morphological transition is shown by TEM images of octanethiol-passivated 4–6-nm-diameter Ag NP LB films shown in Figure 3(b)(i). At low coverage, the domains of NPs form circular islands. Upon compression, however, these islands can be pushed toward each other until each domain is strongly repelled by other domains. At this point, the transition into stripes (Figure 3(b)(ii)) results in a lower energy as the separation between stripes is larger than the separation between domains would be.48
Figure 3.

(a) Merging of domains into a continuous film as shown by SEM (π = 0, 15, and 20 mN/m in (i), (ii), and (iii), respectively). Reprinted with permission from ref (47). Copyright 2004 American Chemical Society. (b) Transition of circular islands into stripes upon compression as shown by TEM. Reprinted with permission from ref (48). Copyright 1999 American Physical Society. (c) AFM phase output of Au38(SC2H4Ph)24 cluster LB film showing an extended domain of hexagonally packed clusters. Adapted with permission from ref (21). Copyright 2021 Wiley Materials.
Folding into Bi-/Trilayers
Further film area reduction below the monolayer regime results in a gradual transition of the monolayer into a multilayer (bilayer or trilayer depending on the NP) via an intermediate hash-like phase characterized by multiple crisscross stripes aligned in a quasi-parallel fashion in the general compression direction (Figure 2(b)(ii)).33,37
For a long time, various groups tried to find the reason some noble metal NP films form bilayers when the barriers are compressed above the monolayer regime (e.g., 5.5 nm Ag NPs, 40% PD or 2 nm Au NPs, 25% PD) while others transitioned from a monolayer to a trilayer via the hash-like phase without showing any evidence of bilayers in XRR (e.g., 6 nm Au NPs, 8% PD). The mechanism leading to the trilayer was subsequently proposed based on an S-fold shown in Figure 4(a)(i). Nevertheless, apart from not being able to explain the bilayer formation in small Au NP and highly polydisperse Ag NP films, the S-folding mechanism would require two adjacent layers to slide over each other as the fold expands, which would be unfavorable kinetically.33
Figure 4.
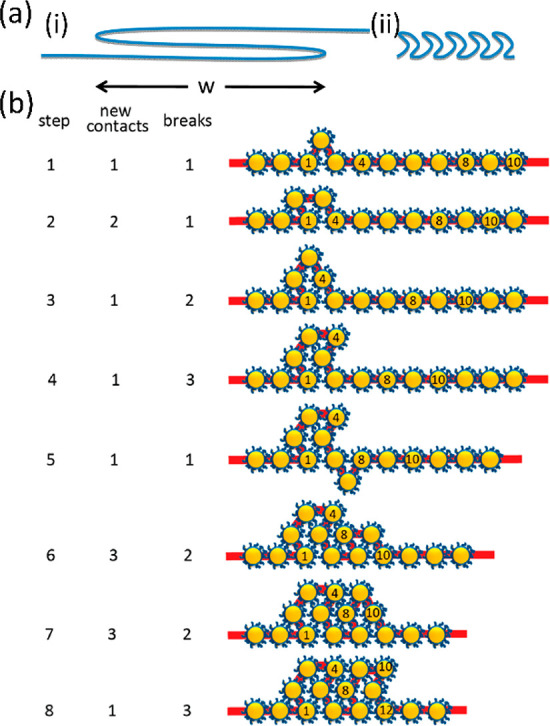
(a) Schematics of the proposed mechanisms of bi/trilayer formation: (i) S-fold mechanism and (ii) crinkle fold mechanism. (b) Details of the crinkle fold mechanism with the number of particle–particle contacts formed and particle–water contacts broken with each step. Reprinted with permission from ref (33). Copyright 2013 American Chemical Society.
However, a recent paper by Dai et al. demonstrated the use of grazing incidence X-ray off-specular scattering (GIXOS) to elucidate the evolution details of a Au NP film that is known to form trilayers. Like conventional XRR, GIXOS yields electron density profiles along the normal to the water surface but it can provide subminute temporal resolution, which is crucial for studying nonequilibrium phases as the films are continuously compressed. The study revealed the coexistence of a transient partial double layer and a partial trilayer which form after the completion of the initial monolayer. Following this observation, the group has hypothesized a new “crinkle” mechanism depicted in Figure 4(a)(ii),(b), which unlike the S-fold can explain the formation of double layers and is more plausible kinetically. One of the assumptions of the crinkle folding is that NPs have a negligible bending energy and behave as freely rotating links.45 The mechanism is based on the local nucleation of bilayers over length scales comparable to the size of the NPs (step 1), which subsequently expand laterally (step 2) or fold into trilayers (steps 3–8). This folding process is favorable kinetically because new particle–particle contacts are formed (short-range attraction) at the expense of losing the attraction between particles and the water subphase, which is relatively weaker. The number of particle–particle contacts formed and particle–water contacts broken associated with each hypothesized step is included in Figure 4(b). Furthermore, with each folding step some free energy is gained as a result of the compression of the layer by one particle width. Although overall this mechanism is kinetically favorable, each nucleation event must involve a transient state, in which water energy has been lost but the interparticle contact energy has not yet been gained. Therefore, an activation barrier is imposed on the crinkling event, which can explain why transient bi- and trilayers are initially formed locally rather than globally.33
Furthermore, the crinkle fold mechanism can provide a plausible reason that smaller Au and Ag NPs tend to form bilayers rather than trilayers: higher polydispersity results in weaker particle–particle interaction, which might suppress further crinkling into trilayers. Similarly, in the case of monodisperse Au NP films that form well-defined trilayers, the formation of the fourth layer is prevented as doing so would require the simultaneous movement of many particles, thus presenting a sizable kinetic barrier. Therefore, according to the proposed crinkle mechanism, following step 8 in Figure 4(b), the trilayer gets expanded by repeating steps 5–8 rather than nucleating into a fourth layer.33
Meso-/Macroscopic Wrinkles
Further compression beyond the bilayer or trilayer regime, as determined predominantly by interactions between the particles, leads to the formation of macroscopic wrinkles discernible by optical microscopy (Figure 2(b)(iii)). These wrinkles are oriented orthogonally to the direction of compression, and unlike the hash phase (Figure 2(b)(ii)), they are aligned parallel with respect to each other. It should be noted that the periodicity of the wrinkles does not depend on the applied surface pressure, although the wrinkles dissipate upon decompression.
Even further deflection of the wrinkled layer can finally lead to the formation of a vertical fold into the subphase. These V-folds can extend from hundreds of micrometers to a few centimeters in the y direction (Figure 2(b)(iv)), but they are no longer periodic like the wrinkles tend to be.45
Deposition on a Surface
The last optional stage of a Langmuir experiment is the transfer of the film onto solid substrates (by either the LB or LS method). For typical amphiphiles, it has been reported that under optimized conditions the deposition can occur without any changes in morphology between the film on the water surface and the solid substrate.49,50 However, in our opinion, not enough attention has been devoted to this topic when it comes to pure films of Au and Ag NPs, where groups typically tend to focus on either Langmuir films on the water subphase or postdeposition LB films, without comparing the differences between the two using the same characterization technique. In fact, in some cases a strong aggregation between NPs upon dewetting has been reported, for example, by Tim et al., who observed the formation of dendritic structures oriented quasi-parallel to the substrate extraction direction.51 Moreover, Kundu and Bal have shown using XRR measurements that the hydrophilicity of a substrate can play a crucial role in the final morphology of an Au NP LB film, as directed by substrate–nanoparticle (Es–n) and nanoparticle–nanoparticle (En–n) interactions upon dewetting. LB transfers onto hydrogen-terminated Si substrates tended to favor the Es–n interactions with both the substrate and the NP being hydrophobic, which resulted in the NPs forming a monolayer on top of Si with a partial second layer. However, Si surfaces terminated with hydrophilic groups (Br and OH) reduced the Es–n interaction, meaning that the En–n interactions dominated, thus leading to the formation of trilayers of Au NPs upon dewetting. The coverage of each successive layer increased with the hydrophilicity of the substrate.52
Finally, although some attempts have been made to elucidate the long-term stability properties of LB films of Au NPs, they are typically restricted to morphology measurements by AFM, which do not show the changes in the internal structure of the film. However, an XRR study by Das and Kundu has concluded that after 12 months the multilayers of the dodecanethiol-capped 3.4-nm-diameter Au NP film that had been clearly discernible initially (and after 2 months) have merged into a single thick electron-rich layer. The XRR curves are shown in Figure 5. In the more intuitive SLD profiles shown in the insets, z = 0 corresponds to the air/film interface and z = ∞ is the bulk substrate, while the variations in the electron density indicate the denser metal cores and the less-dense ligand shells. With a strong red shift of the SPR being observed, the group concluded that the metallic cores coalesce into larger NPs after a prolonged reorganization on the surface,53 exacerbating the importance of more stringent stability studies should the LB films of noble metal NPs become more common in industrial applications.
Figure 5.
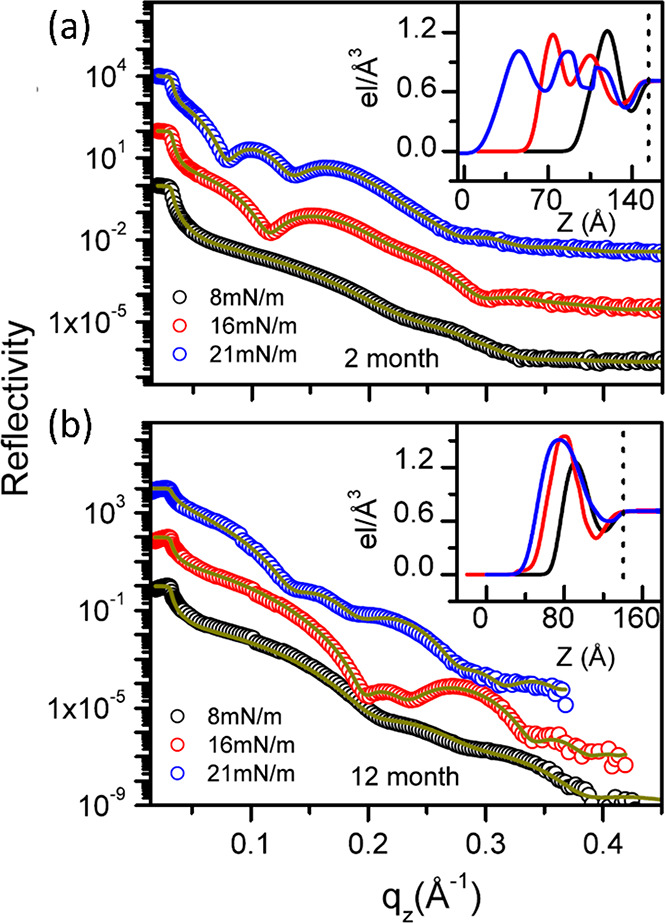
XRR curves and the corresponding SLD profiles of Au NP films transferred onto H-terminated Si at three different surface pressures recorded (a) 2 months and (b) 12 months after the deposition. Reprinted with permission from ref (53). Copyright 2014 AIP Publishing.
NP Structure and Spreading Solution
Core:Ligand Size Ratio
Given the interplay between weak interactions directing the assembly and evolution of nonamphiphilic metal NP films described in the previous sections, it is understandable that subtle differences in the structure of the NPs as well as the experimental conditions will be crucial in determining the properties of the film. On top of the surface pressure already discussed in more detail, the relevant factors include the metal core size, length and chemical nature of the capping ligand, solvent used, NP concentration, volume of the injected solution, temperature, speed of compression, speed and mode of transfer onto a substrate, and hydrophilicity of the substrate. We discuss some of these aspects here, paying particular attention to publications that consider one parameter while keeping as many of the other factors constant as possible.
We start this discussion by expanding on the work of Heath et al., who compared Langmuir films of Ag and Au NPs in the size range of 2–7.5 nm capped with alkanethiol ligands based on 9, 12, and 18 carbon atoms.14 Although their work is over two decades old now and NP synthetic procedures and size-selection techniques have certainly improved since then,54 the authors reach prominent conclusions on the significance of the core:ligand size ratio by considering the concept of excess conical volume (Ve). As linear ligands extend from the surface of the metal core, Ve available to the ligand increases with increasing distance from the core, as shown in Figure 6(e). With f being the footprint of the ligand which is assumed to be independent of the NP size (21.4 Å2), for a spherical particle Ve is proportional to L3/R2, where L is the length of the ligand and R is the core radius. Ve determines the ability of the ligands to interpenetrate with those of the neighboring NPs in order to maximize the dispersion interactions between the ligands. Depending on Ve, the group classified three distinct types of particles which are reflected in the π–A isotherms and the morphology of the films: case I particles with small cores and long ligands (Ve > 350 Å3), case III particles with large cores and short ligands (Ve < 150 Å3), and case II particles lying between the two extremes, with their exemplary isotherms shown in Figure 6(a)–(c) in order of decreasing Ve.14
Figure 6.

Isotherms of exemplary films with Ve decreasing along the series: (a) case I NPs and (b) case II NPs. Reprinted with permission from ref (37). Copyright 2011 AIP Publishing. (c) Case III NPs. Reprinted with permission from ref (43). Copyright 2001 American Vacuum Society. (d) Phase diagram of a case II NP film. (e) Diagram explaining the concept of Ve. (f) TEM micrograph of a case II NP film. Panels a, d, e, and f reprinted with permission from ref (14). Copyright 1997 American Chemical Society.
The isotherms of typical Langmuir amphiphiles tend to proceed from a highly compressible gaseous phase at high area to gradually less compressible liquid and solid phases upon compression. Films of NPs with small cores and long ligands (case I) such as 2-nm-diameter Au cores protected by oleyamine ligands (C18H35NH2) exhibited unusually low compressibility in the region between 3 and 5% coverage, which was followed by a higher compressibility region above 8% coverage (Figure 6(a)). To explain this surprising behavior, Heath et al. suggested that as two NPs approach each other and their ligands interpenetrate, the tied-up ligand structure effectively blocks the binding site that would maximize the dispersion interactions with the third approaching particle (equilateral triangle). Thus, TEM micrographs of this low-compressibility phase are dominated by 1D structures (cyclic or linear) that adopt a more open trimer structure with the angle among the three NPs being 90–180°. Upon further compression, the tied-up ligand pairs are “untangled”, resulting in higher compressibility and the formation of a “foam-like” phase which has an overall low coverage and does not exhibit long-range order. However, it should be noted that case I isotherms are fully reversible upon expansion and recompression.14
At the other extreme are particles with very low Ve (case III) such as dodecanethiol-capped 17-nm-diameter Au NPs, an isotherm of which is shown in Figure 6(c). These large-core NPs irreversibly aggregate into nonequilibrium structures due to strong dispersion interactions and do not form closely packed structures or relax upon barrier expansion.43
Out of the isotherms discussed here, the typical Langmuir compression behavior most closely resembles the isotherm of a case II NP film shown in Figure 6(b) (dodecanethiol-capped 5-nm-diameter Au core), albeit with less-distinct transitions than normally observed.37 Indeed, NPs with small–medium core size and short–medium-length ligands were shown to be able to form a highly crystalline 2D phase, provided the monodispersity was sufficiently high. With such a core:ligand size ratio, the dispersive forces are sufficient to induce particle aggregation (unlike case I NPs) but they are weak enough to avoid irreversible aggregation into nonequilibrium structures that occurs for case III NPs with large cores. A TEM micrograph of a highly crystalline film based on dodecanethiol-capped 2.8-nm-diameter Ag NPs is shown in Figure 6(f).14
To gain further insight into the behavior of NP Langmuir films, Heath et al. constructed π/T phase diagrams by compressing the films at a range of temperatures and using the local minima from the isotherm derivative plots. A phase diagram of a dodecanethiol-capped 4-nm-diameter Au film (case II) is shown in Figure 6(d). Drawing a vertical line at a given T shows which phases the film goes through as π is increased.2 The negative slope of the 2D to 3D phase boundary implies that the higher-density phase is more disordered than the lower-density phase (increase in entropy), which is uncommon for typical Langmuir films. In physical terms this result is explained by TEM micrographs of films transferred at high surface pressure, which demonstrates that parts of the film that transition into multilayers are not as ordered as the monolayer.14 Similarly, our AFM study of Au38(SC2H4Ph)24 cluster films leads to the conclusion that in a trilayer the bottom layer is more ordered than the upper layers.21 In fact, the phase diagrams of case I and case III NPs were also shown to exhibit a negative dπ/dT at the boundary between low- and high-density phases. For the case I particle discussed here, this result was explained by a small degree of long-range order exhibited by 1D structures at low π, which diminished further in the “foam-like” phase at higher compression.14
Ligand Length/Chemical Nature
The length of the alkanethiol ligand capping the NP can have important effects on the electron-transfer properties, with the transfer rate decreasing with longer alkanethiols as the metal cores are further away from each other. This trend has been reported to be true until the length of 12 carbon atoms, after which the chain forms bundle-like structures. Yokoyama et al. have measured the vibrational spectra of dodecanethiol-capped Au25 clusters by infrared reflection absorption spectroscopy (IRAS) and noticed a temperature-dependent conformational change (trans to gauche) by inspecting the intensity ratio of the in-plane and out-of-plane modes of the asymmetric terminal methyl group vibration. The group has also studied the effect of changing the chemical nature of the capping ligand on the morphology of the respective LB films they form. While dodecanethiol-capped Au25 clusters can form LB monolayers, films of the Au25(SC2H4Ph)18 cluster (with a much shorter and stiffer phenylethanethiol ligand) had a reticulated structure, which is in line with our discussion about the increase in the dispersive forces and the ease of aggregation as the intermolecular distance decreases.17
On the other hand, a different approach was taken by Brust et al. in order to tune the electron-transfer properties as well as the mobility of the ligand shell, where they deposited LB films of Au NPs capped by alkanethiols, alkaneselenides, and alkanetellurides. Although films containing Se in the ligand shell of the NPs exhibited properties similar to those of thiolated ones, those based on Te showed a high degree of NP coalescence due to their low stability.55
Although in their calculation of Ve Heath et al. assumed a constant footprint of a thiol molecule on the metal core of the NP, experiments by Schultz et al. demonstrated that the footprint can indeed be controlled by varying the thiol concentration in the NP solution, which in turn affects the interparticle spacing between the particles.13 As briefly mentioned before, multiple GIXD or GIWAXS studies have proven that the interparticle separation distance is established during the evaporation of the solvent and does not depend on the surface pressure.13,21 This is shown in Figure 7(a), where the first three diffraction peaks of a GIXD signal integrated along the qz direction of the dodecanethiol-capped 6-nm-diameter Au NP film are the same before and after compression (represented by circles and triangles, respectively).
Figure 7.

GIXD signal integrated along the qz direction of a Au NP film (a) showing the same diffraction peaks for a monolayer and a multilayer and (b) showing an increasing correlation distance in the presence of additional thiol in the spreading solution. Panels a and b are reprinted with permission from ref (13). Copyright 2006 American Chemical Society. (c) SEM images of the islands of domains formed in a Au NP film at three different NP densities on the surface, where the densities of (ii) and (iii) are 5 and 10 times higher than that of (i), respectively. Reprinted with permission from ref (43). Copyright 2001 American Vacuum Society. (d) Diagram of domains formed at (i) low and (ii) high NP concentrations in the solution demonstrating that higher concentration can lead to larger defects. Reprinted with permission from ref (58). Copyright 2001 American Vacuum Society.
Moreover, the group varied the thiol concentration on the Au NPs by repeatedly washing one batch with ethanol to remove excess thiol and by dissolving additional dodecanethiol in the solution prior to injection (labeled as Au and ΔT-Au in Figure 7(b), respectively). A clear shift to lower qxy values was observed for the film with higher thiol concentration. The conclusion was that for NPs with low dodecanethiol density on the surface the ligand shells of neighboring particles can easily rearrange and interdigitate with each other, resulting in an interparticle separation (edge–edge) distance of 1.4 nm. Conversely, the steric repulsion between dodecanethiol ligands densely packed on the NPs’ surface in the ΔT-Au film constricts their degrees or freedom and forces them to extend axially, resulting in a larger interparticle separation of 2.5 nm. Similar conclusions were reported by other groups.35 Consequently, the increase in the interparticle separation results in a reduction of the attractive interactions between the metal cores, leading to a much higher reversibility of a wrinkled film upon expansion in the film with additional dodecanethiol.13 A comparable value of 2.2 nm for the interparticle separation was found by Huang et al. for an LB film of dodecanethiol-capped 8.5-nm-diameter Au NP as measured by SEM (Figure 3(a)).47 On the other hand, the LB films of Au38(SC2H4Ph)24 clusters studied by our group were shown by XRR and GIWAXS to be highly interdigitated, which is perhaps affected by the stabilizing π–π stacking between the phenyl rings and strong vdW interactions caused by the short length of the ligand.21
Although primarily used to calculate the correlation distance between NPs, 2D X-ray techniques can provide further important structural details. For example, Vegso et al. have shown that the collapse of a monolayer of a Ag NP film into a bilayer is associated with the emergence of additional peaks in GISAXS.56 Furthermore, the width of the peaks in a GIXD plot can provide information about the average size of the domains of NPs formed upon solvent evaporation using the following equation
where Δθ is the angular width of the peak, Δk is the width of the inverse space, and LD is the size of the domain. Schultz et al. found the domains to be on the order of 7–9 times the particle diameter,13 which agrees well with the results from Fukuto et al.57 as well as our ultrafast AFM measurements in the liquid of Au38(SC2H4Ph)18 cluster films transferred onto mica.21 Sear et al. have argued that the domain size is primarily determined by the balance between attractive dispersive interactions and long-range repulsions between domains.48
Volume and Concentration of Spreading Solution
On the other hand, Huang et al. have demonstrated that the size of the circular islands formed upon spreading can be increased by increasing the volume of NP solution injected onto the water surface before compressing as shown by SEM, where the islands formed in Figure 7(c)(ii),(iii) had 5 and 10 times more NPs injected onto the surface than the one shown in Figure 7(c)(i), respectively. It should be noted that in these experiments the number of consecutive injections of a small volume at regular intervals increased but the volume injected each time remained constant. With the initial spreading and evaporation of the solvent, small domains were formed, but with the successive addition of NPs, the size of the nuclei increased through convective mechanisms described before and shown in Figure 2(a). Moreover, the size of islands did not increase linearly with the increased NP density but increased considerably faster, suggesting the coalescence of multiple neighboring nucleus sites at high particle densities. Although increasing the domains or island size might be beneficial as it improves the crystallinity of the film, it can also lead to the emergence of large defects due to poor accommodation of particle domains as they coalesce.43
Similar conclusions were reported in another publication by the same authors, whereby the concentration of the solution was varied. SEM images of the dodecanethiol-capped 8.3-nm-diameter Au NP film showed a higher crystallinity and lower extent of defects between merged domains at lower concentration than at a 10× higher concentration. At low concentration, the islands were formed as described above, i.e., with the formation of small domains and their successive growth by agglomeration. In this scenario, small defects may arise due to sample polydispersity, as shown in Figure 7(d)(i). However, when higher concentrations of the NP solution were used, many small domains or islands form in close proximity, which again can lead to larger defects due to the nonaccommodation of domains upon merging, as seen in Figure 7(d)(ii).58 Sear et al. also investigated the change in concentration of their octanethiol-capped Ag NP film shown in Figure 3(b) and reported that increasing the concentration of the spreading solution in the range of 0.1–1 mg/mL had a similar effect to that of increasing surface pressure; i.e., circular islands transitioned into stripes.48 Although low concentrations of the solution may lead to fewer defects and more crystalline films, it should be noted that higher concentrations can shorten the manufacturing time so that finding an optimal concentration might be crucial for industrial applications.
Solvent Used for Spreading
Another factor whose importance cannot be overstated is the solvent used for spreading the NPs on the water surface. Huang et al. observed higher crystallinity in their system when using chloroform than they did with benzene. The authors explain this difference by suggesting that with the slower evaporating benzene, the crystalline domains that form have enough time to coalesce together, leading to aforementioned defects due to poor domain accommodation.58
The evaporation speed of the solvent, however, cannot explain the results observed by Pei et al., who compared LB films of dodecanethiol-capped 4.8-nm-diameter Au NPs spread in hexane and toluene. TEM micrographs of the film injected in the slower evaporating toluene exhibited highly ordered arrays of NPs without any compression. The use of faster evaporating hexane, on the other hand, resulted in a random arrangement of NPs in the film.36 The authors propose the effect of Hansen’s solubility parameter (δ) theory to explain their results.59,60 Here, δ determines the extent of affinity between the solvent and the capping ligand of the NP. More broadly, it is used as a framework to break down the cohesive energy density into the intermolecular force components due to dispersive (δd), polar (δp), and hydrogen bond (δh) interactions, according to the equation
With the δ value of toluene (18.7) being much closer to that of dodecanethiol (17.6) than δ of hexane is (14.9), toluene can penetrate easily into the ligand shell of the NP, allowing the ligands to extend uniformly in all directions as shown in Figure 8(a). Hexane, however, has a poor affinity toward dodecanethiol molecules, meaning that the chains interact directly with each other, resulting in the deformations shown in Figure 8(b). Effectively, the toluene-solvated NPs are all spherical and monodisperse, leading to a high degree of crystallinity upon spreading, while hexane-solvated NPs form defects in the particle arrangement due to their twisted shape. Interestingly, at very high compressions (36 mN/m) the group observed the fusion of NPs into nanowires, which were straight and aligned with respect to each other in the case of toluene but polycrystalline and “worm-like” in shape in the case of hexane.36
Figure 8.
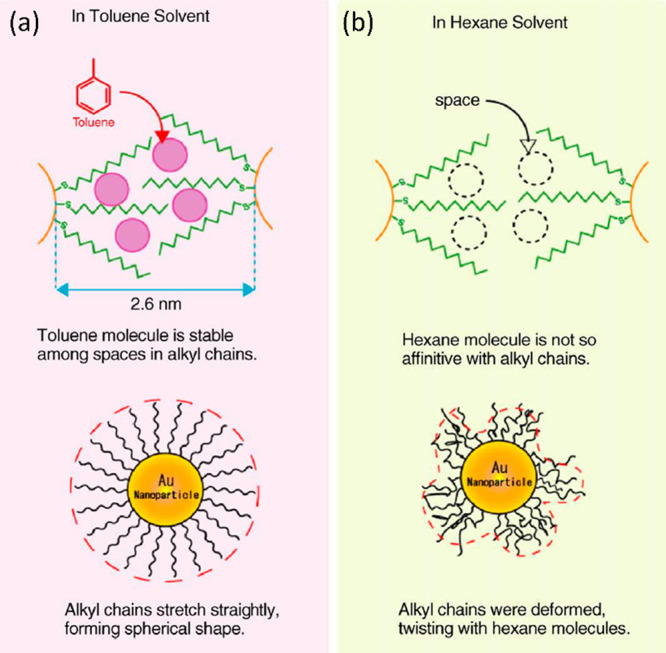
Diagram showing how ligand shells of the NP orient themselves in the presence of a solvent with (a) similar and (b) different solubility parameters δ. Adapted with permission from ref (36). Copyright 2006 Elsevier.
Nevertheless, returning to the results of Huang et al., chloroform and benzene have very similar δ values (18.7 and 18.4, respectively), both of which are close to that of dodecanethiol. On the other hand, the results by Heath et al. discussed previously have shown that another pair of solvents with similar δ values (hexane and heptane) resulted in higher crystallinity with the more slowly evaporating solvent (heptane),14 contrary to the results of Huang et al. The group suggested an annealing effect to explain the higher crystallinity. Therefore, it appears that the evaporation speed becomes a determining factor for successful crystalline phase formation when the δ values of both solvents are similar, but the directionality of this change is poorly understood.61
Further consideration of the interfacial self-assembly model proposed by Bigioni et al. suggests why faster evaporating solvents might lead to films with higher crystallinity. In this model, the requirement for successful 2D nucleation at the air/water interface is a sufficiently high concentration of NPs on the solvent droplet surface, which can be achieved when the interface is moving downward (i.e., evaporates) faster than the NPs can diffuse away. Experiments with slowly evaporating solvents were shown to nucleate into 3D crystals in the bulk drop rather than at the interface, leading to the formation of far-from-equilibrium percolating structures.11
Another key solvent property that determines the quality of the Langmuir and LB film is the ability of the solvent to spread on the water surface. When the solvent spreads, the area available to NPs is maximized, which reduces NP aggregation upon solvent evaporation. Although in most cases the equation for the spreading coefficient mentioned briefly in the Introduction is valid, there are examples of low-surface-tension liquids typically used in Langmuir experiments that contradict this equation. For instance, the surface tensions (γs) of toluene and dichloromethane are within 0.1 mN m–1 of one another, but the difference in polarity between the two solvents results in dichloromethane being able to spread on water while toluene cannot. A model that captures this intermolecular interaction at the water–solvent interface is again Hansen’s solubility parameter theory described above. The spreading behavior of various commonly used solvents calculated using Hansen’s model are summarized in the paper by Large et al.59
An often-neglected detail that leads to challenges in directly comparing results between any two experiments is that the presence of the film on the water subphase increases its surface tension, which lowers the threshold for spreading. This means that in Langmuir experiments where spreading requires the injection of multiple small-volume drops (as is the case for chloroform and dichloromethane, which are both good solvents for NPs that spread well but require careful injection due to being denser than water)7 each successive drop spreads less than the previous one until the spreading ceases completely.59 This effect could further lead to difficulties in drawing conclusions about the impact of parameters such as the solvent evaporation speed or injection volume on the film quality, and hence, it might partially explain the disparities in conclusions reported by different groups.
Mixed Systems and Applications
Recent years have seen a shift of interest from fundamental properties of pure Langmuir and LB films of noble metal NPs toward mixed films or functionalized NPs that aim either to enhance the mechanical stability of the film or to increase the potential use of the NP films in their prospective applications. In this section, we give a brief summary of the most interesting reports and assess how in our opinion they shape the future of Langmuir and LB films of noble metal NPs.
Due to the abundance of LB reports of Ag and Au NPs capped with purely hydrophobic ligands, the discussion of fundamental properties in this feature article is based on them. In fact, to our surprise, very few LB films of NPs functionalized with amphiphilic ligands have been reported to date. The primary aim of this approach is to improve the mechanical stability of the film through the formation of hydrogen bonds between the hydrophilic groups and the water subphase as well as to suppress the relative contribution of the attractive dispersion interactions between the metallic cores. This is well achieved in the case of 2-nm-diameter Au NPs functionalized with long amphiphilic polybutadiene-poly(ethylene glycol) chains, whereby the overall diameter of the NP including the ligand shell can vary from 11 to 30 nm depending on the solvent.62 Another NP that has been functionalized with amphiphilic molecules in multiple studies is the Au55 cluster. Although the Au55(PPh3)12Cl6 cluster with its 1.4 nm core diameter was the first example of such a small NP to be successfully transferred by the LB technique in 1998, the film exhibited a low degree of local order and a lack of long-range order.63 However, since then the mechanical stability and ordering of the film have been improved by derivatizing the cluster with p-N-methylcarboxamidophenylphosphine groups that can form an amide bond to palmitic acid (C14H29COOH), granting the NP amphiphilic character.64 In another strategy, Schmid et al. greatly improved the mechanical properties of the film by cross-linking Au55 clusters on the water surface either by cospreading with dithiol linker molecules with different lengths (such as 1,4-benzenedithiol or 4,4′-thiobis(benzenethiol)) or by derivatizing Au55 clusters with (vinyl)8Si8O11(OH)2 and inducing polymerization by irradiation. Given the higher rigidity of the cross-linked film, the transfer was facilitated by extracting the substrate by LB at an angle.65 On the other hand, Bourgoin et al. have shown that LB films of dodecanethiol-capped Au NPs (2.9 nm diameter) can be dipped in a solution of a rigid bisthiol (2,5′′-bis(acetylthio)-5,2′,5′,2′′-terthienyl) which, following a ligand exchange, interconnects the NPs, thus greatly increasing the conductivity of the monolayer without significantly affecting the morphology.66 Another postdeposition method employed to alter the properties of the film was reported by Lemineur and Ritcey, where NPs in a deposited LB film were exposed to a solution of a gold salt and a mild reducing agent, which induced a growth of the NPs up to a diameter of 40 nm without modifying the superstructure organization of the film. This method is particularly relevant for applications based on the plasmonic properties since increasing the particle size without altering the location of the NP can affect the interparticle coupling.67 The separation between particles can be further controlled by transferring the Langmuir layer onto a thermosensitive polymer substrate that shrinks with increasing temperature, as shown by Lu et al. for a film of Ag NPs.68
Mixing molecular systems is an established strategy in LB film preparation whereby an amphiphile is used to stabilize the layer of a compound that would not form a monolayer by itself.2 Recently, Chen et al. have demonstrated by XRR that mixing octanethiol-capped Au NPs with charged surfactants such as sodium dodecyl sulfate (SDS) or dipalmitoylphosphatidylcholine (DPPC) can lead to the lipid monolayer supporting the NP film on top of itself, thereby preventing the hydrophobic moiety from being in contact with water (Figure 9(a)). Through interdigitation of the surfactant tail with the NPs’ ligand shell, the collapse of the NP film can be suppressed at higher surface pressures. Furthermore, by adjusting the concentration of SDS and DPPC in the mixed layer, the morphology of the film as well as the interparticle separation between Au NPs was tuned, which can prove to be crucial to controlling the optical and electronic properties of the film.69 Other groups have also reported mixed systems of Au NPs and amphiphiles such as lipids, fatty acids, or polymers affecting the morphology of the film.70−72 Mayya et al. have shown that negatively charged Au NPs derivatized with carboxylic acid dispersed in the subphase can be immobilized on a monolayer of positively charged fatty amine with the degree of incorporation being controlled by the subphase pH. Repeated downward and upward LB strokes lead to the formation of multilayers of up to 20 layers.73
Figure 9.
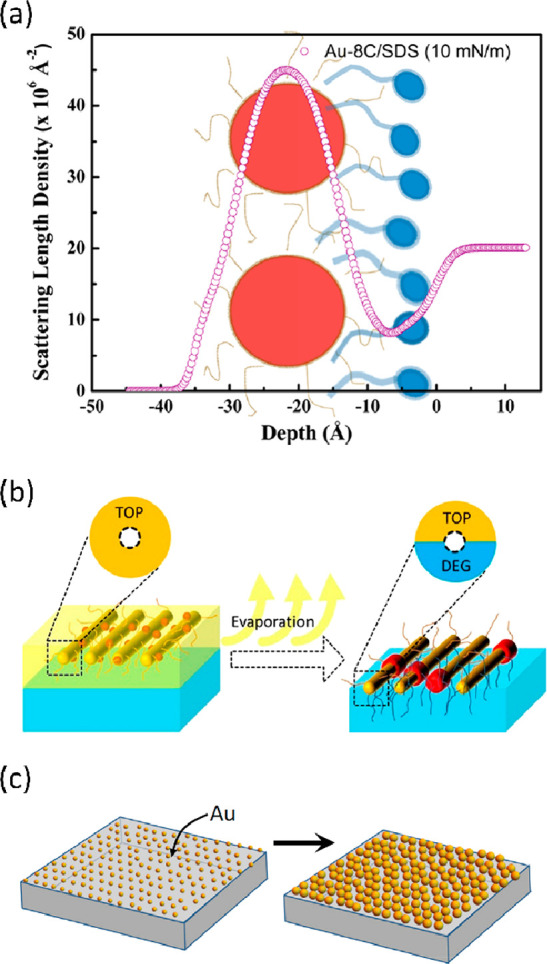
(a) XRR SLD profile of a Au NP film cospread with a surfactant (SDS) preventing the contact of NP with water. Reprinted with permission from ref (69). Copyright 2017 American Chemical Society. (b) Schematic of the formation of a Janus-type structure, whereby the hydrophobic ligand TOP was exchanged by the DEG subphase only from the bottom of the NP. Reprinted with permission from ref (77). Copyright 2020 American Chemical Society. (c) Spherical NPs used for SERS grown by electroless deposition. Reprinted with permission from ref (85). Copyright 2018 Elsevier.
Electrochemical impedance spectroscopy and cyclic voltammetry have been used to show that doping 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) with small concentrations (0–2%) of Au25(SC4)18 clusters can mediate the electron-transfer properties of the lipid layer, which may be useful for applications in novel biosensors and biomolecular electronics.74 Furthermore, Paul et al. have used the LB technique to fabricate a charge storage device by incorporating a monolayer of Au NPs and layers of cadmium arachidate into a metal–insulator–semiconductor structure, which is promising for the future fabrication of field-effect transistor devices based on Au NPs.75
The LB technique can also be used to transfer previously doped NPs onto solid supports. Palladium has a specific reactivity toward hydrogen based on the reversible conversion to palladium hydride. Colloids of palladium, however, are very challenging to prepare. Rajoua et al. have shown that Au NPs can be decorated with a Pd shell and that the LB films of the resulting Pd@Au NPs can be used to detect H2 over a wide range of concentrations.76 On the other hand, Tang and Li demonstrated that semiconductor nanorods can be decorated with multiple Au NPs (2 to 3 nm), which when injected onto a Langmuir through can ripen on the nanorod surface into larger NPs (8 to 9 nm). Moreover, the hydrophobic trioctylphosphine (TOP) capping the hybrid nanorod was found by Fourier transform infrared (FTIR) spectroscopy to presumably be exchanged by diethylene glycol (DEG) used as a subphase uniquely from the bottom side of the nanorod (Figure 9(b)).77 This would indicate the formation of a “Janus”-like structure comprising both hydrophobic and hydrophilic regions, which might be an efficient way to combine unique noble metal NP properties with the high mechanical stability of typical LB amphiphiles. Similarly, Pradhan et al. prepared spherical Janus NPs by injecting hydrophilic thiol derivatives into the water subphase under a compressed monolayer of alkanethiolate-capped NPs.78 Furthermore, Sashuk et al. have demonstrated that amphiphilic Janus-type Au or Ag NPs can be prepared by ligand exchange with a mixture of hydrophobic and charged hydrophilic (negative or positive) ligands in solution and successive spreading on a Langmuir layer. The electrostatic repulsions between the charged ligand shells provide a strong stabilizing effect. A combination of contact angle measurements and IR spectroscopy showed that a rearrangement of ligands occurs during the spreading of the solution, resulting in orienting all of the charged ligands toward the water subphase.79
Last but not least, Solanki et al. demonstrated LB films of MoS2 and Au NPs composites, onto which antibodies specific to dengue NS1 antigen were immobilized. The resulting immunosensor showed detection performance suitable for clinical applications and laid foundations to utilize this biosensing platform for a wider range of analytes relevant for medical diagnostics.80
Although the fundamental part of this feature article was focused on LB films of spherical NPs, nanorods cannot be omitted when discussing arguably the single most advanced application of Au and Ag NPs, namely, the substrates for surface-enhanced Raman spectroscopy (SERS). SERS is a commonly used method for molecule identification with high sensitivity and specificity. The efficiency of molecule detection by SERS substrates can be increased by a few orders of magnitude in the presence of hot spots, which are the sites where the light is trapped by local plasmonic effects.51 The LB technique allows high control over the surface coverage as well as the separation between nanorods, both of which are key factors determining the surface density of hot spots and hence the SERS efficiency. In contrast to the spherical NPs discussed in this feature article, the ordering of nanorods is favorable as it maximizes the entropy of the system by minimizing the excluded volume per particle, i.e., the volume that is not available to other molecules due to the presence of the first molecule.81 Consequently, depending on the aspect ratio of the nanorod, the transitions among isotropic, 2D nematic, 2D smectic, and 3D nematic can be observed with increasing surface pressure.82 Multiple strategies have been employed to optimize the SERS performance of various NPs to detect molecules relevant in chemical and biological warfare, medical diagnostics, or environmental catastrophes. These include polymer-functionalized Au nanorods,51 Ag nanowires,83 urchin-shaped Au NPs,84 and spherical NPs whose size was increased by electroless gold deposition (Figure 9(c)).85
Finally, there are a number of studies reporting the directed self-assembly of Au NPs by immobilizing them by immersion on a patterned LB film of another molecule. Although this method does not involve Langmuir compression of NPs and hence does not exactly follow the topic of this feature article, we feel that this technique deserves a mention as it can lead to structures that cannot be achieved by the simple LB deposition of noble metal NPs. For instance, Watanabe et al. have shown that a micropatterned surface can be obtained by spreading a mixed LB film of icosanoic acid (C19H39COOH) and C8F17C2H4SiCl3 which undergoes phase separation into disks made of the former and the hydrophobic surroundings composed of the latter compound. Icosanoic acid can be selectively removed and the hydrophilic exdomain can be functionalized with amino groups that immobilize Au NPs capped with carboxyl groups using electrostatic forces. By varying the components of the initial mixed LB film, the group demonstrated a number of patterns that are possible to fabricate using this method.86 On the other hand, Ikegami et al. have shown that citrate-stabilized Au NPs can be adsorbed onto an LB film of n-octadecanamine which results in the NPs being partially decorated with hydrophobic groups. Upon desorption, the NPs self-assembled into dimers and larger oligomers through hydrophobic interactions.87 Langmuir monolayers of phospholipids are often used as a model biological membrane. Phan et al. have demonstrated that Au NPs coated with a cell-penetrating peptide (HIV-1-derived trans-activator of transcription or TAT) can strongly bind to a monolayer of phospholipids, which can be crucial for future drug delivery systems in pharmaceutics.88
Outlook: Clusters
Another group of compounds that we think has a lot of untapped potential in the future development of LB films of noble metal NPs is gold clusters, which we briefly mentioned already in this feature article. Thiolate-protected gold clusters are a subgroup of small Au NPs and typically consist of tens to hundreds of metal atoms.9 Some configurations of Aum(SR)n clusters are stabilized geometrically or electronically due to attaining a complete electron shell, thus exhibiting particular stability. Therefore, after careful size-exclusion techniques, atomic precision can be achieved.89 This leads to a very high monodispersity, which can be extremely relevant for fundamental studies of NPs described in this feature article, where the varying degree of polydispersity among different NP samples complicated the determination of the effect of changing parameters, such as the NP size or type of metal, on the Langmuir film quality.
As much as the study of clusters has to offer to the field of LB in terms of elucidation of fundamental properties, arguably even higher benefits can be presented to the field of clusters by using LB as a deposition technique and highly reducing their prohibitively expensive synthetic costs. Reaching atomic precision requires multiple size-exclusion steps on top of thermal etching, meaning that the overall yields of cluster synthesis tend to be rather low even for NPs standards. Given that optimized LB deposition requires micrograms of material to cover areas of tens of cm2 of substrate, the transition to the industrial-scale production of cluster films can be much cheaper than when using other methods such as drop casting or spin-coating. This is particularly true for clusters that underwent ligand exchange or metal doping. In fact, given the atomic precision and well-defined structure of clusters, high-performance liquid chromatography (HPLC) not only allows the separation of species with a given number of exchanges (ligand or metal exchanges) but also in some cases it is possible to isolate structural isomers of exchanged clusters (e.g., doping atoms exchanged on the surface vs those exchanged in the core of a cluster)90,91 or enantiomers,92,93 albeit in very small amounts. This, on the other hand, can lead to the elucidation of fundamental differences through a degree of control unprecedented for larger NPs. Alternatively, although LB films of NPs with charged ligands have been reported before,79 the effect of charge located on the metal core can be studied with the help of the Au25(SC2H4Ph)18 cluster, which has been synthesized with charge states of −1, 0, and +1.94
Moreover, while the optical properties of larger NPs are dominated by the SPR based on quasi-continuous electronic energy levels, clusters exhibit more molecular-like behavior with their distinct absorption features originating from discrete electronic energy levels.95,96 This has led to a number of potential optical applications, which can be further enhanced through their collective action in close-packed 2D assemblies. With some Au cluster systems already demonstrating their usefulness in prospective applications, for example, as glucose sensors97 or in liver disease detection,98 we believe that many of them as well as countless new platforms would benefit highly from being deposited as LB films which offer low production costs, high coverage, and high durability.
At the same time, a number of fundamental properties of noble metal LB films that are still not clear after decades of studying large NPs could be elucidated with the help of atomically precise clusters. Nevertheless, to date few examples of LB films based on clusters have been reported, owing to the relatively recent discovery and the development of better synthetic routes of clusters but also to the challenging optimization of Langmuir conditions stemming from the weak interactions between particles and stability issues. To date the reported LB films of clusters include Au25(SR)18,17 Au38(SR)24,21 and Au55,65 with the potential lying in countless other configurations waiting to be explored.
Conclusions
In this feature article, we addressed the formation, evolution, and applications of Langmuir and LB/LS films of noble metal NPs, particularly focusing on spherical Au- and Ag-based systems. Although numerous excellent reviews describe in detail the theory and report recent advancements in the field of Langmuir layers of long amphiphilic molecules, very few concentrate on purely hydrophobic molecules and nanomaterials, and to the best of our knowledge, none direct their entire focus toward NPs despite the vast number of research studies reported over the years. To our surprise, some assumptions that are true for typical LB amphiphiles are wrongly concluded to apply to Langmuir films of NPs. The most common misconception that we came across is the presumption that the separation between NPs changes with applied surface pressure (as it does for long amphiphilic molecules). In fact, numerous surface-specific X-ray techniques (GIXD, GIXOS, GIWAXS, etc.) have proved repeatedly that the interparticle gap is established upon solvent evaporation and it is the interdomain separation that changes upon compression.21,27,33 One of the primary targets of this feature article is to familiarize researchers with the notion that there are fundamental differences between the interactions directing the layer formation between hydrophobic NPs and long amphiphiles. In the absence of hydrogen bonds between the polar head and the subphase, multiple weaker interactions such as dispersive forces, long-range repulsion, and entropic effects are in competition.
The fact that various hypotheses have been proposed by multiple groups to explain the development of seemingly similar morphologies highlights the importance of further fundamental research in the field. In our opinion, the process that would benefit most from additional studies is the dewetting upon LB/LS deposition as hardly any efforts have been made to observe the morphology of the same film on the water surface and following the transfer onto a solid substrate using similar analytical methods. Useful structure-determining techniques that have already been applied separately on water and on hard surfaces (but not in tandem) include XRR, GIXD (and other related 2D X-ray techniques), and even AFM with recent advancements allowing the operation on the water surface.99,100 Furthermore, we believe that particular attention should be devoted to directly comparing the dewetting processes of the same NP solution in Langmuir films on water and in drop-casted films on solid substrates, for which a wide array of literature is available and the interfacial self-assembly is better understood. Although the two processes are fundamentally different in the sense that the assembly in the former typically occurs on a nonwetting layer on a static surface while the latter wets a fluid subphase, some similarities in the convective flows and the interfacial self-assembly can be expected.
While there was a general consensus when comparing the effect on the film quality regarding some factors, such as the NP size or the length of the capping thiol, others such as the effect of the solvent on ordering showed conflicting conclusions between different groups, likely due to the difficulties in decoupling the influences of evaporation speed and the solubility parameter. Furthermore, comparative studies of this type all suffer from the fact that various NPs will inevitably exhibit different degrees of polydispersity, which has an imperative effect on efficient particle ordering. This issue should be alleviated in the future by more efficient size-selection techniques.
Following the description of techniques improving the stability of Langmuir films of NPs, we believe that future efforts in the field should be aimed at combining these strategies with the functionalization employed in the reported examples of potential applications. For instance, the preparation of Janus-type NPs can in theory allow the functionalized hydrophobic or hydrophilic group to be exposed on the surface depending on the deposition method (LB or LS, respectively). Finally, we believe that combining the fields of metal clusters, which to date are relatively unstudied, and LB offers great potential to facilitate their transition to being useful in applications while at the same time elucidating fundamental properties of LB films of NPs that are still unresolved.
Acknowledgments
The lengthy discussions regarding Langmuir–Blodgett technique and fruitful collaboration with Dr. Lay-Theng Lee are greatly appreciated.
Glossary
Abbreviations
- AMFM,
amplitude modulated–frequency modulated
- AFM
atomic force microscopy
- GISAXS
grazing incidence small-angle X-ray scattering
- GIWAXS
grazing incidence wide-angle X-ray scattering
- GIXD
grazing incidence X-ray diffraction
- GIXOS
grazing incidence X-ray off-specular scattering
- FTIR
Fourier transform infrared spectroscopy
- LB
Langmuir–Blodgett
- LS
Langmuir–Schaefer
- NP
nanoparticle
- SEM
scanning electron microscopy
- SLD
scattering-length density
- SPR
surface plasmon resonance
- TEM
transmission electron microscopy
- XRR
X-ray reflectivity
Biographies

Michal Swierczewski received his M.Sc. degree in chemistry with nanotechnology at Heriot Watt University in Edinburgh, Scotland (2017). Since then, he has been working as a Ph.D. candidate under the supervision of Prof. Thomas Bürgi in the Department of Physical Chemistry at the University of Geneva, Switzerland. His research focuses primarily on the formation of gold nanocluster assemblies and the deposition of ultrathin films on surfaces with their subsequent analysis by AFM and other surface-specific techniques.

Thomas Bürgi studied chemistry and obtained his Ph.D. (1995) at the University of Berne (Switzerland). After a postdoctoral stay at MIT, he did his habilitation at ETH, Zürich. He became an assistant professor at the University of Neuchâtel (Switzerland, 2005) and a full professor at the University of Heidelberg (2008). In 2010, he moved to the University of Geneva, where he is a professor of physical chemistry. His research focuses on fundamental aspects and applications of chiral metal clusters and the development of in situ and chiroptical spectroscopy.
Generous support by the University of Geneva and the Swiss National Science Foundation (grant number CRSII5_173720) is kindly acknowledged.
The authors declare no competing financial interest.
References
- Oliveira O. N.; Caseli L.; Ariga K. The Past and the Future of Langmuir and Langmuir–Blodgett Films. Chem. Rev. 2022, 122 (6), 6459–6513. 10.1021/acs.chemrev.1c00754. [DOI] [PubMed] [Google Scholar]
- Petty M. C.Langmuir-Blodgett Films: An Introduction; Cambridge University Press: 1996. [Google Scholar]
- Ariga K.; Yamauchi Y.; Mori T.; Hill J. P. 25th Anniversary Article: What Can Be Done with the Langmuir-Blodgett Method? Recent Developments and its Critical Role in Materials Science. Adv. Mater. 2013, 25 (45), 6477–6512. 10.1002/adma.201302283. [DOI] [PubMed] [Google Scholar]
- Akutagawa T.; Kakiuchi K.; Hasegawa T.; Nakamura T.; Christensen C. A.; Becher J. Langmuir–Blodgett Films of Amphiphilic Bis(tetrathiafulvalene) Macrocycles with Four Alkyl Chains. Langmuir 2004, 20 (10), 4187–4195. 10.1021/la049950e. [DOI] [PubMed] [Google Scholar]
- Sieling T.; Christoffers J.; Brand I. Langmuir–Blodgett Monolayers of Partially Fluorinated Ionic Liquids as Two-Dimensional, More Sustainable Functional Materials and Coatings. ACS Sustain. Chem. Eng. 2019, 7 (13), 11593–11602. 10.1021/acssuschemeng.9b01496. [DOI] [Google Scholar]
- Stankovic N.; Todorović-Marković B.; Marković Z. Self-assembly of carbon based nanoparticles films by Langmuir-Blodgett method. J. Serb. Chem. Soc. 2020, 85, 1095–1127. 10.2298/JSC191225008S. [DOI] [Google Scholar]
- Bodik M.; Jergel M.; Majkova E.; Siffalovic P. Langmuir films of low-dimensional nanomaterials. Adv. Colloid Interface Sci. 2020, 283, 102239. 10.1016/j.cis.2020.102239. [DOI] [PubMed] [Google Scholar]
- Ariga K. Don’t Forget Langmuir–Blodgett Films 2020: Interfacial Nanoarchitectonics with Molecules, Materials, and Living Objects. Langmuir 2020, 36 (26), 7158–7180. 10.1021/acs.langmuir.0c01044. [DOI] [PubMed] [Google Scholar]
- Chakraborty I.; Pradeep T. Atomically Precise Clusters of Noble Metals: Emerging Link between Atoms and Nanoparticles. Chem. Rev. 2017, 117 (12), 8208–8271. 10.1021/acs.chemrev.6b00769. [DOI] [PubMed] [Google Scholar]
- Tang J.; Ge G.; Brus L. E. Gas–Liquid–Solid Phase Transition Model for Two-Dimensional Nanocrystal Self-Assembly on Graphite. J. Phys. Chem. B 2002, 106 (22), 5653–5658. 10.1021/jp014559c. [DOI] [Google Scholar]
- Bigioni T. P.; Lin X.-M.; Nguyen T. T.; Corwin E. I.; Witten T. A.; Jaeger H. M. Kinetically driven self assembly of highly ordered nanoparticle monolayers. Nat. Mater. 2006, 5 (4), 265–270. 10.1038/nmat1611. [DOI] [PubMed] [Google Scholar]
- Narayanan S.; Wang J.; Lin X.-M. Dynamical Self-Assembly of Nanocrystal Superlattices during Colloidal Droplet Evaporation by in situ Small Angle X-Ray Scattering. Phys. Rev. Lett. 2004, 93 (13), 135503. 10.1103/PhysRevLett.93.135503. [DOI] [PubMed] [Google Scholar]
- Schultz D. G.; Lin X.-M.; Li D.; Gebhardt J.; Meron M.; Viccaro J.; Lin B. Structure, Wrinkling, and Reversibility of Langmuir Monolayers of Gold Nanoparticles. J. Phys. Chem. B 2006, 110 (48), 24522–24529. 10.1021/jp063820s. [DOI] [PubMed] [Google Scholar]
- Heath J. R.; Knobler C. M.; Leff D. V. Pressure/Temperature Phase Diagrams and Superlattices of Organically Functionalized Metal Nanocrystal Monolayers: The Influence of Particle Size, Size Distribution, and Surface Passivant. J. Phys. Chem. B 1997, 101 (2), 189–197. 10.1021/jp9611582. [DOI] [Google Scholar]
- Gupta R.; Manjuladevi V.. Molecular Interactions at Interfaces; InTech: 2012. [Google Scholar]
- Rivière S.; Hénon S.; Meunier J.; Schwartz D. K.; Tsao M. W.; Knobler C. M. Textures and phase transitions in Langmuir monolayers of fatty acids. A comparative Brewster angle microscope and polarized fluorescence microscope study. J. Chem. Phys. 1994, 101 (11), 10045–10051. 10.1063/1.467993. [DOI] [Google Scholar]
- Yokoyama T.; Hirata N.; Tsunoyama H.; Eguchi T.; Negishi Y.; Nakajima A. Vibrational Spectra of Thiolate-Protected Gold Nanocluster with Infrared Reflection Absorption Spectroscopy: Size- and Temperature-Dependent Ordering Behavior of Organic Monolayer. J. Phys. Chem. C 2020, 124 (1), 363–371. 10.1021/acs.jpcc.9b07550. [DOI] [Google Scholar]
- Dynarowicz-Łątka P.; Dhanabalan A.; Oliveira O. N. Modern physicochemical research on Langmuir monolayers. Adv. Colloid Interface Sci. 2001, 91 (2), 221–293. 10.1016/S0001-8686(99)00034-2. [DOI] [PubMed] [Google Scholar]
- Möhwald H. Direct characterization of monolayers at the air-water interface. Thin Solid Films 1988, 159 (1), 1–15. 10.1016/0040-6090(88)90612-8. [DOI] [Google Scholar]
- Vegso K.; Siffalovic P.; Jergel M.; Weis M.; Benkovicova M.; Majkova E.; Luby S.; Kocsis T.; Capek I. Silver Nanoparticle Monolayer-to-Bilayer Transition at the Air/Water Interface as Studied by the GISAXS Technique: Application of a New Paracrystal Model. Langmuir 2012, 28 (25), 9395–9404. 10.1021/la301577a. [DOI] [PubMed] [Google Scholar]
- Swierczewski M.; Maroni P.; Chenneviere A.; Dadras M. M.; Lee L.-T.; Bürgi T. Deposition of Extended Ordered Ultrathin Films of Au38(SC2H4Ph)24 Nanocluster using Langmuir–Blodgett Technique. Small 2021, 17 (27), 2005954. 10.1002/smll.202005954. [DOI] [PubMed] [Google Scholar]
- Geuchies J. J.; Soligno G.; Geraffy E.; Hendrikx C. P.; Overbeek C. v.; Montanarella F.; Slot M. R.; Konovalov O. V.; Petukhov A. V.; Vanmaekelbergh D. Unravelling three-dimensional adsorption geometries of PbSe nanocrystal monolayers at a liquid-air interface. Commun. Chem. 2020, 3 (1), 28. 10.1038/s42004-020-0275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born M.; Wolf E.. Principles of Optics: Electromagnetic Theory of Propagation, Interference and Diffraction of Light; Cambridge University Press: 1975. [Google Scholar]
- Bian K.; Gerber C.; Heinrich A. J.; Müller D. J.; Scheuring S.; Jiang Y. Scanning probe microscopy. Nat. Rev. Methods Primers 2021, 1 (1), 36. 10.1038/s43586-021-00033-2. [DOI] [Google Scholar]
- Jalili N.; Laxminarayana K. A review of atomic force microscopy imaging systems: application to molecular metrology and biological sciences. Mechatronics 2004, 14 (8), 907–945. 10.1016/j.mechatronics.2004.04.005. [DOI] [Google Scholar]
- Athanasopoulou E.-N.; Nianias N.; Ong Q. K.; Stellacci F. Bimodal atomic force microscopy for the characterization of thiolated self-assembled monolayers. Nanoscale 2018, 10 (48), 23027–23036. 10.1039/C8NR07657J. [DOI] [PubMed] [Google Scholar]
- Swierczewski M.; Chenneviere A.; Maroni P.; Lee L.-T.; Bürgi T. Nanomechanical and Structural Study of Au38 Nanocluster Langmuir-Blodgett Films Using Bimodal AFM and X-Ray Reflectivity. J. Colloid Interface Sci. 2023, 630, 28–36. 10.1016/j.jcis.2022.10.081. [DOI] [PubMed] [Google Scholar]
- Gisbert V. G.; Garcia R. Accurate Wide-Modulus-Range Nanomechanical Mapping of Ultrathin Interfaces with Bimodal Atomic Force Microscopy. ACS Nano 2021, 15 (12), 20574–20581. 10.1021/acsnano.1c09178. [DOI] [PubMed] [Google Scholar]
- Merle B.; Nicholson K. S.; Herbert E. G.; Göken M. An improved method for point deflection measurements on rectangular membranes. Mater. Des. 2016, 109, 485–491. 10.1016/j.matdes.2016.07.054. [DOI] [Google Scholar]
- Martins P.; Delobelle P.; Malhaire C.; Brida S.; Barbier D. Bulge test and AFM point deflection method, two technics for the mechanical characterization of very low stiffness free standing film. EPJ. Appl. Phys. 2009, 45, 10501. 10.1051/epjap:2008187. [DOI] [Google Scholar]
- Bertolazzi S.; Brivio J.; Kis A. Stretching and Breaking of Ultrathin MoS2. ACS Nano 2011, 5 (12), 9703–9709. 10.1021/nn203879f. [DOI] [PubMed] [Google Scholar]
- Rojewska M.; Skrzypiec M.; Prochaska K. The wetting properties of Langmuir–Blodgett and Langmuir–Schaefer films formed by DPPC and POSS compounds. Chem. Phys. Lipids 2019, 221, 158–166. 10.1016/j.chemphyslip.2019.04.004. [DOI] [PubMed] [Google Scholar]
- Dai Y.; Lin B.; Meron M.; Kim K.; Leahy B.; Witten T. A.; Shpyrko O. G. Synchrotron X-ray Studies of Rapidly Evolving Morphology of Self-Assembled Nanoparticle Films under Lateral Compression. Langmuir 2013, 29 (46), 14050–14056. 10.1021/la403252d. [DOI] [PubMed] [Google Scholar]
- Bera M. K.; Sanyal M. K.; Pal S.; Daillant J.; Datta A.; Kulkarni G. U.; Luzet D.; Konovalov O. Reversible buckling in monolayer of gold nanoparticles on water surface. EPL 2007, 78 (5), 56003. 10.1209/0295-5075/78/56003. [DOI] [Google Scholar]
- Reik M.; Calabro M.; Griesemer S.; Barry E.; Bu W.; Lin B.; Rice S. A. The influence of fractional surface coverage on the core–core separation in ordered monolayers of thiol-ligated Au nanoparticles. Soft Matter 2019, 15 (43), 8800–8807. 10.1039/C9SM01579E. [DOI] [PubMed] [Google Scholar]
- Pei L.; Mori K.; Adachi M. Investigation on arrangement and fusion behaviors of gold nanoparticles at the air/water interface. Colloids Surf. A Physicochem. Eng. Asp. 2006, 281 (1), 44–50. 10.1016/j.colsurfa.2006.02.029. [DOI] [Google Scholar]
- Kim K.; Leahy B. D.; Dai Y.; Shpyrko O.; Soltau J. S.; Pelton M.; Meron M.; Lin B. Governing factors in stress response of nanoparticle films on water surface. J. Appl. Phys. 2011, 110 (10), 102218. 10.1063/1.3661988. [DOI] [Google Scholar]
- Alfrey T. Fr.; Bradford E. B.; Vanderhoff J. W.; Oster G. Optical Properties of Uniform Particle-Size Latexes. J. Opt. Soc. Am. 1954, 44, 603. 10.1364/JOSA.44.000603. [DOI] [Google Scholar]
- Jensen T. R.; Østergaard Jensen M.; Reitzel N.; Balashev K.; Peters G. H.; Kjaer K.; Bjørnholm T. Water in contact with extended hydrophobic surfaces: direct evidence of weak dewetting. Phys. Rev. Lett. 2003, 90 (8), 086101. 10.1103/PhysRevLett.90.086101. [DOI] [PubMed] [Google Scholar]
- Jensen M. Ø.; Mouritsen O. G.; Peters G. H. The hydrophobic effect: Molecular dynamics simulations of water confined between extended hydrophobic and hydrophilic surfaces. J. Chem. Phys. 2004, 120 (20), 9729–9744. 10.1063/1.1697379. [DOI] [PubMed] [Google Scholar]
- Tkachenko A. V.; Rabin Y. Fluctuation-Stabilized Surface Freezing of Chain Molecules. Phys. Rev. Lett. 1996, 76 (14), 2527–2530. 10.1103/PhysRevLett.76.2527. [DOI] [PubMed] [Google Scholar]
- Tabe Y.; Yamamoto T.; Nishiyama I.; Aoki K. M.; Yoneya M.; Yokoyama H. Can Hydrophobic Oils Spread on Water as Condensed Langmuir Monolayers?. J. Phys. Chem. B 2002, 106 (47), 12089–12092. 10.1021/jp026724+. [DOI] [Google Scholar]
- Huang S.; Tsutsui G.; Sakaue H.; Shingubara S.; Takahagi T. Formation of a large-scale Langmuir–Blodgett monolayer of alkanethiol-encapsulated gold particles. J. Vac Sci. Technol. B Microelectron Nanometer Struct Process Meas Phenom 2001, 19 (1), 115–120. 10.1116/1.1335678. [DOI] [Google Scholar]
- Ge G.; Brus L. Evidence for Spinodal Phase Separation in Two-Dimensional Nanocrystal Self-Assembly. J. Phys. Chem. B 2000, 104 (41), 9573–9575. 10.1021/jp002280a. [DOI] [Google Scholar]
- Leahy B. D.; Pocivavsek L.; Meron M.; Lam K. L.; Salas D.; Viccaro P. J.; Lee K. Y. C.; Lin B. Geometric Stability and Elastic Response of a Supported Nanoparticle Film. Phys. Rev. Lett. 2010, 105 (5), 058301. 10.1103/PhysRevLett.105.058301. [DOI] [PubMed] [Google Scholar]
- Collier C. P.; Saykally R. J.; Shiang J. J.; Henrichs S. E.; Heath J. R. Reversible Tuning of Silver Quantum Dot Monolayers Through the Metal-Insulator Transition. Science 1997, 277 (5334), 1978–1981. 10.1126/science.277.5334.1978. [DOI] [Google Scholar]
- Huang S.; Minami K.; Sakaue H.; Shingubara S.; Takahagi T. Effects of the Surface Pressure on the Formation of Langmuir–Blodgett Monolayer of Nanoparticles. Langmuir 2004, 20 (6), 2274–2276. 10.1021/la0302293. [DOI] [PubMed] [Google Scholar]
- Sear R. P.; Chung S. W.; Markovich G.; Gelbart W. M.; Heath J. R. Spontaneous patterning of quantum dots at the air-water interface. Phys. Rev. E Stat Phys. Plasmas Fluids Relat Interdiscip Topics 1999, 59 (6), R6255–8. 10.1103/PhysRevE.59.R6255. [DOI] [PubMed] [Google Scholar]
- Gehlert U.; Fang J.; Knobler C. M. Relating the Organization of the Molecular Tilt Azimuth to Lateral-Force Images in Monolayers Transferred to Solid Substrates. J. Phys. Chem. B 1998, 102 (15), 2614–2617. 10.1021/jp9811124. [DOI] [Google Scholar]
- Martín-García B.; Velázquez M. M. Block copolymer assisted self-assembly of nanoparticles into Langmuir–Blodgett films: Effect of polymer concentration. Mater. Chem. Phys. 2013, 141 (1), 324–332. 10.1016/j.matchemphys.2013.05.017. [DOI] [Google Scholar]
- Tim B.; Błaszkiewicz P.; Nowicka A. B.; Kotkowiak M. Optimizing SERS performance through aggregation of gold nanorods in Langmuir-Blodgett films. Appl. Surf. Sci. 2022, 573, 151518. 10.1016/j.apsusc.2021.151518. [DOI] [Google Scholar]
- Kundu S.; Bal J. K. Reorganization of Au nanoparticle Langmuir-Blodgett films on wet chemically passivated Si(001) surfaces. J. Appl. Phys. 2011, 110 (11), 114302. 10.1063/1.3664696. [DOI] [Google Scholar]
- Das K.; Kundu S. Variation in surface plasmonic response due to the reorganization of Au nanoparticles in Langmuir-Blodgett film. J. Appl. Phys. 2014, 116 (2), 024316. 10.1063/1.4890034. [DOI] [Google Scholar]
- Anu Mary Ealia S.; Saravanakumar M. P. A review on the classification, characterisation, synthesis of nanoparticles and their application. IOP Conf. Ser. Mater. Sci. Eng. 2017, 263, 032019. 10.1088/1757-899X/263/3/032019. [DOI] [Google Scholar]
- Brust M.; Stuhr-Hansen N.; Nørgaard K.; Christensen J. B.; Nielsen L. K.; Bjørnholm T. Langmuir–Blodgett Films of Alkane Chalcogenide (S,Se,Te) Stabilized Gold Nanoparticles. Nano Lett. 2001, 1 (4), 189–191. 10.1021/nl015506+. [DOI] [Google Scholar]
- Vegso K.; Siffalovic P.; Majkova E.; Jergel M.; Benkovicova M.; Kocsis T.; Weis M.; Luby S.; Nygård K.; Konovalov O. Nonequilibrium phases of nanoparticle Langmuir films. Langmuir 2012, 28 (28), 10409–14. 10.1021/la301764t. [DOI] [PubMed] [Google Scholar]
- Fukuto M.; Heilmann R. K.; Pershan P. S.; Badia A.; Lennox R. B. Monolayer/bilayer transition in Langmuir films of derivatized gold nanoparticles at the gas/water interface: An x-ray scattering study. J. Chem. Phys. 2004, 120 (7), 3446–3459. 10.1063/1.1640334. [DOI] [PubMed] [Google Scholar]
- Huang S.; Tsutsui G.; Sakaue H.; Shingubara S.; Takahagi T. Experimental conditions for a highly ordered monolayer of gold nanoparticles fabricated by the Langmuir-Blodgett method. J. Vac. Sci. Technol. B 2001, 19, 2045–2049. 10.1116/1.1410943. [DOI] [Google Scholar]
- Large M. J.; Ogilvie S. P.; King A. A. K.; Dalton A. B. Understanding Solvent Spreading for Langmuir Deposition of Nanomaterial Films: A Hansen Solubility Parameter Approach. Langmuir 2017, 33 (51), 14766–14771. 10.1021/acs.langmuir.7b03867. [DOI] [PubMed] [Google Scholar]
- Mathieu D. Pencil and Paper Estimation of Hansen Solubility Parameters. ACS Omega 2018, 3 (12), 17049–17056. 10.1021/acsomega.8b02601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orbulescu J.; Leblanc R. M. Importance of the Spreading Solvent Evaporation Time in Langmuir Monolayers. J. Phys. Chem. C 2009, 113 (13), 5313–5315. 10.1021/jp9012587. [DOI] [Google Scholar]
- Genson K. L.; Holzmueller J.; Jiang C.; Xu J.; Gibson J. D.; Zubarev E. R.; Tsukruk V. V. Langmuir-Blodgett monolayers of gold nanoparticles with amphiphilic shells from V-shaped binary polymer arms. Langmuir 2006, 22 (16), 7011–5. 10.1021/la061163p. [DOI] [PubMed] [Google Scholar]
- Chi L. F.; Rakers S.; Hartig M.; Fuchs H.; Schmid G. Preparation and characterization of Langmuir monolayers and Langmuir–Blodgett films of nanosized Au55-clusters. Thin Solid Films 1998, 327–329, 520–523. 10.1016/S0040-6090(98)00701-9. [DOI] [Google Scholar]
- Brown J. J.; Porter J. A.; Daghlian C. P.; Gibson U. J. Ordered Arrays of Amphiphilic Gold Nanoparticles in Langmuir Monolayers. Langmuir 2001, 17 (26), 7966–7969. 10.1021/la010691s. [DOI] [Google Scholar]
- Schmid G.; Vidoni O.; Torma V.; Pollmeier K.; Rehage H.; Vassiliev A. Polymer Two-Dimensional Networks of Au55 Clusters. Z. anorg. allg. Chem. 2005, 631 (13–14), 2792–2799. 10.1002/zaac.200500148. [DOI] [Google Scholar]
- Bourgoin J.-P.; Kergueris C.; Lefèvre E.; Palacin S. Langmuir–Blodgett films of thiol-capped gold nanoclusters: fabrication and electrical properties. Thin Solid Films 1998, 327–329, 515–519. 10.1016/S0040-6090(98)00700-7. [DOI] [Google Scholar]
- Lemineur J.-F.; Ritcey A. M. Controlled Growth of Gold Nanoparticles Preorganized in Langmuir–Blodgett Monolayers. Langmuir 2016, 32 (46), 12056–12066. 10.1021/acs.langmuir.6b02595. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Liu G. L.; Lee L. P. High-Density Silver Nanoparticle Film with Temperature-Controllable Interparticle Spacing for a Tunable Surface Enhanced Raman Scattering Substrate. Nano Lett. 2005, 5 (1), 5–9. 10.1021/nl048965u. [DOI] [PubMed] [Google Scholar]
- Chen Y.-T.; Su H.-S.; Hung C.-H.; Yang P.-W.; Hu Y.; Lin T.-L.; Lee M.-T.; Jeng U. S. X-ray Reflectivity Studies on the Mixed Langmuir–Blodgett Monolayers of Thiol-Capped Gold Nanoparticles, Dipalmitoylphosphatidylcholine, and Sodium Dodecyl Sulfate. Langmuir 2017, 33 (41), 10886–10897. 10.1021/acs.langmuir.7b01559. [DOI] [PubMed] [Google Scholar]
- Hansen C. R.; Westerlund F.; Moth-Poulsen K.; Ravindranath R.; Valiyaveettil S.; Bjørnholm T. Polymer-Templated Self-Assembly of a 2-Dimensional Gold Nanoparticle Network. Langmuir 2008, 24 (8), 3905–3910. 10.1021/la703479w. [DOI] [PubMed] [Google Scholar]
- Jayaraman S.; Yu L. T.; Srinivasan M. P. Polythiophene–gold nanoparticle hybrid systems: Langmuir–Blodgett assembly of nanostructured films. Nanoscale 2013, 5 (7), 2974–2982. 10.1039/c3nr33385j. [DOI] [PubMed] [Google Scholar]
- De Barros A.; Constantino C. J. L.; Bortoleto J. R. R.; Da Cruz N. C.; Ferreira M. Incorporation of gold nanoparticles into Langmuir-Blodgett films of polyaniline and montmorillonite for enhanced detection of metallic ions. Sens. Actuators B Chem. 2016, 236, 408–417. 10.1016/j.snb.2016.06.022. [DOI] [Google Scholar]
- Mayya K. S.; Patil V.; Sastry M. Lamellar Multilayer Gold Cluster Films Deposited by the Langmuir–Blodgett Technique. Langmuir 1997, 13 (9), 2575–2577. 10.1021/la962057y. [DOI] [Google Scholar]
- Wieckowska A.; Jablonowska E.; Dzwonek M.; Jaskolowski M.; Bilewicz R. Tailored Lipid Monolayers Doped with Gold Nanoclusters: Surface Studies and Electrochemistry of Hybrid-film-covered Electrodes. ChemElectroChem. 2022, 9 (9), e202101367 10.1002/celc.202101367. [DOI] [Google Scholar]
- Paul S.; Pearson C.; Molloy A.; Cousins M. A.; Green M.; Kolliopoulou S.; Dimitrakis P.; Normand P.; Tsoukalas D.; Petty M. C. Langmuir–Blodgett Film Deposition of Metallic Nanoparticles and Their Application to Electronic Memory Structures. Nano Lett. 2003, 3 (4), 533–536. 10.1021/nl034008t. [DOI] [Google Scholar]
- Rajoua K.; Baklouti L.; Favier F. Electronic and Mechanical Antagonist Effects in Resistive Hydrogen Sensors Based on Pd@Au Core-Shell Nanoparticle Assembles Prepared by Langmuir-Blodgett. J. Phys. Chem. C 2015, 119, 10130. 10.1021/acs.jpcc.5b01636. [DOI] [Google Scholar]
- Tang X.; Li X. Ripening of Gold Clusters into a Single Domain on Semiconductor Quantum Rods during Langmuir–Blodgett Deposition. Crystal Growth Des 2020, 20 (9), 5890–5895. 10.1021/acs.cgd.0c00582. [DOI] [Google Scholar]
- Pradhan S.; Xu L.; Chen S. Janus Nanoparticles by Interfacial Engineering. Adv. Funct. Mater. 2007, 17 (14), 2385–2392. 10.1002/adfm.200601034. [DOI] [Google Scholar]
- Sashuk V.; Hołyst R.; Wojciechowski T.; Fiałkowski M. Close-packed monolayers of charged Janus-type nanoparticles at the air–water interface. J. Colloid Interface Sci. 2012, 375 (1), 180–186. 10.1016/j.jcis.2012.02.057. [DOI] [PubMed] [Google Scholar]
- Solanki S.; Soni A.; Pandey M.; Biradar A.; Sumana G. Langmuir-Blodgett nanoassemblies of MoS2-Au composite at the air-water interface for dengue detection. ACS Appl. Mater. Interfaces 2018, 10, 3020–3028. 10.1021/acsami.7b14391. [DOI] [PubMed] [Google Scholar]
- Onsager L. The effects of shape on the interaction of colloidal particles. Ann. N.Y. Acad. Sci. 1949, 51 (4), 627–659. 10.1111/j.1749-6632.1949.tb27296.x. [DOI] [Google Scholar]
- Kim F.; Kwan S.; Akana J.; Yang P. Langmuir–Blodgett Nanorod Assembly. J. Am. Chem. Soc. 2001, 123 (18), 4360–4361. 10.1021/ja0059138. [DOI] [PubMed] [Google Scholar]
- Tao A.; Kim F.; Hess C.; Goldberger J.; He R.; Sun Y.; Xia Y.; Yang P. Langmuir–Blodgett Silver Nanowire Monolayers for Molecular Sensing Using Surface-Enhanced Raman Spectroscopy. Nano Lett. 2003, 3 (9), 1229–1233. 10.1021/nl0344209. [DOI] [Google Scholar]
- Tahghighi M.; Janner D.; Ignés-Mullol J. Optimizing Gold Nanoparticle Size and Shape for the Fabrication of SERS Substrates by Means of the Langmuir–Blodgett Technique. Nanomater 2020, 10 (11), 2264. 10.3390/nano10112264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahghighi M.; Mannelli I.; Janner D.; Ignés-Mullol J. Tailoring plasmonic response by Langmuir–Blodgett gold nanoparticle templating for the fabrication of SERS substrates. Appl. Surf. Sci. 2018, 447, 416–422. 10.1016/j.apsusc.2018.03.237. [DOI] [Google Scholar]
- Watanabe S.; Shibata H.; Sakamoto F.; Azumi R.; Sakai H.; Abe M.; Matsumoto M. Directed self-assembly of gold nanoparticles and gold thin films on micro- and nanopatterned templates fabricated from mixed phase-separated Langmuir-Blodgett films. J. Mater. Chem. 2009, 19 (37), 6796–6803. 10.1039/b905963f. [DOI] [Google Scholar]
- Ikegami S.; Yamaguchi K.; Tanaka T.; Takeyasu N.; Kaneta T. Hydrophobic assembly of gold nanoparticles into plasmonic oligomers with Langmuir–Blodgett film. Jpn. J. Appl. Phys. 2018, 57 (12), 120311. 10.7567/JJAP.57.120311. [DOI] [Google Scholar]
- Phan M.; Kim H.; Lee S.; Yu C.-J.; Moon B.; Shin K. HIV Peptide-Mediated Binding Behaviors of Nanoparticles on Lipid Membrane. Langmuir 2017, 33 (10), 2590–2595. 10.1021/acs.langmuir.6b04234. [DOI] [PubMed] [Google Scholar]
- Qian H.; Zhu M.; Wu Z.; Jin R. Quantum Sized Gold Nanoclusters with Atomic Precision. Acc. Chem. Res. 2012, 45 (9), 1470–1479. 10.1021/ar200331z. [DOI] [PubMed] [Google Scholar]
- Kazan R.; Müller U.; Bürgi T. Doping of thiolate protected gold clusters through reaction with metal surfaces. Nanoscale 2019, 11 (6), 2938–2945. 10.1039/C8NR09214A. [DOI] [PubMed] [Google Scholar]
- Kazan R.; Zhang B.; Bürgi T. Au38Cu1(2-PET)24 nanocluster: synthesis, enantioseparation and luminescence. Dalton Trans 2017, 46 (24), 7708–7713. 10.1039/C7DT00955K. [DOI] [PubMed] [Google Scholar]
- Knoppe S.; Bürgi T. Chirality in Thiolate-Protected Gold Clusters. Acc. Chem. Res. 2014, 47 (4), 1318–1326. 10.1021/ar400295d. [DOI] [PubMed] [Google Scholar]
- Dolamic I.; Knoppe S.; Dass A.; Bürgi T. First enantioseparation and circular dichroism spectra of Au38 clusters protected by achiral ligands. Nat. Commun. 2012, 3 (1), 798. 10.1038/ncomms1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M.; Eckenhoff W. T.; Pintauer T.; Jin R. Conversion of Anionic [Au25(SCH2CH2Ph)18]– Cluster to Charge Neutral Cluster via Air Oxidation. J. Phys. Chem. C 2008, 112 (37), 14221–14224. 10.1021/jp805786p. [DOI] [Google Scholar]
- Zhou M.; Zeng C.; Chen Y.; Zhao S.; Sfeir M. Y.; Zhu M.; Jin R. Evolution from the plasmon to exciton state in ligand-protected atomically precise gold nanoparticles. Nat. Commun. 2016, 7 (1), 13240. 10.1038/ncomms13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R. Quantum sized, thiolate-protected gold nanoclusters. Nanoscale 2010, 2 (3), 343–362. 10.1039/B9NR00160C. [DOI] [PubMed] [Google Scholar]
- Kwak K.; Kumar S. S.; Pyo K.; Lee D. Ionic Liquid of a Gold Nanocluster: A Versatile Matrix for Electrochemical Biosensors. ACS Nano 2014, 8 (1), 671–679. 10.1021/nn4053217. [DOI] [PubMed] [Google Scholar]
- Li Z.; Xiao W.; Huang R.; Shi Y.; Fang C.; Chen Z. A Gold Nanoclusters Film Supported on Polydopamine for Fluorescent Sensing of Free Bilirubin. Sensors 2019, 19 (7), 1726. 10.3390/s19071726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa L.; Li-Destri G.; Thomson N. H.; Konovalov O.; Pontoni D. Real Space Imaging of Nanoparticle Assembly at Liquid–Liquid Interfaces with Nanoscale Resolution. Nano Lett. 2016, 16 (9), 5463–5468. 10.1021/acs.nanolett.6b01877. [DOI] [PubMed] [Google Scholar]
- Dubreuil F.; Daillant J.; Guenoun P. Direct Topographic Measurement of Multilayers on Water by Atomic Force Microscopy. Langmuir 2003, 19 (20), 8409–8415. 10.1021/la0343268. [DOI] [Google Scholar]