

Abstract
Aim
Mass spectrometry (MS)-based proteomics, particularly with the development of nano-ESI, have been invaluable to our understanding of altered proteins related to human disease. Niemann–Pick, type C1 (NPC1) disease is a fatal, autosomal recessive, neurodegenerative disorder. The resulting defects include unesterified cholesterol and sphingolipids accumulation in the late endosomal/lysosomal system resulting in organ dysfunction including liver disease.
Materials & methods
First, we performed MS analysis of a complex mammalian proteome using both nano- and standard-flow ESI with the intent of developing a differential proteomics platform using standard-flow ESI. Next, we measured the differential liver proteome in the NPC1 mouse model via label-free quantitative MS using standard-flow ESI.
Results
Using the standard-flow ESI approach, we found altered protein levels including, increased Limp2 and Rab7a in liver tissue of Npc1-/- compared to control mice.
Conclusion
Standard-flow ESI can be a tool for quantitative proteomic studies when sample amount is not limited. Using this method, we have identified new protein markers of NPC1.
Keywords: AJS-ESI, biomarker, ion funnel, mass spectrometry, Niemann–Pick type C1, NPC, proteomics, thermal gradient focusing
Graphical abstract
Mass spectrometry (MS)-based proteomics has become the analytical tool of choice for the identification and quantification of proteins in complex biological systems since the availability of genome sequence databases. During MS analysis, analytes are ionized in the gas phase, followed by m/z determination via a mass analyzer, and registration of ions at each m/z value via a detector. ESI is one of the most common techniques used for the ionization of peptides and proteins prior to MS analysis [1]. In this experimental strategy, analytes are ionized from solution making it highly compatible for the integration of liquid-based chromatographic and electrophoretic separation systems. Furthermore, the implementation of nanoflow (10–1000 nl/min) ESI has provided a platform for improving sensitivity and therefore increasing protein identification yields, thereby becoming a routine approach for large-scale proteomic studies [2]. The introduction of a standard-flow (>100 μl/min) ESI source (Agilent Jet Stream thermal gradient focusing electrospray ionization, AJS-ESI) has shown promise for the high-throughput analysis of complex biological systems where it has been used for proteomic [3], metabolomic [4–6] and lipidomic studies [7,8].
The AJS-ESI source is unique in that its design offers improved desolvation and spatial focusing of ions. Studies have shown that utilization of this source enables a five- to ten-fold increase in sensitivity over a traditional ESI source when considering the analysis of pesticides, cosmetics and illicit drugs in biological fluids at standard-flow rates [9]. The enhanced desolvation and increase in sensitivity is made possible by the use of a super-heated nitrogen sheath gas which is positioned orthogonally 90° to the entrance of the MS. Here, the superheated sheath gas is used to collimate the nebulizer spray creating a higher density of ions in front of the capillary, thereby allowing more ions to be sampled by the MS. It is important to note that in ESI ions are created at atmospheric pressure and ultimately detected at lower pressure. Due to joule expansion, ions can be lost as they traverse from higher to lower pressure. Markedly, the use of ion funnel technology refocuses ions efficiently as they are transmitted from areas of high to low pressure by the use of a series of ring electrodes, sequentially decreasing in size which facilitates precise shaping of the ion beam [10]. Moreover, dual ion funnels which consist of a high-pressure funnel (source region) and a slightly off-axis low pressure funnel has been shown to reduce source contamination and interference, thereby further increasing the transmission of charged particles into the MS [11,12]. Therefore, significantly increased signal-to-noise is observed in standard-flow ESI experiments when both thermal gradient focusing and dual ion funneling technologies are combined.
Niemann–Pick, type C1 (NPC1) disease is a fatal, autosomal recessive, neurodegenerative disorder caused by mutations in the NPC1 gene. As a result of the genetic defect, accumulation of unesterified cholesterol and sphingolipids is observed in the late endosomal/lysosomal system [13]. This accumulation of lipids results in a cascade of pathophysiological events including hepatomegaly, vertical gaze palsy, and progressive cerebellar degeneration, among others (as reviewed in [14,15]). In addition, many NPC1 patients develop chronic liver disease which could result in liver failure [16]. Currently, there is no the US FDA-approved therapy for NPC1, therefore additional studies are needed to develop therapeutic options. The NPC1 null mouse model, hereafter referred to as Npc1-/- mice, recapitulates the human disease and was therefore used in these studies. In addition to liver disease, the lifespan of Npc1-/- mice includes a presymptomatic neurological phenotype time up to 3 weeks of age followed by the development of tremors at 5 weeks, ataxia at 7 weeks and is terminal at approximately 3 months of age. Progressive loss of cerebellar Purkinje neurons is observed in this model which is a hallmark of the disease and supports the clinical phenotype.
The measurement of multiple NPC1 biomarkers has been made possible using MS as an analysis tool. One study revealed increases in oxidative stress and fatty acid binding proteins in cerebellar tissue by combining 2D gel electrophoresis (2D-GE) and MS (2D-GE-MS) [17]. Another study using 2D-GE-MS revealed alterations in the glutamate and GABA transporters in the hippocampal region of Npc1-/- mice [18]. Targeted MS-based assays for sphingolipids [19,20], bile acids [21,22] and 24(S)-hydroxycholesterol [23] in biological fluids have also been developed. Additionally, the use of GC–MS has been employed to measure cholestane-3β,5α,6β-triol in NPC patients [24]. Most recently, the use of isobaric peptide labels was used to carry out a large-scale quantitative proteomics study in NPC1 patient fibroblasts to evaluate the differential proteome as a result of treatment with 2-hydroxypropyl-β-cyclodextrin or histone deacetylase inhibitors [25,26]. Additionally, there are numerous studies reported in the literature that would benefit from MS-based monitoring of biomarkers parallel to disease progression [27–29]. Notably, few MS-based proteomic studies have been carried out with focus on the NPC1 liver proteome. Interestingly, microarray analysis indicated numerous liver protein biomarkers which were evaluated across all ages of the Npc1-/- mouse lifespan and found secreted lysosomal enzymes altered in both the mouse model and in NPC1 patients [30]. Enzymes associated with drug metabolism and related toxicity have also been identified regarding the liver [31]. Other studies confirming alterations of GM2 gangliosides [32] and sphingosine and sphinganine in the liver of Npc1-/- mice have been reported [33].
The goals of this study were twofold. First, we sought to compare the depth of proteome coverage obtained upon analysis of a complex mammalian proteome using both nano- and standard-flow ESI approaches; the standard-flow ESI analysis was done using the AJS-ESI source interface. Here, the proteome coverage and technical reproducibility were accessed for both approaches using a HeLa digest. Second, we implemented the standard-flow ESI method to carry out a large-scale label-free quantitative proteomics analysis of liver tissue from the null NPC1 mouse model to determine whether this approach could be used to identify new biomarkers. Based on reported NPC1 proteomic changes in the literature, we then evaluated the standard-flow ESI approach for reliable label-free quantification via orthogonal methods.
Materials & methods
Reagents & chemicals
All reagents were acquired from Sigma-Aldrich (MO, USA) and used as received unless otherwise noted.
Preparation of HeLa digest for LC–MS analysis
HeLa cells were purchased from ATCC (VA, USA), cultured as recommended, and lysed in RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1% Triton-X 100, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate, with protease inhibitors). Protein concentration was determined via the BCA assay (Pierce-Thermo, CA, USA) and 100 μg was processed via filter-aided sample preparation (FASP) method, which has been described [34]. Samples were desalted using a C18 TopTip (PolyLC Inc., MD, USA), dried via vacuum centrifugation and resuspended in 0.1% formic acid (v/v) prior to MS analysis.
Analysis of HeLa digest via nano & standard-flow ESI
An aliquot of the HeLa digest was analyzed via nano- and standard-flow ESI tandem MS. In initial pilot studies (data not shown), we reviewed the raw MS data and based the chromatographic gradients for both methods on the average number of possible precursors observed per MS1 scan. The gradient was adjusted until a similar number of precursors were observed per MS1 scan in both the nano- and standard-flow ESI approaches. The resulting methods were used for this study. For the analysis via nano-flow ESI, an Agilent 1260 HPLC Chipcube was interfaced to an Agilent 6550 iFunnel Q-TOF LC/MS system. Two micrograms of peptides were loaded onto a High Performance Chip (G4240–62030, Agilent Technologies Inc., CA, USA) at 2 μl per min in 0.1% formic acid and trapped on the 360 nl enrichment column using a sample flush volume of 5 μl. Peptide separation was performed on the chip’s 150 mm × 75 μM column using the following gradient: 3% B at 0 min, 3–25% B at 105 min, 40% B at 120 min, 90% B at 125 min, hold at 90% B until 130 min at a flow rate of 300 nl/min. Source parameters were: gas temperature (225°C), drying gas flow (13 l/min), VCap (1800 V) and Oct 1 RF Vpp (750 V). For the analysis via standard-flow ESI, the AJS-ESI ion source was interfaced to an Agilent 6550 iFunnel Q-TOF LC/MS system. The mobile phase A was 0.1% formic acid and mobile phase B was acetonitrile. Two micrograms of peptides were loaded onto a 2.1 × 100 mm Poroshell 120 EC-18, 2.7 μm column (Agilent Technologies Inc.) at 200 μl per min and separation performed on an Agilent 1290 UPLC system using the following gradient: 3% B at 0–5 min, 30% B at 45 min, 60% B at 50 min, 90% B at 53 min, hold at 90% B until 60 min. Source parameters were as follows: drying gas temperature (250°C), drying gas flow (14 l/min), nebulizer (35 Ψ), sheath gas temperature (250°C), sheath gas flow (11 l/min), VCap (3,500 V), and Oct 1 RF Vpp (750 V). Acquisition parameters for MS analysis for both nano- and standard-flow ESI analyses were as follows: 8 MS scans/sec (300–1700 m/z), 3 MS/MS scans/sec (50–1700 m/z) and maximum precursor selection of the top 20 ions of charge states ≥2 with collision energy maintained with slope = 3 and offset = 2 for +2 charge state species and slope = 3.6 and offset = -4.8 for species with charge state ≥3.
Data were acquired in technical triplicates for both the nano- and standard-flow ESI experiments and all raw MS data were deposited and is publicly available via MassIVE (ftp://massive.ucsd.edu/MSV000082874). LC–MS/MS data were analyzed using Spectrum Mill Version B.06.00.200 (Agilent Technologies Inc.) against the UniprotKB/SwissProt database (2017), which contained 20,198 sequences for Homo sapiens. Search parameters were as follows: precursor mass tolerance was 15 ppm and fragment mass tolerance was 0.02 Da, enzyme was trypsin with two missed cleavages and carbamidomethylation (C) as a fixed modification and oxidation (M) and deamidation (NQ) as variable modifications. Auto thresholds were used for peptide identification to achieve a target 1% false discovery rate. The number of unique peptides and protein groups for the triplicate analysis of both the nano- and standard-flow ESI approaches were analyzed to access peptide and protein identification reproducibility.
Animal breeding & tissue collection
The animals were housed in conditions of controlled light (12 h light/dark cycle) and temperature (20–24°C) where they received food and water as needed. All animal experiments were performed according to UIC institutional animal care committee-approved protocols (UIC 18–163). Npc1+/+ and Npc1-/- mice (BALB/cNctr-Npc1m1N/m1N, Jackson Laboratories (ME, USA) were euthanized at 3, 5, 7, 9 and 11 weeks of age (N = 3 for each genotype and age) via CO2 asphyxiation, decapitated, and liver, cerebella, and cerebral cortex tissues collected, flash frozen on dry ice, and stored at -80°C, as previously described [35].
Preparation of liver tissue for LC–MS analysis
Protein lysate pools for liver tissue from 11-week Npc1+/+ and Npc1-/- mice were made for the control and mutant animals by combining equal amount of protein from each animal (1.2 mg total protein for each genotype). Next, a recombinant green fluorescent protein (GFP) internal standard was spiked into the pools at a concentration of 100 fmol per 1 μg protein extract prior to sample preparation via filter-aided sample preparation (as previously described above) where 200 μg was digested per filter (6 filters per genotype). The digest for each genotype was combined, desalted and concentrated via a C18 SPE cartridge. The resulting peptides for each genotype were resolved into 20 fractions using a Waters XBridge BEH130 C18 3.5 μm 4.6 × 250 mm column on a Lab Alliance HPLC system. All fractions were dried and resuspended in 45 μl of 0.1% (v/v) formic acid prior to analysis.
Protein identification, label-free quantification & pathway analysis of liver tissue
Ten microliters of each fraction were injected for MS analysis using the standard-flow ESI method described above. Analysis of each fraction was acquired in technical triplicates. LC–MS/MS data for each fraction was merged for each technical replicate and proteins identified in the same manner as the HeLa digest by searching against the UniprotKB/SwissProt database (2017), which contained 16,935 sequences for Mus musculus. Additionally, an in-house GFP database was created in Spectrum Mill and incorporated into the search. Protein identification results from Spectrum Mill were imported into Mass Profiler Professional (Agilent Technologies Inc.) for label-free quantitation using the median peptide abundance for each protein. Retention times were evaluated prior to quantitative analysis. Only peptides with charges +2 to +4 with two or more isotopic peaks were included. Protein identifications were further filtered to include those containing a minimum of two unique peptides. A list of differential proteins for the liver tissue (fold change ≥2 and p ≤ 0.05) was exported and KEGG pathways enriched using the STRING software (https://string-db.org/) [36].
Western blot analysis
Protein lysates for each individual animal and tissue type (liver, cerebellum and cerebral cortex) were subjected to separation by electrophoresis and western blotting for evaluation of selected protein biomarkers. The samples were thermally denatured in Laemmli buffer containing DTT at 70°C for 10 min then loaded onto a 12% NuPage Bis-Tris gel (Novex Life Technologies, CA, USA). Separation was carried out by SDS-PAGE (130 V for ∼120 min) and transferred to nitrocellulose paper using a Pierce Power blotting system (Thermo Pierce Biotechnology, IL, USA) at 25 V for 7 min. Blocking was carried out in 5% (wt/v) dry milk with 0.1% (v/v) Tween-20 (PBS-T) overnight before addition of primary antibody followed by the addition of the secondary antibody in 5% (wt/v) milk PBS-T for 1 h. Excess secondary antibody was removed by washing with PBS-T three-times. Protein bands were visualized using Pico West Blotting Substrate (Thermo Pierce Biotechnology) and imaged on an Azure c300 imaging system (Azure Biosystems, CA, USA). Densitometry analysis was performed using ImageJ (http://rsb.info.nih.gov/ij/index.html) [37]. GraphPad Prism (v5.01) was used to determine statistical significance between groups. Details regarding western blot reagents are provided in the Supplementary Table 1.
Immunohistochemistry
Npc1+/+ and Npc1-/- mice (BALB/cNctr-Npc1m1N/m1N; Jackson Laboratories, ME, USA) at 7-weeks of age (N = 1 for each genotype) were subject to vascular perfusion with saline followed by 4% (w/v) paraformaldehyde. The liver tissue was dissected and fixed for 24 h in 4% (w/v) paraformaldehyde. Sections (50 μm) were processed using the free-floating technique and blocked (10% fetal bovine serum and 0.3% Titon-X 100) for 1 h. The tissue sections were washed, incubated overnight with primary antibodies at 4°C, washed again, and incubated for 1 h at room temperature with the appropriate Alexa Fluor-conjugated secondary antibodies. After extensive washing, the tissue sections were subsequently mounted on positively charged, coated glass slides and covered with mounting media where nuclei were counterstained with DAPI. The slides were analyzed using a DM5500 Q Microscope with a Leica DFC 500 Camera. Details regarding immunofluorescence reagents are provided in the Supplementary Table 1.
Results & discussion
The goals of this study were twofold: to compare the depth of proteome coverage from the analysis of a complex proteomic sample via nano- and standard-flow ESI approaches and to carry out a large-scale bottom-up proteomics analysis of liver tissue using the standard-flow ESI approach to identify new liver protein biomarkers in the null NPC1 mouse model.
Comparative analysis of nano & standard-flow ESI approaches
Discovery-based proteomics has been largely dominated by the use of nano-flow LC (nLC) coupled to MS due to increased sensitivity and detection of peptides that can be achieved [38]. In experiments where sample amount is limited nLC-MS is ideal, but trade-offs include technical challenges in operation and reproducibility in chromatography [12]. A more attractive approach is the use of a standard-flow chromatography coupled with an ESI source that can offer high throughput, reproducible chromatography. The AJS-ESI source is one option that allows for increased ion transfer into the MS compared with traditional ESI sources owing to improved desolvation and enhanced special focusing of ions, particularly when coupled with ion funneling technology [10]. The increased ion transfer into the MS of the AJS-ESI source compared with traditional electrospray sources, is also advantageous when considering implementation of retention time alignment for label-free proteomic studies.
To compare the depth of proteome coverage obtained via nano- and standard-flow electrospray analysis of complex proteomic samples, we analyzed the same amount of HeLa digest using identical MS parameters. Figure 1A shows representative basepeak chromatograms from the analysis of 2 μg of HeLa digest via nanoflow (top) and standard-flow (bottom) ESI approaches. Notably, both approaches reveal well-resolved chromatograms, with the nano-flow ESI approach having double the MS analysis time. Proteomic analysis for unique peptides (Supplementary Table 2) and protein groups (Supplementary Table 3) of 2 μg of HeLa digest via nano- and standard-flow ESI approaches was performed. Analysis of the HeLa digest via nano-flow ESI analysis resulted in 6165 ± 14 unique peptides and 1178 ± 15 protein groups. Analysis of an aliquot for the same HeLa digest via standard-flow ESI resulted in 4634 ± 28 unique peptides and 826 ± 14 protein groups. Here, we observed that by using the nano-flow ESI approach we were able to identify an average of 1531 more peptides, corresponding into an increase in an average of 352 protein group identifications. Next, we accessed our technical reproducibility from each ESI approach. Figure 1B shows Venn diagram analysis of the number of unique peptides and protein groups from a triplicate technical analysis from both the nano-flow ESI (top) and standard-flow ESI (bottom) approaches, respectively. In this experiment, we observed an overlap of 3771 and 2557 unique peptides and 953 and 616 protein groups from the nano- and standard-flow ESI approaches, respectively. Overall, when considering the number of unique peptides and protein identified, we observed an approximate 30% loss in the number of identifications when using the standard-flow ESI approach.
Figure 1. . Representative base peak chromatograms and overlap of peptides and protein groups for 2 μg of HeLa protein digest obtained via nano- and standard-flow ESI.

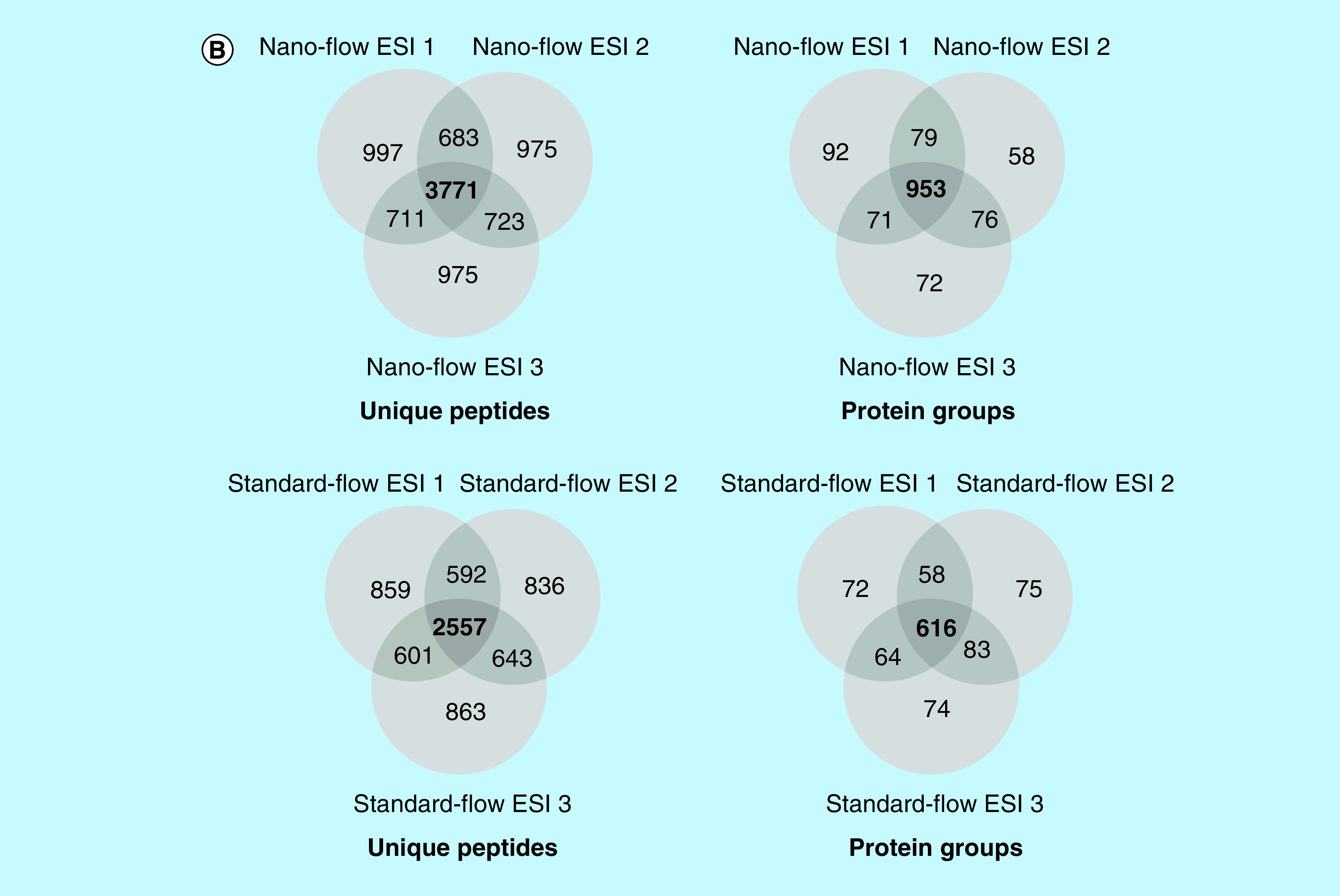
(A) Analysis of 2 μg of HeLa protein digest via nano-flow (top) and standard-flow (bottom) ESI revealed well-resolved chromatograms in both approaches, with the nano-flow ESI method having double the analysis time. (A) Proteomic analysis resulted in 6165 ± 14 and 4634 ± 281653 ± 37 peptides and 1178 ± 15 and 826 ± 14 protein groups, respectively. (B) Venn diagram analysis revealed a technical replicate overlap of 3771 peptides and 953 protein groups for 2 μg of HeLa protein digest via nano-flow ESI (top) and a technical replicate overlap of 2577 peptides and 616 protein groups for 2 μg of HeLa protein digest via standard-flow ESI (bottom).
Traditionally, large-scale bottom-up proteomic studies are carried out using nano-flow ESI due to its increased sensitivity. This is due to the smaller droplet size produced via the nano-flow ESI approach resulting in enhanced desolvation and ion formation, allowing for more ions to be detected, a particularly advantageous approach when the amount of sample is limited. A disadvantage of the nano-flow ESI approach is the increase in analysis time, nearly double that of the standard-flow ESI approach. This can become costly as the number of samples to be analyzed increases. With this, the use of the AJS-ESI source interface may not be appropriate if there are proteins of interest known to be of lower abundance. Alternatively, more sample preparation steps may need to be incorporated such as subcellular fractionation prior to enzymatic digestion or peptide fractionation prior to MS analysis. On the other hand, when samples are not limited (as is the case in many proteomic studies involving tissue) the standard-flow ESI approach could be advantageous as it offers decreased analysis time and a potential for decreased cost.
Differential proteomic analysis in liver tissue
To address whether we could use the standard-flow ESI approach to identify new biomarkers in the NPC1 mouse model, we carried out a label-free, quantitative MS analysis of Npc1-/- and Npc1+/+ liver samples. Previous experiments have shown that by injecting 10 μg of protein digest the proteome coverage can be significantly increased when employing the standard-flow ESI approach [39]. With this, the following experiment was designed such that after fractionation approximately 10 μg of protein would be analyzed on column per injection. A triplicate technical replicate analysis for each peptide fraction for each genotype was performed and protein identifications assigned (Supplementary Table 4). Next, the identification results were filtered for a minimum of two unique peptides, resulting in 4008 protein identifications (Supplementary Table 5). At last, differential analysis was performed revealing 950 significantly altered proteins (≥twofold change and p ≤ 0.05) (Supplementary Table 6). The standard-flow ESI differential analysis approach presented in this study proved to be highly robust when subject to in depth statistical evaluation (Figure 2). The median intensity of proteins in each replicate was compared where very little deviation was observed between replicates shown. In addition, review of the intensity of the GFP internal standard (Supplementary Figure 1) indicated that no normalization was needed between replicates. Furthermore, principle component analysis of altered proteins from each geneotype also reveals only one main component of separation, suggesting little technical variability. With this, we concluded that the standard-flow ESI approach is highly reproducible upon evaluation of technical replicates from each genotype.
Figure 2. . Statistical analysis of the fractionated liver proteome for 11-week Npc1+/+ and Npc1-/- mice.
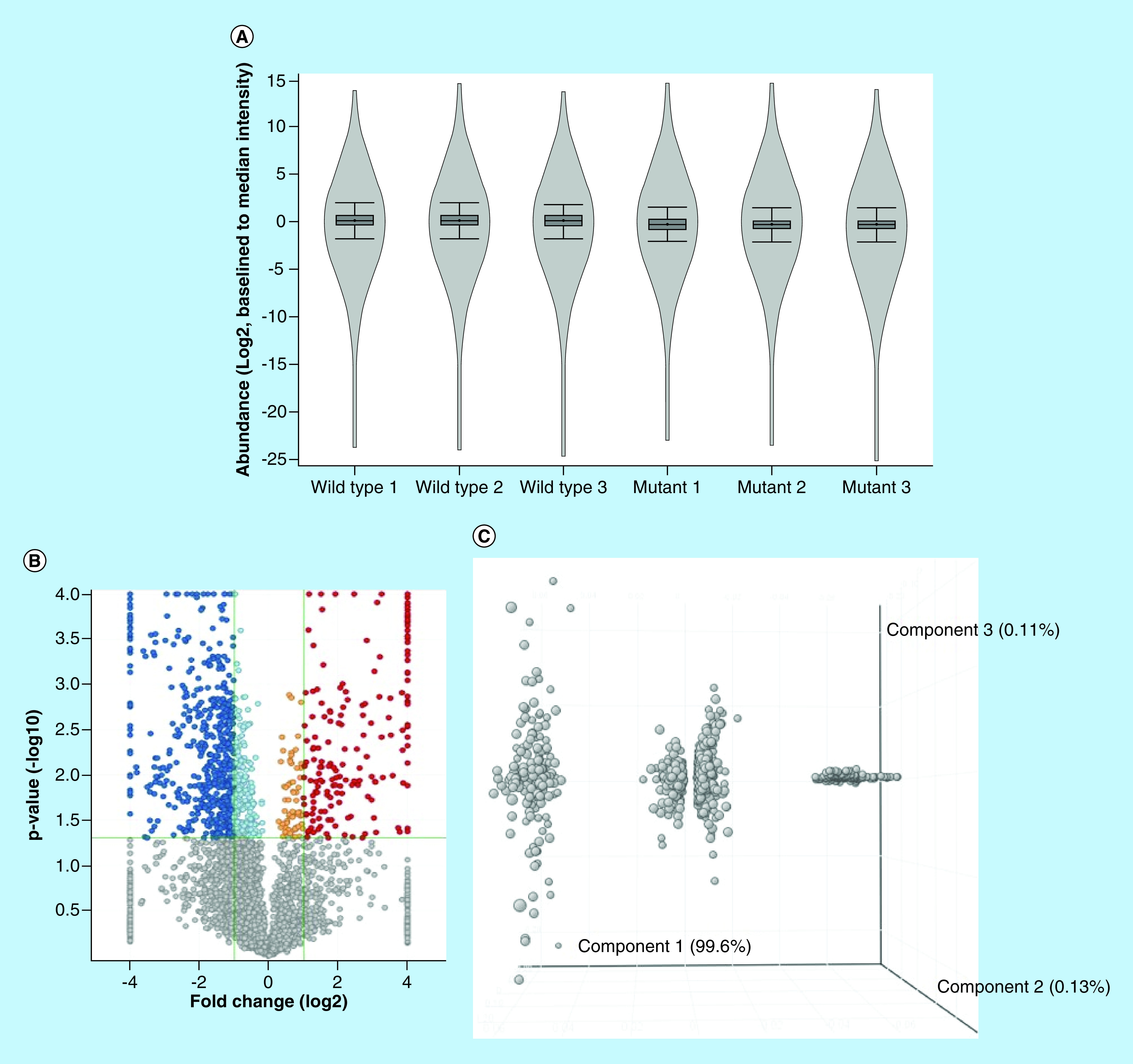
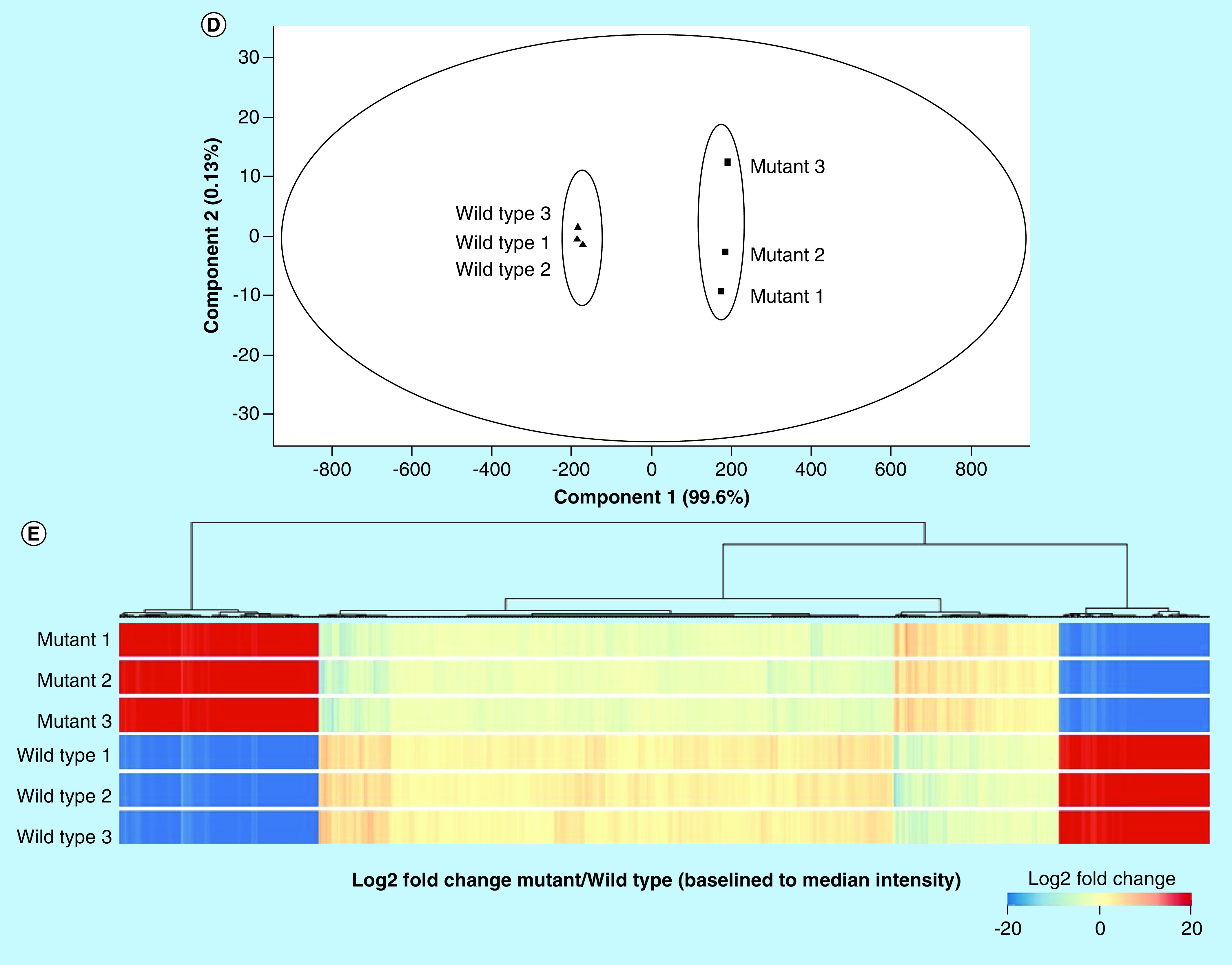
(A) Violin plot showing the distribution and density of measured proteins, (B) Volcano plot depicting the distribution of significantly altered proteins (dark blue is upregulated fold change ≥2, light blue is upregulated fold change <2, orange is upregulated fold change <0.5 and dark red is downregulated fold change ≥0.5, grey is p ≥ 0.05), (C) Principle component analysis of statistically differential proteins indicating that there is only one main component (disease) driving the alteration of the measured proteins, (D) principle component analysis of Npc1-/- and Npc1+/+samples, indicating that there is only one main component (disease) driving the separation between the two groups and (E) Heatmap of all measured proteins. All fold changes are noted as Npc1-/- relative to Npc1+/+.
To evaluate the standard-flow ESI approach for reliable label-free quantification, we evaluated protein lysates via Western blot from each individual animal of each genotype for the expression of the Npc1 protein as well as others previously shown to be altered in NPC1 based on previous accounts in the literature. Here, we observed that the standard-flow ESI approach was able to detect only very large changes in protein expression. Western blot analysis confirmed no expression of Npc1 in Npc1-/- mice animals (Figure 3A), as expected. These data correspond with the MS analysis where we observed a Log2 fold change of -21.2 in Npc1-/- compared with Npc1+/+ mice. Next, we evaluated fatty acid binding protein 5 (Fabp5) and fatty acid binding protein 7 (Fabp7) which have previously been shown to be altered in brain tissue the disease [17]. Figure 3A shows increased expression of Fabp5 and Fabp7 in Npc1-/- compared with Npc1+/+ mice. The Western blot analysis of Fabp5 is consistent with the MS analysis where we observed a Log2 fold change of 26.7 in Npc1-/- compared with Npc1+/+ mice. Using the standard-flow ESI method, we were able to identify Fabp7 (Supplementary Table 2), but we were not able to assign it to be statistically altered. This is the first study confirming altered expression of Fabp5 and Fabp7 at the protein level in liver tissue of Npc1-/- mice. Traditionally, label-free quantification is well suited to detect larger changes in peptide intensity, whereas other relative quantification strategies such as isobaric labeling are better suited for smaller changes in peptide intensity [40]. With this, we hypothesized that the standard-flow ESI approach would result in the detection of very reliable biomarkers in the NPC1 disease process. Moreover, the standard-flow ESI method shows potential to be beneficial for the analysis of NPC1 patient samples for disease biomarkers in a high-throughput manner in cases where sample amount is not limited.
Figure 3. . Method evaluation and analysis of the differential liver proteome from an 11-week symptomatic NPC1 mouse model.

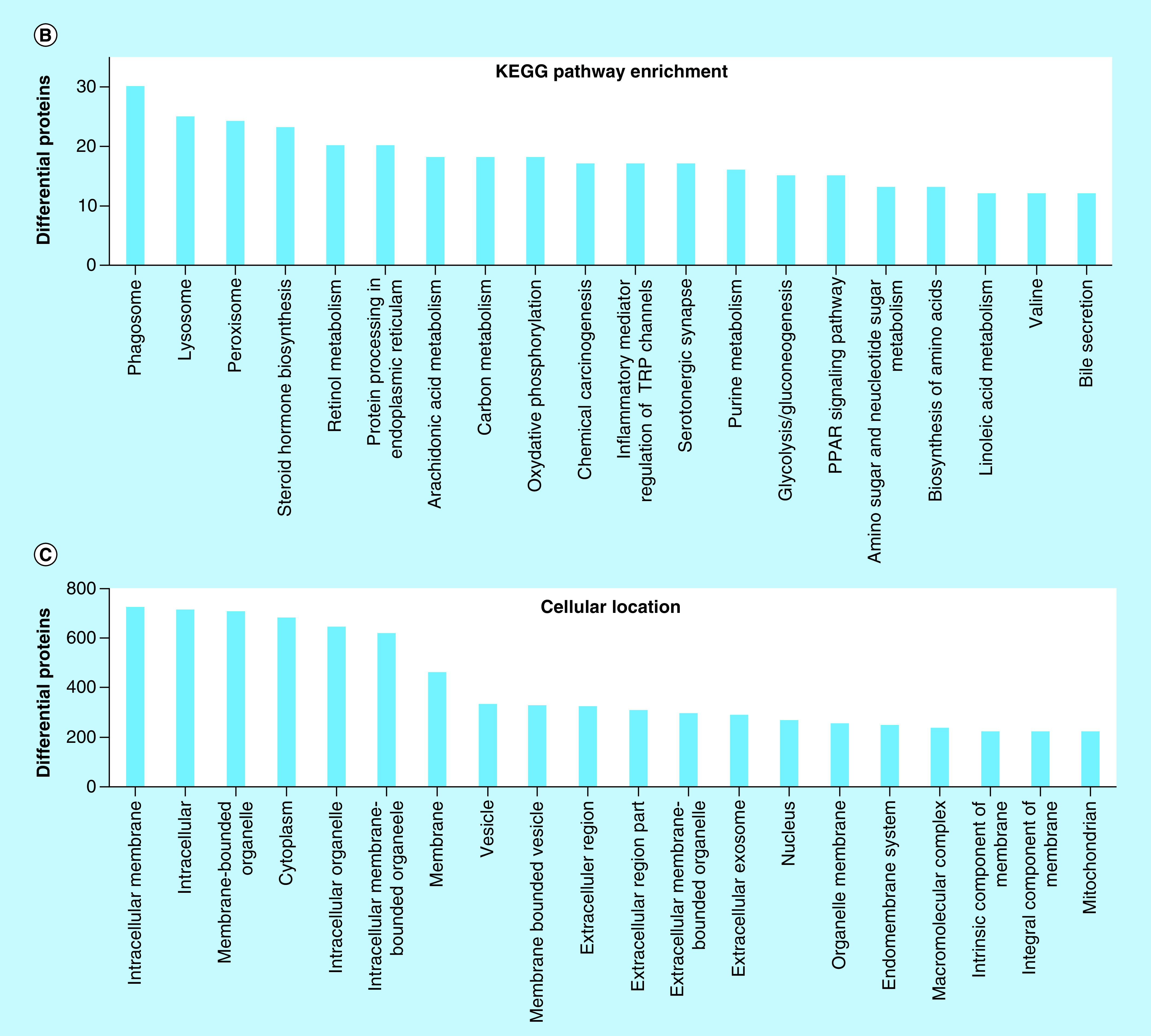
(A) Individual liver lysates (N = 3 for each genotype) were subject to electrophoresis and western blotting to evaluate the sensitivity of the standard-flow ESI approach for measuring the differential proteome in the liver tissue of 11-week Npc1+/+ and Npc1-/- mice. Niemann–Pick, type c1, (NpC1), fatty acid binding protein 5 (Fabp5), and fatty acid binding protein 7 (Fabp7) were observed to be increased in Npc1-/- mice relative to Npc1+/+ mice. Intensities are reported as relative to Gapdh. Significance was determined using an unpaired t-test with Welch’s correction. (B) Differential proteins (y-axis) from the liver proteome where enriched for the top 20 matching KEGG pathways (x-axis). (C) Differential proteins (y-axis) from the liver proteome where enriched for the top cellular locations (x-axis).
Pathway analysis results of the differential proteome of liver tissue from the 11-week symptomatic NPC1 mouse model suggest that several biological processes are altered in the liver. A complete list of all altered pathways is available for review (Supplementary Table 4), although only top altered pathways were considered in this study. Figure 3B shows that the most top significantly altered pathways correspond to differential proteins generally related to the lysosome (Supplementary Figure 2) and phagosome (Supplementary Figure 3). Since NPC1 is a lysosomal storage disorder, this is consistent with many studies documented in the primary literature. Specifically, lysosomal proteins involved in sphingolipid metabolism were observed to be altered including: glucosylceramidase (GCase, Log2 = -2.9) and sphingomyelin phosphodiesterase (Smpd1, Log2 = -21.6). Evaluation of the proteins mapped to the biogenesis of mature phagosomes, particularly those involved in the fusion of the lysosome and late endosomes were also altered. These include increased expression of lysosome membrane protein 2 (Limp2, Log2 = 1.8), lysosome-associated membrane glycoprotein (Lamp1, Log2 = 2.1), as well as various v-type proton ATPase subunits in Npc1-/- compared with Npc1+/+ mice. Notably, we identified the early endosome marker Rab5b, but did not determine it to be significantly altered via our standard-flow ESI approach. Other altered proteins involved in the formation of the phagolysosome included: rab-interacting lysosomal protein (Log2 = -1.4), tubulin β-4B chain (Log2 = -1.1), tubulin α-8 chain (Log2 = 26.2) and protein transport protein Sec61 subunit α isoform 1 (Log2 = -1.2). Figure 3C shows that the cellular location of the proteins determined to be significantly altered have a broad range of cellular locations including membranes, cytosol and vesicles.
Alterations in lysosome-related proteins
A series of orthogonal studies were carried out to confirm the proteomic findings observed in the standard-flow ESI approach for differential proteomic analysis of liver tissue. We evaluated Lamp1 and Limp2 expression via Western blot in the same liver tissue lysates used for the MS analysis (Figure 4A). Comparative analysis of lysosome membrane proteins Lamp1 and Limp2 shows increased expression in Npc1-/- compared with Npc1+/+ mice. This result is consistent with the increased levels of Lamp1 and Limp2 observed in the MS analysis (Log2 = 1.8 and 2.1, respectively). To determine whether these observations were specific to the liver tissue, we evaluated protein lysates from the cerebellum (Figure 4B) and cerebral cortex (Figure 4C) of the 11-week Npc1+/+ and Npc1-/- mice. Comparative analysis of Lamp1 and Limp2 in both the cerebellum and cerebral cortex are consistent with the protein expression trends observed in the liver, suggesting these changes are not tissue specific. Additionally, we evaluated Limp2 expression in liver tissue across the progression of the NPC1 disease (Figure 5) and show that Limp2 is increased even at early stages.
Figure 4. . Western blot analysis of lysosome and phagosome/endosome markers in liver, cerebellar, and cerebral cortex tissue from 11-week mice from the symptomatic NPC1 mouse model.
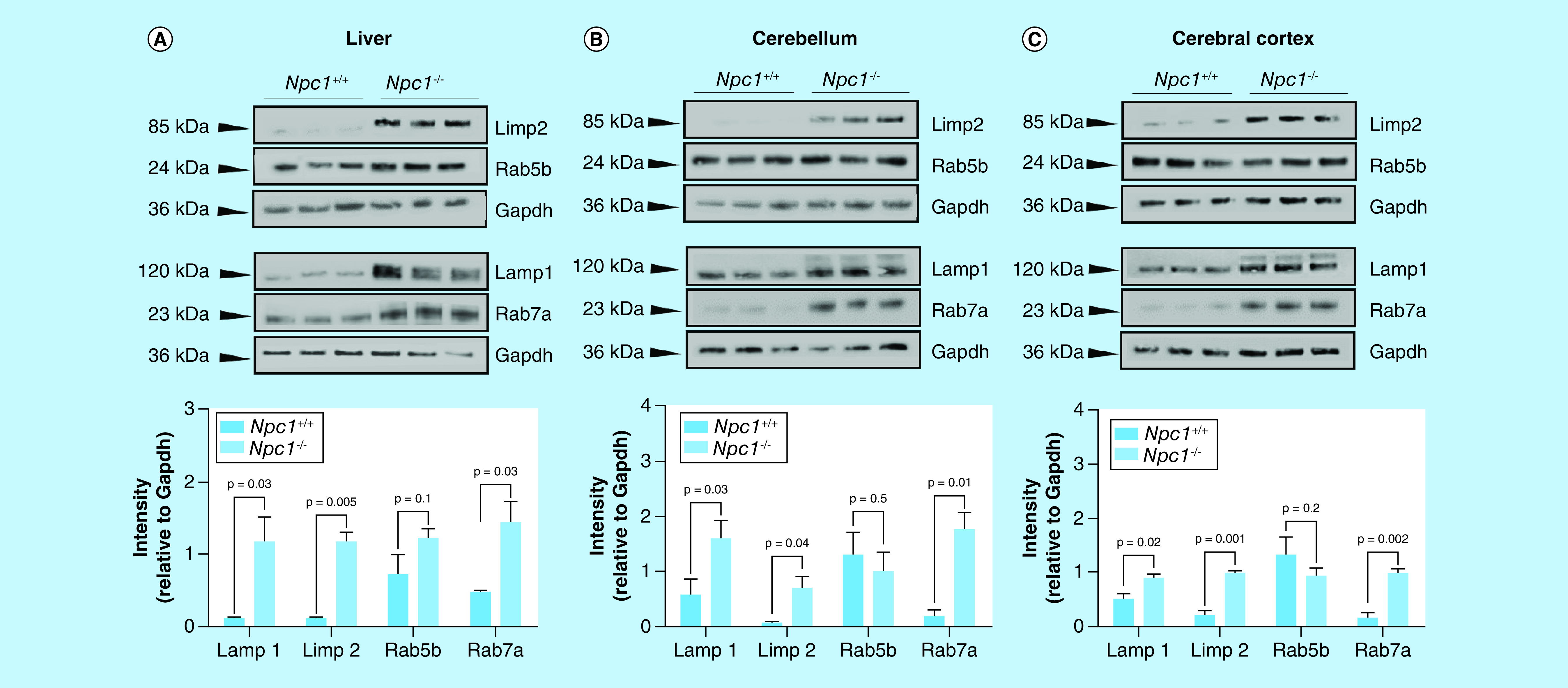
Individual lysates (N = 3 for each genotype) from 11-weelk Npc1+/+ and Npc1-/- animals from the (A) liver, (B) cerebella, and cerebral cortex (C) were subject to electrophoresis and western blotting of ras-related binding protein 5b (Rab5b), ras-related binding protein 7a (Rab7a), lysosome membrane protein 2 (Limp2) and lysosome-associated membrane glycoprotein 1 (Lamp1). Intensities are reported as relative to Gapdh. Significance was determined using an unpaired t-test with Welch’s correction.
Figure 5. . Western blot evaluation of lysosome membrane protein 2 and ras-related binding protein 7a at different stages of Npc1.
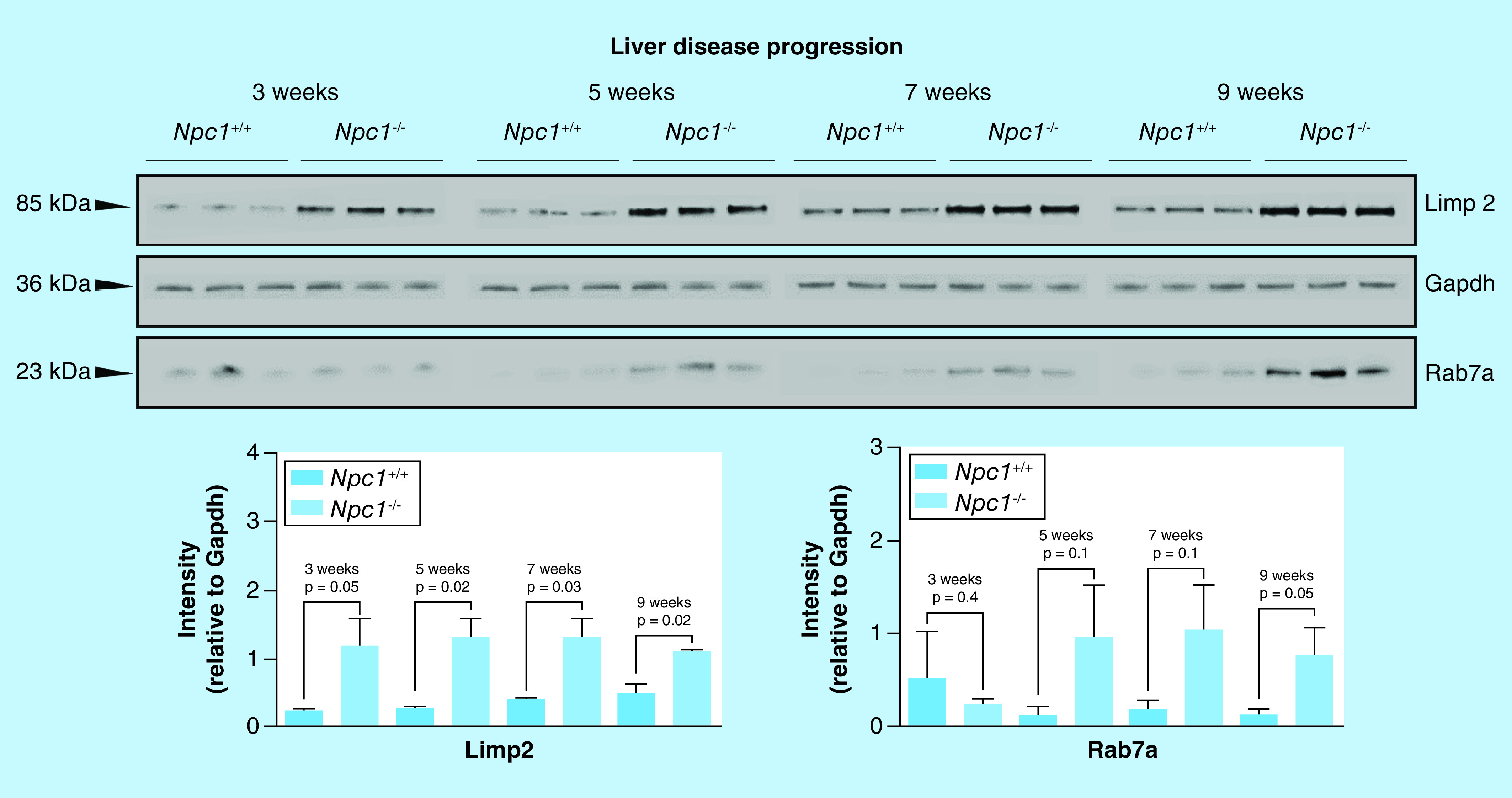
Individual liver lysates (N = 3 for each genotype) 3-, 5-, 7- and 9-week Npc1+/+ and Npc1-/- animals were subject to electrophoresis and Western blotting of Limp2 and Rab7a. Limp2 shows increased expression at all stages of the disease while Rab7a is only significantly altered at 9 and 11 weeks. Intensities are reported as relative to Gapdh. Significance was determined using an unpaired t-test with Welch’s correction.
Accumulation of unesterified cholesterol and sphingolipids in the late endosomal/lysosomal system in NPC1 disease intuitively results in alterations in lysosome structure and function. Observations from previous studies of cholesterol accumulation on lysosomes include: atypical lysosome buoyancy [41], abnormal inclusion distributions [42], increased volume [43] and altered size [44]. In this study, we observed alterations in multiple lysosomal membrane proteins (Supplementary Figure 2). Previous studies have suggested that increased expression of Lamp proteins may be an indicator of cholesterol storage defects [45]. Interestingly, Limp2 is a multifunctional lysosomal membrane protein that contains multiple binding sites for various ligands including cholesterol and phosphatidylcholine (as reviewed in [46–48]). Furthermore, Limp2 has been proposed as a marker of Gaucher and Parkinson disease [49]. Studies overexpressing Limp2 have shown cholesterol containing lysosomal-type vesicles [50]. Here, we observed an increase in Limp2 expression across all tissue types (liver, cerebellum and cerebral cortex) of Npc1-/- mice. This may simply reflect a compensatory mechanism to transport cholesterol out of the lysosome; however, it is possible that the increased expression of Limp2 may be a result of the increased number and size of the lysosomes in Npc1-/- mice. Previous studies in brain tissue have shown that Limp2 is altered at early time points in NPC1 in cerebellar tissue [39] which is consistent to our observations in the liver of Npc1-/- mice. Additional studies are needed to elucidate the functional consequence of increased expression in the presence of cholesterol accumulation and to determine whether high levels of Limp2 are detrimental to the cell, further compounding the biochemical phenotype.
We also observed alterations in proteins involved in sphingolipid metabolism which is consistent with previous observations in NPC1. Specifically, we observed decreased expression of both GCase and Smpd1 in the liver tissue of Npc1-/- mice. Previous studies in NPC fibroblasts revealed that increased cholesterol storage affects the stability of GCase and that reduction of cholesterol increased the level of GCase to nearly normal values [51]. Additionally, GCase expression was decreased in NPC fibroblasts due to the enhanced processing of the enzyme through the cholesterol induced physical dissociation of GCase and its activating/stabilizing factors [52]. The most notable function of Limp2 is that it serves as a lysosomal sorting receptor for GCase; Limp2 binds to GCase in the endoplasmic reticulum, transports it through the Golgi network and endosomal compartments, and delivers the enzyme to the lysosome after an acidic pH-modulated dissociation of the receptor and ligand in the late endosomal compartment [49]. With this, it is reasonable to infer that the increased expression of Limp2 in Npc1-/- mice may be a compensatory mechanism to increase the levels of GCase in the lysosome. We also observed decreased expression in Smpd1 in Npc1-/- mice. The most notable function of Smpd1 is the conversion of sphingomyelin to ceramide. Previous studies have suggested that sphingosine storage in NPC1 alters calcium homeostasis, leading to the secondary storage of cholesterol and various sphingolipids, such as sphingomyelin [53]. Taken together, these data suggest that utilization of the standard-flow ESI approach in analyzing the differential proteome of the liver tissue was able to identify multiple lysosome-related proteins documented to be altered in NPC1.
Rab7a is increased in Npc1-/- mice
Intracellular degradation pathways are fundamental for cellular function. Phagocytosis is one of these degradation pathways that work by forming a vesicle around engulfed particles. The phagosome has membrane-bound proteins to recruit and fuse with lysosomes to form mature phagolysosomes. As the phagosome matures, it sequentially acquires different proteins (e.g., the Rab GTPases) eventually leading to fusion of the acidic lysosome structure (phagolysosome) [54]. Rab5b has been detected in early phagosomes [55], whereas Rab7a is present at later stages in the phagosome maturation process [56]. Specifically, Rab5b is present in sorting vesicles and directs the fusion of early endosomes [57,58] where it is subsequently exchanged for Rab7a, termed the ‘Rab conversion’ process [59]. Compromised degradation pathways such as phagocytosis can result in the accumulation of toxic cellular products, leading to a cascade to pathophysiological effects.
Pathway analysis of the differential liver proteome revealed alterations in various proteins involved in the biogenesis of phagolysosomes. This includes the lysosomal proteins mentioned above (Limp2, Lamp1 and v-ATPase) [60]. Evaluation of Rab5b (early endosome/early phagosome specific) revealed no significant change in expression in NPC1 while analysis of Rab7a (late endosome/late phagosome/phagolysosome specific) shows increased expression in Npc1-/- compared with Npc1+/+ mice (Log2 = 1.1). This is the first reports of alterations of Rab7a in NPC1 liver tissue. To further confirm these data, we evaluated the expression of Rab5b and Rab7a via orthogonal methods.
Comparative western blot analysis of Rab5b shows no statistically significant differential expression in either genotype while Rab7a shows increased expression in Npc1-/- compared with Npc1+/+ mice, consistent with the MS analysis western blot analysis (Figure 4A). To determine whether these observations were specific to the liver tissue, we also evaluated protein lysates from the cerebellum (Figure 4B) and cerebral cortex (Figure 4C) of the 11-week Npc1+/+ and Npc1-/- mice. Remarkably, we observed an increased expression of Rab7a only at the late stage of the NPC1 disease process (Figure 4). Overall, comparative analysis of Rab5b and Rab7a in both the cerebellum and cerebral cortex are consistent with the protein expression trends observed in the liver, suggesting these changes are not tissue specific. Further, we also visualized the expression of (Supplementary Figure 4). In both experiments, we observed a striking increase in Lamp1 in the Npc1-/- mice, consistent with MS and western blot results. Visual comparison of Rab5b initially suggested an increase in Npc1-/- mice; however, further evaluation of the tissue (liver parenchyma specifically) (Supplementary Figure 5) revealed that this initial observation may be animal specific or localized to areas around the central vein. To the best of our knowledge, this is the first study to show alterations in Rab7a in the NPC1 null mouse model. Here, we propose that Rab7a may be a candidate marker of NPC1 disease progression, and specific to late-stage NPC1 disease.
Conclusion
This study aims to evaluate the use of a standard-flow ESI MS-based approach to identify new disease biomarkers. Here, we report a comparative tandem MS analysis of a complex mammalian proteome using both nano- and standard-flow ESI approaches. Next, we report observed alterations in the differential liver proteome in the null NPC1 mouse model including, increased Limp2 and Rab7a expression. To this end, we propose that the standard-flow ESI approach presented in this study can be used to successfully identify new disease biomarkers in a robust and high through-put manner in cases where sample amount is not limited.
Future perspective
Traditionally, large-scale bottom-up proteomic studies are carried out using nano-flow ESI due to increased sensitivity. A disadvantage of nano-flow ESI is an increase in analysis time. With this in mind, the use of an efficient standard-flow ESI source, such as the AJS-ESI source interface, has proven to be advantageous as it offers decreased analysis time, little technical variability and a potential for decreased cost. Further, coupling this technology to other ion focusing technologies, such as MS systems that contain ion funnel technology, can further make the standard-flow ESI approach appealing for efficient biomarker analyses.
Summary points.
This study reports a proteomic analysis via standard-flow ESI approach and elucidates altered liver proteins in late stage Niemann–Pick, type C1 disease.
-
Experimental:
We performed MS analysis of a complex mammalian proteome using both nano- and standard-flow ESI with the intent of developing a differential proteomics platform using standard-flow ESI.
We measured the differential liver proteome the NPC1 mouse model via label-free quantitative MS using standard-flow ESI.
-
Results:
The standard-flow-ESI approach can provide sufficient proteome coverage to carry out large-scale differential proteomic studies when sample is not limited.
By employing the commercially available AJS-ESI source interface, we were able to obtain a depth of proteome coverage needed to carry out MS-based proteomic studies comparable to a nano-flow ESI approach.
Analysis of the differential proteome of the liver of Npc1 -/- mice, revealed new disease biomarkers specific alterations in lysosomal and phagosomal membrane proteins, among others.
Fabp5, Fabp7, Limp2, Lamp1 and Rab7a were observed to be altered in the liver of Npc1 -/- mice.
Limp2, Lamp1 and Rab7a were also observed to be altered in the cerebellum and cerebral cortex of Npc1 -/- mice.
-
Conclusion:
We have characterized the differential liver proteome at a late-stage time point in Niemann–Pick Disease, type C1 disease identifying new biomarkers which may be an indicator of disease progression using a nontraditional, standard-flow ESI experimental setup.
Supplementary Material
Acknowledgments
The authors would like to acknowledge SU Walkley, Albert Einstein College of Medicine, for providing animal tissue supported by the National Institutes of Health (R01 NS053677). MR Pergande would like to acknowledge the UIC DFI Fellowship Program.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: https://www.futuremedicine.com/doi/suppl/10.4155/bio-2018-0232
Financial & competing interests disclosure
MR Pergande is currently an intern for Agilent Technologies. This work was supported by the Department of Chemistry, College of Liberal Arts and Sciences, University of Illinois at Chicago. Additional support was received from the Ara Parseghian Medical Research Foundation at Notre Dame and the National Institutes of Health (R01 NS065808 to ER Bongarzone). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 246(4926), 64–71 (1989). [DOI] [PubMed] [Google Scholar]; • The fundamental concept of ESI for biomolecules is decribed here.
- 2.Wilm M, Shevchenko A, Houthaeve T et al. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379(6564), 466–469 (1996). [DOI] [PubMed] [Google Scholar]; • The advantages of using nano-flow ESI are highlighted here.
- 3.Gonzalez Fernandez-Nino SM, Smith-Moritz AM, Chan LJ, Adams PD, Heazlewood JL, Petzold CJ. Standard flow liquid chromatography for shotgun proteomics in bioenergy research. Front. Bioeng. Biotechnol. 3, 44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kronstrand R, Forsman M, Roman M. Quantitative analysis of drugs in hair by UHPLC high resolution mass spectrometry. Forensic Sci. Int. 283, 9–15 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Åstrand A, Vikingsson S, Lindstedt D et al. Metabolism study for CUMYL-4CN-BINACA in human hepatocytes and authentic urine specimens: free cyanide is formed during the main metabolic pathway. Drug Test. Anal. (2018) (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 6.Wohlfarth A, Vikingsson S, Roman M et al. Looking at flubromazolam metabolism from four different angles: metabolite profiling in human liver microsomes, human hepatocytes, mice and authentic human urine samples with liquid chromatography high-resolution mass spectrometry. Forensic Sci. Int. 274, 55–63 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Rashid R, Cazenave-Gassiot A, Gao IH et al. Comprehensive analysis of phospholipids and glycolipids in the opportunistic pathogen Enterococcus faecalis. PLoS ONE 12(4), e0175886 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cajka T, Garay LA, Sitepu IR, Boundy-Mills KL, Fiehn O. Multiplatform mass spectrometry-based approach identifies extracellular glycolipids of the yeast Rhodotorula babjevae UCDFST 04–877. J. Nat. Products 79(10), 2580–2589 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Mordehai A, Fjeldsted J. Agilent jet stream thermal gradient focusing technology. Agielnt Technologies Technical Note (2009). www.agilent.com/cs/library/technicaloverviews/public/5990-3494en_lo%20CMS.pdf [Google Scholar]; •• The fundamental concept of the AJS-ESI source interface is highlighted.
- 10.Kelly RT, Tolmachev AV, Page JS, Tang K, Smith RD. The ion funnel: theory, implementations, and applications. Mass Spectrom. Rev. 29(2), 294–312 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The advantages of using ion focusing technology is higlighted here.
- 11.Ibrahim Y, Belov ME, Tolmachev AV, Prior DC, Smith RD. Ion funnel trap interface for orthogonal time-of-flight mass spectrometry. Analyt. Chem. 79(20), 7845–7852 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian WJ, Jacobs JM, Liu T, Camp DG 2nd, Smith RD. Advances and challenges in liquid chromatography–mass spectrometry-based proteomics profiling for clinical applications. Mol. Cell. Proteomics 5(10), 1727–1744 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanier MT, Millat G. Niemann–Pick disease type C. Clin. Genet. 64(4), 269–281 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Sturley SL, Patterson MC, Balch W, Liscum L. The pathophysiology and mechanisms of NP-C disease. Biochim. Biophys. Acta 1685(1-3), 83–87 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Ory DS. Niemann–Pick type C: a disorder of cellular cholesterol trafficking. Biochim. Biophys. Acta 1529(1–3), 331–339 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Kelly DA, Portmann B, Mowat AP, Sherlock S, Lake BD. Niemann–Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 123(2), 242–247 (1993). [DOI] [PubMed] [Google Scholar]
- 17.Cologna SM, Jiang XS, Backlund PS et al. Quantitative proteomic analysis of Niemann–Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS ONE 7(10), e47845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byun K, Kim J, Cho SY et al. Alteration of the glutamate and GABA transporters in the hippocampus of the Niemann–Pick disease, type C mouse using proteomic analysis. Proteomics 6(4), 1230–1236 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Pettazzoni M, Froissart R, Pagan C et al. LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: a novel tool for the screening of sphingolipidoses and Niemann–Pick type C disease. PLoS ONE 12(7), e0181700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuchar L, Sikora J, Gulinello ME et al. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann–Pick A/B and C diseases. Analyt. Biochem. 525, 73–77 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Jiang X, Sidhu R, Mydock-McGrane L et al. Development of a bile acid-based newborn screen for Niemann–Pick disease type C. Sci. Transl. Med. 8(337), 337ra–363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazzacuva F, Mills P, Mills K et al. Identification of novel bile acids as biomarkers for the early diagnosis of Niemann–Pick C disease. FEBS Lett. 590(11), 1651–1662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sidhu R, Jiang H, Farhat NY et al. A validated LC-MS/MS assay for quantification of 24(S)-hydroxycholesterol in plasma and cerebrospinal fluid. J. Lipid Res. 56(6), 1222–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kannenberg F, Nofer JR, Schulte E, Reunert J, Marquardt T, Fobker M. Determination of serum cholestane-3beta,5alpha,6beta-triol by gas chromatography–mass spectrometry for identification of Niemann–Pick type C (NPC) disease. J. Steroid Biochem. Mol. Biol. 169, 54–60 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Rauniyar N, Subramanian K, Lavallee-Adam M, Martinez-Bartolome S, Balch WE, Yates JR 3rd. Quantitative proteomics of human fibroblasts with I1061T mutation in Niemann–Pick C1 (NPC1) protein provides insights into the disease pathogenesis. Mol. Cell. Proteomics 14(7), 1734–1749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian K, Rauniyar N, Lavallee-Adam M, Yates JR 3rd, Balch WE. Quantitative analysis of the proteome response to the histone deacetylase inhibitor (HDACi) vorinostat in Niemann–Pick Type C1 disease. Mol. Cell. Proteomics 16(11), 1938–1957 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ribas GS, Pires R, Coelho JC et al. Oxidative stress in Niemann–Pick type C patients: a protective role of N-butyl-deoxynojirimycin therapy. Int. J. Develop. Neurosci. 30(6), 439–444 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Ribas GS, Souza HM, De Mari J et al. Selective screening of Niemann–Pick type C Brazilian patients by cholestane-3beta,5alpha,6beta-triol and chitotriosidase measurements followed by filipin staining and NPC1/NPC2 gene analysis. Clin. Chim. Acta 459, 57–62 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Hammerschmidt TG, De Oliveira Schmitt Ribas G, Saraiva-Pereira ML et al. Molecular and biochemical biomarkers for diagnosis and therapy monitorization of Niemann–Pick type C patients. Int. J. Develop. Neurosci. 66, 18–23 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Cluzeau CV, Watkins-Chow DE, Fu R et al. Microarray expression analysis and identification of serum biomarkers for Niemann–Pick disease, type C1. Hum. Mol. Genet. 21(16), 3632–3646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicoli ER, Al Eisa N, Cluzeau CV et al. Defective cytochromeP450-catalysed drug metabolism in Niemann–Pick Type C disease. PLoS ONE 11(3), e0152007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan M, Sidhu R, Fujiwara H et al. Identification of Niemann–Pick C1 disease biomarkers through sphingolipid profiling. J. Lipid Res. 54(10), 2800–2814 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldin E, Roff CF, Miller SP et al. Type C Niemann–Pick disease: a murine model of the lysosomal cholesterol lipidosis accumulates sphingosine and sphinganine in liver. Biochim. Biophys. Acta 1127(3), 303–311 (1992). [DOI] [PubMed] [Google Scholar]
- 34.Wisniewski JR. Quantitative evaluation of filter aided sample preparation (FASP) and multienzyme digestion FASP protocols. Analyt. Chem. 88(10), 5438–5443 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Tobias F, Olson MT, Cologna SM. Mass spectrometry imaging of lipids: untargeted consensus spectra reveal spatial distributions in Niemann–Pick disease type C1. J. Lipid Res. 59(12), 2446–2455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szklarczyk D, Morris JH, Cook H et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45(D1), D362–d368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9(7), 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sestak J, Moravcova D, Kahle V. Instrument platforms for nano liquid chromatography. J. Chromatogr. A 1421, 2–17 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Pergande MR, Nguyen TTA, Haney-Ball C, Davidson CD, Cologna SM. Quantitative, label-free proteomics in the symptomatic Niemann–Pick, Type C1 mouse model using standard flow liquid chromatography and thermal focusing electrospray ionization. Proteomics 19(9), 1800432 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Megger DA, Pott LL, Ahrens M et al. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim. Biophys. Acta 1844(5), 967–976 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Lange Y, Ye J, Steck TL. Circulation of cholesterol between lysosomes and the plasma membrane. J. Biol. Chem. 273(30), 18915–18922 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Kamensky E, Philippart M, Cancilla P, Frommes SP. Cultured skin fibroblasts in storage disorders. An analysis of ultrastructural features. Am. J. Pathol. 73(1), 59–80 (1973). [PMC free article] [PubMed] [Google Scholar]
- 43.Te Vruchte D, Speak AO, Wallom KL et al. Relative acidic compartment volume as a lysosomal storage disorder-associated biomarker. J. Clin. Investig. 124(3), 1320–1328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kosicek M, Gudelj I, Horvatic A et al. N-glycome of the lysosomal glycocalyx is altered in Niemann–Pick Type C disease model cells. Mol. Cell. Proteomics 17(4), 631–642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appelqvist H, Nilsson C, Garner B, Brown AJ, Kagedal K, Ollinger K. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am. J. Pathol. 178(2), 629–639 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coutinho MF, Prata MJ, Alves S. A shortcut to the lysosome: the mannose-6-phosphate-independent pathway. Mol. Genet. Metabolism 107(3), 257–266 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez A, Valeiras M, Sidransky E, Tayebi N. Lysosomal integral membrane protein-2: a new player in lysosome-related pathology. Mol. Genet. Metabolism 111(2), 84–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dibbens L, Schwake M, Saftig P, Rubboli G. SCARB2/LIMP2 deficiency in action myoclonus-renal failure syndrome. Epileptic Disord. 18(S2), 63–72 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Liou B, Haffey WD, Greis KD, Grabowski GA. The LIMP-2/SCARB2 binding motif on acid beta-glucosidase: basic and applied implications for Gaucher disease and associated neurodegenerative diseases. J. Biol. Chem. 289(43), 30063–30074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuronita T, Eskelinen EL, Fujita H, Saftig P, Himeno M, Tanaka Y. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J. Cell Sci. 115(Pt 21), 4117–4131 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Salvioli R, Scarpa S, Ciaffoni F et al. Glucosylceramidase mass and subcellular localization are modulated by cholesterol in Niemann–Pick disease type C. J. Biol. Chem. 279(17), 17674–17680 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Reczek D, Schwake M, Schroder J et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131(4), 770–783 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Lloyd-Evans E, Morgan AJ, He X et al. Niemann–Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14(11), 1247–1255 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Smith AC, Heo WD, Braun V et al. A network of Rab GTPases controls phagosome maturation and is modulated by Salmonella enterica serovar Typhimurium. J. Cell Biol. 176(3), 263–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Desjardins M, Celis JE, Van Meer G et al. Molecular characterization of phagosomes. J. Biol. Chem. 269(51), 32194–32200 (1994). [PubMed] [Google Scholar]
- 56.Desjardins M, Huber LA, Parton RG, Griffiths G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124(5), 677–688 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bucci C, Parton RG, Mather IH et al. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70(5), 715–728 (1992). [DOI] [PubMed] [Google Scholar]
- 58.Langemeyer L, Frohlich F, Ungermann C. Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. 28(11), 957–970 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122(5), 735–749 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Goyette G, Boulais J, Carruthers NJ et al. Proteomic characterization of phagosomal membrane microdomains during phagolysosome biogenesis and evolution. Mol. Cell. Proteomics 11(11), 1365–1377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.