

Abstract
We have measured and analyzed the first combined 151Eu and 57Fe nuclear resonant vibrational spectroscopy (NRVS) for naturally abundant KEu(III)[Fe(II)(CN)6] and Eu(III)[Fe(III)(CN)6] complexes. Comparison of the observed 151Eu vs. 57Fe NRVS spectroscopic features confirms that Eu(III) in both KEu(III)[Fe(II)(CN)6] and Eu(III)[Fe(III)(CN)6] occupies the position outside the [Fe(CN)6] core and coordinates to the N atoms of the CN- ions, whereas Fe(III) or Fe(II) occupies the site inside the [Fe(CN)6]4- core and coordinates to the C atoms of the CN- ions. In addition to the spectroscopic interests, the results from this study provides invaluable insights for the design and evaluation of the nanoparticles of such complexes as potential cellular contrast agents for their use in magnetic resonance imaging. The combined 151Eu and 57Fe NRVS measurement is also among the few first expedition on bi-isotopic NRVS experiments.
Graphical Abstract
1. Introduction
Magnetic resonance imaging (MRI) has become a prominent non-invasive tool for the diagnosis of various diseases 1. Frequently, a contrast enhancement agent is required for obtaining sufficient image contrast in the clinical diagnosis 1–4. One of the most popular commercial MRI agents in use today is Gd-DTPA (Magnevist®, Schering AG, Germany), which contains a single paramagnetic gadolinium (III) (Gd3+) ion coordinated by the octadentate ligand and a water molecule [Figure 1(a)] 5. Although the complex has a high formation constant, even a trace amount of dissociated Gd(III) 5 can disrupt the normal function of calcium ion channels, as Gd3+ and Ca2+ have a similar ionic radius. In extreme cases, disruption of calcium channels by Gd (III) can lead to nephrogenic systemic fibrosis (NSF) 6, 7 8, 9, a serious and sometimes fatal disease. Therefore, use of Gd-DTPA and other Gd-based complexes as MRI contrast agents is considered harmful in general 6, 7, 10 8, 9, and the development of new-generation MRI contrast agents remains an unmet medical need.
Figure 1.

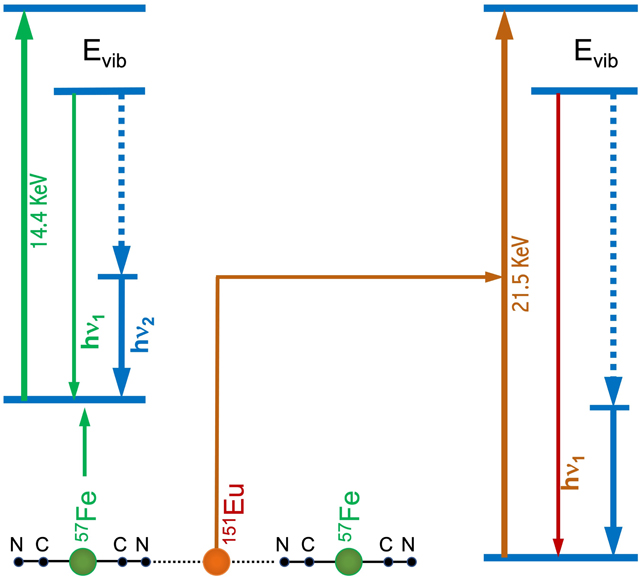
(a) Molecular structure of the commercial MRI contrast agent Gd-DTPA; (b) Unit-cell structure of the prototypical PB; (c) Comparison of electronic spin centres in a mononuclear metal complex vs. a nanoparticulated cluster with a single magnetic domain; (d) simplified one-dimensional diagram of KEu(III)Fe(II)(CN)6 and Eu(III)Fe(III)(CN)6; (e, f) nuclear resonant vibrational spectroscopic transitions of 57Fe (e) and 151Eu (f).
The toxic sides effects of these paramagnetic ions (e.g. Gd or Eu) released from their complexes into the human bloodstream can be addressed via the incorporation of such metal ions into the crystal lattice of Prussian blue (PB) to form water-dispersible nanoparticles 11, 12. The prototypical PB with the chemical formula of Fe(III)4[Fe(II)(CN)6]3.xH2O is a dark blue pigment that is currently listed as one of the essential medicines by the World Health Organization 13. In the structure of PB, the Fe(II)(CN)6 core has a low-spin (ls) Fe(II) (S=0) coordinated to the C atoms of the six CN- ligands (i.e., the inner Fe(II) hereafter). Notably, PB also has a high-spin (hs) Fe(III) (S=5/2) located outside the core that is coordinated to the N atoms of six CN- ligands (i.e., the outer Fe(III) hereafter). Together, the inner and outer Fe centres along with the CN- ligands form the three-dimensional coordination network structure as shown in Figure 1(b). Please note that the inner vs. outer positions are in reference of the core [Fe(CN)6] structure. For example, the inner Fe(II)s are illustrated with grey color while the outer Fe(III)s are illustrated with dark red in Figure 1(b). The inner Fe(II) and outer Fe(III) occupy alternate points of a cubic lattice, while the CN- ion is in the connecting line to link two different Fe sites. In order to maintain electroneutrality, an Fe(III):[Fe(II)(CN)6]4- ratio of 4:3 is required. As a result, a quarter of [Fe(II)(CN)6]4- units must be missing from the structure with the vacancies filled by H2O [Figure 1(b)].
In the PB analogues 14–16, the inner Fe(II) inside the Fe(II)(CN)6 core can be substituted by a different transition metal ion to form M(CN)6 (e.g. M = Co or Mn). On the other hand, the outer Fe(III) can be substituted by many different metal ions from the main group, d-block and f-clock as long as the 1:1 ratio of M to Fe is maintained. Additional monovalent ions, such as Li+, Na+, K+ Rb+, Cs+, and Tl+, are sometimes required to balance the charge. For example, in KEu(III)Fe(II)(CN)6, one K+ ion is necessary to balance the residual −1 charge for KEu(III)Fe(II)(CN)6, while there is no K+ ion in Eu(III)Fe(III)(CN)6.
It is intuitive to suggest that the strong inorganic core of [Fe(II)(CN)6]4-, provided from the starting material K4[Fe(II)(CN)6], can retain its original Fe(II) (S=0) state, while it is possible to tune the total spin of each PB-like complex via varying the electronic spin of the “introduced” metal. Suitable metal candidates for forming potential MRI agents must have a high number of unpaired electrons, a long electronic relaxation time, and a stable oxidation state under physiological conditions concomitantly. Some of such candidates include Gd(III) (S=7/2), Eu(II) (S=7/2), Eu(III) (S=3), Mn(II) (S=5/2), Fe(II) (S= 3) and Fe(III) (S=5/2), with Eu(II) (S=7/2) or its counterpart Eu(III) (S=3) being the closest to Gd(III) (S=7/2).
For example, KEu(III)[Fe(II)(CN)6] has a total spin of S=3 because the [Fe(II)(CN)6]4- core has a total spin of S=0 and the “added” Eu(III) has a total spin of S=3. For similar reasons, the ls-Fe(III) in [Fe(III)(CN)6]3- has a spin of S=1/2, whereas the whole molecule of Eu(III)[Fe(III)(CN)6], has a total spin of S=7/2 because the magnetic coupling does not readily occur between an f-block metal ions and a transition metal ion. Then the Eu(S=3) and Fe(S=1/2) are paramagnetically aligned to form a S=7/2 in a magnetic field at the human body temperature (37Co). In addition, nanoparticulated materials of PB can be best viewed as multinuclear clusters, and thus each superparamagnetic nanoparticle (i.e., a single magnetic domain) has a much higher total spin than the chemical formula suggests, as illustrated in Figure 1(c). Meanwhile, such nanoparticles are small enough to be transported within human blood vessels to be delivered to the intended organ for the diagnostic applications. Consequently, these nanoparticulated complexes are potential candidates for the next-generation of MRI contrast agents as their PB-like structure 11, 12 is extremely stable and their total electronic spin per magnetic domain are high. Although it seems obvious with theoretical intuition, use of modern X-ray spectroscopies 17–27 to clarify whether the “introduced” Eu(III) takes the outer Fe(III) position or the inner Fe(II) position and to confirm their oxidation states remains an important but unfulfilled task.
Nuclear resonant vibrational spectroscopy (NRVS) is a synchrotron radiation (SR)-based modern X-ray spectroscopic technique that measures the vibrational modes associated with Mössbauer-active nuclei 17–19, 28. In comparison with conventional vibrational spectroscopies, such as infrared absorption spectroscopy 29, Raman spectroscopy 29 or laser induced fluorescence spectroscopy 30 31 32, 33, NRVS has several distinct advantages. The most prominent advantages include (but are not limited to) being isotope-specific for studying complicated systems 20–24, 34, having almost zero background 22, 23, 28, and having a partial vibrational density of states (PVDOS) that are deductible from the measured raw NRVS spectra 17–20, 28, 35. This modern X-ray spectroscopic technique has become more widely available since the mid-1990s due to the development of third-generation SR sources, advanced X-ray optics, and modern detectors. Overall, the development has made NRVS a valuable tool in physics, geophysics, chemistry, biochemistry, and materials science, for more than 25 years 17–24. It becomes the third most popular modern X-ray method for biochemical research, following X-ray crystallography and X-ray absorption spectroscopy [for example, extended X-ray absorption fine structures (EXAFS) 27].
Since the nuclear scattering spectroscopy (including NRVS) is isotope-specific, and therefore site-specific 28 36 23, it can be used to survey specific metal sites within the same system. For NRVS, one pioneering example is the 57Fe/125Te NRVS for a 125Te-modified iron-sulphur complex - (Et4N)3[57Fe4125Te4(SPh)4], which was published in 2019 37. In addition to highlighting the potential of sulfur/tellurium exchange as a method to isolate the iron-only motion in enzymatic systems, this study revealed and well-demonstrated the differences and the correlation between 57Fe and 125Te NRVS profiles – providing insights from two points of view. In another example, a combined bi-isotopic (163Dy and 57Fe) NRVS on DyFe2 material 38 was successfully measured at APS 03ID. However, no measurement on unenriched chemical complexes has been reported thus far.
Figure 1(d) provides a simplified one-dimensional “structure” for KEu(III)[Fe(II)(CN)6] or Eu(III)[Fe(III)(CN)6], in which the Fe and Eu can be studied alternately with 57Fe and 151Eu NRVS. The energy diagrams for 57Fe and the 151Eu NRVS transitions are shown in Figure 1(e) and Figure 1(f), respectively. The 57Fe NRVS is the most frequently studied NRVS. It has been utilized to reveal Fe-S/P/Cl 20, 21, 39, 40, Fe-CO/CN/NO 22, 24, 41 42 and Fe-H/D 22, 23 vibrational modes inside various chemical complexes and enzymes. Nevertheless, NRVS with other isotopes 28 is also possible as long as their nuclear transitions have “low” transition energies and “long” excited state lifetimes 28, 36. For example, the isotope 151Eu 43 has a nuclear transition energy of 21.5 keV, that can be obtained using a third generation SR source, and an excited state lifetime of 13.8 ns that is long enough for detection with avalanche photodiode (APD) detectors (which have a time resolution ~ 1 ns). Another preferred feature of 151Eu is that it has a natural abundance of 48%, making enrichment with 151Eu unnecessary during sample preparation for most cases. In comparison, 57Fe only has a natural abundance of 2%. As a result, 151Eu is one of the most active isotopes researched with NRVS and other synchrotron-based nuclear scattering techniques in recent years for a variety of applications ranging from simple complexes to high pressure systems 44–51.
For the two complexes used in this study, it is important to compare the information obtained via 57Fe and 151Eu NRVS to understand, for example, whether the “introduced” Eu(III) occupies the outer Fe(III) site or the inner Fe(II) site in the PB-like structure, and to observe any subtle differences in the complexes containing either Fe(II) or Fe(III). These issues are the central tasks of this publication.
2. Experimental Aspects
2.1. Sample preparation and characterization
Samples of KEu(III)[Fe(II)(CN)6] (1), Eu(III)[Fe(III)(CN)6] (2), and K57Fe(II)[Co(III)(CN)6] (3) were prepared at Kent State University with a standard procedure similar to the one previously published 52. Samples of 1 and 2 were not enriched with either 57Fe or 151Eu. Sample of 3 was enriched with 57Fe.
The following samples were prepared or purchased and studied as references for comparison. The 57Fe-enriched PB (4) and Fe2O3 (6) were purchased from Isoflex, USA and used without further purification. (NH4)2Mg(II)[57Fe(II)(CN)6] (5) was the calibration standard normally used at ESRF ID18. Aqueous solutions of K4[Fe(II)(CN)6] (7) and K3[Fe(III)(CN)6] (8) were prepared at 680 mM (corresponding to ~14 mM 57Fe by assuming a natural abundance of 2%) and 1 M (~20 mM 57Fe), respectively, at The University of Alabama. The Eu(III)2O3 (99.99% in purity but non-enriched) (9) was purchased from Sigma Aldrich. All the samples 1–6 and 9 were delivered in solid state (powders) while the samples 7 and 8 were prepared as water solution.
2.2. 57Fe NRVS measurements
57Fe NRVS spectra were recorded using a published procedure 20, 21, 28, 39, 41, 53 26, 54 at SPring-8 BL09XU 55 (for samples 1-3) / BL19LXU 56 (for 7, 8), APS 03ID 36 (for 4 and 6) and ESRF ID18 19 (for 5). A high heat load monochromator (HHLM) produced 14.4 keV radiation with ~1.0 eV energy resolution, and a high energy resolution monochromator (HRM) for further monochromatizing the beam bandwidth to 0.8 meV (SPring-8 BL09XU and BL19LXU), 1.0 meV (APS 03ID) or 0.6 meV (ESRF ID18 at time of our measurement). During the NRVS measurements, the samples were maintained at a cryogenic temperature using a liquid helium (LHe) cryostat maintained at 10K. However, the real sample temperatures 57 were derived from the anti-Stoke/Stoke intensity ratios and often fell in the range of 40–80K depending on the measurements and the measuring locations. As illustrated in Figure 1(e), while an incident X-ray beam (green) scans through an interested energy region to cover the nuclear transition (e.g., 14.4 keV for 57Fe) and the associated vibrations (e.g. Fe-CN), the extremely narrow linewidth for the nuclear fluorescence (hν1) can be used as an excellent intrinsic “spectrometer” to “define” the scattered energy with precision. NRVS thus does not need a low-throughput spectrometer for filtering the electronic scattering beam and has a much higher photon in and photon out efficiency, e.g.in comparison with inelastic X-ray scattering (IXS) 28 58.
The total intensities collected from both the direct nuclear fluorescence at hν1 and the internally converted electron K shell fluorescence at hν2 were recorded with a single element APD detector and a dome-shaped vacuum window (at APS or ESRF) or a 2×2 APD array and a flat vacuum window (at SPring-8) 28. A detector with a dome-shaped window is easier to optimize the measurement geometry while an APD array has a higher limit for the detection saturation. The delayed (nuclear) scattering signal vs. the incident X-ray energy forms a raw NRVS spectrum that can be further processed via PHOENIX software package or a web tool at spectra.tools (a variant form of using PHOENIX) to obtain a partial vibrational density of state (PVDOS) vs. the vibrational energy (Evib) 28, 35, 59. The detailed information about NRVS instruments, measurement and analysis were the same as those published previously 17–24, 28, 35.
Samples of 1 and 2, both are not enriched with 57Fe, have very weak signals (2% vs. 100% for the enriched samples) in their NRVS spectra, thereby precluding a full NRVS scan. Instead, selected narrow regions were measured around the nuclear resonant peak (±40 cm−1 or ±5 meV) and around the possible peaks for [Fe(II)(CN)6]4- and [Fe(III)(CN)6]3- (380–700 cm−1). We therefore were unable to convert such measured NRVS spectra for 1 and 2 to PVDOS because the full scans were not obtained. Instead, their absorption peaks in the raw NRVS were used in the analysis or presentations.
2.3. 151Eu NRVS measurements
151Eu NRVS spectra (for 1, 2, 9) were recorded using a published procedure at APS 03ID 44–47, 51. A HHLM produced 21.5 keV radiation with ~1.0 eV energy resolution, and a HRM with a “nested” Si(4,4,0)-Si(15,11,3)x2-Si(4,4,0) structure subsequently produced 21.5 keV radiation with 1.0 meV energy resolution, perfect for measuring vibrations 60. The beam flux was ~0.7×109 photons s−1 60. During NRVS measurements, the samples were maintained at a cryogenic temperature using a LHe cryostat maintained at 10K. The analyzed real sample temperature was about 80K. The total intensities collected from both the direct nuclear fluorescence at hν1 and the internally converted electron L shell fluorescence at hν2 [as shown in Figure 1(f)] were recorded with a single-element APD detector with a dome-shaped vacuum window. As for 57Fe NRVS, a dome shaped beryllium window was used to separate the sample at the cryogenic temperature and the APD at room temperature (RT) and to allow for a free adjustment of the detection angle of the APD detector. The delayed (nuclear) scattering signal vs. the incident X-ray energy forms a raw NRVS spectrum, which was then transferred to PVDOS vs. the vibrational energy (Evib) with the PHOENIX software at the beamline 28, 35. The detailed information about 151Eu NRVS instruments, measurement and analysis were the same as those published widely for other 151Eu measurements 44–50.
3. Results and discussion, Experimental
3.1. 57Fe NRVS results
Similar to other vibrational spectroscopies 24, 26, 30, 32, 61, 62, NRVS is sensitive to the oxidation states and the bonding environment of the isotopes measured. For example, NRVS has been widely used to assign the oxidation states for 57Fe inside various complexes and enzymes 22–24, 28, 34, 39–41, 63, 64. It has also provided a wealth of information regarding the ligands and coordination symmetry around the 57Fe site(s) in these samples.
Due to its distinct Fe(II, S=0) and Fe(III, S=5/2) sites, PB serves as one classical example for the evaluation of new high resolution spectroscopies. For example, almost 20 years ago, Glatzel et al. published an article 65 demonstrating site-selective EXAFS using high-resolution Kβ fluorescence detection. The Kβ fluorescence lines arising from the ls-Fe(II) and hs-Fe(III) sites have slight differences in energy that can be resolved using a 1 eV resolution energy analyser. Fluorescence-detected absorption spectra using different emission energies represent different linear combinations of the pure ls-Fe(II) and hs-Fe(III) EXAFS. A numerical method was used to extract the EXAFS spectrum from the raw experimental data for each individual site.
For the prototypical PB sample, NRVS is also capable of indicating whether a particular NRVS feature is from the [Fe(II)(CN)6]4- core or from the loose interaction between the Fe(III) site and the [Fe(II)(CN)6]4- core 28. Natural PB (4) has two major peaks [the black curve in Figure 2(a)]: a sharp peak at 594 cm−1, arising from strong interactions between the ls-Fe(II) ion and the six (CN)6 ligands, and a broad hump between 120 – 280 cm-1. Please note that the sharp peak position was mentioned at 602 cm−1 in several previous publications 28, 66, 67. The NRVS for PB and all associated samples were “calibrated” using the 602 cm−1 peak position 67 measured for (NH4)2Mg(II)[Fe(II)(CN)6] at ESRF ID18. According to our calibration procedure, assigning the Fe-Cl stretching peak from (Et4N)[FeCl4] as 380 cm−1 (IR peak position) 28 68 20 36, the scaling factor for ESRF ID18 is 0.986, which shifts the peak at 602 cm−1 to 594 cm-1. Although we acknowledge that there is no absolute standard and we do not intend to calibrate the standards used by other researchers, it is justifiable to calibrate energies within one research group with one standard. We therefore “re-calibrate” the sharp peak position in 4 and 5 to 594 cm−1 and will use this assignment for the future publications from this research team.
Figure 2.

(a) NRVS spectra for (NH4)2Mg(II)[57Fe(II)(CN)6] (5) (green), K57Fe(II)[Co(III)(CN)6] (3) (purple) and 57Fe(III)4[57Fe(II)(CN)6]3.xH2O (PB) (4) (black); (b) NRVS spectra for K3[Fe(III)(CN)6] (8) (red) and the Fe(II)-CN NRVS peak position for K4[Fe(II)(CN)6] (7) (green bar): the NRVS spectrum for 7 is shown as in Figure S1; (c) partial NRVS spectra for Eu(III)[Fe(III)(CN)6] (2) (red) and KEu(III)[Fe(II)(CN)6] (1) (green) in the characteristic Fe-CN peaks region.
To further illustrate the origins of the sharp peak at 594 cm−1 and the hump between 120–280 cm−1, the inner ls-Fe(II) or the outer hs-Fe(III) site in PB can be “substituted” with another metal, leaving only one site labelled with 57Fe. This approach was realized via synthesizing two site–specific complexes. The first complex is K57Fe(II)[Co(III)(CN)6] (3), which has a [Co(III)(CN)6]3- core and an “outer” 57Fe(II) loosely bound to the [Co(III)(CN)6]3- core. This complex lacks the sharp peak at 594 cm−1 observed for the natural PB and only has the lower energy hump at 120 – 280 cm−1 [purple curve in Figure 2(a)]. The second complex is (NH4)2Mg(II)[Fe(II)(CN)6] (5) 67, which has an “outer” Mg ion loosely bound to the [57Fe(II)(CN)6]4- core. Notably, this complex has one strong NRVS peak at 594 cm−1 [green curve in Figure 2(a)], but it lacks intensity in the lower energy region. The profound difference among samples 3, 4, and 5 [Figure 2(a), purple, black and green curves] demonstrates: 1) the hump between 120 – 280 cm−1 is due to the loose interaction between the core and the outer iron; 2) the sharp peak at 594 cm−1 is from the Fe-CN interaction inside the [57Fe(II)(CN)6]4- core; and 3) the weak “very low energy” features in 5 might be due to a large-scale motion – the lattice phonons inside the structure. 57Fe-labelled PB 28 has a hump at 120 – 280 cm−1 region and a sharp peak at 594 cm−1, overlapping with the above two peaks 28.
Our NRVS spectra in Figure 2(a) have well-illustrated the specific feature which is attributable to the specific iron site – i.e., the ls-Fe(II) inside the [Fe(II)(CN)6]4- core leads to a sharp peak at 594 cm−1 (green), whereas the hs-Fe(III) outside the [Fe(II)(CN)6] core produces a hump at 120 – 280 cm-1. Since no PB or PB analogues with an [Fe(III)(CN)6]3- core have been synthesized, two more comparison samples were prepared and measured. Figure 2(b) illustrates the NRVS spectrum for the natural abundance K3[Fe(III)(CN)6] (8) (red) with a green bar to indicate the sharp peak position for the natural abundance K4[Fe(II)(CN)6] (7). Figure S1 further illustrates the differences between NRVS collected for 7 (green) and 8 (red). The characteristic peak for the K4[Fe(II)(CN)6] is at 593 cm−1 (Figure S1), similar to that observed for PB (4) and (NH4)2Mg(II)[Fe(II)(CN)6] (5) (at 594 cm−1). On the other hand, the characteristic peak from the K3[Fe(III)(CN)6] is at 516 cm−1, 78 cm−1 lower than that for 4, 5, 7.
The down shift in peak position for the oxidized form is actually common in other CN or CO containing structures due to strong back bonding interactions. For example, in the NRVS spectra for hydrogenase from Desulfovibrio vulgaris Miyazaki F (DvMF), the Fe-CO vibrations in the oxidized sample were found to shift to lower energy positions rather than shift to the higher energy positions 24. The authors of that work explained that the trend is consistent with the well-known inverse correlation between Fe-C and C-O in FeCO complexes 29, 69–71. In a different example, Spiro found that the Fe-CO frequency for myoglobin-CO increases by a factor of 0.75 times the magnitude of the decrease of νCO 69. The Fe(III)-CN also has a wider peak in comparison with Fe(II)-CN. Although the mechanism behind is not clear, Figure 2(a and b) [as well as Figure S1] establishes the NRVS standard for [Fe(III)(CN)6]4- vs. [Fe(II)(CN)6]3- cores for our next work.
Since complexes of KEu(III)[Fe(II)(CN)6] (1) and Eu(III)[Fe(III)(CN)6] (2) are not enriched with 57Fe, their NRVS have very weak signals (2% vs. 100% for the enriched samples). After evaluating several 57Fe enriched PB and other comparison samples, we decided to scan narrow regions around the nuclear resonant peak at Evib=0 and around the characteristic regions in 380–700 cm−1 where the possible characteristic peaks for [Fe(II)(CN)6]4- and [Fe(III)(CN)6]3- exist. The peak positions for both 1 and 2 are slightly higher than their corresponding reference counterparts (594 cm−1 in 4, 5 as a reference for 1 and 516 cm−1 in 8 for 2). This can be understood with the concept proposed in a recent manuscript about NRVS energy re-calibration 72. In a sectional scan or a skip scan, the energy should be calibrated point by point according to their accumulated scanning time, rather than scaled with a universal factor 72. When a universal scaling factor is used to calibrate the energy axis for a sectional (skip) scan, which takes very different time at different data point, the energy axis can be over-scaled, resulting in a slightly higher energy positions for the characteristic peaks 72.
Although the exact peak position cannot be accurately determined due to the skip scan operation, the obtained peak features are sharp and clear as shown in Figure 2(c). The peak position for 2 is at a lower energy position and with a wider width in comparison with those for 1. These features are consistent with the observation for their reference samples 8 [in Figure 2(b)] and vs. 4,5 [Figure 2(a)] or 7 [Figure 2(b) mark]. This observation shows that 1 has an ls-Fe(II) [Figure 2(c) (green) vs. Figure 2(a) (green)], 2 has an ls-Fe(III) [Figure 2(c) (red) vs. Figure 2(b) (red)] and both contain the inner Fe centres inside the [Fe(CN)6] cores. In addition, the difference between the peak centroids for 1 and 2 (80 cm−1) is almost the same as the difference between their reference samples (78 cm−1 between 7 and 8).
3.2. 151Eu NRVS results
The discussion in Sec. 3.1 illustrates the existence of Fe(II) or Fe(III) inside the [Fe(CN)6] cores for KEu(III)[Fe(II)(CN)6] or Eu(III)[Fe(II)(CN)6]. However, one may still be unable to rule out the possibility of Fe ions scrambling between the [Fe(CN)6] core and the “outer” Fe(III) site in the prototypical PB. Since our elemental analysis confirms the 1:1 ratio of Fe:Eu, all Fe ions should be found in the [Fe(CN)6] core if the Eu is completely confined in the loosely bound location of Eu…[Fe(CN)6] (Figure 1(d)). Therefore, the feature and the peak location information obtained from the 151Eu NRVS can provide additional insights into the information about the Fe locations.
The 151Eu NRVS [the NRVS derived PVDOS] for complexes KEu(III)[Fe(II)(CN)6] (1, blue) and Eu(III)[Fe(III)(CN)6] (2, red) are shown in Figure 3(a). Intuitively, if Eu(III) takes the Fe position inside the [Fe(CN)6] core, it should have a similar (sharp) peak but the heavier relative mass should locate it at a lower energy position, assuming both have the same interaction force constant. As for the degree of such shift, it depends on how the vibrator is composed, which will be discussed in detail in the next paragraph. Since we did not observe any sharp peak in the 151Eu NRVS, the existence of a possible [Eu(CN)6] core can be essentially ruled out, leaving all of the core “locations” for Fe.
Figure 3.

(a) 151Eu NRVS for complexes KEu(III)[Fe(II)(CN)6] (1) (blue) and Eu(III)[Fe(III)(CN)6] (2) (red); (b) 151Eu NRVS for Eu2O3 (9).
According to previous analysis of the [Fe(II)(CN)6] vibrational modes 67, the sharp peak at 594 cm−1 belongs to two degenerate stretching modes (μ4), which are illustrated in Figure 4(a and b) or Figure S2. Inside a [Fe(CN)6] core, (CN)6 can be considered as six point charges around the Fe ion in an octahedral arrangement, with each CN acting as one point charge of −1. In vibrational mode (a) 67, the two axial CNs move along (in phase) with the Fe’s motion, while the four equatorial CNs move opposite to (out of phase with) the Fe’s motion. We can further simplify this model into a “diatomic” stretching vibrator X—Y, where X = Fe + 2CN and Y =4CN. In (b) 67, the four equatorial CNs are in phase with Fe motion, while the two axial CNs are out of phase with Fe motion, resulting in the X—Y model of X = Fe + 4CN and Y = 2CN. Per the analysis in SI.2, the relative masses are different for the two modes, meaning their force constants must be different to maintain the degeneracy between them. Further details relating to this analysis are, however, out of the scope of this publication.
Figure 4.

The two degenerated stretching vibrational mode (μ4) for [M(CN)6]4-: M=metal.
The analysis in SI.2 also shows that the relative mass for the X-Y vibrator in modes (a) and (b) for the [57Fe(CN)6] is 53.2 and 39.3, respectively. For a conceptual [151Eu(CN)6], these relative masses increase to 68.8 (a) and 43.9 (b) respectively. Please note, although neither 1 or 2 is isotope-enriched, we still used 57Fe and 151Eu to calculate the relative masses since the NRVS signal derives solely from the naturally abundant 57Fe and 151Eu in each sample. With this analysis, when 57Fe inside [57Fe(II)(CN)6] is replaced with 151Eu to form [151Eu(CN)6], the resulting NRVS can have a peak at: 594·(53.2/68.8)1/2 = 594·0.879 = 522 cm−1 or 594·(39.3/43.9)1/2 = 594·0.945 = 561 cm−1 using a simplified calculation from the diatomic vibrator model X—Y. More realistically, starting from the [57Fe(III)(CN)6] peak at 516 cm−1, the [151Eu(CN)6] can have a peak at: 516·(53.2/68.8)1/2 = 516·0.879 = 454 cm−1 or 516·(39.3/43.9)1/2 = 594·0.945 = 487 cm-1. The overall possible region will therefore be between 454 cm−1 and 561 cm−1, as shown with the two vertical green bars in Figure 3(a). This calculation might not be entirely accurate, but it does provide a guideline to looking for a peak from [151Eu(CN)6]. Importantly, we did not observe any peak in this region, nor in an extended region between 300 and 700 cm-1. Therefore, we conclude that there be no [Eu(CN)6] structure in complexes 1 or 2 - the observation is consistent with chemical intuition.
On the other hand, if Eu occupies the “outer” Fe(III) location in PB, the 151Eu NRVS should have a hump in the region scaled by a factor of 0.713 as described in SI.2. There will be no difference between the modes (a) and (b) because the vibration discussed here is M…[(NC)6Fe] while M = Fe or Eu ( SI.2). This makes the search region from 85.6 to 199.6 cm−1 [120*0.713 = 85.6 and 280*0.713 = 199.6 cm−1] as illustrated in Figure 3(a) (horizontal green bar). The observed 151Eu NRVS for 1 and 2 have humps below 260 cm−1 which is qualitatively consistent with the above estimation. The mis-match between the observed and the estimated upper edges (260 vs. 199.6 cm−1) may indicate the estimation model is too simple or may suggest the large Eu mass bring in more distortion to the complexes (1 and 2). The lower limit for the hump, on the other hand, is hard to judge in the observed spectra because it may overlap with lattice-phonon features. However, the above estimated value (85.6 cm−1) is still reasonable.
For reference, 151Eu NRVS for Eu2O3 (9) [Figure 3(b)] and 57Fe NRVS for Fe2O3 (6) [Figure S3] were also measured and compared with their corresponding Eu (9 vs. 1, 2) and Fe NRVS in (6 vs. 1, 2, 3, 4) complexes.
3.3. Combined Discussion on 151Eu and 57Fe NRVS
All the results from our 151Eu NRVS show that the Eu(III) takes the outer hs-Fe(III) location in a PB structure. No spectral feature for a possible [Eu(CN)6] structure was found. Since Eu:Fe = 1:1, Fe then must all be inside the [Fe(CN)6] core. In other words, the [Fe(II)(CN)6]4- or [Fe(III)(CN)6]3- core is intact from the starting materials during the synthesis of either 1 or 2. This conclusion is confirmed with the existence of sharp 57Fe-CN peaks around 594 cm−1 (for 1) and a relative sharp peak 516 cm−1 (for 2).
The above conclusion clearly illustrates the power of using combined bi-isotope (e.g. 151Eu and 57Fe) NRVS to resolve practical issues. If 151Eu were not measured, and only 57Fe NRVS were measured in the narrow region around the sharp peak, it could have only confirmed that there is Fe inside the [Fe(II)(CN)6]4- or [Fe(III)(CN)6]3- core, and cannot rule out the situation that Fe may also exist outside the core. Since there is only 2% 57Fe in natural abundance, measuring full NRVS scan for the unenriched 1 and 2 will take a tremendous amount of beamtime. In addition, as 57Fe NRVS for unenriched samples has a low signal-to-noise ratio, it will be almost impossible to distinguish the subtle difference between the case with a partial outer Fe and the one without it at all (e.g. between samples 1 and 2, or unenriched versions of 5 and 4).
The 48% nature abundance and thus the available full scans for 151Eu NRVS provided a clear advantage in pinpointing the Eu is (only) at the outer ls-Fe(III) location in PB structure. This rules out a possible [Eu(CN)6] (or a possible Fe outside the core) since Eu: Fe = 1:1.
In addition, the minor but clear difference in the lowest energy peaks in the 151Eu NRVS between KEu(III)[Fe(II)(CN)6] (1) and Eu(III)[Fe(III)(CN)6] (2) shows even the skeleton structural motion (lattice phonons) can be affected by the oxidation states of the Fe ions (II vs. III) inside the [Fe(CN)6] core structure.
Summary
We have measured and analyzed the first combined 151Eu and 57Fe NRVS for naturally abundant KEu(III)[Fe(II)(CN)6] and Eu(III)[Fe(III)(CN)6]. All of the observed NRVS spectroscopic features confirm that Eu(III) in both 1 and 2 occupies the position outside the [Fe(CN)6] core and coordinates the N atoms of the CN- ions in Figure 1(d), whereas Fe(III) or Fe(II) occupies the site inside the [Fe(CN)6] core. This notion is also consistent with the stepwise synthetic approach employed for preparing both 1 and 2. Specifically, K4[Fe(II)(CN)6] and K3[Fe(III)(CN)6] are used as the complex ligands to coordinate to Eu(III) using the N-donor atoms from the six CN- molecules, while the C-bound Fe(II) or Fe(III) remains intact during the synthesis. In other words, Eu(III) takes the outer Fe(III) position only, while Fe(II) or Fe(III) takes the position inside the [Fe(CN)6]4- core.
In addition to the practical evaluation of complexes 1 and 2, this study well illustrates the many significant advantages of NRVS (especially the bi-isotope NRVS on one sample) in revealing site-specific information for nanoparticulated PB-like complexes, which may very well serve as prime candidates for the next-generation MRI agents.
In the future, it will be interesting to observe whether the so-called coordination isomers of 1 and 2 (with the formulas KFe(II)[Eu(III)(CN)6] and Fe(III)[Eu(III)(CN)6]) or certain types of mixtures where Eu(III) ions are partitioned between the two possible positions can be prepared if one uses a one-pot self-assembly synthesis involving the stoichiometric amounts of Fe2+ (or Fe3+), Eu3+, and CN-.
Supplementary Material
Acknowledgements
We thank Dr. Stephen P. Cramer at SETI Institute (USA) for his overall support for this project via US NIH grants GM-65440, beamtimes as well as other assistance, and Dr. Aleksandr I. Chumakov at ESRF for providing the NRVS spectrum of (NH4)2Mg(II)[57Fe(II)(CN)6] (5). 57Fe NRVS was measured at SPring-8 BL09XU and BL19LXU (via JASRI proposal 2021A1436, 2018B1379, RIKEN proposal 20210033), APS 03ID and ESRF ID18. 151Eu NRVS was measured at APS 03ID. APS is a user facility operated by Argonne National Laboratory for the U.S. Department of Energy Office of Science under Contract No. DE-AC02-06CH11357.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Bibliography
- 1.Geraldes CFGC and Laurent S, Contrast Media & Molecular Imaging, 2009, 4, 1–23. [DOI] [PubMed] [Google Scholar]
- 2.Wahsner J, Gale EM, Rodríguez-Rodríguez A. and Caravan P, Chemical Reviews, 2019, 119, 957–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao Y-D, Paudel R, Liu J, Ma C, Zhang Z-S and Zhou S-K, Int J Mol Med, 2016, 38, 1319–1326. [DOI] [PubMed] [Google Scholar]
- 4.Strijkers JG, Mulder JWM, van Tilborg AGF and Nicolay K, Anti-Cancer Agents in Medicinal Chemistry, 2007, 7, 291–305. [DOI] [PubMed] [Google Scholar]
- 5.Do C, DeAguero J, Brearley A, Trejo X, Howard T, Escobar GP and Wagner B, Kidney360, 2020, 1, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.High WA, Ayers RA, Chandler J, Zito G. and Cowper SE, Journal of the American Academy of Dermatology, 2007, 56, 21–26. [DOI] [PubMed] [Google Scholar]
- 7.Grobner T. and Prischl FC, Kidney International, 2007, 72, 260–264. [DOI] [PubMed] [Google Scholar]
- 8.Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S. and LeBoit PE, The Lancet, 2000, 356, 1000–1001. [DOI] [PubMed] [Google Scholar]
- 9.George SJ, Webb SM, Abraham JL and Cramer SP, British Journal of Dermatology, 2010, 163, 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.FDA, FDA Drug Safety Communication: FDA warns that gadolinium-based contrast agents (GBCAs) are retained in the body; requires new class warnings, https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-gadolinium-based-contrast-agents-gbcas-are-retained-body). [Google Scholar]
- 11.Shokouhimehr M, Soehnlen ES, Hao J, Griswold M, Flask C, Fan X, Basilion JP, Basu S. and Huang SD, Journal of Materials Chemistry, 2010, 20, 5251–5259. [Google Scholar]
- 12.Kandanapitiye MS, Valley B, Yang LD, Fry AM, Woodward PM and Huang SD, Inorganic Chemistry, 2013, 52, 2790–2792. [DOI] [PubMed] [Google Scholar]
- 13.Organization WH, Journal, 2019. [Google Scholar]
- 14.Chen J, Wei L, Mahmood A, Pei Z, Zhou Z, Chen X. and Chen Y, Energy Storage Materials, 2020, 25, 585–612. [Google Scholar]
- 15.Huang T, Du G, Qi Y, Li J, Zhong W, Yang Q, Zhang X. and Xu M, Inorganic Chemistry Frontiers, 2020, 7, 3938–3944. [Google Scholar]
- 16.Yi H, Qin R, Ding S, Wang Y, Li S, Zhao Q. and Pan F, Advanced Functional Materials, 2021, 31, 2006970. [Google Scholar]
- 17.Seto M, Yoda Y, Kikuta S, Zhang X. and Ando M, Physical Review Letters, 1995, 74, 3828–3831. [DOI] [PubMed] [Google Scholar]
- 18.Sturhahn W, Toellner TS, Alp EE, Zhang X, Ando M, Yoda Y, Kikuta S, Seto M, Kimball CW and Dabrowski B, Physical Review Letters, 1995, 74, 3832–3835. [DOI] [PubMed] [Google Scholar]
- 19.Chumakov AI, Rffer R, Grnsteudel HF, Grnsteudel HF, Gr¸bel G, Metge J, Leupold O. and Goodwin HA, EPL, 1995, 30, 427–432. [Google Scholar]
- 20.Xiao Y, Wang H, George SJ, Smith MC, Adams MW, Jenney FE Jr., Sturhahn W, Alp EE, Zhao J, Yoda Y, Dey A, Solomon EI and Cramer SP, Journal of the American Chemical Society, 2005, 127, 14596–14606. [DOI] [PubMed] [Google Scholar]
- 21.Xiao YM, Koutmos M, Case DA, Coucouvanis D, Wang HX and Cramer SP, Dalton Transactions, 2006, DOI: 10.1039/b513331a, 2192–2201. [DOI] [PubMed] [Google Scholar]
- 22.Ogata H, Kramer T, Wang H, Schilter D, Pelmenschikov V, van Gastel M, Neese F, Rauchfuss TB, Gee LB, Scott AD, Yoda Y, Tanaka Y, Lubitz W. and Cramer SP, Nat Commun, 2015, 6, 7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Yoda Y, Ogata H, Tanaka Y. and Lubitz W, Journal of Synchrotron Radiation, 2015, 22, 1334–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamali S, Wang H, Mitra D, Ogata H, Lubitz W, Manor BC, Rauchfuss TB, Byrne D, Bonnefoy V, Jenney FE, Adams MWW, Yoda Y, Alp E, Zhao J. and Cramer SP, Angew. Chem. Int. Ed, 2013, 52, 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeBeer George S, Metz M, Szilagyi RK, Wang H, Cramer SP, Lu Y, Tolman WB, Hedman B, Hodgson KO and Solomon EI, Journal of the American Chemical Society, 2001, 123, 5757–5767. [DOI] [PubMed] [Google Scholar]
- 26.Wang HX, Ge PH, Riordan CG, Brooker S, Woomer CG, Collins T, Melendres CA, Graudejus O, Bartlett N. and Cramer SP, Journal of Physical Chemistry B, 1998, 102, 8343–8346. [Google Scholar]
- 27.Gu WW, Jacquamet L, Patil DS, Wang HX, Evans DJ, Smith MC, Millar M, Koch S, Eichhorn DM, Latimer M. and Cramer SP, Journal of Inorganic Biochemistry, 2003, 93, 41–51. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Braun A, Cramer SP, Gee LB and Yoda Y, Crystals, 2021, 11, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamoto K, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th ed., Wiley-Interscience, New York, 1997. [Google Scholar]
- 30.Wang H, Salzberg AP and Weiner BR, Applied Physics Letters, 1991, 59, 935–937. [Google Scholar]
- 31.Wang H, Chen X. and Weiner BR, The Journal of Physical Chemistry, 1993, 97, 12260–12268. [Google Scholar]
- 32.He M, Wang H. and Weiner BR, Chemical Physics Letters, 1993, 204, 563–566. [Google Scholar]
- 33.Chen X, Asmar F, Wang H. and Weiner BR, The Journal of Physical Chemistry, 1991, 95, 6415–6417. [Google Scholar]
- 34.Scheidt WR, Li J. and Sage JT, Chemical Reviews, 2017, 117, 12532–12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sturhahn W, Hyperfine Interact., 2000, 125, 149–172. [Google Scholar]
- 36.Wang H, Alp E, Yoda Y. and Cramer S, in Metalloproteins, eds. Fontecilla-Camps JC and Nicolet Y, Humana Press, 2014, vol. 1122, ch. 9, pp. 125–137. [Google Scholar]
- 37.Wittkamp F, Mishra N, Wang H, Wille H-C, Steinbrügge R, Kaupp M, Cramer SP, Apfel U-P and Pelmenschikov V, Chemical Science, 2019, 10, 7535–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown DE, Toellner TS, Sturhahn W, Alp EE, Hu M, Kruk R, Rogacki K. and Canfield PC, Hyperfine Interact., 2004, 153, 17–24. [Google Scholar]
- 39.Xiao YM, Fisher K, Smith MC, Newton WE, Case DA, George SJ, Wang HX, Sturhahn W, Alp EE, Zhao JY, Yoda Y. and Cramer SP, Journal of the American Chemical Society, 2006, 128, 7608–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cramer SP, Xiao Y, Wang H, Guo Y. and Smith MC, Hyperfine Interact., 2006, 170, 47–54. [Google Scholar]
- 41.Tinberg CE, Tonzetich ZJ, Wang H, Do LH, Yoda Y, Cramer SP and Lippard SJ, Journal of the American Chemical Society, 2010, 132, 18168–18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birrell JA, Pelmenschikov V, Mishra N, Wang H, Yoda Y, Tamasaku K, Rauchfuss TB, Cramer SP, Lubitz W. and DeBeer S, Journal of the American Chemical Society, 2020, 142, 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmida M, J. Applied Mathematicsand Physics, 2019, 7, 254–270. [Google Scholar]
- 44.Kobayashi H, Yoda Y, Shirakawa M. and Ochiai A, Journal of the Physical Society of Japan, 2006, 75, 034602. [Google Scholar]
- 45.Tsutsui S, Kobayashi Y, Kobayashi Y, Higashitaniguchi S, Yoda Y, Seto M. and Takabatake T, Journal of Physics: Conference Series, 2010, 217, 012123. [Google Scholar]
- 46.Sergueev I, Glazyrin K, Herrmann MG, Alexeev P, Wille H-C, Leupold O, May AF, Pandey T, Lindsay LR, Friese K. and Hermann RP, Journal of Synchrotron Radiation, 2019, 26, 1592–1599. [DOI] [PubMed] [Google Scholar]
- 47.Alp EE, Sturhahn W, Toellner TS, Zhao J, Hu M. and Brown DE, Hyperfine Interact., 2002, 144, 3–20. [Google Scholar]
- 48.Nowik I, Felner I, Ren Z, Cao GH and Xu ZA, New Journal of Physics, 2011, 13, 023033. [DOI] [PubMed] [Google Scholar]
- 49.Wortmann G, Ponkratz U, Bielemeier B. and Rupprecht K, High Pressure Research, 2008, 28, 545–551. [Google Scholar]
- 50.Baggio‐Saitovitch E, Passamani EC, Mibu K. and Shinjo T, Journal of Applied Physics, 1994, 75, 6483–6485. [Google Scholar]
- 51.Long GJ, Hermann RP, Grandjean F, Alp EE, Sturhahn W, Johnson CE, Brown DE, Leupold O. and Rüffer R, Physical Review B, 2005, 71, 140302. [Google Scholar]
- 52.Perera VS, Yang LD, Hao J, Chen G, Erokwu BO, Flask CA, Zavalij PY, Basilion JP and Huang SD, Langmuir, 2014, 30, 12018–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo Y, Wang H, Xiao Y, Vogt S, Thauer RK, Shima S, Volkers PI, Rauchfuss TB, Pelmenschikov V, Case DA, Alp EE, Sturhahn W, Yoda Y. and Cramer SP, Inorganic Chemistry, 2008, 47, 3969–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilbert-Wilson R, Siebel JF, Adamska-Venkatesh A, Pham CC, Reijerse E, Wang H, Cramer SP, Lubitz W. and Rauchfuss TB, Journal of the American Chemical Society, 2015, 137, 8998–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoda Y, Imai Y, Kobayashi H, Goto S, Takeshita K. and Seto M, Hyperfine Interact., 2012, 206, 83. [Google Scholar]
- 56.Yabashi M, Mochizuki T, Yamazaki H, Goto S, Ohashi H, Takeshita K, Ohata T, Matsushita T, Tamasaku K, Tanaka Y. and Ishikawa T, Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 2001, 467–468, 678–681. [Google Scholar]
- 57.Wang H, Yoda Y, Kamali S, Zhou Z-H and Cramer SP, Journal of Synchrotron Radiation, 2012, 19, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dong W, Wang H, Olmstead MM, Fettinger JC, Nix J, Uchiyama H, Tsutsui S, Baron AQR, Dowty E. and Cramer SP, Inorganic Chemistry, 2013, 52, 6767–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gee LB, Wang H. and Cramer SP, in Methods in Enzymology, ed. S. S. David, Academic Press, 2018, vol. 599, pp. 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toellner TS, Hu MY, Bortel G, Sturhahn W. and Shu D, Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 2006, 557, 670–675. [Google Scholar]
- 61.Wang HX, Peng G, Miller LM, Scheuring EM, George SJ, Chance MR and Cramer SP, Journal of the American Chemical Society, 1997, 119, 4921–4928. [Google Scholar]
- 62.Wang H, Friedrich S, Li L, Mao Z, Ge P, Balasubramanian M. and Patil DS, Physical Chemistry Chemical Physics, 2018, 20, 8166–8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pelmenschikov V, Birrell JA, Pham CC, Mishra N, Wang HX, Sommer C, Reijerse E, Richers CP, Tamasaku K, Yoda Y, Rauchfuss TB, Lubitz W. and Cramer SP, Journal of the American Chemical Society, 2017, 139, 16894–16902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiao Y, Tan M-L, Ichiye T, Wang H, Guo Y, Smith MC, Meyer J, Sturhahn W, Alp EE, Zhao J, Yoda Y. and Cramer SP, Biochemistry, 2008, 47, 6612–6627. [DOI] [PubMed] [Google Scholar]
- 65.Glatzel P, Jacquamet L, Bergmann U, de Groot FMF and Cramer SP, Inorganic Chemistry, 2002, 41, 3121–3127. [DOI] [PubMed] [Google Scholar]
- 66.Wang H, Yoda Y, Dong W. and Huang SD, Journal of Synchrotron Radiation, 2013, 20, 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chumakov AI, Rüffer R, Leupold O. and Sergueev I, Structural Chemistry, 2003, 14, 109–119. [Google Scholar]
- 68.Smith MC, Xiao Y, Wang H, George SJ, Coucouvanis D, Koutmos M, Sturhahn W, Alp EE, Zhao J. and Cramer SP, Inorganic Chemistry, 2005, 44, 5562–5570. [DOI] [PubMed] [Google Scholar]
- 69.Spiro TG and Wasbotten IH, Journal of Inorganic Biochemistry, 2005, 99, 34–44. [DOI] [PubMed] [Google Scholar]
- 70.Li XY and Spiro TG, Journal of the American Chemical Society, 1988, 110, 6024–6033. [DOI] [PubMed] [Google Scholar]
- 71.Galinato MGI, Whaley CM and Lehnert N, Inorganic Chemistry, 2010, 49, 3201–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J, Yoda Y. and Wang H, Review of Scientific Instruments, 2022, 93, 095101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.