

Abstract
Congenital bilateral absence of the vas deferens (CBAVD) is observed in 1%–2% of males presenting with infertility and is clearly associated with cystic fibrosis transmembrane conductance regulator (CFTR) mutations. CFTR is one of the most well-known genes related to male fertility. The frequency of CFTR mutations or impaired CFTR expression is increased in men with nonobstructive azoospermia (NOA). CFTR mutations are highly polymorphic and have established ethnic specificity. Compared with F508Del in Caucasians, the p.G970D mutation is reported to be the most frequent CFTR mutation in Chinese patients with cystic fibrosis. However, whether p.G970D participates in male infertility remains unknown. Herein, a loss-of-function CFTR p.G970D missense mutation was identified in a patient with CBAVD and NOA. Subsequent retrospective analysis of 122 Chinese patients with CBAVD showed that the mutation is a common pathogenic mutation (4.1%, 5/122), excluding polymorphic sites. Furthermore, we generated model cell lines derived from mouse testes harboring the homozygous Cftr p.G965D mutation equivalent to the CFTR variant in patients. The Cftr p.G965D mutation may be lethal in spermatogonial stem cells and spermatogonia and affect the proliferation of spermatocytes and Sertoli cells. In spermatocyte GC-2(spd)ts (GC2) Cftr p.G965D cells, RNA splicing variants were detected and CFTR expression decreased, which may contribute to the phenotypes associated with impaired spermatogenesis. Thus, this study indicated that the CFTR p.G970D missense mutation might be a pathogenic mutation for CBAVD in Chinese males and associated with impaired spermatogenesis by affecting the proliferation of germ cells.
Keywords: azoospermia, congenital bilateral absence of the vas deferens, CFTR mutation, male infertility, spermatogenesis
INTRODUCTION
Infertility affects approximately 15% of couples worldwide,1 and about 7% of men are infertile.2 Dysregulated spermatogenesis is considered the main cause of male infertility,3,4 which is a complex multifactorial pathological state with heterogeneous phenotypic presentations, from the absence of spermatozoa in the testes to distinct alterations in sperm quality.5,6 Altered genetics are found in approximately 15% of affected men.6
Azoospermia is classified as obstructive azoospermia (OA) or nonobstructive azoospermia (NOA). Congenital bilateral absence of the vas deferens (CBAVD) occurs in 1%–2% of infertile men.7 Among patients with CBAVD, 78% have at least one cystic fibrosis transmembrane conductance regulator (CFTR) gene mutation.8 The first CFTR mutation identified in patients with CBAVD in 1990 showed that patients with CBAVD typically carry one or more mutation of the gene.9
The frequency of CFTR mutations and impaired CFTR expression increased in men with NOA or OA.10–13 Some CFTR gene mutations are associated with NOA, such as W1282X and K1351R.14–17 Recent semen analysis from a study of 71 infertile males with two (biallelic) CFTR mutations revealed azoospermia in 70 of 71 (99%) patients, and severe oligozoospermia in one patient.7 Therefore, some researchers propose that CFTR mutations may affect male fertility through mechanisms other than reproductive tract obstruction.18 In contrast, other investigators have not found a significant relationship between NOA and CFTR mutations.19–21 We previously found that CFTR is expressed on human sperm and that it plays critical roles in sperm capacitation and fertilizing capability by regulating ion transport.22 Although CFTR may function during the later stages of spermatogenesis, its role in early spermatogenesis awaits clarification. Therefore, the exact role of CFTR in male reproductive physiology requires further investigation.
Compared with relatively higher morbidity rates in Caucasians, traditional studies have found fewer patients with cystic fibrosis (CF) in Asia.23,24 However, recent studies have found that p.G970D, which was first reported in 1999,25 is the most frequent CFTR mutation in Chinese patients with CF.26–28 However, whether p.G970D participates in male infertility remains unknown.
This study found that among 122 Chinese patients with CBAVD, five had CFTR p.G970D heterozygous mutations. Whole-exome sequencing (WES) and Sanger sequencing of one infertile patient with CBAVD and NOA who had no CF-like phenotype-related symptoms revealed the presence of the homozygous missense CFTR p.G970D variant. Furthermore, we verified the effects of this mutation on CFTR expression and cellular proliferation in a germ cell line model in vitro. Thus, the present study discovered that the CFTR p.G970D missense mutation might cause CBAVD and be associated with impaired spermatogenesis.
PATIENTS AND METHODS
Patients
A male Han Chinese patient with CBAVD and NOA was recruited at the Reproductive Medical Center, Chengdu Xi’nan Gynecology Hospital (Chengdu, China). The parents of this patient also participated in this study. The 122 patients with CBAVD in the retrospective analysis were recruited from the Reproductive and Genetic Hospital of CITIC-Xiangya (Changsha, China) admitted between January 2014 and December 2017.
Ethical approval
This study was approved by the Ethics Committee of West China Second University Hospital, Sichuan University (approval No. K2018089; Chengdu, China). All the patients and family members included in the study provided written informed consent to the evaluation, use, and publication of their clinical data for scientific purposes.
WES
Genomic DNA was isolated from peripheral blood samples of the proband using whole blood DNA purification kits (QIAGEN, Hilden, Germany). We captured exons by WES using Agilent SureSelect Human All Exon V6 kits (Agilent Technologies Inc., Santa Clara, CA, USA) and an Illumina HiSeq 4000 sequencer (Illumina Inc., San Diego, CA, USA). Variants were prioritized and etiological involvement was evaluated using the medical resequencing analysis pipeline, which initially filtered all identified variants by comparisons with disease-associated variants in the Human Gene Mutation Database (www.hgmd.cf.ac.uk) and the Online Mendelian Inheritance in Man (OMIM; www.omim.org), to collect known disease-causing variants. Pathogenic and likely pathogenic genes/variants were defined according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) using the 5-tier classification.29
CFTR exon and Sanger sequencing
Mutations in CFTR from the proband and his parents and sister were validated by Sanger sequencing. Target sequences were amplified by polymerase chain reaction (PCR) using 2× Rapid Taq Master Mix (P222-01, Vazyme Biotech Co., Ltd., Nanjing, China) and Dyad Polymerase (Bio-Rad Laboratories Inc., Hercules, CA, USA); the primers are listed in Supplementary Table 1. The PCR amplicons were analyzed by Sanger sequencing (Tsingke Biological Technology Co., Ltd., Beijing, China).
Supplementary Table 1.
Primers for verifying cystic fibrosis transmembrane conductance regulator variations by Sanger sequencing
| Primers | Sequences (5′–3′) |
|---|---|
| CFTR 970-sanger-F | TTATTTGGGTTCTGAATGCGTC |
| CFTR 970-sanger-R | ATGTCACACTTTGTGGCCAG |
| CFTR 470-sanger-F | CTTCTGCTTAGGATGATAATTGG |
| CFTR 470-sanger-R | GCTTTGATGACGCTTCTGTA |
| CFTR 508-sanger-F | CCAGACTTCACTTCTAATGG |
| CFTR 508-sanger-R | CTAACCGATTG AATATGGAG |
| CFTR 117-sanger-F | CCTTTTGTAGGAAGTCACC |
| CFTR 117-sanger-R | CTGTGCAAGGAAGTATTAC |
| CFTR 5T-sanger-F | TGGGCAAATATCTTAGTTT |
| CFTR 5T-sanger-R | GAGTACCAAGAAGTGAGAA |
CFTR: cystic fibrosis transmembrane conductance regulator
Genomic DNA from the 122 patients with CBAVD was extracted, then 27 CFTR exons were amplified by PCR using the primers listed in Supplementary Table 2. The products were then sequenced (BioSune Biotechnology [Shanghai] Co., Shanghai, China) and compared with the CFTR exon sequences to screen out the sites with mutations.
Supplementary Table 2.
Primers for Sanger sequencing all cystic fibrosis transmembrane conductance regulator exons
| Primers | Sequences (5′–3′) |
|---|---|
| CFTR-1F | GACATCACAGCAGGTCAG |
| CFTR-1R | ACTGCTTATTCCTTTACCC |
| CFTR-2F | CCAAATCTGTATGGAGACCA |
| CFTR-2R | TATGTTGCCCAGGCTGGTAT |
| CFTR-3F | CTTGGGTTAATCTCCTTGGA |
| CFTR-3R | ATTCACCAGATTTCGTAGTC |
| CFTR-4F | ATGGTATGACCCTCTATATAAAC |
| CFTR-4R | CTACCTAATTTATGACATT |
| CFTR-5F | ATTTCTGCCTAGATGCTGGG |
| CFTR-5R | AACTCCGCCTTTCCAGTTGT |
| CFTR-6aF | TTAGTGTGCTCAGAACCACG |
| CFTR-6aR | CTATGCATAGAGCAGTCCTG |
| CFTR-6bF | GACTTAAAACCTTGAGCAG |
| CFTR-6bR | GAGGTGGAAGTCTACCATG |
| CFTR-7F | CAGATCTTCCATTCCAAG |
| CFTR-7R | TAGAACTGATCTATTGACTG |
| CFTR-8F | AGATGTAGCACAATGAGAG |
| CFTR-8R | AGACCGGGTTTTACCATG |
| CFTR-9F | TATACAGTGTAATGGATCATGG |
| CFTR-9R | CTTCCAGCACTACAAACTAG |
| CFTR-10F | GCAGAGTACCTGAAACAGGA |
| CFTR-10R | CATTCACAGTAGCTTACCCA |
| CFTR-11F | CTGTGGTTAAAGCAATAGTGTG |
| CFTR-11R | CAGATTCTGAGTAACCATA |
| CFTR-12F | GTGAATCGATGTGGTGACCA |
| CFTR-12R | CTGGTTTAGCATGAGGCGGT |
| CFTR-13F | TGCTAAAATACGAGACATATTGCA |
| CFTR-13R | GCTACATATTGCATTCTACTC |
| CFTR-14aF | GATGTGAATTTAGATGTGGG |
| CFTR-14aR | GGATCTCACTATATTGTCCA |
| CFTR-15F | GTGCATGCTCTTCTAATGC |
| CFTR-15R | AACAAAGGGCACATGCCTC |
| CFTR-16F | GTCTACTGTGATCCAAACT |
| CFTR-16R | GACAGGACTTCAACCCTC |
| CFTR-17aF | CACTGACACACTTTGTCC |
| CFTR-17aR | TAGATTAACAATAAAGAATCTC |
| CFTR-18F | GTGCCCTAGGAGAAGTG |
| CFTR-18R | CTTCCTCATGCTATTACTC |
| CFTR-19F | ACAAATAGCAAGTGTTGCA |
| CFTR-19R | GCTTCAGGCTACTGGGATTC |
| CFTR-20F | GGTCAGGATTGAAAGTGT |
| CFTR-20R | ATGAGAAAACTGCACTGGA |
| CFTR-21F | AAGCAGACATGATAAAATAT |
| CFTR-21R | AGGTCCAGTCAAAAGTACC |
| CFTR-22F | GCTTTCAGAACTCCTGTG |
| CFTR-22R | TGTCACCATGAAGCAGGC |
| CFTR-23F | CAATAGACATATTATCAAGGT |
| CFTR-23R | AGGAACTATCACATGTGAG |
| CFTR-24F | CTGTGCCAGTTTCTGTCC |
| CFTR-24R | CCATGAGGTGACTGTCCC |
CFTR: cystic fibrosis transmembrane conductance regulator
Hematoxylin–eosin (HE) staining
Briefly, testicular tissue samples were processed according to the standard protocol by immersion in 4% Bouin’s fixative solution (PH0976; Phygene, Hong Kong, China) overnight and dehydration in ethanol. The tissues were embedded in paraffin, then sliced into 5-µm sections. The sections were stained routinely with hematoxylin (G1150; Solarbio, Beijing, China) and eosin (G1100; Solarbio) for histological examination.
Immunofluorescence staining
Paraffin-embedded tissue sections were prepared according to standard protocols. Briefly, the slices were conventionally dewaxed, immersed in 0.01 mol l−1 citrate solution, microwave-heated to boiling, washed once after 5 min, cooled at room temperature, washed three times with phosphate-buffered saline (PBS), and permeabilized with 0.3% Triton X-100 (T8200; Solarbio). Nonspecific protein binding was blocked with 5% bovine serum albumin fraction V (BSA; 4240GR500; Biofroxx, Guangzhou, China), and then, the slices were incubated with the following primary antibodies: 1:100-diluted CFTR (MAB25031; R&D Systems Inc., Minneapolis, MN, USA), 1:50-diluted SOX9 (K003355P; Solarbio), and 1:50-diluted DDX4 (ab13840; Abcam, Cambridge, UK) overnight at 4°C. The sections were washed three times with PBS, incubated with 1:1000-diluted donkey anti-mouse IgG Alexa-Fluor 488 and donkey anti-rabbit IgG Alexa-Fluor 594 (A21202 and A21207, respectively; Invitrogen, Carlsbad, CA, USA) for 1 h at 4°C, and then counterstained with 4’,6-diamidino-2-phenylindole (DAPI; Beyotime, Shanghai, China) to label nuclei. The sections were visualized, and images were acquired using an FV-1000 laser scanning confocal microscope (Olympus Optical Co., Ltd., Tokyo, Japan).
Cell culture
The GC-2(spd)ts (GC2) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Mouse spermatogonial stem cell line (C18-4) cells were donated by Prof. Zhong-Han Li from the School of Life Sciences, Sichuan University, and GC-1spg (GC1) cells and mouse Sertoli cells (TM4) were a gift from Prof. Jin-Peng Sun from the Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Shandong University (Jinan, China). GC1, GC2, and C18-4 cell lines were cultured under sterile conditions in Dulbecco’s modified Eagle’s medium (DMEM; 11965092; Thermo Fisher Scientific Inc., Waltham, MA, USA), containing 10% fetal bovine serum (FBS; 10270-106; Thermo Fisher Scientific Inc.), and TM4 cells were cultured in Dulbecco’s modified Eagle’s medium/F12 GlutaMax (DMEM/F12; 31331028, Thermo Fisher Scientific Inc.), containing 100 U ml−1 penicillin, 100 mg ml−1 streptomycin, 2.5% FBS, and 5% horse serum (S9050; Solarbio). All cells were harvested using 0.25% trypsin-ethylenediaminetetraacetic acid (Trypsin-EDTA; 2520056; Thermo Fisher Scientific Inc.).
Generation of Cftr p.G965D-mutated cell models
The clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) gene editing system was used to knock-in Cftr p.G965D. Exon 18 of Cftr was targeted using a guide RNA (gRNA). Primers are described in Supplementary Table 3. Knock-in was performed using the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector (Addgene, Watertown, MA, USA). The ligated vector was inserted into Tran5α chemically competent cells (TSC-C01; Tsingke Biological Technology Co., Ltd.). Plasmids were constructed as described by the manufacturer and confirmed by sequencing. All cell lines were electro-transfected with the Cftr p.G965D knock-in plasmid using an Selleck Electroporator (Selleck, Houston, TX, USA) as described by the manufacturer. Then, the transfected C18-4, GC1, GC2, and TM4 cells were selected with puromycin (1.5 ng µl−1, 3.0 ng µl−1, 9.0 ng µl−1, and 4.5 ng µl−1, respectively) 48 h after transfection. Thereafter, cells were individually cloned in 96-well plates and sequenced to detect knock-in efficiency.
Supplementary Table 3.
Primer sequences of single guide RNA used to construct cystic fibrosis transmembrane conductance regulator p.G965D and cystic fibrosis transmembrane conductance regulator p.G970D mutation
| Primers | Sequences (5′–3′) |
|---|---|
| Cftr-gRNA | F: CACCGTATTTATTTGTATCACAGG |
| R: AAACCCTGTGATACAAATAAATAC | |
| Cftr-mut965 | F: TTATTTGTATCACAGATGGAATTCTTAACAGATTCTCCAAAG |
| R: CTGTTAAGAATTCCATCTGTGATACAAATAAATATTCTC | |
| CFTR-gRNA | F: CACCGTCATCTTGTATATTATAGG |
| R: AAACCCTATAATATACAAGATGAC | |
| CFTR-mut970 | F: TCTTGTATATTATAGATGGGATTCTTAATAGATTCTCCAAAG |
| R: CTTTGGAGAATCTATTAAGAATCCCATCTATAATATACAAGA |
CFTR: cystic fibrosis transmembrane conductance regulator; gRNA: guide RNA
Western blot
Proteins were extracted from cultured cells in radioimmunoprecipitation assay buffer (RIPA; P0013B; Beyotime) containing a protease inhibitor cocktail (B14001; Bimake, Shanghai, China). Denatured proteins were transferred to polyvinylidene difluoride membranes (PVDF; MilliporeSigma Co., Ltd., Burlington, MA, USA). The membranes were incubated with the primary antibodies (at 1 μg ml−1; mouse anti-CFTR [MAB25031, R&D Systems Inc.] and beta-tubulin [1E1] mouse mAb [200608; Zen-Bio, Inc., Chengdu, China]) at 4°C overnight. The membranes were then incubated with secondary antibodies (goat anti-mouse IgG or goat anti-rabbit IgG; ZB-2305 and ZB-2301, respectively; ZSGB-BIO, Beijing, China). Proteins on the membranes were visualized using a chemiluminescent horseradish peroxidase (HRP) substrate (WBLUF0500; MilliporeSigma Co., Ltd.).
Reverse transcription (RT)-PCR
Total RNA was extracted using TRIzol Reagent (15596026; Invitrogen); then, complementary DNA (cDNA) was synthesized by reverse transcription PCR using PrimeScript RT reagent kit (RR047A; Takara Bio Inc., Kusatsu, Japan) as described by the manufacturer (Cmax Plus, Shanghai, China).
Cell proliferation assays
We seeded 4 × 103 cells per well in triplicate in 96-well plates, then determined the numbers of viable cells 24 h, 48 h, 72 h, and 96 h later using a Cell Counting Kit-8 (CCK8; B34302; Bimake). After adding CCK8 solution, plates were incubated at 37°C for 2 h, and then absorbance was determined at 450 nm using a Molecular plate reader.
RESULTS
Clinical characteristics of the proband
This study investigated an infertile male diagnosed with idiopathic NOA at the Reproductive Medical Center, Chengdu Xi’nan Gynecology Hospital. The findings of palpation and ultrasound confirmed a diagnosis of CBAVD, and microsurgical testicular sperm extraction did not recover any spermatozoa. The wife of the patient had not become pregnant after two years of marriage. Neither the patient nor his wife had a medical history of chronic diseases.
The proband had normal general parameters and hormone profiles except for smaller testes, lower semen volume, and a lower pH. The karyotype was 46,XY. No inherited or de novo deletions or duplications with clearly pathogenic effects on spermatogenesis were detected in the genome of the patient, either on autosomes or the X chromosome. Microdeletions of azoospermia factor a (AZFa), AZFb, and AZFc were undetectable (Table 1). Other causes of infertility, such as cryptorchidism, varicocele, testicular trauma, recent febrile illness, drugs, and exposure to toxic substances, were excluded.
Table 1.
Clinical characteristics of the proband at clinical assessment
| Clinical characteristic | Proband | Reference valuea |
|---|---|---|
| General parameters | ||
| Age (year) | 29 | |
| Height (cm) | 175 | |
| Weight (kg) | 65 | |
| BMI (kg m−2) | 21.2 | 18.5–24.9 |
| SBP/DBP (mmHg) | 120/70 | <140/90 |
| Fertility parameters | ||
| Total testicular volume (ml) | 24 | 50.0 (33.0–70.0)b |
| Testicular volume: left (ml) | 12 | |
| Testicular volume: right (ml) | 12 | |
| Varicocele testes (both sides) | No | |
| FSH (IU l−1) | 8.4 | 1.3–19.3 |
| LH (IU l−1) | 1.9 | 1.2–8.6 |
| Prolactin (mIU l−1) | 63.6 | 56–278 |
| Testosterone (ng dl−1) | 476.7 | 175–781 |
| Estradiol (pmol l−1) | 43 | <159 |
| Semen volume (ml) | 0.4 | >1.5 |
| Semen pH | 6.3 | >7.2 |
| Total sperm count (×106 per ejaculate) | 0 | >39 |
| Karyotype | 46,XY | |
| AZFa, AZFb, and AZFc microdeletions | No |
aSourced from Andrology Laboratory of Maternity and Child Health Hospital of Jinjiang District and World Health Organization Laboratory Manual for the Examination and Processing of Human Semen (5th). bBaltic young male cohort with data presented as medians (5th–95th percentiles). AZFa–c: azoospermia factor a–c; BMI: body mass index; FSH: follicle-stimulating hormone; LH: luteinizing hormone; SBP/DBP: systolic blood pressure/diastolic blood pressure
In normal men, HE staining of the cone-shaped Sertoli cells is arranged on the basement membrane, germ cells are embedded in intercellular spaces, and spermatids can be seen in the lumen. In contrast, analysis of the patient’s testes detected Sertoli cells, but germinal epithelium and sperm were absent (Figure 1a). Diameters of 137 spermatogenic tubules were measured. The seminiferous tubules of the patient were significantly smaller than those of normal male (P < 0.01; Figure 1b). The CFTR costaining with DEAD-box helicase 4 (DDX4; a marker for spermatogonia) or SRY-box transcription factor 9 (SOX9; a marker of Sertoli cells) showed that SOX9 and CFTR were detected, but DDX4 was not detected in the patient’s samples, providing evidence that the seminiferous tubules did not contain spermatogonia (Figure 1c and 1d).
Figure 1.
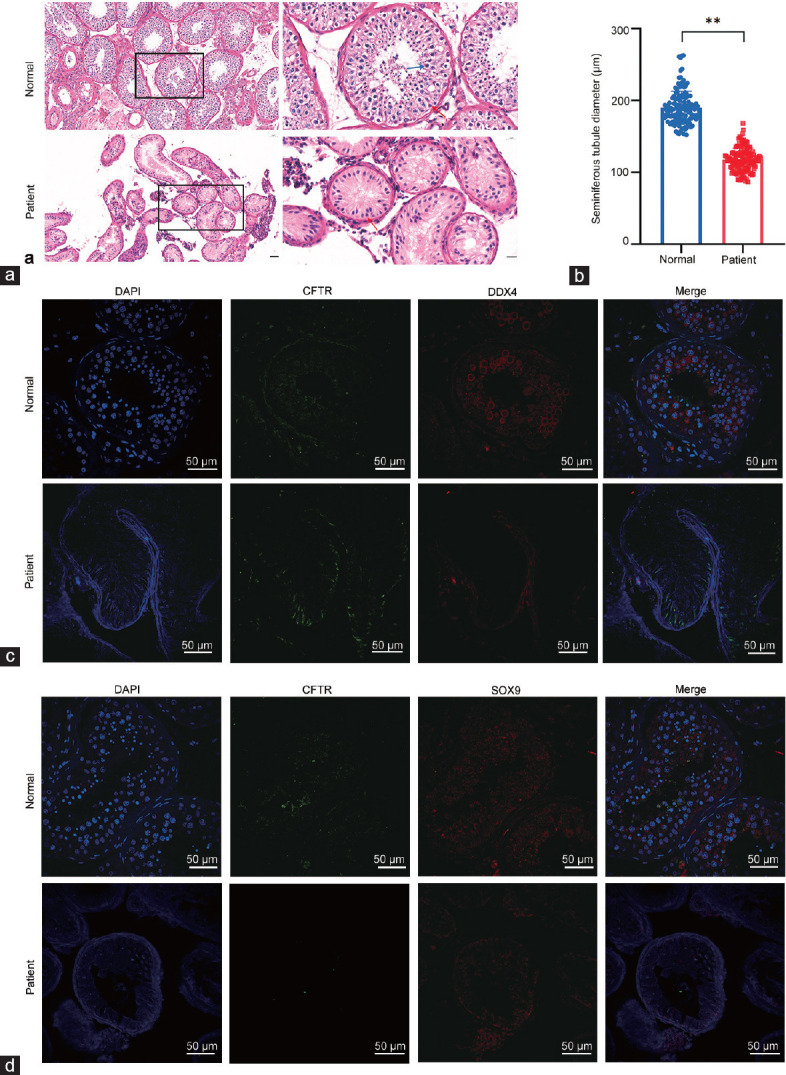
Patient with CBAVD and NOA. (a) Hematoxylin–eosin staining of testicular tissue sections shows normal spermatogenesis. Scale bars = 50 µm (left) and 20 µm (right), respectively. Red arrow: Sertoli cells; blue arrow: germ cells. (b) The diameter of the seminiferous tubules of the patient was significantly smaller than that in the normal male. **P < 0.01. (c) Immunofluorescence staining shows spermatogenesis in testicular tissue sections representing normal spermatogenesis and SCOS. Staining for CFTR (green) is weak, and staining for DDX4 (red) is absent, in testicular tubules from the proband (blue: DAPI; scale bars = 50 µm). (d) Immunofluorescence staining shows spermatogenesis in normal and SCOS afflicted testicular tissue sections. Weak CFTR (green) and SOX9 (red) staining indicates Sertoli cells in testicular tubules from the proband (blue: DAPI; scale bars = 50 µm). SCOS: Sertoli cell-only syndrome; DDX4: DEAD-box helicase 4; SOX9: SRY-box transcription factor 9; DAPI: 4’,6-diamidino-2-phenylindole; CFTR: cystic fibrosis transmembrane conductance regulator; CBAVD: congenital bilateral absence of vas deferens; NOA: nonobstructive azoospermia.
Homozygous missense mutation in CFTR in the proband determined by WES
We analyzed genomic DNA extracted from blood sample of the proband using WES to detect the causative mutation(s) of the infertility. After excluding frequent variants and using technical and biological filters (Supplementary Table 4), a homozygous missense mutation c.2909G>A p.G970D found in CFTR was the presumed cause of azoospermia in the proband (Table 2). We investigated the CFTR mutation in the proband and relatives by Sanger sequencing. His parents (I:1 and I:2) had delivered a daughter and a son (Figure 2a). The results showed that he inherited the homozygous missense mutation from both parents (Figure 2b).
Supplementary Table 4.
Screening strategy to identify the disease-causing genes by whole-exome sequencing
| Screening procedure | Proband |
|---|---|
| Filter rare variants (MAF <5%) in public databasesa | 2207 |
| Filter variants with functional impacta | 204 |
| Homozygous | 2 |
| Relevancy for phenotypeb | 1 |
| Sanger sequencing/cosegregation | 1 |
aRepresent variants predicted to be deleterious; bRepresent variants associated with phenotype, including expression in testis or the existence of a mouse KO model with infertility phenotype. MAF: minor allele frequency
Table 2.
Homozygous CFTR mutation identified in the proband
| Gene information | CFTR |
|---|---|
| Variant coordinates | Chr7:117246728G>A |
| Transcript | NM_000492 |
| cDNA mutation | c.2909G>A |
| Protein alternation | p.G970D |
| Mutation type | Missense, homozygous |
| PROVEAN score | −5.9 |
| PROVEAN prediction (cut-off=−2.5) | Deleterious |
Variant coordinate is based on the human genome assembly GRCh37/h19. CFTR: cystic fibrosis transmembrane conductance regulator; cDNA: complementary DNA; PROVEAN: Protein Variation Effect Analyzer
Figure 2.
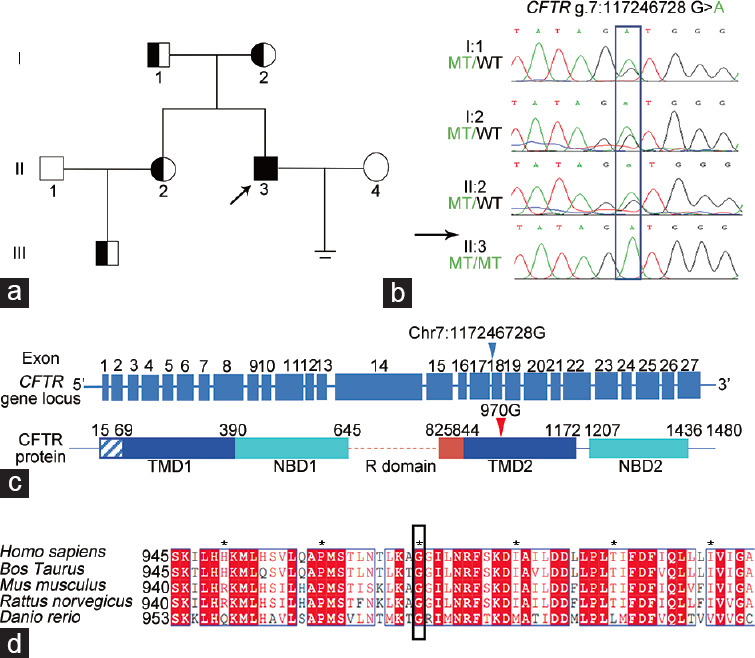
Identification of homozygous CFTR mutation in the patient with CBAVD and NOA. (a) Pedigree of family with inherited CFTR mutation. Unfilled squares and circles denote unaffected male and female family members, respectively. Filled squares denote affected males; half black and white symbols denote heterozygous members; black arrows indicate proband. (b) The CFTR mutation was verified by Sanger sequencing. Homozygous CFTR mutation in the proband (II:3) was inherited from heterozygous parental carriers (I:1 and I:2). Rectangle indicates position of mutation. (c) The CFTR p.G970D mutation at exon 18 causes G-to-A substitution at cDNA (NCBI reference sequence: NM_000492.4) nucleotide position 2909, replacing glycine (G) with aspartic acid (D) at amino acid 970 in the CFTR protein. Arrowheads indicate the mutation site. (d) Sequence alignment shows conservation of affected amino acid (glycine) across various species. The asterisks are defined for amino acid number interval. WT: wild-type; MT: mutation; CFTR: cystic fibrosis transmembrane conductance regulator; TMD: transmembrane domains; NBD: nucleotide binding domains; cDNA: complementary DNA; NCBI: National Center for Biotechnology Information; R domain: disordered area - regulatory area; CBAVD: congenital bilateral absence of vas deferens; NOA: nonobstructive azoospermia.
CFTR p.G970D mutation is a common pathogenic mutation of CBAVD in Chinese
We retrospectively analyzed 122 patients with CBAVD at the Reproductive and Genetic Hospital of CITIC-Xiangya from January 2014 to December 2017. The CFTR all-exon test results showed that five of these patients harbored heterozygous CFTR p.G970D mutations (4.1%). The mutation frequency was higher than other mutations excluding polymorphic sites, and the mutation was considered a common CFTR pathogenic mutation. The karyotype of all the patients was 46,XY. Microdeletions of AZFa, AZFb, and AZFc were undetectable. The median age at diagnosis of the five patients was 29 (range: 24–36) years. Only one patient underwent testicular sperm extraction, which revealed a small number of immotile sperms.
Missense Cftr p.G965D mutation in spermatogonia affects cell proliferation
The amino acid of CFTR altered by the mutation was located in the membrane-spanning domain 2, which is a transmembrane domain that forms selective chloride channels (Figure 2c). The amino acid sequence is conserved, along with the expression and localization of CFTR between humans and mice (Figure 2d). The immunoblotting results showed that CFTR was differentially expressed in spermatogonia cells (Figure 3a). To further understand the role of p.970 in CFTR in spermatogenesis, we generated mouse cell lines harboring a homozygous Cftr c.G1128A[p.G965D] mutation equivalent to CFTR c.G2909A[p.G970D] in patients, using the CRISPR/Cas9-mediated genome editing (Figure 3b). The target mutation was assessed in all knock-in clones by DNA sequencing (Supplementary Table 5). The Cas9 mutation was repeated twice in the C18-4 and GC1 cell lines, and the single-cell clones proliferated for a few generations, then died. We could not obtain enough cells to analyze proliferation. We produced one homozygous clone among 32 GC2 single-cell clones and three among 43 homozygous single-cell clones of mouse Sertoli cell (TM4). Preliminary verification proceeded in homozygous clones named GC2-mutN6 and TM4-mut29#.
Figure 3.
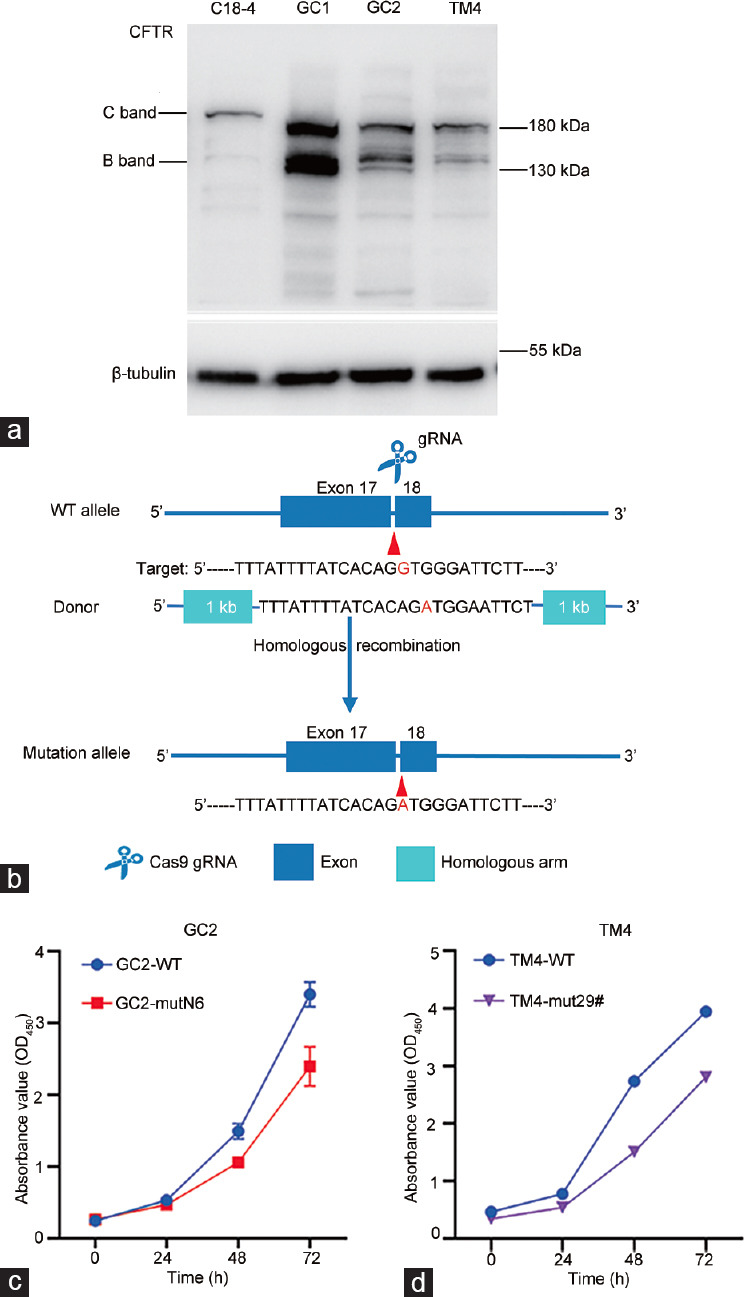
Missense mutation in CFTR affects proliferation of germ and Sertoli cells. (a) Endogenous CFTR protein expression in four types of cells. Loading control was β-tubulin. C band: mature CFTR; B band: immature CFTR. (b) Schematic illustration of targeting strategy for generating Cftr p.G965D knock-in cell lines. Mutated nucleotides are shown in red. The nucleotide in blue indicates a mutation that does not affect the amino acid sequence in the protospacer adjacent motif. (c) Proliferation of wild-type (WT) and point mutant GC2 cell lines. (d) Proliferation of wild-type and point mutant TM4 cell lines. Cftr p.G965D mutation affected GC2 cell proliferation and slowed TM4 cell growth. CFTR: cystic fibrosis transmembrane conductance regulator; gRNA: guide RNA; Cas9: CRISPR-associated protein 9; CRISPR: clustered regularly interspaced short palindromic repeats; OD: optical density.
Supplementary Table 5.
Primer sequence used for identifying monoclonal cells
| Primer names | Primer sequences (5′–3′) |
|---|---|
| Cftr mut-JD | F: ATGGCAGAAAACTCCACAGG |
| R: CAGGGCATGCCTGAAATAGA | |
| CFTR mut-JD | F: TGGGTTCTGAATGCGTCTACT |
| R: TGTGGCCAGTGATTTTGGGA |
CFTR: cystic fibrosis transmembrane conductance regulator
The effects of the Cftr p.G965D mutation on cellular proliferation were assessed using CCK8 assays. Figure 3c and 3d show that the mutation adversely affected the proliferation of GC2-mutN6 and TM4-mut29# cells.
These results indicated that Cftr p.G965D impacted the proliferation of germ cells. We could not obtain Cftr p.G965D cell groups in C18-4 and GC1, suggesting that Cftr p.G965D is detrimental to spermatogonial stem cells and spermatogonia. Considering the variable effects of CFTR mutations on spermatogonial stem cells, spermatogonia, spermatocytes, and Sertoli cell lines, the absence of sperm in testes of the proband was likely due to the lethal effect of the mutation on spermatogonial stem cells.
Missense mutation Cftr p.G965D might affect CFTR expression and alter CFTR RNA splicing in spermatocytes
Cell proliferation was affected; therefore, we assessed the functional consequences of the Cftr p.G965D mutation at the protein level. The results revealed reduced CFTR in GC2-mutN6 compared with wild-type cells, indicating decreased CFTR on the cell membrane. In contrast, TM4-mut29# did not significantly alter CFTR expression (Figure 4a).
Figure 4.

Missense mutation Cftr p.G965D might affect CFTR function by decreasing its expressing and altering Cftr RNA splicing. (a) Immunoblots of CFTR protein in wild-type (WT) GC2 cells and TM4 cells with point mutations. Loading control was β-tubulin. Cftr p.G965D mutation reduced mature CFTR protein in GC2 but did not significantly affect TM4 Sertoli cells. C band: mature CFTR; B band: immature CFTR. (b) Splicing variant of Cftr mRNA produced in GC2-mutN6 cells. Sanger sequencing indicated RNA splicing variants. Red arrowhead indicates position of mutation and the sequence in the rectangular box indicates the abnormal splicing product. (c) mRNA splicing profiles in GC2-mutN6 cells. Rectangular boxes: exons; dotted lines: introns; red letters: mutation sites. CFTR: cystic fibrosis transmembrane conductance regulator; Pre-RNA: heterogeneous nuclear RNA.
Total RNA was extracted from GC2-WT and GC2-mutN6 cells, and the region encompassing exon 18 was analyzed using RT-PCR of mRNA (the primers are listed in Supplementary Table 6). Sequencing of amplificons of GC2-mutN6 revealed one normal product and an abnormal splicing product (Figure 4b). We cloned the two RT-PCR products of GC2-mutN6 into the T-vector, and sequencing results showed that 7 of 17 ligated plasmids had truncated mRNA missing exon 18. These results indicated that the mutation promotes an alteration in CFTR pre-mRNA splicing in GC2-mutN6 cells (Figure 4c).
Supplementary Table 6.
Primers for verifying cystic fibrosis transmembrane conductance regulator messenger RNA by Sanger sequencing
| Primers | Sequences (5′–3′) |
|---|---|
| Cftr ex18-mRNA-F | GCCGCTGGTGCATACGTTAA |
| Cftr ex18-mRNA-R | ATGAAGGAAGTAGGCCCTCAG |
CFTR: cystic fibrosis transmembrane conductance regulator; mRNA: Messenger RNA
Combined with previous findings of GC2 spermatocytes, the Cftr p.G965D mutation significantly reduced CFTR protein expression, and the decrease may be due to splicing variants, which may alter the maturation and transport of CFTR.
In conclusion, the CFTR p.G970D missense mutation might be a pathogenic mutation for CBAVD in Chinese males and associated with impaired spermatogenesis by reducing germ cell CFTR expression and proliferation. We recommend that CFTR mutations should be fully evaluated in Chinese infertile male patients.
DISCUSSION
Until November 2021, the National Center for Biotechnology Information (NCBI) has published more than 2000 CFTR gene variants that are highly polymorphic and have established ethnic specificity. The most prevalent mutant alleles in Caucasian patients with CBAVD are p.F508del, IVS9-10 5T, and p.R117H, with frequencies of 17%, 25%, and 3%, respectively.8
Studies on CFTR exon mutations in Chinese patients with CBAVD have found a very low exon mutation rate in CFTR, and the most common mutations (p.F508del and p.R117H) in Caucasians have not been found in Chinese males.30 This indicates that there is an ethnic-specific CFTR mutation profile in the Chinese population. We retrospectively analyzed 122 Chinese patients with CBAVD, and CFTR exon sequencing revealed that 5 (4.1%) of them carried CFTR p.G970D mutations. One CFTR p.G970D homozygous mutation carrier was diagnosed with CBAVD and had impaired spermatozoa. These data indicated that this mutation is a common pathogenic mutation in Chinese patients with CBAVD.
Isolated CBAVD has been regarded as a primarily genital form of CF.31,32 No other CF phenotypes and common mutations in CFTR were identified in the proband patient (Supplementary Figure 1 (195.4KB, tif) ). It has been reported that epididymal sperm quality and in vitro fertilization rate were decreased in patients with CBAVD harboring CFTR mutations,33,34 suggesting that CFTR mutations may directly or indirectly affect sperm quality and quantity in addition to CBAVD. Seminiferous tubules did not contain spermatogonia in the proband, indicating severely impaired spermatogenic function. This finding provides evidence that CFTR may be related to early spermatogenesis. Importantly, in vitro data demonstrated that the Cftr p.G965D mutation had mild-to-adverse effects on the proliferation of spermatocyte GC2 cells, and that the mutation was likely to have more serious effects on spermatogonial stem cells and spermatogonia, which may be unable to differentiate to the spermatocyte stage. Proportional RNA splicing variants at the mRNA level in GC2 Cftr p.G965D cells indicated that splicing variants may contribute to the phenotypes associated with impaired spermatogenesis.
Since no stable human spermatogenic cell lines are available for in vitro analysis, we constructed a HEK293 CFTR p.G970D cell line to further explore the effects of the CFTR mutation on human cells. We found significantly reduced cell proliferation and a high risk of cell death in the HEK293 CFTR p.G970D 293-mutK5 cells. Furthermore, a 150-kDa CFTR protein accumulated in the cytoplasm of 293-mutK5 cells. Moreover, the inhibited cell proliferation was partially restored by the CFTR potentiator ivacaftor (VX-770). Sequencing results showed that no splicing variant was detected in the mRNA of 293-mutK5 cells. We analyzed pre-mRNA splicing in our homozygous proband using a minigene vector constructed by DNA extracted from blood samples (Supplementary Materials and Methods). Agarose gel electrophoresis revealed one fragment in wild-type (CFTR c.2909G) and mutant (CFTR c.2909A) samples, indicating that the mutation did not perturb splicing in the proband. Sequencing the amplified band revealed a single nucleotide substitution corresponding to the c.2909G>A variant (Supplementary Figure 2 (170.4KB, tif) ). Considering that the minigene vector was constructed by DNA extracted from blood samples, whether RNA splicing variants exist in the testes of the patient with homozygous mutations awaits clarification. Alternative splicing in GC2-N6 cells may be species- or tissue-specific.
Among the six types of mutations affecting CFTR function, Tian et al.28 grouped CFTR p.G970D within the category of mutations exhibiting reduced conduction, which are generally associated with mild CF disease. In our study, ivacaftor (VX-770) partially restored the phenotype and 150-kDa CFTR protein accumulated in the cytoplasm of 293-mutK5 cells, suggesting that CFTR p.G970D may impact not only the function of the CFTR ion channel but also the trafficking of CFTR protein.
Different mutations of CFTR have heterogeneous phenotypes, and the CFTR p.G970D mutation has different effects on different species and cell lines, indicating that CFTR mutations are highly polymorphic- and tissue-specific. How CFTR mutations participate in different stages of male infertility remains unknown.35,36 Previous analyses of CFTR mutations found a broader indication than simply the absence of vasa deferens.7 Our results indicated that CFTR mutations are also associated with different aspects of spermatogenic defects.
Our study has some limitations. Because of low mutation rates in the Chinese population, the insufficient number of biological samples from patients harboring homozygous CFTR p.G970D has limited the generalization of our findings. Therefore, the role of this mutation in azoospermia requires confirmation in a larger patient cohort. Recent research reported that among 263 Chinese patients with CBAVD, 5 (1.9%) patients were detected for copy number variants in the region of the CFTR gene (four of them carried partial deletions and one carried partial duplication of CFTR),37 but other studies did not find copy number variations of CFTR in males with CBAVD.38,39 A larger number of samples and more sensitive gene detection methods are essential for further research. Although a specific knock-in mouse model supported our findings, further investigations are needed to clarify the function of CFTR in spermatogenesis in animal models in vivo. However, considering that the Cftr p.F508del mice are fertile40,41 and that CFTR functions differ among species, primate models may be considered if a mouse model is unavailable.
To conclude, our results revealed that CFTR p.G970D mutations might associate with disrupted spermatogenesis, especially during the early stage, in a Chinese patient. Furthermore, the Cftr p.G965D mutation altered Cftr RNA splicing in GC2-mutN6 cells, suggesting a specific role in germ cells. Further investigations are required to determine the mechanisms of CFTR p.G970D actions associated with spermatogenesis.
AUTHOR CONTRIBUTIONS
WMX and QTL conceived and designed the experiments. JWH drafted the manuscript. JWH and XLL performed the experiments. LW, EPT, CLD, and YQT collected the samples. NL analyzed the WES data. WMX and XHJ reviewed and revised the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declare no competing interests.
SUPPLEMENTARY MATERIALS AND METHODS
Minigene
The RHCglo vector (a gift from Professor Chen Lu from the School of Life Sciences, Sichuan University) was constructed by PCR using oligonucleotide primers containing restriction sites that were unique to the plasmid. An exon was derived from oligonucleotides that generated 5 overhangs compatible with BamHI and XhoI restriction sites. Exons were amplified by PCR using forward primers (5′-GGATCCTTTGGGTTCTGAATGCGTCT-3′) and reverse primers (5′-CTCGAGCCAATGCCAAAATGTCACAC-3′) from healthy human and patient genomic DNA of blood samples. The PCR products and RHCglo were digested with BamHI and XhoI, ligated, and sequenced to identify clones. The plasmid constructs were transfected into HEK293 cells for 48 h, then RNA was extracted. Splicing variants were verified by reverse transcription (RT) PCR. The RT and PCR upstream primer was RSV5U (5′-CATTCACCACATTGGTGTGC-3′), and the downstream primers were TNIE4 (5′-AGGTGCTGCCGCCGGGCGGTGGCTG-3′) or RTRHC (5′-GGGCTTTGCAGCAACAGTAAC-3′), which anneal upstream and downstream of the PpuMI site, respectively.
Other common mutations of CFTR or other typical CF symptoms were not found in proband. (a) Sanger sequencing results of proband show p.F508 and p.R117 wild-type alleles, TG12-T7-V470, but no other common CFTR mutations. (b) Mucosa of left middle nasal turbinate stained with hematoxylin-–eosin shows no obvious inflammatory phenotype in proband. CFTR: cystic fibrosis transmembrane conductance regulator; CF: cystic fibrosis.
The proliferation and matured CFTR of CFTR p.G970D mutant 293-mutK5 cells are seriously affected. (a) Expression of mature CFTR protein (180 kDa) in total protein of 293-mutK5 cells is decreased compared with wild-type 293 cells, and immature CFTR protein (150 kDa) has accumulated. (b) Ivacaftor which potentiates CFTR, partially restores inhibited proliferation of CFTR p.G970D mutated cells. (c) Sanger sequencing indicated no RNA splicing variant in 293-mutK5 cells. (d) Homozygous CFTR p.G970D missense mutation in patients does not affect pre-mRNA splicing. RSV5U-TNIE4 and RSV5U-RTRHC primers verified cDNA in HEK293 cells transfected with minigene vector. Results of agar gel electrophoresis show that compared with normal controls, minigene vector constructed from template DNA from a patient did not detect small bands caused by splicing changes. (e) Sequencing results of RT-PCR products of HEK293 cells after minigene transfection. Results of Sanger sequencing show intact exon 18, except for single nucleotide substitution corresponding to c.2909G>A variant; that is, first nucleotide of exon 18 is changed from G to A. Arrow, mutation site. RHCglo-Exon1/2 represents exon sequence of vector. CFTR: cystic fibrosis transmembrane conductance regulator; RT-PCR: reverse transcription polymerase chain reaction.
ACKNOWLEDGMENTS
We thank all patients and control individuals for their participation. We are grateful to the support from the National Key Research and Developmental Program of China (No. 2018YFC1003603) and the National Natural Science Foundation of China (No. 81971445).
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Agarwal A, Mulgund A, Hamada A, Chyatte MR. A unique view on male infertility around the globe. Reprod Biol Endocrinol. 2015;13:37. doi: 10.1186/s12958-015-0032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol. 2018;15:369–84. doi: 10.1038/s41585-018-0003-3. [DOI] [PubMed] [Google Scholar]
- 3.Ibtisham F, Wu J, Xiao M, An L, Banker Z, et al. Progress and future prospect of in vitro spermatogenesis. Oncotarget. 2017;8:66709–27. doi: 10.18632/oncotarget.19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Flynn O'Brien KL, Varghese AC, Agarwal A. The genetic causes of male factor infertility: a review. Fertil Steril. 2010;93:1–12. doi: 10.1016/j.fertnstert.2009.10.045. [DOI] [PubMed] [Google Scholar]
- 5.Tournaye H, Krausz C, Oates RD. Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol. 2017;5:544–53. doi: 10.1016/S2213-8587(16)30040-7. [DOI] [PubMed] [Google Scholar]
- 6.Krausz C. Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab. 2011;25:271–85. doi: 10.1016/j.beem.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Rudnik-Schoneborn S, Messner M, Vockel M, Wirleitner B, Pinggera GM, et al. Andrological findings in infertile men with two (biallelic) CFTR mutations: results of a multicentre study in Germany and Austria comprising 71 patients. Hum Reprod. 2021;36:551–9. doi: 10.1093/humrep/deaa348. [DOI] [PubMed] [Google Scholar]
- 8.Yu J, Chen Z, Ni Y, Li Z. CFTR mutations in men with congenital bilateral absence of the vas deferens (CBAVD): a systemic review and meta-analysis. Hum Reprod. 2012;27:25–35. doi: 10.1093/humrep/der377. [DOI] [PubMed] [Google Scholar]
- 9.Dumur V, Gervais R, Rigot JM, Lafitte JJ, Manouvrier S, et al. Abnormal distribution of CF ΔF508 allele in azoospermic men with congenital aplasia of epididymis and vas deferens. Lancet. 1990;336:512. doi: 10.1016/0140-6736(90)92066-q. [DOI] [PubMed] [Google Scholar]
- 10.Xu WM, Chen J, Chen H, Diao RY, Fok KL, et al. Defective CFTR-dependent CREB activation results in impaired spermatogenesis and azoospermia. PLoS One. 2011;6:e19120. doi: 10.1371/journal.pone.0019120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomaiuolo R, Fausto M, Elce A, Strina I, Ranieri A, et al. Enhanced frequency of CFTR gene variants in couples who are candidates for assisted reproductive technology treatment. Clin Chem Lab Med. 2011;49:1289–93. doi: 10.1515/CCLM.2011.637. [DOI] [PubMed] [Google Scholar]
- 12.Li CY, Jiang LY, Chen WY, Li K, Sheng HQ, et al. CFTR is essential for sperm fertilizing capacity and is correlated with sperm quality in humans. Hum Reprod. 2010;25:317–27. doi: 10.1093/humrep/dep406. [DOI] [PubMed] [Google Scholar]
- 13.Kanavakis E, Tzetis M, Antoniadi T, Pistofidis G, Milligos S, et al. Cystic fibrosis mutation screening in CBAVD patients and men with obstructive azoospermia or severe oligozoospermia. Mol Hum Reprod. 1998;4:333–7. doi: 10.1093/molehr/4.4.333. [DOI] [PubMed] [Google Scholar]
- 14.Sharma H, Mavuduru RS, Singh SK, Prasad R. Increased frequency of CFTR gene mutations identified in Indian infertile men with non-CBAVD obstructive azoospermia and spermatogenic failure. Gene. 2014;548:43–7. doi: 10.1016/j.gene.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 15.Hojati Z, Heidari S, Motovali-Bashi M. Exon 10 CFTR gene mutation in male infertility. Iran J Reprod Med. 2012;10:315–20. [PMC free article] [PubMed] [Google Scholar]
- 16.Gallati S, Hess S, Galie-Wunder D, Berger-Menz E, Bohlen D. Cystic fibrosis transmembrane conductance regulator mutations in azoospermic and oligospermic men and their partners. Reprod Biomed Online. 2009;19:685–94. doi: 10.1016/j.rbmo.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Stuppia L, Antonucci I, Binni F, Brandi A, Grifone N, et al. Screening of mutations in the CFTR gene in 1195 couples entering assisted reproduction technique programs. Eur J Hum Genet. 2005;13:959–64. doi: 10.1038/sj.ejhg.5201437. [DOI] [PubMed] [Google Scholar]
- 18.Fedder J, Jorgensen MW, Engvad B. Prevalence of CBAVD in azoospermic men carrying pathogenic CFTR mutations –evaluated in a cohort of 639 non-vasectomized azoospermic men. Andrology. 2021;9:588–98. doi: 10.1111/andr.12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slezak R, Szczepaniak M, Pasinska M, Czemarmazowicz H. [The analysis of CFTR mutations in men with azoospermia, oligozoospermia and asthenozoospermia. Ginekol Pol. 2007;78:605–10. [Article in Polish] [PubMed] [Google Scholar]
- 20.Ravnik-Glavac M, Svetina N, Zorn B, Peterlin B, Glavac D. Involvement of CFTR gene alterations in obstructive and nonobstructive infertility in men. Genet Test. 2001;5:243–7. doi: 10.1089/10906570152742308. [DOI] [PubMed] [Google Scholar]
- 21.Tuerlings JH, Mol B, Kremer JA, Looman M, Meuleman EJ, et al. Mutation frequency of cystic fibrosis transmembrane regulator is not increased in oligozoospermic male candidates for intracytoplasmic sperm injection. Fertil Steril. 1998;69:899–903. doi: 10.1016/s0015-0282(98)00050-8. [DOI] [PubMed] [Google Scholar]
- 22.Xu WM, Shi QX, Chen WY, Zhou CX, Ni Y, et al. Cystic fibrosis transmembrane conductance regulator is vital to sperm fertilizing capacity and male fertility. Proc Natl Acad Sci U S A. 2007;104:9816–21. doi: 10.1073/pnas.0609253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashiro Y, Shimizu T, Oguchi S, Shioya T, Nagata S, et al. The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nutr. 1997;24:544–7. doi: 10.1097/00005176-199705000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Powers CA, Potter EM, Wessel HU, Lloyd-Still JD. Cystic fibrosis in Asian Indians. Arch Pediatr Adolesc Med. 1996;150:554–5. doi: 10.1001/archpedi.1996.02170300108024. [DOI] [PubMed] [Google Scholar]
- 25.Wagner JA, Vassilakis A, Yee K, Li M, Hurlock G, et al. Two novel mutations in a cystic fibrosis patient of Chinese origin. Hum Genet. 1999;104:511–5. doi: 10.1007/s004390050996. [DOI] [PubMed] [Google Scholar]
- 26.Liu K, Xu W, Xiao M, Zhao X, Bian C, et al. Characterization of clinical and genetic spectrum of Chinese patients with cystic fibrosis. Orphanet J Rare Dis. 2020;15:150. doi: 10.1186/s13023-020-01393-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo X, Liu K, Liu Y, Situ Y, Tian X, et al. Clinical and genetic characteristics of cystic fibrosis in Chinese patients: a systemic review of reported cases. Orphanet J Rare Dis. 2018;13:224. doi: 10.1186/s13023-018-0968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian X, Liu Y, Yang J, Wang H, Liu T, et al. p. G970D is the most frequent CFTR mutation in Chinese patients with cystic fibrosis. Hum Genome Var. 2016;3:15063. doi: 10.1038/hgv.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Wen Q, Li H, Zhao L, Zhang X, et al. Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) in Chinese patients with congenital bilateral absence of vas deferens. J Cyst Fibros. 2012;11:316–23. doi: 10.1016/j.jcf.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Timmreck LS, Gray MR, Handelin B, Allito B, Rohlfs E, et al. Analysis of cystic fibrosis transmembrane conductance regulator gene mutations in patients with congenital absence of the uterus and vagina. Am J Med Genet A. 2003;120 A:72–6. doi: 10.1002/ajmg.a.20197. [DOI] [PubMed] [Google Scholar]
- 32.Stuhrmann M, Dork T. CFTR gene mutations and male infertility. Andrologia. 2000;32:71–83. doi: 10.1046/j.1439-0272.2000.00327.x. [DOI] [PubMed] [Google Scholar]
- 33.van der Ven K, Messer L, van der Ven H, Jeyendran RS, Ober C. Cystic fibrosis mutation screening in healthy men with reduced sperm quality. Hum Reprod. 1996;11:513–7. doi: 10.1093/humrep/11.3.513. [DOI] [PubMed] [Google Scholar]
- 34.Patrizio P, Ord T, Silber SJ, Asch RH. Cystic fibrosis mutations impair the fertilization rate of epididymal sperm from men with congenital absence of the vas deferens. Hum Reprod. 1993;8:1259–63. doi: 10.1093/oxfordjournals.humrep.a138237. [DOI] [PubMed] [Google Scholar]
- 35.Bieth E, Hamdi SM, Mieusset R. Genetics of the congenital absence of the vas deferens. Hum Genet. 2021;140:59–76. doi: 10.1007/s00439-020-02122-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H, Ruan YC, Xu WM, Chen J, Chan HC. Regulation of male fertility by CFTR and implications in male infertility. Hum Reprod Update. 2012;18:703–13. doi: 10.1093/humupd/dms027. [DOI] [PubMed] [Google Scholar]
- 37.Ma C, Wang R, Li T, Li H, Wang B. Analysis of CNVs of CFTR gene in Chinese Han population with CBAVD. Mol Genet Genomic Med. 2020;8:e1506. doi: 10.1002/mgg3.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, An M, Liu Y, Hu K, Jin Y, et al. Genetic diagnosis and sperm retrieval outcomes for Chinese patients with congenital bilateral absence of vas deferens. Andrology. 2020;8:1064–9. doi: 10.1111/andr.12769. [DOI] [PubMed] [Google Scholar]
- 39.Yuan P, Liang ZK, Liang H, Zheng LY, Li D, et al. Expanding the phenotypic and genetic spectrum of Chinese patients with congenital absence of vas deferens bearing CFTR and ADGRG2 alleles. Andrology. 2019;7:329–40. doi: 10.1111/andr.12592. [DOI] [PubMed] [Google Scholar]
- 40.Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, et al. A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest. 1995;96:2051–64. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Philp AR, Riquelme TT, Millar-Buchner P, Gonzalez R, Sepulveda FV, et al. Kcnn4 is a modifier gene of intestinal cystic fibrosis preventing lethality in the Cftr-F508del mouse. Sci Rep. 2018;8:9320. doi: 10.1038/s41598-018-27465-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Other common mutations of CFTR or other typical CF symptoms were not found in proband. (a) Sanger sequencing results of proband show p.F508 and p.R117 wild-type alleles, TG12-T7-V470, but no other common CFTR mutations. (b) Mucosa of left middle nasal turbinate stained with hematoxylin-–eosin shows no obvious inflammatory phenotype in proband. CFTR: cystic fibrosis transmembrane conductance regulator; CF: cystic fibrosis.
The proliferation and matured CFTR of CFTR p.G970D mutant 293-mutK5 cells are seriously affected. (a) Expression of mature CFTR protein (180 kDa) in total protein of 293-mutK5 cells is decreased compared with wild-type 293 cells, and immature CFTR protein (150 kDa) has accumulated. (b) Ivacaftor which potentiates CFTR, partially restores inhibited proliferation of CFTR p.G970D mutated cells. (c) Sanger sequencing indicated no RNA splicing variant in 293-mutK5 cells. (d) Homozygous CFTR p.G970D missense mutation in patients does not affect pre-mRNA splicing. RSV5U-TNIE4 and RSV5U-RTRHC primers verified cDNA in HEK293 cells transfected with minigene vector. Results of agar gel electrophoresis show that compared with normal controls, minigene vector constructed from template DNA from a patient did not detect small bands caused by splicing changes. (e) Sequencing results of RT-PCR products of HEK293 cells after minigene transfection. Results of Sanger sequencing show intact exon 18, except for single nucleotide substitution corresponding to c.2909G>A variant; that is, first nucleotide of exon 18 is changed from G to A. Arrow, mutation site. RHCglo-Exon1/2 represents exon sequence of vector. CFTR: cystic fibrosis transmembrane conductance regulator; RT-PCR: reverse transcription polymerase chain reaction.