

Abstract
The past two centuries have witnessed an unprecedented rise in human life expectancy. Sustaining longer lives with reduced periods of disability will require an understanding of the underlying mechanisms of ageing, and genetics is a powerful tool for identifying these mechanisms. Large-scale genome-wide association studies have recently identified many loci that influence key human ageing traits, including lifespan. Multi-trait loci have been linked with several age-related diseases, suggesting shared ageing influences. Mutations that drive accelerated ageing in prototypical progeria syndromes in humans point to an important role for genome maintenance and stability. Together, these different strands of genetic research are highlighting pathways for the discovery of anti-ageing interventions that may be applicable in humans.
Ageing is a seemingly universal biological phenomenon; yet, it is surprisingly difficult to define. In essence, ageing is a term used to describe a correlated set of declines in functioning with advancing chronological age, which generally begins after sexual maturity1,2. Characteristic functional changes occur at the molecular and cellular levels, known as biological “hallmarks” of ageing2, through to declining physiological homeostasis at the organism level as well as lessening abilities to perform everyday physical and cognitive tasks. Ageing also increases the susceptibility to many common diseases, with death rates rising approximately exponentially with advancing age3. The main scientific effort in human ageing, termed ‘geroscience’4, sees ageing as the primary cause of many chronic diseases of later life, including Alzheimer disease, chronic kidney disease, coronary artery disease (CAD), osteoarthritis, stroke, type 2 diabetes mellitus (T2DM) and common cancers, such as breast, prostate and colorectal cancer. Slowing ageing might prevent many of these diseases simultaneously4, as seen in various experimental interventions in laboratory models that slow and partially reverse features of ageing (BOX 1).
Box 1 |. Slowing ageing in laboratory models.
Interventions in laboratory models can delay or reverse specific aspects of ageing. These models provide robust insights into ageing mechanisms, which might be conserved across species.
Targeting nutrient sensing and dietary restriction
Various genetic manipulations can markedly prolong survival in Caenorhabditis elegans. Long-lived genetic mutants may result from modulation of different, independent biological pathways. For example, daf-2 affects nutrient sensing through modulation of insulin–insulin-like growth factor 1 (IGF1) signalling and upregulation of autophagy147; eat-2 codes for a defective nicotinic acetylcholine receptor, causing impaired pharyngeal pumping and a genetic form of caloric restriction148,149; knockdown of ife-2 increases lifespan by downregulating protein translation150; and knockdown of the clock gene clk-1 affects longevity by impairing mitochondrial electron transport chain activity, thereby reducing oxidative stress151,152. However, only the modulation of insulin–IGF1 (via daf-2) shows an increased healthspan and, in all four long-lived mutants, the period of frailty is disproportionally extended compared with lifespan153.
Restricting calorie intake while maintaining necessary micronutrient intakes produces lean animals and improves functioning and lengthens life in many but not all mouse strains154. Dietary restriction increases longevity by modulating the expression of growth factors, such as IGF1, and affecting the mammalian target of rapamycin (mTOR) and ribosomal protein S6 kinase (S6K) signalling pathway155. The effectiveness of dietary restriction in promoting healthspan and lifespan seems to be evolutionarily conserved in yeast, worms, flies and some mouse strains154. However, in primates, the effect of caloric restriction varied between experiments, with some detectable positive effects on metabolic disease prevention, which may be partially related to the effect of the intervention on body composition156. Nevertheless, whether such positive effects of metabolic controls are strong enough to affect longevity remains unclear.
Caloric restriction in healthy, non-obese humans has beneficial effects on multiple cardiometabolic risk factors, might prevent decline of memory and is associated with the slowing down of a proxy biomarker of biological ageing157–159, although these findings need confirmation with longer follow-up times. Several nutritional interventions are under development to delay ageing, including different forms of caloric restriction, fasting and calorie restriction mimetics, agents that mimic the beneficial effects of dietary restriction without necessarily restricting calories, thus avoiding detrimental impact160,161.
Targeting mTOR
The mTor signalling pathway, a central regulator of cell metabolism, growth, proliferation and survival162, has been extensively implicated in ageing. Rapamycin, a licensed immunosuppressive drug targeting mTOR, extended healthspan and lifespan, even in already older mice, in randomized trials in three different studies163. A human trial of a low-dose rapamycin analogue reported enhanced immune function and a reduction in infections but the potential effect on longevity in humans is unknown164. The effects of rapamycin include increasing autophagy, which may counteract immune senescence by affecting energy metabolism and organelle recycling as well as the fate and function of immune cells165.
Targeting senescent cells
A wide range of biological stresses that cause cell damage can lead to cellular senescence, in which cells become unable to replicate, secrete large quantities of cytokines, chemokines, proteases and growth factors, and develop resistance to apoptosis82. A targeted removal of senescent cells expressing p16ink4a resulted in the reversal of several features of ageing in mouse models81. The development of senolytic molecules that can selectively induce apoptosis of senescent cells is an active area of investigation, with a ‘first in man’ clinical trial in idiopathic pulmonary fibrosis recently reporting efficacy and safety166.
Although declining function is characteristic of ageing, the rates of decline vary enormously. Whereas some people die of age-related disease in their 60s, a few are still active at 100 years of age and beyond. Understanding the genetic factors driving this variability in ageing between people is the main focus of this Review. The scope for experimental studies in human ageing is limited and conventional observational studies are often distorted by confounding factors such as smoking or obesity. However, over the past decade, results of genome-wide association studies (GWAS)5 for many phenotypes relevant to ageing have started to emerge. Inherited (that is, germline) variant associations are minimally affected by confounding and can provide unique biological insights to clarify whether observations in laboratory animals apply to humans. Furthermore, drugs with support from human genetic studies for related effects (reported in GWAS and single-gene disorder databases) succeed from phase I trials to final approval twice as often as those without such evidence6. Indeed, even genes harbouring variants with small effect sizes can provide successful drug targets5. Therefore, pinpointing the genetic variation that influences human ageing could help identify suitable targets for anti-ageing interventions in humans.
Genome-wide association studies.
(GWAS). A study that involves genotyping large numbers of participants to identify statistical associations between genetic variants and traits of interest.
In this Review, we discuss recent results from the most robust genetic association studies relevant to human ageing, with a focus on ‘multi-trait’ variants, that is, variants that affect several ageing-related phenotypes simultaneously, reflecting the view that correlated changes of ageing have common mechanisms. We describe three key accelerated ageing (progeroid) genetic disorders in humans and review evidence on the accumulation of somatic mutations with advancing age, with these two sections providing insights into underlying DNA damage processes that increase cancer risk and may also contribute to functional declines in ageing. Evidence is accumulating for age-related epigenetic7, gene expression and splicing8 changes; however, as the causal roles of these changes are uncertain, we do not discuss these topics.
Somatic mutations.
Changes to the genetic code arising from errors during DNA damage repair, DNA replication or mitosis, occurring in somatic (non-germline) tissues.
Heritability of ageing traits
Ideal human ageing phenotypes would measure the underlying biological mechanisms and minimize ‘extraneous’ environmental factors9. At the molecular level, hallmark biological characteristics of ageing include genomic instability (especially DNA damage), telomere attrition, epigenetic alterations, loss of protein homeostasis and deregulated nutrient sensing2. At the cellular level, ageing is associated with stem cell exhaustion, mitochondrial dysfunction with falling energy outputs and increased oxidative stress, as well as altered intercellular communications2. Unfortunately, few of these hallmarks can currently be measured directly in large enough samples to be analysed in human genetic association studies and therefore other age-related phenotypes must serve as proxies (BOX 2).
Box 2 |. Proxy phenotypes of ageing.
Lifespan
Lifespan, the time from birth to death, is an often-measured ageing proxy. Longevity (used here to mean relatively long lifespans, combining a variety of specific definitions) is a particularly popular ageing measure, as some long-lived people, especially centenarians, experience delayed onset of age-related diseases, with compression of morbidity into a smaller proportion of their lifespans12. For example, in one study of centenarians, 32% of men and 15% of women had no age-associated diagnoses at age 100 years167. Longevity has been defined in terms of arbitrary age cut-off points (for example, ≥85 years or centenarians) or by, for example, the longest 10% or 1% of lifespans within a specified cohort168. Some studies have focused on exceptional longevity in the context of long-lived families, as there is evidence of increased heritability of longevity in this context44.
Disease and physiological markers
Clinical diagnoses of age-associated chronic diseases and cancers can provide accessible proxy measures of ageing. A summary measure of healthspan62 can be made from the duration of life spent without major age-associated conditions, including dementia, myocardial infarction, stroke and type 2 diabetes mellitus. Other elements can be included in healthspan, such as physical disabilities, cognitive impairments and mental wellbeing.
Biomarkers — currently mostly of clinical measures such as lipid levels, inflammation, and kidney and liver function169,170 — can be combined into indices that predict later clinical outcomes in ageing and can serve as proxy ageing phenotypes. Various physiological characteristics that decline with age are measurable, for example, muscle strength171, cognitive function172 and gait speed173. Measures of disability and frailty — the age-associated increase in vulnerability to stresses and adverse outcomes174 — have also been developed.
Challenges in studying ageing
In addition to variations in study design and the vast range of ageing-related phenotypes studied, ageing research is bedevilled by a lack of standard terminology9. More fundamentally, the validity of essentially all available measures of ageing itself is debateable. results of analyses can depend on the specific combination of causal factors driving each phenotype; for example, including smoking-induced cancers in disease measures results in smoking-related genes emerging as important, whereas including common skin cancers will increase the importance of DNA damage pathways related to UV light exposure. Ultimately, human ageing always occurs in the context of common environmental exposures in studied populations, and what to include and exclude in proxy measures of ageing will likely remain a matter of debate until direct measures of the hallmarks of ageing become available.
To what extent are these proxy ageing phenotypes driven by genetic variation between people? Twin- or family-based estimates indicate substantial inherited genetic influences. Heritability estimates the ratio of the genetic component to the sum of genetic and other (termed ‘environmental’) factors, and is specific to each studied population and the environmental exposures current in that population10. Widely cited heritability estimates for human longevity from twin studies range from 20% to 30%11, whereas in families with a centenarian, estimates increase to 48% in men and 33% in women12. However, a recent analysis of millions of family trees estimated the heritability of longevity to be 16% (standard error 0.4%), suggesting that previous studies may have overestimated the heritability of longevity13. A second family tree study found evidence for extensive assortative mating inflating heritability estimates and concluded that the true heritability of longevity is below 10%14.
Heritability.
The proportion of variance in a phenotype that can be attributed to genetic differences among individuals in a given population. Narrow-sense heritability estimates additive genetic effects. Broad-sense heritability includes both additive and dominance effects.
Common age-related diseases also have substantial heritable components; for example, hip osteoarthritis is estimated to be 68% heritable15, T2DM heritability is estimated at 61–78%16, the heritable component of Alzheimer disease is estimated to be 58–79% (depending on the model used)17, and cardiovascular disease heritability has been estimated at 45–69%18. Individual genetic variants that are associated with many ageing phenotypes have now been identified, mostly by GWAS, and the amount of variation in the trait attributed to these specific genetic differences can be estimated. These genotyped variants cumulatively explain 51.9% of the variation in osteoarthritis of the hip19 and 18% in T2DM20 but only 8.5% of the variation in parental age at death21 and 7.1% in Alzheimer disease22. The gap between the higher twin study-based heritability estimates for each phenotype and the lower estimates from GWAS, often termed the ‘missing heritability’, is attributed partly to a lack of coverage of several types of genetic variation, including rare large-effect variants in GWAS, but also to possible overestimates in pedigree-based analyses23.
Nonetheless, many ageing-related diseases and traits seem to have substantial heritable components. However, heritability estimates do not help identify pathways or mechanisms. For genetics to help reveal biological mechanisms, find new drug targets and potentially identify individuals for precision medicine treatments, specific human genetic variants associated with ageing phenotypes must be determined. Such variants have recently been identified even for traits with modest heritability.
Candidate gene studies and GWAS
Ageing-related genetic variants have been identified mostly in GWAS, which test statistical associations between millions of germline variants and a phenotype, often in several hundred thousand people5. Smaller-scale candidate studies test a subset of variants, yet both approaches include multiple statistical tests. As more variants are compared, it becomes increasingly likely that some will be statistically significant merely by chance5. To guard against false-positive findings, stringent statistical significance thresholds are used, and ‘significant’ associations need to be replicated in independent samples. In reviewing ageing studies, we therefore prioritize the strongest available associations (preferably at a genome-wide statistical significance24 of P < 5 × 10–8), especially those with independent replication.
Parental lifespan
The most robust identifications of lifespan-related variants currently come from recent, very large cohorts such as the UK Biobank25, which includes 500,000 community volunteers. These studies have focused on parental age at death, as offspring health status and survival are associated with parental lifespan. For example, analysis of data from the University of Michigan Health and Retirement Study, a longitudinal study of approximately 20,000 participants aged 51–61 years at baseline who were followed up for 18 years, found that all-cause mortality consistently declined by 19% for each decade that participants’ mothers had survived beyond 65 years of age (14% per decade for fathers)26. Additionally, study participants had progressively lower incidences of cardiovascular disease and cancers26 as well as reduced rates of cognitive decline27, with increasing parental survival. A study of 186,151 non-adopted UK Biobank participants followed up for up to 6 years produced similar results, with declines in all-cause mortality of 16% per decade of mothers’ survival ≥70 years of age (HR 0.84, 95% CI 0.79–0.89) and 17% for fathers’ survival (HR 0.83, 95% CI 0.78–0.89). Cause-specific mortality declined with advancing parental ages, especially for coronary heart disease (20% per decade with decades of mothers’ age ≥70 years: HR 0.80, 95% CI 0.68–0.95; and 21% for fathers’ age: HR 0.79, 95% CI 0.63–0.98), but declines in cancer mortality (HR 0.92, 95% CI 0.90–0.95) were also present28.
Several GWAS have been reported for parental age at death (or attained age thus far) either in the UK Biobank cohort alone21,29,30 or in meta-analyses combining UK Biobank data with data from other cohorts; the LifeGen consortium included data on approximately 160,000 study participants from 25 cohorts in addition to UK Biobank data31, and a separate analysis included pedigree data from AncestryDNA32 on 300,000 individuals for a meta-analyses with UK Biobank data. The ages at death of the parents (or current age if still alive) studied in the LifeGen analysis31 varied from 40 to 107 years, whereas the AncestryDNA32 lifespans ranged from 40 to 120 years. Six genetic loci were identified in both studies for longer parental lifespan (TABLE 1), with 12 additional loci identified only in one or the other study. Many of the implicated variants have been linked previously to cardiometabolic conditions (mostly myocardial infarction and T2DM), with some linked to Alzheimer disease, autoimmunity and cancer risk31, thus reflecting the more common causes of death in older people.
Table 1 |.
Genetic variants associated with parental lifespan
| rsID (effect allele) | Effecta | Mapped genes | Gene name | Variant position | Associated disease | |
|---|---|---|---|---|---|---|
| Loci significant in bothb GWAS meta-analyses31,32 | ||||||
| rs429358 (T) | 1.06 | APOE | Apolipoprotein E | Missense | Cardiometabolic, dementia | |
| rs10455872 (A) | 0.76 | LPA | Lipoprotein A | Intronic | Cardiometabolic | |
| rs8042849 (T) c | 0.44 | CHRNA3/5 | Cholinergic receptor nicotinic α3/5 subunit | Intronic | Smoking related | |
| rs142158911 (A) | 0.36 | LDLR | Low-density lipoprotein receptor | Intergenic | Cardiometabolic | |
| rs11065979 (C) d | 0.28 | SH2B3, ATXN2 | SH2B adaptor protein 3, ataxin 2 | Intergenic | Cardiometabolic, cancer, autoimmunitye | |
| rs1556516 (G) | 0.25 | CDKN2B-AS1 | CDKN2B antisense RNA 1 | Intronic | Cardiometabolic, cancere | |
| Loci significant only in the UK Biobank and LifeGen cohorts 31 | ||||||
| rs34967069 (T) | 0.56 | HLA-DQA1 | Major histocompatibility complex, class II, DQ alpha 1 | Intergenic | Autoimmune | |
| rs1230666 (G) | 0.32 | MAGI3 | Membrane associated guanylate kinase, WW and PDZ domain containing 3 | Intronic | Autoimmune | |
| rs12924886 (A) | 0.28 | HP | Haptoglobin | Intergenic | Cardiometabolic | |
| rs1275922 (G) | 0.26 | KCNK3 | Potassium two pore domain channel subfamily K member 3 | Intronic | Cardiometabolic | |
| rs6224 (G) f | 0.25 | FURIN/FES | Furin, paired basic amino acid cleaving enzyme | Intronic | Cardiometabolic | |
| rs61348208 (T) | 0.23 | HTT | Huntingtin | Intronic | NR | |
| Loci significant only in the UK Biobank and AncestryDNA cohorts 32 | ||||||
| rs7844965 (G) g | 0.25 | EPHX2 | Epoxide hydrolase 2 | intronic | NR | |
| rs4774495 (G) g | 0.23 | SEMA6D | Semaphorin 6D | intronic | NR | |
| rs599839 (G)g | 0.21 | CELSR2, PSRC1 | Cadherin EGF LAG seven-pass G-type receptor 2, proline and serine rich coiled-coil 1 | intergenic | Cardiometabolic | |
| rs3131621 (G) g | 0.20 | MICA/B | MHC class I polypeptide-related sequence A/B | intergenic | NR | |
| rs15285 (G) g | 0.18 | LPL | Lipoprotein lipase | 3’ UTR | Cardiometabolic | |
| rs9872864 (G) h | 0.14 | IP6K1 | Inositol hexakisphosphate kinase 1 | intronic | NR | |
GWAS, genome-wide association study; NR , none reported; rsID, Reference SNP cluster ID; UTR , untranslated region.
Effect, years added to lifespan of parents (from LifeGen analysis31).
rsID (effect allele) and ‘Effect’ information from LifeGen analysis31. The AncestryDNA analysis may have reported a different lead SNP for the same locus.
Intron variant of the HYKK gene in the CHRNA3/5 locus.
Variant located between ATXN2 and BRAP but is correlated (R2 > 0.8) with missense variant in SH2B3.
Not exhaustive list.
Located in the intron of gene FURIN.
Significantly associated with fathers’ lifespan, not mothers’.
Significantly associated with mothers’ lifespan, not fathers’.
Gene–environment interactions were evident for some variants. For example, the variant rs1051730 lies in an exon of CHRNA3, which encodes a nicotinic acetylcholine receptor subunit, and the lifespan-reducing allele was correlated with rs8042849, which increases susceptibility to nicotine dependence33 and likely reduces lifespan by increasing smoking exposure. Interestingly, this association was stronger for fathers’ age at death than mothers’ age at death, possibly owing to gender differences in smoking in the parental generation21. Effect sizes for all lifespan-associated variants were modest, with the largest per allele effect for the APOE ɛ4 variant accounting for 1.06 years of parental lifespan31. The smallest-effect variant identified by LifeGen was intronic in the HTT gene (also known as Huntingtin), although the relationship of this variant to Huntington disease mutations is unclear. Of the 18 variants identified, only one (in APOE) was exonic and affected the coding sequence, suggesting mainly regulatory effects, as is common for polygenic traits34.
In a UK Biobank GWAS, sub-analysis of the participants’ genotypes in the top 10% of parental survival (with survival to ≥90 years in mothers and ≥87 years in fathers) produced results similar to overall lifespan analyses29: four loci remained associated at genome-wide significance (APOE, CHRNA3, LPA and CDKN2B-AS1), with the others remaining nominally significant (all with P values <0.002). Two additional loci (MC2R and USP2-AS1) were significant for the top 10% survival in the analysis of parental lifespan. These results were consistent with an analysis of centenarian parents, with similar genotype– lifespan effect sizes29, although numbers here were small, with only 1,181 participants having at least one centenarian parent, meaning that only the APOE locus reached genome-wide significance29. Thus, lifespan-associated variants can also be important for longevity, with some variants likely being specific to longevity.
Genetic associations with longevity.
GWAS have also directly compared long-lived individuals (aged ≥90 years) to younger control individuals (aged <65 years, although definitions vary35–38, as recently reviewed elsewhere3). The most recent meta-analysis included 11,262 participants who lived beyond the 90th percentile38. The most robust findings have been for the APOE haplotypes, with ɛ4 being less common in long-lived participants and APOE ɛ2 more common (versus the ɛ3 haplotype). The two APOE haplotypes have similarly inverse associations with Alzheimer disease22 and cardiovascular disease39. Apolipoprotein E (APOE) is involved in the transport of cholesterol and other lipids to cells; in the brain, this function is important in neural cell membrane and synapse maintenance and repair40, although the full mechanisms causing Alzheimer disease remain elusive. A recent study showed that the APOE ɛ4 haplotype was associated with excess mortality even within the longest-lived 1% of survivors, whereas the ɛ2/ɛ2 or ɛ2/ɛ3 haplotypes were associated with modestly decreased mortality within the longest-lived 1% of survivors41. The recent meta-analysis of longevity GWAS38 identified a new locus, GPR78; variants in this locus associated with longevity were not previously linked in GWAS with other traits, but the gene, which encodes G protein-coupled receptor 78, has been implicated in traits such as lung function42 in the GWAS Catalog43.
The study of extreme longevity has been refined recently by the finding that heritability is higher in those who are part of long-lived families and that environmental factors seem to be more important in sporadic longevity44. A GWAS in 583 families of the Long Life Family Study cohort (covering long-lived individuals and offspring, which unusually also included predicted longevities) confirmed associations at the APOE locus and also identified a variant (rs1927465) between the genes MYOF (which encodes myoferlin) and CYP26A1 (which encodes cytochrome P450 family 26 subfamily A member 1) at genome-wide significance45. At the time of writing, rs1927465 has not been reported in the GWAS Catalog for other phenotypes.
A much studied set of extreme longevity-associated variants has been reported in the FOXO3A gene, which encodes a transcription factor that influences energy metabolism, cell cycle regulation and inflammation, and is important in modulating the effect of calorie restriction on longevity in model organisms46. In the longest-lived 1% of survivors, 17 FOXO3A variants were more common than in controls (n = 2,072 aged ≥96 years versus <96 years); the strongest association was found for the variant rs4946935 (OR 1.20 for extreme longevity, P = 3.2 × 10−5)47. However, none of these 17 variants affected death rates for the younger 99% of lifespans, which is consistent with no FOXO3A variants reaching genome-wide significance in the large parental lifespan GWAS discussed above29,31.
The evidence on mostly candidate gene variants has also been reviewed, comparing groups aged ≥85 years, including centenarians, versus those aged <85 years, with most aged <60 years48. Overall, seven variants claimed to be associated with longevity were found to be weakly or not associated with survival to age ≥85 years. It has been argued that different populations may have different exceptional longevity variants due to particular environmental exposures and ancestry-specific genetic differences49, providing a possible explanation for the limited replication of variants linked to exceptional longevity. Another explanation might be the fairly modest sample sizes (often fewer than 10,000 long-lived individuals) or potential false-positive findings of some associations.
None of the variants identified thus far as being associated with (extreme) longevity seems essential (that is, not all long-lived people harbour them) and none seems sufficient to achieve longevity (all are fairly common in groups who die earlier). This finding is consistent with the notion that the heritable component in the longest 10% for survival is a quantitative trait44 likely affected by large numbers of small effect variants.
Reproductive lifespan in women
Women are unusual compared with other female mammals in having a total lifespan that is substantially longer than their reproductive lifespan. In a GWAS of age at menopause50, 56 variants were identified, with approximately two-thirds of loci implicated in the DNA damage response (DDR)51. As discussed below, unrepaired DNA damage might be a major driver of overall ageing. Moreover, some of the menopause-associated variants have effects on the hypothalamic– pituitary axis, which controls many hormone levels. A polygenic risk score for each individual in a study population can be calculated by summing the number of risk-increasing alleles (weighted by published effect size) that each participants carries. However, a genetic risk score for age at menopause was not associated with parental lifespan in the UK Biobank21. Menopause-associated variants may therefore have limited effects on human ageing more generally.
Polygenic risk score.
Individual-level scores that summarize genetic risk (or protection) for a given phenotype. For each person, a score is computed by counting the number of effect alleles (genetic variants, weighted by their effect) that the person carries. A polygenic score is computed by summing scores from a large number, potentially all, of the variants in the genome.
Muscle strength
Decreasing muscle strength is a common feature of ageing and is associated with increased risks of cardiovascular disease and mortality, even in those aged <60 years52. A GWAS of the full range of strength (measured as grip strength) identified several lead variants in or near genes implicated in the structure and function of skeletal muscle fibres, neuronal maintenance and signal transduction in the central and peripheral nervous systems53. A targeted study of the human leukocyte antigen (HLA)-mediated autoimmunity-associated region reported associations with low muscle strength in those aged 60–70 years54 without autoimmune disease, but whether low muscle strength in older people is driven by the same variants is unclear.
Cognitive impairment
Normal ageing is often associated with impairment in some cognitive tests, even in the absence of dementia. Cognitive impairment has a number of risk factors, such as hypertension, some of which are treatable if caught early55. The largest recent genetic study of general cognitive function in >300,000 people identified >100 loci, implicating genes expressed in the brain but also including loci associated with traits such as hypertension, suggesting systemic effects on cognition56. A recent genetic analysis of decline in cognitive ability in 1,091 people found that the APOE ɛ4 allele alone was the strongest predictor compared with polygenic risk scores for CAD, educational attainment and other traits57. Another study in 1,176 men aged in their 50s58 found that genetic risk of Alzheimer disease was associated with mild cognitive impairments. Although these studies were limited by small sample sizes, cognitive impairment is evidently a complex multifactorial process in which inherited variation has a role in addition to lifestyle and other health-related factors59.
Age-related disease variants
Many GWAS of age-related diseases have been reported, with thousands of variants now identified5. One of the earliest successful GWAS identified variants that influence age-related macular degeneration60, the most common cause of blindness in the western world. Two large-effect loci were found: one mapped to CFH, which encodes complement factor H in the complement inflammation cascade, and one to ARMS2 (also known as HTRA1), which is involved in extracellular matrix turnover. Both variants were associated with more than 2.5-fold differences in age-related macular degeneration risk in a recent large study61. The effect sizes of these variants contrast with many other common disease-associated variants for which effect sizes are typically small, often with <10% differences in risk5.
Genetic variation across common diseases
Composite measures of healthspan
The duration of the period of life free from disease and functional limitations (that is, ‘healthspan’62) is an important measure of the physical health aspects of ageing well and was recently studied in UK Biobank participants aged 37–73 years62. All available age-associated diagnoses were included and a notable variant found to be associated with healthspan had previously been implicated in skin cancers63. As skin cancer represents the most common cancer type64, the prominence of skin cancer variants in the results of this composite healthspan measure is unsurprising. However, skin cancer risk is correlated to high levels of sun exposure65, which is a highly variable behavioural exposure, especially in older people. Thus, composite measures may highlight common exposures rather than necessarily identifying shared ageing mechanisms.
Multi-trait variants associated with disease
An alternative to examining composite measures of disease is to examine common loci associated with several different diseases of ageing. This approach is in line with the geroscience view that biological mechanisms of ageing underlie many of the diseases that occur in later life. The most common diseases of ageing, including Alzheimer disease, CAD, chronic kidney disease, osteoarthritis, stroke, T2DM and common cancers, have been extensively studied in GWAS. Of note, this list excludes diseases with known dominant environmental risk factors, such as lung cancer and chronic obstructive pulmonary disease, the risks of which are strongly related to smoking. Based on data from the GWAS Catalog, 961 genome-wide significant variant associations have been reported for common age-related conditions to date (Supplementary Tables 1 and 2). As biological pathways of ageing likely affect susceptibility to many of these diseases of later life, finding genetic variants associated with several pathways simultaneously should help reveal underlying ageing mechanisms.
Genetic correlation between multiple traits.
One approach to explore shared genetic effects is to estimate genetic correlation — the degree of genetic overlap between pairs of traits — using GWAS results66,67 (FIG. 1; Supplementary Table 3). This method uses linkage disequilibrium (LD) information in conjunction with the variant–trait associations to compute cross-trait LD score regression estimates of shared heritability (genetic correlation) and is well suited to analyses of complex traits with many thousands of small-effect variants that do not necessarily reach genome-wide significance66. Analyses of previously published data show that CAD, osteoarthritis, T2DM and stroke GWAS results correlate negatively with parental longevity, with 40–60% overlap (Supplementary Table 3), suggesting strong shared mechanisms. Alzheimer disease GWAS results had a moderate negative correlation with lifespan, but breast cancer and prostate cancer variants were not correlated with lifespan (Supplementary Table 3), suggesting that variants shared across diseases of ageing are important, albeit not for all phenotypes.
Fig. 1 |. Genetic overlap between age-related chronic diseases and parental longevity , based on correlations between whole-genome association results.
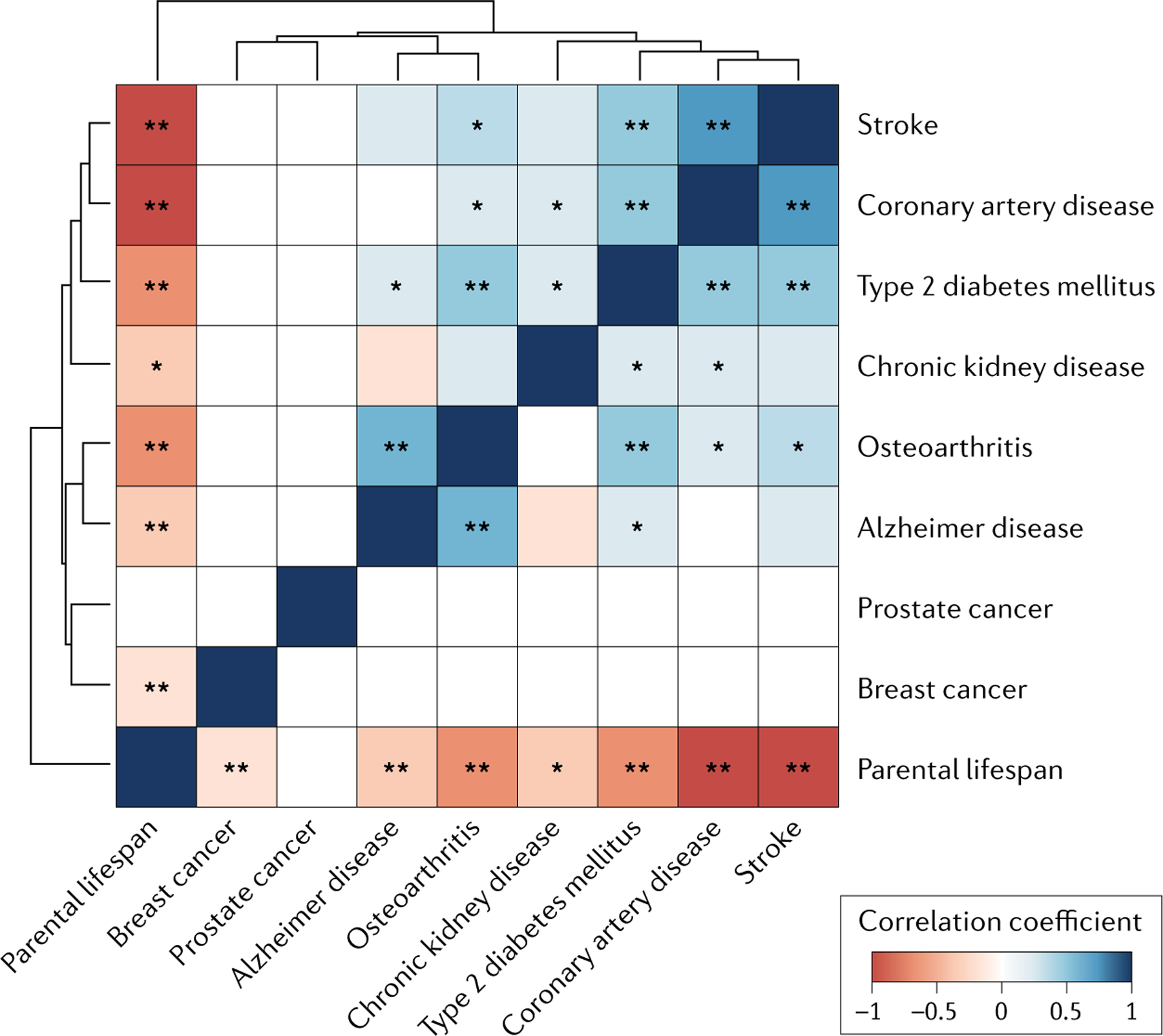
Genetic correlations are from linkage disequilibrium score regression methods using available genome-wide association study summary statistics20,22,31,39,76,77,83,91,184,185. Statistically significant correlations are indicated with an asterisk (single for nominal P < 0.05 significance, double indicates significant after Bonferroni correction for 36 tests in this analysis). The diseases and parental lifespans are ordered by similarity in patterns of genetic correlations using hierarchical clustering (see Supplementary Table 3 for details).
Linkage disequilibrium.
(LD). Non-random associations between alleles at different loci.
Associations with polygenic risk scores.
An alternative approach to revealing shared mechanisms is to test whether a polygenic risk score for a biomarker is associated with a phenotype. For example, increasing parental lifespan was associated with lower genetic risk of raised LDL-cholesterol levels and systolic blood pressure21. Additionally, a 7-month shortening in parental lifespan per unit of genetically determined increasing body mass index (BMI) has been reported30. This adverse effect of increased BMI contrasts with claims that being obese or overweight is beneficial in older people68 but is consistent with the success of calorie restriction in lengthening survival in animal models (BOX 1). Furthermore, high numbers of senescent cells accumulate in adipose tissue with age, especially around internal organs69,70.
The paradoxical obesity claims seem to be generated in part by weight loss resulting from serious disease in older people. In other words, older people with obesity must be fairly healthy to maintain their obesity. Longer-term observational analyses minimizing the effects of diseases that cause weight loss showed that those aged 65–74 years with obesity are at higher risk of dementia71 and have higher death rates72. However, whether avoiding obesity is important to reach centenarian status remains unclear73.
Shared genetic effects on ageing phenotypes
As noted, there were 961 genome-wide significant variant–trait associations across selected age-related diseases (Supplementary Table 2). Given our focus on variants that affect several age-related diseases simultaneously — as these are more likely to reveal underlying ageing mechanisms — we searched for loci associated with three or more of the selected age-related traits (with variants separated by <250 kb). We found 22 such ageing multi-trait loci; of these, 12 loci had variants in LD (R2 > 0.6) with each other (Supplementary Tables 4 and 5). One of those loci was APOE, which, as described above, has been associated with Alzheimer disease and CAD as well as with exceptional longevity and parental lifespan. Two other loci (LPA and LDLR) contain variants that affect blood lipid levels74 and cardiovascular traits39. The disease associations of the remaining nine multi-trait loci are more diverse (FIG. 2), as we discuss below.
Fig. 2 |. Selected loci with correlated variants associated with three or more age-related diseases or lifespan.
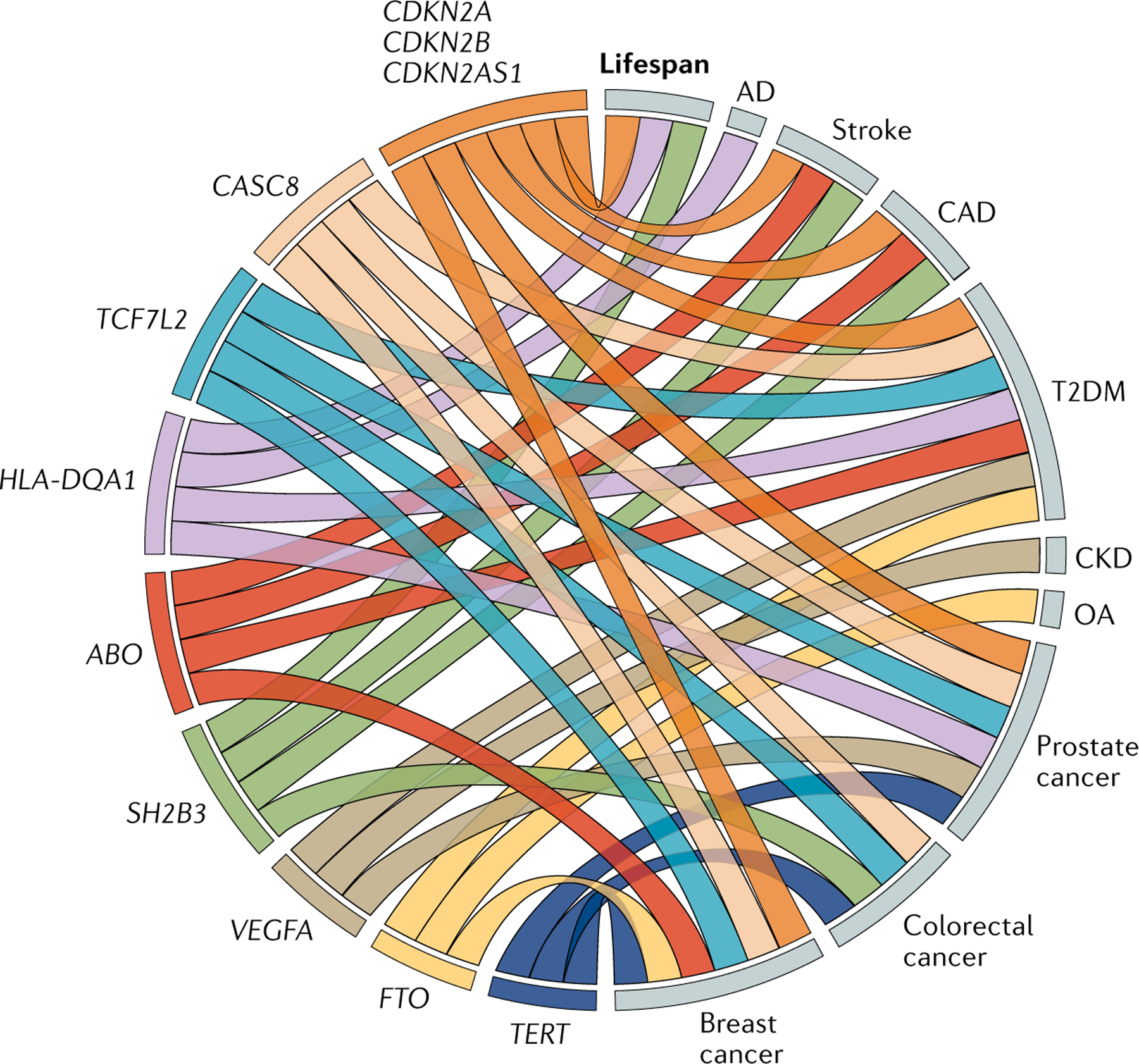
Nine loci were identified as hot spots for at least three major age-related diseases or lifespan in genome-wide association studies (GWAS) that included multiple correlated (R2 > 0.6) genetic variants20,22,31,39,76,77,83,91,184,185 (see Supplementary Tables 4 and 5 for details). The lipid-related variants LPA, LDLR and APOE were excluded for simplicity. Genes are shown on the left, with each ‘link’ to a disease on the right indicating a GWAS-identified signal. Lifespan refers to parental lifespan. Circos Table Viewer186 was used for visualization. AD, Alzheimer disease; CAD, coronary artery disease; CKD, chronic kidney disease; OA , osteoarthritis; T2DM, type 2 diabetes mellitus.
The CDKN2A/B and CAS8 loci
The CDKN2A/B locus (also known as the 9p21 locus) contains genes that produce the p16ink4a, p14arf and p15ink4b tumour suppressor proteins (FIG. 3) and the long non-coding RNA (lncRNA) CDKN2B-AS1 (also known as ANRIL), which regulates CDKN2A/B expression75. Variants in or near these genes have been associated with multiple diseases, including cardiovascular conditions39, T2DM20, cancers76,77, endometriosis78, glaucoma and related optic disc traits79 as well as blood cell count80 and parental lifespan31 (see Supplementary Table 6 for further details and references). Cell senescence is often accompanied by expression of p16ink4a (CDKN2A), and clearance of CDKN2A-expressing cells in mice attenuated age-related deterioration in the eye, kidney, heart and fat, with no overtly adverse effects81 (BOX 1). Efforts to develop interventions to remove senescent cells in humans are being actively pursued82.
Fig. 3 |. Disease-associated genetic variants in the 9p21.3 locus by effect size of association with parental lifespan.
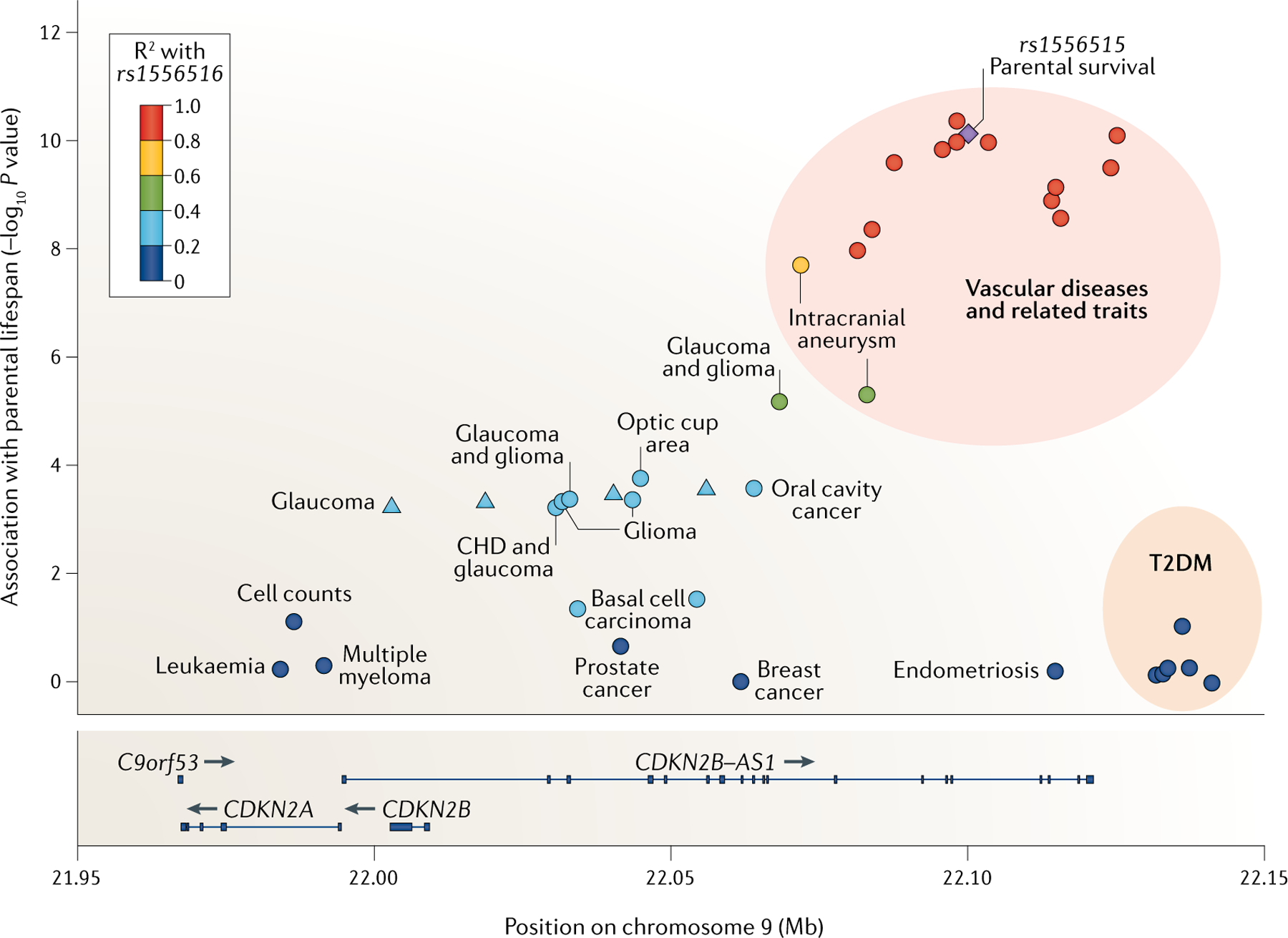
LocusZoom187 plot of known disease-associated variants in the 9p21.3 locus, which harbours the genes CDKN2A (encodes p16ink4a), CDKN2B (encodes p15ink4b) and CDKN2B-AS1 (encodes the long non-coding RNA ANRIL). Genetic variants are labelled with the traits they are reported to be associated with in published genome-wide association studies (GWAS) (see Supplementary Table 6 for details). The y-axis shows the association (−log10 P value) with parental lifespan from the 2019 UK Biobank and LifeGen cohort GWAS meta-analysis31. The variant in purple (rs1556516) is the lead signal at this locus from the parental lifespan analysis. The other variants are coloured according to their correlation with rs1556516 in 1000 Genomes European ancestry data (v. Nov 2014). CHD, coronary heart disease; T2DM, type 2 diabetes mellitus.
Interestingly, the disease-associated variants in the CDKN2A/B locus are not in coding areas (FIG. 3; Supplementary Table 6) and there is limited overlap in variants associated with different traits. For example, variants associated with vascular disease, such as rs944797 (REF.39), are not associated with T2DM20 and vice versa. This locus may therefore be an example of genetic variation influencing the cell type-specific regulation of genes important in human ageing.
Another multi-trait locus of potential importance for ageing is CASC8 (cancer susceptibility candidate 8), which is a lncRNA containing variants associated with breast76, prostate77 and colorectal83 cancers. A T2DM-associated variant is located nearby (<250 kb) in CASC11 (REF.20). Both CASC8 and CASC11 are upstream of MYC, an oncogene known to be regulated by lncRNAs84. This observation could imply a similar mechanism to that of the lncRNA CDKN2B-AS1, whereby genetic variants affect regulation of senescence-related genes via lncRNAs.
Telomere-related genetic variants
Four unique genetic variants mapped to the telomerase gene TERT have been associated with breast, colorectal and prostate cancer risk in GWAS76,77,83. Telomerase is involved in telomere maintenance, the end fragments of chromosomes that shorten with each cell cycle. Telomere shortening is a major contributor to replicative senescence85 and therefore a biological hallmark of ageing (BOX 2). In humans, telomere length measured in blood has a strong inherited genetic component, with heritability estimates of 34–82%86. GWAS have linked 16 inherited variants to human leukocyte telomere length87, including variants in the genes encoding telomerase and telomere-protective protein genes (TERC, TERT, NAF1, OBFC1 and RTEL1)88. A Mendelian randomization study of genetic variants associated with longer telomeres reported reduced risks of CAD and interstitial lung disease but increased risks of several forms of cancer87, suggesting a trade-off between risks of cancer and chronic age-related disease. No association was found between the genetic predisposition to longer telomeres and parental longevity21, which raises the possibility that the positive and negative effects on health cancel themselves out for overall survival. A recent analysis suggested that telomere variants were also not associated with the top 1% of parental longevity or measures of cognitive or physical function in older UK Biobank participants89.
Mendelian randomization.
A method that uses single-nucleotide polymorphisms associated with an exposure as instruments to probe the causal nature of the relationship between this exposure and an outcome of interest.
SH2B3: a partial Drosophila homologue
In humans, the SH2B3 gene encodes lymphocyte adaptor protein LNK, which is an intracellular modulator of the erythropoietin receptor, the stem cell factor receptor c-Kit and JAK2 (REF.90). GWAS have implicated the SH2B3 locus and nearby genes (ATXN2 and BRAP) in many diseases39,83,91. The likely causal variant in SH2B3 associated with shorter parental longevity29,92 is a missense variant (rs3184504-T) predicted to disrupt LNK protein functioning. The T allele is more common in autoimmune and cardiovascular conditions93 and myeloproliferative cancers90 as well as breast, colorectal and lung cancers94 (compared with the ‘normal’ functioning C allele). By contrast, the C allele of rs3184504 is more common in the offspring of longer-lived parents (those reaching the longest-lived 10%29 and 1% of lifespans95). A GWAS of human blood protein levels96 found that the rs3184504 C allele is associated with reduced levels of vascular cell adhesion protein 1 (VCAM1), which functions in leukocyte recruitment in the cellular immune response and in angiogenesis96, and influences the development and spread of cancers97. Interestingly, the rs3184504 C allele is also associated with an increased cardiovascular risk39,91 and yet with a decreased cancer risk83, again suggesting a trade-off between chronic disease and cancer mechanisms in ageing (Supplementary Table 2).
In Drosophila melanogaster, the SH2B gene is an insulin-like growth factor 1 (IGF1) and energy balance signalling modulator. An SH2B loss-of-function mutant was shown to be long-lived under starvation conditions as a result of increased carbohydrate stores98. Modifications to the IGF pathway in model organisms, from worms to mice, produce dramatic lifespan extensions akin to those seen in dietary restriction experiments99,100 (BOX 1). However, whereas the human SH2B1 and SH2B2 homologues of SH2B are important for IGF1 signalling, SH2B3 is not an important IGF1 regulator101. The SH2B3 locus is therefore clearly important in human ageing but likely through more specialized mechanisms than those targeted in SH2B model organisms of ageing, underlining the need for caution in generalizing from ancestral genes in model organisms to humans.
The HLA region and ABO blood groups
The histocompatibility complex gene group on chromosome 6 encodes HLA genes, which mediate chronic inflammatory pathways in autoimmune and infectious diseases102. Genetic variants near HLA-DQA1 are associated with Alzheimer disease22, lifespan31, prostate cancer77 and T2DM20, and also alter levels of the circulating inflammation marker C-reactive protein103. An HLA variant has also been linked to low muscle strength54, suggesting a role in the development of physical impairments in human ageing. These associations suggest that overlaps exist between autoimmune inflammatory mechanisms and the chronic inflammation commonly seen in ageing.
Somewhat similarly, genetic variants in the ABO locus have been associated with T2DM20, breast cancer76 and stroke91 as well as altered levels of the circulating inflammation marker C-reactive protein103. ABO antigens, which determine blood group types, are expressed on a wide range of tissues and cell surfaces and are important in infectious disease susceptibility, tumori-genesis and cardiovascular disease104. In each study, the O (recessive) blood type was protective later in life (compared with the other blood groups) but also seemed to increase the risk of haemorrhages at younger ages104, an apparent example of antagonistic pleiotropy (BOX 3).
Box 3 |. Evolutionary theories of ageing.
Lifespans of different animal species vary enormously, from less than 1 day in mayflies to more than 400 years in ocean quahog clams175, and this variation must ultimately arise from evolved differing abilities to adapt to the surrounding environment and respond to stress, both of which are likely genetically encoded.
The early theory of group selection argued that ageing is a genetically encoded adaptive trait that increases mortality in individuals with declining reproductive potential, thereby freeing up resources for the younger generation to reproduce176. This theory was soon abandoned by the original author because of evidence that natural selection is most effective at the individual level even when it may conflict with the interests of the species177. Additionally, no organism-level ‘programmed death gene’ has been found. Currently, the most widely accepted theories of ageing are the mutation accumulation theory, the antagonistic pleiotropy theory and the disposable soma theory.
The mutation accumulation theory proposes that organisms accumulate damaging germline mutations that are expressed only in the post-reproductive period of life, as these mutations would not be eliminated by earlier selective pressures178. This theory interprets variations in lifespan between different mammal species as related to differences in age of sexual maturity; Huntington disease provides a prominent example. However, the rapid post-reproduction increase in death rates that would be expected according to this theory has not been detected in humans or in animal species179.
The antagonistic pleiotropy theory argues that some mutations selected because they are beneficial to early fitness become harmful late in life, causing ageing180. Cell senescence pathways may provide examples of antagonistic pleiotropy: programmed senescence occurs during normal mammalian development, protects against cancer and promotes wound healing at younger ages but contributes to degenerative chronic disease at older ages181.
The disposable soma theory states that given the availability of limited resources, ageing arises from evolutionary trade-offs between growth and reproduction, on the one hand, and repair mechanisms on the other182. This theory is consistent with evidence that long-lived species, such as humans, evolved by developing more sophisticated and effective, albeit not unlimited, repair mechanisms. For example, comparative studies have found that the capacity to recycle deteriorated macromolecules and organelles by autophagy correlates with lifespan across species183. This theory is also consistent with the current consensus that ageing results from the accumulation of unrepaired cellular and molecular damage2,183. Human resilience mechanisms were likely selected to be robust enough to match the high environmental pressures that drove human evolution. The very recent dramatic decline of environmental pressures has extended survival beyond the previous resilience ‘warranty period’. Contemporary environments allow lengthy survival after the reproductive period, with an eventual increasing predominance of damage over repair.
Antagonistic pleiotropy.
Theory arguing that some mutations are selected because they are beneficial to early-life fitness but become harmful later in life, thus causing ageing.
Obesity, T2DM and cancer loci
Variants in the TCF7L2 locus are linked to T2DM20, breast cancer76 and colorectal cancer83. TCF7L2 is involved in β-cell proliferation and insulin production105, and variants in this locus are also associated with increased obesity106, which itself is a driver of T2DM and many cancers107. Other multi-trait loci are also linked to T2DM and multiple cancers, including ZMIZ1 (REFS20,76,83), VEGFA20,77 and FTO20,76 (Supplementary Table 5). These loci highlight the importance of adiposity as an accelerator of ageing, which is consistent with the accumulation of senescent cells in adipose tissue69 and the success of caloric restriction in lengthening the lifespan in many laboratory models (BOX 1).
Inferring ageing mechanisms from mutations
The integrity and maintenance of the genome seems to be strongly connected with ageing and longevity. Mutations in genes that affect DNA repair capacity and DNA maintenance cause progerias, and there is evidence that the accumulation of somatic DNA mutations is associated with phenotypes of ageing and contributes to chronic diseases.
Single-gene disorders of accelerated ageing
Segmental progerias are genetic diseases that exhibit many clinical manifestations similar to those that develop with ageing but that manifest earlier in life108. Prototypical examples of these syndromes are Werner syndrome (Online Mendelian Inheritance of Man (OMIM) #277700), Hutchinson–Gilford progeria (OMIM #176670) and Cockayne syndrome (OMIM #216400). The study of these conditions can provide important indications on the genetic and biological mechanisms that drive the ageing process.
Clinical phenotypes.
Patients with Werner syndrome develop normally but lack a pubertal growth spurt and later develop skin atrophy, loss of subcutaneous adipose tissue, loss and greying of hair, T2DM, cataracts, osteoporosis, early loss of fertility, severe arteriosclerosis, peripheral neuropathy and cancer109. Interestingly, cells from patients with Werner syndrome exhibit a shortened replicative lifespan110. Patients with Hutchinson–Gilford progeria are normal at birth but then grow slowly and develop baldness, loss of eyelashes and eyebrows, prominent eyes, convex nasal bridge and a small jaw, loss of subcutaneous fat, musculoskeletal abnormalities and premature cardiovascular pathology111. Patients with Cockayne syndrome display photosensitivity and developmental failure after 1 year of age, under-represented subcutaneous adipose tissue, premature decline of cognitive function and cardiovascular pathology, leading to a median survival of approximately 12 years112.
Genetics.
Interestingly, all three syndromes arise from genetic mutations that affect genomic structure and function through DNA repair, nuclear architecture and the fidelity of DNA replication113. In particular, Werner syndrome is caused by mutations of the WRN gene, which encodes a member of the RecQ family of helicases that is involved in DNA recombination, replication and telomere maintenance109. More than 90% of Hutchinson– Gilford progeria cases are caused by de novo heterozygous mutations in the LMNA gene, which encodes the proteins lamin A and lamin C through alternative splicing114. Lamin A and C are assembled to form a matrix on the inner surface of the nuclear membrane, the integrity of which is important for DNA maintenance mechanisms such as DNA double-strand break repair114. Of note, in Hutchinson–Gilford progeria, a point mutation within LMNA exon 11 leads to the production of progerin, a mutant protein responsible for the fragility of the nuclear envelope, with rearrangement of hetero-chromatin similar to what is often seen in fibroblast nuclei from older persons115. Small amounts of progerin accumulate with normal ageing and may contribute to age-related cardiovascular diseases116. Finally, Cockayne syndrome is caused by mutations in the ERCC8 and ERCC6 genes, respectively, encoding the CSA and CSB proteins, which play central roles in the transcription-coupled nucleotide excision repair that occurs at specific secondary DNA structures117.
Overall, prototypical progerias suggest that progressive loss of genetic and genomic integrity contributes to the ageing process. So far, data on progerias indicates that mechanisms of DNA repair and maintenance are important for health conservation, but whether they underlie true accelerated ageing syndromes remains uncertain. A key question that follows is whether common variants mapped to these same progeria genes are important for the common traits in the general population. The GWAS Catalog43 reports that variants in the Werner syndrome-associated WRN gene are associated with cognitive function and educational attainment118, variants in the Hutchinson–Gilford-associated LMNA gene are associated with white blood cell counts42 and height42, and variants in the Cockayne syndrome-associated ERCC8 gene affect age at smoking initiation119. No variants in ERCC6 have reached genome-wide significance in studies published in the GWAS Catalog (as of 19 July 2019).
Hereditary haemochromatosis.
Other lower penetrance genetic disorders also provide insights into the mechanisms of ageing. A prototypic example is iron overload due to hereditary haemochromatosis, which causes widespread oxidative damage120. The main homozygous HFE p.C282Y mutation, which is present in approximately 1 in 150 people of Northern European ancestry, is associated with increased incidence of arthritis, T2DM and liver disease121, typically after the age of 40 years, as well as low muscle strength and chronic pain in those aged 60–70 years122. Treatment (withdrawing blood) is effective and safe but often delayed because early manifestations are mistaken for ‘normal’ ageing. This mutation probably became common during the transition from hunter–gatherer to agricultural living, when low meat intake made increased dietary iron absorption advantageous, especially for pregnant women. If so, this disease supports the antagonistic pleiotropy theory of ageing, being a variant selected to enhance reproduction but that has adverse effects later in life (BOX 3). More work is needed to identify other mutations with later life effects123, including rare mutations.
Somatic mutations in human ageing
As several of the progerias (and age at menopause) are caused by genes involved in DNA repair and genomic stability, an obvious related question is whether accumulation of somatic mutations are important drivers of ageing124. There is now increasing evidence from single-cell and small sample size sequencing studies that unrepaired or incompletely repaired somatic mutations accumulate with ageing, including in oncogenes, and can lead to clonal expansion of mutated cells125. Stem cells generate new cells for tissue repair or remodelling, and somatic mutations in stem cells therefore have the highest potential for clonal expansion. A study of small intestine, colon and liver samples from human donors aged 3–87 years showed an accumulation of approximately 40 somatic mutations per year in stem cells126.A study of human B lymphocyte somatic mutations, in both coding and regulatory regions, found <500 mutations per cell in neonates, rising to >3,000 per cell in centenarians127. Similar accumulations of somatic mutations with age have been reported for satellite cells in muscle128. Moreover, sequencing of oesophageal micro-samples showed age-associated accumulation of mutations, with middle-aged and elderly donors having cancer-associated mutation clones (including in the TP53 cancer control gene and NOTCH1, which encodes a tissue development regulator) covering a majority of the epithelium, with evidence that the burden of mutations is higher in heavy drinkers and smokers129,130. There is some evidence that clonal expansion of somatic mutations in haematopoietic stem cells is associated with an increased risk of cardiovascular diseases and leukaemia131, but whether it is also linked to biological ageing has not been demonstrated. However, it remains unclear how important these somatic mutations are for longevity; one study of 864 people aged 80–105 years found that somatic mutations in genes linked to clonal expansion of haematopoietic stem cells did not compromise 10-year survival132.
Clonal expansion.
The production of daughter cells from a single parent cell, all sharing a particular characteristic or trait.
Acquired mitochondrial genetic variants.
Mitochondria have their own DNA (mtDNA), a circular 16.5 kb double-stranded loop of DNA that encodes 13 protein subunits of the electron transport chain and is uniquely inherited from mother to offspring133. Each cell contains many mitochondria and each one contains many mtDNA copies; therefore, multiple mutations can coexist in the same cell, a phenomenon known as hetero-plasmy134. Maternally inherited mtDNA variants can cause mitochondrial diseases135. Interestingly, inherited mitochondrial diseases are often characterized by progeroid characteristics, such as neurodegenerative and neuromuscular manifestations136, suggesting that mitochondrial dysfunction contributes to ageing. Indeed, de novo mtDNA mutations occur at random and accumulate unrepaired in several tissues at a rate substantially higher than nuclear DNA, probably because of the direct exposure to reactive oxygen species (ROS), lack of true histones and limited DNA repair mechanisms137–139. If the mutational load reaches a certain threshold it can affect the efficiency of oxidative phosphorylation and increase the production of ROS, especially in individuals who already have inherited mtDNA mutations140. This accumulation of mtDNA variants may cause many of the phenotypes of ageing, including metabolic, neurodegenerative and neoplastic diseases140. In support of this hypothesis, a knock-in mouse mutation that compromises the proofreading ability of mtDNA polymerase and results in massive accumulation of mtDNA mutations with age was associated with a reduced lifespan and early development of ageing phenotypes141. Indeed, the frequency and quantity of mtDNA mutations is higher in organs of patients with chronic diseases that specifically affect those organs but is particularly evident in neurodegenerative diseases such as Alzheimer, Parkinson and Huntington diseases142,143.
Conclusions and future perspectives
Despite still being in the early phases of genetic discovery, available evidence supports an emerging picture of human ageing being driven by a balance of damage and repair processes, influenced by both environmental exposures and genetic variation between people (FIG. 4). Although the hallmark pathways of ageing have been identified mainly in animal models144, a role for some of these mechanisms is evident in GWAS results: several multi-trait loci, including CDKN2A/B, SH2B3 and TERT, as well as inflammation-related and obesity-related variants, are linked to hallmark pathways of ageing. In some cases, variants in these loci result in trade-offs between cancer and chronic disease risks (FIG. 4). DNA repair mechanisms have emerged as important for female reproductive ageing as well as being key in progeria syndromes, and evidence is accumulating that unrepaired somatic mutations may be of fundamental importance in human ageing.
Fig. 4 |. Diagram of the major influences and mechanisms of human ageing.
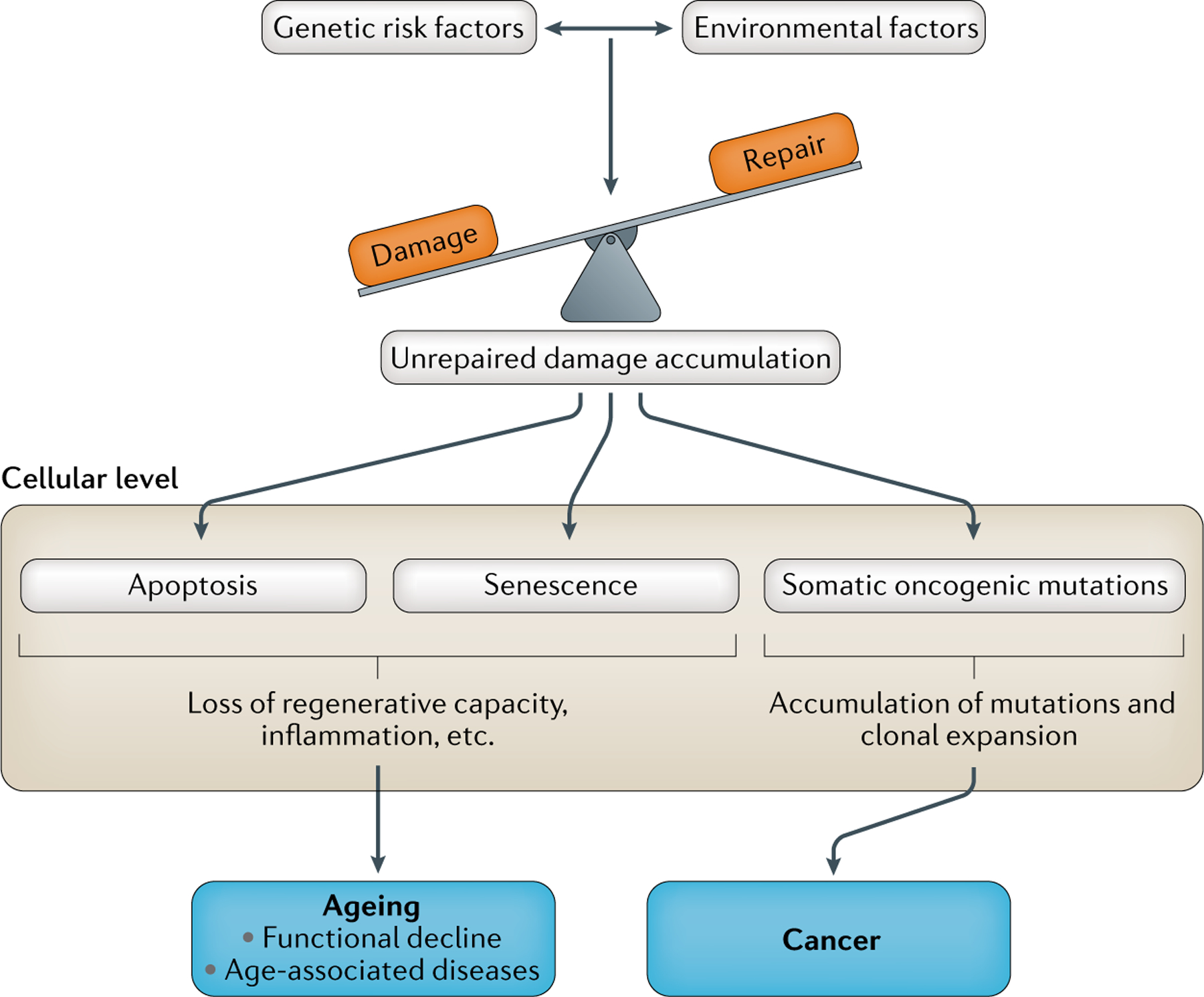
The emerging picture from genetic studies of human ageing supports the hypothesis that ageing is driven by the balance of damage and repair processes. There is genetic evidence for the importance of several damage pathways in humans. Damage can be intrinsic, for example, through somatic mutations arising during cell division. Also important are health behavioural risk factors such as smoking and obesity , which are also influenced by gene–environment interactions. The net impact of damage depends on the activity of repair and response mechanisms. At the cellular level, complete repair can yield undamaged cells (not shown). By contrast, unrepaired damage can lead to cell death (apoptosis), preventing cancers but leading to the depletion of stem cells and loss of regenerative capacity. Cells with somatic oncogene mutations can survive and replicate, sometimes leading to tumour development. Alternatively , damaged cells can enter senescent states and produce a secretory senescence phenotype (SASP), resulting in inflammation and reduced repair that contributes to degenerative diseases582. These mechanisms can result in reduced repair and increasing incidence of chronic diseases of ageing but with decreased cancer risks, or vice versa. This ageing versus cancer trade-off is evident for several of the loci described, notably in the 9p21 cell cycle and senescence-related locus, telomere variation and in the SH2B3 locus.
In addition to multi-trait loci, many variants that exhibit disease-specific effects are important in how humans age. For example, the vascular disease effects of lipid-altering variants or the cartilage-specific variants involved in osteoarthritis19 suggest that both ageing mechanisms and disease-specific mechanisms are important in human ageing. Disease-specific genetic and environmental risks help explain the great variability of ages at disease onset and comorbidities seen across older populations.
Will knowledge of ageing-related genetic variation ever yield personal predictions for later life? A whole-genome risk score from the 1 million lifespan parental longevity GWAS, comparing individuals in the top and bottom deciles, was associated with an increase of 3–5 years in life expectancy31. Although this observation is promising, this variation constitutes only a small proportion of the variation seen in the human lifespan. Additionally, better control of potentially modifiable health risks that have an important role in ageing, such as obesity, blood pressure and cholesterol levels, could alter genetic predictions.
Genetic studies of human ageing are currently limited to the study of proxy measures of biological ageing. For many variants implicated in genetic association studies, exact effect mechanisms are unproven, and the general assumption that effects are mediated through the nearest gene is not always true145. The effects of some of the multi-trait loci highlighted may be through distinct, ‘tagged’ variants and/or different pathways. Much work is needed to establish the biological mechanisms influenced by GWAS-identified variants, including in loci that affect multiple traits.
In the coming years, much more will be learned about human ageing, hopefully with better phenotyping of the biological hallmarks of ageing. Larger sample sizes as well as DNA sequencing and linked studies of proteomics, gene expression and epigenetics will capture more of the genetic variation between individuals and will help identify the mechanisms of effect of these genetic variations. Thus far, most of the evidence is from European ancestry groups, and studies of others — especially African ancestry groups, who have greater genetic heterogeneity — are likely to extend findings146. It seems probable that much more evidence of ageing pathways will be found, including novel pathways that could provide intervention targets or offer new prevention opportunities. There remains ample scope for using inherited variants to understand human ageing mechanisms. Overall, human genetics will likely continue to provide growing insights into how we age and play a major role in identifying ways in which we might slow ageing, thus helping more people to age well.
Supplementary Material
Acknowledgements
D.M. and L.C.P. are supported by the University of Exeter Medical School and additionally by the University of Connecticut School of Medicine. This work is supported in part by the UK Medical Research Council (grants MR/M023095/1 and MRS009892/1). This work was supported in part by the Intramural Research Program at the National Institute on Aging.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Genetics thanks P. K. Joshi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41576-019-0183-6.
References
- 1.Partridge L & Mangel M Messages from mortality: the evolution of death rates in the old. Trends Ecol. Evol 14, 438–442 (1999). [DOI] [PubMed] [Google Scholar]
- 2.López-Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reviews the evidence for nine key mechanisms that are characteristic of mammalian ageing.
- 3.Partridge L, Deelen J & Slagboom PE Facing up to the global challenges of ageing. Nature 561, 45–56 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Kennedy BK et al. Geroscience: linking aging to chronic disease. Cell 159, 709–713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides an overview of geroscience, which argues that many chronic diseases share underlying ageing mechanisms that should be targeted to improve overall health in later life.
- 5.Tam V et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet 20, 467–484 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Nelson MR et al. The support of human genetic evidence for approved drug indications. Nat. Genet 47, 856–860 (2015). [DOI] [PubMed] [Google Scholar]; This study found that development of drugs supported by genetic evidence of mechanism could double the success rate.
- 7.Declerck K & Vanden Berghe W Back to the future: epigenetic clock plasticity towards healthy aging. Mech. Ageing Dev 174, 18–29 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Harries LW et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 10, 868–878 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carnes BA What is lifespan regulation and why does it exist? Biogerontology 12, 367–374 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Tenesa A & Haley CS The heritability of human disease: estimation, uses and abuses. Nat. Rev. Genet 14, 139–149 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Herskind AM et al. The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870–1900. Hum. Genet 97, 319–323 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Sebastiani P & Perls TT The genetics of extreme longevity: lessons from the New England centenarian study. Front. Genet 3, 277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplanis J et al. Quantitative analysis of population-scale family trees with millions of relatives. Science 360, 171–175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruby JG et al. Estimates of the heritability of human longevity are substantially inflated due to assortative mating. Genetics 210, 1109–1124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skousgaard SG et al. Probability and heritability estimates on primary osteoarthritis of the hip leading to total hip arthroplasty: a nationwide population based follow-up study in Danish twins. Arthritis Res. Ther 17, 336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willemsen G et al. The concordance and heritability of type 2 diabetes in 34,166 twin pairs from international twin registers: the discordant twin (discotwin) consortium. Twin Res. Hum. Genet 18, 762–771 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Gatz M et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Zdravkovic S et al. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J. Intern. Med 252, 247–254 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Tachmazidou I et al. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet 51, 230–236 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahajan A et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pilling LC et al. Human longevity is influenced by many genetic variants: evidence from 75,000 UK Biobank participants. Aging 8, 547–560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vinkhuyzen AA, Wray NR, Yang J, Goddard ME & Visscher PM Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu. Rev. Genet 47, 75–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barsh GS, Copenhaver GP, Gibson G & Williams SM Guidelines for genome-wide association studies. PLOS Genet 8, e1002812 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutta A et al. Longer lived parents: protective associations with cancer incidence and overall mortality. J. Gerontol. A Biol. Sci. Med. Sci 68, 1409–1418 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutta A et al. Aging children of long-lived parents experience slower cognitive decline. Alzheimers Dement 10 (5 Suppl), 315–322 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Atkins JL et al. Longer-lived parents and cardiovascular outcomes: 8-year follow-up in 186,000 U.K. Biobank participants. J. Am. Coll. Cardiol 68, 874–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilling LC et al. Human longevity: 25 genetic loci associated in 389,166 UK Biobank participants. Aging 9, 2504–2520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi PK et al. Genome-wide meta-analysis associates HLA-DQA1/DRB1 and LPA and lifestyle factors with human longevity. Nat. Commun 8, 910 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Timmers PR et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. eLife 10.7554/eLife.39856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This LifeGen study is a GWAS meta-analysis of parental lifespan using UK Biobank plus 25 independent cohorts, and including data on 1 million parents’ lifespans.
- 32.Wright KM et al. A prospective analysis of genetic variants associated with human lifespan. G3 9, 2863–2878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keskitalo K et al. Association of serum cotinine level with a cluster of three nicotinic acetylcholine receptor genes (CHRNA3/CHRNA5/CHRNB4) on chromosome 15. Hum. Mol. Genet 18, 4007–4012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ & Richards JB Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat. Rev. Genet 19, 110–124 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Deelen J et al. Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell 10, 686–698 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deelen J et al. Genome-wide association meta-analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum. Mol. Genet 23, 4420–4432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broer L et al. GWAS of longevity in CHARGE consortium confirms APOE and FOXO3 candidacy. J. Gerontol. A. Biol. Sci. Med. Sci 70, 110–118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deelen J et al. A meta-analysis of genome-wide association studies identifies novel longevity genes. Nat. Commun 10, 3669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the largest GWAS directly studying long-lived participants (aged ≥90 years) compared with shorter-lived (<65) controls to date.
- 39.Nikpay M et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet 47, 1121–1130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belloy ME, Napolioni V & Greicius MD A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 101, 820–838 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sebastiani P et al. APOE alleles and extreme human longevity. J. Gerontol. A Biol. Sci. Med. Sci 74, 44–51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kichaev G et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet 104, 65–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van den Berg N et al. Longevity defined as top 10% survivors and beyond is transmitted as a quantitative genetic trait. Nat. Commun 10, 35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yashin AI et al. Genetics of human longevity from incomplete data: new findings from the long life family study. J. Gerontol. A Biol. Sci. Med. Sci 73, 1472–1481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martins R, Lithgow GJ & Link W Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15, 196–207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bae H et al. Effects of FOXO3 polymorphisms on survival to extreme longevity in four centenarian studies. J. Gerontol. A Biol. Sci. Med. Sci 73, 1439–1447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Revelas M et al. Review and meta-analysis of genetic polymorphisms associated with exceptional human longevity. Mechanisms Ageing Dev 175, 24–34 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Giuliani C, Garagnani P & Franceschi C Genetics of human longevity within an eco-evolutionary nature-nurture framework. Circ. Res 123, 745–772 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Day FR et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet 47, 1294–1303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruth K & Murray A Lessons from genome-wide association studies in reproductive medicine: menopause. Semin. Reprod. Med 34, 215–223 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Cooper R, Kuh D & Hardy R, Mortality Review Group; FALCon and HALCyon Study Teams. Objectively measured physical capability levels and mortality: systematic review and meta-analysis. BMJ 341, c4467 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willems SM et al. Large-scale GWAS identifies multiple loci for hand grip strength providing biological insights into muscular fitness. Nat. Commun 8, 16015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones G et al. Sarcopenia and variation in the human leukocyte antigen complex. J. Gerontol. A. Biol. Sci. Med. Sci 10.1093/gerona/glz042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morley JE et al. Brain health: the importance of recognizing cognitive impairment: an IAGG consensus conference. J. Am. Med. Dir. Assoc 16, 731–739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davies G et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat. Commun 9, 2098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritchie SJ et al. Polygenic predictors of age-related decline in cognitive ability. Mol. Psychiatry 10.1038/s41380-019-0372-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Logue MW et al. Use of an Alzheimer’s disease polygenic risk score to identify mild cognitive impairment in adults in their 50s. Mol. Psychiatry 24, 421–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corley J, Cox SR & Deary IJ Healthy cognitive ageing in the Lothian Birth Cohort studies: marginal gains not magic bullet. Psychol. Med 48, 187–207 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Black JR & Clark SJ Age-related macular degeneration: genome-wide association studies to translation. Genet. Med 18, 283–289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fritsche LG et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet 48, 134–143 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zenin A et al. Identification of 12 genetic loci associated with human healthspan. Commun. Biol 2, 41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barrett JH et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet 43, 1108–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guy GP, Machlin SR, Ekwueme DU & Yabroff KR Prevalence and costs of skin cancer treatment in the U.S., 2002−2006 and 2007−2011. Am. J. Prev. Med 48, 183–187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.American Cancer Society. Cancer Facts & Figures 2019. Cancer https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html (2019).
- 66.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Rheenen W, Peyrot WJ, Schork AJ, Lee SH & Wray NR Genetic correlations of polygenic disease traits: from theory to practice. Nat. Rev. Genet 20, 567–581 (2019). [DOI] [PubMed] [Google Scholar]; This review defines genetic correlations, summarizes methods, and discusses their interpretation and uses with respect to human health and disease.
- 68.Dixon JB, Egger GJ, Finkelstein EA, Kral JG & Lambert GW ‘Obesity paradox’ misunderstands the biology of optimal weight throughout the life cycle. Int. J. Obes 39, 82–84 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Tchkonia T et al. Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu M et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 4, e12997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bowman K, Thambisetty M, Kuchel GA, Ferrucci L & Melzer D Obesity and longer term risks of dementia in 65–74 year olds. Age Ageing 48, 367–373 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bowman K et al. Obesity in older people with and without conditions associated with weight loss: follow-up of 955,000 primary care patients. J. Gerontol. A. Biol. Sci. Med. Sci 72, 203–209 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pignolo RJ Exceptional human longevity. Mayo Clin. Proc 94, 110–124 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Willer CJ et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet 45, 1274–1283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Congrains A, Kamide K, Ohishi M & Rakugi H ANRIL: molecular mechanisms and implications in human health. Int. J. Mol. Sci 14, 1278–1292 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michailidou K et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schumacher FR et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet 50, 928–936 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rahmioglu N et al. Genetic variants underlying risk of endometriosis: insights from meta-analysis of eight genome-wide association and replication datasets. Hum. Reprod. Update 20, 702–716 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Springelkamp H et al. Meta-analysis of genome-wide association studies identifies novel loci associated with optic disc morphology. Genet. Epidemiol 39, 207–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Astle WJ et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baker DJ et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study of a transgenic mouse model was the first to show that removal of senescent cells from an adult animal extended lifespan and improved some aspects of health.
- 82.Tchkonia T & Kirkland JL Aging, cell senescence, and chronic disease emerging therapeutic strategies. JAMA 320, 1319–1320 (2018). [DOI] [PubMed] [Google Scholar]; This manuscript provides an overview of cell senescence, links to ageing and future therapeutics.
- 83.Schmit SL et al. Novel common genetic susceptibility loci for colorectal cancer. J. Natl. Cancer Inst 111, 146–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiang JF, Yang L & Chen LL The long noncoding RNA regulation at the MYC locus. Curr. Opin. Genet. Dev 33, 41–48 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Hayflick L & Moorhead PS The serial cultivation of human diploid cell strains. Exp. Cell Res 25, 585–621 (1961). [DOI] [PubMed] [Google Scholar]
- 86.Broer L et al. Meta-analysis of telomere length in 19,713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur. J. Hum. Genet 21, 1163–1168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Telomeres Mendelian Randomization Collaboration et al. Association between telomere length and risk of cancer and non-neoplastic diseases: a Mendelian randomization study. JAMA Oncol. 3, 636–651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study found that genetic predisposition to longer telomeres increased risk of multiple cancers but reduced risk for some other diseases, including cardiovascular disease.
- 88.Codd V et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet 45, 422–427 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuo C-L, Pilling LC, Kuchel GA, Ferrucci L & Melzer D Telomere length and aging-related outcomes in humans: a Mendelian randomization study in 261,000 older participants. Aging Cell 24, e13017 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maslah N, Cassinat B, Verger E, Kiladjian JJ & Velazquez L The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 31, 1661–1670 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Malik R et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet 50, 524–537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fortney K et al. Genome-wide scan informed by age-related disease identifies loci for exceptional human longevity. PLOS Genet 11, e1005728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Laroumanie F et al. LNK deficiency promotes acute aortic dissection and rupture. JCI Insight 3, e122558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hung RJ et al. Cross cancer genomic investigation of inflammation pathway for five common cancers: lung, ovary, prostate, breast, and colorectal cancer. J. Natl Cancer Inst 107, djv246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kuo C-L et al. The longevity associated sh2b3 (lnk) genetic variant: selected aging phenotypes in 379,758 subjects. J. Gerontol. A. Biol. Sci. Med. Sci 10.1093/gerona/glz191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun BB et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schlesinger M & Bendas G Vascular cell adhesion molecule-1 (VCAM-1) — an increasing insight into its role in tumorigenicity and metastasis. Int. J. Cancer 136, 2504–2514 (2015). [DOI] [PubMed] [Google Scholar]
- 98.Slack C et al. Regulation of lifespan, metabolism, and stress responses by the Drosophila SH2B protein, Lnk. PLOS Genet 6, e1000881 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mair W & Dillin A Aging and survival: the genetics of life span extension by dietary restriction. Annu. Rev. Biochem 77, 727–754 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Junnila RK, List EO, Berryman DE, Murrey JW & Kopchick JJ The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol 9, 366–376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Desbuquois B, Carré N & Burnol AF Regulation of insulin and type 1 insulin-like growth factor signaling and action by the Grb10/14 and SH2B1/B2 adaptor proteins. FEBS J 280, 794–816 (2013). [DOI] [PubMed] [Google Scholar]
- 102.Shiina T, Hosomichi K, Inoko H & Kulski JK The HLA genomic loci map: expression, interaction, diversity and disease. J. Hum. Genet 54, 15–39 (2009). [DOI] [PubMed] [Google Scholar]
- 103.Ligthart S et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am. J. Hum. Genet 103, 691–706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Franchini M & Bonfanti C Evolutionary aspects of ABO blood group in humans. Clin. Chim. Acta 444, 66–71 (2015). [DOI] [PubMed] [Google Scholar]
- 105.Jin T Current understanding on role of the wnt signaling pathway effector TCF7L2 in glucose homeostasis. Endocr. Rev 37, 254–277 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Locke AE et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stone TW, McPherson M & Gail Darlington L Obesity and cancer: existing and new hypotheses for a causal connection. eBioMedicine 30, 14–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martin GM & Oshima J Lessons from human progeroid syndromes. Nature 408, 263–266 (2000). [DOI] [PubMed] [Google Scholar]
- 109.Oshima J, Sidorova JM & Monnat RJ Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev 33, 105–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pignolo RJ et al. Defects in telomere maintenance molecules impair osteoblast differentiation and promote osteoporosis. Aging Cell 7, 23–31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ahmed MS, Ikram S, Bibi N & Mir A Hutchinson–Gilford progeria syndrome: a premature aging disease. Mol. Neurobiol 55, 4417–4427 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Nance MA & Berry SA Cockayne syndrome: review of 140 cases. Am. J. Med. Genet 42, 68–84 (1992). [DOI] [PubMed] [Google Scholar]
- 113.Burla R, La Torre M, Merigliano C, Vernì F & Saggio I Genomic instability and DNA replication defects in progeroid syndromes. Nucleus 9, 368–379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gonzalo S, Kreienkamp R & Askjaer P Hutchinson–Gilford progeria syndrome: a premature aging disease caused by lmna gene mutations. Ageing Res. Rev 33, 18–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scaffidi P & Misteli T Lamin A-dependent nuclear defects in human aging. Science 312, 1059–1063 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jiang Y & Ji JY Understanding lamin proteins and their roles in aging and cardiovascular diseases. Life Sci 212, 20–29 (2018). [DOI] [PubMed] [Google Scholar]
- 117.Scheibye-Knudsen M et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl Acad. Sci. USA 113, 12502–12507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee JJ et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet 50, 1112–1121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu M et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet 51, 237–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yun S & Vincelette ND Update on iron metabolism and molecular perspective of common genetic and acquired disorder, hemochromatosis. Crit. Rev. Oncol. Hematol 95, 12–25 (2015). [DOI] [PubMed] [Google Scholar]
- 121.Pilling LC et al. Common conditions associated with hereditary haemochromatosis genetic variants: cohort study in UK Biobank. BMJ 364, k5222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tamosauskaite J et al. Hereditary hemochromatosis associations with frailty, sarcopenia and chronic pain: evidence from 200,975 older UK biobank participants. J. Gerontol. A. Biol. Sci. Med. Sci 74, 337–342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Telenti A, Perkins BA & Venter JC Dynamics of an aging genome. Cell Metab 23, 949–950 (2016). [DOI] [PubMed] [Google Scholar]
- 124.Zhang L & Vijg J Somatic mutagenesis in mammals and its implications for human disease and aging. Annu. Rev. Genet 52, 397–419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reviews the literature on low-abundance somatic DNA mutations in cells, in human and animal tissues and new technology that allows their assessment, with a focus on their possible functional consequences.
- 125.Risques RA & Kennedy SR Aging and the rise of somatic cancer-associated mutations in normal tissues. PLOS Genet 14, e1007108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Blokzijl F et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang L et al. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl Acad. Sci. USA 116, 9014–9019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Franco I et al. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nat. Commun 9, 800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study investigated somatic mutations in human B lymphocytes. Results indicate that spontaneous somatic mutations that accumulate with age may contribute to both the increased risk for leukemia and the functional decline of B lymphocytes in the elderly.
- 129.Martincorena I et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yokoyama A et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317 (2019). [DOI] [PubMed] [Google Scholar]
- 131.Fuster JJ & Walsh K Somatic mutations and clonal hematopoiesis: unexpected potential new drivers of age-related cardiovascular disease. Circ. Res 122, 523–532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van den Akker EB et al. Uncompromised 10-year survival of oldest old carrying somatic mutations in DNMT3A and TET2. Blood 127, 1512–1515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Andrews RM et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet 23, 147 (1999). [DOI] [PubMed] [Google Scholar]
- 134.Stewart JB & Chinnery PF The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet 16, 530–542 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Hutchison CA, Newbold JE, Potter SS & Edgell MH Maternal inheritance of mammalian mitochondrial DNA. Nature 251, 536–538 (1974). [DOI] [PubMed] [Google Scholar]
- 136.Scheibye-Knudsen M, Scheibye-Alsing K, Canugovi C, Croteau DL & Bohr VA A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging. Aging 5, 192–208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a novel classification algorithm based on qualitative and quantitative characteristics assessing whether a disorder can be characterized as mitochondrial. Using this tool, the authors find that many of the features of normal ageing may be linked to mitochondrial dysfunction.
- 137.Wallace DC et al. Sequence analysis of cDNAs for the human and bovine ATP synthase beta subunit: mitochondrial DNA genes sustain seventeen times more mutations. Curr. Genet. 12, 81–90 (1987). [DOI] [PubMed] [Google Scholar]
- 138.Bua E et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet 79, 469–480 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Greaves LC et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLOS Genet 10, e1004620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Picard M, Wallace DC & Burelle Y The rise of mitochondria in medicine. Mitochondrion 30, 105–116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Trifunovic A et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004). [DOI] [PubMed] [Google Scholar]; This manuscript describes the phenotype of a homozygous knock-in mice that expresses a proof-reading-deficient version of PolgA, the nucleus-encoded catalytic subunit of mtDNA polymerase. The PolgA mouse accumulates point mutations in mitochondrial DNA much faster than wild-type and shows reduced lifespan and premature onset of ageing-related phenotypes.
- 142.Kraytsberg Y et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet 38, 518–520 (2006). [DOI] [PubMed] [Google Scholar]
- 143.Franco-Iborra S, Vila M & Perier C Mitochondrial quality control in neurodegenerative diseases: focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci 12, 342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ferrucci L, Levine ME, Kuo P & Simonsick EM Time and the metrics of aging. Circ. Res 123, 740–744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 146.Campbell MC & Tishkoff SA African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu. Rev. Genomics Hum. Genet 9, 403–433 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kenyon CJ The genetics of ageing. Nature 464, 504–512 (2010). [DOI] [PubMed] [Google Scholar]
- 148.McKay JP, Raizen DM, Gottschalk A, Schafer WR & Avery L eat-2 and eat-18 are required for nicotinic neurotransmission in the Caenorhabditis elegans pharynx. Genetics 166, 161–169 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Greer EL & Brunet A Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 8, 113–127 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gingras A-C, Raught B & Sonenberg N eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem 68, 913–963 (1999). [DOI] [PubMed] [Google Scholar]
- 151.Lakowski B & Hekimi S Determination of life-span in Caenorhabditis elegans by four clock genes. Science 272, 1010–1013 (1996). [DOI] [PubMed] [Google Scholar]
- 152.Liu X et al. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev 19, 2424–2434 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bansal A, Zhu LJ, Yen K & Tissenbaum HA Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc. Natl Acad. Sci. USA 112, E277–E286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mitchell SJ et al. Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab 23, 1093–1112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fontana L, Partridge L & Longo VD Extending healthy life span — from yeast to humans. Science 328, 321–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mattison JA et al. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun 8, 14063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kraus WE et al. 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol 7, 673–683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Belsky DW, Huffman KM, Pieper CF, Shalev I & Kraus WE Change in the rate of biological aging in response to caloric restriction: CALERIE Biobank Analysis. J. Gerontol. A Biol. Sci. Med. Sci 73, 4–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Leclerc E et al. The effect of caloric restriction on working memory in healthy non-obese adults. CNS Spectr 10.1017/S1092852918001566 (2019). [DOI] [PubMed] [Google Scholar]
- 160.Madeo F, Carmona-Gutierrez D, Hofer SJ & Kroemer G Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab 29, 592–610 (2019). [DOI] [PubMed] [Google Scholar]
- 161.Most J, Tosti V, Redman LM & Fontana L Calorie restriction in humans: an update. Ageing Res. Rev 39, 36–45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Laplante M & Sabatini DM mTOR signaling at a glance. J. Cell Sci 122, 3589–3594 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Harrison DE et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mannick JB et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl Med 10, eaaq1564 (2018). [DOI] [PubMed] [Google Scholar]
- 165.Dowling RJ, Topisirovic I, Fonseca BD & Sonenberg N Dissecting the role of mtor: lessons from mtor inhibitors. Biochim. Biophys. Acta 1804, 433–439 (2010). [DOI] [PubMed] [Google Scholar]
- 166.Justice JN et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Evert J, Lawler E, Bogan H & Perls T Morbidity profiles of centenarians: survivors, delayers, and escapers. J. Gerontol. A Biol. Sci. Med. Sci 58, 232 (2003). [DOI] [PubMed] [Google Scholar]
- 168.Sebastiani P et al. Four genome-wide association studies identify new extreme longevity variants. J. Gerontol. A Biol. Sci. Med. Sci 72, 1453–1464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Levine ME Modeling the rate of senescence: can estimated biological age predict mortality more accurately than chronological age? J. Gerontol. A Biol. Sci. Med. Sci 68, 667–674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu Z et al. A new aging measure captures morbidity and mortality risk across diverse subpopulations from NHANES IV: a cohort study. PLOS Med 15, e1002718 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Matteini AM et al. GWAS analysis of handgrip and lower body strength in older adults in the CHARGE consortium. Aging Cell 15, 792–800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gow AJ et al. Stability and change in intelligence from age 11 to ages 70, 79, and 87: the Lothian Birth Cohorts of 1921 and 1936. Psychol. Aging 26, 232–240 (2011). [DOI] [PubMed] [Google Scholar]
- 173.Ben-Avraham D et al. The complex genetics of gait speed: genome-wide meta-analysis approach. Aging 9, 209–246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fried LP et al. Frailty in older adults: evidence for a phenotype. J. Gerontol. A Biol. Sci. Med. Sci 56, M146–M156 (2001). [DOI] [PubMed] [Google Scholar]
- 175.Butler PG, Wanamaker AD, Scourse JD, Richardson CA & Reynolds DJ Variability of marine climate on the North Icelandic Shelf in a 1357-year proxy archive based on growth increments in the bivalve Arctica islandica. Palaeogeogr. Palaeoclimatol. Palaeoecol 373, 141–151 (2013). [Google Scholar]
- 176.Weismann A, Poulton Sir EB, Schönland S & Shipley Sir AE Essays upon heredity and kindred biological problems (Arthur E) v.1 (Clarendon Press, 1891). [Google Scholar]
- 177.Fabian D & Flatt T The evolution of aging. Nat. Educ. Knowl 3, 9 (2011). [Google Scholar]
- 178.Medawar PB An Unsolved Problem of Biology (H. K. Lewis, London, 1952). [Google Scholar]
- 179.Partridge L & Barton NH Optimally, mutation and the evolution of ageing. Nature 362, 305–311 (1993). [DOI] [PubMed] [Google Scholar]
- 180.Williams GC Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411 (1957). [Google Scholar]
- 181.Campisi J Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522 (2005). [DOI] [PubMed] [Google Scholar]
- 182.Kirkwood TB Evolution of ageing. Nature 270, 301–304 (1977). [DOI] [PubMed] [Google Scholar]
- 183.Pride H et al. Long-lived species have improved proteostasis compared to phylogenetically-related shorter-lived species. Biochem. Biophys. Res. Commun 457, 669–675 (2015). [DOI] [PubMed] [Google Scholar]
- 184.Pattaro C et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun 7, 10023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zengini E et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat. Genet 50, 549–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Krzywinski M et al. Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pruim RJ et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.