

Abstract
Perimenopause is a midlife transition state experienced by women that occurs in the context of a fully functioning neurological system and results in reproductive senescence. Although primarily viewed as a reproductive transition, the symptoms of perimenopause are largely neurological in nature. Neurological symptoms that emerge during perimenopause are indicative of disruption in multiple estrogen-regulated systems (including thermoregulation, sleep, circadian rhythms and sensory processing) and affect multiple domains of cognitive function. Estrogen is a master regulator that functions through a network of estrogen receptors to ensure that the brain effectively responds at rapid, intermediate and long timescales to regulate energy metabolism in the brain via coordinated signalling and transcriptional pathways. The estrogen receptor network becomes uncoupled from the bioenergetic system during the perimenopausal transition and, as a corollary, a hypometabolic state associated with neurological dysfunction can develop. For some women, this hypometabolic state might increase the risk of developing neurodegenerative diseases later in life. The perimenopausal transition might also represent a window of opportunity to prevent age-related neurological diseases. This Review considers the importance of neurological symptoms in perimenopause in the context of their relationship to the network of estrogen receptors that control metabolism in the brain.
Introduction
Perimenopause is a midlife transition state that leads to reproductive senescence in women.1–4 Worldwide, >850 million women are currently aged 40–60 years,5 88% of whom will transition through perimenopause at an average age of 51.4 years with a Gaussian distribution of 40–58 years.6 Regardless of ethnicity, geographic location or culture, all women who reach the age of 60 years with their reproductive organs intact will transition through perimenopause to menopause with a duration of 1–5 years from start to completion.1,3 Menopause is the final stage of perimenopause and is complete after 12 months of amenorrhoea. This final stage is associated with a decrease in ovarian secretion of estrogen and progesterone.3,7
Both puberty and perimenopause are developmental transitions that occur in the context of a fully functioning neurological system that undergoes a conversion from one state of developmental ageing to another.8 Puberty is an endocrine transition state that occurs during adolescence and results in the conversion of a system that is incapable of reproduction to one with reproductive competence, in both female and male individuals. As with puberty, the perimenopause can be accompanied by shifts in neurological function that are seemingly unrelated to reproduction.8 This transition from one neurological state to another during perimenopause and its implications for health in later life are the focus of this Review.
Transition states
Transition states in humans that can span several years, such as puberty and the perimenopause, are dependent upon an integrated series of phase-dependent transformations, which involve sequential activation and deactivation of complex regulatory pathways.9 These transition states are associated with the restructuring of regulatory networks, which include systems of genetic, epigenetic and protein factors.9 Owing to the multifactorial nature of these transition states, small perturbations, either of endogenous or exogenous origin, during the restructuring process can directly induce vulnerabilities or unmask existing ones.10
Complex biological systems have critical thresholds, often referred to as tipping points, when a system can shift from one state to the other.10 That complex diseases are characterized by transitions from a healthy state to a predisease (that is, a prodromal phase) and finally a disease is well established.10 The predisease transition is typically defined as the limit of the normal state that precedes the tipping point into disease. The predisease state is often unstable and, therefore, potentially reversible; however, the period of time during which this state could be reversed is limited. The bifurcation point between predisease and disease states is characterized by a critical slowing of the system, in which the system becomes increasingly slow to recover from small perturbations to the equilibrium.10
In neurological transition states, indicators of dysfunction at the limits of normal function can be signals of instability and tipping points. The presence, variability, intensity and duration of perimenopausal symptoms might provide warning signs for an increased risk of adverse health consequences in later life, particularly neurodegenerative diseases. Multiple conditions that emerge during the perimenopause, such as insomnia, depression, subjective memory complaints and cognitive decline (Figure 1) are associated with an increased risk of neurodegenerative diseases, particularly Alzheimer disease.11,12 Multiple neurodegenerative diseases, including Alzheimer disease and multiple sclerosis, have a greater prevalence in postmenopausal women than in premenopausal women, and are characterized by prodromal periods that precede diagnosis of disease.11,12
Figure 1 |.
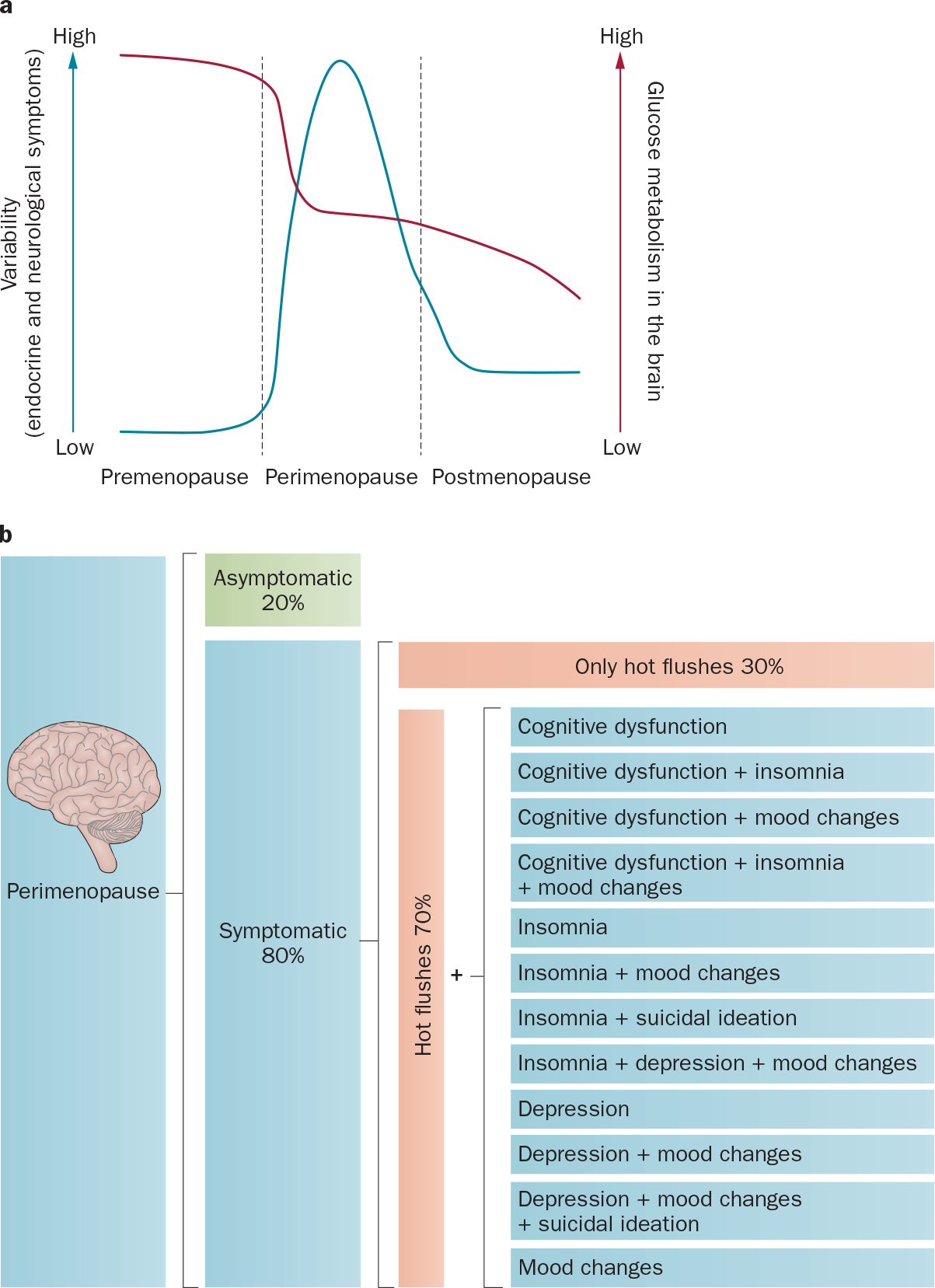
Symptoms during perimenopause. Perimenopause is characterized by heightened variability in neurological symptoms, which is co-incident with a decline in glucose metabolism in the brain. a | Variability of symptoms in premenopause, perimenopause and postmenopause (blue line; left axis); glucose metabolism in the brain (red line; right axis). b | Diversity of neurological symptoms during perimenopause. The majority of women will experience neurological symptoms during perimenopause; however, a small proportion (~20%) of women will transition without symptoms.
Perimenopause
Perimenopause is often referred to as ‘the change’, which is an apt description because the variability in physiological and neurological symptoms increases with the change toward reproductive senescence (Figure 1a). The female reproductive axis comprises the hypothalamic–pituitary–ovarian–uterine axis and undergoes targeted accelerated changes relative to other systems that are otherwise healthy.6 Reproductive senescence in women is defined by oocyte depletion, which begins at birth and continues until menopause is achieved.3 The relatively wide age range (40–58 years3,4) for menopause suggests that women either have a highly variable number of oocytes or that the rate of oocyte loss varies greatly between individuals.
Characteristic symptoms inherent to the perimenopausal transition have been extensively documented.3 The Stages of Reproductive Aging Workshop (STRAW),3 which was an international collaborative effort, along with the Study of Women Across the Nation (SWAN), which studied an ethnically diverse population in the USA,13 have contributed to the classification of the perimenopausal transition and characterized the complexity of symptoms and the ethnic diversity of perimenopausal phenotypes. Principal symptoms include variability in cycle length, vasomotor symptoms (hot flushes) and hormonal fluctuations (Figure 1b).3 Determination of these well-defined endocrine features of the perimenopause has enabled a symptom and indicator classification system, known as the STRAW criteria, that describes the stages of reproductive ageing across the transition from perimenopause to postmenopause.3
From an endocrine system perspective, perimenopause is characterized by increased variability in the length of the menstrual cycle and levels of circulating hormones (Figure 1a). During perimenopause, ovulation occurs irregularly owing to fluctuations in levels of hypothalamic, pituitary and ovarian hormones.2 Variability in the hormonal milieu within and across the stages of perimenopause is high. For example, follicle-stimulating hormone (FSH) levels increase during some cycles but return to premenopausal levels in subsequent cycles.14 The accurate determination of FSH levels is complicated by a pulsatile pattern of secretion; levels of FSH stabilize only when perimenopause is complete.3
Circulating levels of 17β-estradiol are also highly variable throughout perimenopause.1 Estrogen levels can be persistently low, as might be expected, but can also be abnormally high.15 The concentration of 17β-estradiol is stabilized at a low level in late-stage perimenopause when cycling has ceased and the menopausal transition has been completed.3 The hormonal phenotypes of perimenopause have been characterized using trajectory clustering analysis of levels of 17β-estradiol and FSH, which revealed four distinct trajectories of change for 17β-estradiol and three for FSH.16 The trajectories of decline in serum levels of 17β-estradiol included a slow steady decline, a low level that did not change over time, a rise with slow decline and a rise with steep decline.16 Trajectories of hormonal change were highly correlated with ethnicity and BMI.16 Remarkably, the cyclical nature of levels of progesterone seem to remain generally intact, whereas the absolute level of progesterone can vary across normal values, high spikes and undetectable levels.14 This variability in hormone levels creates difficulties in relying upon a single measurement of a hormone level at a single time point to ascertain a diagnosis of perimenopause.17 Despite the well-characterized trajectory of hormone levels across the perimenopausal stages, variability in stage duration, complex symptom profiles and symptomatic severity (Figure 1b) can differ dramatically both across and within different ethnic groups.18
Reproductive senescence occurs in all species with some, such as rodents and nonhuman primates, sharing critical features with the human perimenopausal transition.19 In all mammals studied, numbers of ovarian follicles and oocytes decline exponentially over the course of life, beginning at, or even before, birth.20 As in humans, rodents undergo a decline in the number of ovarian follicles, irregular cycling, irregular fertility, steroid hormone fluctuations and development of insensitivity to estrogen.21,22 However, unlike humans, rodents do not menstruate, but many nonhuman primates do, such as the rhesus monkey.23 High plasma concentrations of estrogen are associated with insensitivity of the hypothalamic–pituitary system to estrogen in humans,24 nonhuman primates and rodents.20
In cultured rat primary hippocampal neurons, the concentration of 17β-estradiol, but not the fluctuating pattern of exposure to this hormone, was the determining factor for neuronal viability.23 Exposure to a low concentration of 17β-estradiol promoted neuronal survival and intracellular calcium homeostasis, whereas exposure to a high concentration was ineffective and resulted in increased cellular vulnerability to neurodegenerative insults.23 These in vitro findings are consistent with in vivo data from mice implanted with pumps that delivered supraphysiologic levels of 17β-estradiol; the mice became permanently acyclic within 1 week of implantation of the pump.24 Insensitivity to estrogen is associated with reactive gliosis in the hypothalamus and ultimately cell death.25 Studies to determine ovarian versus neuroendocrine contributions to reproductive senescence revealed that both the ovaries and the brain are determinants of reproductive life. Notably, neuroendocrine responses (probably because of exposure to high levels of 17β-estradiol24), can limit reproductive system function, whereas the ovarian transplants remain viable.25,26 These data indicate that reproductive senescence is driven by both the ovaries and the brain; persistent high levels of estrogen from the ovaries, as occurs during perimenopause, is a likely candidate for mediating the neural switch to reproductive senescence.
Neurological symptoms
Although the clinical definition of perimenopause focuses on functional changes in the reproductive system, the symptoms of perimenopause are largely neurological in nature (Figure 1b), the documentation of which spans continents, cultures and ethnicities.15,27–43 The complexity of the perimenopausal transition is reflected by a range of neurological symptoms, which include temperature dysregulation, depression, insomnia, pain and cognitive dysfunction.16,31,43 Neurological symptoms tend to cluster together in occurrence and severity44 and, in many instances, persist into postmenopause.34,35 The breadth of neurological symptoms that can occur during the perimenopausal transition is indicative of disruption in multiple systems (Figure 2), whereas their simultaneous emergence during the same transition is indicative of involvement one or more common controlling factors. If common controlling factors exist, then they must meet multiple requirements. Among these requirements would be change from premenopausal levels of the factors, a distributed system of response elements within affected neural systems, mechanisms of action that would account for the breadth of symptoms and evidence for reversal of symptoms in the presence of the controlling factors. Other requirements might of course exist, but these conditions serve as a ‘first pass’ set of considerations. Among potential controlling factors, estrogen fulfils many of these requirements.
Figure 2 |.

Neurological functions affected by the perimenopausal transition. Brain regions and their corresponding functions provide a map of the neural circuits regulated by estrogen and a neurobiological basis for the array of symptoms that can emerge during perimenopause. Nuclear, membrane-associated and mitochondrial estrogen receptors are distributed within each of the neural circuits and can be present in both neurons and glial cells. While the complete distribution of ER-α and ER-β remains to be completely mapped in humans, in rats the location of these receptors is well documented.45–53 Dysregulation of estrogen signalling, either through changes in estrogen concentration or through modifications of estrogen receptor, will affect neural circuit activation and, thus, neurological function. Abbreviations: ER, estrogen receptor; GPER, G protein-coupled estrogen receptor.
Estrogen and the estrogen receptor
Localization in the brain
Neural structures in the brain that control the multiple functions affected during the perimenopausal transition are populated with multiple types of estrogen receptors (Figure 2).45–52 To date, three distinct estrogen receptors have been identified: estrogen receptor 1 (ESR1; commonly known as ER-α), estrogen receptor 2 (ESR2; commonly known as ER-β) and G-protein coupled estrogen receptor 1 (GPER; formerly known as GPR30). For estrogen to be a regulator of the neurological systems that generate the symptoms associated with perimenopause, the locations of estrogen receptors need to be co-incident with the relevant neuroanatomical regions. The abundance of estrogen receptors in the hypothalamic preoptic nucleus, which is the primary thermoregulatory centre, is among the densest in the brain.45,53 Likewise, the suprachiasmatic nucleus of the hypothalamus, which has a central role in regulation of sleep and circadian rhythms, is rich in estrogen receptors.53 In brain regions that are crucial to learning and memory, estrogen receptors are present in the prefrontal cortex, each of the subfields of the hippocampus, the amygdala, the cingulate cortex and the retrosplenial cortex.45,53 The 5-hydroxytryptaminergic neurons of the raphe nucleus are positive for estrogen receptors, as are the adrenergic neurons of the locus coeruleus.45,50,54
Removal of the ovaries before menopause induces the neurological symptoms of the perimenopausal transition, whereas restoration of estrogen prevents or reverses neurological symptoms.6,47,55–57 Progesterone, which is also produced by the ovaries, does not seem to have the same neuronal effects and, in many instances, antagonizes the neural actions of estrogen in both preclinical and clinical analyses.58–60 The ovarian–neural estrogen axis regulates multiple neurological systems and functions in the brain through a network of estrogen receptors; thus, estrogen is a master regulator that ensures the brain can effectively respond within timescales ranging from rapid (seconds to minutes), intermediate (minutes to hours) and long (hours to days) to regulate and coordinate multiple systems (Figures 2 and 3). Moreover, estrogen signalling through this network of receptors ensures that neurons can generate sufficient energy to meet demand.61 Changes in either the availability of estrogen or its receptor network can affect intracellular signalling and neural circuit function. While the intricacies of estrogen receptors and signalling cascades are well characterized, the coordination across the network of estrogen receptors necessary for neural circuit function awaits detailed investigation.
Figure 3 |.

Estrogen receptor network. The estrogen receptor network integrates cellular responses and functions in the brain. Estrogen binding and activation of the membrane-bound receptors, mER-α, mER-β and GPER, leads to activation of signalling pathways that regulate expression of early and intermediate response genes. Estrogen binding to the nuclear estrogen receptors, ER-α and ER-β, results in activation of transcriptional pathways that regulate expression of late response genes. Activation and translocation of ER-β to the mitochondria has been implicated in regulating expression of mitochondrial genes. Furthermore, estrogen can modulate transcriptional gene expression, activated by either rapid signalling or transcriptional pathways, via epigenetic regulation. This network of receptors enables the integration of signals across rapid, intermediate and late response pathways to coordinate a broad spectrum of cellular elements, including substrate transporters, metabolic enzymes and catalytic processes, which ultimately results in generation of energy to fuel neurological function. Abbreviations: ER, estrogen receptor; GPER, G-protein coupled estrogen receptor 1; mER, membrane estrogen receptor; mtER, mitochondrial estrogen receptor.
Cellular localization
Plasma membrane
The localization of estrogen receptors facilitates the sequential activation of estrogen receptors across a network, which initiates rapid-response cascades followed by an engagement of gene expression for long-term responses (Figure 3). Regardless of their intracellular location, all estrogen receptors are activated by the endogenous estrogen, 17β-estradiol. Starting at the plasma membrane, the first estrogen receptor that can be activated is GPER, which traverses the plasma membrane. Activation of GPER triggers rapid signalling cascades.49 The specific localization of GPER remains controversial with some groups reporting localization of GPER at the plasma membrane,62 and others reporting GPER as being localized to the membrane of the endoplasmic reticulum.49 Estrogen activates monomeric GTPase (Ras)-driven signalling cascades through GPER,49 including cAMP, the PI3K–Akt and MAPK–Erk signalling pathways, the latter of which leads to neurite generation in developing hippocampal neurons.63 In the specialized caveolar involutions of the intracellular side of the plasma membrane, membrane-bound ER-α and ER-β (mER-α and mER-β) are associated with activation of fast-acting signalling pathways, such as the PI3K–Akt and MAPK cascades.48,64 Activation of fast-acting signalling cascades is one of several mechanisms by which estrogen regulates gene transcription (Figure 3). In the case of estrogen receptor activation of the PI3K–Akt and MAPK pathways, estrogen binding to its receptor initiates a signalling cascade that leads to the phosphorylation and activation of CREB transcription factors, which upregulate expression of genes encoding proteins involved in learning and memory, synaptic transmission and neural growth factors such as brain-derived neurotrophic factor.47,56,65,66
Mitochondria
ER-β is also found in mitochondria (referred to as mtER-β), where it regulates mitochondrial function and can induce expression of mitochondrial genes (Figure 3).67–70 ER-β is also localized to the mitochondria interface and communicates with the endoplasmic reticulum, a region also known as mitochondria-associated membranes.69 mtER-β within mitochondria is uniquely positioned to regulate mitochondria-specific gene expression,71 which enables estrogen to regulate energy homeostasis throughout the cell.47 This function of estrogen is particularly important to neurons, which require ATP for synaptic transmission.47 75% of all ATP generated in neurons is used to meet the energy demands of the sodium–potassium pump, which actively pumps both Na+ and K+ against their concentration gradients to re-establish the neuronal membrane potential necessary to respond to the next wave of action potentials.47,56 This function is particularly important for maintaining the high frequency synaptic transmissions that are associated with learning and memory function.47,56 Nuclear ER-α can also regulate mitochondrial function via modulating the expression of nuclear genes that encode proteins that are targeted to the mitochondria.69–71
Nucleus
Estrogen-mediated transcription of genes requires translocation of ER-α and ER-β into the nucleus. ER-α and ER-β dimerize and are transported into the nucleus, which can comprise α or β homodimers or heterodimers of ER-α and ER-β proteins (Figure 3).48 Estrogen receptors bind directly to specific estrogen response element sequences on the DNA or are tethered to other transcription factors, such as the activator protein 1 (also known as transcription factor AP-1), to regulate gene expression.48 A 1D view of regulation of gene expression by estrogen receptors is that these transcription factors only promote gene expression.56,71 However, this hypothesis is an over simplification, as demonstrated by estrogen-mediated regulation of the bioenergetic system of the brain; both ER-α and ER-β in the brain promote expression of nuclear encoded genes that are required for glucose transport, glucose metabolism and mitochondrial functions, while simultaneously suppressing expression of genes required for ketone body metabolism, inflammation and β-amyloid generation.59,71
Splice variants
The complexity of estrogen receptors is extended by splice variants of both ER-α and ER-β, which typically have functional differences.72 Three splice variants of ER-α and one variant of ER-β have been described in the brain.73–75 Some particular splice variants are generated during the perimenopausal transition. Splice variants of ER-α have been detected in the hypothalamus and hippocampus of the human female brain.75 The ER-α splice variant MB1 has been detected in the brain of perimenopausal women (49–50 years) and has increased expression throughout the transition, with greatest expression in postmenopause (58–94 years of age).75 This variant is expressed in neurons, astrocytes and endothelia, where it is associated with nitric oxide production required for vasodilation.75 ER-β splice variants are also expressed in select brain regions of the rat, including the hypothalamus, hippocampus and cerebral cortex.76,77 A well-characterized rodent ER-β variant, ER-β2, contains an additional 18 amino acids in the ligand-binding domain, which considerably reduces the binding affinity of 17β-estradiol.72 In ovariectomized rats, protein levels of the ER-β2 splice variant were increased in the hippocampus, which was associated with reduced neurogenesis and an increase in behavioural indicators of depression.77 Expression of ER-β2 was reversed when estrogen was administered soon after removal of the ovaries (6 days), as demonstrated by restoration of neurogenesis and amelioration of depressive behaviours in these animals.77 However, the restorative potential of estrogen was time-dependent, as delay in estrogen replacement (180 days after removal of the ovaries) showed no therapeutic benefits for the ovariectomy-induced decline in neurogenesis and increase in depressive behaviours.77
The reduced binding affinity of some ER-α and ER-β splice variants might partially account for the perimenopausal loss in sensitivity to 17β-estradiol.22 In addition to splice variants, several polymorphisms in ESR1 are associated with an increased risk of Alzheimer disease in women, particularly when co-occurring with the APOE*e4 allele.61
Post-translational modifications
In addition to splice variants and polymorphisms, estrogen signalling is modulated by ubiquitination and subsequent proteasomal degradation of estrogen receptors.78 Long-term estrogen deprivation results in increased interactions between ER-α and the heat shock 70 kDa protein 1A/1B, which leads to ubiquitination and proteasomal degradation of ER-α.78 Furthermore, ubiquitination of ER-α was specific to the hippocampal neurons and could be prevented when 17β-estradiol treatment was administered before, but not after, ovariectomy.78
Epigenetic regulation
A fairly unexplored area of research in the brain is the control of epigenetic modifications by estrogen during perimenopause and, in turn, epigenetic regulation of expression of the genes encoding the estrogen receptors. In young female mice, 17β-estradiol induced epigenetic changes, such as acetylation and methylation.79 Histone 3 seems to be a key protein involved in the epigenetic regulation mediated by 17β-estradiol; both acetylation and methylation of histone 3 in neurons of the mouse dorsal hippocampus regulate 17β-estradiol-driven enhancement of memory consolidation.80 17β-estradiol induces acetylation of histone 3, but not of histone 4, via activation of the ERK signalling pathway in the mouse brain.80 Moreover, administration of 17β-estradiol to female mice reduced protein expression of histone deacetylase 2, which sustains a permissive chromatin state for transcription of genes that are required for hippocampal synaptic plasticity and memory consolidation.80,81 DNA methylation of ER-α at Lys26678 within a critical 3-h period also seems to be necessary for 17β-estradiol to enhance memory consolidation.80,81 The idea that menopause and ageing might induce a transcriptionally repressive chromatin state that decreases transcription of memory-related genes has been proposed.80 Epigenetic control of genes encoding estrogen receptors, a mechanism well-described in breast cancer cell lines,82 remains undetermined in the female brain under normal and disease states. Given that onset of puberty can be determined by epigenetic regulation,83,84 these modifications might also be key drivers of the onset and emergent symptoms of perimenopause.
Adaptation in perimenopause
Collectively, these findings indicate that the perimenopausal transition is a dynamic state for signalling via estrogen receptors and is associated with the emergence of estrogen receptor splice variants, changes in protein expression and alterations in receptor degradation, as well as possible epigenetic reconfigurations. An estrogen response network is activated within and across neural networks through a series of membrane-associated, mitochondrial and nuclear receptors. Although this distributed network of estrogen receptors forms the basis of the remarkable effects of estrogen in brain, it can also be a point of vulnerability for the female brain. In many women, the brain compensates for the changes in levels of estrogen and its receptors during perimenopause. However, for some women this adaptive compensation is diminished, lacking or only expressed in some estrogen-regulated neural networks, which gives rise to the complex phenotype of neurological symptoms of menopause (Figure 2).18,27–43 The extent to which these changes in the estrogen regulatory network are drivers of the perimenopausal transition or an outcome of the transition remain to be fully explored. Elucidating the spectrum of compensatory responses that can be induced by the perimenopausal transition is still in the early stages.
Estrogen and neural bioenergetics
Estrogen and its network of receptors are strong candidates for being regulators of bioenergetic systems in neural circuits of the brain, while simultaneously being affected by the perimenopausal transition.6,47,56,61 Estrogen utilizes a complex repertoire of receptors and signalling pathways to activate and regulate molecular and genomic responses required for neurological function at the cellular, organismic and, ultimately, whole-body level.61 Moreover, biochemical, genomic and bioinformatic data indicate a synergistic interaction between estrogen and insulin signalling pathways exists in the brain.85 Regulating the bioenergetic capacities within and across neural circuits could both support the functions of multiple circuits and, concurrently, create an energetic connectome between and across neural circuits.47
Within the brain, estrogen signalling contributes to processes within the entire bioenergetic system, including glucose transport, glucose metabolism, mitochondrial respiration and ATP production (Figure 4).47,56,67,71,86,87 A portion of the bioenergetic system in the mouse brain is linked to estrogen, as evidenced by the fact that loss of estrogen by surgical removal of the ovaries or reproductive endocrine ageing results in a 15–25% decline in metabolic function in brain.87,88 During the early stages of reproductive senescence or perimenopause in the rodent, the neuronal bioenergetic system undergoes a transformation, which results in a persistent decline in glucose metabolism in the brain.86–89 In female mice, hypometabolism in the brain was demonstrated by a decline in glucose uptake and hexokinase activity at 6–9 months of age (immediately before the transition into reproductive senescence).86 These metabolic changes were accompanied by inactivation of pyruvate dehydrogenase and complex IV activity,86 which led to impairment of the mitochondrial energy-conservation systems in these animals. Lactate transport and utilization also decreased in parallel to the decline in glucose transport,86 which indicates that lactate (which is generated from glucose in astrocytes) is not used as an alternative fuel source during the transition to reproductive senescence. Signs of oxidative stress and free radical damage to mitochondrial lipid membranes are not evident until the animals have transitioned through perimenopause to reproductive senescence.86,89 In the periphery of perimenopausal aged female mice and rats, dysregulation of the metabolic system was demonstrated by impaired glucose tolerance, which is indicative of insulin resistance.86,89
Figure 4 |.

Estrogen-mediated regulation of the bioenergetic system. Estrogen signalling supports and sustains glucose metabolism in the brain by regulating expression of glucose transporters, which results in increased glucose uptake, and by stimulating glucose metabolism, mitochondrial oxidative phosphorylation and ATP generation—collectively referred to as aerobic glycolysis. Glucose (1) is the primary metabolic fuel for the brain. Estrogen regulates the bioenergetic system in brain the through the estrogen receptors, GPER, ER-α and ER-β, and their activation of PI3K and downstream Akt and MAPK–ERK signalling pathways. When the glucose pathway is compromised, for example, during starvation, acetyl-CoA can be generated from ketone bodies via ketogenesis in the liver and transported through the blood to the brain through monocarboxylate transporters (2) or from fatty acid via β-oxidation (3). During the perimenopausal transition, neuronal levels of glucose transporters decline, which is co-incident with the appearance of hypometabolism in the brain.86 The brain adapts to this decline in glucose availability by increasing reliance on ketone bodies as an alternative fuel to generate acetyl-CoA required for entry to the TCA cycle (4) and ultimately generation of ATP via complexes of the mitochondrial redox carriers (5). Initially, ketone bodies are derived from the periphery by lipid metabolism in the liver. Depletion of peripheral sources of ketone bodies can result in metabolism of brain-derived fatty acids to generate ketone bodies via β-oxidation in glia cells (3). Abbreviations: GP, glucose-6-phosphate; GPER, G protein-coupled estrogen receptor 1; ER-α, estrogen receptor α; ER-β, estrogen receptor β; HK, hexokinase; α-KGDH, α-ketoglutarate dehydrongenase, IGF-1, insulin growth factor-1; IRS, insulin receptor substrate; MCT, monocarboxylate transporter; PDH, pyruvate dehydrogenase; PI3K, phosphoinositide 3-kinase; SCOT, succinyl-CoA:3-ketoacid CoA transferase; SDH, succinate dehydrogenase; TCA, tricarboxylic acid cycle.
Removal of ovaries in premenopausal mice results in bioenergetic deficits that are similar in magnitude to those that occur during reproductive ageing.88 However, critical distinctions exist between natural endocrine ageing (that is, changes associated with endocrine function rather than chronological ageing) and surgically induced menopause, and are evident in the differences in the bioenergetic systems of animal models of these processes. In ovariectomized rodents and nonhuman primates, endocrine and ageing programmes seem to be compressed or accelerated, as induction of hypometabolism, mitochondrial dysfunction and oxidative stress occur simultaneously.88,89 The adverse effects of loss of ovarian hormones on the bioenergetic system can be prevented by administration of 17β-estradiol at the time of ovariectomy.71,88–90 In ovariectomized rhesus macaque, changes to mitochondrial morphology were frequently indicative of development of oxidative stress and occurred with a concomitant decline in number of mitochondria within the presynaptic bouton; these boutons were typically smaller than those of normal ageing in non-ovariectomized monkeys.91 The appearance of mitochondrial oxidative stress morphology was associated with a decline in cognitive function mediated by the prefrontal cortex in ovariectomized animals.91 By contrast, natural endocrine ageing of the nonhuman primate prefrontal cortex led to mitochondrial morphology that was associated with increased respiratory capacity, an increased number of presynaptic boutons containing mitochondria, an enlargement of presynaptic boutons and preservation of cognitive function.91 Administration of estrogen at the time of ovariectomy preserved the normal ageing mitochondrial phenotype and cognitive function in nonhuman primates, as well as preventing conversion of mitochondria to a stress-associated phenotype.91
In response to glucose deprivation, the perimenopausal brain shifts to a bioenergetic response that is indicative of long-term fasting—that is, the use of ketone bodies as an alternative energy fuel.86,87,92–94 This response is indicated by a rise in peripheral levels of ketone bodies and their neuronal and astrocytic monocarboxylate transporters, as well as an increase in levels of mitochondrial succinyl-CoA:3-ketoacid CoA transferase (SCOT), which is necessary to metabolize ketone bodies.86,87,94 In addition to use of peripheral ketone bodies as an alternative fuel source, a rise in the levels of proteins required to metabolize fatty acids in the brain was observed in both mice and rats.87,88,93 These proteins, CPT1-L (carnitine O-palmitoyltransferase 1, liver isoform) and HADHA (trifunctional enzyme subunit alpha, mitochondrial), are required for fatty acid transport and metabolism by the mitochondria, respectively.87,88 HADHA converts long-chain fatty acids to acetyl-CoA, which is the primer for ketone body synthesis. As neither cholesterol nor long-chain fatty acids can pass through the blood–brain barrier, an increase fatty acid metabolism in the brain is indicative of increased catabolism of brain-derived lipids, which can disrupt cell membranes and lipid-rich reservoirs, such as myelin, in the brain.93
A shift in metabolism during perimenopause is also evident in women and has been associated with dysregulated glucose metabolism and insulin resistance.13,61 Collectively, preclinical data in rodent models of perimenopause and clinical analyses of the human female brain indicate that the perimenopausal transition is an ordered and sequential process, during which the brain seems to undergo dismantling of the connections between the estrogen receptor network and the bioenergetic system.13,20,89,95 The disengagement of the estrogen receptor network from the bioenergetic system in the brain is also associated with a release of suppression of enzymes required to metabolize ketone bodies.86–89 This loss of suppression enables the emergence of an adaptive compensatory response in the brain that is necessary to utilize ketone bodies as an alternative fuel (Figure 4).86–89
Hypometabolism and neurological systems
If a decline in brain metabolism is a causative factor in the development of neurological symptoms of the perimenopausal transition, then hypometabolism in neuronal circuits is expected to proceed or be co-incident with these symptoms. Moreover, if estrogen is a controlling factor in this process, then women receiving estrogen replacement therapy should exhibit a metabolic profile that is associated with preservation of glucose metabolism in the brain. Evidence to support these hypotheses in affected neurological systems does not yet exist; however, multiple studies of brain metabolism suggest an association between hypometabolism in the brain and development of neurological symptoms in women.60,95–97 Notably, the preponderance of available data have been derived from studies of women who have transitioned through the menopause,60,95,96 with only a few studies specifically targeting women of perimenopausal age.38,60 In studies of women after menopause, those receiving estrogen replacement therapy have a different pattern of brain metabolism to women not receiving estrogen therapy.60,95 After the menopause, women who began estrogen replacement therapy during perimenopause or their menopausal year, had preserved glucose metabolism in brain regions with estrogen-dependent neurological functions, such as hippocampus, entorhinal cortex, medial temporal cortex and posterior cingulate.60,95,96 After the menopause, glucose metabolism declined in estrogen-dependent regions of the brain, but increased in regions, such as pons, caudate and precuneus in women who did not receive estrogen therapy during perimenopause and menopause.96 An important caveat to these studies is that the women who were enrolled were symptomatic and often compared to women who were premenopausal and or women of a similar age who were asymptomatic. Consequently, the data derived from these studies are probably only relevant to symptomatic women and not those undergoing the perimenopausal transition who do not have neurological symptoms. Taken together, these data highlight the adaptive capacity of the perimenopausal female brain and the need for additional detailed analyses of brain metabolism during perimenopause, with a particular focus on genotype and phenotype of this transition.
Hot flushes
The most frequent, and often defining, feature of the perimenopausal transition is the hot flush, which occurs in 75–80% women in the perimenopausal period and can continue for several decades in ~5% of women.3,98,99 Hot flushes can be the only symptom experienced by women undergoing perimenopause (Figure 1b). However, in many women, hot flushes co-occur with other neurological symptoms, including decline in cognitive function,34,100 insomnia, pain and depression (Figure 1b).44 The clinical manifestations of the hot flush have been well documented and are characterized by the onset of a disturbing and intense rise in body temperature, which is primarily experienced in the upper body (chest, neck and head), as well as being accompanied by visible flushing.99 The hot flush is a transient event with sudden and rapid onset, and dissipation within seconds to minutes, but can also last up to 60 min. Hot flushes can range from mild to severe in intensity and with a frequency of up to 50 throughout the day and night; increased severity and frequency during the sleep cycle has also been reported, which can disrupt sleep and daily activities and cause severe stress and anxiety.101
The hypothalamus controls thermoregulation and is, therefore, thought to initiate the hot flush in response to a rise in core temperature (Figure 2).102 The prevailing conceptualization of the hot flush is that a compression of the hypothalamic thermoneutral zone leads to episodic releases of heat in response to a slight increase in core body temperature.99 However, although the hypothalamus is undoubtedly involved in the thermoregulatory response, functional MRI analyses of women in the perimenopausal period indicate activation of the insula and anterior cingulate cortex during hot flushes, which is coupled to activation of the superior frontal gyrus and is associated with sweating.102 Activation of the insular cortex is associated with the sensation of ‘rush of heat’.99 These findings advance our understanding of the perimenopausal hot flush beyond the temperature regulatory zones of the hypothalamus to include an expanded neural network that includes subcortical and cortical structures.
Increasing evidence also indicates an association of the hot flush with altered glucose metabolism.86,103,104 Preclinical studies of bioenergetics using animal models of the female brain during perimenopause indicate that the rise in skin temperature is co-incident with the onset of reproductive variability and senescence.86 Levels of glucose metabolism in the brain also decline. Furthermore, glucose tolerance is compromised in the periphery (which is indicative of insulin resistance) and increased use of ketone bodies and fatty acid metabolism occurs.86,87,94 Consistent with these preclinical findings, analysis in humans using 18F-FDG-PET indicate a decline in brain glucose metabolism in the brain occurs during late perimenopause and continues into postmenopause.95 In the SWAN study,104 hot flushes or night sweats were strongly associated with dysregulation in glucose metabolism, which was indicated by considerably increased levels of fasting blood glucose and raised HOMA scores. The relationship between estrogen receptors and insulin receptors in the brain is well established and provides additional evidence for the contribution of decline in estrogen levels to dysregulated glucose metabolism.85 Moreover, an association between impairment of the adipokine profile and occurrence of hot flushes early in the perimenopausal transition has been described, which also supports a link between metabolic dysregulation and hot flushes.61,105 Decreased adiponectin and increased leptin levels were associated with increased risk of hot flushes in early, but not late, stages of the perimenopause transition.105 This altered adipokine profile parallels BMI measurements of women with stage-dependent risk of hot flashes, such that a high BMI was associated with increased risk of hot flushes in early, but not late, in the perimenopausal transition.105 Increased serum levels of inflammatory cytokine monocyte chemotactic protein 1 (also known as C-C motif chemokine 2) were associated with an increased risk of night sweats, regardless of menopause stage.105 Collectively, these data indicate a strong association between hot flushes and impaired glucose homeostasis in both the brain and the periphery of women undergoing perimenopause.61
Insomnia
Insomnia is a prevalent symptom of the perimenopausal transition and is frequently associated with hot flushes, night sweats, depression and cognitive deficits (Figure 1b).44,106,107 The hypothalamus is the key nodal complex that regulates sleep–wake cycles (Figure 2). Interestingly, in the suprachiasmatic nucleus (located within the hypothalamus and a key determinant of diurnal rhythms), ER-β expression follows a diurnal rhythm, which is evident in young (aged 3–4 months) and middle aged (aged 11–12 months) rats but not in old rats (aged 19–24 months).108 In the SWAN study, prevalence of sleep disturbance varied with ethnicity, with the highest prevalence (40%) occurring in white women, intermediate prevalence (31–38%) in Chinese, Hispanic and African American women and the lowest prevalence in Japanese women (28%).107 The SWAN data indicate that sleep difficulties are maximal during late perimenopause and persist into postmenopause, irrespective of vasomotor symptoms.107 Brain imaging studies in women during the perimenopausal stage who have sleep disturbances remain to be conducted. Having increased understanding of the effects of insomnia in this population is particularly important given the emerging body of data that are indicative of long-term adverse consequences of sleep deprivation.109 Multiple studies have indicated an association between insomnia and reduced brain volume in regions that include the hippocampus and orbitofrontal and parietal grey matter.109 Furthermore, in many prospective studies, poor sleep quality is associated with an increased likelihood of cognitive impairment and, in some studies, an increased risk of dementia.109 Sleep deprivation can also induce β-amyloid accumulation in the brain.110
Cognition
Subjective and objective memory deficits during the perimenopause have been well documented (Figure 1).35 In 18F-FDG-PET analyses, indicators of hypometabolism in brain regions required for learning and memory function have been demonstrated.60,95,96 Over a 2-year observation period, hypometabolism was apparent in women undergoing menopause in the hippocampus, parahippocampal gyrus and temporal lobe,96 as well as in the medial prefrontal cortex and the posterior cingulate cortex.60,95 Estrogen replacement therapy prevented hypometabolism in each of these brain regions and preserved memory function.60,95 In a study that assessed regional cerebral blood flow, women in the postmenopausal period who were receiving estrogen had increased resting state cerebral blood flow in brain regions that are now recognized as part of the default mode network; this effect did not occur in women who did not receive estrogen.96 The default mode network, or resting state network, is a system of brain regions that includes the medial prefrontal cortex, the posterior cingulate cortex and lateral parietal cortices. These regions have a high degree of functional connectivity during rest, which consumes a substantial amount of ATP in its continuous activation.111 However, women in the postmenopausal period who were not treated with hormone therapy have increased cerebral blood flow to brain regions that are required for motor and spatial function.96,112
Women in perimenopause who received hormone therapy (estrogen plus a progestin) also had enhanced activation in the hippocampus combined with decreased parahippocampal activation, which suggests that this treatment administered during perimenopause increases state-dependent and memory-dependent processes to promote verbal recognition performance.113 Analyses of the default mode network in women in postmenopause who were neurologically healthy but at risk of Alzheimer disease indicated that high blood insulin levels were associated with diminished network connectivity.111 Protective metabolic effects of hormone therapy were most evident in women with low insulin resistance, whereas the same therapy was ineffective in women with increased insulin resistance.114 Performance in working memory in both groups of women also paralleled the metabolic response to hormone therapy.114 Glucose metabolism in the brain is established as being critical to neurological function and that evidence of hypometabolism is apparent several decades before diagnosis of neurodegenerative diseases such as Alzheimer disease.115–117
Depression
Increased risk of depression in perimenopausal women is supported by evidence from several longitudinal studies that have documented odds ratios ranging from 1.8 to 2.9 of increased risk compared with women in premenopause.33,35,36 During perimenopause, both hot flushes and depression can occur early in the transition in women with no previous history of these symptoms although depression was more likely to precede hot flushes.36 In perimenopausal women who had depression, short-term 17β-estradiol treatment had antidepressant efficacy.33 Increased risk of major depression has been linked to variants of ER-α. For example, in one study, women homozygous for the ESR1 rs9340799 variant G had a 1.6-fold increased lifetime risk of major depressive disorder.118
Glucose metabolism in brain regions that regulate depression and anxiety is complex, with different metabolic phenotypes in the pons (where the serotonergic raphe nucleus and locus coeruleus adrenergic systems originate) and frontal cortices (Figure 2).31,97 Compared with women in postmenopause without depression, those with depression have hypometabolism in the pons and hypermetabolism in the middle and inferior (Broca’s) frontal gyrus.97 In women in postmenopause aged >65 years without depression, regional cerebral blood flow was increased in the left pons, which suggests a link to metabolism in the pons and an absence of depressive symptoms.96 Timing of the administration of estrogen therapy seems to be critical to the effectiveness of treatment for depression, as depression scores were not improved in women who received estrogen treatment 5–10 years after perimenopause.31 The long-term effects of perimenopausal depression remain to be determined; however, extensive epidemiological analyses indicate that depression is also a risk factor for Alzheimer disease.119,120
Interventions
Although the outcome of the perimenopause (that is, reproductive senescence) is inevitable for all women, the experience of the perimenopausal transition can be highly variable (Figure 1). Following completion of the perimenopause transition, the neurological system is transformed. Notably, for some women intervention is not warranted, whereas for others, intervention is appropriate and required.
Determining the best strategy to sustain neurological bioenergetic capacity in women during the perimenopausal and menopausal transition is crucial to sustain brain function. Given that estrogen regulates bioenergetic system in brain regions that contribute to perimenopausal symptoms, estrogen replacement therapy is a logical choice. However, many women do not choose this treatment, because the data on the benefits and risks of estrogen replacement therapy are complicated, controversial and confusing.51,52 Many factors contribute to this confounding state, including unresolved issues regarding the timing of intervention, hormone formulation, dose and route of administration. Optimizing and personalizing hormone therapy remains an unmet need in women’s health but is a central issue for precision medicine to treat neurological symptoms that develop during, and persist beyond, the perimenopause. Given the complexity of the perimenopausal transition, women can be grouped into categories according to their symptoms, and clinical trials of new interventions might, therefore, be improved by targeting specific perimenopausal phenotypes rather than simply enrolling a heterogeneous population of women.
Conclusions
The perimenopausal transition occurs within healthy women and initiates a targeted and systematic disassembly of the reproductive system and the neural substrates controlling its function. As a transition state, the perimenopause is similar to developing neurological dysfunction at puberty.8,121 In this regard, the perimenopause satisfies the criteria for a critical period in the neuroadaptive landscape of ageing in the female brain. Many women transition through perimenopause without long-term adverse effects and are likely to remain healthy. However, a substantial proportion of women are vulnerable to the neurological shifts that can occur during this transition and are at increased risk of neurological decline. The association between hypometabolism and the neurological symptoms of the perimenopause suggests that, for some women, this critical transition period could be a tipping point for neurological disease in later life. Whether the neurological vulnerabilities that can emerge during the perimenopause lead to increased risks of neurological disease in later life still needs to be determined; however, multiple neurological symptoms that emerge during the perimenopause are also risk factors for neurodegenerative disease. The ability to detect both peripheral and neurological indicators of metabolic dysfunction might enable the prevention of neurological disease in later life, and might be achieved by sustaining bioenergetic capacity during this critical midlife transition in women.
Key points.
The clinical definition of perimenopause focuses on functional changes in the reproductive system; however, the symptoms of the perimenopause are largely neurological in nature
Most women transition through perimenopause without long-term adverse effects; however, a substantial proportion of women emerge from this transition with an increased risk of neurological decline
Estrogen functions as a master regulator to ensure the brain responds appropriately to coordinate signalling and transcriptional pathways that regulate energy metabolism
The estrogen receptor network becomes uncoupled from the bioenergetic system during the perimenopausal transition and a hypometabolic state associated with neurological dysfunction emerges
In neurological transition states, indicators of dysfunction at the limits of those normally seen can signal tipping points for neurological diseases
The presence, variability, intensity and duration of neurological perimenopausal symptoms could be warning signs for increased risk of neurodegenerative diseases later in life
Acknowledgements
R.D.B. would like to acknowledge the support of grants from the National Institute on Aging (P01AG026572 and R01-AG032236).
Footnotes
Competing interests
The authors declare no competing interests.
Contributor Information
Roberta D. Brinton, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, 1985 Zonal Avenue, Los Angeles, CA 90089, USA.
Jia Yao, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, 1985 Zonal Avenue, Los Angeles, CA 90089, USA..
Fei Yin, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, 1985 Zonal Avenue, Los Angeles, CA 90089, USA..
Wendy J. Mack, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, 1975 Zonal Avenue, Los Angeles, CA 90089, USA.
Enrique Cadenas, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, 1985 Zonal Avenue, Los Angeles, CA 90089, USA..
References
- 1.Brinton RD in Brockelhurst’s Textbook of Geriatric Medicine and Gerontology (eds Fillit H, Rockwood K & Young J) 163–171 (Saunders, 2010). [Google Scholar]
- 2.Butler L & Santoro N The reproductive endocrinology of the menopausal transition. Steroids 76, 627–635 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harlow SD et al. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. Menopause 19, 387–395, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greendale GA, Ishii S, Huang MH & Karlamangla AS Predicting the timeline to the final menstrual period: the study of women’s health across the nation. J. Clin. Endocrinol. Metab. 98, 1483–1491 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.United States Census Bureau. World population by age and sex, [online], http://www.census.gov/population/international/data/worldpop/tool_population.php (2014).
- 6.Brinton RD, Gore AC, Schmidt PJ & Morrison JH in Hormones, Brain and Behavior 2nd edn (eds Pfaff DW et al. ) 2199–2222 (Elsevier, 2009). [Google Scholar]
- 7.Burger HG, Dudley EC, Robertson DM & Dennerstein L Hormonal changes in the menopause transition. Recent Prog. Horm. Res. 57, 257–275 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Paus T, Keshavan M & Giedd JN Why do many psychiatric disorders emerge during adolescence? Nat. Rev. Neurosci. 9, 947–957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petricka JJ & Benfey PN Reconstructing regulatory network transitions. Trends Cell Biol. 21, 442–451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Liu R, Liu ZP, Li M & Aihara K Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2, 342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson RS, Leurgans SE, Boyle PA & Bennett DA Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch. Neurol. 68, 351–356 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramagopalan SV, Dobson R, Meier UC & Giovannoni G Multiple sclerosis: risk factors, prodromes, and potential causal pathways. Lancet Neurol. 9, 727–739 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Santoro N & Sutton-Tyrrell K The SWAN song: Study of Women’s Health Across the Nation’s recurring themes. Obstet. Gynecol. Clin. North Am. 38, 417–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burger H et al. Nomenclature and endocrinology of menopause and perimenopause. Expert Rev. Neurother. 7 (11 Suppl.), S35–S43 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Genazzani AR et al. Endocrinology of menopausal transition and its brain implications. CNS Spectr. 10, 449–457 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Tepper PG et al. Trajectory clustering of estradiol and follicle-stimulating hormone during the menopausal transition among women in the Study of Women’s Health across the Nation (SWAN). J. Clin. Endocrinol. Metab. 97, 2872–2880 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bastian LA, Smith CM & Nanda K Is this woman perimenopausal? JAMA 289, 895–902 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Avis NE et al. Is there a menopausal syndrome? Menopausal status and symptoms across racial/ethnic groups. Soc. Sci. Med. 52, 345–356 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Brinton R Minireview: translational animal models of human menopause: challenges and emerging opportunities. Endocrinology 153, 3571–3578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finch CE The menopause and aging, a comparative perspective. J. Steroid Biochem. Mol. Biol. 142, 132–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker ML, Gordon TP & Wilson ME Menstrual cycle characteristics of seasonally breeding rhesus monkeys. Biol. Reprod. 29, 841–848 (1983). [DOI] [PubMed] [Google Scholar]
- 22.Weiss G, Skurnick JH, Goldsmith LT, Santoro NF & Park SJ Menopause and hypothalamic-pituitary sensitivity to estrogen. JAMA 292, 2991–2996 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Nilsen J & Brinton RD Dose and temporal pattern of estrogen exposure determines neuroprotective outcome in hippocampal neurons: therapeutic implications. Endocrinology 147, 5303–5313 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Mobbs CV et al. Estradiol-induced adult anovulatory syndrome in female C57BL/6J mice: age-like neuroendocrine, but not ovarian, impairments. Biol. Reprod. 30, 556–563 (1984). [DOI] [PubMed] [Google Scholar]
- 25.Finch CE, Felicio LS, Mobbs CV & Nelson JF Ovarian and steroidal influences on neuroendocrine aging processes in female rodents. Endocr. Rev. 5, 467–497 (1984). [DOI] [PubMed] [Google Scholar]
- 26.Mobbs CV, Gee DM & Finch CE Reproductive senescence in female C57BL/6J mice: ovarian impairments and neuroendocrine impairments that are partially reversible and delayable by ovariectomy. Endocrinology 115, 1653–1662 (1984). [DOI] [PubMed] [Google Scholar]
- 27.Maki PM et al. Objective hot flashes are negatively related to verbal memory performance in midlife women. Menopause 15, 848–856 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maki PM et al. Depressive symptoms are increased in the early perimenopausal stage in ethnically diverse human immunodeficiency virus-infected and human immunodeficiency virus-uninfected women. Menopause 19, 1215–1223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greendale GA et al. Effects of the menopause transition and hormone use on cognitive performance in midlife women. Neurology 72, 1850–1857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greendale GA et al. Menopause-associated symptoms and cognitive performance: results from the study of women’s health across the nation. Am. J. Epidemiol. 171, 1214–1224 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rasgon N, Shelton S & Halbreich U Perimenopausal mental disorders: epidemiology and phenomenology. CNS Spectr. 10, 471–478 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Schmidt PJ & Rubinow DR Reproductive ageing, sex steroids and depression. J. Br. Menopause Soc. 12, 178–185 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Schmidt PJ & Rubinow DR Sex hormones and mood in the perimenopause. Ann. NY Acad. Sci. 1179, 70–85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber MT, Rubin LH & Maki PM Cognition in perimenopause: the effect of transition stage. Menopause 20, 511–517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber MT, Maki PM & McDermott MP Cognition and mood in perimenopause: a systematic review and meta-analysis. J. Steroid Biochem. Mol. Biol. 142, 90–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freeman EW, Sammel MD & Lin H Temporal associations of hot flashes and depression in the transition to menopause. Menopause 16, 728–734 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bromberger JT et al. Longitudinal change in reproductive hormones and depressive symptoms across the menopausal transition: results from the Study of Women’s Health Across the Nation (SWAN). Arch. Gen. Psychiatry 67, 598–607 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thurston RC, Santoro N & Matthews KA Adiposity and hot flashes in midlife women: a modifying role of age. J. Clin. Endocrinol. Metab. 96, E1588–E1595 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson HD Menopause. Lancet 371, 760–770 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Usall J et al. Suicide ideation across reproductive life cycle of women. Results from a European epidemiological study. J. Affect Disord. 116, 144–147 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Genazzani AR, Gambacciani M & Simoncini T Menopause and aging, quality of life and sexuality. Climacteric 10, 88–96 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Genazzani AR, Pluchino N, Luisi S & Luisi M Estrogen, cognition and female ageing. Hum. Reprod. Update 13, 175–187 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Cray LA, Woods NF & Mitchell ES Identifying symptom clusters during the menopausal transition: observations from the Seattle Midlife Women’s Health Study. Climacteric 16, 539–549 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Cray LA, Woods NF, Herting JR & Mitchell ES Symptom clusters during the late reproductive stage through the early postmenopause: observations from the Seattle Midlife Women’s Health Study. Menopause 19, 864–869 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shughrue PJ, Lane MV & Merchenthaler I Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J. Comp. Neurol. 388, 507–525 (1997). [DOI] [PubMed] [Google Scholar]
- 46.O’Dowd BF et al. Discovery of three novel, G-protein-coupled receptor genes. Genomics 47, 310–313 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Brinton RD Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci. 30, 212–222 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nilsson S, Koehler KF & Gustafsson JA Development of subtype-selective estrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 10, 778–792 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Prossnitz ER & Barton M The, G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 7, 715–726 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki H et al. Involvement of estrogen receptor β in maintenance of serotonergic neurons of the dorsal raphe. Mol. Psychiatry 18, 674–680 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Brailoiu E et al. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J. Endocrinol. 193, 311–321 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Naugle MM et al. G-protein coupled estrogen receptor, estrogen receptor α, and progesterone receptor immunohistochemistry in the hypothalamus of aging female rhesus macaques given long-term estradiol treatment. J. Exp. Zool. A Ecol. Genet. Physiol 321, 399–414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McEwen BS, Akama KT, Spencer-Segal JL, Milner TA and Waters EM Estrogen effects on the brain: actions beyond the hypothalamus via novel mechanisms. Behav. Neurosci. 126, 4–16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bethea CL, Mirkes SJ, Su A & Michelson D Effects of oral estrogen, raloxifene and arzoxifene on gene expression in serotonin neurons of macaques. Psychoneuroendocrinology 27, 431–445 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Maki PM The timing of estrogen therapy after ovariectomy--implications for neurocognitive function. Nat. Clin. Pract. Endocrinol. Metab. 4, 494–495 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Brinton RD The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 31, 529–537 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rocca WA, Grossardt BR & Shuster LT Oophorectomy, estrogen, and dementia: a 2014 update. Mol. Cell. Endocrinol. 389, 7–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brinton RD et al. Progesterone receptors: form and function in brain. Front. Neuroendocrinol. 29, 313–339 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao L et al. Continuous versus cyclic progesterone exposure differentially regulates hippocampal gene expression and functional profiles. PLoS ONE 7, e31267 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rasgon NL et al. Prospective randomized trial to assess effects of continuing hormone therapy on cerebral function in postmenopausal women at risk for dementia. PLoS ONE 9, e89095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rettberg JR, Yao J & Brinton RD Estrogen: a master regulator of bioenergetic systems in the brain and body. Front. Neuroendocrinol. 35, 8–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tiano JP & Mauvais-Jarvis F Importance of estrogen receptors to preserve functional β-cell mass in diabetes. Nat. Rev. Endocrinol. 8, 342–351 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Ruiz-Palmero I, Hernando M, Garcia-Segura LM & Arevalo MA G protein-coupled estrogen receptor is required for the neuritogenic mechanism of 17β-estradiol in developing hippocampal neurons. Mol. Cell. Endocrinol. 372, 105–115 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Levin ER Extranuclear estrogen receptor’s roles in physiology: lessons from mouse models. Am. J. Physiol. Endocrinol. Metab 307, E133–E140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mannella P & Brinton RD Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J. Neurosci. 26, 9439–9447 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scharfman HE & Maclusky NJ Similarities between actions of estrogen and BDNF in the hippocampus: coincidence or clue? Trends Neurosci. 28, 79–85 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Simpkins JW, Yi KD, Yang SH & Dykens JA Mitochondrial mechanisms of estrogen neuroprotection. Biochim. Biophys. Acta 1800, 1113–1120 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milner TA et al. Ultrastructural localization of estrogen receptor β immunoreactivity in the rat hippocampal formation. J. Comp. Neurol. 491, 81–95 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Irwin RW et al. Selective estrogen receptor modulators differentially potentiate brain mitochondrial function. J. Neuroendocrinol. 24, 236–248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arnold S, Victor MB & Beyer C Estrogen and the regulation of mitochondrial structure and function in the brain. J. Steroid Biochem. Mol. Biol. 131, 2–9 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Nilsen J, Irwin RW, Gallaher TK & Brinton RD Estradiol in vivo regulation of brain mitochondrial proteome. J. Neurosci. 27, 14069–14077 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heldring N et al. Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 87, 905–931 (2007). [DOI] [PubMed] [Google Scholar]
- 73.NCBI AceView. Homo sapiens gene ESR1, encoding estrogen receptor 1 [online], http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?db=human&term=ESR1 (2010).
- 74.NCBI AceView. Homo sapiens complex locus ESR2, encoding estrogen receptor 2 (ER beta). [online], http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?db=human&term=ESR2 (2010).
- 75.Ishunina TA & Swaab DF Age-dependent ERα MB1 splice variant expression in discrete areas of the human brain. Neurobiol. Aging 29, 1177–1189 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Chung WC et al. Detection and localization of an estrogen receptor beta splice variant protein (ERβ2) in the adult female rat forebrain and midbrain regions. J. Comp. Neurol. 505, 249–267 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Wang JM et al. A dominant negative ERβ splice variant determines the effectiveness of early or late estrogen therapy after ovariectomy in rats. PLoS ONE 7, e33493 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang QG et al. C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-α and the critical period hypothesis of estrogen neuroprotection. Proc. Natl Acad. Sci. USA 108, E617–E624 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frick KM Epigenetics, estradiol and hippocampal memory consolidation. J. Neuroendocrinol. 25, 1151–1162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fortress AM & Frick KM Epigenetic regulation of estrogen-dependent memory. Front. Neuroendocrinol. 35, 530–549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Z, Fan L, Fortress AM, Boulware MI & Frick KM Hippocampal histone acetylation regulates object recognition and the estradiol-induced enhancement of object recognition. J. Neurosci. 32, 2344–2351 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang X et al. Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl Acad. Sci. USA 110, 17284–17289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lomniczi A et al. Epigenetic control of female puberty. Nat. Neurosci. 16, 281–289, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCarthy MM A piece in the puzzle of puberty. Nat. Neurosci. 16, 251–253 (2013). [DOI] [PubMed] [Google Scholar]
- 85.Mendez P, Wandosell F & Garcia-Segura LM Cross-talk between estrogen receptors and insulin-like growth factor-I receptor in the brain: cellular and molecular mechanisms. Front. Neuroendocrinol. 27, 391–403 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Ding F, Yao J, Rettberg JR, Chen S & Brinton RD Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS ONE 8, e79977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yao J, Hamilton RT, Cadenas E & Brinton RD Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim. Biophys. Acta 1800, 1121–1126 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yao J et al. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta-amyloid. Neurobiol. Aging 33, 1507–1521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yin F et al. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol. Aging 10.1016/j.neurobiolaging.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao L, Mao Z, Chen S, Schneider LS & Brinton RD Early intervention with an estrogen receptor β-selective phytestrogenic formulation prolongs survival, improves spatial recognition memory, and slows progression of amyloid pathology in a female mouse model of Alzheimer’s disease. J. Alzheimers Dis. 37, 403–419 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hara Y et al. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc. Natl Acad. Sci. USA 111, 486–491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cahill GF Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 26, 1–22 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Yao J, Rettberg JR, Klosinski LP, Cadenas E & Brinton RD Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol. Aspects Med. 32, 247–257 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yao J et al. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 106, 14670–14675 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rasgon NL et al. Estrogen use and brain metabolic change in postmenopausal women. Neurobiol. Aging 26, 229–235 (2005). [DOI] [PubMed] [Google Scholar]
- 96.Maki PM & Resnick SM Longitudinal effects of estrogen replacement therapy on PET cerebral blood flow and cognition. Neurobiol. Aging 21, 373–383 (2000). [DOI] [PubMed] [Google Scholar]
- 97.Rasgon NL, Kenna HA, Geist C, Small G & Silverman D Cerebral metabolic patterns in untreated postmenopausal women with major depressive disorder. Psychiatry Res. 164, 77–80 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Santoro N Symptoms of menopause: hot flushes. Clin. Obstet. Gynecol. 51, 539–548 (2008). [DOI] [PubMed] [Google Scholar]
- 99.Freedman RR Menopausal hot flashes: mechanisms, endocrinology, treatment. J. Steroid Biochem. Mol. Biol. 142, 115–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maki PM Minireview: effects of different HT formulations on cognition. Endocrinology 153, 3564–3570 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Avis NE et al. Change in health-related quality of life over the menopausal transition in a multiethnic cohort of middle-aged women: Study of Women’s Health Across the Nation. Menopause 16, 860–869 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Freedman RR, Benton MD, Genik RJ 2nd & Graydon, F. X. Cortical activation during menopausal hot flashes. Fertil. Steril. 85, 674–678 (2006). [DOI] [PubMed] [Google Scholar]
- 103.Simpkins JW, Katovich MJ & Millard WJ Glucose modulation of skin temperature responses during morphine withdrawal in the rat. Psychopharmacology (Berl.) 102, 213–220 (1990). [DOI] [PubMed] [Google Scholar]
- 104.Thurston RC et al. Vasomotor symptoms and insulin resistance in the study of women’s health across the nation. J. Clin. Endocrinol. Metab. 97, 3487–3494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thurston RC, Chang Y, Mancuso P & Matthews KA Adipokines, adiposity, and vasomotor symptoms during the menopause transition: findings from the Study of Women’s Health Across the Nation. Fertil. Steril. 100, 793–800 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Campbell IG et al. Evaluation of the association of menopausal status with delta and beta EEG activity during sleep. Sleep 34, 1561–1568 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kravitz HM & Joffe H Sleep during the perimenopause: a SWAN story. Obstet. Gynecol. Clin. North Am. 38, 567–586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wilson ME et al. Age differentially influences estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) gene expression in specific regions of the rat brain. Mech. Ageing Dev. 123, 593–601 (2002). [DOI] [PubMed] [Google Scholar]
- 109.Yaffe K, Falvey CM & Hoang T Connections between sleep and cognition in older adults. Lancet Neurol. 13, 1017–1028 (2014). [DOI] [PubMed] [Google Scholar]
- 110.Ju YE, Lucey BP & Holtzman DM Sleep and Alzheimer disease pathology-—a bidirectional relationship. Nat. Rev. Neurol 10, 115–119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kenna H et al. Fasting plasma insulin and the default mode network in women at risk for Alzheimer’s disease. Neurobiol. Aging 34, 641–649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yee LT, Roe K & Courtney SM Selective involvement of superior frontal cortex during working memory for shapes. J. Neurophysiol. 103, 557–563 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maki PM et al. Perimenopausal use of hormone therapy is associated with enhanced memory and hippocampal function later in life. Brain Res. 1379, 232–243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rasgon NL et al. Insulin resistance and medial prefrontal gyrus metabolism in women receiving hormone therapy. Psychiatry Res. 223, 28–36 (2014). [DOI] [PubMed] [Google Scholar]
- 115.Mosconi L et al. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 72, 513–520 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rasgon NL et al. Insulin resistance and hippocampal volume in women at risk for Alzheimer’s disease. Neurobiol. Aging 32, 1942–1948 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mosconi L et al. Brain imaging of cognitively normal individuals with 2 parents affected by late-onset AD. Neurology 82, 752–760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ryan J et al. Estrogen receptor α gene variants and major depressive episodes. J. Affect Disord. 136, 1222–1226 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Yaffe K et al. The epidemiology of Alzheimer’s disease: laying the foundation for drug design, conduct, and analysis of clinical trials. Alzheimers Dement. 8, 237–242 (2012). [DOI] [PubMed] [Google Scholar]
- 120.Norton S, Matthews FE, Barnes DE, Yaffe K & Brayne C Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 13, 788–794 (2014). [DOI] [PubMed] [Google Scholar]
- 121.Deecher D, Andree TH, Sloan D & Schechter LE From menarche to menopause: exploring the underlying biology of depression in women experiencing hormonal changes. Psychoneuroendocrinology 33, 3–17 (2008). [DOI] [PubMed] [Google Scholar]