

Abstract
Mitochondria generate second messengers, such as H2O2, that are involved in the redox regulation of cell signalling and their function is regulated by several cytosolic signalling pathways. IIS [insulin/IGF1 (insulin-like growth factor 1) signalling] in the brain proceeds mainly through the PI3K (phosphatidylinositol 3-kinase)–Akt (protein kinase B) pathway, which is involved in the regulation of synaptic plasticity and neuronal survival via the maintenance of the bioenergetic and metabolic capacities of mitochondria. Conversely, the JNK (c-Jun N-terminal kinase) pathway is induced by increased oxidative stress and JNK translocation to the mitochondrion results in impairment of energy metabolism. Moreover, IIS and JNK signalling interact with and antagonize each other. This review focuses on functional outcomes of a metabolic triad that entails the co-ordination of mitochondrial function (energy transducing and redox regulation), IIS and JNK signalling, in the aging brain and in neurodegenerative disorders, such as Alzheimer’s disease.
Keywords: brain aging, c-Jun N-terminal kinase (JNK), energy metabolism, insulin/insulin-like growth factor-1 signalling, mitochondrion, neurodegeneration
Introduction
A decrease in energy metabolism, leading to a hypometabolic state, is associated with brain aging and the early stages of neurodegenerative disorders, such as Alzheimer’s disease [1]. Neurons require energy to support action potentials, neuronal plasticity and neurotransmission; thus, the age-related neuronal energy deficits contribute to the cognitive decline associated with aging and to the pathogenesis of several neurodegenerative disorders [2,3]. Age-related changes in mitochondrial bioenergetics cannot be viewed independently of their redox-modulating function, as mitochondria are sources of H2O2 and, as such, are actively involved in the regulation redox-sensitive signalling pathways. Although high levels of H2O2 have been associated with the detrimental effects of oxidative stress, low to intermediate levels of H2O2 are involved in the regulation of redox-sensitive signalling and transcriptional pathways and the resulting cellular responses [4,5].
Hence, mitochondrial function cannot be viewed secluded from either the co-ordinated signalling responses that they trigger or the cytosolic signalling molecules they are recipients of, such as MAPKs (mitogen-activated protein kinases) and the PI3K (phosphatidylinositol 3-kinase)–Akt (protein kinase B) pathway of IIS [insulin/IGF-1 (insulin-like growth factor 1) signalling]. Both the IIS and JNK (c-Jun N-terminal kinase) signalling pathways elicit profound changes in mitochondrial function, thus establishing an intricate signalling network with close connections to mitochondrial bioenergetics and biogenesis and redox homoeostasis. From a cell signalling perspective, brain aging is associated with the activation of JNK pathways and reduced levels of insulin/IGF-1 and their receptors [6,7]. Therefore a co-ordinated metabolic triad encompassed by mitochondria, IIS and JNK signalling acquires further significance in brain aging and the progression of neurodegenerative disorders.
IIS and mitochondrial function
Insulin and IGF-1 bind to their receptors on the cell surface, leading to the phosphorylation of tyrosine residues on the IR (insulin receptor) and the IRS (IR substrate). It further activates PI3K–Akt signalling, which regulates several cellular processes, such as inhibition of the pro-apoptotic GSK3β (glycogen synthase kinase 3β), inactivation of FoxO1 (forkhead box O1) transcription factor, and increased coupling of glucose metabolism by promoting the VDAC (voltage-dependent anion channel)–hexokinase-2 interaction. Accumulating evidence suggests that declined levels of circulating IGF-1 and impairments of IIS in the brain contribute to the age-dependent cognitive decline and Alzheimer’s disease and that insulin resistance observed in diabetes constitutes a risk factor for Alzheimer’s disease [8].
In the brain, IIS signalling occurs mainly through the PI3K–Akt pathway, which is involved in the regulation of synaptic plasticity and neuronal survival via energy metabolism, inactivation of the pro-apoptotic machinery, and the induction of long-term potentiation and depression [9]. Brain glucose uptake and utilization is regulated by IIS. GLUT (glucose transporter) 4, present in neurons, is sensitive to insulin signalling: Akt phosphorylates AS160, thus facilitating its dissociation from GLUT4 storage vesicles, preventing inactivation of Rab-GTP [10] and enhancing translocation of the GLUT4 to the plasma membrane. A similar effect was observed for GLUT3 trafficking, which was induced by increased neuronal activity and mediated by the NMDAR (N-methyl-d-aspartate receptor)/Akt-dependent nNOS (neuronal nitric oxide synthase)–cGMP–PKG (protein kinase G) pathway [11]. The enhanced biosynthesis of GLUT3 in rat neurons is also induced by chronic insulin administration [12].
The PI3K–Akt pathway exhibits neuroprotective properties by mechanisms entailing inhibition of pro-apoptotic Bcl-2 family members (Bad) and phosphorylation of FoxO factors, that results in shuttling of phosphorylated FoxO from the nucleus to cytosol, thereby preventing the transcription of FoxO-driven pro-apoptotic and haem degradation genes [13]. Also, the anti-apoptotic effect of PI3K–Akt signalling is a consequence of phosphorylation and inhibition of GSK3β that, in its active form, phosphorylates anti-apoptotic Bcl-2, Bcl-xL and Mcl-1 [14]. Insulin prevents the release of cytochrome c in the reperfused brain in a PI3K-dependent way [15]. The role of IIS in astrocytes is less well-described, although it promotes glycogenesis and proliferation in astrocytes [16]. Delivery of the IGF1 gene to astrocytes reduces their inflammatory response to lipopolysaccharide [17]. Insulin increases the total and surface expression of glutamate transporter GLT1 in astrocytes by a pathway involving the PI3K–Akt/mTOR (mammalian target of rapamycin) signalling cascade, thus playing a role in astrocyte reactivation after CNS (central nervous system) injuries [18]. Given their essential role in removing excessive neurotoxic glutamate from the extrasynaptic space, increased GLT1 expression by insulin could be involved in the attenuation of excitotoxicity.
Among its various effects, the IIS impinges on mitochondrial function directly: insulin signalling was shown to support the functional integrity of the electron-transfer chain by suppressing the FoxO1–HMOX1 (haem oxygenase 1) pathway. IIS is believed to regulate mitochondrial biogenesis through alterations in NAD+ /NADH values and subsequent activation of the SIRT1 (sirtuin 1)–PGC1α (peroxisome-proliferator-activated receptor co-activator 1α) pathway [13]; conversely, another study provided evidence that insulin and IGF-1 attenuated SIRT1 activation induced by caloric restriction [19]. Upon insulin stimulation, active Akt is also able to translocate to mitochondria in human neuroblastoma cells where it phosphorylates a constitutive form of GSK3β and the β subunit of ATPase [20], a viable mechanism to stimulate the energy-transducing capacity of mitochondria.
Mitochondrial H2O2 originates from the stoichiometric disproportionation of O2•− [21] and is regulated by four main determining factors [22]: the ratios of reduced/oxidized pyridine nucleotides, reduced/oxidized coenzyme Q, the local mitochondrial (O2) and the inner membrane potential. Mitochondrial H2O2 is involved in the regulation of insulin signalling, owing to the redox-sensitive cysteine residues in the IR, IGF-1 receptor and IRS. Oxidation of cysteine residues to Cys-OH, Cys-SG (S-glutathionylation) or disulfides promotes tyrosine autophosphorylation of the IR [23], as well as the inhibition of tyrosine phosphatase [e.g. PTP1B (protein tyrosine phosphatase non-receptor type 1)] and lipid phosphatase (PTEN; phosphatase and tensin homologue deleted on chromosome 10), which are both negative regulators of IIS through the dephosphorylation of IR/IRS and PIP3 (phosphatidylinositol-3,4,5-trisphosphate) respectively.
Insulin induces the generation of low levels of H2O2 directly, which is essential for the initial activation of insulin pathway (redox priming) [24]. H2O2 generated by mitochondria plays a significant role in the activation of the IR (tyrosine autophosphorylation) in neurons upon insulin stimulation: the mitochondrial uncoupler FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) diminishes not only mitochondrial respiration, but also insulin-induced H2O2 production and phosphorylation of IR [23]. Therefore the activation of IIS without the involvement of ligand-receptor binding through the redox priming mechanism, suggests that endogenous H2O2 generated by mitochondria could have important roles in the regulation of a wide variety of cellular functions through the IIS. However, this concept needs to include the spatial regulation of H2O2 signals, such as its formation at specific cellular compartments, the occurrence of H2O2 gradients and the modulation of H2O2 removal systems close to the site of generation [25,26]. Furthermore, in NIH 3T3 cell lines, mitochondrial H2O2 is involved in the translocation and incorporation of cytosolic Akt (phosphorylated at Ser473) into mitochondria; once in mitochondria, Akt is further phosphorylated at Thr308, a step required for its further shuttling to the nuclei [27]. These results indicate a functional role of mitochondrial energetic and redox function in the modulation of the IIS.
JNK signalling and mitochondrial function
JNK is involved in the regulation of several cellular responses including transcription, survival and apoptosis upon diverse stimuli [28]. As a pro-apoptotic kinase, activated JNK phosphorylates 14-3-3 proteins, a cytosolic anchor of Bax, thereby facilitating the translocation of Bax to mitochondria, which further initiate mitochondrion-dependent apoptosis [29]. JNK signalling is also implicated in inflammatory responses in astrocytes: JNK is activated in primary glia cultures in response to TNF (tumour necrosis factor) α/β, IL-1 (interleukin 1), protein synthesis inhibitors, bacterial sphingomyelinase, cell-permeable ceramide, UV light, heat shock and mechanical injury [30]. JNK activation in the brain is associated with intracellular β-amyloid accumulation and neuronal death in animal models of Alzheimer’s disease and in patients [31].
Anisomycin- or H2O2-mediated activation of JNK in the cytosol of primary cortical neurons results in its translocation to mitochondria. Upon association of active JNK with the outer mitochondrial membrane, several mitochondrial proteins are phosphorylated [32], among them the anti-apoptotic proteins Bcl-2 and Bcl-xL, thereby regulating mitochondrion-driven apoptotic signalling. The association of JNK with the outer mitochondrial membrane initiates a phosphorylation cascade that leads to the phosphorylation of the E1α subunit of the PDH (pyruvate dehydrogenase) complex (and inactivation of the complex activity) [33]. PDH bridges cytosolic anaerobic and mitochondrial aerobic energy metabolism and its inhibition results in a bioenergetic deficit expressed as a decrease in cellular ATP levels and an increase in lactate formation; the latter suggests a compensatory effect by anaerobic glycolysis. The phosphorylation cascade triggered upon association of JNK with the outer mitochondrial membrane is likely to be mediated by pyruvate dehydrogenase kinase-2 [34]. JNK bisphophorylation (activation) and its association with the outer mitochondrial membrane increased as a function of age in rat brain [34]; this was associated with a decreased in PDH activity and subsequent deficit in energy metabolism. Thus JNK is an important negative regulator of mitochondrial metabolic function. Of note, PDH activity was reduced in the cortex of Alzheimer’s disease patients, suggesting a mitochondrial energy deficit and impairment of calcium homoeostasis [35]. Also RCAN1 (regulator of calcineurin 1) is overexpressed in Alzheimer’s disease and other neurodegenerative disorders and this overexpression results in a shift of cellular bioenergetics from aerobic respiration to glycolysis as well as induction of mitochondrial autophagy [36]. nNOS expression and activity also increase in brain as a function of age and the increased levels of nitric oxide diffusing into mitochondria, the cellular site of superoxide anion generation, results in peroxynitrite formation and subsequent nitration of ATPase at Tyr269 that results in its inactivation [37].
JNK is redox sensitive. Oxidative stress conditions, entailing also enhanced generation of mitochondrial H2O2, result in its activation. JNK activation by elevated mitochondrial H2O2 production can be induced by the knockdown of the mitochondrial NNT (nicotinamide nucleotide transhydrogenase) [38]. NNT is an important source of mitochondrial NADPH, which is required for the maintenance of the glutathione and thioredoxin systems and, hence, NNT is involved in the regulation of mitochondrial H2O2 steady-state levels. H2O2-mediated activation of JNK (and p38) may be effected upon dissociation of thioredoxin from the ASK1 (apoptosis signal-regulating kinase 1) complex [39] or of the GST (glutathione transferase)–JNK complex [40], or inhibition of MAPK phosphatases [41]. The interaction between JNK activity and mitochondrial metabolic-redox status may partly control the cellular energy and redox environments (Figure 1) and be involved in processes leading to the loss or decline of cell function that occurs with aging.
Figure 1 |. Regulation of energy metabolism by the dynamic interaction between the redox-sensitive JNK signalling pathway and mitochondria.
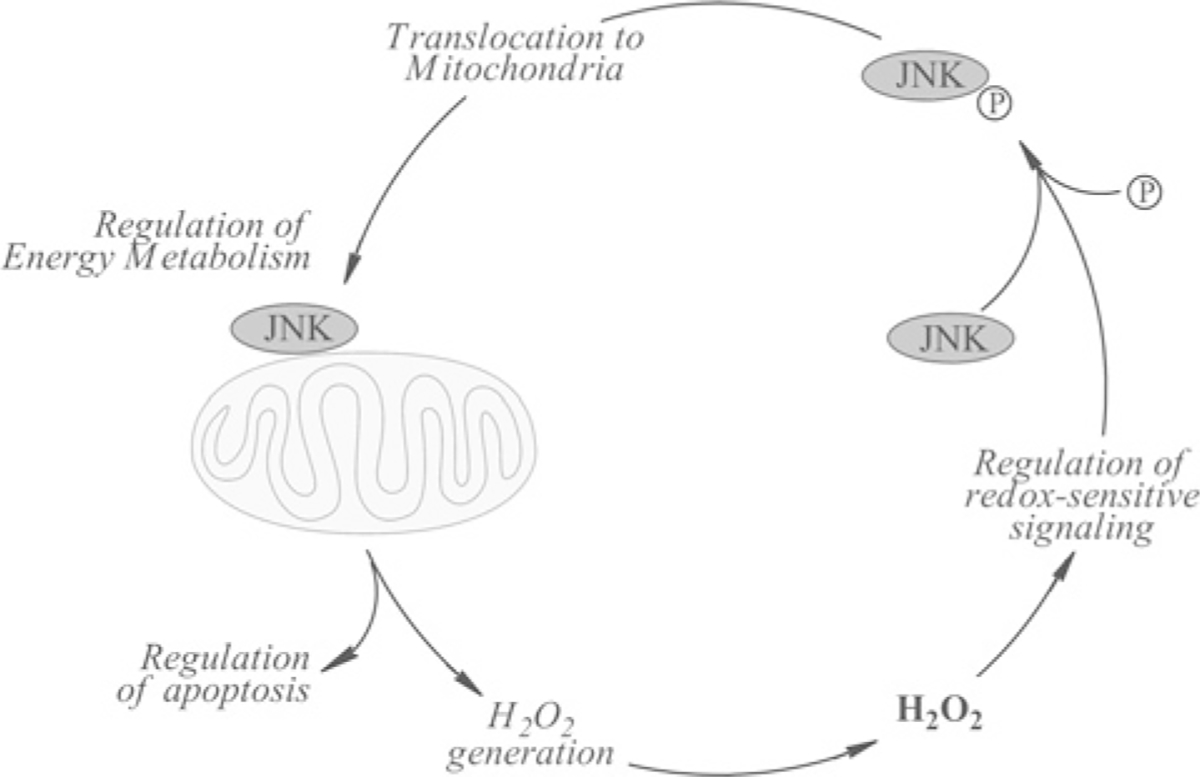
Activation (bisphosphorylation) of redox-sensitive JNK by stress conditions or mitochondrionally generated H2O2 translocates to the mitochondrion. The association of JNK with the outer mitochondrial membrane triggers a phosphorylation cascade (partly mediated by PDH kinase-2) that results in phosphorylation and inhibition of PDH, impairment of energy metabolism and greater generation of H2O2.
IIS–JNK interactions
The JNK pathway has been identified as a negative feedback regulator of the IIS signalling and is involved in the regulation of insulin resistance, thus linking environmental stress challenges to metabolic function. JNK phosphorylates the IRS at Ser307 (Ser312 in human IRS1), thus counteracting the insulin-mediated phosphorylation of IRS, a tyrosine residue [42]. Activated JNK decreases Akt activity and glucose transport activity [43]. Brain tissue from Alzheimer’s disease patients exhibit impaired IRS-mediated signalling in terms of elevated levels of IRS1 phosphorylation at serine residues and activated JNK. In hippocampal neurons, Aβ oligomers induce phosphorylation of several serine residues of IRS1, and inhibit physiological IRS1 phosphorylation at tyrosine through the JNK–TNF-α pathway [44]. This suggests the pivotal role of the IIS–JNK interactions in regulating neuronal function and provides potential therapeutic targets for multiple age-related neurodegenerative disorders.
FoxO, which is negatively regulated by the PI3K–Akt pathway, can be activated by JNK through direct phosphorylation or indirectly through the phosphorylation of 14-3-3 protein [42]. The release of FoxO from this complex promotes the translocation of FoxO to the nucleus. Mice with inactivated JNK1 in the hypothalamus and pituitary glands exhibited improved insulin sensitivity as well as improved glucose metabolism with high-fat feeding [45]. Systemically, JNK inhibits IIS activity by repressing insulin and insulin-like peptide expression and secretion in β cells [46], and could represent the interaction between the neuronal function and systemic environment.
Conversely, Akt inhibits JNK activation through the in vitro and in vivo phosphorylation of MLK3 (mixed lineage kinase 3) [47]. In primary neurons, Akt regulates JNK signalling by binding to the JIP1 (JNK-interacting protein 1): the Akt–JIP1 interaction prevents the binding of JIP1 to specific JNK targets, thereby reducing apoptosis elicited by excitotoxicity [48]. In addition, Akt is found to inhibit ASK1 activity and its downstream activation of JNK [49].
In ischaemic brain injury, Akt and JNK phosphorylate Bad at two distinct sites, the Akt-mediated phosphorylation leads to neuronal survival whereas JNK-mediated phosphorylation promotes apoptosis. The balance between the survival and death signals determines the fate of neurons [50]. At a systemic level, overactive IIS may lead to increased stress sensitivity and decreased lifespan, whereas reduced IIS may result in metabolic dysfunction [42]; conversely, excessive JNK activity leads to insulin resistance and neurodegeneration. Therefore an appropriate balance between the IIS and JNK signalling ought to be maintained in order to achieve optimal metabolic and redox homoeostasis in the brain and peripheral tissues. Both PI3K–Akt and JNK signalling are redox sensitive. As PI3K–Akt and JNK signalling elicit distinct downstream responses, the imbalance of these two pathways (partly induced by different cellular levels and spatial distributions of H2O2) is expected to disrupt cellular co-ordinated responses. These disparate effects of mitochondrial H2O2 require quantitative and spatial assessments in connection with redox-sensitive IIS and JNK signalling.
Perspective
The interactions between IIS and JNK signalling pathways in the brain, their regulation by mitochondrially generated signals, and how these signals have as impact on mitochondrial function constitute co-ordinated responses essential to neuronal function and the cognitive decline associated with aging as well as with several neurodegenerative disorders. As an early event that occurs in brain aging and neurodegeneration, the decline in brain energy metabolism could potentially be attenuated by therapeutic/nutriceutical approaches that affect IIS and JNK signalling pathways. However, the complexity of the antagonism and cross-talk between IIS and JNK signalling and how they converge on mitochondrial function requires a thorough understanding of the mechanistic basis of the mitochondria–IIS–JNK triad, a prerequisite for the design of efficacious and safe approaches to restoring the delicate balance between these pathways.
Funding
This review was supported by National Institutes of Health [grant numbers RO1AG016718 (to E.C.) and PO1AG026572 (to Roberta Díaz Brinton and E.C. as co-investigator in Project 1)].
Abbreviations used:
- ASK1
apoptosis signal-regulating kinase 1
- FoxO
forkhead box O
- GLT1
glutamate transporter 1
- GLUT
glucose transporter
- GSK3β
glycogen synthase kinase 3β
- IGF1
insulin-like growth factor-1
- IIS
insulin/IGF-1 signalling
- IR
insulin receptor
- IRS
insulin receptor substrate
- JNK
c-Jun N-terminal kinase
- JIP1
JNK-interacting protein 1
- MAPK
mitogen-activated protein kinase
- nNOS
neuronal nitric oxide synthase
- NNT
nicotinamide nucleotide transhydrogenase
- PDH
pyruvate dehydrogenase
- PI3K
phosphatidylinositol 3-kinase SIRT1, sirtuin 1
- TNF
tumour necrosis factor
References
- 1.Kapogiannis D and Mattson MP (2011) Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 10, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yap LP, Garcia JV, Han D and Cadenas E (2009) The energy–redox axis in aging and age-related neurodegeneration. Adv. Drug Delivery Rev. 61, 1283–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin F, Boveris A and Cadenas E (2012) Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signaling, doi: 10.1089/ars.2012.4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rigoulet M, Yoboue ED and Devin A (2011) Mitochondrial ROS generation and its regulation: mechanisms involved in H2O2 signaling. Antioxid. Redox Signaling 14, 459–468 [DOI] [PubMed] [Google Scholar]
- 5.Yin F, Sancheti H and Cadenas E (2012) Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signaling 17, 1714–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A and Hoyer S et al. (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 105, 423–438 [DOI] [PubMed] [Google Scholar]
- 7.Trejo JL, Carro E, Lopez-Lopez C and Torres-Aleman I (2004) Role of serum insulin-like growth factor I in mammalian brain aging. Growth Horm. IGF Res. 14 (Suppl. A), S39–S43 [DOI] [PubMed] [Google Scholar]
- 8.de la Monte SM (2012) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 9, 35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Heide LP, Ramakers GM and Smidt MP (2006) Insulin signaling in the central nervous system: learning to survive. Prog. Neurobiol. 79, 205–221 [DOI] [PubMed] [Google Scholar]
- 10.Packer L and Cadenas E (2011) Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J. Clin. Biochem. Nutr. 48, 26–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreira JM, Burnett AL and Rameau GA (2011) Activity-dependent regulation of surface glucose transporter-3. J. Neurosci. 31, 1991–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uehara Y, Nipper V and McCall AL (1997) Chronic insulin hypoglycemia induces GLUT-3 protein in rat brain neurons. Am. J. Physiol. 272, E716–E719 [DOI] [PubMed] [Google Scholar]
- 13.Cheng Z, Tseng Y and White MF (2010) Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 21, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maurer U, Charvet C, Wagman AS, Dejardin E and Green DR (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21, 749–760 [DOI] [PubMed] [Google Scholar]
- 15.Sanderson TH, Kumar R, Sullivan JM and Krause GS (2008) Insulin blocks cytochrome c release in the reperfused brain through PI3-K signaling and by promoting Bax/Bcl-xL binding. J. Neurochem. 106, 1248–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide AM, Krebs N, Machicao F, Fritsche A, Haring HU and Staiger H (2011) Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS ONE 6, e21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellini MJ, Herenu CB, Goya RG and Garcia-Segura LM (2011) Insulin-like growth factor-I gene delivery to astrocytes reduces their inflammatory response to lipopolysaccharide. J. Neuroinflammation 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, Kihara T, Akaike A, Niidome T and Sugimoto H (2010) PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem. Biophys. Res. Commun. 393, 514–518 [DOI] [PubMed] [Google Scholar]
- 19.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R and Sinclair DA (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392 [DOI] [PubMed] [Google Scholar]
- 20.Bijur GN and Jope RS (2003) Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 87, 1427–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cadenas E, Boveris A, Ragan CI and Stoppani AO (1977) Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 180, 248–257 [DOI] [PubMed] [Google Scholar]
- 22.Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Storozhevykh TP, Senilova YE, Persiyantseva NA, Pinelis VG and Pomytkin IA (2007) Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons. BMC Neurosci. 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT and Goldstein BJ (2001) Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J. Biol. Chem. 276, 48662–48669 [DOI] [PubMed] [Google Scholar]
- 25.Veal E and Day A (2011) Hydrogen peroxide as a signaling molecule. Antioxid. Redox Signal. 15, 147–151 [DOI] [PubMed] [Google Scholar]
- 26.Antunes F and Cadenas E (2000) Estimation of H2O2 gradients across biomembranes. FEBS Lett. 475, 121–126 [DOI] [PubMed] [Google Scholar]
- 27.Antico Arciuch VG, Galli S, Franco MC, Lam PY, Cadenas E, Carreras MC and Poderoso JJ (2009) Akt1 intramitochondrial cycling is a crucial step in the redox modulation of cell cycle progression. PLoS ONE 4, e7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vallerie SN and Hotamisligil GS (2010) The role of JNK proteins in metabolism. Sci. Transl. Med. 2, 60rv65. [DOI] [PubMed] [Google Scholar]
- 29.Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N and Gotoh Y (2004) JNK promotes Bax translocation to mitochondria through phosphorylation of 14–3-3 proteins. EMBO J. 23, 1889–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang P, Miller BS, Rosenzweig SA and Bhat NR (1996) Activation of C-jun N-terminal kinase/stress-activated protein kinase in primary glial cultures. J. Neurosci. Res. 46, 114–121 [DOI] [PubMed] [Google Scholar]
- 31.Shoji M, Iwakami N, Takeuchi S, Waragai M, Suzuki M, Kanazawa I, Lippa CF, Ono S and Okazawa H (2000) JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res. Mol. Brain Res. 85, 221–233 [DOI] [PubMed] [Google Scholar]
- 32.Schroeter H, Boyd CS, Ahmed R, Spencer JP, Duncan RF, Rice-Evans C and Cadenas E (2003) c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochem J. 372, 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Q, Lam PY, Han D and Cadenas E (2008) c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J. Neurochem. 104, 325–335 [DOI] [PubMed] [Google Scholar]
- 34.Zhou Q, Lam PY, Han D and Cadenas E (2009) Activation of c-Jun-N-terminal kinase and decline of mitochondrial pyruvate dehydrogenase activity during brain aging. FEBS Lett. 583, 1132–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheu KF, Kim YT, Blass JP and Weksler ME (1985) An immunochemical study of the pyruvate dehydrogenase deficit in Alzheimer’s disease brain. Ann. Neurol. 17, 444–449 [DOI] [PubMed] [Google Scholar]
- 36.Ermak G, Sojitra S, Yin F, Cadenas E, Cuervo AM and Davies KJ (2012) Chronic expression of RCAN1–1L protein induces mitochondrial autophagy and metabolic shift from oxidative phosphorylation to glycolysis in neuronal cells. J. Biol. Chem. 287, 14088–14098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lam PY, Yin F, Hamilton RT, Boveris A and Cadenas E (2009) Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radical Res. 43, 431–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin F, Sancheti H and Cadenas E (2012) Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim. Biophys. Acta 1817, 401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K and Ichijo H (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adler V, Funchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ and Ronai Z (1999) Regulation of JNK signaling by GSTp. EMBO J. 18, 1321–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foley TD, Armstrong JJ and Kupchak BR (2004) Identification and H2O2 sensitivity of the major constitutive MAPK phosphatase from rat brain. Biochem. Biophys. Res. Commun. 315, 568–574 [DOI] [PubMed] [Google Scholar]
- 42.Karpac J and Jasper H (2009) Insulin and JNK: optimizing metabolic homeostasis and lifespan. Trends Endocrinol. Metab. 20, 100–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos FR, Diamond-Stanic MK, Prasannarong M and Henriksen EJ (2012) Contribution of the serine kinase c-Jun N-terminal kinase (JNK) to oxidant-induced insulin resistance in isolated rat skeletal muscle. Arch. Physiol. Biochem. 118, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL et al. (2012) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J. Clin. Invest. 122, 1339–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC and Bruning JC (2010) Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc. Natl. Acad. Sci. U.S.A. 107, 6028–6033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S and Weir GC (2002) Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 277, 30010–30018 [DOI] [PubMed] [Google Scholar]
- 47.Barthwal MK, Sathyanarayana P, Kundu CN, Rana B, Pradeep A, Sharma C, Woodgett JR and Rana A (2003) Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J. Biol. Chem. 278, 3897–3902 [DOI] [PubMed] [Google Scholar]
- 48.Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, Birnbaum MJ and Chao MV (2002) Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron 35, 697–709 [DOI] [PubMed] [Google Scholar]
- 49.Kim AH, Khursigara G, Sun X, Franke TF and Chao MV (2001) Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 21, 893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang XT, Pei DS, Xu J, Guan QH, Sun YF, Liu XM and Zhang GY (2007) Opposing effects of Bad phosphorylation at two distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell. Signalling 19, 1844–1856 [DOI] [PubMed] [Google Scholar]