

Abstract
Viral infections of the central nervous system (CNS) are a significant cause of neurological impairment and mortality worldwide. As tissue resident macrophages, microglia are critical initial responders to CNS viral infection. Microglia seem to coordinate brain-wide antiviral responses of both brain resident cells and infiltrating immune cells. This review discusses how microglia may promote this antiviral response at a molecular level, from potential mechanisms of virus recognition to downstream cytokine responses and interaction with antiviral T cells. Recent advancements in genetic tools to specifically target microglia in vivo promise to further our understanding about the precise mechanistic role of microglia in CNS infection.
Keywords: microglia, viral infection, neuroimmunology, central nervous system, innate immune signaling, antiviral immunity
Graphical Abstract
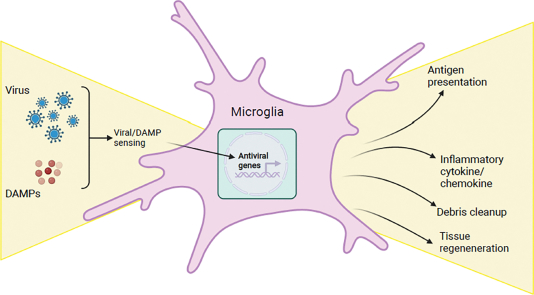
1. Introduction
Viral infections in the central nervous system (CNS) represent a significant cause of neurologic impairment [1], driven by both the primary infection and the subsequent inflammatory reaction. Understanding CNS antiviral immunity has never been more important, as the novel coronavirus pandemic continues to highlight the acute and chronic consequences of systemic infection on neurological and psychiatric function [2–4].
Microglia constitute 5–10% of brain cells, and are the only resident immune cell present in the brain parenchyma, making them a central figure in CNS viral infection [5,6]. Unlike peripheral bone marrow-derived macrophages, microglia are derived from early embryonic yolk sac progenitor cells, and once seeded in the embryonic neural tissue, they self-renew throughout life without significant contribution from bone-marrow derived macrophages [7,8]. In addition to their homeostatic roles in surveying the brain environment [9,10] and synaptic pruning during development [11–13], microglia also rapidly respond to acute CNS damage by migrating to sites of injury and secreting inflammatory cytokines [14,15]. Their embryonic origin, coupled with their relative isolation from the peripheral immune system, make microglia highly specialized for the brain environment.
In general, microglial dysfunction and inflammatory responses contribute to pathological outcomes in brain disease [15,16], but may also facilitate repair after injury [17]. Microglia are a central player in the inflammatory response to CNS infections, serving as first responders to viral invaders and essential coordinators of the antiviral responses of both CNS resident cells and peripheral immune cells. In this review, we discuss the current understanding of the roles microglia play in the antiviral response, focusing on how viruses enter the CNS, microglial recognition of viral infection, their inflammatory responses, and interactions with the peripheral immune system. We also discuss limitations to existing studies and how recent technological advances will help uncover the specific mechanistic roles of microglia in CNS viral immunity.
2. Viral invasion of the CNS
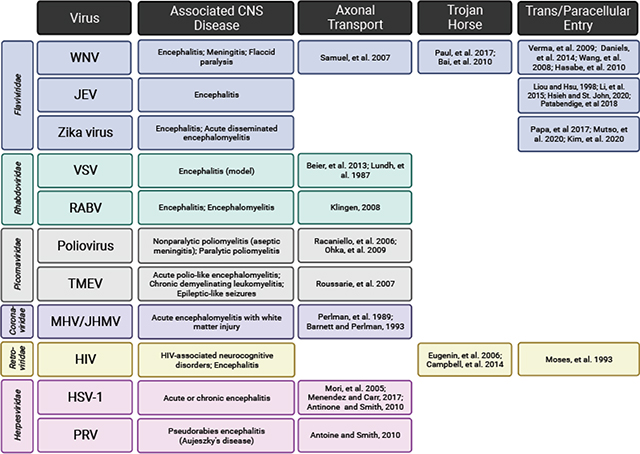
The first step in appreciating how microglia contribute to antiviral responses is understanding how they first encounter a virus. There are two major routes of entry viruses use to infiltrate the CNS – through bypassing the blood-brain barrier (BBB) or blood-cerebrospinal fluid (CSF) barrier from the circulation, or by retrograde axonal transport following primary transduction of peripheral or olfactory neurons. Viral entry can occur without frank breakdown of cellular barriers associated with the BBB by directly transducing brain endothelial cells or pericytes to infiltrate the brain parenchyma. For example, the flaviviruses West Nile Virus (WNV), Zika virus, and Japanese encephalitis virus (JEV), as well as the retrovirus human immunodeficiency virus (HIV) can transduce brain microvascular endothelial cells to access the brain parenchyma [18–24]. Zika virus can also transduce pericytes in the choroid plexus, which promotes blood-CSF barrier compromise [25]. After initial CNS invasion, subsequent inflammation can cause breakdown of BBB components such as endothelial tight junctions and the basal lamina, which facilitates increased viral infiltration through either paracellular entry or “hijacked” immune cells – known as the “Trojan horse” method of entry. WNV promotes overt BBB breakdown early after initial invasion [26,27], allowing for infiltration of WNV-infected immune cells into the CNS [28–30]. Similarly, in vitro human BBB models show that HIV-infected leukocytes traverse the endothelium in response to chemokines such as CCL2 [31,32].
Alternatively, some encephalitogenic viruses first transduce peripheral or olfactory neurons with central synapses to efficiently bypass protective barriers. Herpes simplex virus (HSV)-1 and a related herpesvirus, pseudorabies virus (PRV), gain access via migration along these neurons into the CNS and/or following reactivation of latent infection in central neurons [33–35]. Conversely, viruses from many families first have tropism for non-neuronal cells but then manage to co-opt axonal transport for CNS access. For example, both rabies virus (RABV) and poliovirus enter the CNS via retrograde transport along lower motor neurons, though they initially infect myocytes and mucosal epithelial cells respectively [36–38]. Several respiratory viruses, including mouse hepatitis virus (MHV), vesicular stomatitis virus (VSV), and Theiler’s murine encephalomyelitis virus (TMEV) can use initial transduction of olfactory neurons as a method of entry into the CNS [39–43]. Some viruses (such as WNV), use both the circulatory route and peripheral neuron transduction as a means to access the CNS [44,45].
Once the barriers protecting the CNS are breached, a coordinated immune response between CNS resident cells and infiltrating immune cells from the periphery must be raised to fight the invading virus and limit its spread. In a delicate, largely non-regenerative tissue like the CNS, this response must be balanced - too much inflammation may clear the virus but can also cause irreparable damage, while too little inflammation may allow the virus to run rampant. As the only resident immune cell in the brain parenchyma, microglia likely play important roles at the center of this inflammatory balance, though their exact roles in this process have long been debated. A summary of known microglial responses are discussed in further detail below.
3. A protective role for microglia during CNS viral infection
As tissue resident immune cells, microglia rapidly respond to invading viral pathogens, and are predicted to serve protective roles by clearing virus particles and coordinating the early inflammatory response. The strongest in vivo evidence that microglia protect the CNS from viral infection relies on studying infection severity in mouse microglia depletion models. Two main methods are used to deplete microglia rapidly and efficiently: 1) pharmacologic inhibition of the microglial survival receptor, colony stimulating factor 1 receptor (CSF1R) or 2) a genetic model that inducibly deletes the CSF1R receptor (Cx3cr1CreER/+::Csf1rFlox/Flox) in microglia [46,47].
Depletion studies indicate that for multiple families of neurotropic viruses, absence of microglia leads to more severe CNS damage and correlates with higher viral titers. CNS infection with the coronavirus MHV typically results in mild encephalitis and spinal cord white matter injury. Pharmacological depletion of microglia before or within the first 3 days of MHV infection is associated with delayed viral clearance, increased demyelination, and increased mortality [48–50]. Infection with flaviviruses WNV or JEV or the rhabdovirus VSV similarly show uncontrolled viral spread in the CNS and increased mortality following microglial depletion [51–53]. Using super resolution and transmission electron microscopy, one group found that microglial depletion during herpesvirus PRV infection resulted in significant increases in extracellular virus protein, infected neurons, and neuronal cell death [54]. This increased neuronal death was linked to worse neurological outcomes, including seizures and muscle spasms [54]. Finally, infection of C57BL/6J mice with the picornavirus TMEV typically leads to hippocampal damage, causing clinical seizures. In addition to increased viral titers throughout the hippocampus and spinal cord, microglia-depleted mice had significantly more neuronal cell death in the brain and spinal cord as measured by neuronal cell death [55]. Clinical outcomes were also worse, as these mice had increased incidence of seizures early in the course of TMEV infection, hind limb paralysis, and increased mortality [55,56]. Taken together, these studies point to microglia being crucial in the antiviral response to many neurotropic viruses, both limiting their spread and preventing virus-induced neuropathology.
While these depletion studies implicate microglia as central players in controlling CNS viral infection, without orthogonal methods it is difficult to determine if it is the loss of a direct protective function or lack of inhibition of a destructive inflammatory response that leads to increased morbidity. For example, several studies observe a compensatory peripheral monocyte response in the brain following depletion of microglia. In MHV encephalomyelitis, increased numbers of blood-derived CD45hiCD11b+ monocyte/macrophages infiltrated the brain following microglia depletion, which correlated with increased RNA expression of type I interferons and IL-6. Interestingly, despite this increased peripheral monocyte/macrophage infiltration, viral load was increased and these mice had worse outcomes [48]. Similar findings occurred in TMEV infection, where increased monocyte/macrophage inflammatory responses in the brain following microglia depletion were insufficient to limit viral replication and mortality [55]. These observations support a model where microglia are necessary players in limiting viral dissemination in the CNS, and further suggest that the antiviral role(s) of microglia are non-redundant with those of other macrophage populations, though it is difficult to decouple the effect of increased peripheral macrophage inflammatory response from the absence of microglia.
One proposed mechanism for how microglia provide protection against viral infection is by supporting the infiltration of T cells. Several studies using CSF1R signaling inhibition to deplete microglia also report decreased T cell responses in the brain [48–51,55], which suggests that microglia act as local antigen presenting cells to support CNS-infiltrating T cells. A significant caveat of this approach, however, is that inhibiting CSF1R signaling systemically can affect maturation of peripheral antigen presenting cells [48,49,51]. During MHV infection, for example, microglia depletion was associated with decreased CD4+ T cell infiltration and decreased interferon (IFN)-γ production. Though this could be due to absent local restimulation by microglia, peripheral monocyte/macrophages also expressed less MHC class II and more Ly6C suggesting an alternative defect [48]. Additionally, in a mouse model of WNV encephalitis, systemic CSF1R inhibition led to decreased costimulatory molecule expression by antigen-presenting cells (APCs) in lymph nodes, blood, and the CNS [51]. CSF1R signaling inhibition during JHM mouse hepatitis virus (JHMV) infection, also decreased costimulatory molecule expression on peripheral macrophages and dendritic cells [49,51]. Altogether, depletion studies suggest that microglia limit viral spread and promote survival, but effects of CSF1R antagonists on other macrophages make differentiating their mechanistic role from that of peripheral monocyte/macrophages difficult.
Genetic approaches to specifically deplete microglia provide additional insight into microglia-specific roles during CNS infections. One study used the Cx3cr1CreER/+;Csf1rFlox/Flox mouse model [57], in which tamoxifen administration inducibly depletes microglia, which highly express CX3CR1. Tamoxifen-induced depletion of microglia before HSV-1 infection resulted in increased viral titers in the brain, rapid weight loss, and increased mortality [58]. These findings support those from pharmacological depletion studies pointing to microglia as essential protective players during CNS infection, however, this genetic approach is also somewhat confounded by CX3CR1 expression by Ly6Clo monocytes and CNS border macrophages. Tamoxifen-induced depletion resulted in fewer CD45hiCD11b+Ly6C+ peripheral monocytes recruited to the brain during HSV-1 infection, suggesting that either microglia are important for recruitment of peripheral immune cells, or that depletion of CSF1R-dependent CX3CR1+ peripheral monocytes occurs in this model.
Overall, existing evidence suggests that microglia play a crucial antiviral role that is not compensated for by infiltrating monocyte/macrophages, however, it is important to consider additional variables such as viral strain and potential differences between murine and human microglial responses. For example, though seemingly rare, there are reports of neuroinvasion of human coronaviruses SARS-CoV, MERS-CoV, and the novel coronavirus SARS-CoV2 [59,60]. Though mice are typically resistant to infection with these human coronaviruses, a transgenic mouse model expressing human angiotensin-converting enzyme 2 (ACE2), the receptor that mediates cellular entry of SARS-CoV and SARS-CoV2, allows for productive infection of SARS-CoV2, including variable neuroinvasion in anywhere from 0–60% of mice [61–64]. One study using this mouse model found that, in contrast to studies using murine coronavirus MHV, pharmacological depletion of microglia had no effect on weight loss or viral dissemination in the brain, though inflammatory cytokine production in the brain decreased [65]. These results suggest the potential for some variability in microglial responses dependent on viral strain that should be considered. On the whole, though it has been well established that microglia play an important protective role in many CNS viral infections, limitations in existing tools have prevented dissection of the precise mechanisms and contribution of microglia in CNS infection. Recent developments in microglia-specific genetic tools, such as microglia-specific (Tmem119CreERT2 or Sall1CreER) [66–68] or monocyte-specific Cre lines (Ms4a3CreERT2) [69] can help delineate the antiviral roles of microglia versus infiltrating macrophages, as well as provide mechanistic insight into antiviral functions of microglia.
4. Microglial recognition of viral infection
A key to understanding the role of microglia in antiviral immunity is defining the molecular mechanisms by which they recognize viruses. Viral recognition by microglia occurs through innate immune sensors known as pattern recognition receptors (PRRs), that recognize highly conserved pathogen motifs known as pathogen-associated molecular patterns (PAMPs). There are several major classes of PRRs including Toll-like receptors (TLRs), the retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing (NLRPs), and C-type lectin receptors (CLRs), as well as the cytosolic DNA sensor cGAS. Of these, TLRs, RLRs, NLRPs, and cGAS recognize viral PAMPs, and are discussed in the context of microglia responses to CNS viral infection in more detail below.
4.1. Toll-like Receptors (TLRs)
One of the best characterized groups of PRRs are TLRs, which consist of TLRs1–10 in humans and TLRs1–9 and TLRs12–13 in mice. TLRs are proteins localized to the cell membrane or intracellular compartments such as endosomes and lysosomes. They recognize a wide array of PAMPs including viral DNA and RNA, and are major contributors to antiviral cytokine responses [70]. Evidence that TLRs are involved in antiviral immunity in human CNS infections is provided by GWAS studies, in which mutations in TLR3 and the type I interferon pathway (activated downstream of TLR signaling) is linked to greater risk of the developing encephalitogenic HSV [71–76]. MyD88 is an important adaptor molecule recruited downstream of TLR activation, and is required for much of the TLR-mediated cytokine and chemokine response during infection [77]. MyD88-deficient mice are generally more susceptible to encephalitogenic viruses [78–80], suggesting that TLR signaling contributes to antiviral immune responses in the CNS. Mouse microglia constitutively express all known murine TLRs, except TLR8, and stimulation with a variety of TLR agonists leads to changes in TLR expression, suggesting that microglia are sensitive to a variety of pathogenic stimuli through members of the TLR family [66,81]. Following transduction with TMEV in vitro, microglia specifically upregulate TLRs 2, 3, 5, and 9, all of which recognize viral components. Increased expression of TLRs in this system also correlated with increased cytokine expression, chemokine production, and APC function, suggesting that this family of innate immune sensors are a mechanism by which microglia recognize and respond to viruses [81]. Additionally, microglia, though not permissive to HSV-1 replication themselves, are the main producers of inflammatory cytokines in the presence of the virus in vitro [82]. Many of these cytokines (TNFα, IL-1, IL-6, and IL-12) were subsequently found to be dependent on TLR2 signaling ex vivo, as microglia isolated from TLR2−/− mice did not produce these cytokines in response to HSV-1 [83]. Interestingly, TLR2−/− mice are not more susceptible to HSV infection in vivo [78,84,85]. As with many innate immune signaling pathways, this is likely due to significant redundancy in TLR activation and signaling in vivo. Supporting this hypothesis, TLR2 and TLR9, by sensing viral particles and genomic DNA, respectively, act synergistically during HSV infection in vivo to induce microglial inflammatory cytokine production and control viral replication in the CNS [84–86].
TLR3, which recognizes viral RNA, is expressed by multiple cell types in the brain [87], but both in vitro and in vivo studies have shown that microglia respond to the TLR3 agonist Poly I:C by producing a wide array of inflammatory cytokines [88]. TLR3 has also been implicated in microglial responses to JEV in vitro. Using a microglial cell line, JEV induced TLR3-mediated inflammatory cytokine expression in cultured microglia, which was decreased with TLR3 knockdown [89].
Taken together, these reports show that microglia can respond through TLR activation to CNS viral pathogens. The use of in vitro microglia culture systems and global knockouts make conclusions about the role of microglia-specific TLR recognition of viruses difficult, as microglia downregulate signature genes in culture, and often do not fully recapitulate in vivo microglia phenotypes [90,91]. The use of microglia-specific TLR knockouts could further clarify how TLRs contribute to microglia antiviral responses in vivo.
4.2. RIG-I like receptors (RLRs)
RLRs are cytosolic innate immune sensors that recognize cytoplasmic viral RNA and contribute to antiviral cytokine and type I interferon responses. There are three known RLR family members: RIG-I, MDA5, and LGP2 [92]. Of these, RIG-I and MDA5 are the signaling effectors that respond to viral RNA. [93]. Cultured murine microglia constitutively express both RIG-I and MDA5 [94,95]), and upregulate their expression following exposure to VSV and JEV [89,94,95]. Additionally, RIG-I is necessary for optimal inflammatory cytokine production by microglia in response to VSV and HSV-1 in vitro [95]. In response to JEV, cultured murine microglia relied on both TLR3 and RIG-I signaling for cytokine production, suggesting that multiple innate immune sensing pathways act in concert to promote antiviral immunity in microglia [89]. In vivo studies interrogating the role of RLR signaling in virus encephalitis are extremely limited, and none specifically focus on microglia. Interestingly, RIG-I/MDA5 double knockout mice are more susceptible to WNV infection, and unable to control viral replication in the periphery and the brain compared to wildtype (WT) mice [96]. Similarly, RIG-I-deficient mice are more susceptible to JEV infection [97]. Together, these studies suggest important roles for RLR signaling in CNS viral infection, though more specific manipulation of microglia RLR signaling would further specify their function in microglial antiviral responses.
4.3. cGAS-STING
Another pathway by which microglia respond to viral pathogens is through recognition of viral nucleic acids by the cytoplasmic DNA sensor cyclic GMP-AMP synthase (cGAS), and its downstream mediator Stimulator of Interferon Genes (STING) [98]. In HSV-1 infection, mice deficient in either cGAS or STING had impaired control of viral replication in the CNS and were highly susceptible to HSV encephalitis [99]. Interestingly, while neurons and astrocytes rely on TLR3 signaling to mount an antiviral defenses against HSV-1, microglia depended on cGAS-STING signaling to produce a potent type I interferon response to limit viral dissemination in the brain [99,100]. Microglia were able to prime antiviral programs in both neurons and astrocytes in a STING-dependent manner, as neurons treated with conditioned media from HSV-1 infected WT microglia, but not STING-deficient microglia, limited viral replication. Similarly, astrocytes exposed to conditioned media from HSV-1 infected WT microglia, but not STING-deficient microglia, upregulated TLR3 signaling, which was linked to increased IFNβ expression upon subsequent infection with HSV-1. These results suggest that microglia may be unique in their reliance on cGAS-STING to recognize HSV-1 infection, and that this pathway may be central to coordinating the brain-wide response to viral infection. One caveat of this study, however, is the use of global knockouts of cGAS and STING, which again leaves open the possibility that peripheral monocyte/macrophages may also rely on cGAS-STING to influence antiviral functions in this context.
Human microglia also express cGAS and STING [101]. One study found that a human microglial cell line heterozygous for cGAS was unable to produce normal levels of IFNβ in response to B- and Y- form DNA (known ligands for cGAS), suggesting cGAS activation may play an important role in amplifying type I interferon in response to DNA viruses [102]. With HSV-1 transduction, however, the same human microglia cell line did not produce detectable levels of IFNβ in either WT or cGAS heterozygous cells [102]. This result, coupled with the caveats of studying microglia in culture and new findings in mouse that cGAS-STING antiviral effects in response to HSV1 may be interferon-independent [103] leave unanswered the question of whether cGAS contributes to antiviral responses of human microglia. The study of cGAS-STING in microglia is a relatively nascent field, and there remain many important unanswered questions regarding the role of cGAS-STING in microglial recognition of of viral infections, as well as its contribution to cytokine responses and coordination of brainwide antiviral responses.
4.4. Recognition of damage-associated molecular patterns (DAMPs)
In addition to viral detection, another critical aspect of the microglial response is recognition of tissue damage. In a model of nasal VSV infection, microglia cluster in the olfactory bulb, an action thought to “wall off” further spread of the virus in the CNS [53]. Loss of type I interferon (IFNAR) signaling specifically in neurons and astrocytes (using Nestin-Cre), but not microglia (using CX3CR1-Cre), decreased VSV-associated microglial accumulation in the olfactory bulb, increased viral dissemination, and increased mortality [53]. The IFNAR-dependent effectors released by neurons and astrocytes in this context remain unidentified, but this study emphasizes the importance of crosstalk between CNS cells to coordinate microglial responses and limit viral spread.
P2RY12 is a highly expressed purinergic receptor that recognizes ADP [10,104], and is the predicted mechanism by which microglia sense neuronal injury that occurs during CNS viral infection. During infection with PRV, P2RY12 was necessary for the recruitment of microglia to PRV-infected neurons. Loss of P2RY12 decreased microglia contact with PRV-infected neurons and increased accumulation of disintegrated neurons and extracellular virus particles, suggesting that microglia detect infected neurons through P2RY12 to limit viral spread [54]. P2RY12+ microglia also cluster around and engulf HSV-1-infected neurons in the human brain, consistent with mouse models, though the dependence of this process on P2RY12 is unknown [54]. As microglia are poor replicative reservoirs for many CNS-infecting viruses [34,105–110], understanding how they respond to viral damage, independent of direct viral recognition, is critical to clarify their response to viral infection.
4.5. Inflammasome
The inflammasome complex allows innate immune cells, such as microglia, to sense PAMPs and DAMPs [111]. It is a subcellular signaling complex with three main components: 1) a PRR (mainly NLRP1, NLRP3, NLRC3, NLRC4, and AIM2), 2) an adaptor molecule (the most common one being apoptosis-associated spec-like protein containing a caspase recruitment domain (ASC-CARD)), and 3) a caspase [112]. Activation of the inflammasome complex leads to the production of IL-1β and IL-18, which occurs in microglia in several models of neurodegeneration [111,113–115].
Polymorphisms in the NLRP3 gene are associated with increased susceptibility to HIV infection [116,117]. Microglia, along with peripheral monocyte/macrophages, are well-documented reservoirs for HIV replication [118–124], suggesting that inflammasome activation in microglia may be key to understanding retroviral responses in the brain. Transcripts for downstream inflammasome pathway components, such as IL-1β, IL-18, ASC, and caspase-1, are expressed by microglia in HIV-infected individuals [125]. Additionally, following exposure to HIV in vitro, primary human microglia form ASC specks (indicating formation of the inflammasome complex), activate caspase-1, and release IL-1β, all of which is dependent on NLRP3 recognition of HIV viral proteins [125–127]. Overall, as the inflammasome can recognize both PAMPs and DAMPs, it has the potential to play an important role in microglial recognition of CNS infection, and remains a critical understudied mechanism regulating the microglial response to CNS viral infection.
5. What is the outcome of microglial response to CNS viral infection?
5.1. Cytokine responses
Microglial cytokines likely limit viral spread and coordinate the antiviral responses of other cells. Following recognition of viral infection,either through viral detection or in response to DAMPs, many cells produce type I interferons (IFN) that limit viral replication and dissemination [48,128–130]. One study found that microglial production of type I IFN was required to upregulate antiviral immune responses in astrocytes and neurons to control infection [99]. Interestingly, CSF1R inhibitor-mediated (PLX5622) depletion of microglia in MHV did not affect type I IFN production [48], suggesting that other cells might compensate for microglial loss of type I IFN. These conflicting results highlight the context-dependent nature of the microglial cytokine response For example, La Crosse virus produces a nonstructural protein that specifically inhibits type I IFN responses in infected astrocytes, but not microglia [128]. Thus, microglia may have unique molecular responses to particular viruses that are crucial to tissue antiviral responses.
Microglia produce other proinflammatory cytokines that have both antiviral and potentially toxic effects. JEV infection promotes microglial production of TNFα, IL-6, IL-1β, nitric oxide (NO) and glutamate, which limit viral replication, but are also neurotoxic [131–133]. Similarly, microglial production of NO during HSV-1 infection is implicated in infection-induced oxidative tissue damage [134], and in WNV infection, microglial production of antiviral cytokines can induce neuronal cell death [135]. Another consequence of these cytokine responses is microglial apoptosis: in response to direct transduction by HSV-1 or phagocytosis of HSV-1-infected cells, microglia undergo cGAS-STING-dependent apoptosis. This is hypothesized to limit type I IFN responses and associated immunopathology in cases of high viral load [136,137]. Finally, microglia may play a regenerative role in some cases of CNS viral infection. Microglial depletion after MHV infection leads to decreased transcripts encoding proteins known to promote remyelination, such as Cst7, Igf1, and Lpl [49], suggesting that microglia may be important in promoting myelin regeneration and recovery. Overall, it is clear that microglia significantly contribute to inflammatory cytokine responses in the brain that are necessary to control and limit viral pathogenesis, yet these same inflammatory responses can also have deleterious effects on CNS cells. In a delicate tissue like the CNS, it is crucial to better understand how this balance is maintained, and what role microglia play.
5.2. Phagocytosis
As resident parenchymal macrophages, a primary role of microglia is phagocytosis of cellular debris and myelin [15]. In response to CNS insult, microglia are the main cells involved in clearing tissue debris, for example in demyelinating disease and spinal cord injury [138–140]. Microglial phagocytosis of virally infected cells occurs in several models. During WNV infection of spinal cord slice cultures, microglia phagocytose WNV-transduced neurons and astrocytes [141]. In vivo and in vitro models of PRV infection demonstrate microglial reliance on P2RY12 to recognize and engulf dying PRV-infected neurons, thereby limiting viral spread [54]. Phagocytosis may be central to microglial antiviral responses, as active uptake of virally-infected cells may be an important mechanism by which microglia initially recognize infection, leading to PRR activation, and amplifying downstream antiviral responses. Clearance of dead and damaged cells may decrease inflammation and help promote tissue regeneration. Further studies of microglial phagocytosis are crucial to fully characterize the role of microglia in antiviral immunity.
5.3. Coordination with the peripheral immune response
Microglia are well situated to orchestrate the antiviral responses of infiltrating peripheral immune cells to limit viral replication and spread. First, as mentioned, microglia produce chemokines in response to viral infection, which may be crucial in recruiting peripheral immune cells, though the necessity of microglia-derived chemokines and cytokines remains unclear [52,81,83,105,141,142]. Second, microglia may provide local antigen presentation to CNS-infiltrating T cells. Several studies report decreased virus-induced T cell responses and worse infections following microglial depletion with pharmacological inhibitors of CSF1R. During active MHV infection, microglia depletion led to fewer CD4+ T cells and IFNγ-producing T cells in the brains but not the spleen, suggesting that microglia are necessary for T cell recruitment [48]. In WNV infection, though similar numbers of CD4+ T cells were found in the CNS following microglia depletion, a smaller fraction expressed markers of full activation [51]. This decreased T cell activation was associated with decreased CD80 and CD86 costimulatory molecule expression by remaining microglia, suggesting that microglia provide local costimulation to fully activate T cells [51]. An important caveat is that peripheral monocyte/macrophages had lower MHCII and costimulatory molecule expression in response to systemic blockade of CSF1R signaling, suggesting that non-microglia macrophages might also contribute to these deficient T cell responses [48,51].
To more specifically interrogate the importance of microglia-T cell interactions during CNS viral infections, a recent study used a bone marrow chimera strategy to specifically label microglia. Using CX3CR1-GFP mice reconstituted with unlabeled WT bone marrow allowed for the differentiation of GFP+ microglia from GFP− infiltrating monocyte/macrophages. Intravital imaging during active VSV infection revealed that CX3CR1-GFP+ microglia interacted extensively with infiltrating virus-specific CD8+ T cells. These interactions resulted in T cell calcium fluxes, a sign of T cell activation. In addition, microglia engulfed infected neuronal debris, and both MHCI ablation on CNS resident hematopoietic cells (largely microglia) and microglial depletion decreased activation of antigen-specific CD8+ T cells, leading to increased viral titer in the brain [143]. These results provide strong evidence that microglia locally activate T cells by ingesting infected cellular debris and cross-presenting viral antigen to induce an antiviral T cell response.
6. Latent and persistent viral infection in the CNS
Two potential outcomes of incomplete viral clearance are latent infection or long-term persistence of the virus in tissues. Latency usually follows retention of the viral genome in the host cell, where small amounts of viral antigens may induce low-level inflammation to keep the virus in check [144]. In latent CNS infections, reactivation of virus poses a significant threat, particularly for immunocompromised or aged individuals. In contrast, persistent viral infections are characterized by continued, active viral replication and transduction of cells over a long period of time [144]. Immune responses typically limit viral spread enough to prevent mortality and morbidity, yet complete viral clearance is not achieved. In the long term, ongoing inflammatory responses in viral persistence can be damaging, particularly to largely non-regenerative tissues like the CNS. HSV [145–148], JC virus [149–151], Epstein-Barr virus (EBV) [152], and HIV [122,153–155] are some of the most commonly characterized encephalitogenic viruses that can become either latent or persistent in the CNS and cause potentially severe disease. Due to minimal disease in immunocompetent individuals and difficulty translating chronic stages of these viral infections into mouse models, studies focused specifically on the immune response to latent or persistent viral infections are limited.
Chronic HSV encephalitis is rare, but can cause severe progressive disease in a small proportion of patients that survive the acute infection [145,146,148]. Neuropathological findings of chronic HSV encephalitis describe long-term microglial activation that is associated with neuronal cell death and cognitive defects [145–147,156]. Additionally, high expression of MHCII by chronically activated microglia is associated with retention of virus-specific T cells in the CNS [156], suggesting an important role for microglia in maintaining an active antiviral T cell responses in the CNS. JC virus is a classical example of latent CNS viral infection, which is asymptomatic in immunocompetent individuals. Immunosuppression can result in reactivation which underlies a devastating demyelinating disease called progressive multifocal leukoencephalopathy (PML) [157]. PML from JC virus is famously linked to immunosuppression that limits T-cell surveillance of the CNS, such as in the use of monoclonal antibodies against α4β1-integrin, which led to JC virus screening of patients with multiple sclerosis prior to starting natalizumab [158]. JC virus reactivation is also associated with reactive microglial state changes, which form clusters around focal areas of demyelination [159–161]. Like in other latent CNS infections, JC virus reactivation can also occur in AIDS-associated immunosuppression [162]. JC virus-associated PML lesions in HIV+ patients contain large numbers of HIV-infected microglia/macrophages [159], while inflammatory cytokines known to be produced by microglia, like IL-1β, TNFα, and IFNγ, activate transcription of JC virus promoter genes [163–165]. These findings together suggest that both the immunosuppression associated with HIV infection, along with an inflammatory cytokine response specifically from HIV-infected microglia/macrophages may play a role in the reactivation of JC virus and lead to the development of PML.
How HIV is reactivated, particularly in the CNS, is a critical piece in understanding HIV-associated neurocognitive disorders (HAND). Unlike most encephalitogenic viruses that transduce neurons, astrocytes, and oligodendrocytes, HIV transduces microglia, which likely serve as viral reservoirs during chronic infection [166]. In vitro, HIV induces widespread apoptosis in both human microglia and macrophages, but a small percentage of infected cells survive [167]. Signs of microglial activation in HIV+ patients correlate with decreased cognitive function [168–171]. This persists even in patients receiving antiretroviral therapies [168,171], suggesting a mechanism by which low-level, chronic neuroinflammation impacts cognitive function even in otherwise healthy HIV+ patients on modern therapies. Intriguingly, co-culture of healthy neurons with HIV-infected human microglia silences viral replication, though the neuron-derived factors that induce this response are still unknown [172]. Co-culture with damaged or dying neurons induces HIV reactivation, an effect further amplified with TLR3 agonism [172,173]. HIV inhibits the phosphorylation of interferon regulatory factor 3 and 7 (IRF3 and IRF7) downstream of TLR3 in latently infected microglia, thereby preventing the normal antiviral effects of TLR3 activation [174]. Interestingly, the strong TLR3 agonist Poly I:C induces HIV replication selectively within microglia, but not bone marrow-derived macrophages or T cells [173]. In addition, combination TNFα and IFNγ treatment, LPS, and methamphetamine all also promote reactivation of HIV replication in latently infected microglia [167], suggesting a wide array of inflammatory factors influence HIV latency. Together, these studies point to the importance of microglia in maintaining and reactivation of latent CNS infections, the dynamics of which likely underlie the long-term morbidity of chronic viral infection. Understanding how microglia carry out these roles at a molecular level is key to improving future therapies for latent and chronic CNS infections.
7. Conclusions and future perspectives
Microglia play a central and unique role in CNS viral infections, but major questions remain about the mechanisms underlying microglia-specific antiviral immunity. Using new tools that specifically target microglia will be key in determining exactly how important microglia are to viral infection, and whether their responses can be harnessed to develop better antiviral therapeutics and reduce morbidity. he discoveries described in this review and the new tools described below provide great potential to answer four pressing and long-standing questions:
7.1. How do microglia sense viral infection?
PRRs are likely key microglial sensors of viral infection, though which PRRs are involved and how PRR-mediated innate immune signaling leads to microglial antiviral responses remains uncertain. Microglia are singularly suited to the CNS by their unique developmental origins, and their long-term residence. Microglia may have different PRR expression patterns or distinct signaling from other macrophage populations, and understanding these unique aspects is crucial to build a more complete picture of CNS viral immunity.
7.2. What mechanisms do microglia use to clear and/or evade virus?
With the exception of HIV, there is minimal in vivo evidence that microglia are productively infected by viruses. Possible explanations are that microglia are more efficient at clearing viruses than other CNS resident cells or other macrophages or that they are not transduced in the first place. A better understanding of the molecular antiviral mechanisms microglia use to clear or evade viruses may lead to more robust antiviral therapies.
7.3. How unique is the microglial response to viral encephalitis compared to that of peripheral monocyte/macrophages?
To date, one of the largest hurdles in identifying microglial-specific responses has been distinguishing them from peripheral monocyte-derived macrophages that infiltrate the brain.. Though microglia have distinct developmental history and transcriptomic features, unique markers and fate mapping tools to differentiate them from infiltrating macrophages are relatively new [66,91]. As such, prior in vivo models did not distinguish the individual contributions of infiltrating macrophages compared to microglia. In vitro infection studies using either primary microglia or microglial cell lines have helped identify potentially important and unique molecular pathways involved in recognition of virus and subsequent downstream antiviral responses, but are limited by known changes in microglial transcriptomic identity once removed from the brain environment. Tools such as brain organoids and brain slice cultures allow for isolation of microglia from peripheral macrophage infiltration while still leaving them in a brain tissue environment and may provide greater insight into molecular antiviral pathways used by microglia. Essential to understanding the in vivo role of microglia during CNS viral infection will be new microglia-[67,175] and monocyte-specific inducible Cre lines [69] that allow specific manipulation of each macrophage population and will delineate those antiviral responses unique to either microglia or brain-infiltrating monocytes. As it is likely that both microglia and monocyte/macrophages are involved in antiviral immunity during viral infection, defining the similarities and differences in their responses is crucial to providing a clearer picture of antiviral macrophage responses in the CNS.
7.4. How do human microglia respond to viral infection?
A major limitation in studying human microglial responses is the relative difficulty of acquiring primary microglia for culture, as well as the loss of microglial identity associated with cultured microglia. Human induced pluripotent stem cells (iPSCs) can be isolated from more accessible tissue sources, and iPSC-derived microglia recapitulate much of the phenotypic signature of primary human microglia, including in response to HIV [176–178], iPSC-derived microglia can potentially be coupled with current gene-editing technology such as CRISPR-Cas9. Additionally, replacement of endogenous microglia with xenografted iPSC-derived microglia in mouse models (reviewed in [179]) can provide much needed in vivo insight into antiviral functions of human microglia in response to viral infection.
Microglia are poised to coordinate the CNS antiviral response. In this review, we covered existing literature about how microglia recognize invading viruses and initiate responses, while also shaping the antiviral activity of other brain resident and infiltrating peripheral immune cells. There is also evidence that microglia participate in tissue repair following viral infection. With the use of newly developed tools to specifically target microglia, the field can better interrogate these diverse roles, allowing for a more complete understanding of antiviral responses in the brain.
Figure 1.

Proposed mechanisms of microglial recognition of viral infection. (1) TLR recognition of viral DNA or RNA, either through direct sensing of virus or following phagocytosis of virally-infected cells, leads to the recruitment and activation of downstream modulators such as MyD88 or TRIF. MyD88-dependent signaling leads to the activation and nuclear localization of the major mediator of inflammatory responses, NFκB, or interferon regulatory factor 7 (IRF7), while activation of TRIF leads to activation and nuclear localization of another critical mediator of innate immune signaling, interferon regulatory factor 3 (IRF3). (2) Recognition of cytoplasmic viral RNA by RLRs RIG-I or MDA5 results in activation of downstream mediator MAVS, which in turn leads to activation and nuclear localization of IRF3. (3) cGAS serves recognizes cytoplasmic viral nucleic acids, leading toSTING activation which in turn activates both NFκB and IRF3, leading to their nuclear localization. (4) Viral components and DAMPs released by transduced or damaged cells activate the inflammasome, leading to the recruitment and activation of caspase-1 (casp-1), which cleaves pro-IL-1β and pro-IL-18 to their active forms, IL-1β and IL-18. Microglia likely integrate these signaling pathways following recognition of viral infection directly or by virally-induced signals from other cells to produce a wide array of antiviral genes such as inflammatory cytokines and chemokines that both serve to clear virus and coordinate the antiviral responses of other cells.
Table 1.
Common encephalitogenic viruses and their differing mechanisms of viral entry into the CNS.
|
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].John CC, Carabin H, Montano SM, Bangirana P, Zunt JR, Peterson PK, Global research priorities for infections that affect the nervous system, Nature. 527 (2015) S178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Li Y, Hu B, Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China, JAMA Neurol. 77 (2020) 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Spudich S, Nath A, Nervous system consequences of COVID-19, Science. 375 (2022) 267–269. [DOI] [PubMed] [Google Scholar]
- [4].Taquet M, Geddes JR, Husain M, Luciano S, Harrison PJ, 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records, Lancet Psychiatry. 8 (2021) 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lawson LJ, Perry VH, Dri P, Gordon S, Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain, Neuroscience. 39 (1990) 151–170. [DOI] [PubMed] [Google Scholar]
- [6].Aguzzi A, Barres BA, Bennett ML, Microglia: scapegoat, saboteur, or something else?, Science. 339 (2013) 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M, Fate mapping analysis reveals that adult microglia derive from primitive macrophages, Science. 330 (2010) 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, García-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M, Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes, Immunity. 38 (2013) 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nimmerjahn A, Kirchhoff F, Helmchen F, Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo, Science. 308 (2005) 1314–1318. [DOI] [PubMed] [Google Scholar]
- [10].Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan W-B, ATP mediates rapid microglial response to local brain injury in vivo, Nat. Neurosci. 8 (2005) 752–758. [DOI] [PubMed] [Google Scholar]
- [11].Tremblay M-È, Lowery RL, Majewska AK, Microglial interactions with synapses are modulated by visual experience, PLoS Biol. 8 (2010) e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B, Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner, Neuron. 74 (2012) 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT, Synaptic Pruning by Microglia Is Necessary for Normal Brain Development, Science. 333 (2011) 1456–1458. [DOI] [PubMed] [Google Scholar]
- [14].Ransohoff RM, El Khoury J, Microglia in Health and Disease, Cold Spring Harb. Perspect. Biol. 8 (2015) a020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Q, Barres BA, Microglia and macrophages in brain homeostasis and disease, Nat. Rev. Immunol. 18 (2017) 225–242. [DOI] [PubMed] [Google Scholar]
- [16].Salter MW, Stevens B, Microglia emerge as central players in brain disease, Nat. Med. 23 (2017) 1018–1027. [DOI] [PubMed] [Google Scholar]
- [17].Rawji KS, Yong VW, The benefits and detriments of macrophages/microglia in models of multiple sclerosis, Clin. Dev. Immunol. 2013 (2013) 948976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR, West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier, Virology. 385 (2009) 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Papa MP, Meuren LM, Coelho SVA, de CG, Lucas O, Mustafá YM, Lemos Matassoli F, Silveira PP, Frost PS, Pezzuto P, Ribeiro MR, Tanuri A, Nogueira ML, Campanati L, Bozza MT, Paula Neto HA, Pimentel-Coelho PM, Figueiredo CP, de Aguiar RS, de Arruda LB, Zika Virus Infects, Activates, and Crosses Brain Microvascular Endothelial Cells, without Barrier Disruption, Front. Microbiol. 8 (2017) 2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Moses AV, Bloom FE, Pauza CD, Nelson JA, Human immunodeficiency virus infection of human brain capillary endothelial cells occurs via a CD4/galactosylceramide-independent mechanism, Proc. Natl. Acad. Sci. U. S. A. 90 (1993) 10474–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mutso M, St John JA, Ling ZL, Burt FJ, Poo YS, Liu X, Žusinaite E, Grau GE, Hueston L, Merits A, King NJC, Ekberg JAK, Mahalingam S, Basic insights into Zika virus infection of neuroglial and brain endothelial cells, J. Gen. Virol. 101 (2020) 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Patabendige A, Michael BD, Craig AG, Solomon T, Brain microvascular endothelial-astrocyte cell responses following Japanese encephalitis virus infection in an in vitro human blood-brain barrier model, Mol. Cell. Neurosci. 89 (2018) 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liou ML, Hsu CY, Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain, Cell Tissue Res. 293 (1998) 389–394. [DOI] [PubMed] [Google Scholar]
- [24].Hsieh JT, St John AL, Japanese encephalitis virus and its mechanisms of neuroinvasion, PLoS Pathog. 16 (2020) e1008260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kim J, Alejandro B, Hetman M, Hattab EM, Joiner J, Schroten H, Ishikawa H, Chung D-H, Zika virus infects pericytes in the choroid plexus and enters the central nervous system through the blood-cerebrospinal fluid barrier, PLoS Pathog. 16 (2020) e1008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS, Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals, MBio. 5 (2014) e01476–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang P, Dai J, Bai F, Kong K-F, Wong SJ, Montgomery RR, Madri JA, Fikrig E, Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain, J. Virol. 82 (2008) 8978–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Paul AM, Acharya D, Duty L, Thompson EA, Le L, Stokic DS, Leis AA, Bai F, Osteopontin facilitates West Nile virus neuroinvasion via neutrophil “Trojan horse” transport, Sci. Rep. 7 (2017) 4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bai F, Kong K-F, Dai J, Qian F, Zhang L, Brown CR, Fikrig E, Montgomery RR, A paradoxical role for neutrophils in the pathogenesis of West Nile virus, J. Infect. Dis. 202 (2010) 1804–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Roe K, Kumar M, Lum S, Orillo B, Nerurkar VR, Verma S, West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases, J. Gen. Virol. 93 (2012) 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW, CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS, J. Neurosci. 26 (2006) 1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Campbell JH, Hearps AC, Martin GE, Williams KC, Crowe SM, The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure, AIDS. 28 (2014) 2175–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mori I, Nishiyama Y, Yokochi T, Kimura Y, Olfactory transmission of neurotropic viruses, J. Neurovirol. 11 (2005) 129–137. [DOI] [PubMed] [Google Scholar]
- [34].Menendez CM, Carr DJJ, Defining nervous system susceptibility during acute and latent herpes simplex virus-1 infection, J. Neuroimmunol. 308 (2017) 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Antinone SE, Smith GA, Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis, J. Virol. 84 (2010) 1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Racaniello VR, One hundred years of poliovirus pathogenesis, Virology. 344 (2006) 9–16. [DOI] [PubMed] [Google Scholar]
- [37].Klingen Y, Conzelmann K-K, Finke S, Double-labeled rabies virus: live tracking of enveloped virus transport, J. Virol. 82 (2008) 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ohka S, Sakai M, Bohnert S, Igarashi H, Deinhardt K, Schiavo G, Nomoto A, Receptor-dependent and -independent axonal retrograde transport of poliovirus in motor neurons, J. Virol. 83 (2009) 4995–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Perlman S, Jacobsen G, Afifi A, Spread of a neurotropic murine coronavirus into the CNS via the trigeminal and olfactory nerves, Virology. 170 (1989) 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barnett EM, Perlman S, The olfactory nerve and not the trigeminal nerve is the major site of CNS entry for mouse hepatitis virus, strain JHM, Virology. 194 (1993) 185–191. [DOI] [PubMed] [Google Scholar]
- [41].Lundh B, Kristensson K, Norrby E, Selective infections of olfactory and respiratory epithelium by vesicular stomatitis and Sendai viruses, Neuropathol. Appl. Neurobiol. 13 (1987) 111–122. [DOI] [PubMed] [Google Scholar]
- [42].Beier KT, Borghuis BG, El-Danaf RN, Huberman AD, Demb JB, Cepko CL, Transsynaptic tracing with vesicular stomatitis virus reveals novel retinal circuitry, J. Neurosci. 33 (2013) 35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roussarie J-P, Ruffié C, Brahic M, The role of myelin in Theiler’s virus persistence in the central nervous system, PLoS Pathog. 3 (2007) e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cho H, Diamond MS, Immune responses to West Nile virus infection in the central nervous system, Viruses. 4 (2012) 3812–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS, Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 17140–17145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Green KN, Crapser JD, Hohsfield LA, To Kill a Microglia: A Case for CSF1R Inhibitors, Trends Immunol. 41 (2020) 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wu W, Li Y, Wei Y, Bosco DB, Xie M, Zhao M-G, Richardson JR, Wu L-J, Microglial depletion aggravates the severity of acute and chronic seizures in mice, Brain Behav. Immun. 89 (2020) 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wheeler DL, Sariol A, Meyerholz DK, Perlman S, Microglia are required for protection against lethal coronavirus encephalitis in mice, J. Clin. Invest. 128 (2018) 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mangale V, Syage AR, Ekiz HA, Skinner DD, Cheng Y, Stone CL, Brown RM, O’Connell RM, Green KN, Lane TE, Microglia influence host defense, disease, and repair following murine coronavirus infection of the central nervous system, Glia. 68 (2020) 2345–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brown DG, Soto R, Yandamuri S, Stone C, Dickey L, Gomes-Neto JC, Pastuzyn ED, Bell R, Petersen C, Buhrke K, Fujinami RS, O’Connell RM, Stephens WZ, Shepherd JD, Lane TE, Round JL, The microbiota protects from viral-induced neurologic damage through microglia-intrinsic TLR signaling, Elife. 8 (2019). 10.7554/eLife.47117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Funk KE, Klein RS, CSF1R antagonism limits local restimulation of antiviral CD8+ T cells during viral encephalitis, J. Neuroinflammation. 16 (2019) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Seitz S, Clarke P, Tyler KL, Pharmacologic Depletion of Microglia Increases Viral Load in the Brain and Enhances Mortality in Murine Models of Flavivirus-Induced Encephalitis, J. Virol. 92 (2018). 10.1128/JVI.00525-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chhatbar C, Detje CN, Grabski E, Borst K, Spanier J, Ghita L, Elliott DA, Jordão MJC, Mueller N, Sutton J, Prajeeth CK, Gudi V, Klein MA, Prinz M, Bradke F, Stangel M, Kalinke U, Type I Interferon Receptor Signaling of Neurons and Astrocytes Regulates Microglia Activation during Viral Encephalitis, Cell Rep. 25 (2018) 118–129.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fekete R, Cserép C, Lénárt N, Tóth K, Orsolits B, Martinecz B, Méhes E, Szabó B, Németh V, Gönci B, Sperlágh B, Boldogkői Z, Kittel Á, Baranyi M, Ferenczi S, Kovács K, Szalay G, Rózsa B, Webb C, Kovacs GG, Hortobágyi T, West BL, Környei Z, Dénes Á, Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms, Acta Neuropathol. 136 (2018) 461–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Waltl I, Käufer C, Gerhauser I, Chhatbar C, Ghita L, Kalinke U, Löscher W, Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage, Brain Behav. Immun. 74 (2018) 186–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sanchez JMS, DePaula-Silva AB, Doty DJ, Truong A, Libbey JE, Fujinami RS, Microglial cell depletion is fatal with low level picornavirus infection of the central nervous system, J. Neurovirol. 25 (2019) 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S, Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis, Immunity. 38 (2013) 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Uyar O, Laflamme N, Piret J, Venable MC, An Early Microglial Response Is Needed To Efficiently Control Herpes Simplex Virus Encephalitis, (2020). https://journals.asm.org/doi/abs/10.1128/jvi.01428-20 (accessed July 16, 2021). [DOI] [PMC free article] [PubMed]
- [59].Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, Laue M, Schneider J, Brünink S, Greuel S, Lehmann M, Hassan O, Aschman T, Schumann E, Chua RL, Conrad C, Eils R, Stenzel W, Windgassen M, Rößler L, Goebel H-H, Gelderblom HR, Martin H, Nitsche A, Schulz-Schaeffer WJ, Hakroush S, Winkler MS, Tampe B, Scheibe F, Körtvélyessy P, Reinhold D, Siegmund B, Kühl AA, Elezkurtaj S, Horst D, Oesterhelweg L, Tsokos M, Ingold-Heppner B, Stadelmann C, Drosten C, Corman VM, Radbruch H, Heppner FL, Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19, Nat. Neurosci. 24 (2021) 168–175. [DOI] [PubMed] [Google Scholar]
- [60].Khateb M, Bosak N, Muqary M, Coronaviruses and Central Nervous System Manifestations, Front. Neurol. 11 (2020) 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, Fox JM, Chen RE, Earnest JT, Keeler SP, Ritter JH, Kang L-I, Dort S, Robichaud A, Head R, Holtzman MJ, Diamond MS, SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function, Nat. Immunol. 21 (2020) 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zheng J, Wong L-YR, Li K, Verma AK, Ortiz ME, Wohlford-Lenane C, Leidinger MR, Knudson CM, Meyerholz DK, McCray PB Jr, Perlman S, COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice, Nature. 589 (2021) 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, Wei Q, Yu P, Xu Y, Qi F, Qu Y, Li F, Lv Q, Wang W, Xue J, Gong S, Liu M, Wang G, Wang S, Song Z, Zhao L, Liu P, Zhao L, Ye F, Wang H, Zhou W, Zhu N, Zhen W, Yu H, Zhang X, Guo L, Chen L, Wang C, Wang Y, Wang X, Xiao Y, Sun Q, Liu H, Zhu F, Ma C, Yan L, Yang M, Han J, Xu W, Tan W, Peng X, Jin Q, Wu G, Qin C, The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice, Nature. 583 (2020) 830–833. [DOI] [PubMed] [Google Scholar]
- [64].Olivarria GM, Cheng Y, Furman S, Pachow C, Hohsfield LA, Smith-Geater C, Miramontes R, Wu J, Burns MS, Tsourmas KI, Stocksdale J, Manlapaz C, Yong WH, Teijaro J, Edwards R, Green KN, Thompson LM, Lane TE, Microglia do not restrict SARS-CoV-2 replication following infection of the central nervous system of K18-hACE2 transgenic mice, bioRxiv. (2021). 10.1101/2021.11.15.468761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Olivarria GM, Cheng Y, Furman S, Pachow C, Hohsfield LA, Smith-Geater C, Miramontes R, Wu J, Burns MS, Tsourmas KI, Stocksdale J, Manlapaz C, Yong WH, Teijaro J, Edwards R, Green KN, Thompson LM, Lane TE, Microglia Do Not Restrict SARS-CoV-2 Replication following Infection of the Central Nervous System of K18-Human ACE2 Transgenic Mice, J. Virol. 96 (2022) e0196921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bennett ML, Chris Bennett F, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA, New tools for studying microglia in the mouse and human CNS, Proc. Natl. Acad. Sci. U. S. A. 113 (2016) E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kaiser T, Feng G, Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia, eNeuro. 6 (2019). 10.1523/ENEURO.0448-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, Greter M, Sall1 is a transcriptional regulator defining microglia identity and function, Nat. Immunol. 17 (2016) 1397–1406. [DOI] [PubMed] [Google Scholar]
- [69].Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, Shin A, Huang W, Dress RJ, Dutertre C-A, Schlitzer A, Chen J, Ng LG, Wang H, Liu Z, Su B, Ginhoux F, Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells, Cell. 178 (2019) 1509–1525.e19. [DOI] [PubMed] [Google Scholar]
- [70].Kawasaki T, Kawai T, Toll-like receptor signaling pathways, Front. Immunol. 5 (2014) 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Andersen LL, Mørk N, Reinert LS, Kofod-Olsen E, Narita R, Jørgensen SE, Skipper KA, Höning K, Gad HH, Østergaard L, Ørntoft TF, Hornung V, Paludan SR, Mikkelsen JG, Fujita T, Christiansen M, Hartmann R, Mogensen TH, Functional IRF3 deficiency in a patient with herpes simplex encephalitis, J. Exp. Med. 212 (2015) 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Herman M, Ciancanelli M, Ou Y-H, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Pérez de Diego R, Abhyankar A, Israelsson E, Guo Y, Cardon A, Rozenberg F, Lebon P, Tardieu M, Heropolitanska-Pliszka E, Chaussabel D, White MA, Abel L, Zhang S-Y, Casanova J-L, Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood, J. Exp. Med. 209 (2012) 1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, Herman M, Cardon A, Durandy A, Bustamante J, Vallabhapurapu S, Bravo J, Warnatz K, Chaix Y, Cascarrigny F, Lebon P, Rozenberg F, Karin M, Tardieu M, Al-Muhsen S, Jouanguy E, Zhang S-Y, Abel L, Casanova J-L, Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis, Immunity. 33 (2010) 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sancho-Shimizu V, Pérez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, Fabrega S, Cardon A, Maluenda J, Tatematsu M, Mahvelati F, Herman M, Ciancanelli M, Guo Y, AlSum Z, Alkhamis N, Al-Makadma AS, Ghadiri A, Boucherit S, Plancoulaine S, Picard C, Rozenberg F, Tardieu M, Lebon P, Jouanguy E, Rezaei N, Seya T, Matsumoto M, Chaussabel D, Puel A, Zhang S-Y, Abel L, Al-Muhsen S, Casanova J-L, Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency, J. Clin. Invest. 121 (2011) 4889–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang S-Y, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku C-L, Casrouge A, Zhang X-X, Barreiro L, Leonard J, Hamilton C, Lebon P, Héron B, Vallée L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova J-L, TLR3 deficiency in patients with herpes simplex encephalitis, Science. 317 (2007) 1522–1527. [DOI] [PubMed] [Google Scholar]
- [76].Lafaille FG, Pessach IM, Zhang S-Y, Ciancanelli MJ, Herman M, Abhyankar A, Ying S-W, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova J-L, Studer L, Notarangelo LD, Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells, Nature. 491 (2012) 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Medzhitov R, Toll-like receptors and innate immunity, Nat. Rev. Immunol. 1 (2001) 135–145. [DOI] [PubMed] [Google Scholar]
- [78].Mansur DS, Kroon EG, Nogueira ML, Arantes RME, Rodrigues SCO, Akira S, Gazzinelli RT, Campos MA, Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1, Am. J. Pathol. 166 (2005) 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, Anderson JF, Flavell RA, Fikrig E, Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing, Immunity. 30 (2009) 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhou S, Halle A, Kurt-Jones EA, Cerny AM, Porpiglia E, Rogers M, Golenbock DT, Finberg RW, Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses, J. Neuroimmunol. 194 (2008) 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Olson JK, Miller SD, Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs, J. Immunol. 173 (2004) 3916–3924. [DOI] [PubMed] [Google Scholar]
- [82].Lokensgard JR, Hu S, Sheng W, vanOijen M, Cox D, Cheeran MC, Peterson PK, Robust expression of TNF-alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus, J. Neurovirol. 7 (2001) 208–219. [DOI] [PubMed] [Google Scholar]
- [83].Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR, Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus, J. Immunol. 175 (2005) 4189–4193. [DOI] [PubMed] [Google Scholar]
- [84].Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, Finberg RW, Kurt-Jones EA, Role of specific innate immune responses in herpes simplex virus infection of the central nervous system, J. Virol. 86 (2012) 2273–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sørensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR, TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain, J. Immunol. 181 (2008) 8604–8612. [DOI] [PubMed] [Google Scholar]
- [86].Lima GK, Zolini GP, Mansur DS, Freire Lima BH, Wischhoff U, Astigarraga RG, Dias MF, das Graças Almeida Silva M, Béla SR, do Valle Antonelli LR, Arantes RM, Gazzinelli RT, Báfica A, Kroon EG, Campos MA, Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection, Am. J. Pathol. 177 (2010) 2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Aravalli RN, Peterson PK, Lokensgard JR, Toll-like receptors in defense and damage of the central nervous system, J. Neuroimmune Pharmacol. 2 (2007) 297–312. [DOI] [PubMed] [Google Scholar]
- [88].Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA, Microglia recognize double-stranded RNA via TLR3, J. Immunol. 176 (2006) 3804–3812. [DOI] [PubMed] [Google Scholar]
- [89].Jiang R, Ye J, Zhu B, Song Y, Chen H, Cao S, Roles of TLR3 and RIG-I in mediating the inflammatory response in mouse microglia following Japanese encephalitis virus infection, J Immunol Res. 2014 (2014) 787023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA, Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures, Neuron. 94 (2017) 759–773.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, Barres BA, A Combination of Ontogeny and CNS Environment Establishes Microglial Identity, Neuron. 98 (2018) 1170–1183.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Takeuchi O, Akira S, Pattern recognition receptors and inflammation, Cell. 140 (2010) 805–820. [DOI] [PubMed] [Google Scholar]
- [93].Rehwinkel J, Gack MU, RIG-I-like receptors: their regulation and roles in RNA sensing, Nat. Rev. Immunol. 20 (2020) 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Furr SR, Chauhan VS, Sterka D Jr, Grdzelishvili V, Marriott I, Characterization of retinoic acid-inducible gene-I expression in primary murine glia following exposure to vesicular stomatitis virus, J. Neurovirol. 14 (2008) 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Crill EK, Furr-Rogers SR, Marriott I, RIG-I is required for VSV-induced cytokine production by murine glia and acts in combination with DAI to initiate responses to HSV-1, Glia. 63 (2015) 2168–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Stone AEL, Green R, Wilkins C, Hemann EA, Gale M Jr, RIG-I-like receptors direct inflammatory macrophage polarization against West Nile virus infection, Nat. Commun. 10 (2019) 3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C-S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S, Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses, Nature. 441 (2006) 101–105. [DOI] [PubMed] [Google Scholar]
- [98].Sun L, Wu J, Du F, Chen X, Chen ZJ, Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway, Science. 339 (2013) 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Reinert LS, Lopušná K, Winther H, Sun C, Thomsen MK, Nandakumar R, Mogensen TH, Meyer M, Vægter C, Nyengaard JR, Fitzgerald KA, Paludan SR, Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS, Nat. Commun. 7 (2016) 13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Sato R, Kato A, Chimura T, Saitoh S-I, Shibata T, Murakami Y, Fukui R, Liu K, Zhang Y, Arii J, Sun-Wada G-H, Wada Y, Ikenoue T, Barber GN, Manabe T, Kawaguchi Y, Miyake K, Combating herpesvirus encephalitis by potentiating a TLR3-mTORC2 axis, Nat. Immunol. 19 (2018) 1071–1082. [DOI] [PubMed] [Google Scholar]
- [101].Jeffries AM, Marriott I, Human microglia and astrocytes express cGAS-STING viral sensing components, Neurosci. Lett. 658 (2017) 53–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jeffries AM, Nitika AW Truman, I. Marriott, The intracellular DNA sensors cGAS and IFI16 do not mediate effective antiviral immune responses to HSV-1 in human microglial cells, J. Neurovirol. 26 (2020) 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung J-YJ, Chen KJ, Bateup HS, Szpara ML, Lee AY, Cox JS, Vance RE, Interferon-independent STING signaling promotes resistance to HSV-1 in vivo, Nat. Commun. 11 (2020) 3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan W-B, Julius D, The P2Y 12 receptor regulates microglial activation by extracellular nucleotides, Nat. Neurosci. 9 (2006) 1512–1519. [DOI] [PubMed] [Google Scholar]
- [105].Cheeran MC-J, Hu S, Sheng WS, Rashid A, Peterson PK, Lokensgard JR, Differential responses of human brain cells to West Nile virus infection, J. Neurovirol. 11 (2005) 512–524. [DOI] [PubMed] [Google Scholar]
- [106].Shrestha B, Gottlieb D, Diamond MS, Infection and injury of neurons by West Nile encephalitis virus, J. Virol. 77 (2003) 13203–13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Samuel MA, Morrey JD, Diamond MS, Caspase 3-Dependent Cell Death of Neurons Contributes to the Pathogenesis of West Nile Virus Encephalitis, (2007). https://journals.asm.org/doi/abs/10.1128/JVI.02311-06 (accessed July 2, 2021). [DOI] [PMC free article] [PubMed]
- [108].Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, Gorelik L, Goldman SA, Human glial chimeric mice reveal astrocytic dependence of JC virus infection, J. Clin. Invest. 124 (2014) 5323–5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Wüthrich C, Koralnik IJ, Frequent infection of cortical neurons by JC virus in patients with progressive multifocal leukoencephalopathy, J. Neuropathol. Exp. Neurol. 71 (2012) 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chucair-Elliott AJ, Conrady C, Zheng M, Kroll CM, Lane TE, Carr DJJ, Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells, Glia. 62 (2014) 1418–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P, Dostert C, NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes, PLoS One. 10 (2015) e0130624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Lamkanfi M, Dixit VM, Mechanisms and functions of inflammasomes, Cell. 157 (2014) 1013–1022. [DOI] [PubMed] [Google Scholar]
- [113].Gold M, El Khoury J, β-amyloid, microglia, and the inflammasome in Alzheimer’s disease, Semin. Immunopathol. 37 (2015) 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, Son J, Yu J-W, MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration, Cell Death Differ. 26 (2019) 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Deora V, Lee JD, Albornoz EA, McAlary L, Jagaraj CJ, Robertson AAB, Atkin JD, Cooper MA, Schroder K, Yerbury JJ, Gordon R, Woodruff TM, The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins, Glia. 68 (2020) 407–421. [DOI] [PubMed] [Google Scholar]
- [116].Pontillo A, Brandão LA, Guimarães RL, Segat L, Athanasakis E, Crovella S, A 3′ UTR SNP in NLRP3 gene is associated with susceptibility to HIV-1 infection, JAIDS Journal of Acquired Immune Deficiency Syndromes. 54 (2010) 236–240. [DOI] [PubMed] [Google Scholar]
- [117].Pontillo A, Oshiro TM, Girardelli M, Kamada AJ, Sergio Crovella P, Duarte AJS, Polymorphisms in inflammasome’genes and susceptibility to HIV-1 infection, JAIDS Journal of Acquired Immune Deficiency Syndromes. 59 (2012) 121–125. [DOI] [PubMed] [Google Scholar]
- [118].Cosenza MA, Zhao M-L, Si Q, Lee SC, Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis, Brain Pathol. 12 (2002) 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D, CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia, Nature. 385 (1997) 645–649. [DOI] [PubMed] [Google Scholar]
- [120].Lavi E, Strizki JM, Ulrich AM, Zhang W, Fu L, Wang Q, O’Connor M, Hoxie JA, González-Scarano F, CXCR-4 (Fusin), a co-receptor for the type 1 human immunodeficiency virus (HIV-1), is expressed in the human brain in a variety of cell types, including microglia and neurons, Am. J. Pathol. 151 (1997) 1035–1042. [PMC free article] [PubMed] [Google Scholar]
- [121].Ghorpade A, Nukuna A, Che M, Haggerty S, Persidsky Y, Carter E, Carhart L, Shafer L, Gendelman HE, Human immunodeficiency virus neurotropism: an analysis of viral replication and cytopathicity for divergent strains in monocytes and microglia, J. Virol. 72 (1998) 3340–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].González-Scarano F, Martín-García J, The neuropathogenesis of AIDS, Nat. Rev. Immunol. 5 (2005) 69–81. [DOI] [PubMed] [Google Scholar]
- [123].Suh H-S, Zhao M-L, Choi N, Belbin TJ, Brosnan CF, Lee SC, TLR3 and TLR4 are innate antiviral immune receptors in human microglia: role of IRF3 in modulating antiviral and inflammatory response in the CNS, Virology. 392 (2009) 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM, HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation, Virology. 373 (2008) 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, Broadhurst DI, Power C, Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS, Retrovirology. 11 (2014) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Mamik MK, Hui E, Branton WG, McKenzie BA, Chisholm J, Cohen EA, Power C, HIV-1 Viral Protein R Activates NLRP3 Inflammasome in Microglia: implications for HIV-1 Associated Neuroinflammation, J. Neuroimmune Pharmacol. 12 (2017) 233–248. [DOI] [PubMed] [Google Scholar]
- [127].Chivero ET, Guo M-L, Periyasamy P, Liao K, Callen SE, Buch S, HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation, J. Neurosci. 37 (2017) 3599–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, Staeheli P, Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice, J. Virol. 86 (2012) 11223–11230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK, Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells, J. Neuroinflammation. 11 (2014) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Syage AR, Ekiz HA, Skinner DD, Stone C, O’Connell RM, Lane TE, Single-Cell RNA Sequencing Reveals the Diversity of the Immunological Landscape following Central Nervous System Infection by a Murine Coronavirus, J. Virol. 94 (2020). 10.1128/JVI.01295-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P, Sen E, Basu A, Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis, Glia. 55 (2007) 483–496. [DOI] [PubMed] [Google Scholar]
- [132].Chen C-J, Ou Y-C, Lin S-Y, Raung S-L, Liao S-L, Lai C-Y, Chen S-Y, Chen J-H, Glial activation involvement in neuronal death by Japanese encephalitis virus infection, J. Gen. Virol. 91 (2010) 1028–1037. [DOI] [PubMed] [Google Scholar]
- [133].Chen C-J, Ou Y-C, Chang C-Y, Pan H-C, Liao S-L, Chen S-Y, Raung S-L, Lai C-Y, Glutamate released by Japanese encephalitis virus-infected microglia involves TNF-α signaling and contributes to neuronal death, Glia. 60 (2012) 487–501. [DOI] [PubMed] [Google Scholar]
- [134].Marques CP, Cheeran MC-J, Palmquist JM, Hu S, Lokensgard JR, Microglia are the major cellular source of inducible nitric oxide synthase during experimental herpes encephalitis, J. Neurovirol. 14 (2008) 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA, Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis, Nat. Med. 10 (2004) 1366–1373. [DOI] [PubMed] [Google Scholar]
- [136].Aravalli RN, Hu S, Lokensgard JR, Toll-like receptor 2 signaling is a mediator of apoptosis in herpes simplex virus-infected microglia, J. Neuroinflammation. 4 (2007) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Reinert LS, Rashidi AS, Tran DN, Katzilieris-Petras G, Hvidt AK, Gohr M, Fruhwürth S, Bodda C, Thomsen MK, Vendelbo MH, Khan AR, Hansen B, Bergström P, Agholme L, Mogensen TH, Christensen MH, Nyengaard JR, Sen GC, Zetterberg H, Verjans GM, Paludan SR, Brain immune cells undergo cGAS/STING-dependent apoptosis during herpes simplex virus type 1 infection to limit type I IFN production, J. Clin. Invest. 131 (2021). 10.1172/JCI136824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jack C, Ruffini F, Bar-Or A, Antel JP, Microglia and multiple sclerosis, J. Neurosci. Res. 81 (2005) 363–373. [DOI] [PubMed] [Google Scholar]
- [139].Kroner A, Rosas Almanza J, Role of microglia in spinal cord injury, Neurosci. Lett. 709 (2019) 134370. [DOI] [PubMed] [Google Scholar]
- [140].Diaz-Aparicio I, Beccari S, Abiega O, Sierra A, Clearing the corpses: regulatory mechanisms, novel tools, and therapeutic potential of harnessing microglial phagocytosis in the diseased brain, Neural Regeneration Res. 11 (2016) 1533–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Quick ED, Leser JS, Clarke P, Tyler KL, Activation of intrinsic immune responses and microglial phagocytosis in an ex vivo spinal cord slice culture model of West Nile virus infection, J. Virol. 88 (2014) 13005–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Marques CP, Hu S, Sheng W, Lokensgard JR, Microglial cells initiate vigorous yet non-protective immune responses during HSV-1 brain infection, Virus Res. 121 (2006) 1–10. [DOI] [PubMed] [Google Scholar]
- [143].Moseman EA, Blanchard AC, Nayak D, McGavern DB, T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection, Sci Immunol. 5 (2020). 10.1126/sciimmunol.abb1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].McGavern DB, Kang SS, Illuminating viral infections in the nervous system, Nat. Rev. Immunol. 11 (2011) 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Taskin BD, Tanji K, Feldstein NA, McSwiggan-Hardin M, Akman CI, Epilepsy surgery for epileptic encephalopathy as a sequela of herpes simplex encephalitis: case report, J. Neurosurg. Pediatr. 20 (2017) 56–63. [DOI] [PubMed] [Google Scholar]
- [146].Love S, Koch P, Urbach H, Dawson TP, Chronic granulomatous herpes simplex encephalitis in children, J. Neuropathol. Exp. Neurol. 63 (2004) 1173–1181. [DOI] [PubMed] [Google Scholar]
- [147].Armien AG, Hu S, Little MR, Robinson N, Lokensgard JR, Low WC, Cheeran MC-J, Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis, Brain Pathol. 20 (2010) 738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Asenbauer B, McEntagart M, King MD, Gallagher P, Burke M, Farrell MA, Chronic Active Destructive Herpes Simplex Encephalitis with Recovery of Viral DNA 12 Years After Disease Onset, Neuropediatrics. 29 (2007) 120–123. [DOI] [PubMed] [Google Scholar]
- [149].Major EO, Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies, Annu. Rev. Med. 61 (2010) 35–47. [DOI] [PubMed] [Google Scholar]
- [150].Tan CS, Koralnik IJ, Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis, Lancet Neurol. 9 (2010) 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Tan CS, Ellis LC, Wüthrich C, Ngo L, Broge TA Jr, Saint-Aubyn J, Miller JS, Koralnik IJ, JC virus latency in the brain and extraneural organs of patients with and without progressive multifocal leukoencephalopathy, J. Virol. 84 (2010) 9200–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, Elledge SJ, Niebuhr DW, Scher AI, Munger KL, Ascherio A, Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis, Science. 375 (2022) 296–301. [DOI] [PubMed] [Google Scholar]
- [153].Achim CL, Wang R, Miners DK, Wiley CA, Brain viral burden in HIV infection, J. Neuropathol. Exp. Neurol. 53 (1994) 284–294. [DOI] [PubMed] [Google Scholar]
- [154].Gray F, Lescs MC, Keohane C, Paraire F, Marc B, Durigon M, Gherardi R, Early brain changes in HIV infection: neuropathological study of 11 HIV seropositive, non-AIDS cases, J. Neuropathol. Exp. Neurol. 51 (1992) 177–185. [DOI] [PubMed] [Google Scholar]
- [155].Wiley CA, Achim CL, Christopherson C, Kidane Y, Kwok S, Masliah E, Mellors J, Radhakrishnan L, Wang G, Soontornniyomkij V, HIV mediates a productive infection of the brain, AIDS. 13 (1999) 2055–2059. [DOI] [PubMed] [Google Scholar]
- [156].Marques CP, Cheeran MC-J, Palmquist JM, Hu S, Urban SL, Lokensgard JR, Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis, J. Immunol. 181 (2008) 6417–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Weissert R, Progressive multifocal leukoencephalopathy, J. Neuroimmunol. 231 (2011) 73–77. [DOI] [PubMed] [Google Scholar]
- [158].Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, Lee S, Plavina T, Scanlon JV, Sandrock A, Bozic C, Risk of natalizumab-associated progressive multifocal leukoencephalopathy, N. Engl. J. Med. 366 (2012) 1870–1880. [DOI] [PubMed] [Google Scholar]
- [159].Vazeux R, Cumont M, Girard PM, Nassif X, Trotot P, Marche C, Matthiessen L, Vedrenne C, Mikol J, Henin D, Severe encephalitis resulting from coinfections with HIV and JC virus, Neurology. 40 (1990) 944–948. [DOI] [PubMed] [Google Scholar]
- [160].Achim CL, Wiley CA, Expression of major histocompatibility complex antigens in the brains of patients with progressive multifocal leukoencephalopathy, J. Neuropathol. Exp. Neurol. 51 (1992) 257–263. [DOI] [PubMed] [Google Scholar]
- [161].Lach B, Connolly B, Wüthrich C, Koralnik IJ, Inflammatory infratentorial progressive multifocal leukoencephalopathy in a patient with rheumatoid arthritis, Neuropathology. 34 (2014) 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Berger JR, Chauhan A, Galey D, Nath A, Epidemiological evidence and molecular basis of interactions between HIV and JC virus, J. Neurovirol. 7 (2001) 329–338. [DOI] [PubMed] [Google Scholar]
- [163].Raj GV, Khalili K, Identification and characterization of a novel GGA/C-binding protein, GBP-i, that is rapidly inducible by cytokines, Mol. Cell. Biol. 14 (1994) 7770–7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Mayreddy RP, Safak M, Razmara M, Zoltick P, Khalili K, Transcription of the JC virus archetype late genome: importance of the kappa B and the 23-base-pair motifs in late promoter activity in glial cells, J. Virol. 70 (1996) 2387–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Kim S-Y, Choi E-C, Woo Jo Y, Henson JW, Kim H-S, Transcriptional activation of JC virus early promoter by phorbol ester and interleukin-1beta: critical role of nuclear factor-1, Virology. 327 (2004) 60–69. [DOI] [PubMed] [Google Scholar]
- [166].Chen NC, Partridge AT, Sell C, Torres C, Martín-García J, Fate of microglia during HIV-1 infection: From activation to senescence?, Glia. 65 (2017) 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Castellano P, Prevedel L, Eugenin EA, HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim, Sci. Rep. 7 (2017) 12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE, Influence of HAART on HIV-related CNS disease and neuroinflammation, J. Neuropathol. Exp. Neurol. 64 (2005) 529–536. [DOI] [PubMed] [Google Scholar]
- [169].Fischer-Smith T, Croul S, Adeniyi A, Rybicka K, Morgello S, Khalili K, Rappaport J, Macrophage/microglial accumulation and proliferating cell nuclear antigen expression in the central nervous system in human immunodeficiency virus encephalopathy, Am. J. Pathol. 164 (2004) 2089–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Hammoud DA, Endres CJ, Chander AR, Guilarte TR, Wong DF, Sacktor NC, McArthur JC, Pomper MG, Imaging glial cell activation with [11C]-R-PK11195 in patients with AIDS, J. Neurovirol. 11 (2005) 346–355. [DOI] [PubMed] [Google Scholar]
- [171].Garvey LJ, Pavese N, Politis M, Ramlackhansingh A, Brooks DJ, Taylor-Robinson SD, Winston A, Increased microglia activation in neurologically asymptomatic HIV-infected patients receiving effective ART, AIDS. 28 (2014) 67–72. [DOI] [PubMed] [Google Scholar]
- [172].Alvarez-Carbonell D, Ye F, Ramanath N, Garcia-Mesa Y, Knapp PE, Hauser KF, Karn J, Cross-talk between microglia and neurons regulates HIV latency, PLoS Pathog. 15 (2019) e1008249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Alvarez-Carbonell D, Garcia-Mesa Y, Milne S, Das B, Dobrowolski C, Rojas R, Karn J, Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells, Retrovirology. 14 (2017) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Liu H, Zhou R-H, Liu Y, Guo L, Wang X, Hu W-H, Ho W-Z, HIV infection suppresses TLR3 activation-mediated antiviral immunity in microglia and macrophages, Immunology. 160 (2020) 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].McKinsey GL, Lizama CO, Keown-Lang AE, Niu A, Santander N, Larpthaveesarp A, Chee E, Gonzalez FF, Arnold TD, A new genetic strategy for targeting microglia in development and disease, Elife. 9 (2020). 10.7554/eLife.54590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Guo Z, Zhang L, Wu Z, Chen Y, Wang F, Chen G, In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model, Cell Stem Cell. 14 (2014) 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Hammond SL, Leek AN, Richman EH, Tjalkens RB, Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection, PLoS One. 12 (2017) e0188830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Akiyama H, Jalloh S, Park S, Lei M, Mostoslavsky G, Gummuluru S, Expression of HIV-1 Intron-Containing RNA in Microglia Induces Inflammatory Responses, J. Virol. (2020). 10.1128/JVI.01386-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Hasselmann J, Blurton-Jones M, Human iPSC-derived microglia: A growing toolset to study the brain’s innate immune cells, Glia. 68 (2020) 721–739. [DOI] [PMC free article] [PubMed] [Google Scholar]