

Abstract
Cognitive dysfunction is often reported in patients with post-coronavirus disease 2019 (COVID-19) syndrome, but its underlying mechanisms are not completely understood. Evidence suggests that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike protein or its fragments are released from cells during infection, reaching different tissues, including the CNS, irrespective of the presence of the viral RNA. Here, we demonstrate that brain infusion of Spike protein in mice has a late impact on cognitive function, recapitulating post-COVID-19 syndrome. We also show that neuroinflammation and hippocampal microgliosis mediate Spike-induced memory dysfunction via complement-dependent engulfment of synapses. Genetic or pharmacological blockage of Toll-like receptor 4 (TLR4) signaling protects animals against synapse elimination and memory dysfunction induced by Spike brain infusion. Accordingly, in a cohort of 86 patients who recovered from mild COVID-19, the genotype GG TLR4-2604G>A (rs10759931) is associated with poor cognitive outcome. These results identify TLR4 as a key target to investigate the long-term cognitive dysfunction after COVID-19 infection in humans and rodents.
Keywords: cognitive dysfunction, SARS-CoV-2 spike protein, neuroinflammation, microgliosis, synapse loss, synaptic pruning, TLR4, genetic variant, long COVID, post-COVID syndrome
Graphical abstract
Cognitive impairment is frequent in post-COVID-19 syndrome patients, but its underlying mechanisms are unclear. Fontes-Dantas et al. show that Spike brain infusion in mice induces late neuroinflammation and synapse loss, leading to long-term cognitive impairment mediated by TLR4 signaling. In patients, genotype GG TLR4-2604G>A was associated with poor cognitive outcome.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is considered a respiratory pathogen, but the impact of the infection on extrapulmonary tissues is of high concern.1 Coronavirus disease 2019 (COVID-19) is associated with unpredictable and variable outcomes, and while most patients show a positive recovery after the acute stages,2 others experience a myriad of acute2 and long-term dysfunctions.3 , 4 Cognitive impairment is a well-characterized feature of post-COVID-19 syndrome, even in patients with mild symptoms, referred to as “long COVID-19 or post-COVID-19.”5 , 6 , 7 , 8 Mounting evidence suggests that COVID-19-induced neurological symptoms are mediated by multiple events, including direct brain viral infection, brain hypoxia, and/or systemic inflammation,9 , 10 , 11 , 12 , 13 but the central mechanism is still unclear.
SARS-CoV-2 Spike protein plays a pivotal role in COVID-19 pathogenesis and is the main target for vaccine development. Spike protein forms a homotrimer on the virus surface that is cleaved into two fragments, S1 and S2, after virus binding to its cellular receptor, angiotensin-converting enzyme 2 (ACE2).14 The S1 fragment contains the binding to ACE2, while the S2 fragment mediates cellular entry through fusion between the viral and cellular membranes. There is evidence suggesting that, during SARS-CoV-2 infection, Spike protein or its S1 fragment is released from the cells, reaching different tissues, including the central nervous system (CNS), irrespective of the presence of the viral RNA.15 , 16 Additionally, it has been demonstrated that cells expressing the Spike protein release extracellular vesicles containing the full-length protein,17 which would be another means of its circulation in the body. Free S1 has been shown to cross the blood-brain barrier (BBB), reaching different memory-related regions of the brain, suggesting that the protein itself, independent of the viral particles, would affect brain functions.18 Notably, Swank et al.19 detected high levels of circulating Spike protein several months after SARS-CoV-2 infection in patients diagnosed with post-COVID-19 but not in individuals who did not present long-term sequelae. Nevertheless, whether the presence of the Spike protein in the brain is a crucial event for development of cognitive impairment in patients with post-COVID-19 as well as its underlying mechanisms remain poorly known.
Toll-like receptors (TLRs) are activated by different pathogen-associated molecular patterns (PAMPs) and are crucial for evoking the innate immune response to infection, stress, or injury.20 Studies have predicted that SARS-CoV-2 Spike protein binds to TLR4 with higher affinity than to ACE2,21 , 22 and its aberrant signaling is involved in the hyperinflammatory response of patients with COVID-19.23 In vitro studies also demonstrated that SARS-CoV-2 Spike protein activates TLR4 in cultured phagocytic cells, stimulating production of proinflammatory mediators.24 , 25 , 26 Although TLR4 has already been implicated in microglial activation and cognitive dysfunction of Alzheimer’s disease,27 the impact of TLR4 signaling in COVID-19-related neurological dysfunction is still unknown.
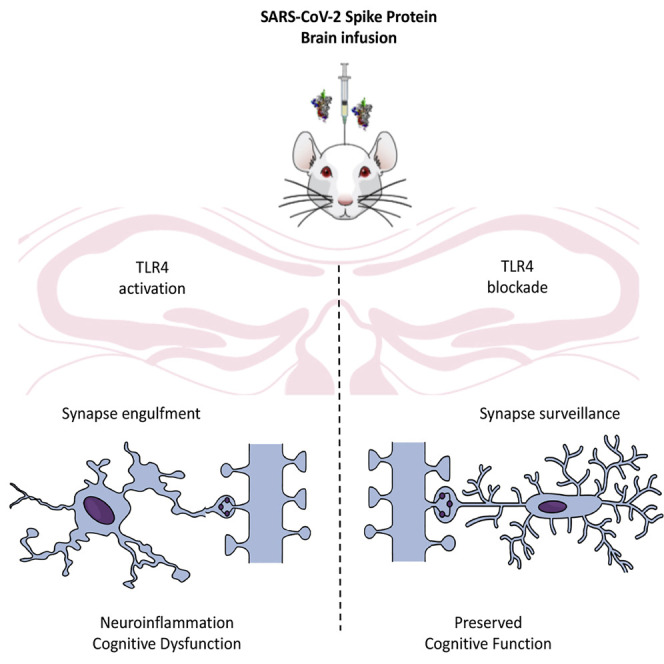
Most experimental studies investigating the effects of SARS-CoV-2 Spike protein on the brain have focused on acute infection.24 , 25 , 28 , 29 , 30 , 31 Also, few studies have used experimental models to evaluate the possible mechanism of post-COVID-19 syndrome.32 , 33 Here, we developed a mouse model of intracerebroventricular (i.c.v.) infusion of Spike to understand the role of this protein in late cognitive impairment after viral infection. We infused Spike protein into mouse brains and demonstrated late cognitive impairment, synapse loss, and microglial engulfment of presynaptic terminals. Early TLR4 blockage prevented Spike-associated detrimental effects on synapse and memory. We also demonstrated that the TLR4 SNP rs10759931 is associated with long-term cognitive impairment in patients who recovered from mild COVID-19. Collectively, these findings show that Spike protein impacts the mouse CNS, independent of virus infection, and identify TLR4 as a key mediator and interesting target to investigate long-term cognitive dysfunction in humans and rodents.
Results
Brain exposure to SARS-CoV-2 Spike protein induces late cognitive impairment and synapse loss in mice
COVID-19 is associated with late cognitive dysfunction.5 To evaluate whether brain exposure to SARS-CoV-2 Spike protein affects cognitive function independent of systemic inflammation, we infused the recombinant protein directly into mouse brains (i.c.v. infusion) and followed behavioral changes in two different time frames, “early and “late” phases, corresponding to assessments performed within the first 7 days and from 30–60 days after Spike protein infusion, respectively (Figure 1 A). The choice of these time points was based on the observations that the acute phase of COVID-1934 comprises a few days or weeks, and late sequelae initiate between 3 and 4 weeks from the onset of acute disease.35 In addition, these time points were similar to those used in our previous studies evaluating long-term cognitive dysfunctions observed in sepsis or Zika virus infection.36 We assessed mouse memory function using the novel object recognition (NOR) test. While vehicle (Veh)-infused mice were able to perform the NOR task, as demonstrated by longer exploration of the novel object over the familiar one (Figures 1B–1E, white bars), mice infused with Spike failed to recognize the novel object when evaluated between 30 and 45 days after injection (Figures 1C and 1D, black bar). Remarkably, memory dysfunction is a late outcome of brain exposure to Spike protein; at the early time point (7 days after infusion), the animals were still able to perform the NOR task (Figure 1B, gray bar). Of note, performance of i.c.v. Spike protein-infused mice in the NOR test returned to normal 60 days after infusion (Figure 1E), showing that memory impairment is reversible. An i.c.v. administration of a 10-fold lower protein amount (0.65 μg) had no impact on memory function in the early and later phase of the model (Figures S1A and S1B). Although the main access route of the virus and its products to the CNS is still under debate,13 , 37 , 38 , 39 , 40 , 41 they may reach the brain from the periphery. Thus, to mimic this possible route by which the protein reaches the CNS, we assessed mouse memory function after subcutaneous (s.c.) Spike protein infusion. The results were similar to those obtained with i.c.v. injected mice, with cognitive dysfunction occurring only at later time points following protein infusion (Figures S1C and S1D).
Figure 1.

Spike protein causes synapse damage and memory impairment in mice
(A) Mice received an i.c.v. infusion of 6.5 μg of SARS-CoV-2 Spike protein (Spike) or vehicle (Veh) and were evaluated at early (up to 7 days) or late time points (from 30–60 days) after infusion using behavioral and molecular approaches.
(B–E) Mice were tested in the NOR test at 6 days (B; t = 2.626, ∗p = 0.0304 for Veh; t = 3.218, ∗p = 0.0105 for Spike), 30 days (C; t = 5.099, ∗p = 0.0014 for Veh; t = 1.645, p = 0.1386 for Spike), 45 days (D; t = 5.122, ∗p = 0.0014 for Veh; t = 1.189, p = 0.2685 for Spike), or 60 days (E; t = 2.913, ∗p = 0.0195 for Veh; t = 2.560, ∗p = 0.0336 for Spike). One-sample Student’s t test compared with the chance level of 50% (n = 8–10 mice per group).
(F and G) Escape latency across 4 consecutive training trials (F) and time spent in the target quadrant during the probe trial (G) of the MWM test performed 45 days after Spike infusion (F, F(3, 45) = 2.857, ∗p = 0.0475, repeated measures ANOVA followed by Tukey’s test G, t = 2.211, ∗p = 0.0442, Student’s t test; n = 7–9 mice per group).
(H) Time spent at the center of the open field arena at early or late stages of the model (early, t = 1.728, p = 0.1021; late, t = 0.5363, p = 0.5348; Student’s t test; n = 8–10 mice per group).
(I) Total distance traveled in the open field arena at early or late stages of the model (early, t = 0.9614, p = 0.3498; late, t = 1.343, p = 0.1993; Student’s t test; n = 8–10 mice per group).
(J, K, O, and P) Representative images of the DG hippocampal region of Veh-infused (J and O) and Spike-infused mice (K and P) in the early (J and K) and late (O and P) stages of the model, immunolabeled for Homer-1 (red) and synaptophysin (SYP; green).
(L–N and Q–S) Number of puncta for Homer-1 (L and Q), SYP (M and R), and colocalized Homer-1/SYP puncta (N and S) in the early (L–N) and late (Q–S) stages of the model. (L, t = 1.202, p = 0.2524; M, t = 0.6648, p = 0.5188; N, t = 0.04952, p = 0.9613; Q, t = 0.7491, p = 0.4711; R, t = 3.400, ∗p = 0.0273; S, t = 4.204, ∗p = 0.0137; Student’s t test; n = 6–7 mice per group).
Scale bar, 20 μm. Symbols represent individual mice. Bars or points represent mean ± SEM. IHC, immunohistochemistry; MWM, Morris water maze; NOR, novel object recognition.
Late cognitive dysfunction induced by Spike protein infusion was confirmed by the Morris water maze (MWM) test, a task widely used to assess spatial memory in rodents.42 Mice infused with Spike protein showed higher latency time to find the submerged platform in sessions 3 and 4 of MWM training compared with control mice (Figure 1F). Also, Spike protein-infused mice showed reduced memory retention, as indicated by the decreased time spent by these animals in the target quadrant during the probe trial (Figure 1G). To rule out the possibility that changes in motivation or motor function eventually induced by Spike protein infusion were influencing NOR or MWM interpretation, mice were also submitted to the open field and rotarod tests. The Spike protein- and Veh-infused groups showed a similar innate preference for the objects in the NOR memory test (Figures S1E, S1F, S1I, S1J, and S2A–S2D), showed similar motivation toward object exploration in the NOR sessions (Figures S1G, S1H, S1K, S1L, and S2E–S2H), and performed similarly in the open field (Figures 1H, 1I, and S1M–S1R) and rotarod (Figure S2I) tests. No differences in swimming speed (Figure S2J) or distance traveled (Figure S2K) were found between groups in the test session of the MWM task. We also found that Spike infusion had no impact on body weight or food intake of mice (Figure S1S, S1T, S2L, and S2M), suggesting that Spike-induced neuroinflammatory modulation is specific to cognitive function rather than to a broader sickness response.
Synapse loss is strongly correlated with the cognitive decline observed in neurodegenerative diseases.43 , 44 Thus, we next investigated whether Spike protein induces synapse damage in the mouse hippocampus, a brain region critical for memory consolidation. Spike protein-infused mice did not show changes in synaptic density at the early stages, as demonstrated by the similar immunostaining for synaptophysin (SYP) and Homer-1 (pre- and postsynaptic markers, respectively) compared with the control group (Figures 1J–1N). Equivalent results were also found for colocalization of these synaptic markers, which indicates no changes in synaptic density (Figures 1J, 1K, and 1N). In contrast, decreased SYP immunostaining (Figures 1O, 1P, and 1R) and synaptic puncta (Figures 1O, 1P, and 1S) were observed in the late stage after protein infusion, indicating that Spike-induced hippocampal synapse damage displays temporal correlation with mouse behavioral phenotype (Figures 1C, 1D, 1F, and 1G). Using Fluoro-Jade staining, we found that Veh- and Spike-infused mouse hippocampal sections had no signal of degenerating neurons in the early and late phases of the model (Figure S3), suggesting that synaptic loss occurs independent of neuronal death. Collectively, these data suggest that a single brain infusion of Spike protein induces late synaptic loss and cognitive dysfunction, mimicking the post-COVID-19 syndrome.5
SARS-CoV-2 Spike protein triggers late neuroinflammation in mice
Neurodegeneration associated with viral brain infection can be mediated either by direct neuronal injury or by neuroinflammation.45 To advance the understanding of the genuine impact of Spike protein on neurons, cultured primary cortical neurons were incubated with the protein for 24 h. Neuron exposure to Spike protein did not affect neuron morphology (Figures S4A–S4E) when the percentage of pyknotic nuclei (Figure S4C), number of primary neurites (Figure S4D), and intensity of β3-tubulin immunostaining (Figure S4E) were similar for Veh- and Spike protein-incubated neurons. Also, Spike protein incubation had no effect on neuronal synaptic density and puncta (Figures S4F–S4J), suggesting that neurons are not directly affected by Spike protein.
Microglia is the primary innate immune cell of the brain and plays a critical role in neuroinflammation-induced cognitive dysfunction.46 To further understand the impact of Spike protein on microglial activation, mouse microglia BV-2 cells were incubated with Spike protein for 24 h. We found that Spike protein stimulation increased ionized calcium binding adaptor molecule 1 (Iba-1; a macrophage/microglia marker) immunoreactivity (Figures S4K–S4M) and upregulated tumor necrosis factor (TNF), interferon β (IFN-β), and interleukin-6 (IL-6) expression (Figures S2N–S2P) without affecting IL-1β and IFNAR2 (Figures S4Q–S4R). To evaluate the time course of microglia activation in vivo, we analyzed cellular features and cytokine production in our mouse model. We found that, at the early stage, i.c.v. injection of Spike protein neither changed the number and morphology of microglia (Figures 2A–2D) nor increased the expression of TNF, IL-1β, IL-6, INF-β, and IFNAR1 genes in hippocampal tissue (Figures 2E–2I). In contrast, the levels of IFNAR2 mRNA decreased significantly at the same time point after Spike protein infusion (Figure 2J).
Figure 2.

Spike protein induces cytokine upregulation and triggers delayed brain inflammation and microgliosis in mice
(A–T) Mice received an i.c.v. infusion of 6.5 μg of Spike or Veh and were evaluated at early (A–J, 3 days) or late (K–T, 45 days) time points. Shown are representative images of Iba-1 immunostaining in the DG hippocampal region of Veh-infused (A and K) or Spike-infused mice (B and L) in the early (A and B) and late (K and L) stages of the model. Scale bar, 25 μm; inset scale bar, 10 μm.
(C and M) Iba-1+ cells in the hippocampi of Veh- or Spike-infused mice in the early (C; t = 1.726, p = 0.1350) and late (M; t = 4.086, ∗p = 0.0035) stages of the model. Student’s t test (n = 4–5 mice per group).
(D and N) Quantification of the proportion of each morphological type of Iba-1+ cells in Veh- or Spike-infused mice evaluated in the in the early (D) and late (N) stages of the model (D: t = 1.383, p = 0.2160 for type I; t = 0.4712, p = 0.6541 for type II; t = 0.8927, p = 0.4064 for type IV; t = 0.8565, p = 0.4246 for type V; N: t = 6.388, ∗p = 0.0002 for type I; t = 4.458, ∗p = 0.0021 for type II; t = 5.513, ∗p = 0.0006 for type IV; t = 8.384, ∗p < 0.0001 for type V). Student’s t test, n = 4–5 mice per group. Type I and type II cells have smaller somata and fewer than 5 thin branches, surveillant microglia. Type III, IV, and V cells have more than 4 branches, thicker branches, and bigger somata, reactive microglia.
(E–J) qPCR analysis of the indicated mRNA isolated from the hippocampus in the early stage of the model: TNF mRNA (E; t = 0.2060, p = 0.8436), IL-1β mRNA (F; t = 0.1601, p = 0.8768), IL-6 mRNA (G; t = 1.555, p = 0.1638), IFNβ mRNA (H; t = 1.091, p = 0.3112), IFNAR1 mRNA (I; t = 0.6806; p = 0.5180), and IFNAR2 (J; t = 4.413, ∗p = 0.0031). Student’s t test, n = 4–5 mice per group.
(O–R) qPCR analysis of the indicated mRNA isolated from the hippocampus in the late stage of the model: TNF mRNA (O; t = 3.189, ∗p = 0.0110), IL-1β mRNA (P; t = 3.322, ∗p = 0.0089), IFN-β mRNA (Q; t = 3.713, ∗p = 0.013), and IFNAR2 mRNA (R; t = 3.743, ∗p = 0.0046).
(S and T) ELISA analysis of TNF (S; t = 2.885, ∗p = 0.0180) and IL-1β (T; t = 3.583, ∗p = 0.0116) protein levels. Student’s t test, n = 4–6 mice per group.
Symbols represent individual mice, and bars represent mean ± SEM.
We next investigated whether gliosis was induced by Spike protein. Mouse hippocampal sections obtained at the early and late stage after Spike infusion were immunolabeled for glial fibrillary acidic protein (GFAP, an astrocyte marker), ionized calcium binding adaptor molecule-1 (Iba-1), and transmembrane protein 119 (TMEM119, a microglia marker). No differences in GFAP immunoreactivity (Figures S5A–S5C and S5F–S5H) or morphology (Figures S5D, S5E, S5I, and S5J) were detected in Spike-infused mice compared with the control group. In contrast, assessments performed at the late time point revealed an increased number of Iba-1+ cells (Figures 2K–2M) and a predominance of cells with ameboid morphology in the hippocampus (Figures 2K, 2L, and 2N). Further indicating that late but not early (Figure S5K–S5M) microgliosis was induced by Spike protein, we found significantly higher TMEM119 immunoreactivity in the dentate gyrus (DG) hippocampal subregion of Spike-infused mice (Figures S5N–S5P). Notably, the mRNA levels of the inflammatory mediators TNF, IL-1β, IFNα, and IFNβ (Figures 2O–2Q) as well as the IFN receptor IFNAR2 (Figure 2R) were higher in the hippocampus of Spike-infused mice at this late time point. The protein levels of TNF and IL-1β (Figures 2S and 2T) were also increased in the hippocampal tissue at the late stage of the model, corroborating the mRNA analysis. Hippocampal expression of IL-6 and IFNγ cytokines and the receptor IFNAR1 were unaffected by Spike protein infusion (Figures S5Q–S5S). We also found increased serum levels of TNF only in the late stage of the model, which returned to control levels 60 days post infusion (Figures S5T–S5V), correlating with cognitive dysfunction (Figures 1B–1E). Altogether, our results indicate that the cognitive impairment induced by Spike protein is accompanied by microglial activation and neuroinflammation.
SARS-CoV-2 Spike protein induces C1q-mediated synaptic phagocytosis by microglia in mice
Synaptic phagocytosis (or synaptic pruning) by microglia has been shown to underlie cognitive dysfunction in dementia and viral encephalitis.36 , 43 , 47 We therefore evaluated whether synaptic phagocytosis by microglia mediates Spike protein-induced synapse damage. Hippocampal three-dimensional image reconstructions of Iba-1+ cells from Spike protein-infused mice showed increased SYP+ terminals inside phagocytic cells (Figures 3A–3D). Complement component 1q (C1q) is known to be involved in the initial tagging of synapses, preceding synaptic engulfment by microglial cells.48 Accordingly, we found that C1q was significantly upregulated in the hippocampus of mice late (but not early) after Spike protein infusion (Figures 3E and 3F). This finding led us to investigate whether blockage of soluble C1q could restore cognitive function in Spike protein-infused mice. For this, the animals were treated by i.c.v. route with a neutralizing anti-C1q antibody immediately after Spike protein infusion and twice a week for 30 days, and the animals were evaluated in the NOR and MWM tasks (Figure 3G). Remarkably, C1q blockage rescued object recognition memory impairment in Spike protein-infused mice (Figure 3H) without any effect on locomotion (Figure 3I) or exploration (Figures S6A and S6B). Similarly, neutralizing C1q antibody treatment also prevented spatial memory dysfunction induced by Spike protein infusion (Figures S6C and S6D), with no changes in the swimming speed (Figure S6E) or distance traveled (Figure S6F) between groups during the MWM test session. We found that C1q blockage also prevented the late decrease in hippocampal synaptic puncta (Figures 3J–3N) and reduced microglial synaptic engulfment (Figures 3O–3R) in mice infused with the Spike protein. Together, these data suggest that C1q-mediated microglial phagocytosis underlies long-term cognitive dysfunction induced by Spike protein, as seen for viral encephalitis.
Figure 3.

C1q neutralization prevents Spike-induced memory impairment in mice
Mice received an i.c.v. infusion of 6.5 μg of SARS-CoV-2 Spike protein (Spike) or Veh, and were evaluated at early (3 days) or late time points (45 days).
(A and B) Representative images of microglia (Iba-1+, green) engulfing pre-synaptic terminals immunolabeled for SYP (red) in the DG hippocampal subregion of Veh-infused (A) or Spike-infused mice (B) in the late stage of the model. Scale bar, 25 μm; inset scale bar, 10 μm.
(C and D) Quantification of microglia-SYP colocalization in CA3 (C; t = 2.949, ∗p = 0.0214) and DG (D; t = 2.271, #p = 0.0574) hippocampal subregions. Student’s t test; n= 4–5 mice per group.
(E and F) C1q mRNA expression in hippocampi of Veh- or Spike-infused mice at early (E; t = 0.7877, p = 0.4567) or late (F; t = 2.425, ∗p = 0.0383) time points. Student’s t test; n = 4–6 mice per group.
(G) Mice received an i.c.v. infusion of 6.5 μg of Spike, were treated with Veh or 0.3 μg anti-C1q antibody (α-C1q; i.c.v., twice a week for 30 days), followed by the NOR test (H; t = 3.438, ∗p = 0.0138 for Spike/α-C1q). One-sample Student’s t test compared with the chance level of 50%; n = 7–8 mice per group.
(I) Total distance traveled in the open field arena at the late time point (t = 1.274, p = 0.2249). Student’s t test; n = 7–8 mice per group.
(J and K) Representative images of the DG hippocampal subregion of Veh/Spike-injected (J) or α-C1q/Spike-injected (K) mice immunolabeled for Homer1 (red) and SYP (green). Scale bar, 20 μm. Number of puncta for Homer-1 (L; t = 0.5215, p = 0.6146), SYP (M; t = 2.881, p = 0.0181) and colocalized Homer-1/SYP puncta (n; t = 2.935, p = 0.0166). Student’s t test; n = 5–6 mice per group.
(O and P) Representative images of microglia (Iba-1+, green) engulfing pre-synaptic terminals immunolabeled for SYP (red) in the DG hippocampal subregion of Veh/Spike (O) or α-C1q/Spike mice (P) in the late stage of the model. Scale bar, 10 μm.
(Q and R) Quantification of microglia-SYP colocalization in the CA3 (Q; t = 3.454, ∗p = 0.0086) and DG (R; t = 2.052, #p = 0.0743) hippocampal subregions. Student’s t test; n = 5 mice per group.
Symbols represent individual mice, and bars represent mean ± SEM.
TLR-4 mediates cognitive dysfunction induced by SARS-CoV-2 Spike protein
Studies have described that Spike protein induces TLR4 activation in cultured immune cells.24 , 25 , 26 , 29 Additionally, TLR4 has been implicated in microglial activation and cognitive dysfunction in degenerative chronic diseases of the CNS, such as Alzheimer’s disease.49 In agreement with these observations, despite no changes in TLR4 expression levels at the early time point after Spike protein infusion (Figure 4 A), we found late upregulation of the TLR4 gene (Figure 4B) in the hippocampus of Spike protein-infused mice that matches the late cognitive dysfunction (Figures 1C, 1D, 1F, and 1G). To evaluate the role of TLR4 in Spike-induced cognitive impairment, we used either a pharmacological approach or a TLR4 knockout mouse model (TLR4 −/−). First, to investigate whether activation of TLR4 is an early event that could impact cognition later on, mice were treated with the TLR4 inhibitor TAK242 1 h before Spike protein brain infusion and once a day for 7 days (Figure 4C). Remarkably, early inhibition of TLR4 greatly prevented late memory dysfunction induced by Spike protein (Figure 4D). Some evidence has shown that high plasmatic levels of neurofilament light chain (NFL) are correlated with poor outcome in patients with COVID-19.50 , 51 , 52 , 53 , 54 Thus, we evaluated the NFL levels in plasma samples of control and Spike protein-infused mice treated or not with TAK242. Like patients with COVID-19, Spike-infused animals presented high serum levels of NFL compared with Veh-infused mice, which was prevented by TAK242 treatment (Figure 4E). Experiments using knockout mice confirmed those using the pharmacological approach. In the early phase after Spike protein infusion, wild-type (WT) and TLR4 −/− mice learned the NOR task (Figure S6G). On the other hand, at a late time point after protein infusion, WT mice had poor performance in the NOR test, while TLR4 −/− animals were able to execute the task (Figure 4F). Also, the absence of TLR4-mediated response in TLR4 −/− mice prevented the reduction of SYP+ terminals inside phagocytic cells later after Spike protein infusion in comparison with WT mice (Figures 4G–4K). Consistent with the previous results, control experiments showed that genetic (Figures S6G–S6M) or pharmacological (Figures S6N–S6P) inhibition of TLR4 had no effect on locomotion or exploratory behavior. Finally, we also found reduced numbers and altered morphology of microglia cells (Figures 4L–4O) as well as fewer microglia-engulfed synapses in the hippocampus of TLR4 −/− mice later after Spike protein brain infusion (Figures 4P–4S). Together, these data suggest that TLR4 activation mediates cognitive deficits and synaptic pruning induced by Spike protein in mice. Importantly, early treatment with the TLR4 inhibitor prevented late neuronal damage, indicating that the TLR4 pathway is central to induce neurodegeneration and long-term cognitive impairment in the present model.
Figure 4.

TLR4 mediates Spike-induced memory impairment in mice and is associated with post-COVID-19 cognitive impairment in a human cohort
(A and B) Mice received an i.c.v. infusion of 6.5 μg of SARS-CoV-2 Spike or Veh, and TLR4 mRNA levels in the hippocampi of Veh- or Spike-infused mice were evaluated at early (A; 3 days, t = 0.8892, p = 0.4034, Student’s t test) or late (B; 45 days, ∗p = 0.0303, Mann-Whitney U test) time points (n = 4–6 mice per group).
(C) Swiss mice received an i.c.v. infusion of 6.5 μg of Spike, were treated with Veh or the TLR4 antagonist TAK-242 (2 mg/kg, intraperitoneal [i.p.], once daily for 7 days), and were tested in the late stage of the model in the NOR test (D; t = 2.713, ∗p = 0.0301 for Spike/TAK-242). One-sample Student’s t test compared with the chance level of 50%; n = 8–9 mice per group.
(E) Plasma NFL levels evaluated in the late stage of the Spike infusion model (F = 6.329, ∗p = 0.0133). One-way ANOVA test followed by Tukey’s test, n = 4–6 mice per group.
(F) Wild-type (WT) and TLR4 knockout (TLR4−/−) mice received an i.c.v. infusion of 6.5 μg of SARS-CoV-2 Spike and were tested in the NOR test in the late stage of the model (F; t = 2.033, p = 0.0883 for WT/Spike and t = 2.744, ∗p = 0.0336 for TLR4−/−/Spike). One-sample Student’s t test compared with the chance level of 50%, n = 7 mice per group.
(G and H) Representative images of the DG hippocampal region of WT/Spike (G) and TLR4−/−/Spike (H) mice immunolabeled for Homer1 (red) and SYP (green). Scale bar, 20 μm.
(I–K) Number of puncta for Homer-1 (I; t = 1.272, p = 0.2506) and SYP (J; t = 1.592, p = 0.1624) and colocalized Homer-1/SYP puncta (K; t = 2.945, ∗p = 0.0258). Student’s t test; n = 4 mice per group.
(L and M) Representative images of Iba-1 immunolabeling in the DG hippocampal subregion of WT (L) and TLR4−/− (M) mice infused with Spike. Scale bar, 25 μm; inset scale bar, 10 μm.
(N) Iba-1+ cells in the DG (t = 5.088, ∗p = 0.0014) hippocampal subregion of WT or TLR4−/− mice infused with Spike.
(O) Quantification of the different morphological types of Iba-1+ cells in the hippocampus of Spike-infused WT and TLR4−/− mice (O; t = 2.229, #p = 0.0611 for type I; t = 3.340, ∗p = 0.0124 for type II; t = 3.277, ∗p = 0.0135 for type IV; t = 3.316, ∗p = 0.0128 for type V). Student’s t test, n = 4–5 mice per group. Type I and type II cells have smaller somata and fewer than 5 thin branches, surveillant microglia. Type III, IV, and V cells have more than 4 branches, thicker branches, and bigger somata, reactive microglia.
(P and Q) Representative images of microglia (Iba-1+, green) engulfing pre-synaptic terminals immunolabeled for SYP (red) in the DG hippocampal subregion of WT (P) and TLR4−/− (Q) mice infused with Spike. Scale bar, 50 μm; inset scale bar, 10 μm.
(R and S) Quantification of microglia-SYP colocalization in the CA3 (R; t = 2.200, #p = 0.0637) and DG (S; t = 4.012, ∗p = 0.0051) hippocampal subregions. Student’s t test; n = 4–5 mice per group. Symbols represent individual mice, and bars represent mean ± SEM.
(T) Pipeline to analyze the impact of TLR4 variants on the cognitive status of patients with post-COVID-19 syndrome.
(U and V) Forest plots showing the OR and 95% confidence interval for risk of cognitive impairment post COVID-19 by genotype for SNPs TLR4 - 2604G>A (U, rs10759931) and TLR4-2272A>G (V, rs2737190). Each square represents the OR for each genotype, and each horizontal line shows the 95% confidence interval.
(W) The expression levels of TLR4 for genotypes of SNP TLR4-2604G>A (rs10759931) was determined from peripheral blood mononuclear cells (PBMCs) treated with 1 μg of Spike protein for 24 h (t = 5.612, ∗p < 0.0001). Student’s t test; n = 7–8 patients per group. Data represents the mean ± SD.
A SNP in the TLR4 gene is associated with increased risk of cognitive dysfunction after COVID-19
Several lines of evidence have suggested that polymorphisms in the TLR4 gene are a risk factor for developing inflammatory diseases, including sporadic Alzheimer’s disease.49 , 55 , 56 , 57 Thus, we sought to extend our findings by investigating whether there is an association between TLR4 gene variants and cognitive outcomes in patients with COVID-19. For this, 86 individuals with a confirmed COVID-19 diagnosis, mostly with mild disease, were included in the study sample (Figures 4T). Characteristics of the sample are displayed in Table S1. Cognition was assessed using the symbol digit modalities test (SDMT) from 1–15 months after onset of acute COVID-19 symptoms (with cognitive deficit mean, 5.88 months; without cognitive deficit mean, 5.9 months). Of interest, nearly half of the patients evaluated (40, 46.51%) presented an important degree of post-COVID-19 cognitive impairment (Table 1 ). Genotyping analysis for two different SNPs (rs10759931 and rs2737190) was performed in all studied subjects. We found that genotypic distributions were in Hardy-Weinberg equilibrium and had no linkage disequilibrium (LD) between the two TLR4 SNPs (D′ > 0.9). Individuals carrying the TLR4-2604G>A (rs10759931) GG homozygous genotype demonstrated a significantly higher risk for developing cognitive impairment following SARS-CoV-2 infection (p = 0.0234, odds ratio [OR] = 1.91), while the GA genotype was associated with a decreased risk (p = 0.0209, OR = 0.50) (Figure 4U; Table 1). Test time was included as a covariate in the logistic regression analyses (p adjusted = 0.0129∗) (Table 1). Conversely, none of the TLR4-2272A>G (rs2737190) genotype variations were associated with increased susceptibility to post-COVID-19 cognitive impairments (Figures 4V; Table 1). Considering our clinical findings demonstrating that the SNP (rs10759931) is associated with poor cognitive function after COVID-19, we performed functional analysis aiming to strengthen the link between this genetic variant and the levels of TLR4 mRNA after Spike stimulation. Spike stimulation of cultured GG genotype cells resulted in increased levels of mRNA TLR4 compared with GA genotype cells (∗p = <0.0001) (Figure 4X). Our findings suggest that polymorphisms in the TLR4 gene are associated with altered Spike-induced host immune responses, increasing the risk of developing long-term cognitive deficits in genetically susceptible individuals.
Table 1.
TLR4 rs10759931 and rs2737190 genotype distribution in patients with or without cognitive deficit following COVID-19
| TLR4-2604G>A (rs10759931) | N (86) | Cognitive deficit (%) | No cognitive deficit (%) | p value | OR (95% CI) | Adjusted p value for SDMT time |
|---|---|---|---|---|---|---|
| GG | 40 | 22 (55) | 18 (39) | 0.0234∗ | 1.91 (1.083–3.301) | 0.0129∗ |
| GA | 35 | 13 (32) | 22 (48) | 0.0209∗ | 0.50 (0.287–0.920) | |
| AA | 11 | 5 (13) | 6 (13) | >0.9999 | 1.00 (0.435–2.294) | |
| MAF (A) |
0.35 |
– |
– |
– |
– |
|
|
TLR4 -2272 A>G (rs2737190) |
N (83) |
Cognitive deficit (%) |
No cognitive deficit (%) |
p value |
OR (95% CI) |
|
| AA | 30 | 14 (37) | 16 (36) | 0.8832 | 1.04 (0.594–1.836) | 0.0809 |
| AG | 35 | 16 (42) | 19 (42) | >0.9999 | 1.0 (0.561–1.781) | |
| GG | 18 | 8 (21) | 10 (22) | 0.8633 | 0.94 (0.483–1.823) | |
| MAF (G) | 0.49 | – | – | – | – |
MAF, minor-allele frequency; OR, odds ratio; CI, confidence interval. Genotype frequency was analyzed by χ2 test (two tailed). Test time was included as a covariate in the logistic regression analyses. ∗Statistical significance (p < 0.05). The reference group in each of the analyses was the most prevalent genotype.
Discussion
Post-COVID-19 syndrome comprises a myriad of symptoms that emerge after the acute phase of infection, including psychiatric symptoms and dementia-like cognitive dysfunction.5 , 58 , 59 , 60 Clinical studies have largely mapped the spectrum of neurological symptoms in patients with post-COVID-19 syndrome but do not provide a significant advance by describing the molecular mechanisms that trigger this condition or targets for preventive/therapeutic interventions. On the other hand, studies involving COVID-19 preclinical models have focused mostly on the acute impacts of viral infection. Therefore, it is mandatory to develop novel tools to dissect the mechanisms underlying the neurological deficits in post-COVID-19 syndrome, especially the direct effects of the virus and/or viral products on the brain.
Here we speculated that Spike protein plays a central role in neurological dysfunction associated with post-COVID-19 syndrome, independent of SARS-CoV-2 replication in the brain. Notably, our hypothesis is supported by recent findings showing that Spike protein persists in the plasma of patients with long COVID-19 for up to 12 months post diagnosis,19 increasing the probability that it reaches the brain. Previous studies have demonstrated that the hippocampus is particularly vulnerable to viral infection.36 , 47 , 61 Accordingly, brain scans of patients who recovered from COVID-19 showed significant changes in hippocampal volume62 , 63 and hypometabolism;64 both factors are important predictors of cognitive dysfunction in normal aging and Alzheimer’s disease.65 , 66 , 67 Using two hippocampus-dependent behavioral paradigms, we found that brain exposure to Spike protein induces reversible late-onset neuroinflammation and memory dysfunction. Thus, our model recapitulates not only long-term cognitive impairment but also recovery of memory function seen in long COVID-19 syndrome, expanding previous studies that were focused on the short-term effects of S1 exposure.24 , 68 , 69 In contrast to our findings, in these studies, acute neuroinflammation and cognitive impairment were observed, which could be explained by the fact that the protein was infused directly into the hippocampal tissue69 or by their use of aged mice.68 We also cannot rule out that the trimeric ectodomain used in our model may induce later effects than those resulting from direct exposure to the S1 fragment.
Synapse damage is a common denominator in a number of memory-related diseases,70 , 71 often preceding neurodegeneration. It has been shown that neuroinvasive viruses, such as West Nile virus (WNV), Borna disease virus (BDV), and Zika virus (ZIKV), are also associated with synapse impairment.36 , 47 , 72 Likewise, we found that the late cognitive dysfunction induced by Spike protein was accompanied by prominent synapse loss in the mouse hippocampus. Recent data have revealed upregulation of genes linked to synapse elimination in SARS-CoV-2-infected human brain organoids and in postmortem brain samples from patients with COVID-19.73 , 74 In line with these observations, we found that infusion of Spike protein into the mouse brain induces a late elevation in plasma levels of NFL, an axonal cytoskeleton protein identified as a component of pre- and postsynaptic terminals.75 Plasma NFL increase can be employed as a marker of synapse loss and disease progression in neurodegenerative diseases, including Alzheimer’s disease.76 Remarkably, some data showed that plasma NFL levels are higher in patients with severe COVID-19 compared with healthy age-matched individuals as well as inversely correlated with cognitive performance in patients with COVID-19,77 , 78 reinforcing the translational potential of our model. Collectively, these findings suggest that brain exposure to Spike protein induces synapse loss and behavioral alterations typical of viral encephalitis, leading to prolonged neurological dysfunction that can persist long after recovery from the infectious event.
Microglia are the most abundant immune cell type in the CNS and play a critical role in most neuroinflammatory diseases.79 In viral encephalitis, microglial cells have protective and detrimental activities depending on the phase of infection.46 Previous studies have shown that human coronaviruses can reach the CNS and induce neuroinflammation and/or gliosis in mature and immature brain tissues.16 , 61 , 80 Here, we found that the microglial cell line BV-2 was impacted by Spike protein, corroborating recent data showing an increase in proinflammatory mediators in S1-stimulated microglia.25 Because cultured primary cortical neurons were not directly affected by Spike stimulation, our in vitro results indicate that microglia could be seen as the main cell type affected by exposure to SARS-CoV-2 Spike protein.
It is well known that viral infections are often associated with excessive activation of inflammatory and immune responses, which may, in turn, elicit and/or accelerate brain neurodegeneration.81 Here, we found that Spike protein-infused mice presented late microglial activation but not astrocyte reactivity, similar to what has been observed in other animal models of viral encephalitis.36 , 47 Increased levels of proinflammatory mediators in the hippocampus and serum were found only at late time points after Spike infusion, showing a temporal correlation with synaptic loss and cognitive dysfunction. Conversely, we found that downregulation of the IFNAR2 gene occurred shortly after Spike injection, similar to what has been observed in neuronal cells of postmortem samples from patients with COVID-19.74 This finding corroborates recent evidence demonstrating that SARS-CoV-2 may evade innate immunity through modulation of type I IFN responses.82 Altogether, our results show that brain exposure to Spike protein induces early negative modulation of the main receptor involved in type I IFN responses, followed by a late proinflammatory process in the hippocampus.
A complement-microglia axis has emerged as one of the key triggers of synapse loss in memory-related diseases.83 The classical complement cascade, a central player of innate immune pathogen defense, orchestrates synaptic pruning by microglia during physiological and pathological conditions.43 , 48 , 84 , 85 We have reported previously that hippocampal synapses are phagocytosed by microglia during ZIKV brain infection in a process dependent on C1q and C3.36 Moreover, Vasek et al.47 showed hippocampal synapse loss in postmortem samples of patients with WNV neuroinvasive disease as well as complement-dependent microglial synapse engulfment in WNV-infected and recovered mice. Accordingly, we demonstrated that cognitive impairment induced by Spike protein is associated with hippocampal C1q upregulation and microglial engulfment of presynaptic terminals. Additionally, chronic C1q neutralization preserved memory function in Spike-infused mice, supporting a role of C1q-mediated synaptic pruning as an important mediator of post-COVID-19 cognitive impairment.
The pattern recognition receptor TLR4 has been implicated in the neuropathology of viral encephalitis, classically associated with memory impairment, including that caused by WNV, Japanese encephalitis virus (JEV), and BDV,86 , 87 , 88 as well as age-related neurodegenerative diseases.27 , 49 , 89 , 90 Notably, in silico simulations predicted that the Spike protein could be recognized by the TLR4,21 , 22 , 91 with this interaction activating inflammatory signaling independent of ACE2.24 , 25 , 26 , 29 Accordingly, here we found that a single brain infusion of Spike protein induced hippocampal TLR4 upregulation. To gain further insight into the role played by TLR4 in COVID-19-induced brain dysfunction, we first performed pharmacological blockage of TLR4 signal transduction early after Spike protein brain infusion. This strategy significantly prevented the long-term cognitive impairment observed in our model. Likewise, late cognitive impairment induced by Spike protein was absent in TLR4-deficient mice, in accordance with previous findings in animal models of dementia.90 , 92 Remarkably, we also found that Spike-induced plasma NFL increase was dependent on TLR4 activation because early TLR4 inhibition mitigated changes in NFL levels. Together, our findings strongly suggest that brain dysfunction in post-COVID-19 is associated with Spike-induced TLR4 signaling in microglial cells.
The engagement of complement and TLRs in signaling cross-talk has been proposed to regulate immune and inflammatory responses in neurodegenerative diseases.93 Indeed, it has been shown that TLR4 activation induces upregulation of complement components in the mouse hippocampus.27 , 94 , 95 Given the role of complement activation in synaptic pruning, we hypothesized that TLR4 is the molecular switch that regulates microglial synaptic engulfment. Notably, our hypothesis is in agreement with emerging evidence showing a role of TLR4 in Spike-induced microglial responses.24 , 25 Olajide et al.25 found significant inhibition in TNF and IL-6 release in S1 Spike-stimulated BV-2 microglia using the same pharmacological inhibitor used in our study (TAK-242) or in cells transfected with TLR4 small interfering RNA. Similar results using TLR4 pharmacological or genetic blockade have been found in murine and human macrophages. S1 also induces proinflammatory gene expression in primary rat microglia and activates TLR4 signaling in HEK293 transgenic cells.24 In our model, the delayed response to Spike protein is indeed an intriguing phenomenon, and it is not shared by other TLR4 agonists.95 , 96 Our animal model provides evidence of the ability of SARS-CoV-2 Spike protein to induce synapse dysfunction. Using brain organoids, Oliveira et al.73 described that SARS-CoV-2 infection is able to increase microglial engulfment of postsynaptic termini 72 h after virus inoculation. Thus, it is plausible that TLR4 activation can induce either acute or delayed synaptic dysfunction, depending on the agonist/proinflammatory insult. In light of this, we speculate that this possible uncommon ability of SARS-CoV-2 Spike protein to induce delayed synapse loss could account for occurrence of the intriguing delayed-onset post-COVID-19 cognitive impairment.
Finally, and relevantly, we validated our preclinical findings by examining whether TLR4 genetic variants could be associated with poor cognitive outcome in patients with COVID-19 with mild disease. In a cohort of patients with mild COVID-19 carrying the GG genotype of the TLR4-2604G>A (rs10759931) variant, we identified increased expression of TLR4 and high risk for cognitive impairment after SARS-CoV-2 infection compared with the GA genotype. The G allele has already been associated with increased risk for different disorders with immunological basis, including cardiovascular diseases,97 diabetes-associated retinopathy,98 cancer,99 and asthma.100 On the other hand, the A allele can affect the binding affinity of the TLR4 promoter to transcription factors, culminating in lower expression of this gene in allele carriers.101 Taken together, our findings suggest that the complex cross-talk between TLR4, the complement system, and neuroinflammation are important events that determine the development of neurological symptoms in patients with post-COVID-19 syndrome.
The impact of long COVID-19 syndrome is emerging as a major public health concern because of the high prevalence of prolonged neurological symptoms among survivors. Therefore, strategies designed to prevent or treat neurological post-COVID-19 symptoms constitute an unmet clinical need. Cognitive symptoms are common post-acute sequelae of SARS-CoV-2 infection, and although some studies have demonstrated a higher prevalence in severe cases,102 asymptomatic individuals or those with mild or moderate COVID-19 also report persistent cognitive symptoms.103 Among severe cases, COVID-19 severity score, mechanical ventilation, and multiorgan support were predictive factors for poorer cognitive outcomes.102 Because our model was not designed to mimic the respiratory, gastrointestinal, and cardiovascular manifestations that characterize severe acute COVID-19, it may not adequately recapitulate the clinical course of post-COVID-19 syndrome in this population.102 Nonetheless, longitudinal data indicate that mild SARS-CoV-2 infection is associated with persistent cognitive symptoms,5 , 7 , 8 , 59 , 104 , 105 , 106 , 107 with delayed symptom onset not only in individuals with pre-existing cognitive risk factors108 but also in young individuals in the absence of comorbidities.106 Thus, our model better replicates the cognitive dysfunction associated with the mild rather than severe COVID-19 phenotype. We found that Spike-induced cognitive impairment triggers innate immunity activation through TLR4, culminating in microgliosis, neuroinflammation, and synaptic pruning. The translational value of our model is supported by the correlation between increased plasma NFL and behavioral deficits as well as by the association between TLR4 genetic status and SARS-CoV-2 cognitive outcomes of patients who recovered from COVID-19. Altogether, our findings indicate key targets for establishment of interventional strategies for prevention and/or treatment of the long-term brain outcomes of COVID-19.
Limitations of the study
Although we clearly demonstrated that Spike protein can directly trigger an inflammatory cascade that culminates in synaptic dysfunction and cognitive impairment in our model, it is not possible to fully establish the extent of this effect in the context of peripheral or central SARS-CoV-2 infection. Furthermore, our study assessed the effect of Spike protein from the original strain; future studies comparing cognitive disturbances induced by emerging variants are warranted. Also, the effect of subsequent exposure to Spike protein in the absence of vaccination or during breakthrough infection in vaccinated individuals remains to be determined. Last, although our study holds translational potential, our findings are limited by the number of patients and SNPs evaluated and the absence of longitudinal assessments. Thus, in future studies, it will be important to extend these investigations to a larger group of patients with varying degrees of cognitive impairment.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal anti-IBA-1 | Wako Chemicals USA | Cat# 019-19741; RRID:AB_839504 |
| Clone 27G12 anti-Synaptophysin | Vector Laboratories | Cat# S285; RRID:AB_2336747 |
| Rabbit monoclonal [EPR15309] to Homer1 | Abcam | Cat# 184955; RRID:AB_2744679 |
| Mouse monoclonal Anti-GFAP | Sigma | Cat# G3893; RRID:AB_477010 |
| Mouse monoclonal Anti-βIII Tubulin | Promega | Cat# G712A; RRID:AB_430874 |
| Mouse TNF (Mono/Mono) ELISA Set | BD Biosciences | Cat# 555268; RRID:AB_2869055 |
| Mouse IL-1 beta/IL-1F2 Quantikine ELISA Kit | R&D Systems | Cat# MLB00C; RRID:AB_2895547 |
| anti-NFL | Quanterix | Cat #103186 |
| Rabbit monoclonal TMEM-119 | Abcam | Cat#209064; RRID:AB_2728083 |
| Biological samples | ||
| Human blood | Gaffrée and Guinle University Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Trizol® | Invitrogen | Cat# 15596026 |
| FluoroJade B | Histo Chem Inc | Cat #MFCD04974901 |
| Fluoroshield mounting medium with DAPI | Abcam | Cat# ab104139 |
| Critical commercial assays | ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368813 |
| Power SYBR Green Master Mix | Life Technologies | Cat# 4367659 |
| BCA Protein Assay | Thermo Scientific | Cat# 23227 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | Cat# 4304437 |
| PureLink Genomic DNA Mini Kit | ThermoFisher Scientific | Cat# K182002 |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Cat# Q32851 |
| Mouse IL-1 beta/IL-1F2 DuoSet ELISA Kit | R&D Systems | Cat# DY401-05 |
| Mouse TNF ELISA Set II Kit | BD Biosciences | Cat# 558534 |
| Experimental models: Cell lines | ||
| BV-2 | Donation from Fiocruz | None |
| Oligonucleotides | ||
| Primers for qPCR, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| ImageJ v1.53 | NIH | https://imagej.nih.gov/ij/ |
| Simoa SR-X™ Analyzer | Quanterix | https://www.quanterix.com |
| Prism 8.0 | Graphpad | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cláudia P. Figueiredo (claudia@pharma.ufrj.br).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
Eight to twelve-week-old male Swiss mice were used in this study. In some experiments, TLR4−/− mice on the C57BL/6 background were used. Animals were housed in groups of five per cage with free access to food and water, under a 12 h light/dark cycle, with controlled temperature and humidity. All procedures followed the “Principles of Laboratory Animal Care” (US National Institutes of Health) and were approved by the Institutional Animal Care and Use Committee of the Federal University of Rio de Janeiro, Brazil (protocol number 068/2).
Spike infusion
The recombinant Spike protein ectodomain from the original SARS-CoV-2 Wuhan strain (amino acids 1–1208) was produced in HEK293 cells and purified in its trimeric prefusion conformation109 by the Cell Culture Engineering Laboratory (LECC) of COPPE/UFRJ, Brazil.110 For protein intracerebroventricular (i.c.v.) infusion, mice were anesthetized with 2.5% isoflurane (Cristália; São Paulo, Brazil) using a vaporizer system (Norwell, MA), and a 2.5 mm-long needle was unilaterally inserted 1 mm to the right of the midline point equidistant from each eye and parallel to a line drawn through the anterior base of the eye. Using a Hamilton syringe, 0.65 or 6.5 μg Spike protein (in 5 μL) or vehicle (PBS) were slowly infused (freehand). For the peripheral model, mice received one single subcutaneous (s.c.) injection of the protein (10 μg in 5 μL) or vehicle (PBS). The trials were divided into two distinct stages: early phase (assessments performed up to one week after administration) and late phase (between 30 and 60 days after administration). Body weight and food intake of animals were measured every 5 days, until 60 days after Spike infusion.
Pharmacological treatments
For TLR4 blockade, TAK-242 (Millipore) was diluted in sterile saline (vehicle) and injected intraperitoneally (ip; 2 mg/kg). Mice received either vehicle or TAK for 7 days beginning immediately after Spike protein i.c.v. administration. For brain C1q blockade, mice received i.c.v. injections of vehicle (PBS) or an antibody against C1q (0.3 μg; Abcam #11861) twice a week for 30 days after S brain infusion.
Study population and cognitive assessment
Outpatients with post-COVID-19 were evaluated between December 2020 and July 2021 by a multidisciplinary team of neurologists and neuropsychologists at the Gaffrée and Guinle University Hospital (Rio de Janeiro, Brazil). Inclusion criteria included: COVID-19 diagnosis confirmed by PCR or serological diagnosis, fulfilling criteria of mild disease (not requiring hospitalization and symptoms that did not include dyspnea), assessment performed at least 15 days after the end of symptoms, blood collection and neurocognitive evaluation consent. Exclusion criteria included: age under 18 years old; individuals with previously known cognitive impairment or other neuropsychiatrist disorders that could interfere with the test results. All study subjects had their detailed clinical history recorded and were subjected to complete physical and neurological examination. This work was approved by the Brazilian Ethics Committee (CONEP, CAAE 33659620.1.1001.5258), and all participants signed the informed consent term, agreeing to participate in this research.
Neurocognitive status was only assessed using the Symbol Digit Modalities Test (SDMT), a screening test developed to identify individuals with cognitive impairment through the domains of attention, processing speed and motor skills. Considering that regressed scaled scores on age, age-squared, sex, and education were similar between the cohort, patients were divided into two main subgroups, “with cognitive deficit” and “without cognitive deficit”. The raw score of the SDMT is converted to scaled scores (M = 10, SD = 3) using the cumulative frequency distribution of the test in order to normalize test score distributions.111
Method details
Behavioral tests
Open field test
Animals were placed in the center of an arena (30 × 30 × 45 cm) divided in nine imaginary quadrants, and exploration was assessed for 5 min. The arena was thoroughly cleaned with 70% ethanol in between trials to eliminate olfactory cues. Total locomotor activity and time spent at central or peripheral quadrants were analyzed using ANY-maze software (Stoelting Company).
Novel object recognition (NOR) test
The test was carried out in an arena measuring 30 × 30 × 45 cm. Before training, each animal was submitted to a 5-min habituation session in the empty arena. Test objects were made of plastic and had different shapes, colors, sizes, and textures. Innate object preferences or neophobia were excluded in preliminary tests. Mice explored the configuration of two identical objects during a 5-min acquisition trial. After 90 min, mice were submitted to a 5-min retention trial, during which one of the familiar objects was replaced by an unfamiliar new one. Sniffing and touching the object were considered exploratory behavior. Results were expressed as a percentage of time exploring each object during the training or test sessions, or as total exploration during each session. Data were analyzed using a one-sample Student’s t-test comparing the mean exploration percentage time for each object with the chance value of 50%. Animals that recognize the familiar object as such (i.e., learn the task) explore the novel object >50% of the total time.
Morris Water Maze (MWM)
The apparatus used for the water maze task was a circular tank (1.2 m diameter) filled with water maintained at 20 ± 0.5°C. The tank was located in a test room containing prominent visual clues. Mice were trained to swim to a 11 cm diameter circular platform submerged 1.5 cm beneath the surface of the water and invisible to the mice while swimming. The platform was located in a fixed position, equidistant from the center and the wall of the tank. Mice were subjected to four training trials per day (inter-trial interval, 10 min). On each trial, mice were placed into the tank at one of four designated start points in a pseudorandom order. Mice were allowed to find and escape onto the submerged platform. If they failed to find the platform within 60 sec, they were manually guided to the platform and allowed to remain for 10 sec. Mice were trained for four consecutive days. The probe trial was assessed 24 hours after the last training session and consisted of a 60 sec free swim in the pool without the platform. Data were collected using the ANY-maze behavioral tracking software (Stoelting).
Rotarod
The test was performed in a mouse rotarod apparatus (Insight Ltda., Brazil), as previously described. Briefly, mice were individually placed in the apparatus floor for 3 minutes followed by a 2-min habituation session to the cylinder rod. The test phase consisted of tree trials (inter-trial interval, 60 min) in which animals were placed on the top of the rod rotating at increasing speed (minimal speed 16 rpm, maximal speed 36 rpm with acceleration rate 3.7 rpm). Latency to fall was recorded for a 5 min period, and results are expressed as average latency in the test phase.
Tissue collection
Animals were anesthetized (90 mg/kg ketamine and 4.5 mg/kg xylazine, i.p.) before perfusion with ice-cold PBS at different time points. Hippocampal tissues were dissected immediately after perfusion, frozen in liquid nitrogen and stored at −80°C before RNA extraction. For immunofluorescence studies, perfusion was performed with 4% PFA, and brains were fixed for 24 h before paraffin processing. To evaluate the serum levels of cytokine, whole blood was collected, aliquoted, and left at room temperature (RT) to be processed at different time points.112
Cell culture and treatments
Primary neuronal cortical culture was prepared as previously described in Diniz 2012.113 Briefly, dissociated cerebral cortices were harvested from embryonic day 14 Swiss mice and cultured in neurobasal medium (Invitrogen) supplemented with B-27, penicillin, streptomycin, l-glutamine, fungizone and cytosine arabinose, and maintained at 37°C with 5% CO2. Neurons were seeded at a density of 50.000–150.000 neurons/well on a 13 mm diameter poly-D-lysine-coated well (10μg/mL; Sigma). One week after dissociation, neuronal cell cultures were treated with PBS or Spike protein (1 μg/mL) for 24 h. Later, cells were fixed in 4% PFA, 6% sucrose in PBS for 10 min before immunocytochemistry assay.
The murine BV-2 cell line was cultured in DMEM supplemented with 10% FBS, and 1% streptomycin/penicillin, and seeded at a density of 100.000 cells/well on a 13 mm diameter poly-D-lysine-coated well. Next, cells were treated with PBS or Spike protein (1 μg/mL) for 24 h and fixed as mentioned above.
RNA extraction and qPCR
RNA extraction of hippocampal tissue and cell cultures was performed using Trizol® reagent (Invitrogen), in accordance with manufacturer’s instructions. Sample concentration and purity was assessed using a NanoDrop 1000 spectrophotometer (ThermoScientific). Only preparations with absorbance ratios >1.8 and no signs of RNA degradation were used. One μg of total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), according to the manufacturer's instructions. qPCR was performed using a QuantStudio 5 PCR system (Applied Biosystems) with reactions performed in triplicate. Briefly, qPCRs were run using Power SYBR Green PCR Master Mix (Life Technologies), and 10 ng of template cDNA in a 10 μL reaction volume. The primers used are listed in Table S2. Cycle threshold (Ct) values were normalized to a control gene (β-actin) and analyzed using the ΔΔCt method to generate fold change values (2–ΔΔCT).114
Immunofluorescence assay
Slides containing sections from the dorsal hippocampus (Bregma −1.46 to −1.94mm) of mice were deparaffinized, and antigen retrieval was carried out by incubation in citrate buffer solution (pH 6.0) at 95°C for 40 min. Afterwards, permeabilization was performed with 0.025% Triton in PBS, followed by incubation with blocking buffer (PBS containing 0.025% Triton, 3% BSA, and 5% normal goat serum) for 2 h. Next, slides were incubated overnight with primary antibodies against Iba-1 (WAKO; 1:800#019-19741), TMEM119 (Abcam. 1:50#210405) synaptophysin (Vector Laboratories; 1:200 #S285), Homer-1 (Abcam; 1:100 #184955), or GFAP (Sigma; 1:500 #G3893). For analyze of Iba-1, GFAP and TMEM119 in the mice hippocampus, four confocal Z-stack images of each mice hippocampal section (CA3 and DG) were acquired using a Leica TSE-SPE3 confocal microscope (0,35μm/z-stack) or Zeiss Cell Observer Spining Disk Confocal microscope at 630× magnification. Each image comprised 9–12 (0.35μm/z-stack) optical planes, three of which were analyzed independently as previously described115 V. Optical density threshold that best discriminated staining from background was defined using NIH ImageJ and total pixel intensity was determined for each image and data are expressed as integrated optical density. For synaptic puncta, each z-stack was individually analyzed using the ImageJ v1.53 plugin SynQuant automated synapse counter. Microglia morphology was assessed evaluating the number of branches emanating from their soma.116 Briefly, type I and type II cells were described as surveillant microglia and present smaller soma and less than 5 thin branches. Type III, IV and V microglia are characterized as reactive microglia, and present more than 4 branches, and thicker branches and bigger soma are observed.116 For astrocytes morphological analyses, sets of images were acquired using 400× magnification and were segmented using threshold tool (fixed parameters) on FIJI ImageJ followed by sholl analysis, set to form concentric circles within the center of astrocytes with 5μm radius. Ten cells were analyzed per mice and only cells with discernible processes were included. To determine synapse engulfment by microglia, fields containing 3–6 Iba-1 positive cells were chosen and Iba-1/Syp colocalization was normalized by the number of Iba positive cells present in the field. Quantitative colocalization of post- (Homer-1) and presynaptic (synaptophysin) markers, or Iba-1 and synaptophysin in control mice were used to normalize the ratio of preserved synaptic puncta and synaptic engulfment, respectively. In graphics, bars represent means ± SEM and each data point represent average of images analyzed from individual mice.
For immunocytochemistry, wells were washed three times with PBS, and incubated for 1 h with blocking buffer, followed by overnight incubation with primary antibodies against β3-tubulin (Promega; 1:1000 #G712A), Iba-1 (1:1000), synaptophysin or Homer-1. For visualization, sections or wells were incubated with AlexaFluor 488- or 546-conjugated secondary antibodies for 2 h at room temperature, washed with PBS and mounted in Fluoroshield with DAPI (Sigma). The β3-tubulin immunoreactivity in cortical neurons, Iba-1 immunoreactivity in BV-2 cells, as well as microglia density and morphology in Iba-1 immunostained brain sections were photographed using a Slight DS-5-M1 digital camera (Nikon,Melville,NY) connected to an epifluorescence Nikon Eclipse 50i light microscope, under a 20 or 40× objective. Cultured cortical neurons optical density for β3-tubulin and Iba-1 was measured using ImageJ v1.53 and normalized by total DAPI stains. Pyknotic nuclei were analyzed using DAPI stains with 400× magnification and normalized by the total DAPI-stained nuclei observed.
FluoroJade B (FJ) staining
FJ histochemistry was used as indicative of neuronal degeneration. Paraffin-embedded brain tissue sections were sequentially immersed in 100% ethanol for 3 min, 70% ethanol for 1 min, and distilled water for 1 min. Sections were then immersed in 0.06% potassium permanganate for 10 min (to suppress endogenous background signal), and washed with distilled water for 1 min. FJ B staining solution (10 mL of 0.01% FJ aqueous solution added to 90 mL of 0.1% acetic acid in distilled water) was added for 30 min. After staining, sections were rinsed three times in distilled water. Excess water was drained off, and slides were coverslipped with Entellan® mounting medium (Sigma-Aldrich). Sections comprising the hippocampus were imaged on epifluorescence microscopes (Nikon Eclipse 50i) at 200x magnification. Positive neurodegeneration staining controls consisted of sections from the hippocampus of a mouse injected i.c.v. with 36.8 nmol quinolinic acid and euthanized 24 h thereafter.
Enzyme-linked immunosorbent assay (ELISA)
For cytokine measurements, hippocampus was homogenized in cold RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris Base, 2 mM PMSF, pH 8), and supernatant was collected after centrifugation at 14,000 g for 10 min at 4°C. Protein concentration was determined using the BCA Protein Assay (Thermo Scientific). Samples diluted 1:10 in the RIPA buffer were used for the detection of TNF (BD Biosciences) and IL1β (R&D Systems) by ELISA according to manufacturer's instructions. Results were expressed as pg/μg protein.
Neurofilament light chain (NFL) measurements
Mouse plasma NFL concentration was measured in triplicate using ultra-sensitive single molecule array (Simoa) technique on the Simoa SR-X™ Analyzer, using Simoa NF-Light Advantage according to the manufacturer's instructions (Quanterix). Briefly, plasma samples were thawed at room temperature for one hour and then centrifuged at 10,000 RCF for 5 min at 24°C. Samples were diluted 1:4 with sample diluent and applied to the plate in duplicate. Paramagnetic beads coated with capture anti-NFL were incubated with a biotinylated anti-NFL detection antibody, followed by incubation with a streptavidin-β-galactosidase complex. A fluorescent signal proportional to the concentration of NFL was generated after the addition of the substrate resorufin β-D-galactopyranoside. Controls were used to validate the detection limit of 0.0552 pg/mL. All coefficients of variance (CVs) of duplicate measurements were below 20%.
Genotyping and functional analysis
Genotyping
Two promoter region TLR4 SNPs, previously implicated in inflammatory and/or neurological disease, were genotyped. Blood samples were collected and centrifuged at 1.500 g at 4°C for 15 min to separate the buffy coat from plasma. Genomic DNA (gDNA) was extracted using the PureLink Genomic DNA Mini Kit (ThermoFisher Scientific). The quality of the gDNA was determined using NanoDrop 2000 (ThermoFisher Scientific) followed by quantification using the Qubit dsDNA HS Assay Kit (ThermoFisher Scientific) and Qubit Fluorometer 3.0 (Thermo Fisher Scientific). The TLR4 -2604G>A (rs10759931) and TLR4 - 2272A>G (rs2737190) variants were genotyped with allelic discrimination using TaqMan qPCR system (ThermoFisher Scientific). The probes were produced by Applied Biosystems [rs10759931 (C___2704046_10) and rs2737190 (C___2704047_10)]. Briefly, genotyping was performed in a 20 μL reaction mixture containing 10 ng DNA, TaqMan Universal PCR Master Mix (1X), Probe TaqMan Gene Expression Assay (1X), and DNAse-free water for the final volume. The reaction was carried out in the following conditions: an UNG incubation step of 2 min at 50°C, polymerase activation for 10 min at 95°C, followed by 40 cycles of 15 s at 95°C for denaturation and 60 s at 60°C for annealing/extension. The amplification and reading of the plates were performed in the QuantStudio 5 Real-Time PCR System (Applied Biosystems). In order to represent the number of minor allele in the genotype, inheritance model 0, 1, and 2 (AA, Aa, and aa) were applied.
Functional analysis
To understand the difference in expression between the main genotypes of SNP rs10759931, we performed a functional analysis. Randomly, we selected 9 patients with GG and 7 patients with GA genotypes. In total, 15 mL of the peripheral blood sample was collected in EDTA tubes to generate peripheral blood mononuclear cells (PBMCs). Briefly, PBMCs were isolated using density gradient centrifugation using Ficoll-Hypaque according to Helgason 2004.117 The PBMCs were cultured in RPMI-1640 Medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% inactivated autologous serum and 1% of antibiotic. 106 cells were placed into each well of a 6-well plate and stimulated with 1 μg of Spike protein for 24 hours and then the analysis of TLR4 expression was performed by qPCR.
Illustrations
Illustrations in Figures 1, 3 and 4 were created using MindtheGraph (www.mindthegraph.com; under FLFD subscription) and subsequently modified (free culture Creative Commons license).
Quantification and statistical analysis
The software Prism v8 (GraphPad) was used for all statistical tests, and values of p ≤ 0.05 were considered statistically significant. Student’s t-test was applied to analyze qPCR, ELISA, NFL measurements and immunohistochemical data when they fit into the normal distribution of the data. Mann–Whitney U test was used for non-normal distributed data. For NOR experiments, data were analyzed using a one-sample Student’s t-test compared to a fixed value of 50%. Kruskal-Wallis test was used for non-normal distributed data. MWM was analyzed using repeated measures or two-way ANOVA followed by Tukey’s test, respectively. Allelic frequencies were determined by direct count of the alleles. Genotypic distributions in Hardy–Weinberg equilibrium were evaluated by two-tailed χ2-test linkage disequilibrium (LD) were reproduced by Linkage Disequilibrium Calculator - Homo_sapiens (https://grch37.ensembl.org/Homo_sapiens/Tools/LD). The significant differences in allelic and genotypic frequencies were evaluated by Fisher’s exact test and two-tailed χ2-test. Using STATA software (version 71.0; Stata Corporation, College Station, Texas, USA), logistic regression analysis with offset variables was used to control the confounding effects of different times in the SDMT. Comparison of mRNA levels of different SNP rs10759931 genotypes was carry out by exact parametric Student’s t-test.
Acknowledgments
The authors thank Luis F. Fragoso and Ana Claudia Rangel for technical support, Prof. Leda R. Castilho and her team at the Cell Culture Engineering Laboratory (LECC) of COPPE/UFRJ for providing the trimeric Spike protein in the prefusion conformation, and Prof. João B. Calixto (Centro de Inovação e Ensaios Pré-clínicos [CIENP]), Prof. João B. Teixeira da Rocha (Federal University of Santa Maria), and Prof. Andreza F. De Bem (Brazilian Federal University) for thoughtful discussions and critical reading of the manuscript. This work was supported by the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ; to F.L.F.-D., G.G.F., L.S.A., L.C.C., S.M.B.A., L.E.B.S., J.L.S., R.d.C., J.R.C., A.T.D.P., S.V.A.-L., G.F.P., and C.P.F.), the Instituto Nacional de Ciência e Tecnologia (INCT) – Inovação em Medicamentos e Identificação de Novos Alvos Terapêuticos – INCT-INOVAMED 465430/2014-7, Brazil (to R.d.C., G.F.P., and C.P.F.), INCT de Biologia Estrutural e Bioimagem – INBEB, Brazil (to J.L.S. and A.T.D.P.), Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq, Brazil (to G.G.F., H.P.M.-A., L.E.B.S., J.L.S., J.R.C., R.d.C., A.T.D.P., S.V.A.-L., G.F.P., and C.P.F.), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-CAPES, Brazil (to E.V.D.L. and T.N.d.S.), Programa de Pesquisa Para o SUS (PPSUS) E-26/210.456/2021, Ministerio da Saude, Brazil, and Instituto D′or de Pesquisa e Ensino, Brazil.
Author contributions
C.P.F., G.F.P., S.V.A.-L., A.T.D.P., J.R.C., R.d.C., and F.L.F.-D. conceived the study. C.P.F., G.F.P., S.V.A.-L., A.T.P., F.L.F.-D., J.R.C., R.d.C., J.L.S., and L.E.B.S. contributed to the experimental design. F.L.F.-D., G.G.F., E.V.D.L., L.S.A., H.P.M.-A., L.C.C., S.M.B.A., and T.N.d.S. performed experiments in mice and analyzed the data. Molecular experiments and ELISA were performed and analyzed by F.L.F.-D., L.C.C., S.M.B.A., and A.T.D.P. Histological and immunohistochemistry/cytochemistry analyses were performed by G.G.F., C.P.F., and E.V.D.L. F.L.F.-D, L.E.B.S., L.R., and G.G.F. performed experiments in cell culture. L.A.D. and A.L.S. performed Simoa experiments. E.G.G., M.B.H., K.L.P., C.C.F.V., and S.V.A.-L. recruited patients, collected clinical information, and performed neuropsychological evaluation. L.A.A.L. performed molecular and serological diagnosis of COVID-19. F.L.F.-D. and E.G.G. carried out genotype analyses. F.L.F.-D., G.G.F., E.G.G., C.P.F., G.F.P., S.V.A.-L., A.T.D.P., J.R.C., R.d.C., and L.E.B.S. contributed to critical analysis of the data. F.L.F.-D., C.P.F., A.T.D.P., and G.F.P. wrote the manuscript. All authors read and approved the final version.
Declaration of interests
The authors declare no competing interests.
Published: February 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112189.
Supplemental information
Data and code availability
-
•
The original data within the paper will be available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information in this paper is available from the lead contact upon requests.
References
- 1.Lamers M.M., Haagmans B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022;20:270–284. doi: 10.1038/s41579-022-00713-0. [DOI] [PubMed] [Google Scholar]
- 2.Wiersinga W.J., Rhodes A., Cheng A.C., Peacock S.J., Prescott H.C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324:782–793. doi: 10.1001/jama.2020.12839. [DOI] [PubMed] [Google Scholar]
- 3.Crook H., Raza S., Nowell J., Young M., Edison P. Long covid-mechanisms, risk factors, and management. BMJ. 2021;374:n1648. doi: 10.1136/bmj.n1648. [DOI] [PubMed] [Google Scholar]
- 4.Mehandru S., Merad M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022;23:194–202. doi: 10.1038/s41590-021-01104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taquet M., Sillett R., Zhu L., Mendel J., Camplisson I., Dercon Q., Harrison P.J. Neurological and psychiatric risk trajectories after SARS-CoV-2 infection: an analysis of 2-year retrospective cohort studies including 1 284 437 patients. Lancet Psychiatr. 2022;9:815–827. doi: 10.1016/S2215-0366(22)00260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceban F., Ling S., Lui L.M.W., Lee Y., Gill H., Teopiz K.M., Rodrigues N.B., Subramaniapillai M., Di Vincenzo J.D., Cao B., et al. Fatigue and cognitive impairment in Post-COVID-19 Syndrome: a systematic review and meta-analysis. Brain Behav. Immun. 2022;101:93–135. doi: 10.1016/j.bbi.2021.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis H.E., Assaf G.S., McCorkell L., Wei H., Low R.J., Re’em Y., Redfield S., Austin J.P., Akrami A. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine. 2021;38:101019. doi: 10.1016/j.eclinm.2021.101019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subramanian A., Nirantharakumar K., Hughes S., Myles P., Williams T., Gokhale K.M., Taverner T., Chandan J.S., Brown K., Simms-Williams N., et al. Symptoms and risk factors for long COVID in non-hospitalized adults. Nat. Med. 2022;28:1706–1714. doi: 10.1038/s41591-022-01909-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song E., Zhang C., Israelow B., Lu-Culligan A., Prado A.V., Skriabine S., Lu P., Weizman O.-E., Liu F., Dai Y., et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021;218:e20202135. doi: 10.1084/jem.20202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etter M.M., Martins T.A., Kulsvehagen L., Pössnecker E., Duchemin W., Hogan S., Sanabria-Diaz G., Müller J., Chiappini A., Rychen J., et al. Severe Neuro-COVID is associated with peripheral immune signatures, autoimmunity and neurodegeneration: a prospective cross-sectional study. Nat. Commun. 2022;13:6777. doi: 10.1038/s41467-022-34068-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Melo G.D., Lazarini F., Levallois S., Hautefort C., Michel V., Larrous F., Verillaud B., Aparicio C., Wagner S., Gheusi G., et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021;13:eabf8396. doi: 10.1126/scitranslmed.abf8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutkai I., Mayer M.G., Hellmers L.M., Ning B., Huang Z., Monjure C.J., Coyne C., Silvestri R., Golden N., Hensley K., et al. Neuropathology and virus in brain of SARS-CoV-2 infected non-human primates. Nat. Commun. 2022;13:1745. doi: 10.1038/s41467-022-29440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bauer L., Laksono B.M., de Vrij F.M.S., Kushner S.A., Harschnitz O., van Riel D. The neuroinvasiveness, neurotropism, and neurovirulence of SARS-CoV-2. Trends Neurosci. 2022;45:358–368. doi: 10.1016/j.tins.2022.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann M., Kleine-weber H., Schroeder S., Mu M.A., Drosten C., Po S., Hoffmann M., Kleine-weber H., Schroeder S., Kru N. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven article SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko C.J., Harigopal M., Gehlhausen J.R., Bosenberg M., McNiff J.M., Damsky W. Discordant anti-SARS-CoV-2 spike protein and RNA staining in cutaneous perniotic lesions suggests endothelial deposition of cleaved spike protein. J. Cutan. Pathol. 2021;48:47–52. doi: 10.1111/cup.13866. [DOI] [PubMed] [Google Scholar]
- 16.Matschke J., Lütgehetmann M., Hagel C., Sperhake J.P., Schröder A.S., Edler C., Mushumba H., Fitzek A., Allweiss L., Dandri M., et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 2020;19:919–929. doi: 10.1016/S1474-4422(20)30308-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Troyer Z., Alhusaini N., Tabler C.O., Sweet T., de Carvalho K.I.L., Schlatzer D.M., Carias L., King C.L., Matreyek K., Tilton J.C. Extracellular vesicles carry SARS-CoV-2 spike protein and serve as decoys for neutralizing antibodies. J. Extracell. Vesicles. 2021;10:e12112. doi: 10.1002/jev2.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhea E.M., Logsdon A.F., Hansen K.M., Williams L.M., Reed M.J., Baumann K.K., Holden S.J., Raber J., Banks W.A., Erickson M.A. The S1 protein of SARS-CoV-2 crosses the blood–brain barrier in mice. Nat. Neurosci. 2021;24:368–378. doi: 10.1038/s41593-020-00771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swank Z., Senussi Y., Manickas-Hill Z., Yu X.G., Li J.Z., Alter G., Walt D.R. Persistent circulating SARS-CoV-2 spike is associated with post-acute COVID-19 sequelae. Clin. Infect. Dis. 2022;76:e487–e490. doi: 10.1093/cid/ciac722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawasaki T., Kawai T. Toll-like receptor signaling pathways. Front. Immunol. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharya M., Sharma A.R., Mallick B., Sharma G., Lee S.-S., Chakraborty C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect. Genet. Evol. 2020;85:104587. doi: 10.1016/j.meegid.2020.104587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choudhury A., Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020;92:2105–2113. doi: 10.1002/jmv.25987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aboudounya M.M., Heads R.J. COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat. Inflamm. 2021;2021:8874339. doi: 10.1155/2021/8874339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank M.G., Nguyen K.H., Ball J.B., Hopkins S., Kelley T., Baratta M.V., Fleshner M., Maier S.F. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: evidence of PAMP-like properties. Brain Behav. Immun. 2022;100:267–277. doi: 10.1016/j.bbi.2021.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olajide O.A., Iwuanyanwu V.U., Adegbola O.D., Al-Hindawi A.A. SARS-CoV-2 spike glycoprotein S1 induces neuroinflammation in BV-2 microglia. Mol. Neurobiol. 2022;59:445–458. doi: 10.1007/s12035-021-02593-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y., Kuang M., Li J., Zhu L., Jia Z., Guo X., Hu Y., Kong J., Yin H., Wang X., You F. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021;31:818–820. doi: 10.1038/s41422-021-00495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J., Wise L., Fukuchi K.-I. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front. Immunol. 2020;11:724. doi: 10.3389/fimmu.2020.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albornoz E.A., Amarilla A.A., Modhiran N., Parker S., Li X.X., Wijesundara D.K., Aguado J., Zamora A.P., McMillan C.L.D., Liang B., et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol. Psychiatr. 2022 doi: 10.1038/s41380-022-01831-0. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirato K., Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. 2021;7:e06187. doi: 10.1016/j.heliyon.2021.e06187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Theobald S.J., Simonis A., Georgomanolis T., Kreer C., Zehner M., Eisfeld H.S., Albert M.-C., Chhen J., Motameny S., Erger F., et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in COVID-19. EMBO Mol. Med. 2021;13:e14150. doi: 10.15252/emmm.202114150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim E.S., Jeon M.-T., Kim K.-S., Lee S., Kim S., Kim D.-G. Spike proteins of SARS-CoV-2 induce pathological changes in molecular delivery and metabolic function in the brain endothelial cells. Viruses. 2021;13:102021. doi: 10.3390/v13102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frere J.J., Serafini R.A., Pryce K.D., Zazhytska M., Oishi K., Golynker I., Panis M., Zimering J., Horiuchi S., Hoagland D.A., et al. SARS-CoV-2 infection in hamsters and humans results in lasting and unique systemic perturbations after recovery. Sci. Transl. Med. 2022;14:eabq3059. doi: 10.1126/scitranslmed.abq3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dinnon K.H., 3rd, Leist S.R., Okuda K., Dang H., Fritch E.J., Gully K.L., De la Cruz G., Evangelista M.D., Asakura T., Gilmore R.C., et al. SARS-CoV-2 infection produces chronic pulmonary epithelial and immune cell dysfunction with fibrosis in mice. Sci. Transl. Med. 2022;14:eabo5070. doi: 10.1126/scitranslmed.abo5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (London, England) 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nalbandian A., Sehgal K., Gupta A., Madhavan M.V., McGroder C., Stevens J.S., Cook J.R., Nordvig A.S., Shalev D., Sehrawat T.S., et al. Post-acute COVID-19 syndrome. Nat. Med. 2021;27:601–615. doi: 10.1038/s41591-021-01283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Figueiredo C.P., Barros-Aragão F.G.Q., Neris R.L.S., Frost P.S., Soares C., Souza I.N.O., Zeidler J.D., Zamberlan D.C., de Sousa V.L., Souza A.S., et al. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 2019;10:3890. doi: 10.1038/s41467-019-11866-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhea E.M., Logsdon A.F., Hansen K.M., Williams L.M., Reed M.J., Baumann K.K., Holden S.J., Raber J., Banks W.A., Erickson M.A. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat. Neurosci. 2021;24:368–378. doi: 10.1038/s41593-020-00771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butowt R., Meunier N., Bryche B., von Bartheld C.S. The olfactory nerve is not a likely route to brain infection in COVID-19: a critical review of data from humans and animal models. Acta Neuropathol. 2021;141:809–822. doi: 10.1007/s00401-021-02314-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meinhardt J., Radke J., Dittmayer C., Franz J., Thomas C., Mothes R., Laue M., Schneider J., Brünink S., Greuel S., et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021;24:168–175. doi: 10.1038/s41593-020-00758-5. [DOI] [PubMed] [Google Scholar]
- 40.Bilinska K., von Bartheld C.S., Butowt R. Expression of the ACE2 virus entry protein in the nervus terminalis reveals the potential for an alternative route to brain infection in COVID-19. Front. Cell. Neurosci. 2021;15:674123. doi: 10.3389/fncel.2021.674123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patrì A., Vargas M., Buonanno P., Annunziata M.C., Russo D., Staibano S., Servillo G., Fabbrocini G. From SARS-CoV-2 hematogenous spreading to endothelial dysfunction: clinical-histopathological study of cutaneous signs of COVID-19. Diagn. Pathol. 2021;16:16. doi: 10.1186/s13000-021-01075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vorhees C.V., Williams M.T. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong S., Beja-Glasser V.F., Nfonoyim B.M., Frouin A., Li S., Ramakrishnan S., Merry K.M., Shi Q., Rosenthal A., Barres B.A., et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colom-Cadena M., Spires-Jones T., Zetterberg H., Blennow K., Caggiano A., DeKosky S.T., Fillit H., Harrison J.E., Schneider L.S., Scheltens P., et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimer's Res. Ther. 2020;12:21. doi: 10.1186/s13195-020-00588-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shives K.D., Tyler K.L., Beckham J.D. Molecular mechanisms of neuroinflammation and injury during acute viral encephalitis. J. Neuroimmunol. 2017;308:102–111. doi: 10.1016/j.jneuroim.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z., Zhong D., Li G. The role of microglia in viral encephalitis: a review. J. Neuroinflammation. 2019;16:76. doi: 10.1186/s12974-019-1443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vasek M.J., Garber C., Dorsey D., Durrant D.M., Bollman B., Soung A., Yu J., Perez-Torres C., Frouin A., Wilton D.K., et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534:538–543. doi: 10.1038/nature18283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B., et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 49.Miron J., Picard C., Lafaille-Magnan M.É., Savard M., Labonté A., Breitner J., Rosa-Neto P., Auld D., Poirier J., PREVENT-AD research group Association of TLR4 with Alzheimer’s disease risk and presymptomatic biomarkers of inflammation. Alzheimers Dement. 2019;15:951–960. doi: 10.1016/j.jalz.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 50.Edén A., Kanberg N., Gostner J., Fuchs D., Hagberg L., Andersson L.-M., Lindh M., Price R.W., Zetterberg H., Gisslén M. CSF biomarkers in patients with COVID-19 and neurologic symptoms: a case series. Neurology. 2021;96:e294–e300. doi: 10.1212/WNL.0000000000010977. [DOI] [PubMed] [Google Scholar]
- 51.Pilotto A., Masciocchi S., Volonghi I., De Giuli V., Caprioli F., Mariotto S., Ferrari S., Bozzetti S., Imarisio A., Risi B., et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) encephalitis is a cytokine release syndrome: evidences from cerebrospinal fluid analyses. Clin. Infect. Dis. 2021;73:e3019–e3026. doi: 10.1093/cid/ciaa1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun B., Tang N., Peluso M.J., Iyer N.S., Torres L., Donatelli J.L., Munter S.E., Nixon C.C., Rutishauser R.L., Rodriguez-Barraquer I., et al. Characterization and biomarker analyses of post-COVID-19 complications and neurological manifestations. Cells. 2021;10:e020386. doi: 10.3390/cells10020386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hay M., Ryan L., Huentelman M., Konhilas J., Hoyer-Kimura C., Beach T.G., Serrano G.E., Reiman E.M., Blennow K., Zetterberg H., Parthasarathy S. Serum neurofilament light is elevated in COVID-19 positive adults in the ICU and is associated with Co-morbid cardiovascular disease, neurological complications, and acuity of illness. Cardiol. Cardiovasc. Med. 2021;5:551–565. doi: 10.26502/fccm.92920221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prudencio M., Erben Y., Marquez C.P., Jansen-West K.R., Franco-Mesa C., Heckman M.G., White L.J., Dunmore J.A., Cook C.N., Lilley M.T., et al. Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID-19. Sci. Transl. Med. 2021;13:eabi7643. doi: 10.1126/scitranslmed.abi7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y.-C., Yip P.-K., Huang Y.-L., Sun Y., Wen L.-L., Chu Y.-M., Chen T.-F. Sequence variants of toll like receptor 4 and late-onset Alzheimer’s disease. PLoS One. 2012;7:e50771. doi: 10.1371/journal.pone.0050771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J.-T., Miao D., Cui W.-Z., Ou J.-R., Tian Y., Wu Z.-C., Zhang W., Tan L. Common variants in toll-like receptor 4 confer susceptibility to Alzheimer’s disease in a Han Chinese population. Curr. Alzheimer Res. 2012;9:458–466. doi: 10.2174/156720512800492495. [DOI] [PubMed] [Google Scholar]
- 57.Wang L.-Z., Yu J.-T., Miao D., Wu Z.-C., Zong Y., Wen C.-Q., Tan L. Genetic association of TLR4/11367 polymorphism with late-onset Alzheimer’s disease in a Han Chinese population. Brain Res. 2011;1381:202–207. doi: 10.1016/j.brainres.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Monje M., Iwasaki A. The neurobiology of long COVID. Neuron. 2022;110:3484–3496. doi: 10.1016/j.neuron.2022.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu E., Xie Y., Al-Aly Z. Long-term neurologic outcomes of COVID-19. Nat. Med. 2022;28:2406–2415. doi: 10.1038/s41591-022-02001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen A.K., Wang X., McCluskey L.P., Morgan J.C., Switzer J.A., Mehta R., Tingen M., Su S., Harris R.A., Hess D.C., Rutkowski E.K. Neuropsychiatric sequelae of long COVID-19: pilot results from the COVID-19 neurological and molecular prospective cohort study in Georgia, USA. Brain Behav. Immun. Health. 2022;24:100491. doi: 10.1016/j.bbih.2022.100491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jacomy H., Fragoso G., Almazan G., Mushynski W.E., Talbot P.J. Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology. 2006;349:335–346. doi: 10.1016/j.virol.2006.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Douaud G., Lee S., Alfaro-Almagro F., Arthofer C., Wang C., McCarthy P., Lange F., Andersson J.L.R., Griffanti L., Duff E., et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature. 2022;604:697–707. doi: 10.1038/s41586-022-04569-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu Y., Li X., Geng D., Mei N., Wu P.-Y., Huang C.-C., Jia T., Zhao Y., Wang D., Xiao A., Yin B. Cerebral micro-structural changes in COVID-19 patients - an MRI-based 3-month follow-up study. EClinicalMedicine. 2020;25:100484. doi: 10.1016/j.eclinm.2020.100484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guedj E., Campion J.Y., Dudouet P., Kaphan E., Bregeon F., Tissot-Dupont H., Guis S., Barthelemy F., Habert P., Ceccaldi M., et al. (18 F-FDG brain PET hypometabolism in patients with long COVID. Eur. J. Nucl. Med. Mol. Imag. 2021;48:2823–2833. doi: 10.1007/s00259-021-05215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng G.-P., Feng Z., He F.-P., Chen Z.-Q., Liu X.-Y., Liu P., Luo B.-Y. Correlation of hippocampal volume and cognitive performances in patients with either mild cognitive impairment or Alzheimer’s disease. CNS Neurosci. Ther. 2015;21:15–22. doi: 10.1111/cns.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ystad M.A., Lundervold A.J., Wehling E., Espeseth T., Rootwelt H., Westlye L.T., Andersson M., Adolfsdottir S., Geitung J.T., Fjell A.M., et al. Hippocampal volumes are important predictors for memory function in elderly women. BMC Med. Imag. 2009;9:17. doi: 10.1186/1471-2342-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mosconi L., De Santi S., Li J., Tsui W.H., Li Y., Boppana M., Laska E., Rusinek H., de Leon M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging. 2008;29:676–692. doi: 10.1016/j.neurobiolaging.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nuovo G.J., Magro C., Shaffer T., Awad H., Suster D., Mikhail S., He B., Michaille J.-J., Liechty B., Tili E. Endothelial cell damage is the central part of COVID-19 and a mouse model induced by injection of the S1 subunit of the spike protein. Ann. Diagn. Pathol. 2021;51:151682. doi: 10.1016/j.anndiagpath.2020.151682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh J., Cho W.-H., Barcelon E., Kim K.H., Hong J., Lee S.J. SARS-CoV-2 spike protein induces cognitive deficit and anxiety-like behavior in mouse via non-cell autonomous hippocampal neuronal death. Sci. Rep. 2022;12:5496. doi: 10.1038/s41598-022-09410-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peng L., Bestard-Lorigados I., Song W. The synapse as a treatment avenue for Alzheimer’s Disease. Mol. Psychiatr. 2022;27:2940–2949. doi: 10.1038/s41380-022-01565-z. [DOI] [PubMed] [Google Scholar]
- 71.Bossy-Wetzel E., Schwarzenbacher R., Lipton S.A. Molecular pathways to neurodegeneration. Nat. Med. 2004;10:S2–S9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 72.Volmer R., Prat C.M.A., Le Masson G., Garenne A., Gonzalez-Dunia D. Borna disease virus infection impairs synaptic plasticity. J. Virol. 2007;81:8833–8837. doi: 10.1128/JVI.00612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oliveira A.O., Malwade S., de Sousa N.R., Goparaju S.K., Lekander J.G., Orhan F., Steponaviciute L., Schalling M., Sheridan S.D., Perlis R.H., et al. SARS-CoV-2 promotes microglial synapse elimination in human brain organoids. bioRxiv. 2022 doi: 10.1101/2021.07.07.451463. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang A.C., Kern F., Losada P.M., Agam M.R., Maat C.A., Schmartz G.P., Fehlmann T., Stein J.A., Schaum N., Lee D.P., et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature. 2021;595:565–571. doi: 10.1038/s41586-021-03710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yuan A., Sershen H., Basavarajappa B.S., Kumar A., Hashim A., Berg M., Lee J.H., Sato Y., Rao M.V., Mohan P.S., et al. Neurofilament subunits are integral components of synapses and modulate neurotransmission and behavior in vivo. Mol. Psychiatr. 2015;20:986–994. doi: 10.1038/mp.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bridel C., van Wieringen W.N., Zetterberg H., Tijms B.M., Teunissen C.E., NFL Group. Alvarez-Cermeño J.C., Andreasson U., Axelsson M., Bäckström D.C., et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol. 2019;76:1035–1048. doi: 10.1001/jamaneurol.2019.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kanberg N., Ashton N.J., Andersson L.-M., Yilmaz A., Lindh M., Nilsson S., Price R.W., Blennow K., Zetterberg H., Gisslén M. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology. 2020;95:e1754–e1759. doi: 10.1212/WNL.0000000000010111. [DOI] [PubMed] [Google Scholar]
- 78.De Lorenzo R., Loré N.I., Finardi A., Mandelli A., Cirillo D.M., Tresoldi C., Benedetti F., Ciceri F., Rovere-Querini P., Comi G., et al. Blood neurofilament light chain and total tau levels at admission predict death in COVID-19 patients. J. Neurol. 2021;268:4436–4442. doi: 10.1007/s00415-021-10595-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dheen S.T., Kaur C., Ling E.-A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007;14:1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- 80.Vivanti A.J., Vauloup-Fellous C., Prevot S., Zupan V., Suffee C., Do Cao J., Benachi A., De Luca D. Transplacental transmission of SARS-CoV-2 infection. Nat. Commun. 2020;11:3572. doi: 10.1038/s41467-020-17436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chhatbar C., Prinz M. The roles of microglia in viral encephalitis: from sensome to therapeutic targeting. Cell. Mol. Immunol. 2021;18:250–258. doi: 10.1038/s41423-020-00620-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thorne L.G., Bouhaddou M., Reuschl A.-K., Zuliani-Alvarez L., Polacco B., Pelin A., Batra J., Whelan M.V.X., Hosmillo M., Fossati A., et al. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature. 2022;602:487–495. doi: 10.1038/s41586-021-04352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schartz N.D., Tenner A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflammation. 2020;17:354. doi: 10.1186/s12974-020-02024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schafer D.P., Lehrman E.K., Kautzman A.G., Koyama R., Mardinly A.R., Yamasaki R., Ransohoff R.M., Greenberg M.E., Barres B.A., Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Severance E.G., Gressitt K.L., Buka S.L., Cannon T.D., Yolken R.H. Maternal complement C1q and increased odds for psychosis in adult offspring. Schizophr. Res. 2014;159:14–19. doi: 10.1016/j.schres.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han Y.W., Choi J.Y., Uyangaa E., Kim S.B., Kim J.H., Kim B.S., Kim K., Eo S.K. Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog. 2014;10:e1004319. doi: 10.1371/journal.ppat.1004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sabouri A.H., Marcondes M.C.G., Flynn C., Berger M., Xiao N., Fox H.S., Sarvetnick N.E. TLR signaling controls lethal encephalitis in WNV-infected brain. Brain Res. 2014;1574:84–95. doi: 10.1016/j.brainres.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tang T., Guo Y., Xu X., Zhao L., Shen X., Sun L., Xie P. BoDV-1 infection induces neuroinflammation by activating the TLR4/MyD88/IRF5 signaling pathway, leading to learning and memory impairment in rats. J. Med. Virol. 2021;93:6163–6171. doi: 10.1002/jmv.27212. [DOI] [PubMed] [Google Scholar]
- 89.Cui W., Sun C., Ma Y., Wang S., Wang X., Zhang Y. Inhibition of TLR4 induces M2 microglial polarization and provides neuroprotection via the NLRP3 inflammasome in Alzheimer’s disease. Front. Neurosci. 2020;14:444. doi: 10.3389/fnins.2020.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Balducci C., Frasca A., Zotti M., La Vitola P., Mhillaj E., Grigoli E., Iacobellis M., Grandi F., Messa M., Colombo L., et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav. Immun. 2017;60:188–197. doi: 10.1016/j.bbi.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 91.Khan K., Khan S.A., Jalal K., Ul-Haq Z., Uddin R. Immunoinformatic approach for the construction of multi-epitopes vaccine against omicron COVID-19 variant. Virology. 2022;572:28–43. doi: 10.1016/j.virol.2022.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhong Q., Zou Y., Liu H., Chen T., Zheng F., Huang Y., Chen C., Zhang Z. Toll-like receptor 4 deficiency ameliorates β2-microglobulin induced age-related cognition decline due to neuroinflammation in mice. Mol. Brain. 2020;13:20. doi: 10.1186/s13041-020-0559-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Okun E., Griffioen K.J., Lathia J.D., Tang S.-C., Mattson M.P., Arumugam T.V. Toll-like receptors in neurodegeneration. Brain Res. Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hajishengallis G., Lambris J.D. More than complementing Tolls: complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol. Rev. 2016;274:233–244. doi: 10.1111/imr.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xin Y.-R., Jiang J.-X., Hu Y., Pan J.-P., Mi X.-N., Gao Q., Xiao F., Zhang W., Luo H.-M. The immune system drives synapse loss during lipopolysaccharide-induced learning and memory impairment in mice. Front. Aging Neurosci. 2019;11:279. doi: 10.3389/fnagi.2019.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosa J.M., Farré-Alins V., Ortega M.C., Navarrete M., Lopez-Rodriguez A.B., Palomino-Antolín A., Fernández-López E., Vila-Del Sol V., Decouty C., Narros-Fernández P., et al. TLR4 pathway impairs synaptic number and cerebrovascular functions through astrocyte activation following traumatic brain injury. Br. J. Pharmacol. 2021;178:3395–3413. doi: 10.1111/bph.15488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Semlali A., Al Mutairi M., Oqla Alanazi I., Awad Aljohi H., Reddy Parine N., Alhadheq A., Al-Jafari A.A., Mobeirek A.F., Al Amri A., Shaik J.P., et al. Toll-like receptor 4 polymorphisms in Saudi population with cardiovascular diseases. Mol. Genet. genomic Med. 2019;7:e852. doi: 10.1002/mgg3.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh K., Kant S., Singh V.K., Agrawal N.K., Gupta S.K., Singh K. Toll-like receptor 4 polymorphisms and their haplotypes modulate the risk of developing diabetic retinopathy in type 2 diabetes patients. Mol. Vis. 2014;20:704–713. [PMC free article] [PubMed] [Google Scholar]
- 99.Song J., Kim D.Y., Kim C.S., Kim H.J., Lee D.H., Lee H.M., Ko W., Lee G. The association between Toll-like receptor 4 (TLR4) polymorphisms and the risk of prostate cancer in Korean men. Cancer Genet. Cytogenet. 2009;190:88–92. doi: 10.1016/j.cancergencyto.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 100.Kerkhof M., Postma D.S., Brunekreef B., Reijmerink N.E., Wijga A.H., de Jongste J.C., Gehring U., Koppelman G.H. Toll-like receptor 2 and 4 genes influence susceptibility to adverse effects of traffic-related air pollution on childhood asthma. Thorax. 2010;65:690–697. doi: 10.1136/thx.2009.119636. [DOI] [PubMed] [Google Scholar]
- 101.Ferronato S., Gomez-Lira M., Menegazzi M., Diani E., Olivato S., Sartori M., Scuro A., Malerba G., Pignatti P.F., Romanelli M.G., Mazzucco S. Polymorphism -2604G>A variants in TLR4 promoter are associated with different gene expression level in peripheral blood of atherosclerotic patients. J. Hum. Genet. 2013;58:812–814. doi: 10.1038/jhg.2013.98. [DOI] [PubMed] [Google Scholar]
- 102.Hampshire A., Chatfield D.A., MPhil A.M., Jolly A., Trender W., Hellyer P.J., Giovane M.D., Newcombe V.F.J., Outtrim J.G., Warne B., et al. Multivariate profile and acute-phase correlates of cognitive deficits in a COVID-19 hospitalised cohort. EClinicalMedicine. 2022;47:101417. doi: 10.1016/j.eclinm.2022.101417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Graham E.L., Clark J.R., Orban Z.S., Lim P.H., Szymanski A.L., Taylor C., DiBiase R.M., Jia D.T., Balabanov R., Ho S.U., et al. Persistent neurologic symptoms and cognitive dysfunction in non-hospitalized Covid-19 “long haulers”. Ann. Clin. Transl. Neurol. 2021;8:1073–1085. doi: 10.1002/acn3.51350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Manukyan P., Deviaterikova A., Velichkovsky B.B., Kasatkin V. The impact of mild COVID-19 on executive functioning and mental health outcomes in young adults. Healthcare (Basel) 2022;10 doi: 10.3390/healthcare10101891. 1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hellmuth J., Barnett T.A., Asken B.M., Kelly J.D., Torres L., Stephens M.L., Greenhouse B., Martin J.N., Chow F.C., Deeks S.G., et al. Persistent COVID-19-associated neurocognitive symptoms in non-hospitalized patients. J. Neurovirol. 2021;27:191–195. doi: 10.1007/s13365-021-00954-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blomberg B., Mohn K.G.-I., Brokstad K.A., Zhou F., Linchausen D.W., Hansen B.-A., Lartey S., Onyango T.B., Kuwelker K., Sævik M., et al. Long COVID in a prospective cohort of home-isolated patients. Nat. Med. 2021;27:1607–1613. doi: 10.1038/s41591-021-01433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ayoubkhani D., Khunti K., Nafilyan V., Maddox T., Humberstone B., Diamond I., Banerjee A. Post-covid syndrome in individuals admitted to hospital with covid-19: retrospective cohort study. BMJ. 2021;372:n693. doi: 10.1136/bmj.n693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Apple A.C., Oddi A., Peluso M.J., Asken B.M., Henrich T.J., Kelly J.D., Pleasure S.J., Deeks S.G., Allen I.E., Martin J.N., et al. Risk factors and abnormal cerebrospinal fluid associate with cognitive symptoms after mild COVID-19. Ann. Clin. Transl. Neurol. 2022;9:221–226. doi: 10.1002/acn3.51498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alvim R.G.F., Lima T.M., Rodrigues D.A.S., Marsili F.F., Bozza V.B.T., Higa L.M., Monteiro F.L., Abreu D.P.B., Leitão I.C., Carvalho R.S., et al. From a recombinant key antigen to an accurate, affordable serological test: lessons learnt from COVID-19 for future pandemics. Biochem. Eng. J. 2022;186:108537. doi: 10.1016/j.bej.2022.108537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Giménez-Garzó C., Fiorillo A., Ballester-Ferré M.P., Gallego J.-J., Casanova-Ferrer F., Urios A., Benlloch S., Martí-Aguado D., San-Miguel T., Tosca J., et al. A new score unveils a high prevalence of mild cognitive impairment in patients with nonalcoholic fatty liver disease. J. Clin. Med. 2021;10:2806. doi: 10.3390/jcm10132806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Greenfield E.A. Sampling and preparation of mouse and rat serum. Cold Spring Harb. Protoc. 2017;2017 doi: 10.1101/pdb.prot100271. pdb.prot100271. [DOI] [PubMed] [Google Scholar]
- 113.Diniz L.P., Almeida J.C., Tortelli V., Vargas Lopes C., Setti-Perdigão P., Stipursky J., Kahn S.A., Romão L.F., de Miranda J., Alves-Leon S.V., et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor β signaling through modulation of D-serine levels in cerebral cortex neurons. J. Biol. Chem. 2012;287:41432–41445. doi: 10.1074/jbc.M112.380824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 115.Bellesi M., de Vivo L., Chini M., Gilli F., Tononi G., Cirelli C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J. Neurosci. 2017;37:5263–5273. doi: 10.1523/JNEUROSCI.3981-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lopez-Rodriguez A.B., Siopi E., Finn D.P., Marchand-Leroux C., Garcia-Segura L.M., Jafarian-Tehrani M., Viveros M.-P. CB1 and CB2 cannabinoid receptor antagonists prevent minocycline-induced neuroprotection following traumatic brain injury in mice. Cerebr. Cortex. 2015;25:35–45. doi: 10.1093/cercor/bht202. [DOI] [PubMed] [Google Scholar]
- 117.Helgason C.D., Miller C.L., Helgason C.D. 2004. Basic Cell Culture Protocols. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The original data within the paper will be available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information in this paper is available from the lead contact upon requests.