

Abstract
The effects of exposure to Myclobutanil, a triazole fungicide, on the development and progression of nonalcoholic fatty liver disease (NAFLD) are unclear, but activation of nuclear receptors (NRs) is a known mechanism of azole-induced liver toxicity. Farnesoid X receptor (FXR) is a NR and is highly expressed in the liver and intestine. Activation of FXR tightly regulates bile acid (BA), lipid and glucose homeostasis, and inflammation partly through the induction of fibroblast growth factor 15 (FGF15; human ortholog FGF19). FXR activation is downregulated during NAFLD and agonists are currently being explored as potential therapeutic strategy. In this study, we aimed to clarify the effects of Myclobutanil exposure on FXR activation and NAFLD development. Reporter assay showed Myclobutanil treatment, following FXR activation with potent FXR agonist (GW4064), resulted in a dose-dependent decrease of FXR activity. Furthermore, a 10-day study in male mice demonstrated that cotreatment with Myclobutanil led to an 80% reduction of GW4064-induced ileal expression of Fgf15. In a diet-induced NAFLD study, low-fat diet (LFD) fed mice administered myclobutanil displayed decreased FXR activity in the liver and ileum, while high-fat-high-sugar-diet (HFHSD) fed mice showed an increase in hepatic FXR activity and an induction of target genes regulated by constitutive androstane receptor and/or pregnane X receptor. Our work demonstrates Myclobutanil inhibits FXR activity and modulates FXR activity differentially in mice fed LFD or HFHSD. Our studies suggest the importance of understanding how Myclobutanil could contribute to BA dysregulation in disease states such as NAFLD.
Keywords: environmental chemicals, liver, nuclear receptors, nonalcoholic fatty liver disease, pesticides
Triazoles are an emerging class of environmental pollutants that have been detected worldwide and are of concern due to their persistence in the environment and endocrine-disrupting potential (Joint FAO/WHO Meeting on Pesticide Residues (JMPR), Standards & Scientific Advice on Food Nutrition, 2015). Triazoles are broad-spectrum, systemic fungicides that generally contain a 1,2,3-triazole moiety (Schermerhorn et al., 2005). Myclobutanil, the approved common name for (R, S)-2-(4-chlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)hexanenitrile by the International Organization for Standardization, is a systemic chiral fungicide belonging to the triazole chemical family (Joint FAO/WHO Meeting on Pesticide Residues (JMPR), Standards & Scientific Advice on Food Nutrition, 2015; Sun et al., 2014). The mode of action of Myclobutanil is disruption of fungal membrane function, specifically, through the inhibition of sterol 14-demethylase enzyme which produces ergosterol, a vital component necessary for sterol biosynthesis (Georgopapadakou, 1998; Lewis et al., 2016; Yan et al., 2014). Myclobutanil is widely used in the USA for its protective efficacy against powdery mildew of cereal, fruits, and vegetables (Di Filippo et al., 2018; Günther, 2018; Yan et al., 2014). The World Health Organization (WHO) identified a no observed adverse effect level (NOAEL) and low adverse effect level (LOAEL) for Myclobutanil as 13.7 and 70.2 mg/kg/day, respectively (Goldman, 1986; Joint FAO/WHO Meeting on Pesticide Residues (JMPR), Standards & Scientific Advice on Food Nutrition, 2015).
Although animal toxicities associated with Myclobutanil are known to include adverse effects on the liver, kidneys, and the development of male reproductive organs (Günther, 2018), the effects and impact of exposure to known hepatotoxic triazole fungicides, like Myclobutanil, warrant further investigation. This is because there is compelling evidence in mice that exposure induces transcription of genes involved in xenobiotic metabolism, oxidative stress response genes, inflammation, and cell death (Martin et al., 2007; Stellavato et al., 2016). Furthermore, current guidance on exposure limits for consumers and occupational health limits may not be sensitive enough.
Prior studies have shown that Myclobutanil interacts with several transcription factor mediated pathways in the liver. Evidence has been presented that this compound may activate xenobiotic nuclear receptors (NRs), including constitutive androstane receptor (CAR), pregnane X receptor (PXR), and/or nuclear factor erythroid 2-related factor 2 (Nrf2) pathway. For example, wild-type mice administered Myclobutanil at 100, 500, or 2000 ppm for 4, 30, or 90 days showed potential activation of CAR (Lake, 2018; Oshida et al., 2015). A toxicogenomic study evaluated the genomic signatures associated with hepatocellular toxicity, hepatic necrosis, and effects on lipid and cholesterol metabolism of Myclobutanil at 300 mg/kg/day for 1, 3, and 5 days found increases in genomic signatures predicted to cause hepatocellular toxicity (ALT elevation), hepatic necrosis and cholesterol-related signatures (Martin et al., 2007). While these observations may not be indicative of effects on humans, it is still worth investigating because alterations in expression of metabolizing enzymes may interfere with concurrent xenobiotic metabolism.
Farnesoid X receptor (FXR) is a NR activated by bile acids (BAs) that serve as endogenous ligands. FXR is highly expressed in the liver and intestines to regulate BA synthesis, transport, metabolism, and detoxification (Kong et al., 2012; Li et al., 2010; Parks et al., 1999; Thomas et al., 2010; Zhan et al., 2014). FXR is also a pivotal regulator and checkpoint for genes and pathways that regulate and maintain lipid, glucose and energy metabolism, metabolic homeostasis, and inflammation. BAs are the end product of cholesterol catabolism. They are synthesized by 2 main pathways, the predominant classical pathway catalyzed by cholesterol 7α-hydroxylase (CYP7A1), and the alternative pathway catalyzed by sterol 27-hydroxylase (CYP27A1). BA synthesis is regulated by a gut-liver negative feedback loop initiated in the distal intestine where FXR activation induces an endocrine fibroblast growth factor 19 (FGF19/15; human vs mouse) that circulates back to the liver via portal vein to activate fibroblast growth factor receptor 4 (FGFR4)/β-klotho complex. This interaction then leads to the activation of extracellular signal-regulated kinase1/2 (ERK1/2) and c-Jun N-terminal kinase 1/2 pathways to repress the expression of genes involved in classical pathway of BA synthesis.
FXR expression and function have been shown to be downregulated during nonalcoholic steatohepatitis (NASH) development (Armstrong and Guo, 2017; Bjursell et al., 2013; Wang et al., 2008; Zhang et al., 2009a, b). Currently, FXR activation is being explored as a therapeutic treatment of NASH. NASH is a severe disease state within the progression of nonalcoholic fatty liver disease (NAFLD) that is characterized by macrovesicular steatosis, inflammation, and fibrosis in the liver. NASH diagnosis relies on an invasive liver biopsy. To date, there are no FDA-approved therapeutics and the recommended course of treatment is change of lifestyle. Approximately, 24% of U.S. adults are currently living with NAFLD, and it is estimated 1.5–6.5% of U.S. adults have NASH as the global burden continues to rise (Spengler and Loomba, 2015; Younossi et al., 2016). NASH disease etiology is multifactorial and exposure to environmental chemicals has been shown to be a significant contributor to NASH development (Cave et al., 2007). Exposure to triazole fungicides in the context of NASH has not been studied in vivo; however, they have been found to induce lipid accumulation through the induction of de novo lipogenesis and impaired beta-oxidation, oxidative stress, and cell death in models of NAFLD in HepG2 cells (Kwon et al., 2021; Stellavato et al., 2016).
The current study examines the effects of an acute exposure to Myclobutanil in vitro and in vivo on the liver-gut Fxr-Fgf15 signaling pathway and the development of a diet-induced NAFLD in mice.
Materials and methods
Animal experimentation
Eight-week-old male C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, Maine). To investigate in vivo antagonism of FXR by Myclobutanil, mice were fed chow diet containing vehicle or Myclobutanil at 70 mg/kg/body weight for 10 days, and at 6:00 pm of day 9 and 8:00 am of day 10, mice were cotreated with a synthetic FXR agonist, GW4064, or vehicle, via oral gavage. GW4064 was suspended in vehicle (1% Tween-20 and 1% methylcellulose) and administered to mice at 150 mg/kg. Mice were euthanized 2 h after the second GW4064 treatment and were followed by tissue collection. To determine the effects of Myclobutanil exposure on the development of NAFLD, mice were administered Myclobutanil at 13 mg/kg/day in either control low-fat diet (LFD) or high-fat-high-sugar diet (HFHSD) for 4 or 9 weeks, followed by euthanization, and tissue collection. The justification for Myclobutanil doses used in the 2 animal studies was selected as known NOAEL (13.7 mg/kg/day) and LOAEL (70.2 mg/kg/day) as defined by WHO (Goldman, 1986; Joint FAO/WHO Meeting on Pesticide Residues (JMPR), Standards & Scientific Advice on Food Nutrition, 2015). The diets, LFD (TD.08485) and HFHSD (TD.96121) were purchased from Envigo (Indianapolis, Indiana). The LFD contents are modified to reduce fat, sucrose, and remove cholesterol compared to HFHSD containing 21% milk fat, 1.25% cholesterol, and 20% sucrose. All mice were group-housed and maintained under standard 12-h light/dark cycles. Food and water were provided ad libitum. The experiments performed in this study were approved by the Rutgers Institutional Animal Care and Use Committee.
Chemicals
Myclobutanil CAS 88671-89-0 (>98%) was purchased from Chem Service, Inc. (West Chester, Pennsylvania). GW4064 was obtained from the University of Kansas Chemical Engineering Core Laboratory (Kong et al., 2012).
Cell culture and luciferase reporter assay
HEK293 cells were kindly provided by Dr. Guofeng You (Rutgers University, Piscataway, New Jersey) and were cultured in DMEM with 10% fetal bovine serum, 1% penicillin streptomycin, and maintained at 37°C in 5% CO2. Cells were plated and grown to 80% confluence before transfection was performed as previously described (Li et al., 2010; Williams et al., 2012). Briefly, cells were transfected with 3 µg plasmid per well containing RXRα (CMV-RXR), FXR (CMV-FXR), and Renilla (SV40-Rluc), as well as Shp-luc, containing Farnesoid X Receptor Response Element from the gene encoding the NR small heterodimer partner (SHP, Nr0b2), using Lipofectamine 2000 according to the manufacturer’s protocol (Li et al., 2010; Williams et al., 2012). Eight hours following transfection, cells were treated with 1 μM GW4064 to achieve FXR activation or 0.1% DMSO as a control, 12 h later cells were cotreated with Myclobutanil at increasing concentrations (0, 0.1, 1, 10, or 100 μM). Twenty-four hours later, firefly and renilla luciferase activities were measured using dual-luciferase reporter assay kit (Promega, Madison, Wisconsin). All experiments were performed in 6 well plates and repeated 4 times. The firefly luciferase activity of each well was calculated as the ratio to that of renilla luciferase.
RNA isolation and mRNA quantification
Total RNA was extracted from frozen liver and ileum using the TRIzol reagent (Thermo Fisher Scientific; Waltham, Massachusetts). Reverse transcription was performed to generate cDNA. Relative gene expression was determined by real-time polymerase chain reaction (RT-PCR) by SYBR green chemistry using the ViiA7 Real-Time PCR system (Life Technologies; Grand Island, New York). All Ct values were converted to delta delta Ct values and were normalized to β-actin mRNA levels. The sequence of primers used is listed in Supplementary Table 1.
Serum biochemistry
Activities of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP), and levels of serum triglycerides (TGs) and total cholesterol (TC) were measured with the use of commercially available kits (Pointe Scientific; Canton, Michigan).
Hepatic lipid analysis
Hepatic TG and TC analyses were measured as previously described using commercially available assay kits (Pointe Scientific) according to the manufacturer’s instructions (Sun et al., 2017). Hepatic lipid content was presented as mg/g liver weight.
Histochemical analyses
Liver tissue sections were deparaffinized with xylene (4 min, 2×) followed by decreasing concentrations of ethanol (100–50%) and then water. Antigen retrieval was performed using citrate buffer (10.2 mM sodium citrate, pH 6.0). Endogenous peroxidase was quenched with 3% H2O2 for 15 min. Tissue sections were incubated for 2 h at room temperature with 10% goat serum to block nonspecific binding and then overnight at 4°C with rat F4/80 (F4/80; 1:1000; Biorad, Hercules, California) or IgG control. Sections were then incubated with biotinylated secondary antibody (Vector Labs, Burlingame, California) for 30 min at room temperature. Binding was visualized using a peroxidase diaminobenzidine Substrate Kit (Vector Labs) and counterstained with hematoxylin and eosin (Thermo Fisher Scientific, Inc.). Slides were dehydrated in alcohol gradient (50–100%), followed by xylenes. Permount was used to coverslip slides. Image acquisition and expression quantification occurred after coverslips were fully dried. Semiquantitative analysis of macrophage staining was performed on 4 representative images per animal per treatment group imaged at 10× magnification. The antibodies used are listed in Supplementary Table 2.
Statistical analyses
Data are presented as mean ± SD. GraphPad Prism (version 9; GraphPad Software Inc., La Jolla, California) was used for statistical analysis. Differences among individual groups were evaluated by 1-way analysis of variance followed by Tukey’s multiple comparison test. Differences were considered statistically significant at p < .05.
Results
Preliminary data (unpublished/supplemental) generated by our laboratory investigated a dose-dependent effect of Myclobutanil exposure on the Fxr-Fgf15/19 liver-gut BA homeostasis through the administration of vehicle or Myclobutanil at 3, 13.7, or 70.2 mg/kg/body weight for 10 days by oral gavage. These unpublished findings suggest that Myclobutanil exposure dramatically altered Fxr-Fgf15/19 signaling at 13.7 and 70.2 mg/kg/body weight dose. We then performed FXR reporter assays and an acute study using the LOAEL 70.2 mg/kg/body weight by oral gavage for 10 days with and without GW4064, a potent FXR agonist, in mice.
Myclobutanil acts as an FXR antagonist in an in vitro reporter assay utilizing HEK293 cells
To clarify the effects of Myclobutanil in regulating BA homeostasis through the Fxr-Fgf15/19 signaling, we utilized an in vitro reporter assay. Luciferase activity measured following treatment demonstrates successful activation of FXR with treatment of 1 µM GW4064, subsequent cotreatments with Myclobutanil showed significant dose-dependent decreases in FXR activation (Li et al., 2010; Williams et al., 2012) (Figure 1). These data support that Myclobutanil may act as an FXR antagonist and furthermore, contribute to altered BA homeostasis through the Fxr-Fgf15/19 signaling axis.
Figure 1.
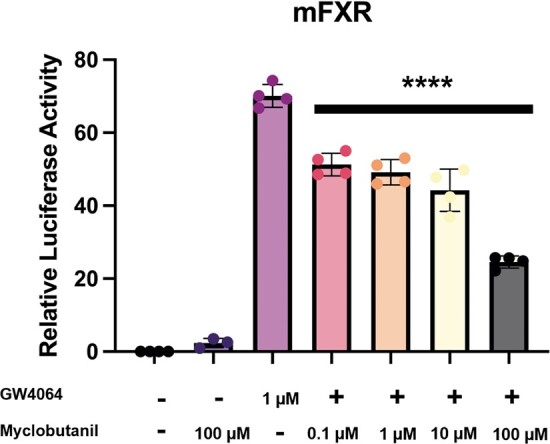
Antagonism of FXR by Myclobutanil significantly decreases the promoter activity of mouse Shp gene activity in the luciferase assay. A symbol * placed on each column identifies significant differences compared to GW4064. Data are presented as mean ± SD (n = 3–4/group), analyzed using 1-way ANOVA followed by a Holm-Sidak’s multiple comparisons test. A p < .05 was considered significant.
Myclobutanil acts as an FXR antagonist in a 10-day LOAEL in vivo mouse model
To delineate the molecular mechanism(s) underlying potential Myclobutanil-mediated modulation of FXR in vivo, mice were administered diets containing vehicle or 500 ppm (70.2 mg/kg/body weight) Myclobutanil for 10 days. All mice were additionally orally gavaged on days 9 and 10 with either vehicle or 150 mg/kg GW4064. Serum activities of (A) ALT, (B) AST, (C) ALP, (D) serum total cholesterol, (E) total BAs, and (F) total triglyceride levels were measured (Figure 2). No significant elevations in serum parameters were observed with either GW4064 or Myclobutanil treatment; however, cotreatment resulted in a significant decrease in ALP activity compared to the vehicle and GW4064 treated group. FXR activity was assessed again by the mRNA levels of the aforementioned FXR target genes. mRNA levels of Cyp7a1 that are negatively regulated by FXR activation, as expected, GW4064 treatment led to significant reductions (Figure 3A), Myclobutanil treatment alone resulted in a significant increase, and cotreatment led to a significant increase compared to GW4064 treatment alone. Myclobutanil treatment and cotreatment resulted in significant reductions in Lcn13 mRNA levels compared to GW4064 treatment alone. In the ileum, GW4064 greatly increased Fgf15 mRNA levels, Myclobutanil alone did not change the Fgf15 mRNA levels; however, cotreatment of Myclobutanil with GW4064 markedly reduced GW4064-mediated Fgf15 induction by 80% (Figure 3B). Similarly, although the degree of alteration was smaller, the ileal mRNA levels of Shp followed the same trend. These results highly suggest that Myclobutanil may act as an intestine FXR antagonist.
Figure 2.

Serum enzyme activities and lipid parameters in an acute treatment with Myclobutanil study with or without GW4064. A, alanine aminotransferase (ALT), B, aspartate aminotransferase (AST) and C, alkaline phosphatase (ALP), and D, serum total cholesterol, E, total BAs and F, total triglyceride levels. A symbol * placed on each column identifies significant differences compared to vehicle, # placed on each column identifies significant differences compared to GW4064. Data are presented as mean ± SD (n = 3–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
Figure 3.

Relative mRNA quantification of FXR target genes in an acute treatment with Myclobutanil study with or without GW4064. A, Hepatic (Cyp7a1, Lcn13, Ostβ, and Shp), B, ileal (Fgf15, Shp, Tgr5, and Ostβ). A symbol * placed on each column identifies significant differences compared to vehicle, # placed on each column identifies significant differences compared to GW4064. Data are presented as mean ± SD (n = 3–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
Effects of Myclobutanil on the development of NAFLD in mice
As a putative intestinal FXR antagonist, we sought to determine the effects of Myclobutanil on NAFLD development in vivo. Mice were fed LFD or HFHSD containing vehicle or 100 ppm (13.7 mg/kg/day) Myclobutanil for 4 or 9 weeks. Shown in Figure 4A, there was a time-dependent increase in body weight gain in mice fed LFD-Myclobutanil. As expected, HFHSD led to a marked increase in body weight compared to LFD-fed group; however, there was a trend of reduced body weight by Myclobutanil when coadministered with the HFHSD. The liver-to-body weight ratio (LBWR) remained unchanged in the LFD groups regardless of Myclobutanil treatment, but there was a slight decrease in the LBWR by Myclobutanil treatment in the HFHSD groups (Figure 4B).
Figure 4.

NAFLD Study. Animal biometrics. A, Terminal percent change body weight, B, Liver: body weight ratio. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat diet in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
The serum ALT activities were unchanged in the LFD groups regardless of Myclobutanil treatment, HFHSD feeding increased the ALT activities, which were reduced by Myclobutanil treatment similarly in both 4- and 9-week groups (Figure 5A). There were no significant alterations in AST, ALP, or serum triglycerides. Serum total cholesterol levels were significantly increased by HFHSD feeding regardless of Myclobutanil treatment (Figure 5D). Lastly, hepatic lipids were quantified, HFHSD feeding resulted in significant increases in hepatic triglycerides, and combination with Myclobutanil did not result in further elevations in hepatic triglycerides. Significant increases in hepatic cholesterol were observed with HFHSD and Myclobutanil combination treatment at 4 and 9 weeks but were not observed in HFHSD vehicle group (Figs. 5F–G).
Figure 5.

Serum liver enzyme activities and lipid parameters. A, alanine aminotransferase (ALT), B, aspartate aminotransferase (AST), C, alkaline phosphatase (ALP), D, total cholesterol, E, total bile acids, F, triglyceride levels and hepatic G, total cholesterol, and H, triglycerides. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat diet in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
FXR activity was assessed through the measurement of relative mRNA levels of classical FXR target genes in the liver (Cyp7a1, Lcn13, Shp, and Bsep) (Figs. 6A–D) and ileum (Fgf15, Fxr, Shp, Ibabp, Asbt, and Ostβ) (Figs. 7A–F). Cyp7a1 mRNA expression was unchanged regardless of diet or presence of Myclobutanil. LCN13 is a FXR activity marker in male mouse livers (data not shown) and known regulator of glucose and lipid metabolism, through the suppression of lipogenesis and promotion of fatty acid β-oxidation (Sheng et al., 2011). Significant increases in Lcn13 expression were observed in HFHSD mice treated with Myclobutanil for 4 and 9 weeks compared to HFHSD alone; however, Lcn13 expression was decreased by Myclobutanil treatment in the LFD-fed mice in a time-dependent manner (Figure 6B). FXR activation induces SHP that suppresses Srepb1c and to subsequently decrease fatty acid synthesis in the liver (Watanabe et al., 2004). In this study, we observed a significant reduction in Shp mRNA expression by HFHSD feeding, which is not affected by Myclobutanil. Myclobutanil reduced Shp mRNA levels in both 4- and 9-week treatment groups (Figure 6C). Bsep expression is found to be inversely correlated with the progression of NAFLD in patients, which can lead to the excessive accumulation of BA in the liver contributing toward inflammation and cellular injury (Okushin et al., 2016). Here, HFHSD vehicle mice had significantly decreased mRNA expression of Bsep compared to LFD vehicle mice, interestingly 4-week Myclobutanil LFD had significantly decreased mRNA expression compared to LFD vehicle, and this was not observed for their HFHSD counterparts. Together, the data suggest that while on LFD, Myclobutanil reduces hepatic FXR activity whereas on HFHSD, Myclobutanil increases FXR activity in the liver.
Figure 6.

Relative mRNA quantification of hepatic FXR target genes in NAFLD Study. A, Relative gene expression of hepatic Fxr target genes; A, Cyp7a1, B, Lcn13, C, Shp, and D, Bsep. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat diet in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
Figure 7.

Relative mRNA quantification of ileal FXR target genes in NAFLD Study. A, Fgf15, B, Fxr, C, Shp, D, Ibabp, E, Asbt, and F, Ostβ. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat diet in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
In general, ileal FXR target gene expression was not changed regardless of diet or Myclobutanil exposure; however, Fgf15 mRNA expression was significantly increased by HFHSD compared to LFD, which is not affected by Myclobutanil treatment. However, Myclobutanil time-dependently increased Fgf15 mRNA levels in the LFD-fed mice (Figure 7A). Overall suggesting under LFD, Myclobutanil suppresses FXR activity in the gut.
To assess inflammation in the liver, relative mRNA expression of inflammatory mediators, Tnfα, Il-6, and Lcn2, were examined (Figure 8). Both HFHSD and Myclobutanil seem to increase Tnfα mRNA expression; however, a synergistic effect was not observed. To our surprise, mRNA expression of Il-6 was significantly decreased by both HFHSD and Myclobutanil, and additive and/or synergistic suppression was not clear (Figure 8B). Lcn2 mRNA expression was increased by HFHSD, which is not additionally induced by Myclobutanil; however, in LFD group, Myclobutanil seemed to decrease Lcn2 mRNA levels. These findings suggest in LFD, Myclobutanil decreases FXR activity and reduces inflammation, presumably due to other unknown mechanisms.
Figure 8.

Relative mRNA quantification of hepatic inflammatory response genes in NAFLD Study. A, Relative gene expression of inflammatory response genes; A, Tnfα, B, Il-6, and C, Lcn2. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat diet in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
To assess hepatic lipid metabolism, we examined the mRNA expression of genes involved in fatty acid synthesis and transport (Figs. 9A–F). CD36 is responsible for long-chain fatty acid transport to the liver, and its expression is generally increased during fatty liver disease state due to increased transcriptional activity of liver X receptor, PXR, and peroxisome proliferator-associated receptor (PPARγ) (Silverstein Roy and Febbraio, 2009). HFHSD and Myclobutanil both increased CD36 mRNA levels, but a synergistic effect was not observed. Furthermore, 4-week Myclobutanil treatment marked increased CD36 mRNA levels in the LFD-fed mice compare to the rest of the groups (Figure 9A). HMGcoA-reductase is encoded by Hmgcr and is the rate-limiting enzyme in synthesizing cholesterol in the liver. As expected, all HFHSD-fed mice had significantly decreased expression of this gene compared to their respective LFD counterparts regardless of Myclobutanil in diet (Figure 9B). Sterol regulatory element-binding protein (Srebp-1c), fatty acid synthase (FAS), and acetyl-CoA carboxylase (ACC) are all involved in de novo lipogenesis, in NAFLD patients, SREBP-1c expression is induced, which leads to an increase in de novo lipogenesis, while FAS and ACC are intermediate enzymes that contribute toward the sequential conversions of acetyl-CoA to generation of intrahepatic lipids. As expected, the expression of Srebp-1c was only increased in HFHSD-fed mice compared to LFD, which is not affected by Myclobutanil treatment (Figs. 9C–E). In addition, ACC and FAS mRNA expression was not increased in any HFHSD-fed mice, which is not a surprise as it is known that HFHSD suppressed de novo fatty acid synthesis in the liver. Microsomal triglyceride transfer protein (MTP) is critical in transporting triglycerides out of the liver in the form of very low-density lipoprotein (VLDL). In the clinic, NAFLD patients MTP mRNA expression can be decreased or increased, a decrease in MTP levels will subsequently lead to decreased VLDL export and thus contribute toward hepatic lipid accumulation (Higuchi et al., 2011; Nakamuta et al., 2009). Both HFHSD and Myclobutanil seem to decrease MTP mRNA levels but did not show a combinational effect (Figure 9F).
Figure 9.

Relative mRNA quantification of hepatic lipogenic genes in NAFLD Study. Relative gene expression of genes involved in lipogenesis; A, CD36, B, Hmgcr, C, FAS, D, Srebp1c, E, ACC, and F, MTP. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
We quantified mRNA expression of xenobiotic metabolism genes regulated by CAR and PXR as a previous research group reported on an increased CAR signature following exposure to Myclobutanil (Oshida et al., 2015). Genes mainly regulated by CAR (Cyp2c55 and Cyp2b10), PXR (Cyp3a11), and PPARα (Cyp4a10) were analyzed (Cui and Klaassen, 2016). In general, either HFHSD or Myclobutanil could increase CAR or PXR target gene expression but only HFHSD increased PPARα target gene expression (Figure 10).
Figure 10.

Relative mRNA quantification of hepatic xenobiotic metabolism genes regulated by CAR, PXR, and PPARα in NAFLD Study. Relative gene expression of genes involved in lipogenesis; A, Cyp2c55, B, Cyp2b10, C, Cyp4a10, and D, Cyp3a11. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat diet in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
Liver histology was examined and showed no evidence of liver injury following treatment with Myclobutanil; however, HFHSD resulted in macro- and macrovesicular steatosis, and combination with Myclobutanil did not lead to liver injury (Figure 11). To determine if macrophages responding to Myclobutanil treatment were activated, we analyzed their expression of proinflammatory macrophages using F4/80 antigen in liver histologic sections. In mice treated with LFD and Myclobutanil for 9 weeks, the percent F4/80 positive area was significantly induced compared to LFD vehicle (Figure 12). In striking contrast, HFHSD and Myclobutanil at 9 weeks were significantly reduced compared to LFD of same treatment duration.
Figure 11.

Liver histology stained with hematoxylin and eosin in NAFLD Study. Scale represents 200 μM section, imaged at 10×.
Figure 12.

Liver immunohistochemistry stained F4/80. A, Representative images of positive staining in NAFLD Study, B, Quantification of percent positive stain area. Scale represents 200 μM section, imaged at 10×. A symbol * placed on each column identifies significant differences compared to vehicle of same diet, # placed on each column identifies significant differences compared to low-fat diet in the same treatment duration. Data are presented as mean ± SD (n = 6–7/group), analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test. A p < .05 was considered significant.
Discussion
Myclobutanil is a widely used environmental contaminant, and its effects on the NR FXR and BA homeostasis are unknown. Since FXR and BAs are important in maintaining liver and extrahepatic functions, in this study, we examined the effects of Myclobutanil on FXR activation in vitro and in vivo, as well as in the development of NAFLD.
Our data suggest that Myclobutanil may serve as a FXR antagonist. We performed a reporter assay study which demonstrates that Myclobutanil acts as an antagonist of FXR in a dose-dependent manner. This novel finding suggests that Myclobutanil may interact with FXR-regulated pathways in vivo regulate BA homeostasis and liver functions. These interesting data led us hypothesize that Myclobutanil similarly behaves as an FXR antagonist in vivo leading to an alteration in Fxr-Fgf15/19-BA homeostasis. We performed a 10-day subacute study in which mice were fed diets containing vehicle or 70.2 mg/kg Myclobutanil and cotreated with vehicle or GW4064 on days 9 and 10. Our findings from this study further support that Myclobutanil’s role as an FXR antagonist in vivo as cotreatment with GW4064 led to significant reductions in the relative mRNA expression levels of hepatic Cyp7a1, Lcn13, and ileal Fgf15, classical FXR target genes in the liver and gut, in comparison to mice treated with GW4064 only.
In the context of NAFLD, FXR is known to be downregulated, as pharmaceutical companies are exploring Fxr-Fgf15/19 signaling pathway as a therapeutic target during NAFLD. Exposure to environmental chemicals such as Myclobutanil is amongst the multitude of possible disease etiologies of NAFLD, therefore, we investigated the impact of exposure to Myclobutanil in the development of NAFLD. The reporter assay experiment and FXR cotreatment study with GW4064 and Myclobutanil both support Myclobutanil inhibits FXR activation especially with a potent activator, therefore, we expected the impact of Myclobutanil exposure during NAFLD development to lead to further Fxr-Fgf15/19 dysregulation of BA homeostasis and signaling. To our surprise, cotreatment of mice on HFHSD and exposed to Myclobutanil led to a decrease in liver inflammation and oxidative stress, and hepatic lipid metabolism was not dramatically altered in the presence of Myclobutanil in HFHSD feeding. Normally, we observe a positive correlation between ALT and cholesterol in NAFLD studies, but in this study, Myclobutanil reduced ALT, which is not associated with a reduction in total cholesterol levels (Schumacher et al., 2017). Lastly, hepatic and ileal FXR signaling was not significantly altered. Classical CAR and PXR xenobiotic metabolism mRNA expression was significantly altered in mice administered Myclobutanil, these findings are consistent with a previous study that reported the induction of CAR and PXR target genes following exposure to Myclobutanil (Oshida et al., 2015). The role of Myclobutanil in hepatic lipid metabolism has been reported in vitro, using HepG2 cells, this compound has been shown to worsen fatty-acid-induced steatosis and cell death, therefore suggesting that Myclobutanil exposure may exacerbate NAFLD development (Stellavato et al., 2016). However, in our in vivo study, we did not observe the worsening of lipid accumulation in the liver after Myclobutanil treatment, and indeed, the mRNA levels of some but not all genes involved in de novo lipid synthesis were reduced following NASH diet/Myclobutanil treatment. The exact mechanism warrants future research efforts.
The observed changes in mRNA expression impact key FXR target genes within the liver and gut which play essential roles in maintaining BA homeostasis and signaling. If these signaling pathways that are tightly regulated by Fxr-Fgf15/19 are dysregulated, there may be implications in specific disease states for humans such as NASH or cholestasis where FXR is known to be downregulated and BA homeostasis is dysregulated. Furthermore, sensitive populations that are working with Myclobutanil in an occupational setting may be at higher risk for exposure and subsequent alterations in BA homeostasis mediated by Fxr-Fgf15/19 signaling.
Further studies are needed to understand how exposure to Myclobutanil may contribute to BA dysregulation and to what extent, this dysregulation may contribute to known disease states that Fxr-Fgf15/19 are known to play major roles in.
Conclusion
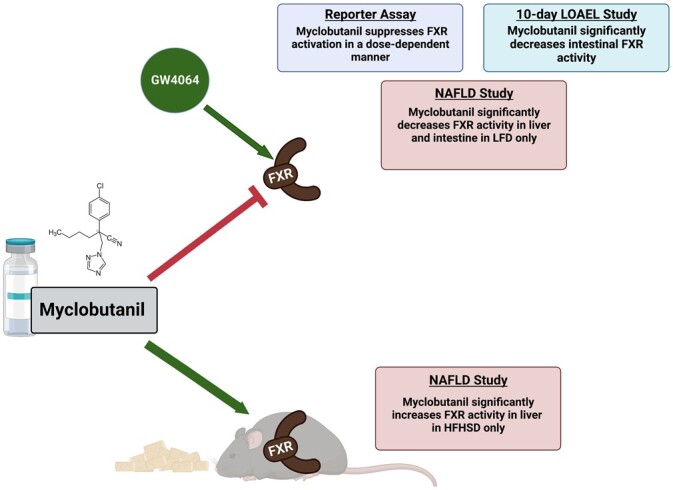
Using an in vitro reporter assay and in vivo mouse experiments, we have shown that Myclobutanil acts as an antagonist of FXR (Figure 13). The studies conducted help understand how exposure to Myclobutanil can contribute to BA dysregulation, and how it may contribute to known disease states such as NASH where FXR is downregulated and the progression of that to end-stage liver disease.
Figure 13.

Summary of findings. Key findings from Reporter Assay, LOAEL, and NAFLD studies are summarized. Figure made with biorender.com.
Supplementary Material
Contributor Information
Rulaiha Taylor, Department of Pharmacology and Toxicology, Rutgers University, Piscataway, NJ 08854, USA; Environmental and Occupational Health Science Institute, Rutgers University, Piscataway, NJ 08854, USA; Rutgers Center for Lipid Research, Rutgers University, New Brunswick, NJ 08901, USA.
Laura Armstrong, Department of Pharmacology and Toxicology, Rutgers University, Piscataway, NJ 08854, USA; Environmental and Occupational Health Science Institute, Rutgers University, Piscataway, NJ 08854, USA; Rutgers Center for Lipid Research, Rutgers University, New Brunswick, NJ 08901, USA.
Anisha Bhattacharya, Department of Pharmacology and Toxicology, Rutgers University, Piscataway, NJ 08854, USA.
Zakiyah Henry, Department of Pharmacology and Toxicology, Rutgers University, Piscataway, NJ 08854, USA; Environmental and Occupational Health Science Institute, Rutgers University, Piscataway, NJ 08854, USA; Rutgers Center for Lipid Research, Rutgers University, New Brunswick, NJ 08901, USA.
Anita Brinker, Environmental and Occupational Health Science Institute, Rutgers University, Piscataway, NJ 08854, USA.
Brian Buckley, Environmental and Occupational Health Science Institute, Rutgers University, Piscataway, NJ 08854, USA.
Bo Kong, Department of Pharmacology and Toxicology, Rutgers University, Piscataway, NJ 08854, USA; Environmental and Occupational Health Science Institute, Rutgers University, Piscataway, NJ 08854, USA; Rutgers Center for Lipid Research, Rutgers University, New Brunswick, NJ 08901, USA.
Grace Guo, Department of Pharmacology and Toxicology, Rutgers University, Piscataway, NJ 08854, USA; Environmental and Occupational Health Science Institute, Rutgers University, Piscataway, NJ 08854, USA; Rutgers Center for Lipid Research, Rutgers University, New Brunswick, NJ 08901, USA; VA New Jersey Health Care System, Veterans Administration Medical Center, East Orange, NJ 07017, USA.
Funding
Foundation for the National Institutes of Health (F32DK116495 to L.A., R21ES029258 and R01GM135258 to G.G., and T32ES007148 to L.A. and R.T.); Center for Integrated Healthcare, U.S. Department of Veterans Affairs (BX002741 to G.G.).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary data
Supplementary data are available at Toxicological Sciences online.
References
- Armstrong L. E., Guo G. L. (2017). Role of FXR in liver inflammation during nonalcoholic steatohepatitis. Curr. Pharmacol. Rep. 3, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell M., Wedin M., Admyre T., Hermansson M., Böttcher G., Göransson M., Lindén D., Bamberg K., Oscarsson J., Bohlooly Y. M. (2013). Ageing Fxr deficient mice develop increased energy expenditure, improved glucose control and liver damage resembling NASH. PLoS One 8, e64721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M., Deaciuc I., Mendez C., Song Z., Joshi-Barve S., Barve S., McClain C. (2007). Nonalcoholic fatty liver disease: Predisposing factors and the role of nutrition. J. Nutr. Biochem. 18, 184–195. [DOI] [PubMed] [Google Scholar]
- Cui J. Y., Klaassen C. D. (2016). RNA-Seq reveals common and unique PXR- and CAR-target gene signatures in the mouse liver transcriptome. Biochim. Biophys. Acta 1859, 1198–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Filippo P., Pomata D., Riccardi C., Buiarelli F., De Gennaro M., Console C., Laurendi V., Puri D. (2018). Determination of pesticides in the respirable fraction of airborne particulate matter by high-performance liquid chromatography-tandem mass spectrometry. Anal. Lett. 51, 600–612. [Google Scholar]
- Georgopapadakou N. H. (1998). Antifungals: Mechanism of action and resistance, established and novel drugs. Curr. Opin. Microbiol. 1, 547–557. [DOI] [PubMed] [Google Scholar]
- Goldman P. H. (1986). Rh-3866: Dietary chronic and oncogenicity study in mice. Unpublished DAS Report No. 84R-023. Rohm and Haas Company.
- Günther H. M.(2018). Toxno sunstance profile: Myclobutanil. Available at: https://www.toxno.com.au/toxins/substance_id_95.html. Accessed December 20, 2022. [Google Scholar]
- Higuchi N., Kato M., Tanaka M., Miyazaki M., Takao S., Kohjima M., Kotoh K., Enjoji M., Nakamuta M., Takayanagi R. (2011). Effects of insulin resistance and hepatic lipid accumulation on hepatic mRNA expression levels of apoB, MTP and L-FABP in non-alcoholic fatty liver disease. Exp. Ther. Med. 2, 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joint FAO/WHO Meeting on Pesticide Residues (JMPR), Standards & Scientific Advice on Food Nutrition. (2015). Pesticide residues in food - 2014: toxicological evaluations. In Joint Meeting of the FAO Panel of Experts on Pesticide Residues in Food and the Environment and the WHO Core Assessment Group on Pesticide Residues, Rome, Italy, 16–25 September 2014. World Health Organization, Geneva.
- Kong B., Wang L., Chiang J. Y., Zhang Y., Klaassen C. D., Guo G. L. (2012). Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H. C., Kim D. H., Jeong C. H., Kim Y. J., Han J. H., Lim S. J., Shin D. M., Kim D. W., Han S. G. (2021). Tebuconazole fungicide induces lipid accumulation and oxidative stress in HepG2 cells. Foods 10, 2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake B. G. (2018). Human relevance of rodent liver tumour formation by constitutive androstane receptor (CAR) activators. Toxicol. Res. (Camb.) 7, 697–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. A., Tzilivakis J., Warner D. J., Green A. (2016). An international database for pesticide risk assessments and management. Hum. Ecol. Risk Asses. 22, 1050–1064. [Google Scholar]
- Li G., Thomas A. M., Hart S. N., Zhong X., Wu D., Guo G. L. (2010). Farnesoid X receptor activation mediates head-to-tail chromatin looping in the Nr0b2 gene encoding small heterodimer partner. Mol. Endocrinol. 24, 1404–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. T., Brennan R. J., Hu W., Ayanoglu E., Lau C., Ren H., Wood C. R., Corton J. C., Kavlock R. J., Dix D. J. (2007). Toxicogenomic study of triazole fungicides and perfluoroalkyl acids in rat livers predicts toxicity and categorizes chemicals based on mechanisms of toxicity. Toxicol. Sci. 97, 595–613. [DOI] [PubMed] [Google Scholar]
- Nakamuta M., Fujino T., Yada R., Yada M., Yasutake K., Yoshimoto T., Harada N., Higuchi N., Kato M., Kohjima M., et al. (2009). Impact of cholesterol metabolism and the LXRα-SREBP-1c pathway on nonalcoholic fatty liver disease. Int. J. Mol. Med. 23, 603–608. [DOI] [PubMed] [Google Scholar]
- Okushin K., Tsutsumi T., Enooku K., Fujinaga H., Kado A., Shibahara J., Fukayama M., Moriya K., Yotsuyanagi H., Koike K. (2016). The intrahepatic expression levels of bile acid transporters are inversely correlated with the histological progression of nonalcoholic fatty liver disease. J. Gastroenterol. 51, 808–818. [DOI] [PubMed] [Google Scholar]
- Oshida K., Vasani N., Jones C., Moore T., Hester S., Nesnow S., Auerbach S., Geter D. R., Aleksunes L. M., Thomas R. S., et al. (2015). Identification of chemical modulators of the constitutive activated receptor (CAR) in a gene expression compendium. Nucl. Recept. Signal. 13, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D. J., Blanchard S. G., Bledsoe R. K., Chandra G., Consler T. G., Kliewer S. A., Stimmel J. B., Willson T. M., Zavacki A. M., Moore D. D., et al. (1999). Bile acids: Natural ligands for an orphan nuclear receptor. Science (New York, NY) 284, 1365–1368. [DOI] [PubMed] [Google Scholar]
- Schermerhorn P. G., Golden P. E., Krynitsky A. J., Leimkuehler W. M. (2005). Determination of 22 triazole compounds including parent fungicides and metabolites in apples, peaches, flour, and water by liquid chromatography/tandem mass spectrometry. J. AOAC Int. 88, 1491–1502. [PubMed] [Google Scholar]
- Schumacher J. D., Kong B., Pan Y., Zhan L., Sun R., Aa J., Rizzolo D., Richardson J. R., Chen A., Goedken M., et al. (2017). The effect of fibroblast growth factor 15 deficiency on the development of high fat diet induced non-alcoholic steatohepatitis. Toxicol. Appl. Pharmacol. 330, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng L., Cho K. W., Zhou Y., Shen H., Rui L. (2011). Lipocalin 13 protein protects against hepatic steatosis by both inhibiting lipogenesis and stimulating fatty acid β-oxidation. J. Biol. Chem. 286, 38128–38135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein Roy L., Febbraio M. (2009). CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2, re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler E. K., Loomba R. (2015). Recommendations for diagnosis, referral for liver biopsy, and treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mayo Clin. Proc. 90, 1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellavato A., Lamberti M., Pirozzi A. V. A., de Novellis F., Schiraldi C. (2016). Myclobutanil worsens nonalcoholic fatty liver disease: An in vitro study of toxicity and apoptosis on HepG2 cells. Toxicol. Lett. 262, 100–104. [DOI] [PubMed] [Google Scholar]
- Sun M., Liu D., Qiu X., Zhou Q., Shen Z., Wang P., Zhou Z. (2014). Acute toxicity, bioactivity, and enantioselective behavior with tissue distribution in rabbits of Myclobutanil enantiomers. Chirality 26, 784–789. [DOI] [PubMed] [Google Scholar]
- Sun R., Yang N., Kong B., Cao B., Feng D., Yu X., Ge C., Huang J., Shen J., Wang P., et al. (2017). Orally administered berberine modulates hepatic lipid metabolism by altering microbial bile acid metabolism and the intestinal Fxr signaling pathway. Mol. Pharmacol. 91, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. M., Hart S. N., Kong B., Fang J., Zhong X. B., Guo G. L. (2010). Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51, 1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. D., Chen W. D., Wang M., Yu D., Forman B. M., Huang W. (2008). Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., Moore D. D., Auwerx J. (2004). Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113, 1408–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. A., Thomas A. M., Li G., Kong B., Zhan L., Inaba Y., Xie W., Ding W. X., Guo G. L. (2012). Tissue specific induction of p62/Sqstm1 by farnesoid X receptor. PLoS One 7, e43961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Zhang P., Wang X., Wang Y., Zhou Z., Zhu W. (2014). Stereoselective degradation of chiral fungicide myclobutanil in rat liver microsomes. Chirality 26, 51–55. [DOI] [PubMed] [Google Scholar]
- Younossi Z. M., Koenig A. B., Abdelatif D., Fazel Y., Henry L., Wymer M. (2016). Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- Zhan L., Liu H. X., Fang Y., Kong B., He Y., Zhong X. B., Fang J., Wan Y. J., Guo G. L. (2014). Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS One 9, e105930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Liu Q., Wang J., Harnish D. C. (2009a). Suppression of interleukin-6-induced C-reactive protein expression by Fxr agonists. Biochem. Biophys. Res. Commun. 379, 476–479. [DOI] [PubMed] [Google Scholar]
- Zhang S., Wang J., Liu Q., Harnish D. C. (2009b). Farnesoid X receptor agonist way-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J. Hepatol. 51, 380–388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.