

Abstract
Tuberculosis, a deadly infectious lung disease caused by Mycobacterium tuberculosis (Mtb), remains the leading cause of bacterial disease-related deaths worldwide. Mtb reprograms and disables key antibacterial response pathways, many of which are regulated by epigenetic mechanisms that control the accessibility of chromatin to the transcriptional machinery. Recent reports suggest that host phosphatases, such as PPM1A, contribute to regulating chromatin accessibility during bacterial infections. However, changes in genome-wide chromatin accessibility during Mtb infection and whether PPM1A plays a role in this process remains unknown. Herein, we use combinatorial chromatin accessibility (ATAC-seq) and transcriptomic (RNA-seq) profiling of wild-type, PPM1A knockout and PPM1A overexpressing macrophages to demonstrate that Mtb infection induces global chromatin remodelling consistent with changes in gene expression. The strongest concordant changes to chromatin accessibility and gene expression triggered by Mtb infection were enriched for genes involved in type I interferon (IFN) signalling pathways. A panel of 15 genes with the strongest concordant changes in chromatin accessibility and gene expression were validated to be significantly upregulated in Mtb-infected human monocyte-derived macrophages. PPM1A expression affects chromatin accessibility profiles during Mtb infection that are reflected in the total number, chromosome location, and directionality of change. Transcription factor binding motif analysis revealed enrichment for transcription factors involved in the type I IFN pathway during Mtb infection, including members of the IRF, MEF2, and AP-1 families. Our study shows that altered type I IFN responses in Mtb-infected macrophages occur due to genome-wide changes in chromatin accessibility, and that PPM1A could influence a subset of these signatures.
Keywords: tuberculosis, Mycobacterium tuberculosis, chromatin accessibility, ATAC-seq, RNA-seq, transcriptome, epigenome, host-directed therapy, type I IFN
Chromatin accessibility and gene expression profiling reveals a concordant type I interferon response in macrophages infected with Mycobacterium tuberculosis.
Introduction
Tuberculosis (TB) is caused by Mycobacterium tuberculosis (Mtb) infection, and until the 2020 coronavirus pandemic, it was the leading cause of death from a single infectious agent, accounting for over 1.5 million deaths in 2021 (World Health Organization 2021). Mtb infects the innate immune cells of the lungs, and the success of infection depends on the re-programming of host cells to evade antibacterial responses, while simultaneously maintaining host cell viability (Roy et al. 2018, Looney et al. 2021). Host immune dysregulation in TB has been demonstrated at the gene expression, or epigenetic level, which is frequently governed by the accessibility of chromatin to the transcriptional machinery. The most common epigenetic mechanisms that regulate gene expression through modulating accessibility of DNA to the transcriptional machinery include DNA methylation, conformation of nucleosomes and histones, and transcription factor (TF) binding. Genome-wide analyses of the DNA methylome of Mtb-infected cells compared to noninfected cells have demonstrated that changes in gene expression are mediated in part by gain or loss of DNA methylation (Zheng et al. 2016, Chen et al. 2020, Karlsson et al. 2021, Looney et al. 2021). Genome-wide histone methylation and acetylation patterns, such as H3K27ac, H3K4me3, H4K20me, H3K14ac, or H3K27me2/3, have also been analysed using ChIP-seq in macrophages infected with Mtb, M. bovis, or blood cells from TB patients (Chen et al. 2017, Hall et al. 2019, Subuddhi et al. 2020, Del Rosario et al. 2022). These studies revealed that Mtb infection or TB disease induced many changes to nucleosome structure that altered gene expression to dampen antibacterial responses. The limitation of these studies is that ChIP-seq analyses specific changes to nucleosome structure and does not simultaneously capture genome-wide changes to chromatin accessibility. There have been a limited number of studies that investigated global chromatin accessibility patterns in macrophages upon Mtb infection (Correa-Macedo et al. 2021, Lin et al. 2022). In both cases, chromatin accessibility was analysed as a complement to the primary goal of the studies, and as such, the authors did not perform an in-depth analysis on the effects of Mtb infection alone. In contrast, multiple studies have revealed key changes in global chromatin accessibility triggered by Mtb infection in dendritic cells, a closely related antigen presenting cell (Pacis et al. 2015, 2019). Studies in dendritic cells revealed that differential DNA methylation patterns upon Mtb infection are correlated with differential chromatin accessibility patterns and identified changes to accessible TF binding motifs. Collectively, these studies provide evidence that Mtb may also trigger similar genome-wide chromatin reorganization in macrophages. Whereas there is a lack of genome-wide chromatin accessibility studies in macrophages, numerous studies have examined the impact of Mtb infection on the transcriptome of macrophages (Madden et al. 2022). These studies predominantly demonstrate that similar type I interferon (IFN) signatures are linked with host-detrimental outcomes in Mtb-infected macrophages, murine TB models in vivo and multiple TB patient cohorts, and concomitantly decrease upon antibacterial therapy or IFN-α/β blockade (Ottenhoff et al. 2012, Moreira-Teixeira et al. 2018, 2020a,b, Zhang et al. 2021). It should be pointed out that under certain conditions, TB patients who received treatment with IFN-α concurrently with antibiotics presented lower bacterial loads than patients receiving antibiotics alone, while some reports show that IFN-α therapy received for other diseases can re-activate TB (Donovan et al. 2017, Moreira-Teixeira et al. 2018).
Several reports have also documented the dysregulated expression of enzymes that mediate histone modifications in macrophages after Mtb infection, such as histone deacetylases HDAC1, HDAC3, and Sirtuins (SIRT) SIRT1, SIRT2, and SIRT3 (Chandran et al. 2015, Cheng et al. 2017, Moores et al. 2017, Kim et al. 2019, Bhaskar et al. 2020, Campo et al. 2021, Madhavan et al. 2021, Smulan et al. 2021). Changes to nucleosome structure that occur during pathogen invasion usually correlated to impaired antibacterial responses, and could be rescued by therapeutically targeting these enzymes (Chandran et al. 2015, Cheng et al. 2017, Moores et al. 2017, Kim et al. 2019, Bhaskar et al. 2020, Campo et al. 2021, Madhavan et al. 2021, Smulan et al. 2021). Similar mechanisms are employed by other intracellular pathogens, such as Listeria monocytogenes. Successful L. monocytogenes infection requires activation of SIRT2 by the host Protein Phosphatase Mg2+/Mn2+-dependent 1A (PPM1A) to deacetylate H3K18, which leads to disabled antibacterial immune responses (Pereira et al. 2018). Interestingly, we have also reported that PPM1A is upregulated in macrophages during Mtb infection to disable multiple antibacterial response pathways including apoptosis, autophagy, and inflammation (Sun et al. 2016, Schaaf et al. 2017, Smith et al. 2018, Berton et al. 2022). Whether the host-detrimental effects of PPM1A activity during Mtb infection extend to the modulation of nucleosome structure or chromatin accessibility, via histone-modifying enzymes or other mechanisms, is unknown.
Here, we use ATAC-seq and RNA-seq to determine the genome-wide changes that occur at the epigenetic (chromatin accessibility) and transcriptomic level in Mtb-infected human macrophages. We identify specific changes to chromatin accessibility and gene expression that are mediated by PPM1A expression. Extensive bioinformatic analysis revealed distinct chromatin accessibility signatures in Mtb-infected wild-type (WT), PPM1A knockout (△PPM1A), and PPM1A overexpressing (PPM1A+) macrophages compared to noninfected controls. The most prominent differences in chromatin accessibility signatures were observed in PPM1A+ macrophages compared to WT and △PPM1A macrophages, under both uninfected and Mtb-infected conditions. The differential chromatin accessibility and gene expression patterns of Mtb-infected compared to noninfected macrophages were highly enriched in genes associated with the type I IFN signalling pathways. A subset of type I IFN response genes where chromatin accessibility changed displayed striking and concordant changes to gene expression. Furthermore, genomic regions where accessibility was altered by Mtb infection were enriched for the binding sites of transcription factors involved in type I IFN responses and TB pathogenicity.
Results
Generation of ATAC-seq libraries from Mtb-infected macrophages
To investigate the impact of Mtb infection on genome-wide chromatin accessibility and gene expression patterns, we performed parallel ATAC-seq and RNA-seq using WT and genetically modified human THP-1 macrophages infected with Mtb mc26206 (Fig. 1A). THP-1 macrophages are routinely used to study innate immune responses to Mtb infection because they are genetically tractable and reproduce key physiological features of primary human monocyte-derived macrophages (hMDM; Tsuchiya et al. 1980, Daigneault et al. 2010, Bosshart and Heinzelmann 2016). Mtb mc26206 is a derivative of virulent Mtb H37Rv (Jain et al. 2014) that retains all virulence genes and has been shown to elicit a comparable host immune response and susceptibility to TB antibiotics, and intramacrophage replication and survival compared to its parent strain (Schaaf et al. 2017, Mouton et al. 2019). We determined that Mtb infection for 48 h at a multiplicity of infection (MOI) of 10 achieved the optimal infection conditions as determined by the highest percentage of infection (70%) and lowest cell death (10%), which abrogates gene regulation (Fig. S1). To determine chromatin accessibility patterns, we used the assay for transposase-accessible chromatin sequencing (ATAC-seq), which uses a hyperactive Tn5 transposase to mark by tagmentation, DNA that is relatively accessible (Buenrostro et al. 2013, Corces et al. 2017). Two independent replicates of Mtb-infected and noninfected THP-1 macrophages (WT, ΔPPM1A, PPM1A +) were processed for ATAC-seq library generation. The OMNI-ATAC version of the ATAC-seq protocol was used to minimize mitochondrial genome representation in DNA libraries (Corces et al. 2017). We maximized the quality of sequencing reads by incorporating dual-end barcoding into the library design, which improves traditional single-end barcoding by limiting barcode hopping and increasing multiplexing (Kircher et al. 2012). Quality control analysis of our ATAC-seq libraries before and after sequencing verified that they conformed to metrics expected of quality DNA accessibility mapping data (Fig. S2A–D). We applied false discovery rate (FDR) cutoffs of [FDR] < 0.05, and peaks were retained if they were present in at least two samples. The number of consensus peaks found in at least two samples was 317537 peaks per sample. Principal component analysis (PCA) of read coverage over all peaks demonstrated that genotype and infection status were the largest and second largest contributor to variance in the dataset, respectively (Fig. S2E). Peaks from technical replicates also clustered together, confirming the reproducibility of our data.
Figure 1.

Mtb infection induces global chromatin remodelling in macrophages. (A) Experimental schematic. WT, △PPM1A, and PPM1A+ THP-1 macrophages were infected with Mtb mc26206 at an MOI of 10 for 48 h. Then, Mtb-infected and noninfected control macrophages were harvested for ATAC-seq (two independent replicates) and RNA-seq (three independent replicates) in parallel from the same wells. (B) MA plots showing log2 fold-change versus average signal (log of counts per million, logCPM) in Mtb-infected samples compared to controls, in each of three genotypes studied. Peaks with significant chromatin accessibility changes are marked in red. Blue lines represent the general signal trend by loess fit. Red-dotted lines mark the location of identical signal. (C) Heatmap of chromatin accessibility in regions that were significantly differentially accessible in WT cells upon Mtb infection. Chromatin accessibility in WT, △PPM1A, and PPM1A+ macrophages before and after Mtb infection is shown. All ATAC-seq peaks were retained if they had >24 counts in at least two samples. Differential peaks between Mtb-infected WT macrophages and noninfected controls with absolute value of log2FC > 1 and FDR < 0.05 were considered significant. (D) PCA showing significant differential ATAC-peaks separate and cluster together based on the first and second principal components, which are genotype and Mtb infection status, respectively. Genotype accounts for 89% of the variance observed in all significant DAR, and Mtb infection accounts for 6%. (E) Total and unique DAR. The first bar displays the total number of all significant differential peaks in each genotype after Mtb infection, separated by whether chromatin accessibility increased or decreased. The second bar displays the total number of peaks that are unique to each genotype, separated by whether chromatin accessibility increased or decreased. (F) Venn diagram showing the overlap of significant differential peaks between Mtb-infected WT, △PPM1A, and PPM1A+ macrophages and noninfected macrophages.
Mtb infection induces global chromatin remodelling in macrophages
Differential analysis of DNA accessibility at peaks showed significant genome-wide changes to chromatin accessibility in WT, △PPM1A, and PPM1A+ cells after Mtb infection (Fig. 1B). Chromatin accessibility profiles in △PPM1A and PPM1A+ cells diverged from WT cells in many regions, before and after Mtb infection (Fig. 1C). There was also a marked increase in the number of significant differentially accessible regions (DAR) in PPM1A+ cells compared to both WT and △PPM1A cells (Fig. 1B and C). PCA of peaks that are differentially accessible in any of the pairwise comparisons demonstrated that all differential peaks also clustered based on genotype and infection (Fig. 1D), resembling the PCA of all peaks (Fig. S2E). Genotype accounted for 89% of the variance observed, while infection accounted for 6%. Taken together, we observed that PPM1A overexpression induced striking changes to chromatin accessibility patterns in macrophages. In WT macrophages, Mtb infection induced 309 significant DAR, where accessibility increased in 160 regions (52%) and decreased in 149 regions (Fig. 1E). Of these loci, 266 (86%) annotated to genes based on their distance from the transcription start site (TSS). The number of significant DAR upon Mtb infection in △PPM1A cells increased to 442, where accessibility increased in 291 regions (66%) and decreased in 151 regions (Fig. 1E). In △PPM1A cells, the proportion of regions where accessibility increased was 14% greater than in WT cells, and 364 (82%) peaks annotated to genes in △PPM1A cells. The number of significant DAR after Mtb infection substantially increased in PPM1A+ cells to 6702 loci, which represents a 22-fold increase in total significant DAR compared to WT cells. The proportion of regions where accessibility increased was higher in PPM1A+ cells, where accessibility increased in 5443 regions, representing 81% of total DAR (Fig. 1E). The accessibility decreased in 1259 regions in PPM1A+ cells and 5372 (80%) of peaks annotated to genes. While all three genotypes shared a set of conserved DAR induced by Mtb infection, each genotype also had a set of unique DAR. There were 172 unique peaks in WT macrophages, where accessibility increased in 68 and decreased in 104 regions. In △PPM1A cells, there were 213 total unique peaks, where accessibility increased in 118 and decreased in 95 regions (Fig. 1E). In PPM1A+ cells, there were 4994 unique peaks where accessibility increased in 3999 and decreased in 995 peaks (Fig. 1E). A total of 57 and 51 DAR were shared between WT and △PPM1A or PPM1A+ cells, respectively, and 79 were shared between △PPM1A and PPM1A+ cells (Fig. 1F). There were 24 DAR shared between all three genotypes. In WT, △PPM1A, and PPM1A+ cells, 16%, 14%, and 13% of significant DAR annotated to promoter regions, while the majority (32%–35%) of significant DAR in all genotypes annotated to regions labelled ‘other introns’ (Fig. S2F).
Chromatin accessibility profiles of Mtb-infected macrophages are enriched in type I IFN response pathways
To identify biological pathways and processes that are associated with Mtb-infection induced changes to chromatin accessibility, we performed gene ontology (GO) analysis of DAR using the genomic regions enrichment annotations tool (GREAT) (McLean et al. 2010). All significant DAR in WT, △PPM1A, and PPM1A+ macrophages upon Mtb infection were analysed separately with GREAT. DAR were annotated to genes based on the association rules provided by GREAT (Fig. S3A–C). For each genotype, GO terms with adjusted P-values < 0.05 were ranked by their enrichment scores and filtered for redundancy. The nonredundant GO terms with the highest enrichment scores from each genotype are reported in Fig. 2A. In WT and △PPM1A cells, GO terms with the highest enrichment scores among terms associated with DAR upon Mtb infection were dominated by type I interferon responses (Fig. 2A). However, only two nonredundant GO terms were statistically significant in PPM1A+ cells (‘Type I interferon signalling pathway’ and ‘Defense response to virus’). Mtb-infected WT cells showed four unique enriched GO terms, whereas there was only one unique enriched GO term in △PPM1A cells, and none in PPM1A+ cells (Fig. 2B). GO terms that were unique to WT cells included ‘2 deoxyribonucleotide biosynthetic process’ and related terms that were enriched by DAR annotated to AK5 and CMPK2. In △PPM1A cells, ‘Positive regulation of neutrophil chemotaxis’ was the sole unique GO term and was enriched by DAR annotated to IL-23, CXCL2, CXCL5, and CXCL8 (Fig. 2B).
Figure 2.

Mtb infection induces changes to chromatin accessibility that are enriched in type I IFN regulatory pathways. (A) GO term enrichment analysis was conducted using GREAT with significant differential peaks in WT, △PPM1A, and PPM1A+ macrophages following Mtb infection. GO terms with FDR < 0.05 were considered significant. GO terms were ranked by enrichment and the top 5 nonredundant terms from each genotype were included. Numbers in bold beside each bar indicate the number of genes that contribute to the enrichment of each pathway. Many of the top 5 nonredundant terms overlapped between genotypes. (B) Venn diagram displaying common significant GO terms between WT, △PPM1A, and PPM1A+ macrophages after Mtb infection. (C) Separate GO enrichment analysis was conducted for chromatin regions where accessibility increased or decreased. Significant DAR in each genotype were split into two groups based on whether chromatin accessibility increased (log2FC > 1) or decreased (log2FC < 1). GO terms were ranked and plotted as in (A). The top 5 nonredundant pathways, where possible, are shown for each genotype. (D) Ingenuity pathway analysis was conducted on all significant DAR for Mtb-infected WT, △PPM1A, and PPM1A+ macrophages. The radial summary graph of DAR in WT macrophages is shown. The nodes and edges are colour coded by predicted activation or inhibition: orange represents ‘predicted activation’ (node) or ‘leads to predicted activation’ (edge), and blue represents ‘predicted inhibition’ (node) or ‘leads to inhibition’ (edge). Arrows represent activation, causation, or modification, and perpendicular lines represent inhibition. Solid lines represent direct interactions, dashed lines represent indirect interactions, and dotted lines represent inferred correlation from machine-based learning. The shapes of the nodes are represented as follows: ovals represent transcription regulators, squares represent cytokines, rectangles represent g-protein-coupled receptors, diamonds represent enzymes, hexagons represent canonical pathways, spheres represent ‘other’. Additional radial summary graphs for △PPM1A and PPM1A+ macrophages are shown in Fig. S5.
Significant DAR from each genotype were split by whether chromatin accessibility increased or decreased upon Mtb infection. GREAT was used to determine the biological pathways that were enriched in each group to associate biological functions with DAR where chromatin accessibility exclusively increased or decreased upon Mtb infection. In all genotypes, type I IFN responses were the most highly enriched GO biological processes in DAR where chromatin accessibility increased. However, enrichment of this term in PPM1A+ cells was 10 to 15-fold less than in WT or △PPM1A cells. In WT cells, the GO terms ‘PERK-mediated unfolded protein response’ and ‘Induction of positive chemotaxis’ were also highly enriched in DAR where chromatin accessibility increased, while the term ‘L-serine metabolic process’ was highly enriched in △PPM1A cells (Fig. 2C). In DAR, where chromatin accessibility decreased, ‘Neural nucleus development’ was the only GO term that was significantly enriched in any genotype, showing up in the PPM1A+ cells (Fig. 2C). The lack of statistically significant pathways enrichment of DAR in PPM1A+ cells was unexpected given the high number of DAR that we observed upon Mtb infection. To rule out any errors, we performed an alternative pathway analysis to validate these results. We performed pathway enrichment analysis with the Human Molecular Signatures Database (MSigDB) using DAR separated by whether chromatin accessibility increased or decreased. Indeed, these results (Fig. S4) were consistent with the GREAT analysis. The only noted differences were some unique pathways enriched in DAR where chromatin accessibility decreased in PPM1A + cells (Fig. S4). Overall, pathway analysis indicated that regions of altered chromatin accessibility induced by Mtb infection are enriched for type I IFN signalling pathway genes. In addition, we used QIAGEN Ingenuity Pathway Analysis (IPA) (Krämer et al. 2014) to analyse all DAR between Mtb-infected and noninfected macrophages. The interaction network that was derived from DAR in WT cells identified the type I IFN transcriptional regulator IRF7 as the central hub, as well as several other genes involved in type I IFN responses such as IRF3, IRF4, IRF5, DDX58/RIGI1, and STAT2 (Fig. 2D). Additional radial summary graphs from the IPA for △PPM1A and PPM1A+ macrophages are shown in Fig. S5.
Chromatin remodelling during Mtb infection may alter transcription factor binding
We performed transcription factor binding motif (TFBM) analysis with ATAC-seq data to identify the transcription factors (TF) for which DNA accessibility is modulated at their predicted binding sites in WT, △PPM1A, and PPM1A+ cells upon Mtb infection. TFBM enrichment analysis revealed that IRF4, MEF2A, ZNF460, and GABPA were significantly enriched following Mtb infection in WT cells, CTCF, GABPA, RFX2, and ZNF143 in △PPM1A cells, and JDP2, TFE3, and TCFL5 in PPM1A+ cells (Fig. S6). Of these, IRF4, MEF2A, ZNF460, JDP2, and TFE3 were enriched in DAR where chromatin accessibility increased, while CTCF, GABPA, MEF2A, RFX2, TCFL5, and ZNF143 were enriched in DAR where chromatin accessibility decreased (Fig. 3A and B). The TFBM with the most abundant changes upon Mtb infection was ZNF460 in WT cells, ZNF143 in △PPM1A cells, and JDP2 in PPM1A+ cells. Interestingly, MEF2A is a regulator of type I IFN responses in macrophages (Cilenti et al. 2021) and TFE3 is a regulator of autophagy that is repressed during Mtb infection (Pastore et al. 2016, Brady et al. 2018). Other than GABPA, significant enrichment for all other TFBM was unique to the genotype. Overall, motifs that were highly enriched in a specific genotype were also specific to the change in chromatin accessibility (i.e. increased or decreased chromatin accessibility), which support the TFBM analysis. The data also show that PPM1A expression modulated the abundance and strength of TFBM enrichment, and whether enriched TFBM were found at DAR where accessibility increased or decreased following Mtb infection.
Figure 3.

Transcription factor binding motif enrichment in Mtb-infected macrophages. TF binding motifs across all genotypes that were significantly enriched in ATAC-seq peaks with increased or decreased chromatin accessibility were identified; those with enrichment score >2.5 and FDR < 0.001 were considered significant. (A) Enrichment and significance of the selected motifs in DAR with increased chromatin accessibility. Grey dots indicate the percentage of regions that have at least one hit to the motif. Bright red indicates motifs that are statistically significant. (B) Enrichment for the same motifs as shown in panel A, but within DAR with decreased chromatin accessibility following Mtb infection. Bright blue indicates motifs that are statistically significant.
Mtb infection alters the transcriptome in WT, △PPM1A, and PPM1A+ macrophages
We performed RNA-seq in parallel to ATAC-seq to correlate changes to gene expression with the DAR induced by Mtb infection. Three independent replicates of Mtb-infected and noninfected THP-1 macrophages (WT, △PPM1A, PPM1A+) were processed for RNA-seq library generation. There were numerous changes to global gene expression in Mtb-infected WT, △PPM1A, and PPM1A+ cells compared to noninfected cells (Fig. 4A and B). As observed with DAR, there was also a significant increase in the total number of differentially expressed genes (DEG) in PPM1A+ cells compared to WT and △PPM1A cells (Fig. 4A and B). Using edgeR, we identified that Mtb infection induced 410 significant DEG (298 up and 112 downregulated) in WT cells, 508 significant DEG (338 up and 170 downregulated) in △PPM1A cells, and 936 significant DEG (713 up and 223 downregulated) in PPM1A+ cells (Fig. 4C). While PPM1A+ cells had an ∼2-fold increase in total DEG compared to WT and △PPM1A cells, this proportion is considerably less than the difference in total DAR between PPM1A+ cells and WT (22-fold) or △PPM1A (15-fold) cells. We identified 33, 94, and 456 unique significant DEG upon Mtb infection in WT, △PPM1A, and PPM1A+ cells, respectively (Fig. 4C and D). In WT and △PPM1A cells, there were more unique DEG that were downregulated (21 and 61, respectively) than upregulated (12 and 33). In contrast, there were more unique DEG that were upregulated (337) than downregulated (119) in PPM1A+ cells (Fig. 4C). There was 326 DEG shared between WT and △PPM1A, 298 between WT and PPM1A+ cells, 330 between △PPM1A and PPM1A+ cells, and 258 DEG shared between all genotypes (Fig. 4D).
Figure 4.

Mtb infection alters gene expression in WT, △PPM1A, and PPM1A+ macrophages. (A) Global gene expression. Heatmap of global gene expression in WT, △PPM1A, and PPM1A+ cells before and after Mtb infection are shown. Genes with log2CPM > 0 counts were considered significantly expressed. (B) Volcano plots displaying DEG in WT, △PPM1A, and PPM1A+ macrophages upon Mtb infection. DEG with absolute value of log2FC > 1 and FDR < 0.05 were considered significant. Gene names of the most striking up and downregulated genes are shown. (C) Graph showing the numbers of total and unique DEG in each genotype. Red or blue bars show the number of DEG that increased or decreased in expression, respectively. (D) Venn diagram showing the overlap of significant DEG between Mtb-infected WT, △PPM1A, and PPM1A+ macrophages and noninfected controls.
Notable DEG unique to WT cells included upregulated C1QA, which is a strong marker of active TB (Dijkman et al. 2020), and downregulated NOXA, which is involved with nitric oxide mechanisms. DEG unique to △PPM1A cells include IL1A, which is important for Mtb clearance, T cell priming, and granuloma formation (Di Paolo et al. 2015, Lovey et al. 2022). In PPM1A+ cells, several interesting genes are upregulated, including IL24 and IL32, which are known as correlates of protection against TB (Ma et al. 2011, Montoya et al. 2014). PCA analysis of total gene expression data also displayed that the largest contributor to variance in the dataset was genotype. Replicates clustered together based on genotype and infection, confirming the changes to gene expression that we observed (Fig. S7).
Gene expression profiles of Mtb-infected macrophages are enriched in type I IFN responses and antiviral signalling pathways
Gene set enrichment analysis (GSEA) was conducted to identify gene sets that were enriched in significant DEG in WT, △PPM1A, and PPM1A+ macrophages upon Mtb infection compared to noninfected cells (Mootha et al. 2003, Subramanian et al. 2005). We focused the GSEA analysis only on GO biological processes to enable a direct comparison to pathway analysis generated by GREAT in the ATAC-seq pipeline (Fig. 2). The most highly enriched GO biological processes in significant DEG in WT cells upon Mtb infection were ‘Defense response to virus’, ‘Response to type I IFN’, ‘Negative regulation of viral processes’, and ‘Response to IFN-γ’ (Fig. 5A). The same GO biological processes were highly enriched in △PPM1A and PPM1A+ cells, as well as ‘Granulocyte chemotaxis’. Interestingly, the most highly enriched GO biological processes in both △PPM1A and PPM1A+ cells were ‘Co-translational protein targeting to membrane’, which along with ‘Establishment of protein localization to the endoplasmic reticulum’, was enriched in downregulated genes (Fig. 5A). There were more common GO terms between △PPM1A cells and PPM1A+ cell genotypes (Fig. 5B). It is important to point out that enrichment of the same GO terms in genotypes does not necessarily indicate these terms are enriched from differential expression of the same genes, as GO terms comprise large gene sets. GSEA plots generated for the GO biological process terms ‘Response to type I IFN’, and ‘Defense Response to Virus’ denoted the leading-edge genes that most influenced the enrichment of these terms in WT cells (Fig. S8A–C and Fig. 5C). Notably, the expression of several leading-edge genes from these GO terms, such as MMP12, ISG20, WNT5A, and HLA-F (type I IFN signalling), BIRC3, ZC3H12A, TLR8, DDX60, andSTAT2 (Defense response to virus) deviated from the mean in opposite directions in PPM1A+ cells compared to WT cells upon Mtb infection. Similarly, in PPM1A+ cells compared to △PPM1A cells, the expression of the leading-edge genes including ERG1, HLA-E/B, IRF2, TRIM56, MX2, NLRC5, OASL, OAS2, IFI16, and APOBEC3F/G (type I IFN signalling), and NT5C3A, MLKL, NLRC5, and IFI6 (Defense response to virus) deviated from the mean in the opposite direction upon Mtb infection. Additionally, all DEG sets between Mtb-infected and noninfected cells were analysed with QIAGEN IPA. The interaction network that was derived from the significant DEG upon Mtb infection in WT cells identified the type I IFN transcriptional regulator IRF7 as the central hub again, as well as several other genes involved in type I IFN responses such as IRF3, TLR3, TLR9, DDX58/RIGI1, and STAT1 (Fig. 5D). Additional radial summary graphs from the IPA for △PPM1A and PPM1A+ macrophages are shown in Fig. S9.
Figure 5.

GSEA identifies a prominent type I IFN signatures in Mtb-infected macrophages. (A) GSEA was conducted to determine gene sets that were enriched in WT, △PPM1A, and PPM1A+ macrophages upon Mtb infection. Results that were GO biological process terms were selected for analysis. GO terms with FDR < 0.05 and a set size of at least 10 genes were considered significant. GO terms were ranked by the absolute value of the normalized enrichment score (NES) and the top 5 nonredundant terms from each genotype were plotted by NES. (B) Venn diagram displaying common significant GO terms between WT, △PPM1A, and PPM1A+ macrophages after Mtb infection. (C) Differentially expressed leading-edge genes. Two highly enriched GO biological process gene sets (‘Response to Type I IFN’ and ‘Defense Response to Virus’) are shown. The corresponding heatmaps display normalized log2FC differential expression values that were median-centred across all samples of the dataset. (D) Ingenuity pathway analysis was conducted on all significant DEG for Mtb-infected WT, △PPM1A, and PPM1A+ macrophages. The radial summary graph of DEG in WT macrophages is shown. The nodes and edges are colour coded by predicted activation or inhibition: orange represents ‘predicted activation’ (node) or ‘leads to predicted activation’ (edge), and blue represents ‘predicted inhibition’ (node) or ‘leads to inhibition’ (edge). Arrows represent activation, causation, or modification, and perpendicular lines represent inhibition. Solid lines represent direct interactions, dashed lines represent indirect interactions, and dotted lines represent inferred correlation from machine-based learning. The shapes of the nodes are represented as follows: ovals represent transcription regulators, squares represent cytokines, rectangles represent g-protein-coupled receptors, diamonds represent enzymes, hexagons represent canonical pathways, spheres represent ‘other’. Radial summary graphs for △PPM1A and PPM1A+ macrophages are shown in Fig. S9.
Mtb infection triggers a type I IFN response that is concordant across chromatin accessibility and gene expression
We investigated whether changes to chromatin accessibility induced by Mtb infection were linked to correlating changes in gene expression in these regions. Associated DAR and DEG were determined by identifying the gene closest to a DAR, meaning that multiple DAR could be associated with one DEG. In WT cells, 34 genes were differentially expressed and located near one or more DAR (11% of total DAR and 8% of total DEG), with 30 genes showing increased chromatin accessibility and gene expression, and 4 genes showing decreased chromatin accessibility and gene expression (Fig. 6A and Fig. S10A). In △PPM1A cells, there were 44 genes that were differentially expressed and located near one or more DAR (10% of total DAR and 9% of total DEG), where chromatin accessibility and gene expression increased in 31 and 34 genes, and decreased in 13 and 10 genes, respectively (Fig. 6A and Fig. S10A). In PPM1A+ cells, there was 347 genes that were differentially expressed and located near one or more DAR (6% of total DAR and 37% of total DEG), where chromatin accessibility and gene expression increased in 283 and 266 genes, and decreased in 63 and 80 genes, respectively (Fig. 6A and Fig. S10A). In all genotypes, the most significant overlapping genes, with the greatest changes to gene expression were all located in peaks where chromatin accessibility increased (Fig. 6A and Fig. S10A). Importantly, in each genotype, the most significant overlapping genes were predominantly type I IFN response genes, and were among the most significant DEG. For further analysis, we selected a single DAR to pair with DEG for genes with multiple DAR annotated to the same gene. We selected DAR with the greatest absolute logFC upon Mtb infection to pair with DEG. We plotted significant overlapping genes by log2FC gene expression versus log2FC chromatin accessibility to determine the correlation of change between DAR and DEG upon Mtb infection. In WT and △PPM1A cells, the log2FC of overlapping genes were highly positively correlated. In PPM1A+ cells, log2FC of overlapping genes was positively correlated but less so than in WT and △PPM1A cells due to lower maximum changes to gene expression, and an increased number of DAR and DEG that decrease (Fig. 6B). Notably, the majority of DAR that overlapped with DEG were annotated to introns in all genotypes (Fig. S10B), compared to all DAR, which were primarily annotated to ‘Other introns’ and ‘Distal intergenic’ regions (Fig. S2F). We compared the changes to chromatin accessibility and gene expression for the top 10 up or downregulated concordant genes identified in WT cells to the corresponding gene in △PPM1A and PPM1A + cells, ranked by log2FC gene expression (Fig. 6C). In all genotypes, changes to gene expression were greater than changes to chromatin accessibility, except for C1S, IFI44, and IFIT1 in PPM1A+ cells. Furthermore, 3 overlapping genes are unique to WT cells, 16 to △PPM1A cells, and 316 to PPM1A+ cells. To validate some of the strongest concordant identified signals in primary macrophages, we measured changes to gene expression in hMDM upon Mtb infection. As shown in Fig. 6D, we observed striking increases to gene expression in the top 10 upregulated concordant genes identified in WT cells (Fig. 6C), suggesting that our THP-1 model has translational potential to primary macrophages. Importantly, these data also corroborate with recent reports investigating the role of these genes in Mtb infection, such as RSAD2 (Zhou et al. 2022), IFITM genes (Ranjbar et al. 2015), IFIT genes (Madhvi et al. 2022), TRIM22 (Lou et al. 2018: 2), CMPK2 (Arumugam et al. 2021), and OAS genes (Leisching et al. 2019). We also confirmed the upregulation of an additional five genes in hMDMs upon Mtb infection (Fig. 6D) that are consistent with strong changes in chromatin accessibility and gene expression in our dataset, but their role in Mtb infection have yet to be studied.
Figure 6.

Differential chromatin accessibility is concordant with differential gene expression. (A) Boxplot of DEG that are concordant with DAR in WT, △PPM1A, and PPM1A+ macrophages upon Mtb infection. All DAR were split by whether chromatin accessibility increased or decreased upon Mtb infection and then matched to the gene that they were closest to. Each data point represents one DAR–DEG pair, such that genes with multiple associated peaks have multiple points in the plot. (B) Correlation of DAR and DEG. The log2FC of all DEG in each genotype was plotted against the log2FC of matching DAR. In cases where multiple peaks annotated to the same gene, the peak with the greatest absolute logFC was used. (C) Highest concordant DEG and DAR. The top 10 highest DEG that are concordant with DAR in WT cells upon Mtb infection were ranked by log2FC of gene expression. Log2FC values are displayed for each genotype, even if they were not significant. Heatmap colour scales represent the magnitude of change (log2FC) in either DEG or DAR.(D) Primary hMDMs were infected with Mtb at an MOI of 10 and quantitative real-time PCR was used to measure the relative expression levels of RSAD2, IFITM, IFIT1, 2, 3, TRIM22, CMPK2, IFI44, OAS, MX1, 2, PARP14, TREML4, and DDX60L mRNA 48 h post-infection. The ΔΔCT method was used for data analysis by normalizing the Cq values of each gene with the reference gene ACTB. Relative fold expression was normalized to noninfected hMDM. Error bars represent the mean ± SD of six technical replicates from two independent experiments (three replicates/experiment).
Discussion
The only previous report to investigate global chromatin accessibility patterns upon Mtb infection in macrophages was a report by Correa-Macedo et al. where the authors investigated chromatin accessibility and gene expression patterns of primary human alveolar macrophages (hAM) infected with Mtb ex vivo (Correa-Macedo et al. 2021). Impressively, Correa-Macedo et al. report over 12 360 DAR and 1 434 DEG in hAM upon Mtb infection, which provided the first evidence that Mtb induces massive global chromatin remodelling in one of the most clinically relevant host cell types for TB. While our study in THP-1 macrophages produced much lower numbers of total DAR and DEG, which is partially due to the application of more stringent log2FC and FDR cut-offs, many of the genes that were annotated to our identified DAR (RSAD2, MX1, 2, IFIT1, 2, 3, and IFITM3) were consistent with identified DAR in the study by Correa-Macedo et al. Pathway analysis by Correa-Macedo et al. also demonstrated enrichment of type I IFN signalling pathways, while IRF9 and ZNF685 motifs, which are involved with type I IFN signalling responses, were significantly enriched in TFBM analysis (Correa-Macedo et al. 2021). They also reported changes to chromatin accessibility at the TSS of genes with the highest log2FC in their study (CXCL10, IFI44L, APOBEC3A, and MX1), which are among concordant genes with high log2FC that we identified in WT cells and PPM1A+ cells. Our data are generally consistent with studies in dendritic cells, where Mtb infection also triggered marked changes to chromatin accessibility (Pacis et al. 2015). Although Pacis et al. did not perform quantitative and pathway analysis of DAR, they demonstrated that increased chromatin accessibility was associated with DNA demethylation and changes to gene expression. Pacis et al. further uncovered that changes to both chromatin accessibility and gene expression precede changes to DNA methylation in dendritic cells upon Mtb infection. Similar to our observations in macrophages, differential accessibility of multiple TFBM, including AP-1 and IRF family members, and GABPA, changed in dendritic cells upon Mtb infection. However, the accessibility of binding motifs for NF-κB/RelA was significantly increased in dendritic cells upon Mtb infection, underlining differences between immune responses of dendritic cells and macrophages to Mtb infection.
We determined that there were significantly more genes that were upregulated than downregulated upon Mtb infection in macrophages. This is consistent with previous transcriptome analyses of Mtb-infected macrophages (Wu et al. 2012, Seshadri et al. 2017, Papp et al. 2018, Roy et al. 2018, Lee et al. 2019, Looney et al. 2021). We also found that there was almost an equal number of DAR where chromatin accessibility increased or decreased upon Mtb infection in macrophages. This is consistent with previous reports that also demonstrate increases in host chromatin accessibility upon Mtb infection, but DAR where host chromatin accessibility decreases is seldom reported (Chandran et al. 2015, Chen et al. 2017, Hall et al. 2019, Subuddhi et al. 2020, Del Rosario et al. 2022). One report demonstrated chromatin accessibility decreasing in macrophages at the IL12B locus due to Mtb-induced upregulation of HDAC1, which resulted in impaired autophagy (Chandran et al. 2015). Limited data on decreasing chromatin accessibility upon Mtb infection could be due to the ChIP-seq methods used in these studies, which indirectly infer chromatin accessibility by the location of a specific histone mark throughout the genome, thereby limiting these analyses to the probe used. Indeed, decreasing chromatin accessibility and gene repression, is the target of many secreted Mtb effector proteins that directly induce histone modifications (Gauba et al. 2021). Therefore, in the present study, we have captured a complementary representation of global changes to chromatin accessibility by evaluating both increases and decreases to chromatin accessibility.
In chromatin accessibility analyses, PPM1A overexpression contributes to large-scale chromatin-remodelling, where DNA accessibility predominantly increases upon Mtb infection. As PPM1A is a phosphatase, this suggests that certain dephosphorylation events may impact the DNA binding of proteins, such as histone-modifying enzymes or TF, that may mediate opening of chromatin upon Mtb infection. The disproportionately large number of DAR in the PPM1A+ cells may be partially a result of artificially high overexpression of PPM1A (Berton et al. 2022), which may induce global nonspecific dephosphorylation effects or compensatory effects in the cell line due to the broad number of pathways influenced by PPM1A activity (Li et al. 2015, 2022, Berton et al. 2022). We also observe this phenotype through differences in baseline chromatin accessibility and gene expression, compared to WT cells, (Figs 1C, 4A and B), and by PCA (Fig. 1D and Fig. S6). Interestingly, despite that PPM1A+ cells have an ∼20-fold increase in the number of DAR compared to WT cells, this only led to a modest ∼2-fold increase in the number of DEGs. This indicates that cells have strict control at various levels of processes leading to transcription and protein translation that can be compensated, consistent with a recent report of protein dosage compensation (Schukken and Sheltzer 2022). However, the subset of DAR and DEG that are unique to △PPM1A cells compared to WT and PPM1A+ cells indicate that PPM1A activity is also involved in gene repression. This is the mechanism illustrated in Listeria monocytogenes infection where PPM1A dephosphorylates the histone deacetylase SIRT2 to induce chromatin remodelling and gene repression, which is required for infection (Pereira et al. 2018). While PPM1A could impact chromatin accessibility directly, by dephosphorylating chromatin-binding proteins or modulating the activity of a chromatin-modifying enzyme such as SIRT2, we cannot rule out that it might additionally affect chromatin accessibility indirectly, by influencing the expression levels of genes that participate in regulating chromatin accessibility. While the impact of PPM1A deletion on genome-wide changes to chromatin accessibility and gene expression upon Mtb infection is modest compared to PPM1A+ cells, we observed interesting changes unique to △PPM1A cells. Concordant changes to chromatin accessibility and gene expression that are unique to △PPM1A cells include IFI16, a cytosolic DNA sensor (Manzanillo et al. 2012), IL-23, important for anti-TB adaptive immune responses (Khader et al. 2011), IRF9, and three unique enriched TFBM (CTCF, RFX2, and ZNF143) (Fig. 3). Interestingly, PPM1A was demonstrated to inhibit DNA and RNA sensing in the cytosol (Li et al. 2015, Xiang et al. 2016), possibly explaining the increased accessibility and expression of IFI16 in △PPM1A cells that we observed. Enrichment of GO terms ‘positive regulation of neutrophil chemotaxis’ and ‘L-serine catabolic process’ was also unique to △PPM1A cells. The proportionately low number of epigenetic changes in △PPM1A cells may also be accounted for by compensations induced by alternative immunoregulation. Nevertheless, we showed that Mtb infection induces changes in WT, △PPM1A, and PPM1A+ macrophages that are predominantly enriched in type I IFN GO pathways for both DAR and DEG.
The type I IFN response triggered by Mtb infection in macrophages and as a transcriptional signature of TB disease is well characterized (Stanley et al. 2007, Dorhoi et al. 2014, Ji et al. 2019). Numerous transcriptome analyses provide evidence that type I IFN response genes are enriched among upregulated genes in Mtb-infected macrophages, including in alveolar macrophages from TB patients (Stanley et al. 2007, Berry et al. 2010, Singhania et al. 2018, Lavalett et al. 2020). However, the role of type I IFN in TB remains incompletely understood as some evidence supports that upregulated type I IFN responses drive host pathology in TB, while others demonstrate a host-protective effect under certain conditions (Donovan et al. 2017, Moreira-Teixeira et al. 2018). Adding to this knowledge, we demonstrated that significant DAR and DEG in macrophages upon Mtb infection are enriched in type I IFN response pathways, but whether this is host detrimental or beneficial will need to be further elucidated.
Pathways such as ‘Type I IFN signaling pathway’, ‘Defense response to virus’, and ‘Response to IFN-α’ were more enriched in DAR where chromatin accessibility increased, indicating that upregulation of genes in these pathways that we and others observe, occurs at the level of chromatin accessibility (Figs 2C and 5A). We uncovered a small subset of genes in type I IFN response pathways that were both the most significant and highly changed DAR and DEG (Fig. 6C), suggesting that these genes are regulated at the level of chromatin accessibility in macrophages upon Mtb infection. Furthermore, changes in chromatin accessibility and gene expression in these genes were highly correlated (Fig. 6C). There were genes within this subset that were unique to either WT, △PPM1A, or PPM1A+ cells, suggesting that PPM1A modulates chromatin accessibility and gene expression even among this small gene subset (Fig. 6F).
Increased expression of several genes in the subset that we identified are known to restrict Mtb burden, highlighting the relevance of our data. Increased expression of OAS1, 2, and 3, (Leisching et al. 2019: 1), IFI44L (Jiang et al. 2021), and IFITM1, 2, and 3 (Ranjbar et al. 2015) restricts Mtb growth in macrophages and promotes inflammatory cytokine secretion. IFITM3 specifically was observed to increase endosomal acidification by associating with Mtb phagosomes (Ranjbar et al. 2015). Expression of IFIT1, 2, and 3 is also significantly associated with reduced Mtb burden in macrophages and is upregulated in blood cells from TB patients compared to latent TB patients and healthy individuals (Madhvi et al. 2022).
Interestingly, type I IFN are also involved in chromatin remodelling and trained immunity in macrophages. IFN-α stimulation in concert with TNF activity was observed to ‘prime’ chromatin by enhancing tri-methylation of H3K4, which blocked downregulation of inflammatory genes and enhanced their responsiveness (Park et al. 2017). Macrophage stimulation with IFN-β has also been shown to remodel 3D chromatin structure and increases chromatin accessibility at many interferon-stimulated genes. Notably, this effect was prominent at Oas, Mx1, Mx2, and Ifit1 genes (Platanitis et al. 2022), which are among the subset of genes we identified (Figs 4B, 5C, and 6C). In fibroblasts, stimulation with IFN-β also leads to accumulation of H3K36me3 at Oas1, Mx1, and Ifit1, termed ‘memory interferon-stimulated genes’, mediating enhanced upregulation upon re-stimulation (Kamada et al. 2018). Our data showing that induction of type I IFN genes occurs at the level of chromatin accessibility upon Mtb infection supports this unique function of type I IFN as modulators of chromatin structure and trained immunity.
Transcription factor binding motif analysis of ATAC-seq data suggests that MEF2A (or another TF with similar DNA sequence binding preference, such as MEF2B, C, or D) binding is modulated upon Mtb infection in WT macrophages (Fig. 3). MEF2A was shown to critically regulate expression of type I IFN genes in murine macrophages upon bacterial infection. MEF2A-regulated genes included Ifit1, Rsad2, Oasl2, Mx1, and Irf7, while MEF2A deficiency reduced chromatin accessibility at enhancers of proinflammatory genes, and impaired Ifnb1 expression upon Mtb infection (Cilenti et al. 2021). This corroborates our findings that chromatin accessibility of the same type I IFN genes increases upon Mtb infection. As such, type I IFN responses induced by Mtb infection could be mediated by increased or decreased activity of genes regulated by MEF2A given that its binding motifs are enriched in both DAR where chromatin accessibility increases and decreases (Fig. 3). Similarly, MEF2C (Zhao et al. 2022) and MEF2D (Pattison et al. 2020) have both been reported to regulate inflammation and macrophage polarization, which corroborates with our observation that enrichment of MEF2A binding motifs occurs in DAR where chromatin accessibility decreased in WT cells upon Mtb infection (Fig. 3).
Our TFBM analysis also revealed that JDP2 activity is modulated by PPM1A expression upon Mtb infection (Fig. 3). It is, however, important to note that multiple other related family members could bind to the same motifs. JDP2 is member of the large AP-1 family of TF, but acts as a repressor of AP-1 TF, such as c-JUN and ATF2, which are repressed through recruitment of HDAC3 (Jin et al. 2006: 2, Nakade et al. 2007). JDP2 is reported to bind to histones and modulates the activity of histone-modifying enzymes (Jin et al. 2006: 2, Nakade et al. 2007). Interestingly, JDP2 is phosphorylated by JNK, which is inactivated by PPM1A (Katz, Heinrich and Aronheim 2001: 2, Schaaf et al. 2017), and therefore corroborates our findings that JDP2 binding sites are enriched in PPM1A+ cells. The AP-1 family member ATF3 is a negative regulator of IFN-β in macrophages, which aligns with our finding that type IFN responses are less enriched in PPM1A+ cells where JDP2 motifs are highly enriched (Labzin et al. 2015: 3). A third AP-1 family member with potential to bind the enriched JDP2 motif, BATF2, is upregulated in proinflammatory (M1) macrophages upon Mtb infection, and induces the expression of several key inflammatory mediators (Roy et al. 2015). Collectively, enrichment of the JDP2 binding motif in PPM1A+ cells provide evidence that PPM1A activity modulates key inflammatory responses in concert with TF that can change the course of antibacterial immune responses.
A limitation of this study is that we broadly analysed chromatin accessibility based on chromatin structure, when it is regulated by many factors including DNA methylation, miRNA, and lncRNA. Therefore, the changes in chromatin accessibility that we have identified may be mediated by another, or likely multiple mechanisms. To account for this, datasets for DNA methylome and miRNA-ome of Mtb infection could be cross-referenced to extend the mechanistic basis of our conclusions. Methylation-specific PCR could be performed to determine the methylation status of the identified genomic regions of interest, although Pacis et al. demonstrated that changes to DNA methylation come after changes to gene expression, at least in dendritic cells (Pacis et al. 2019). Further analysis of the 3D structure of DNA could be investigated using HiChIP, Hi-C, or other related chromosome conformation analysis methods. It is also important to note that this study was primarily conducted with THP-1 macrophages. We validated differential expression of many identified genes in primary hMDM upon Mtb infection (Fig. 6D), which were consistent with the literature for a subset of these genes (Ranjbar et al. 2015, Lou et al. 2018, Leisching et al. 2019: 2, Arumugam et al. 2021, Madhvi et al. 2022, Zhou et al. 2022), but further investigation of other genes in our dataset is required. Further, it would be prudent to confirm key findings in primary hAM, which are the most clinically relevant model for Mtb infection (Madden et al. 2022).
This study reveals that significant genome-wide changes to chromatin accessibility are induced by Mtb infection of macrophages, and that expression of a subset of type I IFN response genes is mediated by these changes. Our results have implications for TB research because of the major role of type I IFN responses in disabling the human immune response to Mtb infection. This provides a novel way that the identified genes and pathways could be developed as biomarkers or targeted for adjunctive therapy. With the rise in cases of multidrug resistant TB and lack of novel antibacterial therapies, developing potent adjunctive therapy is imperative to combatting the world’s deadliest bacterial pathogen.
Materials and methods
Cell culture
THP-1 monocytes (ATCC TIB-202) and human peripheral blood mononuclear cells (PBMCs) were maintained in RPMI 1640 medium (Gibco, Gaithersburg, MD). RPMI 1640 was supplemented with 2 mM l-glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, and 10% heat-inactivated fetal bovine serum purchased from Gibco. THP–PPM1A+ and THP–△PPM1A cells were generated previously as described (Sun et al. 2016, Berton et al. 2022). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2. THP-1 monocytes were differentiated with 100 ng/ml phorbol ester 13-phorbol-12-myristate acetate (PMA, Alfa Aesar, Haverhill, MA) for 72 h. PBMCs were collected according to approved ethics protocol (#2005388-01H) and isolated via density centrifugation method using the Lymphoprep density gradient medium (StemCell Technologies). Positive selection of monocytes was performed using anti-CD14 coated magnetic particles from StemCell Technologies according to the manufacturer’s protocol. Monocytes were differentiated with 5 ng/ml GM-CSF (Gibco) for 9 days to obtain hMDMs.
Bacterial strains and infection
The M. tuberculosis H37Rv-derived auxotroph strain mc26206 (Sampson et al. 2004, Mouton et al. 2019) was grown in Middlebrook 7H9 medium (BD Biosciences, Franklin Lakes, NJ) supplemented with 0.2% glycerol (Fisher Chemical, Waltham, MA), 0.05% Tween-80 (Acros Organics, Fair Lawn, NJ), 10% OADC (BD Biosciences), 24 μg/ml d-pantothenic acid (Alfa Aesar), and 50 μg/ml l-leucine (Alfa Aesar). Liquid Mtb cultures were maintained at 37°C with slow shaking (50 rpm).
Mtb mc26206 growing in log-phase was quantified by optical density measurement at 600 nm using the conversion of OD 1 = 3 × 108 Mtb bacteria per ml. The number of bacteria required for an MOI of 10 was washed and resuspended in RPMI 1640 cell culture media without antibiotics. Bacteria were added to the differentiated THP–WT, THP–△PPM1A, or THP–PPM1A+ macrophages and incubated at 37°C for 16 h. Three phosphate buffered saline (PBS) washes were then performed to remove extracellular, nonphagocytosed bacteria and infection was continued at 37°C for a total infection time of 48 h. Noninfected control cells underwent identical wash and incubation steps.
ATAC-seq library preparation
ATAC-seq of infected and noninfected macrophages was performed according to published OMNI-ATAC protocols (Buenrostro et al. 2013, Corces et al. 2017). Tn5 transposomes were generated following the protocol of Picelli et al., using pTXB1-Tn5 (gift from Rickard Sandberg, (Addgene plasmid # 60240) (Picelli et al. 2014: 5). Transposomes were used with 2× Tagmentation DNA buffer (20 mM Tris-HCl, 10 mM MgCl2, 20% dimethyl formamide) from the OMNI-ATAC protocol. Macrophages were washed with PBS and harvested from cell culture plates using TrypLE Express Enzyme (Gibco). Fifty thousand Mtb-infected or noninfected THP–WT, THP–△PPM1A, or THP–PPM1A+ cells were processed for ATAC-seq in duplicate. Each transposition reaction was cleaned up individually using Qiagen MinElute PCR purification kits. Transposed DNA libraries were amplified for 4–8 cycles using NEBNext High-Fidelity 2× PCR Master Mix. Individual DNA libraries were amplified using unique dual barcoding primers with unique 5′ i5 and 3′ i7 barcode sequences between the flow cell binding sequence and sequencing primer start site. PCR amplification reactions were purified using MagBio HighPrep PCR clean up kits to remove DNA fragments <50 and >1000 bp. Quality control of our ATAC-seq libraries demonstrated that loci known to be accessible in THP-1 cells at baseline (Cuellar et al. 2017, Phanstiel et al. 2017) were enriched for compared to inaccessible loci (Fig. S2A). Prior to sequencing, the quality of the pooled sample library was determined through bioanalyzer analysis where the size of DNA fragments on average were 52 bp and the concentration was 1.57 ng/µl (Fig. S2B). Pooled DNA libraries were sequenced in one lane of an S1 flowcell on the Illumina NovaSeq 6000 platform at The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, Canada. Libraries were sequenced using 50 bp paired-end reads to reach a target of 30–50 million reads per sample. Following sequencing, quality control analysis confirmed that average Tn5 transposome insert sizes ranged from 100 to 1000 bp (Fig. S2C) and that peaks were enriched in regions 5000 bp up or downstream of the transcriptional start site (TSS) (Fig. S2D).The DNA fragments tagmented by the Tn5 transposase were enriched in loci that are accessible ( RPLP0, PRKAB1) in WT THP-1 macrophages under homeostasis, compared to those that are inaccessible (CHRNA1, NANOG) (Cuellar et al. 2017, Phanstiel et al. 2017).
Raw data processing
Fasta sequence files were analysed for quality using FastQC v0.11.9 (FastQC 2015). Remaining adaptor sequences and low-quality sequence fragments were removed using fastp v0.20.1 with the parameters –length_required 25, –cut_tail, –cut_tail_window_size 4, –cut_tail_mean_quality 20, –disable_quality_filtering (Chen et al. 2018). Filtered reads were aligned to the human hg38 genome using STAR v2.7.5a without gene annotation GTF file, with the parameters –alignIntronMax 1, –alignEndsType Extend5pOfReads12 –alignMatesGapMax 2000, –outFilterMatchNminOverLread 0.50, –outFilterScoreMinOverLread 0.50, and filtering for properly paired mates with a mapq score of 40 or above (Dobin et al. 2013) and duplicates were marked using Picard (Broad Institute, 2022). deepTools AlignmentSieve with parameter –ATACshift was used to adjust sequencing pairs by +5 and −4 base pairs to account for the 9 bp shift introduced at Tn5 insert sites (Ramírez et al. 2016). Genome-wide sequencing coverage was calculated using deepTools v3.5.0 bamCoverage function and then converted to bigWig tracks that were normalized by counts per million (CPM) in each sample. ATAC-seq peak calling was conducted separately on each replicate using MACS2 v2 with q-value of 0.00001 cutoffs, and peaks within blacklisted regions were removed with BEDTools (Zhang et al. 2008). We used rmspc to identify consensus peaks, which we defined at peaks that were found in at least two samples (Jalili et al. 2015). Therefore, peaks that were present only in two replicates of one condition would be retained with parameters -r bio -w 1e-5 -s 1e-10 -c 2 and using the P-value calculated by MACS2 as the ‘value’.
Differential accessibility analysis
Differential peak analysis was performed to compare significant differentially accessible chromatin regions in Mtb-infected macrophages versus noninfected macrophages. To qualify for differential accessibility testing, all peaks were required to have at least 20 Tn5 insertions sites in two samples. Qualifying peaks were analysed with trimmed mean of M-values (TMM) normalization and batch effects were removed with RUVseq using the RUVs algorithm. Empirical tests determined that using a k-value of 2 was sufficient to remove unwanted variation (Risso et al. 2014, Peixoto et al. 2015). Differential peak testing was then performed with edgeR and glmQLFTest function to contrast Mtb-infected cells of each genotype with corresponding noninfected cells (Robinson et al. 2010). Differentially accessible peaks with Benjamini–Hochberg adjusted P-value (FDR) < 0.05 and absolute log 2FC value of at least 1 were considered significant. Heatmaps were generated using the ComplexHeatmap package (Gu et al. 2016). Overlapping peaks between each genotype were calculated and plotted with the BioVenn R package (Hulsen et al. 2008).
Peak annotation and pathway analysis
Significant differentially accessible peaks were annotated to the human hg38 genome using ChIPseeker to determine the genes or regulatory elements in proximity to the genomic locations of peaks. Peaks were annotated to transcription start sites (TSS) within 1 kb of peak centres. The GREAT was used for GO pathway analysis of significant differentially accessible regions. GREAT annotates proximal and distal chromatin regions to their target genes based on prior annotations from the literature (McLean et al. 2010). All differentially accessible peaks passing cut-offs were used as a background to calculate the statistical significance of enrichment using Fisher’s exact test. GO biological pathways with adjusted P-value < 0.05 were ranked by enrichment. GO terms were also evaluated for redundancy, where redundancy was defined as 40% overlap of the genes included in the GO term. Finally, the most highly enriched, nonredundant terms were plotted by significance. Significant DAR from each genotype were subgrouped by log2FC > 1 or log2FC < −1. Each subset was analysed with GREAT as previously described, with the exception that all significant GO terms are displayed for differentially accessible regions where chromatin accessibility decreased in PPM1A+ cells. For pathway analysis using the Human Molecular Signatures Database (MSigDB), genes were annotated to DAR by association with the nearest transcription start site within 50 kb. Overrepresentation analysis was performed using five MSigDB collections (Hallmark pathways, C2: Curated gene sets, C3: Regulatory gene sets, C5: Gene Ontology, and C7: immunologic signature gene sets). Only gene sets containing between 10 and 1000 genes were tested. Gene sets were considered overrepresented if they had >2-fold enrichment, adjusted P-value < 0.05, and at least 10 genes contributing to the enrichment. All significantly differentially accessible regions in each genotype were analysed using QIAGEN Ingenuity Pathway Analysis (IPA) software (Krämer et al. 2014). IPA used Fisher’s exact test with the Benjamini–Hochberg correction, and only terms with FDR < 0.05 were retained. Terms classified as ‘Disease’, ‘Function’, or ‘Upstream Regulator’, had absolute z-score > 2.
Transcription factor binding motif analysis
Transcription factor binding motif (TFBM) analysis was conducted on all significant differential ATAC-seq peaks in WT, △PPM1A, and PPM1A + cells upon Mtb infection. Transcription factor motif enrichment within the sequences was analysed using the R/Bioconductor package universalmotif (Tremblay and Nystrom 2021) and the JASPAR 2022 clustered set of nonredundant vertebrate motifs (Castro-Mondragon et al. 2017, 2022). For all sets of sequences tested for enrichment, the background set was a shuffled version of the foreground using k-let of 2. Motif hits on sequences were recorded if they generated a P-value < 0.0001. Enrichment was calculated with the formula
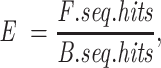 |
where E is the calculated enrichment, F.seq.hits represents the number of foreground sequences with at least one motif hit, and B.seq.hits represents the number of shuffled background sequences with at least one motif hit. This simple formula is appropriate considering that the number and length of sequences in the background and the foreground are equal. Enrichment significance was assessed by Fisher’s exact test and P-values were corrected to generate q-values using the False discovery rate method. Only motifs with E of at least 2.5 and q-value < 0.001 were considered enriched. For motif clusters, the most similar motif from JASPAR was identified using the compare_motifs function of universalmotif.
RNA-seq library preparation
Macrophages were harvested as described for ATAC-seq. A third independent replicate was included for RNA-seq. mRNA extraction was performed using the Aurum™ Total RNA Mini Kit (Bio-Rad) as per the manufacturer’s protocols. All samples were incubated in lysis buffer for 20 min to inactivate bacteria in Mtb-infected samples. After extraction, the quality of mRNA was assessed using the Agilent RNA 6000 Nano LabChip Bioanalyzer. cDNA libraries were generated from mRNA samples with the NEB Ultra II Directional polyA mRNA library prep kit for Illumina. cDNA libraries were then sequenced with the Illumina NovaSeq platform for a total of 100 bp paired-end reads per mRNA sample to reach a target of 30–50 million total reads per sample. cDNA library preparation and sequencing were performed by The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, Canada. Fasta sequence files were analysed for quality using FastQC v0.11.9 (FastQC 2015). Remaining adaptor sequences and low-quality sequence fragments were removed using fastp v0.20.1 with the parameters –length_required 25, –cut_tail, cut_tail_window_size 4, –cut_tail_mean_quality 20, –disable_quality_filtering, and –overrepresentation_analysis (Chen et al. 2018). Filtered reads were aligned to the human hg38 genome using STAR v2.7.5a, with the parameters –outReadsUnmapped Fastx, –outFilterMatchNminOverLread 0.50, –outFilterScoreMinOverLread 0.50 –outSAMmapqUnique 40, –outFilterMultimapNmax 1 (Dobin et al. 2013), and duplicate reads were marked with Picard (Picard Tools). Subread featureCounts was used to assign reads to gene exons and to summarize counts at the gene level (Liao et al. 2014). Represented genes were annotated using biomaRt (Smedley et al. 2009).
Normalization and differential gene expression analysis
Differential expression analysis was performed using edgeR with glmQLFTest function for each comparison as described for ATAC-seq (Robinson et al. 2010). To be retained in the analysis, each gene had counts >10 in at least two samples. Qualifying genes were analysed with TMM normalization and batch effects were removed with RUVseq using RUVr k = 3 (Risso et al. 2014, Peixoto et al. 2015). Genes with log2CPM (counts per million) less than zero were not included for further analysis. Differentially accessible genes with Benjamini–Hochberg adjusted P-value (B–H adj. P-value), or FDR < 0.05 and absolute log 2FC value of at least 1 were considered significant. Heatmaps were generated using the complexHeatmap package and PCA plots were generated with plot PCA function (Gu et al. 2016).
RNA-seq gene set enrichment analysis
GSEA was conducted with all differentially expressed genes, using the R clusterProfiler package with the parameters minGSSize = 10, maxGSSize = 6000, pvalueCutoff = 1.0, pAdjustMethod = ‘BH’, and Eps = 0 (Wu et al. 2021). Only gene sets that were GO biological pathways were considered for further analysis as ATAC-seq pathway analysis was limited to GO biological pathways. GO biological pathways with FDR < 0.05 that contained between 10 and 500 genes were ranked by normalized enrichment score (NES) and considered significant if the NES was >1, or < −1. The GO biological processes that the most significantly enriched in WT cells (‘Type I IFN response’ and ‘Defense Response to Virus’) were selected for further analysis. Gene set enrichment plots were used to determine the leading-edge genes enriched in each pathway. Heatmaps of the leading-edge genes were generated using normalized log 2FC differential expression values were median-centred across all samples of the dataset. All significantly differentially expressed genes in each genotype were also analysed using QIAGEN IPA software, with the same parameters used for ATAC-seq analysis (Krämer et al. 2014).
Integration of ATAC-seq and RNA-seq data
Genes that were both differentially accessible and expressed were determined by matching the genes that chromatin accessibility peaks were annotated to, with DEG. For DEG with multiple chromatin accessibility peaks, the peak with the greatest absolute logFC was selected to be the match for further analysis. A maximum distance of 50 kb between peak centre and the gene TSS was set. Matching genes were considered significant if they had FDR < 0.05 and absolute log 2FC > 1 for both chromatin accessibility and gene expression. A separate dataset including log2FC was generated for all significant overlapping DAR and DEG in WT, △PPM1A, and PPM1A+ cells for comparison. For Fig. 6C overlapping genes were ranked by log2FC gene expression, and the top 10 upregulated or downregulated genes in each genotype were selected. log2FC values were displayed for all of these genes in each genotype, even if they were not significant.
Quantitative real-time PCR
Total RNA was isolated from hMDM using the Aurum Total RNA Mini Kit from Bio-Rad (Hercules, CA). One microgram of total RNA was used in the cDNA synthesis reaction using the iScript Reverse Transcription Supermix (Bio-Rad). Four microliter of synthesized cDNA (out of 20 μl reaction was used to analyse the gene expression of RSAD2, IFITM, IFIT1, 2, 3, TRIM22, CMPK2, IFI44, OAS, MX1, 2, PARP14, TREML4, and DDX60L or the reference gene ACTB by real-time PCR on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Custom primers that were designed according to MIQE guidelines (Bustin et al. 2009) (Table S1) in combination with the Sso Advanced Universal SYBR Green Supermix (Bio-Rad) were used to amplify genes. Thermocycling parameters were 98°C for 3 min, followed by 40 cycles of 98° for 15 s and 51°, 57°, or 65° for 30 s. Gene expression was determined using the △△Cq method (Livak and Schmittgen 2001). Relative fold-expression was estimated as 2^ −ΔΔCq when normalizing to noninfected hMDM as 1.0. Statistical significance was determined using log 2 transformed fold-change paired t-tests for each gene where P< 0.05 considered significant. Analysed by Prism version 9.3.1.
Supplementary Material
Acknowledgements
We thank members of the Sun lab for insightful discussions and the Common Equipment and Technical Service (CETS) at the University of Ottawa for expert technical assistance. We would also like to thank Karen Ho and Sergio Pereira at The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, Canada for assistance with next-generation sequencing, and Compute Canada for access to the Cedar high-performance computing cluster.
Contributor Information
Katrina Madden, Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada; Centre for Infection, Immunity and Inflammation, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada.
Rayan El Hamra, Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada; Centre for Infection, Immunity and Inflammation, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada.
Stefania Berton, Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada; Centre for Infection, Immunity and Inflammation, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada.
Jake Felker, Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada.
Gonzalo G Alvarez, Ottawa Hospital Research Institute, Ottawa, Ontario K1Y 4E9, Canada; School of Public Health, University of Ottawa, Ottawa, Ontario K1G 5Z3, Canada; Division of Respirology, Department of Medicine, The Ottawa Hospital, Ottawa, Ontario K1H 8L6, Canada.
Alexandre Blais, Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada; Centre for Infection, Immunity and Inflammation, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada; Ottawa Institute of Systems Biology, Ottawa, Ontario K1H 8M5, Canada; Éric Poulin Centre for Neuromuscular Disease, Ottawa, Ontario K1H 8M5, Canada.
Jim Sun, Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada; Centre for Infection, Immunity and Inflammation, University of Ottawa, Ottawa, Ontario K1H 8M5, Canada.
Author contributions
K.M., R.E.H., S.B., G.G.A., A.B., and J.S.: conceptualization, methodology, and visualization; K.M., R.E.H., S.B., J.F., and A.B.: investigation; K.M. and J.S.: writing—original draft; K.M., R.E.H., S.B., G.G.A., A.B., and J.S.: writing—review and editing; G.G.A, A.B., and J.S.: supervision, funding acquisition, and resources.
Conflict of interest statement
None declared.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) PJT-162424 and the National Sanitarium Association Scholars Program to J.S., a CIHR operating grant (MOP 119458) to A.B., and the University of Ottawa Faculty of Medicine Translational Research Grant to J.S. and G.G.A. K.M. and R.E.H were supported by graduate scholarships from the University of Ottawa Centre for Infection, Immunity and Inflammation.
Data availability
All ATAC-seq and RNA-seq data are publicly available on the NCBI Gene Expression Omnibus (GEO) under GSE211977.
References
- Arumugam P, Singla M, Lodha R, et al. Identification and characterization of novel infection associated transcripts in macrophages. RNA Biol. 2021;18:604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MPR, Graham CM, McNab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton S, Chen L, Liang YC, et al. A selective PPM1A inhibitor activates autophagy to restrict the survival of Mycobacterium tuberculosis. Cell Chem Biol. 2022;29:1126–39.e12. [DOI] [PubMed] [Google Scholar]
- Bhaskar A, Kumar S, Khan MZ, et al. Host sirtuin 2 as an immunotherapeutic target against tuberculosis. Elife. 2020;9:e55415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosshart H, Heinzelmann M. THP-1 cells as a model for human monocytes. Ann Transl Med. 2016;4:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Martina JA, Puertollano R. Emerging roles for TFEB in the immune response and inflammation. Autophagy. 2018;14:181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute . Picard Tools: a set of command line tools (in Java) for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. 2022. https://broadinstitute.github.io/picard/.
- Buenrostro JD, Giresi PG, Zaba LC, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, et al. The MIQE Guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22. [DOI] [PubMed] [Google Scholar]
- Campo M, Heater S, Peterson GJ, et al. HDAC3 inhibitor RGFP966 controls bacterial growth and modulates macrophage signaling during Mycobacterium tuberculosis infection. Tuberculosis. 2021;127:102062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Mondragon JA, Jaeger S, Thieffry D, et al. RSAT matrix-clustering: dynamic exploration and redundancy reduction of transcription factor binding motif collections. Nucleic Acids Res. 2017;45:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran A, Antony C, Jose L, et al. Mycobacterium tuberculosis infection induces HDAC1-mediated suppression of IL-12B gene expression in macrophages. Front Cell Infect Microbiol. 2015;5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinforma Oxf Engl. 2018;34:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-C, Chao T-Y, Leung S-Y, et al. Histone H3K14 hypoacetylation and H3K27 hypermethylation along with HDAC1 up-regulation and KDM6B down-regulation are associated with active pulmonary tuberculosis disease. Am J Transl Res. 2017;9:1943–55. [PMC free article] [PubMed] [Google Scholar]
- Chen Y-C, Hsiao C-C, Chen T-W, et al. Whole genome DNA methylation analysis of active pulmonary tuberculosis disease identifies novel epigenotypes: pARP9/miR-505/RASGRP4/GNG12 gene methylation and clinical phenotypes. Int J Mol Sci. 2020;21:3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Gutierrez NM, Marzuki MB, et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Science Immunology. 2017;2:eaaj1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilenti F, Barbiera G, Caronni N, et al. A PGE2–MEF2A axis enables context-dependent control of inflammatory gene expression. Immunity. 2021;54:1665–82.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Macedo W, Fava VM, Orlova M, et al. Alveolar macrophages from persons living with HIV show impaired epigenetic response to Mycobacterium tuberculosis. J Clin Invest. 2021;131:148013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueller TL, Herzner A-M, Zhang X et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. The Journal of Cell Biology. 2017;216(11):3535–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigneault M, Preston JA, Marriott HM, et al. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One. 2010;5:e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rosario RCH, Poschmann J, Lim C, et al. Histone acetylome-wide associations in immune cells from individuals with active Mycobacterium tuberculosis infection. Nat Microbiol. 2022;7:312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo NC, Shafiani S, Day T, et al. Interdependence between interleukin-1 and tumor necrosis factor regulates TNF-dependent control of Mycobacterium tuberculosis infection. Immunity. 2015;43:1125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkman K, Lubbers R, Borggreven NV, et al. Systemic and pulmonary C1q as biomarker of progressive disease in experimental non-human primate tuberculosis. Sci Rep. 2020;10:6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma Oxf Engl. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan ML, Schultz TE, Duke TJ, et al. Type I interferons in the pathogenesis of tuberculosis: molecular drivers and immunological consequences. Front Immunol. 2017;8:1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhoi A, Yeremeev V, Nouailles G, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol. 2014;44:2380–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FastQC . FastQC: a quality control tool for high throughput sequence data. 2015. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Gauba K, Gupta S, Shekhawat J, et al. Immunomodulation by epigenome alterations in Mycobacterium tuberculosis infection. Tuberc Edinb Scotl. 2021;128:102077. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinforma Oxf Engl. 2016;32:2847–9. [DOI] [PubMed] [Google Scholar]
- Hall TJ, Vernimmen D, Browne JA, et al. Alveolar macrophage chromatin is modified to orchestrate host response to mycobacterium bovis infection. Front Genet. 2019;10:1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P, Hsu T, Arai M, et al. Specialized transduction designed for precise high-throughput unmarked deletions in mycobacterium tuberculosis. mBio. 2014;5:e01245–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili V, Matteucci M, Masseroli M, et al. Using combined evidence from replicates to evaluate ChIP-seq peaks. Bioinforma Oxf Engl. 2015;31:2761–9. [DOI] [PubMed] [Google Scholar]
- Ji DX, Yamashiro LH, Chen KJ, et al. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat Microbiol. 2019;4:2128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Tsang L, Wang H, et al. IFI44L as a forward regulator enhancing host antituberculosis responses. J Immunol Res. 2021;2021:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Kato K, Chimura T, et al. Regulation of histone acetylation and nucleosome assembly by transcription factor JDP2. Nat Struct Mol Biol. 2006;13:331–8. [DOI] [PubMed] [Google Scholar]
- Kamada R, Yang W, Zhang Y, et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci USA. 2018;115:E9162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson L, Das J, Nilsson M, et al. A differential DNA methylome signature of pulmonary immune cells from individuals converting to latent tuberculosis infection. Sci Rep. 2021;11:19418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz S, Heinrich R, Aronheim A. The AP-1 repressor, JDP2, is a bona fide substrate for the c-Jun N-terminal kinase. FEBS Lett. 2001;506:196–200. [DOI] [PubMed] [Google Scholar]
- Khader SA, Guglani L, Rangel-Moreno J, et al. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol. 2011;187:5402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TS, Jin YB, Kim YS, et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy. 2019;15:1356–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, et al. Causal analysis approaches in ingenuity pathway analysis. Bioinforma Oxf Engl. 2014;30:523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labzin LI, Schmidt SV, Masters SL, et al. ATF3 Is a key regulator of macrophage IFN responses. J Immunol. 2015;195:4446–55. [DOI] [PubMed] [Google Scholar]
- Lavalett L, Ortega H, Barrera LF. Human alveolar and splenic macrophage populations display a distinct transcriptomic response to infection with Mycobacterium tuberculosis. Front Immunol. 2020;11:630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Lee S-G, Kim KK, et al. Characterisation of genes differentially expressed in macrophages by virulent and attenuated Mycobacterium tuberculosis through RNA-Seq analysis. Sci Rep. 2019;9:4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisching G, Cole V, Ali ATet al. OAS1, OAS2, and OAS3 restrict intracellular M. tb replication and enhance cytokine secretion. Int J Infect Dis. 2019;80:S77–84. [DOI] [PubMed] [Google Scholar]
- Li M, Xu X, Su Y, et al. A comprehensive overview of PPM1A: from structure to disease. Exp Biol Med Maywood NJ. 2022;247:453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu G, Sun L, et al. PPM1A Regulates antiviral signaling by antagonizing TBK1-mediated STING phosphorylation and aggregation. PLoS Pathog. 2015;11:e1004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma Oxf Engl. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- Lin D, Xu W, Hong P, et al. Decoding the spatial chromatin organization and dynamic epigenetic landscapes of macrophage cells during differentiation and immune activation. Nat Commun. 2022;13:5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- Looney M, Lorenc R, Halushka MK, et al. Key macrophage responses to infection with Mycobacterium tuberculosis are co-regulated by microRNAs and DNA methylation. Front Immunol. 2021;12:685237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J, Wang Y, Zheng X, et al. TRIM22 regulates macrophage autophagy and enhances Mycobacterium tuberculosis clearance by targeting the nuclear factor–multiplicity κB/beclin 1 pathway. J Cell Biochem. 2018;119:8971–80. [DOI] [PubMed] [Google Scholar]
- Lovey A, Verma S, Kaipilyawar V, et al. Early alveolar macrophage response and IL-1R-dependent T cell priming determine transmissibility of Mycobacterium tuberculosis strains. Nat Commun. 2022;13:884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Chen H-D, Wang Y, et al. Interleukin 24 as a novel potential cytokine immunotherapy for the treatment of Mycobacterium tuberculosis infection. Microbes Infect. 2011;13:1099–110. [DOI] [PubMed] [Google Scholar]
- Madden K, Liang YC, Rajabalee N, et al. Surveying the epigenetic landscape of tuberculosis in alveolar macrophages. Infect Immun. 2022;90:e0052221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan A, Arun KB, Pushparajan AR, et al. Transcription repressor protein ZBTB25 associates with HDAC1-Sin3a complex in Mycobacterium tuberculosis-infected macrophages, and its inhibition clears pathogen by autophagy. mSphere. 2021;6:e00036–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhvi A, Mishra H, Chegou NN, et al. Increased interferon-induced protein with tetracopeptides (IFITs) reduces mycobacterial growth. Front Cell Infect Microbiol. 2022;12:828439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanillo PS, Shiloh MU, Portnoy DA, et al. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe. 2012;11:469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya D, Inkeles MS, Liu PT, et al. IL-32 is a molecular marker of a host defense network in human tuberculosis. Sci Transl Med. 2014;6:250ra114. 10.1126/scitranslmed.3009546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moores RC, Brilha S, Schutgens F, et al. Epigenetic regulation of matrix metalloproteinase-1 and -3 expression in Mycobacterium tuberculosis infection. Front Immunol. 2017;8:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- Moreira-Teixeira L, Mayer-Barber K, Sher A, et al. Type I interferons in tuberculosis: foe and occasionally friend. J Exp Med. 2018;215:1273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira-Teixeira L, Stimpson PJ, Stavropoulos E, et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat Commun. 2020a;11:5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira-Teixeira L, Tabone O, Graham CM, et al. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Nat Immunol. 2020b;21:464–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton JM, Heunis T, Dippenaar A, et al. Comprehensive characterization of the attenuated double auxotroph Mycobacterium tuberculosisΔleuDΔpanCD as an alternative to H37Rv. Front Microbiol. 2019;10:1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade K, Pan J, Yoshiki A, et al. JDP2 suppresses adipocyte differentiation by regulating histone acetylation. Cell Death Differ. 2007;14:1398–405. [DOI] [PubMed] [Google Scholar]
- Ottenhoff THM, Dass RH, Yang N, et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One. 2012;7:e45839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacis A, Mailhot-Léonard F, Tailleux L, et al. Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc Natl Acad Sci. 2019;116:6938–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacis A, Tailleux L, Morin AM, et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015;25:1801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp AC, Azad AK, Pietrzak M, et al. AmpliSeq transcriptome analysis of human alveolar and monocyte-derived macrophages over time in response to Mycobacterium tuberculosis infection. PLoS One. 2018;13:e0198221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Kang K, Giannopoulou E, et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol. 2017;18:1104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore N, Brady OA, Diab HI, et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy. 2016;12:1240–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison MJ, Naik RJ, Reyskens KMSE, et al. Loss of Mef2D function enhances TLR induced IL-10 production in macrophages. Biosci Rep. 2020;40:BSR20201859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto L, Risso D, Poplawski SG, et al. How data analysis affects power, reproducibility and biological insight of RNA-seq studies in complex datasets. Nucleic Acids Res. 2015;43:7664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JM, Chevalier C, Chaze T, et al. Infection reveals a modification of SIRT2 critical for chromatin association. Cell Rep. 2018;23:1124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Van Bortle K, Spacek D, et al. Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol Cell. 2017;67:1037–48.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Reinius B, et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014;24:2033–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanitis E, Gruener S, Ravi Sundar Jose Geetha A, et al. Interferons reshape the 3D conformation and accessibility of macrophage chromatin. Iscience. 2022;25:103840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjbar S, Haridas V, Jasenosky LD, et al. A role for IFITM proteins in restriction of Mycobacterium tuberculosis infection. Cell Rep. 2015;13:874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, et al. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 2014;32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinforma Oxf Engl. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Guler R, Parihar SP, et al. Batf2/Irf1 Induces inflammatory responses in classically activated macrophages, lipopolysaccharides, and mycobacterial infection. J Immunol. 2015;194:6035–44. [DOI] [PubMed] [Google Scholar]
- Roy S, Schmeier S, Kaczkowski B, et al. Transcriptional landscape of Mycobacterium tuberculosis infection in macrophages. Sci Rep. 2018;8:6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson SL, Dascher CC, Sambandamurthy VK, et al. Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in Guinea pigs. Infect Immun. 2004;72:3031–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf K, Smith SR, Duverger A, et al. Mycobacterium tuberculosis exploits the PPM1A signaling pathway to block host macrophage apoptosis. Sci Rep. 2017;7:42101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schukken KM, Sheltzer JM. Extensive protein dosage compensation in aneuploid human cancers. Genome Res. 2022;32:1254–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri C, Sedaghat N, Campo M, et al. Transcriptional networks are associated with resistance to Mycobacterium tuberculosis infection. PLoS One. 2017;12:e0175844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhania A, Verma R, Graham CM, et al. A modular transcriptional signature identifies phenotypic heterogeneity of human tuberculosis infection. Nat Commun. 2018;9:2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Ballester B, et al. BioMart—biological queries made easy. BMC Genomics. 2009;10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SR, Schaaf K, Rajabalee N, et al. The phosphatase PPM1A controls monocyte-to-macrophage differentiation. Sci Rep. 2018;8:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smulan LJ, Martinez N, Kiritsy MC, et al. Sirtuin 3 downregulation in Mycobacterium tuberculosis-infected macrophages reprograms mitochondrial metabolism and promotes cell death. Mbio. 2021;12:e03140–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SA, Johndrow JE, Manzanillo P, et al. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol. 2007;178:3143–52. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subuddhi A, Kumar M, Majumder D, et al. Unraveling the role of H3K4 trimethylation and lncRNA HOTAIR in SATB1 and DUSP4-dependent survival of virulent Mycobacterium tuberculosis in macrophages. Tuberc Edinb Scotl. 2020;120:101897. [DOI] [PubMed] [Google Scholar]
- Sun J, Schaaf K, Duverger A, et al. Protein phosphatase, Mg2+/Mn2+-dependent 1A controls the innate antiviral and antibacterial response of macrophages during HIV-1 and Mycobacterium tuberculosis infection. Oncotarget. 2016;7:15394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay BJ-M, Nystrom S. Universalmotif: import, modify, and export motifs with R. 2021. Bioconductor version: Release (3.13). https://bioconductor.uib.no/packages/3.13/bioc/html/universalmotif.html(1 August 2022, date last accessed).
- Tsuchiya S, Yamabe M, Yamaguchi Y, et al. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. 1980;26:171–6. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Global Tuberculosis Report 2021. Geneva: World Health Organization, 2021. [Google Scholar]
- Wu K, Dong D, Fang H, et al. An interferon-related signature in the transcriptional core response of human macrophages to Mycobacterium tuberculosis infection. PLoS One. 2012;7:e38367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov Camb Mass. 2021;2:100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Zhang Q, Lin X, et al. PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci Adv. 2016;2:e1501889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jiang X, Pfau D, et al. Type I interferon signaling mediates Mycobacterium tuberculosis-induced macrophage death. J Exp Med. 2021;218:e20200887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Di Q, Liu H, et al. MEF2C promotes M1 macrophage polarization and Th1 responses. Cell Mol Immunol. 2022;19:540–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Leung ETY, Wong HK, et al. Unraveling methylation changes of host macrophages in Mycobacterium tuberculosis infection. Tuberc Edinb Scotl. 2016;98:139–48. [DOI] [PubMed] [Google Scholar]
- Zhou X, Zhang Z, Xu H, et al. Viperin impairs the innate immune response through the IRAK1-TRAF6-TAK1 axis to promote Mtb infection. Sci Signal. 2022;15:eabe1621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All ATAC-seq and RNA-seq data are publicly available on the NCBI Gene Expression Omnibus (GEO) under GSE211977.