

Abstract
Regenerative therapeutics hold the promise of self-renewal and repair. Ageing and age-associated neurodegenerative diseases are marked by a decline in self-renewal and repair, but a capacity for regeneration is retained. The challenge faced by researchers developing molecular therapeutics to promote self-renewal in the nervous system is to activate regenerative and repair pathways often in the context of progressive degeneration. Neurosteroids regulate both regeneration and repair systems in the brain, and among this class of molecules, allopregnanolone has been broadly investigated for its role to promote regeneration in both the central and peripheral nervous systems. In the brain, allopregnanolone induced generation and survival of new neurons in the hippocampus of both aged mice and mice with Alzheimer disease, accompanied by restoration of associative learning and memory function. In the brain of mice with Alzheimer disease, allopregnanolone increased liver X receptor and pregnane X receptor expression, reduced amyloid-β and microglial activation, and increased markers of myelin and white matter generation. Therapeutic windows for efficacy of allopregnanolone were evident in the brains of mice with both normal ageing and Alzheimer disease. Allopregnanolone dose and a regenerative treatment regimen of intermittent allopregnanolone exposure were determining factors regulating therapeutic efficacy. Allopregnanolone serves as proof of concept for therapeutics that target endogenous regeneration, windows of therapeutic opportunity for regeneration, and critical system biology factors that will determine the efficacy of regeneration.
Introduction
“Neurosteroids: of the nervous system, by the nervous system, for the nervous system”1 is a maxim of Étienne–Émile Baulieu that has stood the test of time and scientific inquiry. That the brain is a steroidogenic organ is well accepted, and the enzymes required for neurosteroid synthesis have been identified and localized to specific populations of glia and neurons within both the central and peripheral nervous systems.2–4 The wide range of functions of this broad class of molecules spans from the organizational to the activational. Of particular focus here is the neurosteroid allopregnanolone and its capacity to promote regeneration and repair within the ageing mammalian brain and the brain affected by Alzheimer disease. Allopregnanolone promotes neurogenesis and the associated cognitive function, reduces pathology related to Alzheimer disease and promotes regeneration of white matter. The regenerative nature of allopregnanolone action has led to a focus on the potential role of this molecule in prevention, delay and treatment of neurodegenerative diseases such as Alzheimer disease. In consideration of this possibility, this Review will include a discussion of the mechanisms of allopregnanolone action, the key determinants for translating basic understanding of these mechanisms into clinical development, and the challenge of regenerating the brain in a milieu of degeneration. Whilst the focus of this Review is on the neurosteroid allopregnanolone, concepts and considerations herein are relevant to developing regenerative therapeutics for neurodegenerative disease more broadly.
Regenerative potential
Regenerative medicine to prevent, delay and treat age-associated neurodegenerative disease, either by promoting endogenous regenerative systems in the brain or by neural stem cell transplants, holds great therapeutic promise and is rapidly becoming a realistic possibility. Neurodegenerative diseases are complex, dynamic processes that in many instances are woven into the fabric of an ageing brain. Age remains the greatest risk factor for Alzheimer disease; in the vast majority of cases, Alzheimer disease occurs in an ageing brain.5 Neurodegenerative diseases, whilst aetiologically distinct, share multiple features, such as metabolic and mitochondrial defects, protein misfolding and aggregation, dysregulated autophagy and inflammation.5,6 As for all therapeutics, the type, localization and magnitude of pathology influence treatment efficacy. Therapeutic strategies that target and rely on an entire system of regenerative responses to be effective are particularly challenging in the context of an ageing and/or degenerating nervous system.
The regenerative potential of neural stem cells diminishes with increasing age, a phenomenon exacerbated in preclinical models of prodromal and mild Alzheimer disease brains.7–13 In transgenic mouse models of Alzheimer disease, deficits in neurogenesis are readily apparent before the detection of Alzheimer disease pathology, and impairment of neurogenesis progresses in parallel with the ageing process.12–14 Of note, most analyses of neurogenesis during ageing and in neurodegenerative disease have been conducted in male rodents, but the loss of ovarian hormones in the female has a profound effect on the regenerative capacity of the brain and on growth factors that regulate neurogenesis.15,16 Not surprisingly, regenerative capacity during ageing and in disease is multifactorial, in that it relies on integrity of the regenerative system and regulation by niche factors critical to regeneration. Despite decreased neurogenic capacity, the regenerative system of the ageing and diseased brain remains viable until late in life and responsive to molecular interventions, as in the case of allopregnanolone, and to systemic interventions, as in the case of exercise.9,12,13
Among the regenerative factors in decline in the ageing brain is allopregnanolone. In postmortem analyses, concentrations of allopregnanolone were considerably reduced in the brains of humans with Alzheimer disease, and this reduction was correlated with the extent of Alzheimer disease pathology.17 Consistent with this finding in humans, a reduced basal level of allopregnanolone was found in the cerebral cortex of the brain in the triple transgenic mouse model of Alzheimer disease (3×TgAD), which suggests either impairment of allopregnanolone synthesis or accelerated allopregnanolone metabolism.13 Data from comparative analyses of plasma versus cortex levels of allopregnanolone in these mice strongly suggest that reduced allopregnanolone levels in cerebral cortex is a problem specific to the brain and not a problem in peripheral sources of allopregnanolone. The reduced level of allopregnanolone in the cerebral cortex of 3×TgAD mice was evident even in mice treated with allopregnanolone, which was probably due to increased catabolism of allopregnanolone by the multifunctional mitochondrial neurosteroid metabolism enzyme 17-β-hydroxysteroid dehydrogenase (also known as amyloid-β-binding alcohol dehydrogenase) in these mice.18 The plasma versus brain concentration issue becomes critical when considering delivery of regenerative agents to the brain, as plasma levels of an agent do not necessarily predict brain concentrations.
With age, decline in regenerative factors in the neurogenic niche leads to an initial phase in which neural stem cells transition from an actively dividing state to a quiescent state.7,10 The rise in the number of quiescent neural stem cells is driven by a select population, the morphologically distinct horizontal neural stem cells.7,10 Type-1 horizontal cells divide at a greater frequency than the type-1 radial cells and account for a large proportion of the total population of dividing cells within the subgranular zone of the dentate gyrus.7,10 That the quiescent state of horizontal neural stem cells is reversible is important for regenerative therapeutics. Both quiescent horizontal and radial neural stem cells can respond to neurogenic stimuli throughout much of the life span.10 Hippocampal neural stem cells exiting the state of quiescence undergo a series of rapid asymmetric divisions that generate progeny fated to become neurons.7 Subsequent to a limited number of divisions, usually only two, these adult neural stem cells differentiate into mature astrocytes.16 Thus, emerging from the quiescent state into proliferative competence predestines these neural stem cells to exit from ‘sternness’ to become mature astrocytes.7 Eventually, the pool of inducible quiescent neural stem cells diminishes leading to a concomitant decline in the regenerative potential of the ageing brain. Collectively, these data indicate that the ageing brain retains regenerative capacity and this regenerative potential remains responsive to regenerative factors. Whilst the ageing brain and the brain in Alzheimer disease have the capacity to regenerate, factors that promote endogenous regeneration will be limited in their efficacy by stage of the ageing process and/or degeneration.7
A key therapeutic issue to consider is whether it is advantageous to target the quiescent population of neural stem cells to increase their entry into the regenerative pool or to target the neural stem cells that have already exited quiescence to undergo one more round of proliferation before differentiating into mature astrocytes. Both targets have their strengths and liabilities. Increasing the number of neural stem cells exiting quiescence would increase the total number of neurons being generated at any one time at the cost of accelerating the depletion of the neural stem cell pool. Targeting the proliferating population to induce an additional round of proliferation sustains the neural stem cell pool at the risk of losing replicative control or fidelity, thereby increasing the rate of apoptosis.19
Regeneration and restoration
In the central nervous system, allopregnanolone has been identified as a proliferative factor for both neural stem cells and preprogenitor oligodendrocytes.13,20–22 The proliferative action of allopregnanolone extends to the peripheral nervous system where it promotes proliferation of Schwann cells, recovery from spinal cord injury and remyelination of peripheral nerve axons.20,23 In the hippocampus, allopregnanolone-induced mitosis was a serendipitous discovery that was initially observed in cultures of hippocampal neurons and was later replicated in cultures of rat and human neural stem cells.22 In embryonic and adult-derived rat neural stem cells, the magnitude of allopregnanolone-induced proliferation in vitro ranged from 20–30%, whereas a greater magnitude of proliferation, 37–49%, was observed in human-derived neural stem cells.22
Mechanistically, allopregnanolone-induced proliferation activates a well-established regenerative pathway mediated by the GABAA receptor complex (Figure 1). The binding site and mechanism of action involved in allopregnanolone-induced potentiation and activation of the GABAA receptor complex in mature neurons are well known and lead to an influx of chloride (Cl−) to hyperpolarize neurons and reduce excitability. In contrast to mature neurons, allopregnanolone-induced activation of the GABAA receptor complex in neural stem cells leads to depolarization. The expression of the SLC12A2 cotransporter in neural stem cells results in an intracellular concentration of Cl− that is elevated relative to extracellular concentrations of Cl−. As a result of the Cl− concentration gradient being reversed in neural stem cells, activation of the GABAA receptor complex leads to an efflux of Cl−, depolarization of the membrane potential and activation of voltage-dependent L-type calcium channels (Figure 1). Furthermore, the α4, β1 and γ1 subunit composition of the GABAA receptor complex of neural stem cells generates synaptic currents with slow rise and decay kinetics,24 which increases the duration of membrane depolarization leading to activation of voltage-dependent L-type calcium channels.
Figure 1 |.
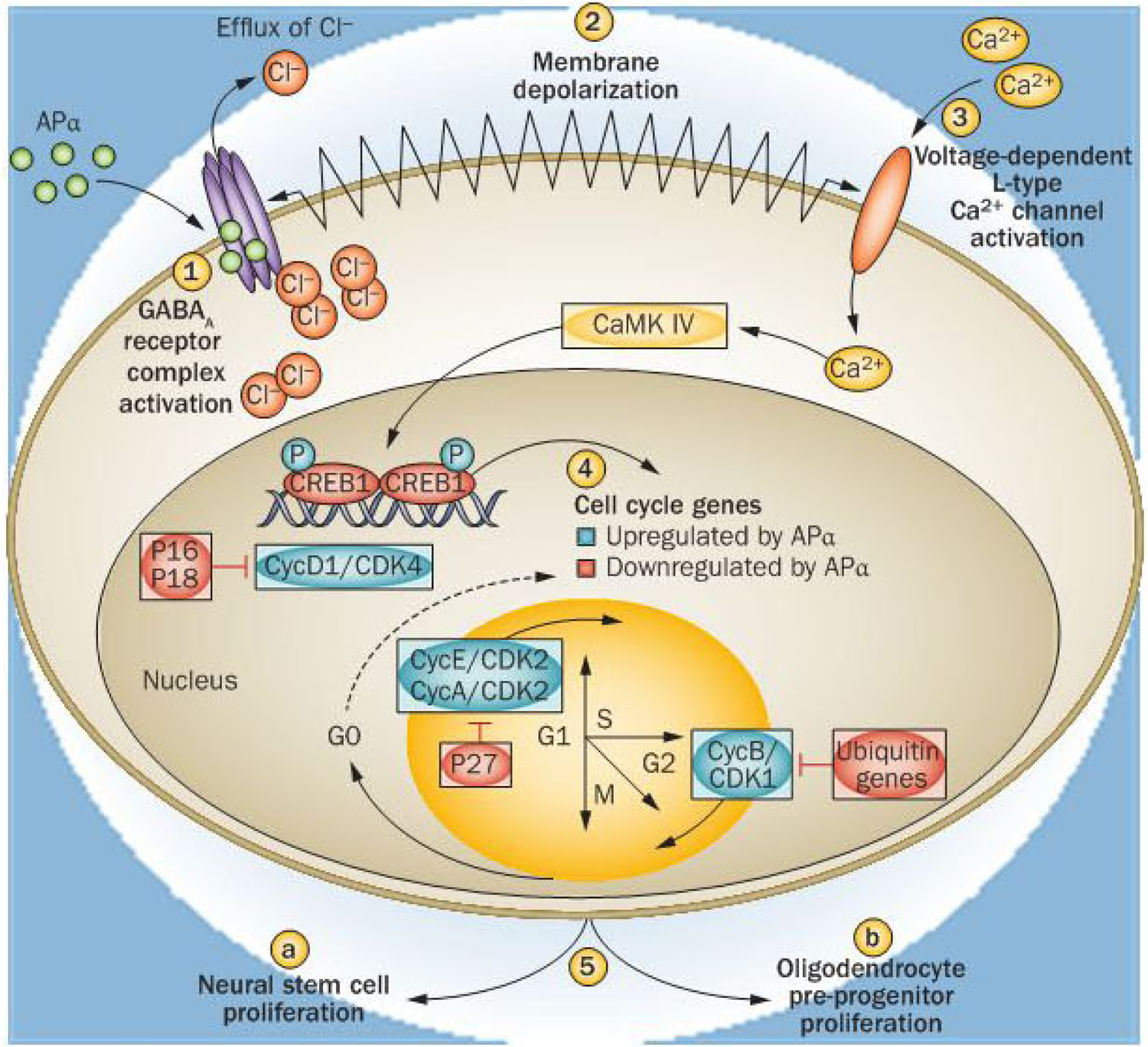
Mechanism of allopregnanolone-induced neural stem cell and oligodendrocyte precursor progenitor mitosis. Allopregnanolone (APα) binds to sites within the transmembrane domains of the GABAA receptor complex to both potentiate and directly activate the GABAA receptor complex (1). APα potentiates GABAA receptor complex responses through binding in a cavity formed by the α-subunit transmembrane domains, whereas direct receptor activation occurs through binding at the interface between the α and β subunits and is enhanced by APα binding to the potentiation site.74 Owing to expression of the SLC12A2 cotransporter in neural stem cells, intracellular Cl− is elevated relative to extracellular Cl−, such that activation of the GABAa receptor complex leads to an efflux of Ch. Efflux of Cl− from the intracellular compartment leads to membrane depolarization (2). Efflux of negatively charged ions leads to membrane depolarization and activation of voltage-dependent L-type calcium (Ca2+) channels β). The subsequent rise in intracellular Ca2+ activates a Ca2+-dependent kinase, CaMK IV, which then phosphorylates and activates the transcription factor cyclic AMP-responsive element-binding protein 1 (CREB1). Through CREB1 activation, APα upregulates expression of cell cycle genes required for transition from GO to S and M phases of the cell cycle (4).22 Simultaneously, genes that express proteins that repress cell division, such as the cyclin-dependent kinase inhibitors P16 and P18 and ubiquitins, are downregulated.22 Successful transition through the cell cycle leads to neural stem cell proliferation in the subgranular zone of the dentate gyrus and oligodendrocyte precursor progenitors in white matter (5). The mechanism of APα-induced neurogenesis takes advantage of the developmentally regulated Cl− gradient to activate a Ca2+-to-CREB signalling cascade to induce mitosis in those cells phenotypically competent to divide whilst not activating this pathway in mature neurons.22,75 Abbreviations: Ca2+, calcium; CDK, cyclin-dependent kinase; Cl−, chloride; Cyc, cyclin; Ub, ubiquitin.
Calcium influx through the L-type calcium channel initiates a downstream signalling cascade to activate the cyclic AMP-responsive element-binding protein 1 and regulation of cell cycle gene expression (Figure 1). Blockade of either the GABAA receptor complex or the L-type Ca2+ channel abolishes allopregnanolone-induced cell cycle protein expression and neural stem cell proliferation,18 whereas SLC12A2 knockdown reduces neural stem cell survival.24 Through this coordinated rapid signalling pathway, allopregnanolone increases expression of genes that promote mitosis and decreases expression of antimitotic genes (Figure 1).22 Activation of the cell cycle by allopregnanolone is also evident in the mouse hippocampus in vivo: allopregnanolone induces an increase in markers of entry into the cell cycle, DNA synthesis and mitosis leading to generation of a considerable increase in the number of newly generated neurons.12,13
In normal ageing mouse brain, allopregnanolone seems to target the state of ageing when the pool of actively proliferating neural stem cells are becoming increasingly quiescent7 to extend the neurogenic capacity of the brain during ageing. In normal young mice, allopregnanolone does not augment an already robust, and perhaps maximal, neurogenic system. A well-described age-related decline in neurogenesis occurs from young to adult mice, during which allopregnanolone does not augment the well-functioning neurogenic system that exists when a fairly small number of the neural stem population is quiescent. However, at 12 months of age, normal mice begin to exhibit a neurogenic response to allopregnanolone that is statistically significant at 15 months of age.12,13 In both aged and 3×TgAD mice (see below), exhaustion of the regenerative system coincides with loss of response to allopregnanolone and probably to other factors that promote endogenous neurogenesis, which indicates a window of opportunity for promoting endogenous neural stem cells for regeneration. Outcomes from analyses in the normal mouse brain indicate a threshold for proliferation that is kept in check and that a deficit in proliferation that occurs in the brain of ageing and 3×TgAD mice is required for allopregnanolone-induced regeneration.
Regenerative deficits have been observed in multiple transgenic mouse models of Alzheimer disease, including the 3×TgAD mice.9,12,14,25 Depending on the transgenic familial model of Alzheimer disease, deficits in neurogenesis are evident in both regenerative zones of the adult rodent brain, the dentate subgranular zone and the subventricular zone. In the case of the 3×TgAD mouse, neurogenic deficits are evident in both the subgranular zone (documented in both males and females) and subventricular zones (documented in males).12,14,25 At each age of 3×TgAD mice, the subgranular zone exhibits a significantly lower magnitude of neurogenesis than that observed in wild-type mice of the same age, such that the 3×TgAD mouse brain seems to be on an accelerated path of regenerative ageing.12,14,25
Allopregnanolone reversed neurogenic deficits in the hippocampus of young and ageing 3×TgAD mice and restored regenerative potential to a magnitude comparable to that of age-matched normal wild-type mice (Figure 2). Importantly, allopregnanolone significantly increased both the number and survival of newly generated cells.12,13 Three features of allopregnanolone regulation of neurogenesis are important to consider. First, allopregnanolone restored the regenerative potential of the brain to normal, not to supra-normal.12,13 Second, the regenerative effect of allopregnanolone is dose dependent, with a classic growth factor inverted-U-shaped dose–response curve,13,22 whereby exceeding the neurogenic dose does not lead to greater response. Both of these characteristics indicate that the regenerative system affected by allopregnanolone is tightly regulated, with closely guarded thresholds for both activation and magnitude of proliferation.
Figure 2 |.
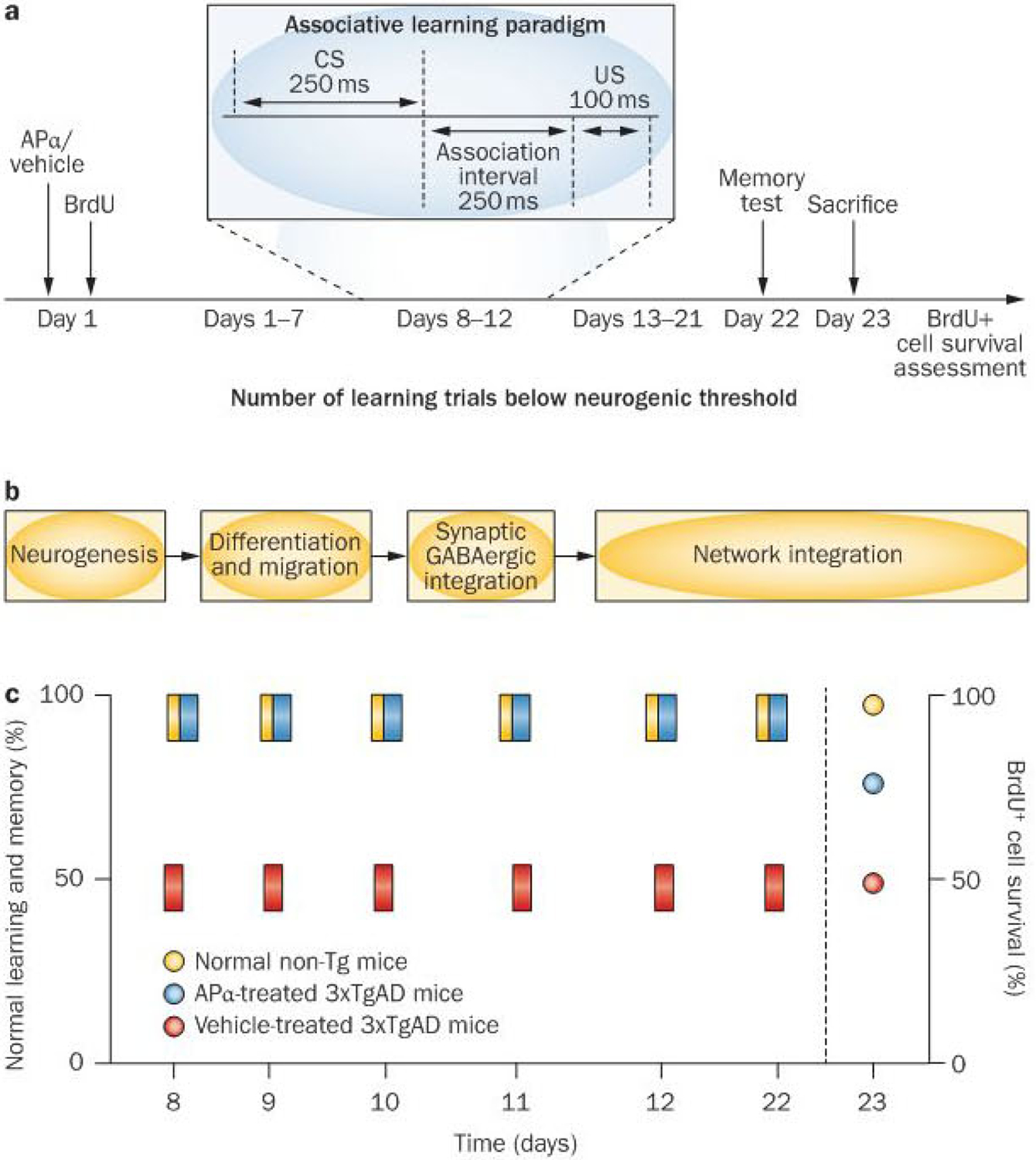
Paradigm of allopregnanolone treatment, behavioural assessments and survival of newly generated neurons in wild-type mice and in the 3×TgAD mice across the life course. APα-induced neurogenesis and the relationship with associative learning and memory was tested using a behavioural paradigm aligned with the time course of proliferation, migration and integration.28 Data summary refers to age-matched mice of 3, 6 and 9 months of age. a | At each age tested, 3×TgAD and wild-type mice received a single injection of APα followed by BrdU on day 1 and returned to their cage for 1 week. Following the associative learning phase, mice were returned to their home cage for a week (days 13–21) followed by a memory trial at day 22 post APα injection.12,13 b | During the week following APα treatment, newly generated cells would have migrated from the subgranular zone into the granule cell layer of the dentate gyrus. Associative learning assessed using the classic trace eye-blink conditioning paradigm was selected because of its hippocampal dependency and well-characterized relationship to neurogenesis.30 To ensure that the learning paradigm did not generate new neurons, association pairings were reduced to one-third of what is required to generate new neurons or to effect their survival.30 During the associative learning phase (days 8–12), cells generated 1 week earlier would have extended dendritic growth into the granule cell layer and molecular layer with an initial emergence of axons into the hilar region of CA3, and would have received GABAergic synaptic input. Also during this time, these cells show increased synaptic plasticity and long-term potentiation.28 c | Summary of learning performance over the days 8–12 of associative conditioning. Vehicle-treated 3×TgAD mice had modest-to-no associative learning. By contrast, learning performance of APα-treated 3×TgAD mice was comparable to that of normal age-matched wild-type mice, which was sustained during the 5 days of training.12,13 APα restored memory function to normal whereas vehicle-treated animals had no improvement in memory. Survival of BrdU-labelled cells generated on day 1 was maximal in the wild-type mice, ~75% of normal value in the 3×TgAD mice and no difference for the vehicle-treated mice. Memory performance and number of surviving BrdU-positive cells were significantly correlated (0.8, P<0.01) in the APα-treated 3×TgAD mice.12,13 Abbreviations: APα, allopregnanolone; BrdU, 5-bromo-2-deoxyuridine; GABA, γ-aminobutyric acid; Tg, transgenic; 3×TgAD, triple transgenic model of Alzheimer disease.
The third feature of allopregnanolone regulation of neurogenesis is that the regenerative effect of allopregnanolone co-varies with age and Alzheimer disease pathology burden. Allopregnanolone was neurogenically active in the hippocampus of 3×TgAD mice at 3, 6 and 9 months of age. At 6 and 9 months of age, intraneuronal amyloid-β is evident and at 9 months of age, extraneuronal amyloid-β plaque can be detected. At 12 months of age, when the hippocampus is heavily burdened with extraneuronal amyloid-β plaque,8 allopregnanolone is no longer effective, which is consistent with earlier reports on amyloid-β plaque inhibition of neurogenesis.26 In contrast to the aged 3×TgAD mouse brain, allopregnanolone promotion of neurogenesis was initially evident at 12 months of age and was statistically significant at 15 months of age in wild-type mice.12
A mounting body of evidence over the past decade indicates a strong relationship between neurogenesis and some, but not all, hippocampal-dependent cognitive functions.27–29 One type of behaviour that seems to be dependent on the generation of new neurons in the dentate gyrus is associative learning and memory across time.26–28 To test the effect of allopregnanolone-induced neurogenesis on cognitive function, a unique treatment paradigm was used. First, 3×TgAD mice were treated once at T0 with allopregnanolone or vehicle, followed by 5-bromo-2-deoxyuridine (BrdU) to label proliferating cells in the subgranular zone of the hippocampus, after which the mice were returned to their cages for 7 days before 5 days of associative learning trials (Figure 2a). Mice were again returned to their cages for another 9 days, followed by a test of their memory function, at which point (22 days after allopregnanolone treatment) the mice were sacrificed for detection of surviving BrdU-positive cells (Figure 2a).
As with the deficit in proliferation, the 3×TgAD mice exhibited deficits in learning and memory (Figure 2c).12,13 In this paradigm, allopregnanolone reversed the cognitive deficits of 3×TgAD mice and restored both learning and memory performance to the level of normal wild-type mice. Furthermore, allopregnanolone-induced survival of neural progenitors was significantly correlated with allopregnanolone-induced memory performance.12,13 Learning and memory function in allopregnanolone-treated 3×TgAD mice was twofold more than that in the age-matched vehicle-treated group and, in general, was comparable to the maximal normal wild-type mouse performance. As with the regenerative response, the effect of allopregnanolone on cognitive function was a restoration of learning and memory to normal, not supranormal, levels. The design of behavioural analyses makes it unlikely that mechanisms other than neurogenesis could account for the restoration of cognition (Figure 2).
The unique properties of newly generated neurons integrating into networks within the dentate gyrus provide a system-level understanding of how a single exposure of allopregnanolone could affect learning and memory function in the weeks that followed the experiment (Figure 2b,c). Within 8 days of birth, neural progenitors within the dentate gyrus receive GABAergic inputs, followed by glutamatergic inputs 10 days later (that is, 18 days after birth).28 During this circuit integration phase, newly arrived neurons have a reduced threshold for induction of long-term potentiation coupled with increased synaptic plasticity, a phenotype long proposed as critical for neural network formation. Activity and experience during the early period of residence within the dentate gyrus is critical for survival and integration of these newly arrived neurons into circuit networks.28 Interruption of the process required for network integration results in reduced survival of newly generated neurons and in deficits in long-term memory.30
Given the timeline for generation and integration of newly generated neurons, allopregnanolone-induced neurogenesis at day 1 would have generated neurons arriving in the dentate gyrus the following week (Figure 2b). During the associative learning phase, these neurons would have received GABAergic inputs during the period of reduced threshold for long-term potentiation and increased synaptic plasticity, which would be manifested as increased learning (Figure 2b,c).28 By day 21, these neurons would have received glutamatergic inputs and integrated into dentate and CA3 neural circuits, which would be manifested as memory of the learned association (Figure 2b,c).28 Allopregnanolone-induced neurogenesis, survival of newly generated neurons and behavioural outcomes are consistent with a conceptual framework that links the unique properties of immature neurons in the dentate gyrus to associative learning across time and with the memory functions of the entire hippocampal circuit.28
White matter regeneration
White matter degeneration occurs with advanced age in persons at risk of Alzheimer disease and is an early event in the pathophysiology of Alzheimer disease.31–34 White matter deficits in preclinical models of Alzheimer disease mirror the early deficits found in human brain.34–36 In vitro, allopregnanolone increased proliferation of oligodendrocyte progenitors and axonal myelination.20 The above-mentioned mechanism by which allopregnanolone promotes neurogenesis in hippocampal-derived neural stem cells (Figure 1) also applies to oligogenesis.20 In both normal wild-type mice and 3×TgAD mice, allopregnanolone increased expression of a marker of myelin generation, CNPase.35 The allopregnanolone-induced increase in CNPase expression was particularly robust in the 3×TgAD mouse brain and occurred within the hippocampus and entorhinal cortex,35 the regions found to be most vulnerable to white matter loss in this Alzheimer disease model.36
Myelin generation is dependent on high levels of cholesterol,37 and synthesis of cholesterol requires expression of HMG-CoA reductase, the enzyme for the rate-limiting step in cholesterol synthesis. In the brains of both normal and 3×TgAD mice, allopregnanolone increased expression of HMG-CoA reductase to an extent necessary to meet the demand for cholesterol required for myelin membrane growth.35,37 In parallel, a feed-forward mechanism occurs in precursor oligodendrocyte progenitors in which intracellular transport of cholesterol into mitochondria through the translocator protein located on the outer mitochondrial membrane and its interaction with the steroidogenic acute regulatory protein to initiate neurosteroidogenesis of allopregnanolone.20,38 The local synthesis of allopregnanolone in oligodendrocytes creates an autocrine/paracrine mechanism that could sustain oligogenesis and cholesterol metabolism. Although the allopregnanolone–FIMG-CoA reductase–CNPase cascade is plausible, it is not yet proven and requires further investigation.
Regeneration in a degenerating brain
A major challenge for regenerative strategies that target age-associated neurodegenerative diseases is that the burden of pathology progressively increases with age and duration of disease. Thus, regenerative therapeutic regimens, while principally targeting the regenerative system of the brain, must also address pathological burden of the disease. Alzheimer disease carries with it a prodromal or preclinical period that can begin 10–20 years before clinical diagnosis.39,40 Deficits in brain metabolism, white matter degeneration and extracellular amyloid-β plaques occur early in the disease process and precede clinical diagnosis.32,41–43 As the disease progresses, additional pathologies develop including neurofibrillary tangles, reactive microgliosis and ultimately loss of neuronal populations.40
Chronic administration of allopregnanolone led to the discovery that this drug reduced the burden of Alzheimer disease pathology in the 3×TgAD mouse brain (Figure 3).35 Allopregnanolone reduced the generation of intracellular amyloid-β oligomers, in particular Aβ*56, the oligomeric form associated with synaptic dysfunction.44 Consistent with the decrease of intracellular amyloid-β oligomers, expression of the mitochondrial protein amyloid-β-binding alcohol dehydrogenase was also significantly reduced.35 Therefore, Allopregnanolone seems to act upstream of amyloid-β plaque generation to reduce it.
Figure 3 |.
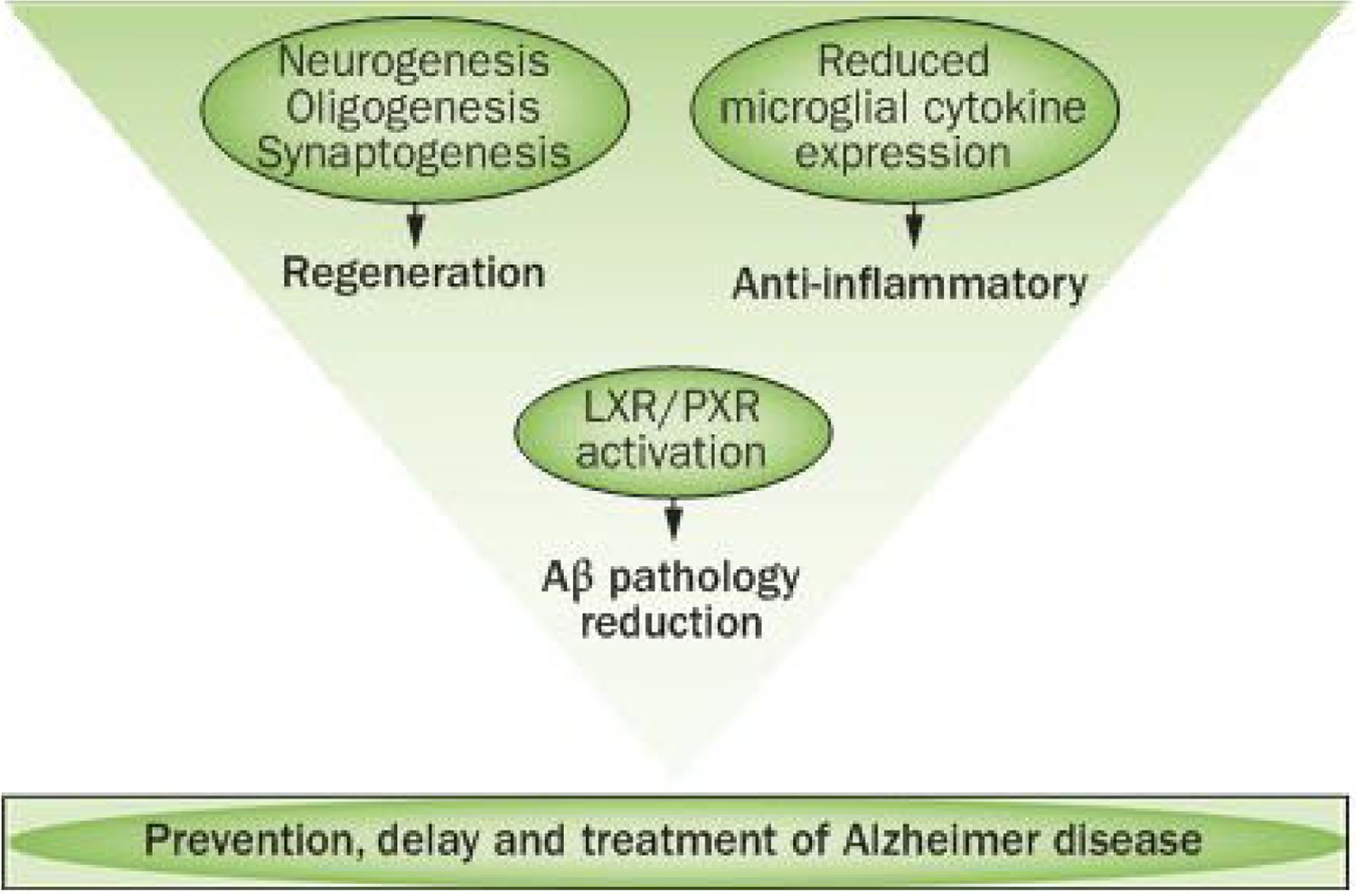
Targeting regeneration and disease mechanisms promotes therapeutic efficacy. Ageing and neurodegenerative diseases such as Alzheimer disease are systems-level changes in brain. As such, therapeutic interventions that regulate systems of responses rather than single targets with single outcomes might be required to achieve a therapeutic benefit. Allopregnanolone functions as a systems-level, pleiotropic therapeutic agent to promote the regenerative system of the brain to renew neuronal and myelinating oligodendrocyte populations, while also activating mechanisms that decrease the burden of pathology. Pleiotropic therapeutic agents that target the endogenous regenerative system of the brain, whilst also activating pathways that reduce neurodegenerative pathology, have the potential to substantially increase the probability of translational success to prevent, delay and treat neurodegenerative disease in the ageing brain. Abbreviations: Aβ, amyloid-β; LXR, liver X receptor; PXR, pregnane X receptor.
The mechanisms underlying allopregnanolone-induced reduction in amyloid-β generation are multifaceted. Evidence is mounting for a pathway involving glial cells and a complex interplay between transcription factors liver X receptor (LXR, also known as NR1H3) and pregnane X receptor (PXR, also known as NR112) to regulate cholesterol homeostasis. As mentioned above, allopregnanolone increases cholesterol synthesis.35 A rise in cholesterol synthesis has the potential to be a doubled-edged sword if not regulated, as cholesterol-laden lipid rafts activate γ-secretase to cleave amyloid precursor protein to generate amyloid-β1–40 and amyloid-β1–42,45 thereby negating the allopregnanolone-induced decline in amyloid-β oligomers.
Allopregnanolone induction of both LXR and PXR provides mechanistic strategies to activate cholesterol transport, metabolism and efflux.35,46 LXR functions as a cholesterol sensor and is activated by cholesterol oxidation products, termed oxysterols. In brain, the primary oxysterol 24(S)-hydroxycholesterol, activates LXR to increase transcription of cholesterol efflux pathways, specifically the ABC transporters ABCA1 and ABCG1, which facilitate transfer of cholesterol to apolipo-protein E for transport between glial and neuronal compartments or across the blood-brain barrier for efflux to the periphery.47–49 Activation of the LXR pathway is associated with significant reduction in amyloid-β generation and recovery of cognitive function in multiple preclinical models of Alzheimer disease.46,48,50,51
Although transport and efflux of cholesterol and its enzymatic oxysterol derivative 24(S)-hydroxycholesterol is primarily driven by LXR-mediated responses in glial cells,51 PXR provides a neuronal protective mechanism against cholesterol oxidation products52 principally generated by free radicals that produce lipid peroxides. PXR is an integral component of the cellular defense mechanisms activated by a broad array of lipophilic molecules, including lipid peroxides.53 Once activated, PXR regulates the expression of cytochromes P450, conjugating enzymes and transporters required for detoxification and clearance.54 Of particular relevance are PXR-regulated gene products that target nonenzymatically derived cholesterol oxidation products formed as a consequence of oxidative stress.55 Within the brain, PXR transcriptionally regulates P450 enzymes including CYP3A4,53 CYP3A11 and CYP3A13. CYP3A11 and CYP3A13 are highly expressed in hippocampal pyramidal and granule neurons.56 An allopregnanolone-induced increase in PXR protein expression and, by extension, transcriptional outcomes would result in an increase of CYP3A13.49 Within the blood–brain barrier, CYP3A expression is present in microvessel smooth muscle cells, ependymal cells and choroid plexus.56 Together, LXR and PXR act synergistically to regulate clearance of both enzymatic and nonenzymatic cholesterol-derived oxysterols from the brain, thereby reducing neurotoxicity associated with both products.
From a therapeutic development perspective, micromolar concentrations of multiple steroids, including pregnenolone and allopregnanolone, activate PXR. High levels of pregnenolone, the first product in the cholesterol–steroid synthesis pathway, are indicative of excessive cholesterol. Thus, a regulatory feedback loop between products of cholesterol metabolism and their clearance is established through activation of PXR. It is highly unlikely that endogenous levels of allopregnanolone reach micromolar concentrations; however, exogenous chronic administration of allopregnanolone could easily reach that level in brain.57 Therefore, as with all therapeutics, including allopregnanolone, it is critical to develop mechanism-based doses and treatment regimens for activators of LXR and PXR to promote therapeutic benefit and to prevent adverse outcomes of excessive exposure.
Inflammation is a feature of neurodegenerative diseases in general and of Alzheimer disease in particular.6 Consistent with increased clearance of cholesterol and reduction in amyloid-β generation, allopregnanolone significantly reduced microglia activation in the 3×TgAD mouse model (Figure 3).35 In multiple preclinical models of neurodegenerative disease, allopregnanolone reduces the inflammatory response. Allopregnanolone reduces microglial activation and proinflammatory cytokine gene expression in preclinical models of Niemann-Pick type C disease,52,58 traumatic brain and spinal cord injury,59 and multiple sclerosis.60 Whether allopregnanolone directly activates anti-inflammatory pathways or whether the decline in inflammatory indicators is a consequence of reduction in pathology is unknown, but the magnitude of the anti-inflammatory effect is more consistent with the extent of decline in pathology.
The capacity of allopregnanolone to modify burden of disease and to activate plausible mechanisms for reducing pathology meets a critical therapeutic challenge for repair to enable self renewal. Collectively, the regenerative processes of neurogenesis, oligogenesis and synaptogenesis (as evidenced by restoration of learning and long-term memory), coupled with reduction in pathological burden, provide multiple lines of preclinical evidence of efficacy that support the potential for allopregnanolone as a safe and efficacious therapeutic agent for regeneration and repair (Figure 3).
Therapeutic hurdles and opportunities
Regeneration is a developmental process that has obligatory temporal and spatial requirements. These obligatory requirements extend to and will define effective regenerative therapeutics. In the case of ageing and Alzheimer disease, regeneration will occur within anatomically defined regions of the adult brain with varying degrees of responsive neural stem cell pools and neuropathology. Therapeutics and regimens of treatment that promote endogenous regeneration and are synergistic to and temporally aligned with renewal processes in vivo are more likely to translate from preclinical to clinical efficacy (Figures 2–4). In parallel, the therapeutically driven regenerative response will occur in the context of disease and pathological burden (Figures 2–4). Translationally, dose and treatment regimens will need to be tailored to simultaneously maximize both a regenerative response and a disease-modifying outcome.35 Furthermore, as neurodegenerative diseases are chronic, it is likely that the treatment will be as well.
Figure 4 |.

Optimization of allopregnanolone treatment regimens for regeneration and repair. The processes of renewal and repair require time and occur in stages in an obligatory sequence. Neurogenesis occurs over the course of hours, in the case of transitioning through the cell cycle, to months in the case of full integration into hippocampal circuitry and networks. Activating both regenerative and repair systems to maximize therapeutic benefit in the ageing or degenerating brain requires simultaneous assessment of both processes and investigation of multiple dosing and regimes to arrive at an optimal course of therapy. In the case of APα, a once per week treatment regimen with a single dose of APα (10 mg/kg) was optimal to promote neurogenesis while also activating systems that reduced Alzheimer disease pathology.35 By contrast, constant infusion treatment regimens over the course of months are antiregenerative and result in adverse outcomes.57 Abbreviation: APα, allopregnanolone.
To this end, our group investigated a treatment regimen to achieve both regeneration and disease-modifying outcomes. Results of these preclinical translational analyses in 3×TgAD mice indicated that an allopregnanolone treatment regimen of once per week over the course of 6 months was optimal (Figure 4).35 Using this treatment regimen, allopregnanolone significantly increased survival of newly generated neurons, simultaneously reduced amyloid-β generation in the hippocampus, cortex, and amygdala, and reduced microglial activation. Remarkably, intermittent allopregnanolone treatment regimens have shown benefit in multiple preclinical neurodegenerative disease models including Niemann-Pick type C,61 traumatic brain injury,59 diabetic neuropathy,23 peripheral nerve crush injury,23 multiple sclerosis60 and Parkinson disease.62 By contrast, continuous infusion of allopregnanolone for several months is not efficacious and can worsen pathology in a preclinical model of Alzheimer disease (Figure 4).57 The benefit of intermittent treatment regimens might apply to other regenerative factors where constant infusion for extended duration has met with adverse outcomes.
An extensive body of safety evidence exists for both acute and chronic exposure to allopregnanolone along with phase I safety data for allopregnanolone in humans.46 In addition, a wide range of allopregnanolone doses, routes of administration and treatment regimens have been studied and outcomes have been reported.46 A sedative hypnotic and/or antiseizure effect of allopregnanolone has long been known; therefore, allopregnanolone has been extensively investigated as a potential therapeutic for epilepsy.63–65 In humans, as in animals, sedation impairs cognitive function, which is consistent with a report of memory impairment in humans 10 minutes following infusion of a sedative dose of allopregnanolone, although the deficit was 0.2 word and no effect was seen on either semantic or working memory functions.66 As with all therapeutics, the dose of allopregnanolone matters. Routes of administration that are intermittent and avoid the ‘first pass’ effect through the liver (that is, non-oral routes) exert long-term efficacy in the brain.46 A clinical trial of cGMP formulated allopregnanolone for traumatic brain injury is ongoing.67
Considerable controversy surrounds reducing amyloid-β once it has been deposited in plaques in the brain as numerous clinical trials using agents that remove these extracellular plaques have failed.68–72 However, the mechanism by which allopregnanolone reduces amyloid-β is upstream of amyloid-β production and deposition into plaques;35 therefore, treatment with allopregnanolone is unlikely to induce the amyloid-β-related abnormalities on imaging that are associated with antibodies to amyloid-β.69 Antibodies against amyloid β bind to and remove this protein through the microvasculature of the brain or from the vascular wall, and are associated with microhaemorrhages.
Relevant to therapeutic development, allopregnanolone passes three of the four criteria of ‘Lipinski rule of five’ for predicting the success of a drug by the oral route of administration (Box 1). Translationally, the poor aqueous solubility of allopregnanolone makes oral administration of the drug challenging but not insurmountable.59 Moreover, formulation of allopregnanolone in β-cyclodextrins, which have been extensively tested in preclinical stages, provides an FDA-approved strategy to deliver allopregnanolone via subcutaneous, intramuscular, intranasal or intravenous routes of administration. Regenerative therapies to prevent, delay and treat age-related neurodegenerative diseases will be administered chronically and, therefore, will require Investigational New Drug (IND)-enabling toxicology to address long-term chronic exposure. Advances in pharmaceutical formulation of steroids coupled with extensive libraries of neurosteroid analogues create a favourable regenerative therapeutic development landscape.
Box 1 |. Allopregnanolone and the Lipinski rule of five78.
Lipinski’s rule of five is a mnemonic developed by Christopher A. Lipinski and colleagues for predicting success of a drug by oral route of administration. They found that four criteria with values close to multiples of five were predictive of oral availability.78 Whilst the rules are based on requirements for oral administration of a drug, they are widely applied as an initial indicator for drug development. The rule of five predicts that poor absorption or permeation are likely if: 1) there are more than five hydrogen donors (allopregnanolone has one); 2) the molecular weight is more than 500 Da (the molecular weight of allopreganolone is 318.49 Da); 3) the Log P value is over five (allopregnanolone is 5.042); and 4) there are more than 10 hydrogen acceptors (allopregnanolone has two). Allopregnanolone meets three of the four Lipinski rules, whereas it does not meet the requirement for a Log P value less than five. The Log P value is an indicator of lipophilicity. Molecules with a high octanol to water partition constant are preferentially distributed to hydrophobic compartments such as lipid bilayers of cells, whereas hydrophilic molecules (low octanol to water partition coefficients) are preferentially distributed in hydrophilic compartments such as blood serum. A Log P value of slightly greater than five indicates that allopreganolone will preferentially distribute to lipophilic compartments which is a feature typical of steroid molecules.
Windows of therapeutic efficacy are evident preclinically and undoubtedly will be true clinically (Figure 5). Factors that promote endogenous regeneration such as allopregnanolone and growth factors require a viable, even if diminished, system of neurogenesis to be therapeutically effective. Once that system is no longer functional, exogenous stem cell transplants are a potential therapeutic strategy (Figure 5), although these exogenous neural stem cells will encounter an advanced stage of pathology in which to repopulate neural circuits.
Figure 5 |.
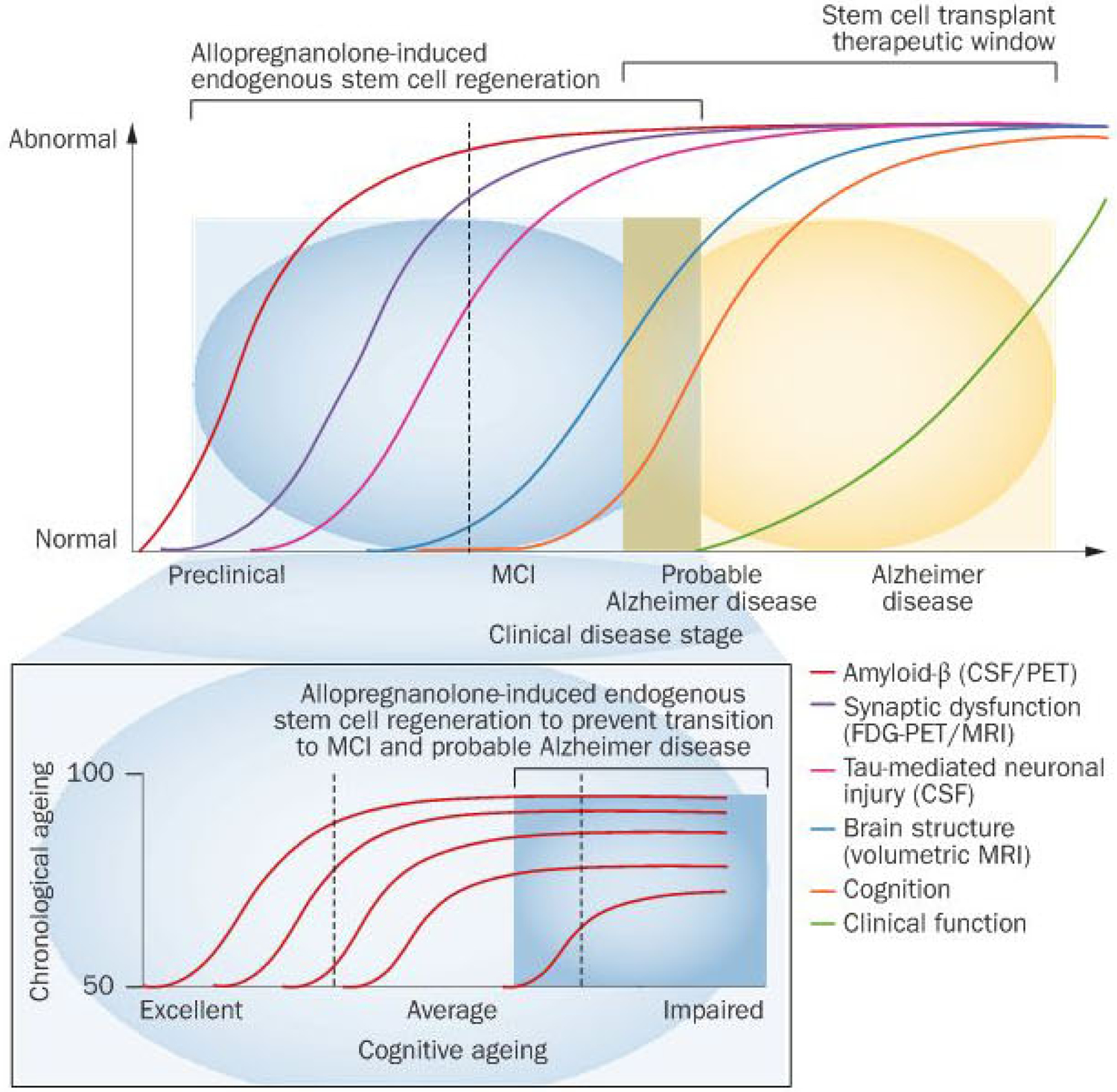
Theoretical window of opportunity for therapeutic efficacy to promote endogenous regeneration of neural stem cells versus stem-cell-based therapies. Age-related neurodegenerative progression to subjective memory complaints, to cognitive impairment, to mild cognitive impairment, to probable Alzheimer disease and ultimately to Alzheimer disease is characterized by sequential development of detectable pathology and clinical symptoms.41 The coloured lines indicate clinical analyses performed at each stage of disease progression: CSF and PET analysis for amyloid-β (red line), FDG-PET/MRI for synaptic dysfunction (purple line), CSF analysis for Tau-mediated neuronal injury (pink line), volumetric MRI for brain structure (blue line), cognition (orange line) and clinical function (green line) analyses. The proposed window of opportunity for promoters of endogenous regeneration is based on preclinical data. Extrapolated to ageing human brain, the window of therapeutic opportunity would be bracketed starting from the onset of age-associated neural stem cell quiescence, coinciding with a decline in cognitive function and extending into the mild cognitive impairment range, and closing with the diminished pool of responsive neural stem cells coinciding with probable Alzheimer disease. Whereas regenerative transitions are fairly well defined in the rodent brain, the transition in the human brain and markers of neurogenesis continue to be at the leading edge of clinical neuroscience.73,76,77 Red lines in inset figure indicate projections of cognitive function relative to chronological age. Abbreviations: CSF, cerebrospinal fluid; FDG-PET, 18F-fluoro-2-deoxyglucose PET; MCI, mild cognitive impairment.
Conclusions
Promoting endogenous regeneration in the ageing and/or degenerating brain is challenging but is likely to be achievable. Investigating the regenerative efficacy of allopregnanolone in preclinical models of ageing and Alzheimer disease has provided insights into pathways to promote endogenous regeneration while also diminishing pathology of Alzheimer disease (Figures 1–4). Targeting systems of endogenous regeneration and repair require dosing and treatment regimens not typically used in clinical practice, but which could be easily adopted (Figure 4). In the case of regeneration, high concentrations and/or unremitting exposure to regenerative therapeutics is likely to exert adverse consequences (Figure 4). By contrast, therapeutic regimens tailored to the temporal and dose requirements for endogenous regeneration and repair are likely to improve probability of success. Windows of opportunity for promoting endogenous regeneration will be governed by the pace of regenerative ageing of the brain (Figure 5). However, existing data from human studies suggest that the window in the ageing human brain could be quite prolonged.73 Currently, allopregnanolone is poised for clinical development as a neurosteroid, “of the nervous system, by the nervous system, for the nervous system”.1
Review criteria.
Initial publications selected for this Review were from the author’s files or identified through a PubMed search for articles in English published from 2000 to 2012 using the terms “allopregnanolone”, “neurogenesis”, “Alzheimer’s disease”, “mild cognitive impairment”, “liver-X-receptor”, “pregnane-X-receptor”, “oligodendrocytes”, “oligogenesis”, “myelin”, and “myelination”. Databases consulted for this Review included Nuclear Receptor Signaling Atlas (NURSA), NCI Cancer Genome Anatomy Project Nuclear Receptors in Lipid Metabolism and Toxicity, Gene Family Database, Alzheimer Research Forum and Clinical Trials.gov. Space limitations restricted citation of many original publications.
Key points.
The neurosteroid allopregnanolone promotes regeneration in the brain, recovery of learning and memory function, and reduces Alzheimer disease pathology in preclinical studies of efficacy
Therapeutic windows of allopregnanolone preclinical efficacy are defined by age and Alzheimer disease burden
Allopregnanolone administered intermittently to synergize with temporal cycles of endogenous regeneration promoted renewal and repair, whereas continuous infusions of allopregnanolone were antiregenerative in mouse models of Alzheimer disease
Allopregnanolone is poised to become the first endogenous regenerative therapeutic to be clinically developed for the prevention, delay or treatment of Alzheimer disease
Acknowledgements
The work that led to this Review was supported by grants from the NIH National Institute on Aging (U01 AG031115). the Alzheimer Drug Development Foundation, the Kenneth T. and Eileen L. Norris Foundation, and The California Institute for Regenerative Medicine (DR2-05410) to R. D. Brinton and by the SC CTSI NIH National Center for Advancing Translational Science (ULI RR031986). The invaluable contributions of J. Wang, S. Chen and R. Irwin of the University of Southern California to our discovery and translational research endeavours described herein are gratefully acknowledged.
Footnotes
Competing interests
The author declares that she has filed applications for patents on therapeutic regimen and method of use of allopregnanolone for neurodegenerative diseases.
References
- 1.Baulieu EE Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog. Horm. Res 52,1–32 (1997). [PubMed] [Google Scholar]
- 2.Baulieu EE & Schumacher M Neurosteroids, with special reference to the effect of progesterone on myelination in peripheral nerves. Mult. Scler 3,105–112 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Melcangi RC & Panzica G Neuroactive steroids: an update of their roles in central and peripheral nervous system. Psychoneuroendocrinology 34 (Suppl 1), S1–S8 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Mellon SH & Griffin LD Neurosteroids: biochemistry and clinical significance. Trends Endocrinol. Metab 13, 35–43 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Bishop NA, Lu T & Yankner BA Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skovronsky DM, Lee. VM & Trojanowski JQ Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu. Rev. Pathol 1,151–170 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Encinas JM et al. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 8, 566–579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhn HG, Dickinson-Anson H & Gage FH Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci 16,2027–2033 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazarov O, Mattson MP, Peterson DA, Pimplikar SW & van Praag H When neurogenesis encounters aging and disease. Trends Neurosci. 33, 569–579 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lugert S et al. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell 6,445–456 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Lugert S & Taylor V Neural stem cells: disposable, end-state glia? Cell Stem Cell 8, 464–465 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Singh C et al. Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3×TgAD and nonTg mice. Neurobiol. Aging 33, 1493–1506 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JM et al. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107,6498–6503 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez JJ et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 3, e2935 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brinton RD The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 31, 529–537 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinton RD Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci 30,212–222 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naylor JC et al. Allopregnanolone levels are reduced in temporal cortex in patients with Alzheimer’s disease compared to cognitively intact control subjects. Biochim. Biophys.Acta 1801, 951–959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang SY, He XY & Schulz H Multiple functions of type 10 17beta-hydroxysteroid dehydrogenase. Trends Endocrinol. Metab 16, 167–175 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Herrup K & Yang Y Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci 8, 368–378 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Schumacher M et al. Progesterone synthesis in the nervous system: implications for myelination and myelin repair. Front. Neurosci 6,10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun C et al. Allopregnanolone increases the number of dopaminergic neurons in substantia nigra of a triple transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res 9, 473–480 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang JM, Johnston RB, Ball BG & Brinton RD The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J. Neurosci 25, 4706–4718 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melcangi RC et al. Role of neuroactive steroids in the peripheral nervous system. Front. Endocrinol. (Lausanne) 2,104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng W, Saxe MD, Gallina IS & Gage FH Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J. Neurosci 29, 13532–13542 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez JJ, Jones VC & Verkhratsky A Impaired cell proliferation in the subventricular zone in an Alzheimer’s disease model. Neuroreport 20, 907–912 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Haughey NJ et al. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med. 1,125–135 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Aimone JB, Deng W & Gage FH Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron 70, 589–596 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng W, Aimone JB & Gage FH New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci 11, 339–350 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y & Gould. E Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus 12, 578–584 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shors TJ et al. Neurogenesis in the adult is involved in the formation of trace memories. Nature 410, 372–376 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Bartzokis G Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 32,1341–1371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuczynski B et al. White matter integrity and cortical metabolic associations in aging and dementia. Alzheimers Dement. 6, 54–62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ringman JM et al. Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain 130,1767–1776 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Yao J, Rettberg JR, Klosinski LP, Cadenas E & Brinton RD Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol. Aspects Med 32, 247–257 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S et al. Allopregnanolone promotes regeneration and reduces beta-amyloid burden in a preclinical model of Alzheimer’s disease. PLoS ONE 6, e24293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desai. MK et al. Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 57, 54–65 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saher G et al. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci 8, 468–475 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Rupprecht R et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov 9, 971–988 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Bateman RJ et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl.J. Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reitz C, Brayne C & Mayeux. R Epidemiology of Alzheimer disease. Nat. Rev. Neurol 7, 137–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jack CR Jr. et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiman EM & Jagust WJ Brain imaging in the study of Alzheimer’s disease. Neuroimage 61, 505–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiner MW et al. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers. Dement 8 (Suppl 1) S1–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lesne S et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357 (2006). [DOI] [PubMed] [Google Scholar]
- 45.De Strooper B, Vassar R & Golde T The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol 6, 99–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irwin RW, Wang JM, Chen S & Brinton RD Neuroregenerative mechanisms of allopregnanolone in Alzheimer’s disease. Front. Endocrinol. (Lausanne) 2, 117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang J & Rivest S Lipid metabolism and neuroinflammation in Alzheimer’s disease: a role for liver X receptors. Endocr. Rev 33, 2011–1049 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Mandrekar-Colucci S, Karlo JC & Landreth GE Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci 32, 10117–10128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Repa JJ et al. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci 27, 14470–14480 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cramer PE et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335,1503–1506 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riddell. DR et al. The LXR agonist T0901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell Neurosci 34 621–628 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Langmade SJ et al. Pregnane X receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc. Natl Acad. Sci. USA 103,13807–13812 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kliewer SA & Willson TM Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J. Lipid Res 43, 359–364 (2002). [PubMed] [Google Scholar]
- 54.Willson TM & Kliewer SA PXR, CAR and drug metabolism. Nat. Rev. Drug Discov 1, 259–266 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Porter FD et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann Pick C1 disease. Sci. Transl. Med 2, 56ra81 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hagemeyer CE, Rosenbrock H, Ditter M, Knoth R & Volk. B Predominantly neuronal expression of cytochrome P450 isoforms CYP3A11 and CYP3A13 in mouse brain. Neuroscience 117, 521–529 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Bengtsson SK, Johansson M, Backstrom. T & Wang M Chronic allopregnanolone treatment accelerates Alzheimer’s disease development in AbetaPP(Swe)PSEN1(DeltaE9) mice. J. Alzheimers Dis 31, 71–84 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Zampieri S et al. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J. Cell. Mol. Med 13, 3786–3796 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He J, Hoffman SW & Stein DG Allopregnanolone, a progesterone metabolite, enhances behavioral recovery and decreases neuronal loss after traumatic brain injury. Restor. Neurol. Neurosci 22,19–31 (2004). [PubMed] [Google Scholar]
- 60.Noorbakhsh F et al. Impaired neurosteroid synthesis in multiple sclerosis. Brain 134, 2703–2721 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffin LD, Gong W, Verot L & Mellon SH Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med 10,704–711 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Adeosun SO et al. Allopregnanolone reinstates tyrosine hydroxylase immunoreactive neurons and motor performance in an MPTP-lesioned mouse model of Parkinson’s disease. PLoS ONE 7, e50040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gee KW, Bolger MB, Brinton RE, Coirini H & McEwen BS Steroid modulation of the chloride ionophore in rat brain: structure-activity requirements, regional dependence and mechanism of action. J. Pharmacol. Exp. Ther 246, 803–812(1988). [PubMed] [Google Scholar]
- 64.Reddy DS & Rogawski MA Neurosteroid replacement therapy for catamenial epilepsy. Neurotherapeutics 6, 392–401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Belelli D & Lambert JJ Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat. Rev. Neurosci 6, 565–575 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Kask. K, Backstrom T, Nilsson LG & Sundstrom Poromaa I Allopregnanolone impairs episodic memory in healthy women. Psychopharmacology (Berl) 199,161–168 (2008). [DOI] [PubMed] [Google Scholar]
- 67.ClinicalTrials.gov. Allopregnanolone for the Treatment of Traumatic Brain Injury [online], http://clinicaltrials.gov/ct2/show/NCT01673828?term=traumatic+brain+injury+and+allopregnanolone&rank=l (2012).
- 68.Sperling R et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 11, 241–249 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sperling RA et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 7, 367–385 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alzheimer Research Forum. CTAD: New Data on Sola, Bapi, Spark Theragnostics Debate [online], http://www.alzforum.org/new/detail.asp?id=3313 (2012).
- 71.Alzheimer Research Forum. Phase 3 Solanezumab Trials “Fail”—Is There a Silver Lining? [online], http://www.alzforum.org/new/detail.asp?id=3254 (2012).
- 72.Alzheimer Research Forum. Drugs In Clinical Trials [online], http://www.alzforum.org/drg/drc/detail.asp?id=108 (2012).
- 73.Eriksson PS et al. Neurogenesis in the adult human hippocampus. Nat. Med 4, 1313–1317 (1998). [DOI] [PubMed] [Google Scholar]
- 74.Hosie AM, Wilkins ME, da Silva. HM & Smart. TG Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Wang JM & Brinton RD Allopregnanolone-induced rise in intracellular calcium in embryonic hippocampal neurons parallels their proliferative potential. BMC Neurosci. 9 (Suppl 2), 2–11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Manganas LN et al. Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science 318,980–985 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pereira AC et al. An in vivo correlate of exercise induced neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. USA 104, 5638–5643 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lipinski CA, Lombardo F, Dominy BW & Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 46, 3–26 (2001). [DOI] [PubMed] [Google Scholar]