

Abstract
The Nrf2-Keap1-ARE pathway, a master regulator of oxidative stress, has emerged as a promising target for cancer therapy. Mutations in NFE2L2, KEAP1, and related genes have been found in many human cancers, especially lung cancer. These mutations lead to constitutive activation of the Nrf2 pathway, which promotes proliferation of cancer cells and their resistance to chemotherapies. Small molecules that inhibit the Nrf2 pathway are needed to arrest tumor growth and overcome chemoresistance in Nrf2 addicted cancers. Here, we identified a novel small molecule, MSU38225, which can suppress Nrf2 pathway activity. MSU38225 downregulates Nrf2 transcriptional activity and decreases the expression of Nrf2 downstream targets, including NQO1, GCLC, GCLM, AKR1C2 and UGT1A6. MSU38225 strikingly decreases the protein level of Nrf2, which can be blocked by the proteasome inhibitor MG132. Ubiquitination of Nrf2 is enhanced following treatment with MSU38225. By inhibiting production of antioxidants, MSU38225 increases the level of reactive oxygen species when cells are stimulated with tert-butyl hydroperoxide. MSU38225 also inhibits the growth of human lung cancer cells in both 2D cell culture and soft agar. Cancer cells addicted to Nrf2 are more susceptible to MSU38225 for suppression of cell proliferation. MSU38225 also sensitizes human lung cancer cells to chemotherapies both in vitro and in vivo. Our results suggest that MSU38225 is a novel Nrf2 pathway inhibitor that could potentially serve as an adjuvant therapy to enhance the response to chemotherapies in lung cancer patients.
Introduction
As a master regulator of homeostasis, the Nrf2-Keap1-ARE pathway plays critical roles in various cellular processes including redox-balancing, detoxification, proliferation, inflammation, and metabolism (1). Nuclear factor erythroid-2-related factor 2 (Nrf2) belongs to a subfamily of basic leucine zipper (bZIP) proteins (2). As a transcription factor, the activity of Nrf2 is tightly controlled. Under basal conditions, Kelch-like ECH-associated protein 1 (Keap1), acting as an adaptor protein, binds to Nrf2 and targets it to the Cullin3-based ubiquitin E3 ligase complex for protein degradation. Upon exposure to cellular stresses, Nrf2 is released from Keap1 and translocates into the nucleus to activate the expression of a network of downstream cytoprotective genes (3).
Because of these cytoprotective roles, activating the Nrf2 pathway historically has been considered an attractive strategy for cancer prevention (4–6). Nrf2 signaling protects cells against both endogenous and exogenous insults. Carcinogenesis is exacerbated in Nrf2-knockout mice vs. wildtype mice in a wide variety of preclinical models, including skin cancer induced by ultraviolet light, liver cancer induced by aflatoxin, cancer in the forestomach induced by polycyclic hydrocarbons, bladder cancer induced by nitrosamines, and colon cancer induced by inflammation (4,7–10). Numerous Nrf2 activators have been tested as chemopreventive agents to prevent or delay tumor development, and the beneficial effects of these agents are greatly dampened in Nrf2 KO mice (9,11).
However, loss-of-function mutations of KEAP1 and gain-of-function mutations of NFE2L2 (gene encoding Nrf2) have been discovered in many human cancers, especially lung cancers. Approximately 28% of lung cancers contain mutations relevant to the Nrf2 pathway, not only in NFE2L2 and KEAP1 but also in genes such as CUL3, BTRC, and MAFG (12–15). These genetic changes in the tumors suggest a growth-promoting role for this pathway. Accumulating evidence has demonstrated that constitutive activation of the Nrf2 pathway favors tumor growth via several mechanisms. Nrf2 acts downstream of key oncoproteins including Kras, Myc, BRAF and PI3K to promote tumor cell survival and growth (16,17). Nrf2 can also regulate cancer metabolism by reprograming cells into anabolic pathways for supporting rapid proliferation. The expression of various metabolic enzymes and transporters is directly regulated by Nrf2 (18,19). Solid tumors with deficiencies in various Krebs cycle enzymes, such as mutations in isocitrate dehydrogenase (IDH) or depletion of succinate dehydrogenase, are sensitive to Nrf2 blockade (20,21). Moreover, the constitutive activation of Nrf2 is associated with chemoresistance, and inhibiting Nrf2 expression sensitizes tumor cells to chemotherapies (22,23).
In contrast to Nrf2 activators, only a few Nrf2 inhibitors have been reported (24). Brusatol was the first Nrf2 inhibitor identified, and it remains the most potent known inhibitor (22). However, a mass spectrometry profiling study suggested that brusatol functions as a global protein synthesis inhibitor rather than a specific inhibitor of Nrf2 (25). High throughput screens also identified AEM1 (26), ML385 (23), and IM3829 (27) as small molecules that inhibit Nrf2 activity. These molecules possess different chemical properties and kinetics to inhibit Nrf2, but all significantly enhanced the susceptibility of Keap1 mutant tumors to radiotherapy or chemotherapies. Tumors with mutations in isocitrate dehydrogenase (IDH) or related metabolic enzymes are also sensitive to Nrf2 inhibition, and several natural compounds have recently been identified that suppress tumor growth by inhibiting the Keap1-Nrf2 pathway (28,29). However, none of these inhibitors has been widely used because of a lack of efficacy or selectivity.
To develop novel Nrf2 inhibitors, we screened a series of small molecules synthesized internally at Michigan State University and identified MSU38225 as a novel inhibitor of the Nrf2 pathway. The inhibition of MSU38225 on the Nrf2 pathway was validated both in vitro and in vivo. In vitro, the expression of Nrf2 and its downstream targets, redox-balancing, and cell proliferation were measured following MSU38225 treatment. In vivo, the efficacy of MSU38225, alone or in combination with carboplatin, for treating KEAP1 mutant tumors in a model of non-small cell lung cancer (NSCLC) was evaluated.
Materials and Methods
Synthesis
MSU38225 was synthesized at Michigan State University as described (30). The compound was dissolved in DMSO to make a 50 mmol/L stock concentration and diluted to the listed concentrations for each experiment. An equivalent amount of DMSO was used as a vehicle control. The purity of the compound used in these studies was confirmed to be >98% by using gas chromatography with flame-ionization detection.
Cell Culture
A NRF2/ARE luciferase reporter stably was transfected into MCF7 cells (purchased from Signosis, Santa Clara, CA). Cells were cultured in DMEM+10% FBS+1% Pen/Strep (Corning Cellgro, Mediatech, Manassas, VA). A549, H460, A427, MCF7, MCF10A, Jurkat cells were purchased from American Type Culture Collection (ATCC, Manassas, VA), and no additional authentication or mycoplasma testing was conducted as cells were only used for 4–5 passages before being discarded. A549 cells were maintained in F12K+10% FBS+1% Pen/Strep. H460 cells were maintained in RPMI+10% FBS+1% Pen/Strep. A427 cells were maintained in DMEM/F12+10% FBS+1% Pen/Strep. MCF10A cells were cultured in DMEM/F12 supplemented with 5% equine serum (Gemini Bio), 0.29 M sodium bicarbonate solution (Sigma-Aldrich), 10 mM Hepes (Sigma-Aldrich), 1% glutamine (Sigma-Aldrich), 10 μg/mL insulin (Sigma-Aldrich), 20 ng/mL EGF (Sigma-Aldrich), 1 mg/mL hydrocortisone (Sigma-Aldrich), 150 μg/mL cholera toxin (Sigma-Aldrich), and 1% Pen/Strep. Jurkat cells were maintained in RPMI+10% FBS+1% Pen/Strep. Keap1 CRISPR KO and βTrCP CRISPR KO Jurkat cells were generated as described (31) and generously provided by the Rockwell lab. NFE2L2 KO A549 cells were generated using the same CRISPR-Cas9 gene editing technology (31). The FBS concentration in media was reduced to 1% when cells were treated with inhibitors. Cells were used within 6–8 passages when thawed.
Nrf2 activity luciferase reporter assay
10,000 NRF2/ARE reporter cells were plated in a 96 well plate (white) with 1% FBS in the media. Cells were treated with different concentrations of MSU38225 for 24 hours; tert-butylhydroquinone (tBHQ) was added 1 hour after MSU38225 to activate the Nrf2 pathway. Cell viability was detected by Celltiter-fluor (Promega) and luciferase activity was detected by Steady-glo (Promega) using Synergy Neo HTS multi-mode microplate reader (BioTek).
RT-qPCR and western blotting
A549 cells were treated with MSU38225 at indicated concentrations for 24 hours. Total RNA was isolated using a RNeasy Mini Kit (QIAGEN). RNA concentrations were determined using NanoDrop. For each sample, 500 ng RNA was used to synthesize cDNA with high capacity cDNA reverse transcription kits (AppliedBiosystems). Primers (sequences listed in Suppl Fig.1) were ordered from IDT. AppliedBiosystems Fast SYBR Green Master Mix and the QuantStudio 7 Flex Real-Time PCR system were used to detect gene expression. The delta-delta Ct method was applied to calculate relative gene expression. Values were normalized to the reference gene GAPDH and expressed as fold change compared to DMSO treated samples.
Western Blotting
A549 or MCF7 cells treated with MSU38225 were lysed in RIPA buffer (5 M NaCl, 1 M Tris-Cl, pH 7.4, 0.5 M EDTA, 25 mM deoxycholic acid, 1% triton-X, 0.1% SDS) containing protease inhibitors (1 mM PMSF, 2 μg/ml aprotinin and 5 μg/ml leupeptin) added just prior to use. Protein concentrations were measured using the BCA assay (Sigma-Aldrich). 20 μg of protein were separated by 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Nrf2 (Novus Biologicals, 1:1000), NQO1 (Abcam, 1:1000), HO-1 (Abcam, 1:1000), Keap1 (Cell Signaling Technology, 1:1000), NF-κB (Cell Signaling Technology, 1:1000), IκBα (Cell Signaling Technology, 1:1000) Cyclin G1 (Santa Cruz, 1:1000), β-TrCP (Cell Signaling Technology, 1:1000), STAT3 (Cell Signaling Technology, 1:1000), Ubiquitin (Cell Signaling Technology, 1:1000), Histone 3 (Cell Signaling Technology, 1:4000), GAPDH (Santa Cruz, 1:4000) and Vinculin (Cell Signaling Technology, 1:4000) primary antibodies were applied to detect the corresponding proteins. Secondary antibodies (anti-rabbit or anti-mouse linked to HRP, 1:1000) were purchased from Cell Signaling Technology. ECL Western blotting substrate (GE Healthcare Life Sciences, UK) was used to detect the signal. Images shown are representative of 3 independent experiments. Protein quantification was done using ImageJ.
Reactive oxygen species (ROS) assay
A549 cells were treated with MSU38225 for 24 hours. DCFDA (10 μM) was added for 2 hrs as an ROS indicator and then cells were treated with tert-butyl hydroperoxide (tBHP, 250 μM) for 15 minutes before harvesting. Then cells were washed with PBS and trypsinized to a single cell suspension. The cell pellet was resuspended in PBS and analyzed using a flow cytometer (Accuri, BD) with the FLA-1 channel. 50,000 events were acquired for each sample. Mean fluorescence intensity (MFI) was calculated using FlowJo software.
Cell growth assay
To evaluate cell proliferation in 2D culture, a MTT assay was performed. Cells were seeded in 96-well plates with 2000 cells/well in their corresponding growth media. Various cell lines were treated with MSU38225 for 72 hrs. To assess cell growth in 3D culture, A549, H460 and A427 cells were plated in 0.6% soft agar for 7 days. A Cytation 3 imaging reader from BioTek with Gen5 3.04 software was used to quantify colonies. Seven pictures were taken every 100 μm and superimposed together by z-projection function. Colonies >50 μm in diameter were quantified for each cell line.
Chemoresistance assay
1000 A549 cells/well were plated in 384 well plates and treated with MSU38225 and chemotherapy drugs using a series of concentrations. Cell viability was detected by Celltiter-glo (Promega) after 72 hours of treatment. Data was normalized to the DMSO control and presented as a percentage of cell viability. Data represent three independent experiments.
In vivo treatment study
All protocols were carried out in accordance to the Regulations for the Management of Laboratory Animals at Michigan State University. All experimental protocols for the ethical use of animal studies were approved by the Institutional Animal Care and Use Committee at Michigan State University (protocol 201800050). Male athymic nude mice (Envigo, 6 weeks-old) were injected in the flank with 5 × 106 A549 cells. Cell line authenticity and absence of pathogens in the A549 cells were confirmed before establishing the model (IDEXX BioAnalytics). Once the tumors reached 4 mm in diameter as measured by a caliper, mice were randomized into four groups (6 mice/group) and treated daily (M-F) i.p. with either vehicle (10% DMSO/10% Cremophor/80% saline), MSU38225 (50 mg/kg, BID), carboplatin (5 mg/kg), or the combination of MSU38225 and carboplatin. All mice were weighed twice a week, and tumors were measured twice a week using a caliper. All mice were sacrificed after 4 weeks of treatment and harvested for analysis of Nrf2 expression and cell proliferation.
Immunohistochemistry
Tumors were harvested and fixed in 10% neutral buffered formalin. Sections were obtained for immunohistochemistry staining. Sodium citrate buffer (10 mM, Vector) was used for antigen retrieval and 3% hydrogen peroxide (15-minute incubation) was used to quench the endogenous peroxidase activity. Sections were stained with PCNA (1:200, Santa Cruz) or Nrf2 (1:200, Novus Biologicals) antibodies for 1 hour at room temperature or overnight at 4°C, respectively. Anti-mouse and anti-rabbit secondary antibodies conjugated to HRP were purchased from Cell Signaling Technology. Signal was detected using a DAB kit (Cell Signaling Technology) and sections were counterstained with hematoxylin (Vector).
Statistical analysis
The in vitro experiments were performed in triplicate and were repeated independently at least three times. Unless noted otherwise, data are presented as mean ± SE. In vitro and in vivo results were analyzed using one-way ANOVA followed by a Tukey test or one-way ANOVA on ranks and the Dunn test if the data did not fit a normal distribution (Prism 6). Tumor volume curves were analyzed using two-way repeated measures ANOVA followed by Tukey’s multiple comparisons test. A p value < 0.05 was considered statistically significant. The combination index (CI) was calculated as: CI = (D)1/(D50)1+(D)2/(D50)2, where (D50)1 or (D50)2 is the concentration of drug 1 or drug 2 that decreased the cell viability by 50% when used alone; and (D)1 or (D)2 is the concentration of drug 1 or drug 2 that decreased the cell viability by 50% when used in combination (32).
Results
MSU38225 inhibits Nrf2 transcriptional activity and suppresses the expression of genes downstream of Nrf2
To identify novel Nrf2 inhibitors, we screened approximately 600 small molecules synthesized at Michigan State University using a cell-based luciferase reporter assay coupled with a cytotoxicity readout. A commercially available NRF2/ARE luciferase reporter construct stably expressed in MCF-7 cells was used for the initial screen. Hit compounds were further validated in KEAP1-mutant cell lines, such as A549 cells (human lung adenocarcinoma cells). MCF-7 cells (human breast cancer cells), stably transfected with a Nrf2 binding site and firefly luciferase coding region, were treated with library compounds at 10 μM for 24 hours. tert-butylhydroquinone (tBHQ), a well-known Nrf2 activator (31), was added 1 hour after the library compounds to stimulate the activation of the Nrf2 pathway in the MCF-7 cells. Before the detection of luciferase activity, cell viability was measured using a cell-titer fluor assay. From the primary screen, MSU38225 (Fig. 1A) was identified as a hit. MSU38225 inhibited Nrf2 transcriptional activity induced by tBHQ in a dose-dependent manner (Fig. 1B) without reducing cell viability (Fig. 1C).
Figure 1. MSU38225 inhibits transcriptional activity of Nrf2.
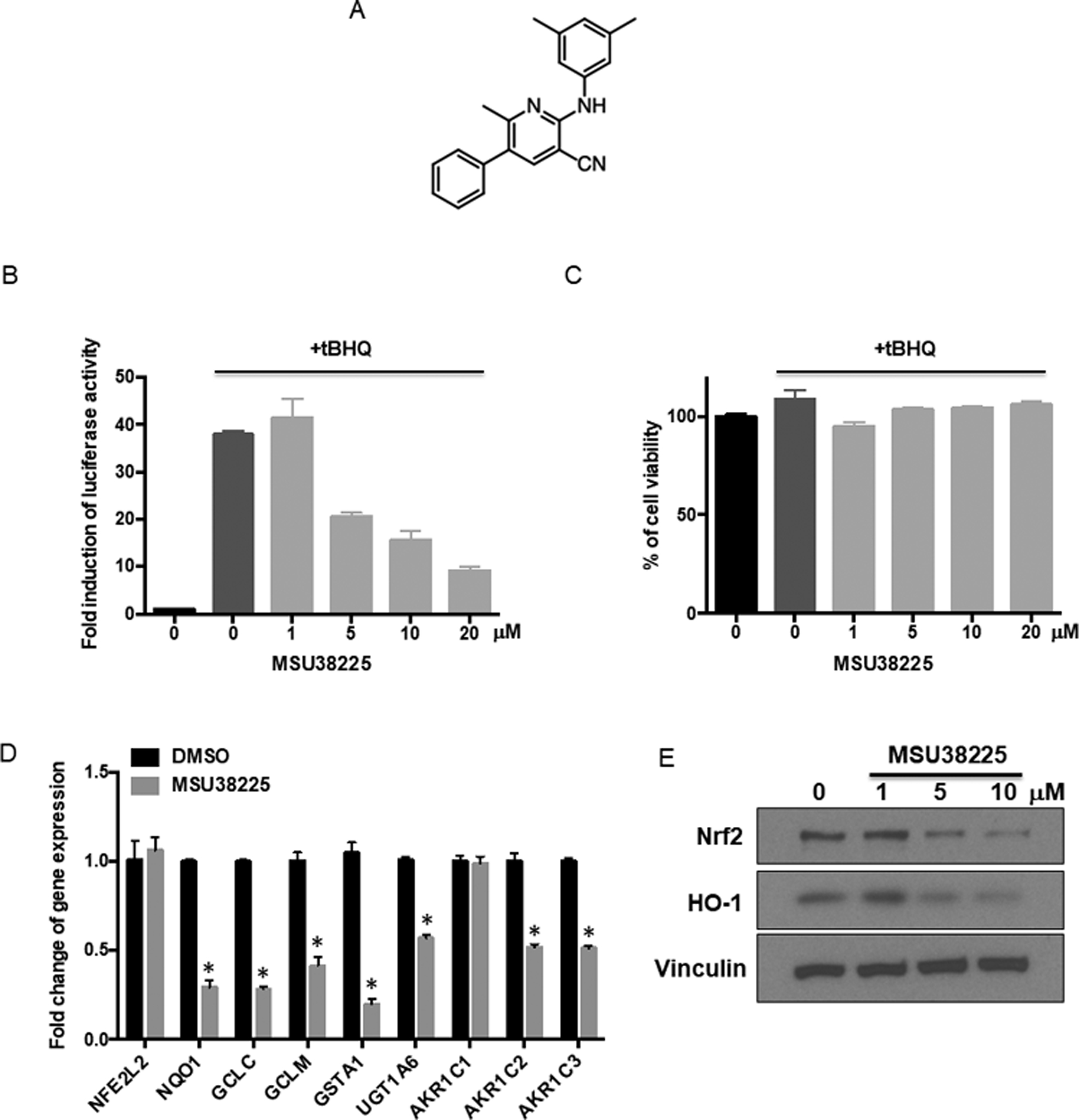
A. Chemical structure of MSU38225. B. MCF-7 reporter cells (B-C) stably transfected with a NRF2/ARE luciferase construct were treated with various concentrations of MSU38225 and stimulated with 20 μM tert-butylhydroquinone (tBHQ) for 24 hours. Luciferase activity was detected and normalized to the DMSO control. C. Cell viability was detected using a CellTiter-Fluor assay just prior to the luciferase assay. D. A549 cells (D-E) were treated with 5 μM MSU38225 for 24 hours, and the mRNA expression of NFE2L2 and target genes downstream of Nrf2 were detected using RT-qPCR. Results were normalized to the reference gene GAPDH and the DMSO control. *, p<0.05 vs. DMSO control. E. A549 cells were treated with 5 μM MSU38225 for 24 hours, and the protein expression of Nrf2 and its downstream target HO-1 was detected using western blotting.
Nrf2 regulates a wide variety of downstream cellular processes, such as detoxification, maintenance of redox homeostasis, and heme metabolism (1). To validate the inhibitory effects of MSU38225 on the Nrf2 pathway in A549 lung cancer cells, mRNA expression of targets downstream of Nrf2 was detected after 24 hours of treatment with MSU38225: NAD(P)H dehydrogenase (quinone 1) (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC), glutamate-cysteine ligase regulatory subunit (GCLM), glutathione S-transferase A1 (GSTA1), UDP-glucuronosyltransferase (UGT1A6), and aldo-keto reductase family 1 members C1–C3 (AKR1C1–3). tBHQ stimulation was not used because of the constitutive activation of the Nrf2 pathway in A549 cells. MSU38225 significantly (p<0.05) decreased the mRNA expression of all these downstream genes except AKR1C1 in A549 cells (Fig. 1D). MSU38225 did not affect the mRNA expression of NFE2L2, the gene that encodes the Nrf2 protein (Fig. 1D). Furthermore, MSU38225 inhibited pathway activity at the protein level, as the protein expression of heme oxygenase-1 (HO-1), another downstream target of Nrf2, was dose-dependently decreased after treatment (Fig. 1E).
MSU38225 enhances Nrf2 degradation through the proteasome
In A549 cells, Nrf2 is constitutively activated because of a mutation in the KEAP1 gene and is expressed at a higher level than in KEAP1 wildtype cells. MSU38225 decreased the protein level of Nrf2 in a dose- (Fig. 1E) and time- (Fig. 2A and B) dependent manner. The largest reduction in Nrf2 protein was found 24 hours post-treatment with MSU38225, which is a different kinetic profile than other Nrf2 inhibitors such as brusatol (22). Nrf2 protein is mainly located in the nucleus in A549 cells because of constitutive activation of the pathway. Although the cytosolic protein level of Nrf2 was very low, MSU38225 did reduce Nrf2 protein expression in both the cytosol and nucleus (Fig. 2C). In addition, the effects of MSU38225 on Nrf2 protein level is independent of the Nrf2 mutation status. In MCF-7 cells, where Keap1 and Nrf2 are wild-type, MSU38225 decreased both tBHQ-induced expression of Nrf2 protein (Fig. 2D) and basal expression of Nrf2 protein (Fig. 2E). NQO1, a downstream target of Nrf2, protein was also decreased after treatment (Fig. 2D). Notably, MSU38225 did not alter the overall expression pattern of abundant cellular proteins (Suppl Fig.2). MSU38225 also did not change levels of NF-κB pathway proteins (IκBα, NF-κB, p-NF-κB) or other fast turnover proteins such as cyclinG1 and STAT3 (Fig. 2F).
Figure 2. MSU38225 decreases Nrf2 protein in cancer cells.
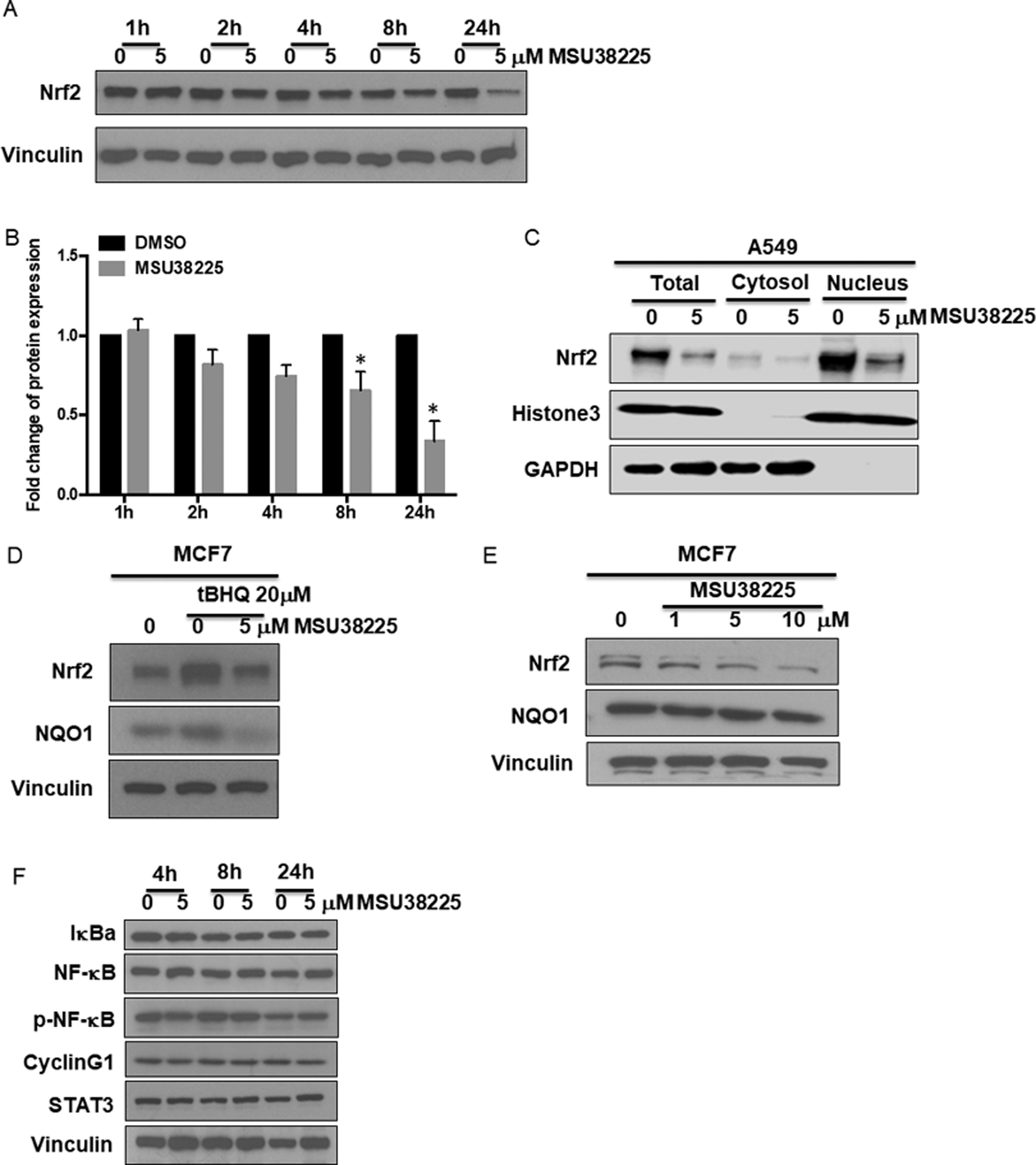
A549 cells were treated with MSU38225 for 1–24 hours. The protein expression of Nrf2 was detected by western blotting (A) and quantified (B) using ImageJ. N=3 independent experiments. *, p<0.05 vs. DMSO control. C. The expression of Nrf2 protein was evaluated in both the cytosol and nucleus after treatment of MSU38225 for 24 hours in A549 cells. MCF7 cells were treated with (D) or without (E) tBHQ +/− MSU38225 and A549 cells (F) were treated with MSU38225 for 24 hours. Protein expression of Nrf2 and its downstream target NQO1 (D, E) or NF-κB family members, cyclinG1 and STAT3 (F) were detected by western blotting.
The protein level of Nrf2 is tightly balanced between synthesis and degradation. To investigate whether MSU38225 affects the degradation of Nrf2 protein, A549 cells were treated with MG132, a proteasome inhibitor. MG132 blocked the decrease in protein expression of Nrf2 when treated with MSU38225 (Fig. 3A), suggesting that MSU38225 enhances the protein degradation of Nrf2 via the proteasome system. Additionally, MSU38225 increased the ubiquitination of Nrf2 (Fig. 3B). In this experiment, A549 cells were treated with either DMSO or MSU38225 for 24 hours, and Nrf2 protein was pulled down using agarose beads. The ubiquitin level was then evaluated on the Nrf2 protein. Because MSU38225 decreased Nrf2 protein level, no change in the ubiquitin level was observed when the same amount of total protein was loaded to compare between the DMSO control and treatment (Fig. 3B left). However, a higher level of ubiquitination of Nrf2 protein was present when the same amount of Nrf2 protein was loaded (Fig. 3B right). The cycloheximide chase assay is widely used to assess the stability of proteins (33). A549 cells were pretreated with DMSO or MSU38225 for 4 hours. After washout with PBS, cells were treated with 1μM cycloheximide for 0–60 minutes before being harvested to detect the protein level of Nrf2. At 30 min, Nrf2 protein levels were lower in cells pretreated with MSU38225 and then cycloheximide compared to the DMSO pretreated controls, suggesting that MSU38225 facilitates the decreased stability of Nrf2 protein (Fig. 3C). Keap1-Cullin3 and βTrCP-Cullin1 are two major pathways that lead to Nrf2 degradation (34,35). However, MSU38225 enhanced Nrf2 degradation in a largely Keap1- and βTrCP- independent manner, as MSU38225 still reduced Nrf2 protein expression in Keap1 CRISPR KO and βTrCP CRISPR KO Jurkat cells (Fig. 3D and E).
Figure 3. MSU38225 enhances the degradation of Nrf2 via the proteasome system.
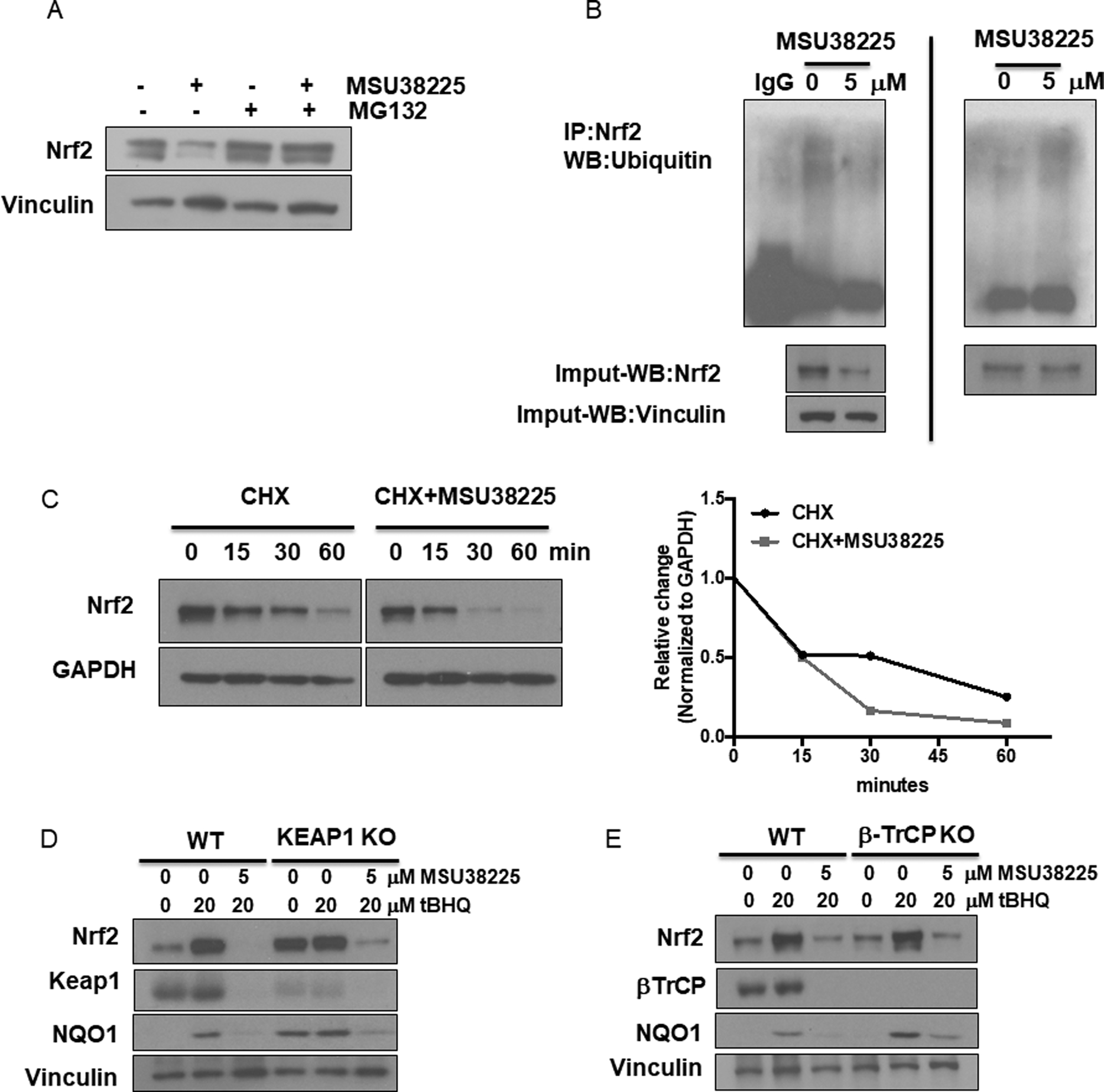
A. A549 cells were treated with 5 μM MSU38225, 5 μM MG132, or the combination for 24 hours, and Nrf2 protein expression evaluated by western blotting. B. A549 cells were treated with either DMSO or MSU38225 for 24 hours, and Nrf2 protein was pulled down using agarose beads followed by immunoblotting with anti-ubiquitin antibodies. On the left, the same amount of total protein was loaded; on the right, the same amount of Nrf2 protein was loaded. C. A549 cells were pretreated with DMSO or 5 μM MSU38225 for 4 hours. After washout with PBS, cells were treated with 1 μM cycloheximide for 0–60 minutes. Nrf2 protein expression was detected and quantified using ImageJ (results were normalized to the respective 0 min time point). Wild type, KEAP1 CRISPR knockout (D) and βTrCP CRISPR knockout (E) Jurkat cells were treated with tBHQ +/− MSU38225 for 24 hours and expression of Nrf2, Keap1, NQO1 and βTrCP was detected by western blotting.
MSU38225 dampens the anti-oxidative response and inhibits cell growth in cancer cells
Nrf2 is a master regulator of anti-oxidative responses and plays critical roles in maintaining redox balancing so Nrf2 activators attenuate oxidative stress (36). To test the effects of MSU38225 on the production of reactive oxygen species (ROS), A549 cells were treated with MSU38225 for 24 hours and then stimulated with tert-butyl hydroperoxide (tBHP) for 15 minutes. MSU38225 dose-dependently increased ROS production in A549 cells (Fig. 4A), which functionally validated the inhibitor as this anti-oxidative response regulated by the Nrf2 pathway was suppressed.
Figure 4. MSU38225 dampens the anti-oxidative response and inhibits cell growth in cancer cells in a Nrf2-dependent manner.

A. A549 cells were treated with MSU38225 for 24 hours. Cells were stimulated with 250 μM tert-butyl hydroperoxide (tBHP) for 15 minutes. Reactive oxygen species (ROS) production was detected using DCFDA as an indicator. Experiment was done in triplicate and repeated in 3 independent assays. *, p<0.05 vs. tBHP+DMSO control. B. Cell viability was analyzed using a MTT assay in four cell lines after 72-hour treatment with MSU38225. C. A549, H460 and A427 cells were plated in 0.6% soft agar for 7 days. Colonies larger than 50 μm in diameter were quantified. The experiment was done in triplicate and repeated twice. *, p<0.05 vs. DMSO control. Data shown as mean±SD. D. The efficacy of Nrf2 deletion in A549 cells was confirmed with western blotting. E. Nrf2 WT and KO A549 cells were treated with 5 μM MSU38225 for 24 hours. Cells were stimulated with 250 μM tert-butyl hydroperoxide (tBHP) for 15 minutes. Reactive oxygen species (ROS) production was detected. Experiment was done in triplicate and repeated in 3 independent assays. *, p<0.05 vs. tBHP+DMSO control; ns, p>0.05 vs. tBHP+DMSO control. F. Cell viability was measured using MTT assay in Nrf2 WT vs. KO A549 cells after 72-hour treatment with MSU38225. *, p<0.05 vs. Nrf2 WT at the same concentration.
In addition to its anti-oxidative properties, Nrf2 activation can contribute to tumor cell growth. Nrf2 not only regulates cell proliferation as a downstream mediator of several onco-proteins but also reprograms cellular metabolism to favor anabolic pathways (37). Cancer cells were treated with MSU38225 for 72 hours and cell viability was detected using the MTT assay. MSU38225 inhibited proliferation in various cancer cells (Fig. 4B), including both Keap1 mutant and Keap1 wild-type cells. Notably, Nrf2-addicted cancer cell lines (A549 and H460 with mutant Keap1) showed a higher sensitivity to MSU38225 treatment compared to non-addicted cells (A427 and MCF-7 with wildtype Keap1). In MCF-10A cells (a non-tumorigenic epithelial cell line), MSU38225 did not have any effects on cell viability at concentrations <20 μM (Suppl Fig. 3). The suppression of MSU38225 on cell growth was also confirmed in 3D cell culture. Three different human lung cancer cell lines, including A549, H460 and A427 cells, were plated in 0.6% soft agar and treated with MSU38225 for 7 days. MSU38225 significantly (p<0.05) suppressed the growth of all three lung cancer cells grown in soft agar in a dose-dependent manner (Fig. 4C).
To determine whether any of these effects are Nrf2 dependent, we knocked out the NFE2L2 gene in A549 cells using CRISPR-Cas9 gene editing technology (Fig. 4D). Unlike in parental A549 cells, MSU38225 lost its ability to further increase ROS production after tBHP stimulation in Nrf2 KO cells (Fig. 4E), indicating its inhibitory effect on the anti-oxidant response is Nrf2 dependent. Nrf2 KO A549 cells also lost their sensitivity to growth inhibition (Fig. 4F), as cell viability of Nrf2 KO cells after MSU38225 treatment was similar to other non-addicted cancer cells (A427 and MCF-7) with wildtype Keap1.
MSU38225 enhances the sensitivity of cancer cells to chemotherapeutic drugs in vitro and in vivo
Drug resistance during chemotherapy remains a major obstacle in cancer treatment. Activation of the Nrf2 pathway is one mechanism that tumor cells hijack to induce chemoresistance. Nrf2 regulates a series of drug metabolizing enzymes and efflux transporters (38) so that drug exposure could be lower in cancer cells with increased Nrf2 expression and activity. Moreover, Nrf2-induced chemoresistance can be redox-mediated. ROS-mediated apoptosis is a common mechanism of action for many chemotherapeutic agents (39). Cancer cells upregulate antioxidants to protect them from high level of ROS by turning on the Nrf2 pathway (40). The Nrf2 pathway also induces drug resistance through the activation of autophagy (41).
To test whether MSU38225 could overcome resistance to chemotherapies, A549 cells were treated with several commonly used chemotherapeutic agents in combination with MSU38225. Combination effects of MSU38225 were observed with four different chemotherapy drugs: carboplatin, doxorubicin, 5-fluorouracil, and topotecan (Fig. 5A). Lower cell viability was observed when chemotherapeutic agents were used in combination with MSU38225 compared to the drugs alone. To determine whether there was synergy between MSU38225 and each chemotherapy agent, isobologram analysis was performed (42). The combination index (CI) was calculated as 0.82, 0.91, 0.54 and 1.01 for carboplatin, doxorubicin, 5-fluorouracil, and topotecan, respectively. A CI of less than 1 indicates synergy, while CI of equal to 1 indicates additivity. Thus, the inhibitory effect of MSU38225 was additive with doxorubicin and topotecan but synergistic with carboplatin and 5-fluorouracil.
Figure 5. MSU38225 enhances the sensitivity of cancer cells to chemotherapies in vitro and in vivo.

A. A549 cells were plated in a 384-well plate and co-treated with MSU38225 and chemotherapeutic agents at various concentrations for 72 hours. Cell viability was detected using CellTiter Glo assay and normalized to the DMSO control. B. A xenograft model was established using A549 cells. Tumors 4 mm in diameter were treated with either vehicle, carboplatin (5 mg/kg), MSU38225 (50 mg/kg, BID) or the combination for 4 weeks. Tumors were measured twice a week using a caliper. N=6 mice/group. **, p<0.01 vs. vehicle control. C. Body weight was measured twice a week during the treatment. Cell proliferation (D, PCNA) and Nrf2 expression (E) in tumors was evaluated by immunohistochemistry. 400x magnification. Scale bar = 60 microns.
Enhanced efficacy was also achieved when chemotherapy was combined with MSU38225 in vivo. A subcutaneous xenograft model was established using A549 cells. Tumors 5 mm in diameter were treated with either vehicle, low dose carboplatin, MSU38225, or the combination for 4 weeks. Carboplatin, a standard of care chemotherapy for treatment of lung cancer (43), failed to arrest tumor growth at this low dose (5 mg/kg) when used alone (Fig. 5B). As a single agent, MSU38225 appeared to slow tumor growth, but the change was not statistically significant. However, the combination of MSU38225 and carboplatin significantly slowed tumor growth compared to the vehicle control (p<0.01) and carboplatin alone (p<0.05) (Fig. 5B). MSU38225 was well-tolerated in the mice and did not induce any changes on body weight over time (Fig. 5C). The combination therapy inhibited cell proliferation as shown by a decrease in PCNA staining in the tumors (Fig. 5D). Consistent with the in vitro results, treatment with MSU38225 decreased the expression of Nrf2 protein in the tumors compared to the vehicle control group (Fig. 5E).
Discussion
In the present study, we identified MSU38225 as a novel Nrf2 pathway inhibitor. MSU38225 decreased Nrf2 protein in KEAP1 mutant cancer cells and suppressed the expression of target genes downstream of Nrf2. These effects are likely because of enhanced degradation of Nrf2 through the proteasome system. MSU38225 not only inhibited the proliferation of cancer cells, especially Nrf2 addicted cancer cells, but also sensitized lung cancer cells to chemotherapies. These results described a novel pharmacological tool compound to study this important signaling pathway and confirmed the relevance of using an Nrf2 pathway inhibitor in combination with chemotherapies in Nrf2 addicted lung cancers.
MSU38225 presents different features compared to other reported Nrf2 pathway inhibitors. Brusatol, the most potent known Nrf2 inhibitor, acts rapidly in reducing Nrf2 protein expression as the maximum reduction of Nrf2 occurs within 2–4 hours. The inhibition of Nrf2 by brusatol is also reversible as Nrf2 protein levels returned to basal levels within 8 hours (22). This fast-acting behavior is consistent with the reported mechanism of brusatol in inhibiting global protein synthesis (25). In contrast, it took 24 hours for MSU38225 to reach peak inhibition. MSU38225 had no effect on Nrf2 transcripts. The different kinetics suggest that MSU38225 regulates the Nrf2 pathway in a mechanism other than inhibiting the synthesis of genes or proteins.
The effect of MSU38225 on Nrf2 protein was largely Keap1- and βTrCP-independent, as MSU38225 maintained its inhibitory effects on Nrf2 in Keap1 and βTrCP knockout cells. This regulation is different than the compound clobetasol propionate, for example, which promoted βTrCP-dependent degradation of Nrf2 (44). MSU38225, with different mechanisms, provides a new pharmacological tool that inhibits the Nrf2 pathway. In the future, we will further explore the molecular mechanisms of how MSU38225 induces the degradation of the Nrf2 protein. SIAH2 and CRIF1 are other negative regulators of Nrf2 beyond Keap1 and βTrCP (45) so MSU38225 will be tested in SIAH2 or CRIF1 deficient lines. Additionally, we are actively working on improving the potency and solubility of MSU38225 in collaboration with our medicinal chemist collaborators. Additional compounds will help establish structure-activity relationships, and the generation of a labeled MSU38225 compound will help determine the direct binding partners of this new class of Nrf2 pathway inhibitors.
MSU38225 showed promising selectivity for targeting the Nrf2 pathway. It did not alter the general expression pattern of enriched cellular proteins. It also did not regulate proteins in the NF-κB pathway, which has a similar mechanism of regulation as Nrf2 (46). When treated with MSU38225, Nrf2 addicted cells (Keap1 mutant) were more sensitive than Nrf2/Keap1 wild type cells. Notably, MSU38225 did not affect proliferation of normal epithelial cells at concentrations that inhibited the growth of tumor cells. All these results suggest an appropriate therapeutic window for MSU38225 to target the Nrf2 pathway in cancer cells without inducing global inhibitory effects. Future studies will be done to evaluate the effects of MSU38225 on the activity of other transcription factors to validate its selectivity.
Targeting the Nrf2 pathway has emerged as a new opportunity to treat tumors with constitutively activated Nrf2 (47). However, patients may not benefit from Nrf2 inhibitors equally. It is critical to determine which patient populations would benefit the most and how to maximize the utility of Nrf2 inhibition. For example, the effects of Nrf2 inhibitors may depend on the status of other genes. Targeting co-dependent vulnerabilities or synthetic lethal partners often enhances efficacy (48). Nrf2-mediated antioxidant activity can be upregulated by oncogenes, such as Kras, B-Raf, and Myc, to detoxify the increased ROS found in tumors (17), as genetic deletion of Nrf2 impairs tumorigenesis driven by activating Kras mutations (17). Loss of Nrf2 has also been shown to impair EGFR signaling, thus inhibiting cell proliferation in pancreatic cancer (49). In KRAS-mutant lung adenocarcinoma patients, KEAP1 mutations often accompany the loss of LKB1. LKB1-deficient cells with Nrf2 activation have enhanced cell survival and better maintenance of energetic and redox homeostasis in a glutamine-dependent manner. They are more sensitive to glutamine inhibitors (50). In future studies, it will be important to study the interactions of the Nrf2 pathway with other cellular signaling and metabolic pathways (such as IDH), so that we can determine how to combine Nrf2 inhibitors with other therapeutic agents and which patient subpopulations are the best candidates for Nrf2 pathway inhibitors.
As immunotherapies have become the first-line therapy in lung cancer, it is also critical to understand the effects of Nrf2 inhibitors on the immune system. The dual role of Nrf2 in cancer has focused on normal epithelial cells vs. cancer cells. In contrast, the effects of Nrf2 activity on immune cells within the tumor microenvironment have not been fully characterized. We have previously identified an unfavorable immune signature in advanced lung tumors from Nrf2 knockout mice in a carcinogen-induced mouse model of lung cancer (51). In contrast, other studies reported that Nrf2 activation promotes the polarization of macrophages to a M2 phenotypes and drives epithelial-mesenchymal transition (52). These differing results suggest that the effects of Nrf2 activation in the tumor microenvironment are likely context dependent. In future studies, we will evaluate the effects of Nrf2 pathway inhibitors in immune competent mice and investigate their modulation of cancer immunotherapies.
Supplementary Material
Acknowledgements
This work was supported by MTRAC for Life Sciences Innovation Hub - Mi-Kickstart Award and NIH R01 grant (RC109542) to Dr. Liby. Di Zhang was supported by the Integrative Pharmacology Sciences Training Program (IPSTP) training fellowship 5T32GM092715-07, a fellowship from the Aitch Foundation, a Penner Fund Fellowship and a NIH NCI F99/K00 fellowship (CA245473). The drug screening was done in the drug discovery core facility (ADDRC) at Michigan State University, and we thank Dr. Thomas Dexheimer for his assistance with this screen. We also thank Dr. Edmund Ellsworth for his input on medicinal chemistry and Jessica Moerland for her assistance with PCR validation. Joseph W. Zagorski, Sheng Liu, Rebekah C. Kennedy, and Cheryl E. Rockwell provided the CRISPR KO Jurkat cells.
Footnotes
Conflict of interest: DZ, ZH, ALO and KTL are named inventors on existing or filed patents assigned to Michigan State University. The other authors declare no competing interests.
References
- 1.Menegon S, Columbano A, Giordano S. The Dual Roles of NRF2 in Cancer. Trends Mol Med 2016;22(7):578–93 doi 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A 1994;91(21):9926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 2013;85(6):705–17 doi 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 4.Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila) 2008;1(3):187–91 doi 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, et al. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res 2007;67(6):2414–9 doi 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 6.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 2012;12(8):564–71 doi 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal 2010;13(11):1713–48 doi 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 8.Hu R, Saw CL, Yu R, Kong AN. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: antioxidant coupled with antiinflammatory. Antioxid Redox Signal 2010;13(11):1679–98 doi 10.1089/ars.2010.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res 2004;64(18):6424–31 doi 10.1158/0008-5472.CAN-04-1906. [DOI] [PubMed] [Google Scholar]
- 10.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res 2006;66(16):8293–6 doi 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 11.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A 2001;98(6):3410–5 doi 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489(7417):519–25 doi 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50 doi 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019;178(2):316–29 e18 doi 10.1016/j.cell.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Lang F, Yang C. NRF2 in human neoplasm: Cancer biology and potential therapeutic target. Pharmacol Ther 2021;217:107664 doi 10.1016/j.pharmthera.2020.107664. [DOI] [PubMed] [Google Scholar]
- 16.Taguchi K, Hirano I, Itoh T, Tanaka M, Miyajima A, Suzuki A, et al. Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol Cell Biol 2014;34(5):900–13 doi 10.1128/MCB.01384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011;475(7354):106–9 doi 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012;22(1):66–79 doi 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 19.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 2015;47(12):1475–81 doi 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Lu Y, Celiku O, Li A, Wu Q, Zhou Y, et al. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J Natl Cancer Inst 2019;111(10):1033–41 doi 10.1093/jnci/djy230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Pang Y, Caisova V, Ding J, Yu D, Zhou Y, et al. Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers (Basel) 2020;12(2) doi 10.3390/cancers12020280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A 2011;108(4):1433–8 doi 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem Biol 2016;11(11):3214–25 doi 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 2019. doi 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 25.Vartanian S, Ma TP, Lee J, Haverty PM, Kirkpatrick DS, Yu K, et al. Application of Mass Spectrometry Profiling to Establish Brusatol as an Inhibitor of Global Protein Synthesis. Mol Cell Proteomics 2016;15(4):1220–31 doi 10.1074/mcp.M115.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bollong MJ, Yun H, Sherwood L, Woods AK, Lairson LL, Schultz PG. A Small Molecule Inhibits Deregulated NRF2 Transcriptional Activity in Cancer. ACS Chem Biol 2015;10(10):2193–8 doi 10.1021/acschembio.5b00448. [DOI] [PubMed] [Google Scholar]
- 27.Lee S, Lim MJ, Kim MH, Yu CH, Yun YS, Ahn J, et al. An effective strategy for increasing the radiosensitivity of Human lung Cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radic Biol Med 2012;53(4):807–16 doi 10.1016/j.freeradbiomed.2012.05.038. [DOI] [PubMed] [Google Scholar]
- 28.Liao H, Zhu D, Bai M, Chen H, Yan S, Yu J, et al. Stigmasterol sensitizes endometrial cancer cells to chemotherapy by repressing Nrf2 signal pathway. Cancer Cell Int 2020;20:480 doi 10.1186/s12935-020-01470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu D, Liu Y, Zhou Y, Ruiz-Rodado V, Larion M, Xu G, et al. Triptolide suppresses IDH1-mutated malignancy via Nrf2-driven glutathione metabolism. Proc Natl Acad Sci U S A 2020;117(18):9964–72 doi 10.1073/pnas.1913633117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dissanayake AA, Staples RJ, Odom AL. Titanium-Catalyzed, One-Pot Synthesis of 2-Amino-3-cyano- pyridines. Advanced Synthesis & Catalysis 2014;356(8):1811–22 doi 10.1002/adsc.201301046. [DOI] [Google Scholar]
- 31.Zagorski JW, Maser TP, Liby KT, Rockwell CE. Nrf2-Dependent and -Independent Effects of tert-Butylhydroquinone, CDDO-Im, and H2O2 in Human Jurkat T Cells as Determined by CRISPR/Cas9 Gene Editing. J Pharmacol Exp Ther 2017;361(2):259–67 doi 10.1124/jpet.116.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984;22:27–55 doi 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 33.Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell 2009;34(2):212–22 doi 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003;8(4):379–91 doi 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 35.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol 2012;32(17):3486–99 doi 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, et al. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res 2005;65(11):4789–98 doi 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 37.Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol 2012;2:200 doi 10.3389/fonc.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai X, Chen Y, Hou X, Huang M, Jin J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab Rev 2016;48(4):541–67 doi 10.1080/03602532.2016.1197239. [DOI] [PubMed] [Google Scholar]
- 39.Yang H, Villani RM, Wang H, Simpson MJ, Roberts MS, Tang M, et al. The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res 2018;37(1):266 doi 10.1186/s13046-018-0909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med 2020;52(2):192–203 doi 10.1038/s12276-020-0384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bao LJ, Jaramillo MC, Zhang ZB, Zheng YX, Yao M, Zhang DD, et al. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int J Clin Exp Pathol 2014;7(4):1502–13. [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao L, Au JL, Wientjes MG. Comparison of methods for evaluating drug-drug interaction. Front Biosci (Elite Ed) 2010;2:241–9 doi 10.2741/e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gronberg BH, Bremnes RM, Flotten O, Amundsen T, Brunsvig PF, Hjelde HH, et al. Phase III study by the Norwegian lung cancer study group: pemetrexed plus carboplatin compared with gemcitabine plus carboplatin as first-line chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 2009;27(19):3217–24 doi 10.1200/JCO.2008.20.9114. [DOI] [PubMed] [Google Scholar]
- 44.Choi EJ, Jung BJ, Lee SH, Yoo HS, Shin EA, Ko HJ, et al. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017. doi 10.1038/onc.2017.153. [DOI] [PubMed] [Google Scholar]
- 45.Lewis KN, Wason E, Edrey YH, Kristan DM, Nevo E, Buffenstein R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc Natl Acad Sci U S A 2015;112(12):3722–7 doi 10.1073/pnas.1417566112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol 2005;7(8):758–65 doi 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panieri E, Saso L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid Med Cell Longev 2019;2019:8592348 doi 10.1155/2019/8592348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 2020;19(1):23–38 doi 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- 49.Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016;166(4):963–76 doi 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res 2019;79(13):3251–67 doi 10.1158/0008-5472.CAN-18-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang D, Rennhack J, Andrechek ER, Rockwell CE, Liby KT. Identification of an Unfavorable Immune Signature in Advanced Lung Tumors from Nrf2-Deficient Mice. Antioxid Redox Signal 2018;29(16):1535–52 doi 10.1089/ars.2017.7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y, et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun Signal 2018;16(1):54 doi 10.1186/s12964-018-0262-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.