

Abstract
Myocardial fibrosis is the heart’s common healing response to injury. While initially seeking to optimize the strength of diseased tissue, fibrosis can become maladaptive, producing stiff poorly functioning and pro-arrhythmic myocardium. Different patterns of fibrosis are associated with different myocardial disease states, but the presence and quantity of fibrosis largely confer adverse prognosis. Current imaging techniques can assess the extent and pattern of myocardial scarring, but lack specificity and detect the presence of established fibrosis when the window to modify this process may have ended. For the first time, novel molecular imaging methods, including gallium-68 (68Ga)-fibroblast activation protein inhibitor positron emission tomography (68Ga-FAPI PET), may permit highly specific imaging of fibrosis activity. These approaches may facilitate earlier fibrosis detection, differentiation of active vs. end-stage disease, and assessment of both disease progression and treatment–response thereby improving patient care and clinical outcomes.
Keywords: myocardial fibrosis, fibrosis imaging, molecular fibrosis imaging, positron emission tomography and cardiovascular magnetic resonance, fibroblast activation protein inhibitor
Graphical Abstract
Graphical Abstract.

Myocardial fibrosis occurs when various forms of myocardial injury affect previously healthy myocardium. Various existing imaging techniques including cardiovascular magnetic resonance, computed tomography, echocardiography, and nuclear imaging (single-photon emission computed tomography and 18F-FDG PET) assess the extent and pattern of myocardium scar. However, they are not specific to fibrosis and detect established fibrosis that may no longer be modifiable with treatment. Novel molecular fibrosis imaging methods may for the first time allow highly specific imaging of fibrosis activity. Benefits over existing modalities may include detection of the earliest stages of fibrogenesis, differentiation between active and end-stage disease, assessment of response to treatment in vivo as well as determination of the anti-fibrotic potential of existing and novel agents. These new techniques remain under investigation to determine their clinical utility. CMR images of myocardial infarction courtesy of Dr Trisha Singh. 68Ga-FAPI-04 Images courtesy of Dr Zohreh Varasteh.
Fibrosis in response to injury is a major mediator of cardiovascular disease. In the myocardium, fibrosis is the final common pathway of virtually all heart muscle disease processes.1–4 While fibrous tissue provides some structural tissue integrity and initial fibrosis can be protective, dysregulated excessive fibrosis causes maladaptive cardiac remodelling, development of heart failure, and adverse cardiovascular events.5–7 Fibrosis therefore represents a key imaging target to improve clinical diagnosis, assess disease activity and provide risk stratification as well as a therapeutic target to halt adverse remodelling and maintain myocardial health.8,9
Several imaging methods can identify and quantify myocardial fibrosis although none are truly specific. Cardiovascular magnetic resonance (CMR) imaging is considered the current reference standard.5,6 However, even CMR only infers the presence of fibrosis by highlighting the presence of increased extracellular space and altered vascularity, two processes accompanying other disease states.7,10,11 Novel molecular approaches now aim to provide more direct and specific assessments of fibrosis activity, potentially allowing the detection and differentiation of the various stages of fibrogenesis.12,13Is the heart scarred or is it scarring? These assessments of fibrosis activity hold promise in improving our understanding of the underlying pathophysiology and identifying the patients most likely to benefit from the array of therapeutic and anti-fibrotic interventions currently in development. Indeed, they may hasten the development of these eagerly anticipated treatments.
In this review, we describe the pathophysiology of myocardial fibrosis and its wide range of potential triggers. We then briefly review current methods for imaging myocardial fibrosis and their clinical applications, before focusing on novel molecular imaging assessments of fibrosis activity and the potential advances that these new approaches might herald.
Normal myocardium
Normal myocardium comprises cardiac myocytes and the surrounding extracellular matrix in a 3:1 ratio.14 Cardiac fibroblasts are also common, maintaining myocardial homeostasis through balanced generation and removal of extracellular matrix components.15 Fibroblasts generate collagen, matrix metalloproteinases, and tissue inhibitors of matrix metalloproteinases to regulate this balance.15
Collagen in its mature form accounts for around 30% of all mammalian protein and maintains the extracellular matrix as a strongly interconnected scaffold, supporting normal tissue architecture.16 In health, the extracellular matrix is comprised of 85% Type I collagen, providing tissue stiffness and structure, and 11% Type III collagen, responsible for tissue elasticity.17 Proline is a pre-collagen constituent that undergoes hydroxylation to hydroxyproline to form the triple helix structure characteristic of mature collagen.16 Unlike other amino acids, proline is exclusive to collagen, making it an excellent fibrosis imaging target.16
Myocardial fibrosis
Fibrosis is the common pathological response to myocardial injury. The initial myocardial insult may arise from a wide range of stimuli leading to different fibrosis patterns (Figure 1). The more common triggers of fibrosis are discussed briefly below.
Figure 1.

Potential triggers for myocardial fibrosis. A variety of cardiovascular conditions can cause myocardial injury and induce fibrogenesis within the myocardium.
Triggers of myocardial fibrosis
Myocardial inflammation
An inflammatory response follows many forms of myocardial injury, including infarction, infection, or toxins. It may be the primary manifestation of the myocardial insult, such as acute myocarditis, or a by-product of that injury, as for myocardial infarction.18,19 The inflammatory process typically includes the recruitment of macrophages, monocytes, mast cells, and lymphocytes.2,18 These inflammatory cells secrete a range of cytokines that are fundamental to the initiation and propagation of myocardial fibrosis activity.2,18,20
Myocardial ischaemia
Coronary artery atherothrombosis leads to Type 1 myocardial infarction and abrupt cessation of blood flow to the myocardium. This results in a wave of myocyte necrosis in the subtended myocardium that extends from the sub-endocardium toward the sub-epicardium and serves as a potent trigger to myocardial fibrosis.19,21 Myocardial fibrosis may also develop in response to recurrent chronic ischaemia with interceding reperfusion, where chemokine-mediated fibroblast and macrophage recruitment are important to its formation.19,22
Pressure overload
In conditions leading to myocardial pressure overload (e.g. hypertension or aortic stenosis), a hypertrophic response is triggered which is initially adaptive restoring wall stress and maintaining cardiac performance. However, with time, the hypertrophied myocardium outgrows its blood supply leading to myocyte cell death, myocardial fibrosis, and the transition to heart failure, symptoms, and adverse events.23 Activation of transforming growth factor-β by Angiotensin-II appears particularly important in driving pressure overload-mediated myocardial fibrosis.19
Volume overload
In conditions such as aortic or mitral regurgitation, volume overload drives maladaptive changes. In contrast to pressure overload, extracellular matrix degradation is a key driver of this transformation, with the up-regulation of proteases and interstitial collagen loss.19 Mast cell numbers increase, contributing matrix metalloproteinases and pro-inflammatory cytokines to aggravate this maladaptive pathological response.19
Genetic mutations
Genetic mutations are responsible for a range of fibrotic myocardial disorders including hypertrophic cardiomyopathy, Fabry disease, Pompé disease, as well as genetically mediated forms of dilated and restrictive cardiomyopathies.24 Of these, hypertrophic cardiomyopathy is the commonest, resulting from sarcomeric protein gene mutations.24 Patients who develop the condition as well as asymptomatic carriers of sarcomeric mutations appear to have a higher risk of heart failure due to the activation of collagen-producing pathways and an early generation of a pro-fibrotic state.24,25 This may be further exacerbated by myocardial ischaemia, again due to hypertrophy and supply–demand mismatch.25
Pathophysiology of myocardial fibrosis
Fibroblast activation and fibrosis formation
Activated fibroblasts are the key cells driving myocardial fibrosis. As a common response to the different forms of myocardial injury, cardiac fibroblasts differentiate into their activated and pro-fibrotic subtype.26
Fibroblast activation protein (FAP) is a membrane-bound serine protease with actions related to matrix remodelling. It is almost exclusively expressed on the surface of activated fibroblasts (termed myofibroblasts in the heart when they also express alpha-smooth muscle actin) and is a marker of fibroblast activation and fibrosis formation.1,26 Seldom seen in healthy adult human tissue, FAP expression is observed in many cardiovascular diseases including acute myocardial infarction, hypertrophic cardiomyopathy, dilated cardiomyopathy, and heart failure, and is considered a marker for myocardial injury, fibrosis activity, and matrix remodelling.1–3 Other markers of activated fibroblasts include alpha-smooth muscle actin and vimentin. Activated fibroblasts participate in myocardial fibrosis via dynamic interactions between collagen, the extracellular matrix, and other cell types involved in fibrosis formation.
Dysregulated collagen formation and its excess deposition from activated fibroblasts are the key changes encountered in pathological fibrosis.19 Collagen deposition causes extracellular matrix expansion, increased tissue stiffness leading to diastolic dysfunction and, when advanced, impaired contractility causing systolic dysfunction. In addition, activated myocardial fibroblasts up-regulate a range of pro-inflammatory and pro-fibrotic mediators, including transforming growth factor-β, interleukin-11, tumour necrosis factor-α, the renin–angiotensin–aldosterone system, the sympathetic nervous system, the endothelin system, and alpha v beta 3 (αvβ3) integrin receptors.15,18,21,26–28 Many of these mediators in turn activate other fibroblasts driving further fibrogenesis.
Forms of myocardial fibrosis
Synthesis of Types I and III collagen are increased in both interstitial and replacement myocardial fibrosis regardless of the underlying aetiology.19 However, the ratio in which they are elevated varies depending on the cause of myocardial fibrosis.19 In myocardial fibrosis caused by myocardial infarction or hypertensive disease, up-regulation of stiff inflexible Type I collagen exceeds that of Type III, but in chronic ischaemic heart disease, Type III collagen predominates.29–31
When considering fibrosis imaging methods, the following two broad categories of fibrosis are often referred: interstitial fibrosis and replacement fibrosis (Figure 2).6 For the purposes of this review, it is convenient to consider these two subtypes separately. However, it must be highlighted that at both the pathological and molecular levels, the highly complex and potentially long-lasting process of fibrogenesis cannot be neatly categorized into one form or another: not only is there a considerable overlap, but they also often coexist.32
Figure 2.

Composition of the normal myocardium and pathological changes of fibrosis. Following myocardial injury, fibroblasts are activated to instigate the transformation from normal myocardium to fibrosis. In interstitial fibrosis, myocyte membranes are not compromised, there is no myocyte death, and a more diffuse pattern of potentially reversible fibrosis ensues. Other more intense forms of injury cause myocyte cell death from the outset, triggering replacement fibrosis seen as focal regions of irreversible cardiac scarring. Interstitial fibrosis can progress to replacement fibrosis in the presence of persistent exposure to myocardial injury. In reality, significant overlap exists between these types but they remain useful when considering the different imaging techniques.
Interstitial fibrosis
Reactive interstitial fibrosis is generally considered a marker of early disease and is associated with more diffuse forms of myocardial injury, such as hypertension and aortic stenosis.2,6 Consequently, interstitial fibrosis is distributed diffusely throughout the myocardium. If the trigger to myocardial injury resolves then interstitial fibrosis has the potential to reverse, leaving behind a fully functional myocardium. However, if the trigger persists leading to myocyte cell death, then interstitial fibrosis can progress to replacement fibrosis.2,6
Replacement fibrosis
Forms of myocardial injury leading to myocyte death and loss of cell membrane integrity, be they abrupt, intense, or insidious, will cause replacement fibrosis. Its molecular and structural components are largely shared with interstitial fibrosis, the main differences being that replacement fibrosis is usually focal rather than diffuse, and is considered irreversible, unlike interstitial fibrosis where regression or resolution is possible.6,11
Current methods for assessing myocardial fibrosis
Non-imaging methods of myocardial fibrosis assessment
Plasma biomarkers and endomyocardial biopsy are the main non-imaging methods of assessing myocardial fibrosis. Plasma biomarkers of fibrosis activity include hydroxyproline, N-terminal propeptide, matrix metalloproteinases, and tissue inhibitors of matrix metalloproteinases.33–35 While elevation of these biomarkers has been described across multiple cardiovascular disorders,34 they are not specific to the heart and can become elevated in fibrotic conditions throughout the body.
Histological assessment of myocardial biopsies is considered to be the gold-standard method of detecting myocardial fibrosis. It is rarely performed apart from post-cardiac transplant where the presence of interstitial fibrosis infers allograft rejection.36 Endomyocardial biopsy confers generic invasive procedural risks alongside serious specific complications, such as myocardial perforation and cardiac tamponade.36 Although prior imaging results can be used to guide sampling, the potential remains for sampling errors causing false-negative results.36 Targeted and specific assessment of myocardial fibrosis therefore can be provided neither by serum biomarkers nor direct tissue sampling.
Imaging
A range of established clinical imaging techniques allows the non-invasive assessment myocardial fibrosis, including echocardiography, computed tomography (CT), CMR, single-photon emission computed tomography (SPECT) imaging, and positron emission tomography (PET). Each has its own advantages and disadvantages, and potential opportunities for development (Table 1).
Table 1.
Conventional imaging modalities for the assessment of established myocardial fibrosis and its surrogates
| Conventional imaging modalities | |||||
|---|---|---|---|---|---|
| Modality | Method | Focus of assessment | Advantages | Disadvantages | Next steps |
| Echocardiography •LVEF •GLS •Diastolic dysfunction | Ultrasound waves directed via probe to detect cardiac structures | LV systolic and diastolic dysfunction and structural change resulting from fibrosis | Cheap, portable, no radiation exposure Available on routine echocardiograms Correlates with CMR fibrosis assessments | Images surrogates of fibrosis rather than direct assessment Less sensitive to detecting smaller or more subtle structural/functional consequences of scar | Establishing clear thresholds to guide patient management and clinical decision-making (GLS) |
| Nuclear •18F-FDG PET | Determines viability of myocytes by visualizing uptake of radiolabelled glucose analogue therefore inferring normal glucose utilization | Detection of viable myocardium | High sensitivity of PET | Ionizing radiation Availability of scanners for cardiovascular imaging Expensive scans Images viable myocardium rather than fibrosis directly | Development of tracers targeting fibrosis more directly |
| ȃ•SPECT | Single-photon emission CT at rest and during stress to detect fixed and reversible perfusion deficits | Detection of irreversible perfusion deficits indicates scar | Widely available imaging technique with well-established protocols and image analysis software Exercise or pharmacological stress options available so suitable for all levels of mobility | Ionizing radiation Caffeine restriction Limited spatial resolution—unable to detect small areas of fibrosis nor to differentiate the different patterns of scarring | Improvements in hardware capability and sensitivity: multiple detectors, high-sensitivity collimation Improvements in software processing: iterative construction, attenuation correction Use of reduced radiation dosage protocols |
| CMR •Native T1 | Maps time course taken by tissues recovering from longitudinal magnetization | Interstitial fibrosis | No GBCA required Well-suited to widespread and diffuse changes Provides prognostic information in several conditions | Lack of consistency in values across scanners and magnetic field strengths Overlap in values between disease states, and with normal myocardium | Standardization of T1 values across scanners Establishment of normal ranges and ranges for specific disease states Further prognostic data |
| ȃ•ECV% | Informs about the percentage of the myocardium comprised by extracellular matrix Uses haematocrit to calculate cell fraction in blood pool and myocardium pre- and post-contrast | Interstitial fibrosis (ECV expansion) | More sensitive in detecting diffuse fibrosis than native T1 Comparable across scanners and magnetic field strengths Prognostic information in several conditions | Requires GBCA Dependent on blood flow and renal clearance Overlap in values between different diseases and with normal myocardium (less so than for native T1) ECV expansion is not synonymous with fibrosis | Establishment of normal ranges and ranges for specific disease states Further prognostic data Multicentre studies |
| ȃ•iECV | Represents the burden of established fibrosis in the LV. Calculated by multiplying indexed LV volume by ECV% | Interstitial fibrosis (ECV expansion) | Tracks progression and regression of fibrosis Good discrimination between disease states Comparable across scanners and magnetic field strengths Prognostic information in AS | Requires GBCA Limited prognostic information ECV expansion is not synonymous with fibrosis | Investigation of prognostic utility in other myocardial disease states Studies investigating change in iECV with anti-fibrotic therapy |
| ȃ•LGE | Slowed GBCA clearance from damaged tissue ECM | Replacement fibrosis | Detection of focal scar tissue Strong prognostic predictor across a wide range of conditions | Requires GBCA Not suited to detecting diffuse interstitial fibrosis Difficulty in detecting fibrosis outside the left ventricle Not specific to fibrosis (increased signal with oedema, infiltration etc.) | Developments to improve detection of atrial and right ventricular fibrosis Clinical trials demonstrating the clinical efficacy of LGE assessments in guiding clinical practice |
| CT •ECV | Uses X-rays to provide cross-sectional imaging. Fibrosis is detected using IV contrast | Interstitial fibrosis (ECV expansion) | Agreement with CMR T1 mapping-derived ECV CT often performed for other clinical indications (e.g. TAVR work up, coronary artery disease) | Requires IV contrast Ionizing radiation Lower contrast intensity compared with CMR | Further comparison with established CMR methods Investigate clinical utility in patients with coronary disease, and patients being considered for TAVR with suspected amyloidosis |
A range of methods are currently available to assess myocardial fibrosis each with different benefits and considerations. CMR, cardiovascular magnetic imaging; LGE, late gadolinium enhancement; ECV, extracellular volume; ECV%, percentage of extracellular volume; iECV, indexed extracellular volume; LV, left ventricular; GBCA, gadolinium-based contrast agent; ECM, extracellular matrix; LVEF, left ventricular ejection fraction; GLS, global longitudinal strain; CT, computed tomography; IV, intravenous; TAVR, transcatheter aortic valve replacement; AS, aortic stenosis; SPECT, single photon emission computed tomography; 18F-FDG, 18F-fluorodeoxyglucose; PET, positron emission tomography.
Echocardiography and nuclear imaging are often employed to provide complementary assessments in myocardial fibrotic conditions, though they detect consequences or surrogates of fibrosis, such as ventricular wall thinning (echocardiography) or lack of radiotracer uptake (SPECT or PET), rather than fibrosis itself (Table 1).37–40
CMR uses both T1 mapping and late gadolinium enhancement (LGE) techniques, the latter of which is considered the gold-standard in myocardial fibrosis assessment and confers important adverse prognostic consequences in a range of myocardial disorders.41–47 T1 mapping records the time course taken by tissues to recover two-thirds from longitudinal magnetization, producing a T1 value measured in milliseconds.10 LGE is a ‘difference test’ which relies on slower clearance of gadolinium-based contrast agents from the extracellular matrix of diseased compared with healthy tissue.5 Extracellular volume can be quantitively measured using techniques that assess T1 values pre- and post-gadolinium contrast administration (Table 1). The percentage of the myocardial volume comprised by the extracellular matrix (ECV%) has been studied across several cardiomyopathic conditions, and is a marker of disease severity and a powerful prognostic indicator in aortic stenosis and dilated cardiomyopathy.11,48–50 Indexed extracellular volume (iECV) is calculated by multiplying the ECV% by the indexed myocardial volume and therefore provides an assessment of the matrix volume and myocardial fibrosis burden (Table 1). It appears well-suited to tracking fibrosis progression and regression.11,48,50
While considerable overlap exists, LGE is predominantly used to detect focal replacement fibrosis whereas T1 techniques image both interstitial and replacement fibrosis (Table 1).5,10 It is also possible to detect areas of focal fibrosis and to measure the extracellular volume using iodinated-contrast-enhanced CT although this is inferior to CMR assessments.51
Current applications of CMR fibrosis imaging
CMR provides a direct assessment of myocardial fibrosis and has now entered widespread clinical use. The pattern and extent of fibrosis detected by CMR have important diagnostic and prognostic implications that might influence clinical management decisions (Table 2, Graphical Abstract).6,11,41–47
Table 2.
Cardiac magnetic resonance assessments of myocardial injury and fibrosis
| Condition | Typical findings in early disease | Typical findings in established disease | T1 mapping appearance | LGE appearance | Current and Emerging Clinical Roles |
|---|---|---|---|---|---|
| Myocardial infarction | • Increased T2, 18F-FDG PET, native T1 and ECV% values • Subendocardial or transmural LGE: reflecting myocyte necrosis and oedema |
• Subendocardial or transmural pattern of LGE reflecting established scar • Normal T1 values in remote myocardium |
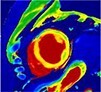
|
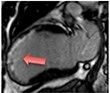
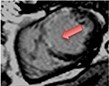
|
Current Diagnosis: Differentiation of ischaemic heart disease vs. dilated cardiomyopathy and in presentations of MINOCA5,53Prognosis: LGE associated with increased all cause and cardiovascular mortality42Viability assessments: Guide consideration of coronary revascularisation |
| Dilated cardiomyopathy | • Interstitial fibrosis generally precedes replacement fibrosis: raised native T1, ECV% | • Linear mid-wall or subepicardial LGE -T1 markers increased interstitial fibrosis commonly co-exist |

|
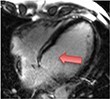
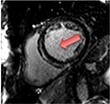
|
Current Diagnosis: Differentiation of ischaemic heart disease vs. dilated cardiomyopathy5Prognosis: • LGE associated with increased mortality43 and dysrhythmia41 • Raised native T1 and ECV% associated with increased all-cause mortality and/or heart failure hospitalisation or heart failure death48,54Emerging Fibrosis assessments to guide ICD implantation: CMR-GUIDE, CMR-ICD |
| Myocarditis | • Increased T2 and 18F-FDG PET (oedema & inflammation) • Focal mid-wall and subendocardial LGE (necrosis and oedema) |
• Mid-wall and subendocardial LGE (established replacement fibrosis) • Patients may go on to develop a dilated cardiomyopathy phenotype |
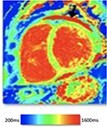
|
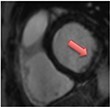
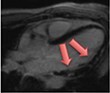
|
Current Diagnosis: Differentiation for other cause of acute chest pain and troponin elevation (e.g. myocardial infarction, Tako-tsubo cardiomyopathy etc.) Prognosis: LGE associated with increased mortality and major adverse cardiovascular events44 |
| Sarcoidosis | • Patchy areas of increased T2 and 18F-FDG PET (oedema & inflammation) • Focal non-infarct pattern of LGE (necrosis, oedema granuloma formation) |
• Patchy non-infarct pattern of LGE often within the subepicardium and mid-wall • Persistent elevated T2 and 18F-FDG activity may or may not be present as evidence of ongoing active disease |

|
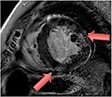
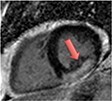
|
Current Diagnosis: Central role in the diagnosis of cardiac sarcoid Prognosis: LGE associated with increased cardiovascular death and ventricular dysrhythmia45 |
| Hypertrophic cardiomyopathy | • Raised native T1 and ECV% compared to normal myocardium but overlap with other disease states | • Mid-wall LGE within hypertrophied segments and ventricular insertion points |

|
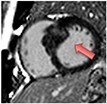
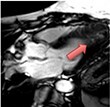
|
Current Diagnosis: Differentiation from phenocopies e.g. hypertension, Fabry’s, amyloid Prognosis: High volume LGE associated with increased mortality and ventricular dysrhythmia/sudden cardiac death46Emerging Assessing the effects of novel medication. Guiding ICD implantation |
| Aortic stenosis | • Raised native T1, ECV% and iECV | • Non-infarct mid-wall LGE |
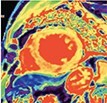
|
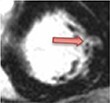
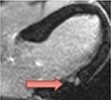
|
Current Diagnosis: Markers of fibrosis provide objective markers of LV decompensation Prognosis: • LGE associated with increased mortality11 • Raised native T1 and ECV% associated with increased mortality,49,55 and native T1 with heart failure hospitalisation55Emerging Optimizing the timing of valve replacement |
| Heart failure with preserved ejection fraction | • Diffusely raised native T1 values | • Non-infarct mid-wall or epicardial LGE has been described |

|
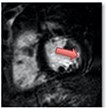
|
Current Diagnosis: Establishing the diagnosis and identifying the underlying cause as well as exclusion of other conditions (e.g. pericardial constriction, pulmonary hypertension) Prognosis: LGE and raised T1 associated with hospitalisation and mortality47 |
The pattern and timing of fibrosis formation and its corresponding imaging findings vary in different cardiomyopathic disorders. Current methods of assessing myocardial fibrosis have several defined and emerging roles in clinical practice. Native T1 values represent interstitial fibrosis with higher values represented by yellow, orange, or red voxels. Late gadolinium enhancement (white areas in the otherwise black myocardium) represents replacement fibrosis. Increased T2 values indicate myocardial oedema suggestive of inflammation.52 Native T1 image in myocardial infarction courtesy of Dr Trisha Singh, in dilated cardiomyopathy, myocarditis, hypertrophic cardiomyopathy, and heart failure with preserved ejection fraction reprinted from Haaf et al.,56 in sarcoidosis from Chang et al.,57 and in aortic stenosis from Lee et al.55 Images depicting late gadolinium enhancement adapted from Bing et al.,5 asides upper sarcoidosis image (Chang et al.)57 and heart failure with preserved ejection fraction (Haaf et al.).5618F-FDG PET, 18F-fluorodeoxyglucose positron emission tomography; ECV%, percentage of extracellular volume; LGE, late gadolinium enhancement; MINOCA, myocardial infarction with non-obstructive coronary arteries; ICD, implantable cardiac defibrillator; iECV, indexed extracellular volume; LV, left ventricular.
Longitudinal CMR studies can inform how myocardial fibrosis changes with time and intervention. The Efficacy and Safety of Pirfenidone in Patients With Heart Failure and Preserved Left Ventricular Ejection Fraction (PIROUETTE) trial investigated the anti-fibrotic effects of pirfenidone, a transforming growth factor-β inhibitor, demonstrating that pirfenidone treatment successfully reduced diffuse interstitial fibrosis as detected with T1 mapping and the percentage of extracellular volume.58 This study paves the way for future mechanistic trials using similar fibrosis imaging endpoints to accelerate the development of novel therapies for patients with heart muscle disease.
Limitations of current fibrosis assessments
All current methods of imaging myocardial fibrosis have limitations (Table 2). Despite being the gold-standard method of fibrosis assessment, CMR-obtained LGE measures extracellular expansion rather than fibrosis itself. Although usually due to fibrosis, it may also reflect other pathological processes depending on the chronicity and the underlying pathology, such as oedema, infiltration, and protein deposition.7,59 The same is true for post-contrast myocardial T1 techniques. Furthermore, native myocardial T1 values vary with multiple intracellular and extracellular factors other than myocardial fibrosis and frequently demonstrate important overlap both between different disease states, and disease states and normal myocardium.10 Native T1 mapping values are also not comparable across different scanners and individuals, although because they are based upon a ratio, extracellular volume assessments can in principle correct for this variability allowing easier comparison between sites.10,11 Fundamentally, while informative about established fibrosis and/or its surrogates, none of these techniques detect the very early stages of fibrogenesis and provide little indication of ongoing disease activity nor whether the disease process is amenable to modification.
Future of molecular myocardial fibrosis imaging
Molecular imaging has the potential to deliver highly specific imaging of fibrogenesis, providing a readout of disease activity to complement the established myocardial fibrosis imaging approaches. The widespread availability of PET imaging for oncology and its increasing application for the assessment of cardiovascular inflammation provides the foundation for molecular fibrosis imaging.60 In principle, molecular PET imaging can measure the activity of any pathological process in the body, subject to the availability of a suitable radiotracer. However, it is only in the last few years that radiotracers specifically targeting fibrosis activity have become available, facilitating for the first time the non-invasive study of myocardial fibrosis activity (Figure 3). Of course, the superiority of cell-specific tracers in detecting molecular markers of fibrosis activity should be expected over conventional methods, which instead detect the less sensitive measures of extracellular expansion or tissue recovery time. When considering the novel radiotracers we will now discuss, a more appropriate comparison would be with a hybrid of CMR and conventional PET imaging with tracers such as fluorine-18-fluorodeoxyglucose (18F-FDG).
Figure 3.

Molecular imaging assessments of myocardial fibrosis activity. Positron emission tomography (PET) radiotracers fused with either computed tomography (CT; PET/CT) or cardiovascular magnetic resonance (CMR; PET/MR) to detect active myocardial damage in a range of conditions. Panels A and B: late gadolinium enhancement (LGE) (left panel) and 18F-fluciclatide PET/CT (right panel) 8 and 13 days following anterior myocardial infarction respectively with intense tracer uptake within the infarct.21 Panel C: intense 68-gallium fibroblast activation protein inhibitor (68Ga-FAPI) uptake on PET/MR within an inferior myocardial infarct in a patient with out-of-hospital cardiac arrest and cardiopulmonary resuscitation. Intense tracer uptake within multiple ribs, representing healing rib fractures (lower panel, 68Ga-FAPI PET image). Panel D: increased 68Ga-FAPI PET/MR uptake within regions of posterior (upper panels) and anteroseptal (lower panels) scar following myocardial infarction in two patients imaged at Days 3 and 8, respectively. Panel E: 68Ga-FAPI PET (top image), CT (middle image), and fused PET/CT image (bottom image) demonstrating right heart 68Ga-FAPI uptake in idiopathic pulmonary artery hypertension and right heart failure. Adapted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature, Wang et al.61 Panel F: 68Ga-FAPI PET (left image), and fused PET/CT (right images) demonstrate left ventricular 68Ga-FAPI uptake in a patient with chemotherapy-induced cardiotoxicity (ejection fraction 41%). Reprinted from Totzeck et al.62
18F-fluciclatide
18F-fluciclatide is a novel arginine–glycine–aspartate tripeptide radiotracer that binds the αvβ3 integrin transmembrane receptor,21 activation of which up-regulates extracellular matrix modification and angiogenesis. 18F-fluciclatide has therefore been proposed as a marker of fibrosis and angiogenesis.21
Increased myocardial 18F-fluciclatide uptake occurs in patients with acute myocardial infarction. This contrasts with the absence of 18F-fluciclatide activity in patients with chronic myocardial infarction (Graphical Abstract, Figure 3).21 Moreover, 18F-fluciclatide uptake is associated with subsequent functional myocardial recovery, suggesting a potential role as a marker of adaptive myocardial remodelling and repair.21 Potential limitations of 18F-fluciclatide are the relatively low myocardial signal and low specificity since uptake may represent fibrosis activity, inflammation, or angiogenesis. The uptake observed in acute but not chronic myocardial infarction, and its association with myocardial recovery could represent any or all of these processes.
18F-proline
Proline is a collagen precursor which is incorporated into mature collagen following hydroxylation to hydroxyproline.16 Proline can also be radiolabelled to form 18F-proline to identify areas of fibrosis activity.63 However, it can exist in cis- or trans-forms, both of which can assume a D- or L-isomer, providing four distinct isomeric configurations, each with different radiotracer properties.
In animal studies, increased 18F-proline uptake occurs in regions of active fibrosis across different organ systems.64 In humans, several 18F-proline isomers have been safely administered to patients in oncological research, with acceptable kinetics and radiation dose.63In vivo stability appears greater for the L-isomers, whereas D-isomers are transported across the blood–brain barrier allowing imaging of brain disease.63 Clinically cis-18F-proline has been investigated in non-cardiac conditions with mixed results65 likely reflecting the challenges of its manufacture. Data in myocardial fibrosis are therefore awaited although pre-clinical studies are ongoing using improved synthetic methods to determine the optimum configuration of this radiotracer for myocardial fibrosis imaging.
Radiotracers of FAP
Given that FAP expression is a marker of myofibroblast activation and active fibrogenesis, it is an ideal target for imaging fibrosis activity. Fibroblast activation protein-specific inhibitors (FAPIs) were originally developed as cancer therapies but have now been radiolabelled to form radiotracers of fibrosis activity that bind but do not affect the function of FAP nor the activated fibroblasts expressing it.12,13 Various FAPIs have been radiolabelled with gallium-68 (68Ga-FAPI) or fluorine-18 aluminium fluoride (18F-AlF-FAPI) and safely administered to humans with rapid uptake by activated fibroblasts and both favourable pharmacokinetics and imaging characteristics at acceptable radiation doses.13 FAPI tracers are highly specific for FAP-positive fibroblasts, becoming internalized within the cell following binding with little subsequent leakage. This leads to a very low background signal in healthy tissue but intense activity in areas of activated fibroblasts with FAP expression. These tracers are being widely investigated in the field of oncology for the imaging of cancer-associated fibroblasts and demonstrate favourable imaging properties compared with 8F-fluorodeoxyglucose, the current standard for tumour imaging. FAPI radiotracers are also increasingly being used to image a wide range of fibrotic disease states including interstitial lung disease, renal fibrosis, liver fibrosis, and IgG4-related disease.66–69
FAP imaging in the myocardium
Initial studies investigating FAPI PET uptake in the cardiovascular system have been largely based on incidental observations in patients with cancer. In the largest study to date, myocardial 68Ga-FAPI uptake was investigated in 229 patients undergoing evaluation for metastatic cancer.70 On multivariable analysis, increased left ventricular 68Ga-FAPI uptake correlated with cardiovascular risk factors (elevated body-mass index, Type II diabetes mellitus), previous platinum-based chemotherapy, and prior radiotherapy to the chest.70 In a sub-group (n = 44) with contemporaneous echocardiography, 68Ga-FAPI uptake was associated with reduced ejection fraction. Similar findings were noted in a smaller observational study of 32 patients who underwent 68Ga-FAPI PET/MR.71 Here, 20% of patients had focal myocardial 68Ga-FAPI uptake, with these patients being older, having lower left ventricular ejection fractions and a higher frequency of coronary artery disease.
Myocardial infarction
68Ga-FAPI PET was first assessed in murine models of myocardial infarction. In a coronary artery ligation model, serial 68Ga-FAPI imaging showed intense radiotracer uptake in and adjacent to the infarct zone which peaked after 6 days.72 Thereafter, myocardial 68Ga-FAPI uptake gradually decreased, returning to near baseline by 2 weeks.72 Interestingly, post-mortem autoradiography demonstrated that 68Ga-FAPI uptake was more intense in the tissues bordering the area of infarction rather than the infarct region itself. No remote myocardial fibrosis activity was detected during the timescale of these experiments. This study highlighted the ability of 68Ga-FAPI imaging to track changes in fibroblast activation and myocardial fibrosis activity over time with histological validation of the signal.72
Single time-point prospective imaging studies have now been performed in patients with recent ST-segment elevation myocardial infarction. Intense 68Ga-FAPI uptake occurs in the infarct zone with excellent signal-to-noise (maximum standardized uptake values (SUVmax) of 5.8 ± 1.6 vs. 2.1 ± 0.5 in the blood pool).73 Interestingly, this uptake again appeared to extend into the peri-infarct zone, being more extensive in size than the infarct zone identified by both nuclear perfusion imaging and CMR LGE.4,73 These findings were confirmed in a recent study of patients imaged within 6 weeks of their acute myocardial infarction, demonstrating consistent intense FAPI uptake within both the infarct and neighbouring border zone.74
Chemotherapy-induced cardiotoxicity
The first report of FAPI imaging in the heart described increased left ventricular 68Ga-FAPI uptake in a patient with chemotherapy-associated cardiotoxicity (Figure 3).62 Subsequent analysis of 229 patients found that patients treated with anthracyclines or alkylating agents had unexpectedly high myocardial 68Ga-FAPI uptake.70 Multivariate modelling also showed an association between increased 68Ga-FAPI uptake and previous radiotherapy.70
Checkpoint inhibitor-related myocarditis is a rare phenomenon in cardio-oncology with a high mortality that is challenging to diagnose. 68Ga-FAPI imaging in three patients with checkpoint inhibitor myocarditis demonstrated focal intense myocardial 68Ga-FAPI uptake in each subject, while no uptake was observed in a further 23 patients who underwent checkpoint inhibitor treatment without myocarditis.75 Together these data suggest 68Ga-FAPI imaging might prove invaluable in imaging both cancer and the cardiotoxicity associated with its treatment.
Cardiomyopathies
In a recent paper by Epstein and colleagues, FAP demonstrated the greatest-fold up-regulation of all fibroblast-specific genes in 116 patients with dilated (n = 89) or hypertrophic (n = 27) cardiomyopathies.8 A subsequent case report reported increased myocardial 68Ga-FAPI uptake in a young male patient with dilated cardiomyopathy.76 There have been no studies investigating FAPI PET in patients with hypertrophic cardiomyopathy at this stage, although recent case reports have demonstrated increased left ventricular 68Ga-FAPI uptake in patients with hypertensive heart disease77 and active cardiac sarcoidosis.78
Right ventricular imaging
In two case reports61,79 and a prospective study80 of patients with chronic thromboembolic pulmonary hypertension, 68Ga-FAPI uptake was seen in the right ventricular free wall which correlated positively with wall thickness and negatively with right ventricular function. The ability to identify fibrosis in the right ventricular wall has always been challenging with CMR and other techniques. FAPI imaging might thus prove of particular value in the investigation of right ventricular fibrosis.
Speculative clinical applications of imaging myocardial fibrosis activity
With the recent emergence of fibrosis radiotracers, the potential applications of an imaging technique for myocardial fibrosis activity should be considered. While somewhat speculative, these discussions are important at this stage so that the optimal prospective studies can be planned and conducted, maximizing the exciting potential of molecular imaging in this field.
Improved sensitivity of detection
A major advantage of PET is its sensitivity, with the ability to detect signal in areas of active disease based on nanomolar concentrations of radiotracer. Existing imaging techniques readily detect myocardial fibrosis within the left ventricle but are unreliable in small thinner structures such as the right ventricle, the atria, valves or arteries, where fibrosis often passes undetected. The clear demonstration of the 68Ga-FAPI uptake in the thinned-walled right ventricle (Figure 3)61–80,76 as well as the aorta81,82 paves the way for investigation of fibrosis activity in conditions, such as arrhythmogenic right ventricular cardiomyopathy, congenital heart disease, pulmonary hypertension, atherosclerosis, vasculitis and atrial fibrillation.
Improved sensitivity will also provide an opportunity to detect early myocardial fibrogenesis before major damage to the heart becomes established and before it is evident on other imaging modalities. The detection of early fibrosis activity may be of diagnostic value in a range of conditions, including chemotherapy cardiotoxicity70,75 and when differentiating the athletic heart from cardiomyopathy. However, assessment of fibrosis activity may also prove valuable in advanced disease to differentiate between burnt-out quiescent disease states and active fibrosis where further disease progression can be expected. Finally, it might provide insights into the ongoing role played by activated fibroblasts in chronic scar maintenance and whether this influences disease progression.
Development of anti-fibrotic therapies
Our previous inability to measure fibrosis activity means that we do not fully understand the anti-fibrotic effects of routinely used heart failure medications even though they improve cardiovascular outcomes in large randomized controlled trials.83,84 Heart failure therapies generally act within the renin–angiotensin–aldosterone and sympathetic nervous systems which are implicated in fibrosis formation (Figure 4). However, data demonstrating their anti-fibrotic properties are largely limited to pre-clinical models or biomarker surrogates. The advent of advanced non-invasive fibrosis imaging presents the opportunity to demonstrate their anti-fibrotic effects of as well as that of newer drugs such as sodium-glucose cotransporter-2 (SGLT2) inhibitors and those currently in development. In this context, 68Ga-FAPI imaging recently demonstrated its ability to assess and track changes in extra-cardiac fibrosis activity with time and in response to therapy.66 In patients with systemic sclerosis, 68Ga-FAPI uptake in areas of lung fibrosis predicted disease progression, demonstrating an independent association with deterioration in forced ventilatory capacity at 6 months. Intriguingly, some patients who received nintedanib therapy demonstrated a reduction in their pulmonary 68Ga-FAPI uptake which correlated with improvements in clinical status.66
Figure 4.

Potential therapeutic targets in myocardial fibrosis. Relevant pathways involved in fibrosis formation include Angiotensin-II, transforming growth factor-β, and interleukin-11 making them key treatment targets. Other targets include the renin–angiotensin–aldosterone system, sympathetic and immune systems, endothelin 1, stem cell therapy, CAR-T therapy, and theranostic FAP inhibitor agents. AngII, angiotensin-II; ACE-i, angiotensin-converting-enzyme inhibitors; ARB, angiotensin receptor blocker; ARNI, angiotensin receptor-neprilysin inhibitor; AT1, Angiotensin receptor; CAR-T, chimeric antigen receptor-modified T cells; ET-1, endothelin-1; FAPI, fibroblast activation protein inhibitor; FAP, fibroblast activation protein; IL, interleukin; PDGF, platelet-derived growth factor; RAAS, renin–angiotensin–aldosterone system; SGLT2i, sodium-glucose cotransporter-2 inhibitor; TGF-β, transforming growth factor (beta).
Companion diagnostic and theranostic applications
FAP-expressing fibroblasts are a key target for therapeutic intervention. In a murine of model of heart failure, targeted destruction of FAP-positive fibroblasts with chimeric antigen receptor-modified T cell (CAR-T) therapy resulted in dramatic reductions in myocardial fibrosis, improved cardiac remodelling, and function, implicating FAP-positive fibroblasts as drivers of fibrogenesis.8 Injected delivery of messenger ribonucleic acid targeted to CD5 cells allows CAR-T cells to be generated entirely in vivo, resulting in the same dramatic reductions in myocardial in a murine model of heart failure.9 There is therefore considerable excitement about using CAR-T therapy to target FAP-positive fibroblasts as a means of reducing myocardial fibrosis in humans. In these circumstances, FAPI imaging would be the ideal agent for patient selection and for assessing treatment efficacy.
Theranostic FAPI tracers are being developed with both the ability to image FAP-positive fibroblasts and to deliver cytotoxic therapy to them.12 The latest generation of FAPI tracers allows conjugation to high-energy, low penetrance beta-emitters, such as 177-Lutetium or 90-Yttrium. These tracers bind and become internalized within myofibroblasts via FAP, thereby delivering their cytotoxic radiation to the cells in a targeted and time-limited manner. 177-Lutetium-FAPI has now been successfully administered to patients, with multiple recent reports describing good initial safety and efficacy data in patients with end-stage cancer.85–87
Hybrid positron emission tomography and CMR imaging
The recent emergence of hybrid PET/MR holds particular promise in the future of myocardial fibrosis imaging. By being able to combine the reference-standard myocardial fibrosis detection by CMR with important and detailed functional and structural assessments with the exquisite molecular-level detail of PET, this novel hybrid imaging method holds considerable promise in the future of myocardial fibrosis imaging.88 Assessment of cardiac involvement in systemic fibrotic disorders as well as identification and characterization of cardiac tumours is also possible using this technique, allowing patients to benefit from the superior tissue characterization CMR possesses beyond what is possible using conventional PET/CT.88 It is possible that the cellular-level detail of fibrosis activity provided by PET will allow fusion with non-contrast-enhanced CMR, minimizing the intervention required for a patient to obtain high-quality information around the presence and activity of myocardial fibrosis.
To date, case studies of patients with myocardial infarction have used combined 68Ga-FAPI PET/MR rather than PET/CT. This has allowed direct comparisons of 68Ga-FAPI uptake with LGE demonstrating good agreement between these techniques albeit with extension of the 68Ga-FAPI signal beyond the infarct zone detected on CMR.89 Further research is currently underway to determine its utility in myocardial infarction which may pave the way for investigation in other myocardial fibrotic conditions (ClinicalTrials.gov Identifier: NCT04723953 and NCT05356923) (Graphical Abstract, Figure 3).90
Limitations
Currently data pertaining to the novel radiotracers has arisen from small retrospective case series. While these novel radiotracers, in particular FAPI, demonstrate ability to detect active myocardial fibrosis in a range of myocardial disorders, their clinical utility has not been established and requires evaluation in dedicated prospective clinical studies. PET imaging, in particular PET/MR, is also an expensive imaging modality available only in select highly specialized centres, meaning the technologies discussed here will not be accessible to many clinicians and their patients. Further prospective work determining the clinical significance and consequences of fibrosis detected by molecular imaging may address many of the limitations of this narrative review based on currently available evidence.
Conclusions
Fibrosis represents the final common response to a wide range of myocardial pathologies, characterized by excess collagen deposition and extracellular matrix expansion mediated by activated fibroblasts and pro-fibrotic mediators. Current imaging methods focus on detecting established fibrosis, providing important diagnostic and prognostic information, but no insights into fibrosis activity. Molecular imaging techniques may address this deficit and prove useful in detecting early disease and monitoring disease progression and response to therapy. The latter in particular may provide the long-awaited impetus to the development of anti-fibrotic medication for patients with the cardiomyopathic disease.
Acknowledgements
The authors wish to acknowledge the library services of the University of Edinburgh for providing access to some of the papers referenced here, and Lynn McKinlay for assisting with copyright requests and permissions for some reproduced elements of the figures for this review. We also wish to acknowledge Clint Waight for his tireless work manufacturing 68Ga-FAPI locally in Edinburgh as well as SOFIE radiopharmaceuticals who have generously provided us with 68Ga-FAPI precursor free of charge which has enabled us to provide some of our own FAPI images within Figure 3 as part of a study ethically approved by the South East Scotland Research Ethics Committee. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
Contributor Information
Anna K Barton, British Heart Foundation Centre for Cardiovascular Science, University of Edinburgh, Chancellor’s Building, Little France Crescent, Edinburgh EH16 4SB, UK.
Evangelos Tzolos, British Heart Foundation Centre for Cardiovascular Science, University of Edinburgh, Chancellor’s Building, Little France Crescent, Edinburgh EH16 4SB, UK.
Rong Bing, British Heart Foundation Centre for Cardiovascular Science, University of Edinburgh, Chancellor’s Building, Little France Crescent, Edinburgh EH16 4SB, UK.
Trisha Singh, British Heart Foundation Centre for Cardiovascular Science, University of Edinburgh, Chancellor’s Building, Little France Crescent, Edinburgh EH16 4SB, UK.
Wolfgang Weber, Department of Nuclear Medicine, Clinikum rechts der Isar, Technical University of Munich, Ismaniger Straße 22, 81675 Munich, Germany.
Markus Schwaiger, Department of Nuclear Medicine, Clinikum rechts der Isar, Technical University of Munich, Ismaniger Straße 22, 81675 Munich, Germany.
Zohreh Varasteh, Department of Nuclear Medicine, Clinikum rechts der Isar, Technical University of Munich, Ismaniger Straße 22, 81675 Munich, Germany.
Riemer H J A Slart, Faculty of Medical Sciences, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands.
David E Newby, British Heart Foundation Centre for Cardiovascular Science, University of Edinburgh, Chancellor’s Building, Little France Crescent, Edinburgh EH16 4SB, UK.
Marc R Dweck, British Heart Foundation Centre for Cardiovascular Science, University of Edinburgh, Chancellor’s Building, Little France Crescent, Edinburgh EH16 4SB, UK.
Funding
No specific funding was sourced for this work. A.K.B. was supported by a British Heart Foundation training scholarship (SS/CH/09/002/26360). T.S. was supported by the Medical Research Council (MR/T029153/1). M.R.D. is supported by the British Heart Foundation (FS/14/78/31020, FS/SCRF/21/32010) and is the recipient of the Sir Jules Thorn Award for Biomedical Research 2015 (15/JTA). D.E.N. is supported by the British Heart Foundation (CH/09/002, RG/16/10/32375, RE/18/5/34216) and is the recipient of a Wellcome Trust Senior Investigator Award (WT103782AIA).
Data availability statement
The majority of data underlying this narrative review article were derived from other previously published manuscripts under licence/by permission as indicated throughout the manuscript. The only original data/analysis included in this article pertains to the clinical MRI and PET/MR images displayed in the Graphical Abstract, panels C and D of Figure 3, and T1 mapping in ischaemic cardiomyopathy image in Table 2, all of which were generated through separate research work performed by at least one of this manuscript’s co-authors and not specifically in support of this review. While the majority of relevant information for these cases is included within this manuscript, request for further de-identified information will be shared on reasonable request to the corresponding author. Otherwise, no new data were generated or analysed in support of this review article.
References
- 1. Avery D, Govindaraju P, Jacob M, Todd L, Monsiow J, Puré E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol 2018;67:90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Travers J, Kamal F, Robbins J, Yutzey K, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res 2016;118:1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagaraju C, Robinson E, Abdesselem M, Trenson S, Dries E, Gilbert Get al. Myofibroblast phenotype and reversibility of fibrosis in patients with end-stage heart failure. J Am Coll Cardiol 2019;73:2267–82. [DOI] [PubMed] [Google Scholar]
- 4. Nagaraju C, Dries E, Popovic N, Singh A, Haemers P, Roderick Het al. Global fibroblast activation throughout the left ventricle but localized fibrosis after myocardial infarction. Sci Rep 2017;7:10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bing R, Dweck M. Myocardial fibrosis: why image, how to image and clinical implications. Heart 2019;105:1832–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mewton N, Liu C, Croisille P, Bluemke D, Lima J. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol 2011;57:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raina S, Lensing S, Nairooz R, Pathineni N, Hakeem A, Bhatti Set al. Prognostic value of late gadolinium enhancement CMR in systemic amyloidosis. JACC Cardiovasc Imaging 2016;9:1267–77. [DOI] [PubMed] [Google Scholar]
- 8. Aghajanian H, Kimura T, Rurik J, Hancock A, Lelbowitz M, Scholler Jet al. Targeting cardiac fibrosis with engineered T cells. Nature 2019;573:430–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rurik J, Tombácz I, Yadegari A, Fernández P, Shewale S, Li Let al. CAR T cells produced in vivo to treat cardiac injury. Science 2022;375:91–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Everett R, Stirrat C, Semple S, Newby D, Dweck M, Mirsadraee S. Assessment of myocardial fibrosis with T1 mapping MRI. Clin Radiol 2016;71:768–78. [DOI] [PubMed] [Google Scholar]
- 11. Everett R, Tastet L, Clavel M, Chin C, Capoulade R, Vassiliou VSet al. Progression of hypertrophy and myocardial fibrosis in aortic stenosis: a multicenter cardiac magnetic resonance study. Circ Cardiovasc Imaging 2018;11:e007451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lindner T, Loktev A, Altmann A, Giesel F, Kratochwil C, Debus Jet al. Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J Nucl Med 2018;59:1415–22. [DOI] [PubMed] [Google Scholar]
- 13. Loktev A, Lindner T, Mier W, Debus J, Altmann A, Jäger Det al. A tumor-imaging method targeting cancer-associated fibroblasts. J Nucl Med 2018;59:1423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moore G, Hutchins G, Bulkey B, Tseng J, Ki P. Constituents of the human ventricular myocardium: connective tissue hyperplasia accompanying muscular hypertrophy. Am Heart J 1980;100:610–6. [DOI] [PubMed] [Google Scholar]
- 15. D’Armiento J. Matrix metalloproteinase disruption of the extracellular matrix and cardiac dysfunction. Trends Cardiovasc Med 2002;12:97–101. [DOI] [PubMed] [Google Scholar]
- 16. Robertson W. Metabolism of collagen in mammalian tissues. Biophys J 1964;4:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weber K. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 1989;13:1637–52. [DOI] [PubMed] [Google Scholar]
- 18. Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med 2009;19:247–52. [DOI] [PubMed] [Google Scholar]
- 19. Kong P, Christia P, Frangogiannis N. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014;71:549–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weber K, Sun Y, Bhattacharya S, Ahokas R, Gerling I. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 2013;10:15–26. [DOI] [PubMed] [Google Scholar]
- 21. Jenkins W, Vesey A, Stirrat C, Connell M, Lucatelli C, Neale Aet al. Cardiac alphaVbeta3 integrin expression following acute myocardial infarction in humans. Heart 2017;103:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker Tet al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 2007;115:584–92. [DOI] [PubMed] [Google Scholar]
- 23. Dweck M, Boon N, Newby D. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol 2012;60:1854–63. [DOI] [PubMed] [Google Scholar]
- 24. Cahill TJ, Ashrafian H, Watkins H. Genetic cardiomyopathies causing heart failure. Circ Res 2013;113:660–75. [DOI] [PubMed] [Google Scholar]
- 25. Olivotto I, Girolami F, Sciagrà R, Ackerman M, Sotgia B, Bos Jet al. Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J Am Coll Cardiol 2011;58:839–48. [DOI] [PubMed] [Google Scholar]
- 26. Lajiness J, Conway S. Origin, development, and differentiation of cardiac fibroblasts. J Mol Cell Cardiol 2014;70:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pepper GS, Lee RW. Sympathetic activation in heart failure and its treatment with β-blockade. Archieves Int Med 1999;159:225–34. [DOI] [PubMed] [Google Scholar]
- 28. Golestani R, Mirfeizi L, Zeebregts C, Westra J, de Haas H, Glaudemans Aet al. Feasibility of [18F]-RGD for ex vivo imaging of atherosclerosis in detection of αvβ3 integrin expression. J Nucl Cardiol 2015;22:1179–86. [DOI] [PubMed] [Google Scholar]
- 29. Mukherjee D, Sen S. Alteration of cardiac collagen phenotypes in hypertensive hypertrophy: role of blood pressure. J Mol Cell Cardiol 1993;25:185–96. [DOI] [PubMed] [Google Scholar]
- 30. Mukherjee D, Sen S. Alteration of collagen phenotypes in ischemic cardiomyopathy. J Clin Invest 1991;88:1141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Whittaker P, Boughner D, Kloner R. Analysis of healing after myocardial infarction using polarized light microscopy. Am J Pathol 1989;134:879–93. [PMC free article] [PubMed] [Google Scholar]
- 32. Treibel T, Badiani S, Lloyd G, Moon J. Multimodality imaging markers of adverse myocardial remodeling in aortic stenosis. JACC Cardiovasc Imaging 2019;12:1532–48. [DOI] [PubMed] [Google Scholar]
- 33. Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 1996;29:225–9. [DOI] [PubMed] [Google Scholar]
- 34. Zile M, Desantis S, Baicu C, Stroud R, Thompson S, McClure Cet al. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ Heart Fail 2011;4:246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zile M, O'Meara E, Claggett B, Prescott M, Solomon S, Swedberg Ket al. Effects of sacubitril/valsartan on biomarkers of extracellular matrix regulation in patients with HFrEF. J Am Coll Cardiol 2019;73:795–806. [DOI] [PubMed] [Google Scholar]
- 36. Ahmed T, Goyal A. Endomyocardial Biopsy Florida, United States of America: StatPearls Publishing; 2022 [Available from: https://www.ncbi.nlm.nih.gov/books/NBK557597/. [PubMed]
- 37. Bulluck H, White SK, Fröhlich GM, Casson S, O’Meera C, Newton Aet al. Quantifying the area at risk in reperfused ST-segment-elevation myocardial infarction patients using hybrid cardiac positron emission tomography-magnetic resonance imaging. Circ Cardiovasc Imaging 2016;9:e003900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gibbons RJ, Miller TD, Christian TF. Infarct size measured by single photon emission computed tomographic imaging with (99 m)Tc-sestamibi: a measure of the efficacy of therapy in acute myocardial infarction. Circulation 2000;101:101–8. [DOI] [PubMed] [Google Scholar]
- 39. Klocke F, Baird M, Lorell B, Bateman T, Messer J, Berman Det al. ACC/AHA/ASNC guidelines for the clinical use of cardiac radionuclide imaging-executive summary. Circulation 2003;108:1404–18. [DOI] [PubMed] [Google Scholar]
- 40. Abbott BG, Case JA, Dorbala S, Einstein AJ, Galt JR, Pagnanelli Ret al. Contemporary cardiac SPECT imaging—innovations and best practices: an information statement from the American society of nuclear cardiology. Circ Cardiovasc Imaging 2018;11:e000020. [DOI] [PubMed] [Google Scholar]
- 41. Marra M P, De Lazzari M, Zorzi A, Migliore F, Zilio F, Calore Cet al. Impact of the presence and amount of myocardial fibrosis by cardiac magnetic resonance on arrhythmic outcome and sudden cardiac death in nonischemic dilated cardiomyopathy. Heart Rhythm 2014;11:856–63. [DOI] [PubMed] [Google Scholar]
- 42. Yang Y, Li W, Zhu H, Pen X YH, Arnott Cet al. Prognosis of unrecognised myocardial infarction determined by electrocardiography or cardiac magnetic resonance imaging: systematic review and meta-analysis. BMJ 2020;369:m1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Becker M, Cornel J, van de Ven P, van Rossum A, Allaart C, Germans T. The prognostic value of late gadolinium-enhanced cardiac magnetic resonance imaging in nonischemic dilated cardiomyopathy: a review and meta-analysis. JACC Cardiovasc Imaging 2018;11:1274–84. [DOI] [PubMed] [Google Scholar]
- 44. Georgiopoulos G, Figliozzi S, Sanguineti F, Aquaro G, Stamatelopoulos K, Chiribiri Aet al. Prognostic impact of late gadolinium enhancement by cardiovascular magnetic resonance in myocarditis: a systematic review and meta-analysis. Circ Cardiovasc Imaging 2021;14:e011492. [DOI] [PubMed] [Google Scholar]
- 45. Hulten E, Agarwal V, Cahill M, Cole G, Vita T, Parrish Set al. Presence of late gadolinium enhancement by cardiac magnetic resonance among patients with suspected cardiac sarcoidosis is associated with adverse cardiovascular prognosis: a systematic review and meta-analysis. Circ Cardiovasc Imaging 2016;9:e005001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weng Z, Yao J, Chan R, He J, Yang X, Zhou Yet al. Prognostic value of LGE-CMR in HCM: a meta-analysis. JACC Cardiovasc Imaging 2016;9:1392–402. [DOI] [PubMed] [Google Scholar]
- 47. Assadi H, Jones R, Swift A, Al-Mohammad A, Garg P. Cardiac MRI for the prognostication of heart failure with preserved ejection fraction: a systematic review and meta-analysis. Magn Reson Imaging 2021;76:116–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schelbert E, Piehler K, Zareba K, Moon J, Ugander M, Messroghli Det al. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc 2015;4:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Everett R, Treibel T, Fukui M, Lee H, Rigolli M, Singh Aet al. Extracellular myocardial volume in patients with aortic stenosis. J Am Coll Cardiol 2020;75:304–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chin C, Everett R, Kwiecinski J, Vesey A, Yeung E, Esson Get al. Myocardial fibrosis and cardiac decompensation in aortic stenosis. JACC Cardiovasc Imaging 2017;10:1320–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dweck MR, Williams MC, Moss AJ, Newby DE, Fayad ZA. Computed tomography and cardiac magnetic resonance in ischemic heart disease. J Am Coll Cardiol 2016;68:2201–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lewis AJM, Burrage MK, Ferreira VM. Cardiovascular magnetic resonance imaging for inflammatory heart diseases. Cardiovasc Diagn Ther 2020;10:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singh T, Chapman A, Dweck M, Mills N, Newby D. MINOCA: a heterogenous group of conditions associated with myocardial damage. Heart 2021;107:1458–64. [DOI] [PubMed] [Google Scholar]
- 54. Puntmann V, Carr-White G, Jabbour A, Yu C, Gebker R, Kelle Set al. T1-Mapping and outcome in nonischemic cardiomyopathy: all-cause mortality and heart failure. JACC Cardiovasc Imaging 2016;9:40–50. [DOI] [PubMed] [Google Scholar]
- 55. Lee H, Park J, Yoon Y, Park E, Kim H, Lee Wet al. Noncontrast myocardial T1 mapping by cardiac magnetic resonance predicts outcome in patients with aortic stenosis. JACC Cardiovasc Imaging 2018;11:974–83. [DOI] [PubMed] [Google Scholar]
- 56. Haaf P, Garg P, Messroghli D, Broadbent D, Greenwood J, Plein S. Cardiac T1 mapping and extracellular volume (ECV) in clinical practice: a comprehensive review. J Cardiovasc Magn Reson 2016;18:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chang S, Lee W, Chun E. Recent update of advanced imaging for diagnosis of cardiac sarcoidosis: based on the findings of cardiac magnetic resonance imaging and positron emission tomography. Investig Magn Reson Imaging 2019;23:100–13. [Google Scholar]
- 58. Lewis GA, Dodd S, Clayton D, Bedson E, Eccleson H, Schelbert Eet al. Pirfenidone in heart failure with preserved ejection fraction: a randomized phase 2 trial. Nat Med 2021;27:1477–82. [DOI] [PubMed] [Google Scholar]
- 59. Andrews JPM, Trivieri MG, Everett R, Spath N, MacNaught G, Moss AJet al. 18F-fluoride PET/MR in cardiac amyloid: A comparison study with aortic stenosis and age- and sex-matched controls. J Nucl Cardiol 2020;29:741–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Slart R, Glaudemans A, Gheysens O, Lubberink M, Kero T, Dweck Met al. Procedural recommendations of cardiac PET/CT imaging: standardization in inflammatory-, infective-, infiltrative-, and innervation (4Is)-related cardiovascular diseases: a joint collaboration of the EACVI and the EANM. Eur J Nucl Med Mol Imaging 2021;48:1016–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang L, Zhang Z, Zhao Z, Yan C, Fang W. 68Ga-FAPI right heart uptake in a patient with idiopathic pulmonary arterial hypertension. J Nucl Cardiol 2022;29:1475–7. [DOI] [PubMed] [Google Scholar]
- 62. Totzeck M, Siebermair J, Rassaf T, Rischpler C. Cardiac fibroblast activation detected by positron emission tomography/computed tomography as a possible sign of cardiotoxicity. Eur Heart J 2020;41:1060. [DOI] [PubMed] [Google Scholar]
- 63. Geisler S, Ermert J, Stoffels G, Willuweit A, Galldiks N, Filss Cet al. Isomers of 4-[18F]fluoro-proline: radiosynthesis, biological evaluation and results in humans using PET. Curr Radiopharm 2014;7:123–32. [DOI] [PubMed] [Google Scholar]
- 64. Wallace W, Gupta N, Hubbs A, Mazza S, Bishop H, Keane Met al. Cis-4-[18F]fluoro-l-proline PET imaging of pulmonary fibrosis in a rabbit model. J Nucl Med 2002;43:413–20. [PubMed] [Google Scholar]
- 65. Sommerauer M, Galldiks N, Barb M, Stoffels G, Willuweit A, Coenen Het al. Cis-4-[18F]fluoro-D-proline detects neurodegeneration in patients with akinetic-rigid parkinsonism. Nucl Med Commun 2019;40:383–7. [DOI] [PubMed] [Google Scholar]
- 66. Bergmann C, Distler J, Treutlein C, Tascilar K, Müller A, Atzinger Aet al. 68Ga-FAPI-04 PET-CT for molecular assessment of fibroblast activation and risk evaluation in systemic sclerosis-associated interstitial lung disease: a single-centre, pilot study. Lancet Rheumatol 2021;3:e185–e94. [DOI] [PubMed] [Google Scholar]
- 67. Luo Y, Pan Q, Yang H, Peng L, Zhang W, Li F. Fibroblast activation protein-targeted PET/CT with (68)Ga-FAPI for imaging IgG4-related disease: comparison to (18)F-FDG PET/CT. J Nucl Med 2021;62:266–71. [DOI] [PubMed] [Google Scholar]
- 68. Rosenkrans Z, Massey C, Bernau K, Ferreira C, Jeffrey J, Schulte Jet al. [(68) ga]ga-FAPI-46 PET for non-invasive detection of pulmonary fibrosis disease activity. Eur J Nucl Med Mol Imaging 2022;49:3705–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhou Y, Yang X, Liu H, Luo W, Liu H, Lv Tet al. Value of [(68)Ga]ga-FAPI-04 imaging in the diagnosis of renal fibrosis. Eur J Nucl Med Mol Imaging 2021;48:3493–501. [DOI] [PubMed] [Google Scholar]
- 70. Heckmann M, Reinhardt F, Finke D, Katus H, Haberkorn U, Leuschner Fet al. Relationship between cardiac fibroblast activation protein activity by positron emission tomography and cardiovascular disease. Circ Cardiovasc Imaging 2020;13:e010628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Siebermair J, Kohler M, Kupusovic J, Nekolla S, Kessler L, Ferdinandus Jet al. Cardiac fibroblast activation detected by ga-68 FAPI PET imaging as a potential novel biomarker of cardiac injury/remodeling. J Nucl Cardiol 2021;28:812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Varasteh Z, Mohanta S, Robu S, Braeuer M, Li Y, Omidvari Net al. Molecular imaging of fibroblast activity after myocardial infarction using a (68)Ga-labeled fibroblast activation protein inhibitor, FAPI-04. J Nucl Med 2019;60:1743–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Diekmann J, Koenig T, Zwadlo C, Derlin T, Neuser J, Thackeray Jet al. Molecular imaging identifies fibroblast activation beyond the infarct region after acute myocardial infarction. J Am Coll Cardiol 2021;77:1835–7. [DOI] [PubMed] [Google Scholar]
- 74. Kessler L, Kupusovic J, Ferdinandus J, Nader H, Lale U, Fadi Zet al. Visualization of fibroblast activation after myocardial infarction using 68Ga-FAPI PET. Clin Nucl Med 2021;46:807–13. [DOI] [PubMed] [Google Scholar]
- 75. Finke D, Heckmann M, Herpel E, Katus H, Haberkorn U, Leuschner Fet al. Early detection of checkpoint inhibitor-associated myocarditis using (68)Ga-FAPI PET/CT. Front Cardiovasc Med 2021;8:614997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shi X, Lin X, Huo L, Li X. Cardiac fibroblast activation in dilated cardiomyopathy detected by positron emission tomography. J Nucl Cardiol 2022;29:881–4. [DOI] [PubMed] [Google Scholar]
- 77. Lin K, Chen X, Xue Q, Yao S, Miao W. Diffuse uptake of [68Ga]Ga-FAPI in the left heart in a patient with hypertensive heart disease by PET/CT. J Nucl Cardiol 2021. doi: 10.1007/s12350-021-02646-2. [DOI] [PubMed] [Google Scholar]
- 78. Siebermair J, Kessler L, Kupusovic J, Rassaf T, Rischpler C. Cardiac fibroblast activation detected by (68)Gallium-FAPI-46 positron emission tomography-magnetic resonance imaging as a sign of chronic activity in cardiac sarcoidosis. Eur Heart J Case Rep 2022;6:ytac005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xing HQ, Gong JN, Chen BX, Guo XJ, Yang YH, Huo Let al. Comparison of 68Ga-FAPI imaging and cardiac magnetic resonance in detection of myocardial fibrosis in a patient with chronic thromboembolic pulmonary hypertension. J Nucl Cardiol 2022;29:2728–30. [DOI] [PubMed] [Google Scholar]
- 80. Chen B, Xing H, Gong J, Guo X, Xi X, Yang Yet al. Imaging of cardiac fibroblast activation in patients with chronic thromboembolic pulmonary hypertension. Eur J Nucl Med Mol Imaging 2022;49:1211–22. [DOI] [PubMed] [Google Scholar]
- 81. Wu M, Ning J, Li J, Lai Z, Shi X, Xing Het al. Feasibility of in vivo imaging of fibroblast activation protein in human arterial walls. J Nucl Med 2022;63:948–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wu S, Pang Y, Zhao L, Zhao L, Chen H. 68Ga-FAPI PET/CT versus 18F-FDG PET/CT for the evaluation of disease activity in takayasu arteritis. Clin Nucl Med 2021;46:847–9. [DOI] [PubMed] [Google Scholar]
- 83. McMurray J, Packer M, Desai A, Gong J, Lefkowitz M, Rizkala Aet al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014;371:993–1004. [DOI] [PubMed] [Google Scholar]
- 84. McMurray J, Solomon S, Inzucchi S, Köber L, Kosiborod M, Martinez Pet al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 2019;381:1995–2008. [DOI] [PubMed] [Google Scholar]
- 85. Ballal S, Yadav M, Moon E, Kramer V, Roesch F, Kumari Set al. First-In-Human results on the biodistribution, pharmacokinetics, and dosimetry of [(177)Lu]lu-DOTA.SA.FAPi and [(177)Lu]lu-DOTAGA.(SA.FAPi)2. Pharmaceuticals 2021;14:1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ballal S, Yadav M, Moon E, Kramer V, Roesch F, Kumari Set al. Biodistribution, pharmacokinetics, dosimetry of [(68)Ga]ga-DOTA.SA.FAPi, and the head-to-head comparison with [(18)F]F-FDG PET/CT in patients with various cancers. Eur J Nucl Med Mol Imaging 2021;48:1915–31. [DOI] [PubMed] [Google Scholar]
- 87. Ballal S, Yadav M, Moon E, Roesch F, Kumari S, Argawal Set al. Novel fibroblast activation protein inhibitor-based targeted theranostics for radioiodine-refractory differentiated thyroid cancer patients: a pilot study. Thyroid 2022;32:65–77. [DOI] [PubMed] [Google Scholar]
- 88. Krumm P, Mangold S, Gatidis S, Nikolaou K, Nensa F, Bamberg Fet al. Clinical use of cardiac PET/MRI: current state-of-the-art and potential future applications. Jpn J Radiol 2018;36:313–23. [DOI] [PubMed] [Google Scholar]
- 89. Yuan T, Wang X. 68Ga-FAPI PET/MRI in Coronary Heart Disease. J Nucl Cardiol 2021. doi: 10.1007/s12350-021-02667-x. [DOI] [PubMed] [Google Scholar]
- 90. Notohamiprodjo S, Nekolla SG, Robu S, Villagran Asiares A, Kupatt C, Ibrahim Tet al. Imaging of cardiac fibroblast activation in a patient after acute myocardial infarction using 68Ga-FAPI-04. J Nucl Cardiol 2022;29:2254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The majority of data underlying this narrative review article were derived from other previously published manuscripts under licence/by permission as indicated throughout the manuscript. The only original data/analysis included in this article pertains to the clinical MRI and PET/MR images displayed in the Graphical Abstract, panels C and D of Figure 3, and T1 mapping in ischaemic cardiomyopathy image in Table 2, all of which were generated through separate research work performed by at least one of this manuscript’s co-authors and not specifically in support of this review. While the majority of relevant information for these cases is included within this manuscript, request for further de-identified information will be shared on reasonable request to the corresponding author. Otherwise, no new data were generated or analysed in support of this review article.