

Abstract
Injectable local anesthetics that can provide a continuous nerve block approximating the duration of a pain state would be a life-changing solution for patients experiencing post-operative pain or chronic pain. Tetrodotoxin (TTX) is a site 1 sodium channel blocker that is extremely potent compared to clinically used local anesthetics. Challengingly, TTX doses are limited by its associated systemic toxicity, thus shortening the achievable duration of nerve blocks. Here, we explore emulsion-induced polymersomes (EIP) as a drug delivery system to safely use TTX for local anesthesia. By emulsifying hyperbranched polyglycerol-poly (propylene glycol)-hyperbranched polyglycerol (HPG-PPG-HPG) in TTX aqueous solution, HPG-PPG-HPG self-assembled into micrometer-sized polymersomes within seconds. The formed polymersomes have microscopically visible internal aqueous pockets that encapsulate TTX with an encapsulation efficiency of up to 94%. Moreover, the polymersomes are structurally stable, enabling sustained TTX release. In vivo, the freshly prepared EIP/TTX formulation can be directly injected and increased the tolerated dose of TTX in Sprague-Dawley rats to 11.5 μg without causing any TTX-related systemic toxicity. In the presence of the chemical penetration enhancer (CPE) sodium octyl sulfate (SOS), a single perineural injection of EIP/TTX/SOS formulation produced a reliable sciatic nerve block for 22 days with minimal local toxicity.
Graphical Abstract
Through a simple physical mixing process, tetrodotoxin and chemical penetration enhancers can be loaded into the polymersomes in seconds. The prepared formulation is ready for use without further processing. In rats, a single injection of this formulation blocked the sciatic nerve for ~22 days without causing systemic toxicity of tetrodotoxin.
1. Introduction
The post-operative duration of pain arising from many surgical interventions typically persists for 5–7 days[1–3]. Not infrequently, inadequate management of these early pain states leads to a chronic pain phenotype that lasts longer than 12 weeks[4, 5]. The prevalence of chronic postsurgical pain, which is severe enough to cause substantial functional impairment, is approximately 10% after all surgeries[6–8]. Chronic postsurgical pain is increasingly recognized as a significant public health problem. More than 320 million people have surgery every year[9]. Poorly controlled acute postoperative pain is a predictor of chronic postsurgical pain development[7]. Opioids are often the mainstay of perioperative and chronic pain management [10–12]. Opioids have many side effects, including nausea, clouding of the sensorium, pruritus, urinary retention, and constipation[13, 14]. More seriously, opioid therapy, as occurs even with uncomplicated post-operative pain management, can lead to tolerance, addiction, diversion, and fatal overdose[15–17].
Local anesthesia is a reversible nerve block that involves injecting local anesthetics as close to the nerve as possible or infiltrating the terminal field of the sensory fiber to block transmission and thereby relieve pain. It is of clinical and scientific interest to establish adequate durations of local anesthesia coincident with the pain state via a single injection of a local anesthetic[18]. This endeavor encounters 3 principal challenges: inadequate duration of action, potential systemic toxicity, and local tissue reaction [19–21]. Conventional amino-ester and amino-amide local anesthetics typically last for a few hours and are intrinsically myotoxic[22, 23], especially when their duration of action is extended by the delivery platform[24]. When cleared from the injection site, conventional local anesthetics will develop a systemic action and such actions, with adequate plasma concentrations, may lead to cardiac dysfunction and neurological syndromes such as seizures[25, 26].
Tetrodotoxin (TTX) is a neurotoxin that has been studied for local anesthesia[27, 28] and treatment of moderate to severe, inadequately controlled cancer-related pain[29]. As a sodium-channel block at site 1, TTX binds to voltage-gated sodium channels on nerve cell membranes to prevent sodium ions from entering neurons, thereby inhibiting the firing of action potentials in neurons[30]. Compared to conventional local anesthetics, TTX has the advantages of high potency, no cardiac and central nervous system toxicity, and no toxicity to local muscles and nerves. Despite these desired characteristics, the clinical use of TTX is hindered by its systemic toxicity, which can cause neural blockade and muscular weakness, resulting in diaphragmatic paralysis that may lead to respiratory failure. For example, as noted, a single injection of 3–4 μg of free TTX can produce a sensory nerve block in rats for 2 hours. Attempts to increase the dose of TTX and in turn increase the duration of the blockade will result in an increased likelihood of systemic toxicity of TTX[31].
Encapsulating TTX in delivery systems is an effective approach to regulating its biodistribution and plasma pharmacokinetics. Delivery systems can enhance its therapeutic effects while diminishing its systemic toxicity. Specifically, a sustained-release TTX system can continuously release a constant amount of TTX over time and provide a stable TTX level sufficient to achieve a therapeutic effect at the target site but below the toxic plasma concentration. Challengingly, TTX is hydrophilic and has a molecular weight of 319.27 g/mol. Therefore, it is difficult to encapsulate TTX in many conventional drug delivery systems, such as poly (lactic-co-glycolic acid) (PLGA) nanoparticles and hydrogels[32–34]. Liposomes, or core-shell structured hollow particles, have been intensively studied as an efficient carrier for TTX administration[35–37]. Liposomes encapsulate TTX into aqueous pockets with loading efficiencies up to 40%. Moreover, the hydrophobic shells of liposomes achieve sustained release by preventing leakage of TTX. A single injection of liposomes containing TTX produced a nerve blockade for up to 2 days, with markedly reduced systemic and local toxicity[35–37]. Polymer-TTX conjugate is another efficient drug delivery approach for TTX. TTX is covalently conjugated onto poly(glycerol sebacate) (PGS) via hydrolyzable ester bonds[31], and it can be released in its native form by hydrolysis of ester bonds. The PGS-TTX conjugate continuously released TTX in a near-linear profile over a 4-week period in vitro and eliminated the burst release of TTX. In vivo, the PGS-TTX conjugate allowed the administration of 80 μg TTX to rats, which is 20-fold higher than the dose tolerance limit[38], and produced a sciatic nerve block lasting for 3 days. Meanwhile, the injection did not cause any adverse side effects including nerve block in the un-injected hind limb, seizures, respiratory distress, terminal apnea or animal death.
Despite this progress, a nerve block lasting for 3 days is not sufficient for many patients with post-operative pain. Instead, a single injection of traditional local anesthesia is judged to be required for clinical applications as it can last a minimum of 7 days without local or systemic toxicity. Furthermore, liposomal TTX formulations and polymer-TTX conjugates suffer additional shortcomings, such as cumbersome preparation procedures, TTX waste, and TTX leakage during storage and transportation. These problems can increase cost, shorten shelf life, and cause depletion of the vehicles’ drug content, which is especially problematic with ultra-potent TTX.
The ideal peripheral nerve block technique would have a duration of action that is (1) of sufficient duration to address the most intense period of postsurgical pain (e.g. 5–7 days) and chronic pain (e.g. 12 weeks), (2) associated with minimal risks of infection, neurological complications, and local anesthetic systemic toxicity, and (3) easy to perform for the physician, convenient for the patient, and easy to manage in the post-operative period[39].
Here, we developed emulsion-induced polymersomes (EIP) to obtain encapsulation and sustained release of TTX. The EIP technique involves a single water-in-oil-in-water (w/o/w) emulsification process to emulsify polymers in aqueous drug solutions to fabricate the polymersomes (Scheme 1). When polymersomes are formed, they encapsulate TTX within their aqueous pockets. The hydrophobic shells of the polymersomes act as diffusion barriers to achieve the sustained release of TTX. Compared to liposomes and traditional polymersomes, a major advantage of EIP is that the formulation is easy to implement, and the preparation can be completed in a few seconds using a common vortex mixer. Moreover, the polymer-to-TTX ratio in the emulsion mixture can be adjusted freely, so an extremely high TTX encapsulation efficiency can be achieved by increasing the polymer-to-drug ratio. Due to the high TTX encapsulation efficiency, the step of removing non-encapsulated TTX can be omitted because the small amount of non-encapsulated TTX is beneficial to rapidly initiate local anesthesia upon in vivo injection. Additionally, the process does not involve organic solvents so no additional purification steps are required. Together, these features enable TTX formulations to be quickly prepared before injection and directly injected from the freshly prepared formation; ultimately, eliminating the problems of TTX waste and early TTX release during storage and transportation.
Scheme 1.
Schematic illustration of emulsion-induced polymersomes encapsulation of hydrophilic small molecules.
2. Results
2.1. Design consideration of HPG-PPG-HPG polymers
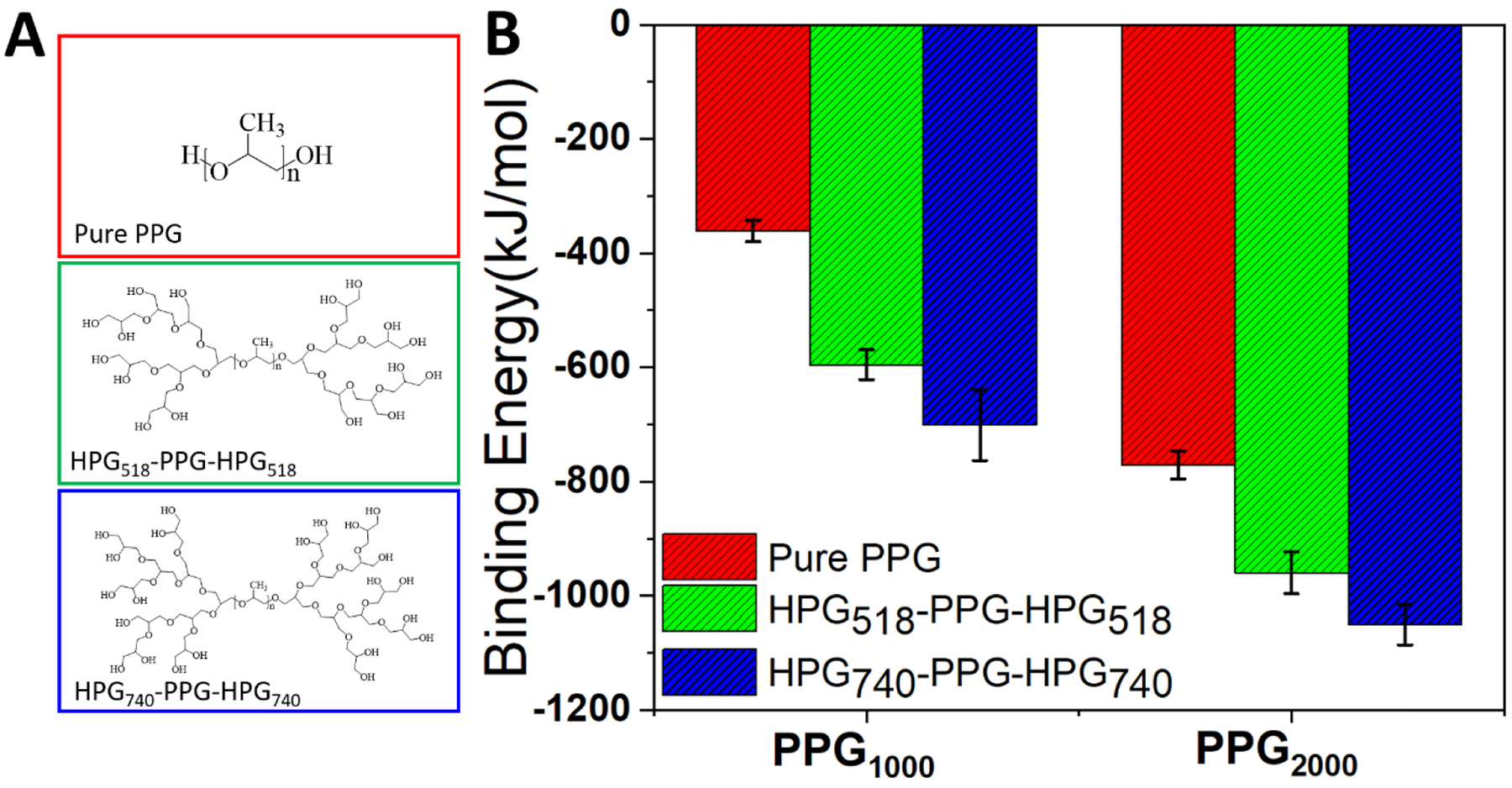
Ideally, polymersomes for TTX administration should have internal aqueous pockets large enough to accommodate TTX, structural stability to maintain their architecture and avoid sudden release of TTX, and suitable shell permeability to achieve sustained TTX release. In the absence of documentation, the “trial and error” method was used to determine candidate polymers for the EIP polymersomes. We hypothesized that in order to prepare polymersomes through one-step emulsification, the candidate polymer should: 1) be liquid and immiscible with water to form an oil-in-water (o/w) emulsion; 2) contain hydrophilic components to form a water-in-oil (w/o) emulsion; 3) be amphiphilic, acting as a surfactant, to stabilize the produced w/o/w emulsion; and 4) have strong interactions between polymer chains to stabilize the formed polymersomes. With these considerations, the first attempt was given to polypropylene glycol (PPG) because 1) PPG is a liquid at room temperature, and its solubility in water decreases rapidly with increasing molecular weight; 2) PPG has two hydroxyl end groups, which have a high affinity for water to form internal water droplets inside the PPG droplets, e.g., forming the w/o emulsion. The simultaneous o/w and w/o emulsification (e.g., w/o/w emulsion) can generate the PPG polymersomes with internal aqueous pockets; 3) the diversity of commercially available PPG (e.g., PPG with different molecular weights of 1000, 2000, 4000Da) allows us to regulate the polymersomes’ size, shell permeability, and physical stability, since the molecular weight determines various physical and chemical properties of polymers[40, 41]; and 4) PPG has low volatility at room temperature and low toxicity[31, 42], enabling the excellent biocompatibility for biomedical engineering applications.
As expected, the results demonstrated that PPGs of all molecular weights tested (Mn = 1000, 2000, 4000Da) can self-assemble into polymersomes with microscopic visible aqueous pockets using the emulsion approach (Supplementary Note 1, Fig. S1). However, the aqueous pockets observed in the polymersomes are limited in size and volume (Fig. S1, S2). Moreover, the formed polymersomes are structurally unstable (Fig. S2, S3). After emulsification, the polymersomes immediately rupture/deconstruct, merge, or coalesce repeatedly until the bulk phase of polymer and water appears (e.g. phase separation).
Encouragingly, we found that the molecular weight of PPG plays an important role in determining the volume of the aqueous pockets and the structural stability of the polymersomes. Specifically, the molecular weight of PPG is inversely proportional to the ability to form aqueous pockets. As the molecular weight of PPG increases, the volume of aqueous pockets observed in PPG polymersomes decreases in the order: PPG (Mn = 1000) > PPG (Mn = 2000) > PPG (Mn = 4000) (Fig. S2A, S4). We suspect this may be due to the changing hydroxyl density. As the molecular weight of PPG increases from 1000 to 4000, the hydroxyl density of PPG (calculated from the molar ratio of the hydroxyl group to monomer propylene glycol, denote as OH/propylene glycol) correspondingly decreases from 0.152 to 0.038. The hydroxyl density determines the water affinity of the polymer and its ability to trap water in the oil droplets during the w/o/w emulsification process. In contrast, the molecular weight of PPG is directly proportional to the physical stability of the formed polymersomes. As the molecular weight of PPG increases, the physical stability of the PPG polymersomes increases in the order: PPG (Mn = 1000) < PPG (Mn = 2000) < PPG (Mn = 4000) (Fig. S2B). In summary, the hydrophobicity of PPG increases with increasing molecular weight, thereby stabilizing the polymersome shell.
We hypothesize that in order to enhance the formation and structural stability of aqueous pockets, the polymer used must have increased hydrophilicity and greater molecular weight. Based on these considerations, we designed HPG-PPG-HPG copolymers. HPG-PPG-HPG has a dumbbell-like shape with two HPG branches grown from the two ends of the linear PPG chain. The molecular weight of the PPG chain can be readily adjusted by selecting PPG polymers with different molecular weights as the reactant. The HPG branch has multiple hydroxyl groups. By grafting HPG branches onto the PPG chain, the OH/propylene glycol molar ratio of the polymer can be increased. Furthermore, the HPG branch is very hydrophilic and thus has a high affinity with water[43]. Therefore, the addition of HPG branches to the PPG chain is expected to facilitate the formation of structurally stable aqueous pockets.
2.2. Synthesis and characterization of HPG-PPG-HPG polymers
HPG-PPG-HPG polymers were synthesized by a one-pot solvent-free hydroxyl group initiated anionic ring-opening polymerization (AROP) of the glycidol using PPG as the initiator and potassium methylate (KOCH3) as the deprotonating agent (Fig. 1A)[43]. After the reaction, all impurities, including unreacted glycidol, KOCH3, and the byproduct (pure HPG), are water-soluble and can be removed by washing with water to obtain high-purity HPG-PPG-HPG. The absence of organic solvents during synthesis and purification avoids the problem of solvent residence in the polymer, which is a considerable challenge for polymer biomaterials because even trace amounts of solvents could cause tissue damage. Furthermore, this one-step reaction could decrease the cost and minimize the batch-to-batch variation.
Figure 1.
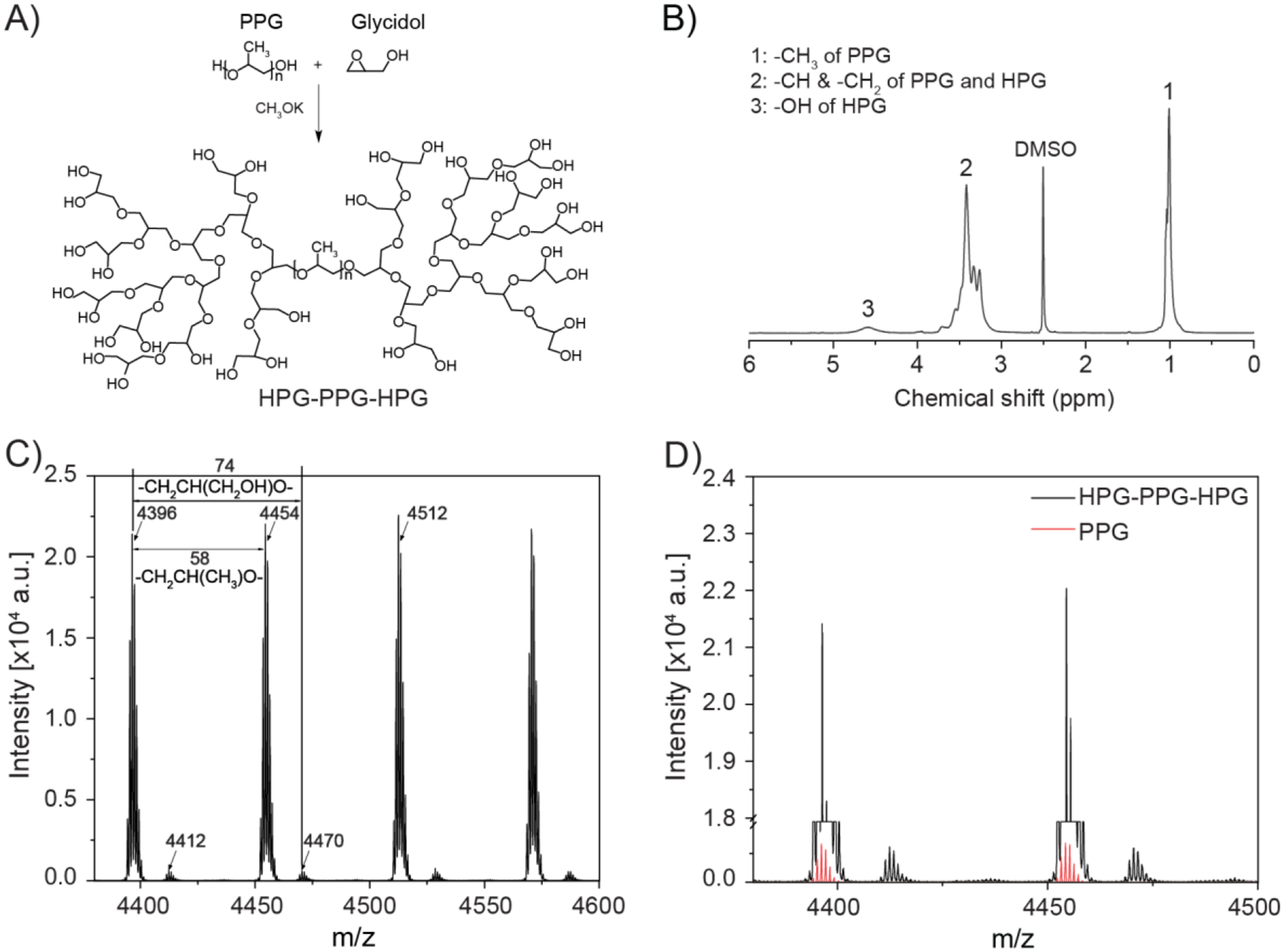
Synthesis and characterization of HPG-PPG-HPG copolymer. A) Synthesis route for HPG-PPG-HPG. B) 1H NMR spectra of HPGL-PPG4000-HPGL. C) MALDI-ToF MS spectra of HPGL-PPG4000-HPGL. D) A comparison of the intensity in MS spectrometry between HPGL-PPG4000-HPGL and pure PPG4000.
By changing the molecular weight of the PPG initiator (Mn = 1000, 2000, 4000) and the ratio of glycidol-to-PPG, seven unique HPG-PPG-HPG polymers were synthesized. The polymers are expressed as HPGL,M,H-PPGMn-HPGL,M,H, where “L”, “M”, and “H” represent the low, middle, and high molecular weights of HPG, respectively.
The chemical structure of the synthesized HPG-PPG-HPG was confirmed by proton nuclear magnetic resonance spectroscopy (1H NMR) (Fig. 1B). The signals of the methyl protons at 1.1 ppm and the methylene protons at 3.5 ppm were due to PPG. The methylene and methine protons of HPG appeared as one broad resonance between 3.3 and 3.9 ppm. Hydroxyl protons gave a broad resonance between 4.4 and 4.8 ppm. The number-average molecular weight of PPG was redeemed as 1000, 2000, and 4000Da since the polymers were used as received. The HPG molecular weight can be readily controlled by adjusting the amount of glycidol feed in the reaction. Based on the integrated intensity analysis of the 1H NMR and 13C NMR spectrum, the Mn of HPG was calculated (Table 1). The hydrophilic fraction (fphil) of the polymers, defined as the weight percentage of HPG within the polymers, was calculated from the molecular weight of the HPG and PPG blocks.
Table 1.
Synthesis and characterization of HPG-PPG-HPG polymers
| Polymer | PPG Mn | PPG Mass (g) | Glycidol Mass (g) | Chemical structure determined by NMR | fphilc (%) |
|---|---|---|---|---|---|
| HPGL-PPG1000-HPGL | 1000 | 9.2 | 1.02 | HPG45-PPG1000-HPG45a | 8.3 |
| HPGH-PPG1000-HPGH | 1000 | 9.2 | 4.09 | HPG724-PPG1000-HPG724b | 59.2 |
| HPGL-PPG2000-HPGL | 2000 | 9.2 | 1.02 | HPG370-PPG2000-HPG370a | 27.0 |
| HPGH-PPG2000-HPGH | 2000 | 9.2 | 4.09 | HPG591-PPG2000-HPG591b | 37.1 |
| HPGL-PPG4000-HPGL | 4000 | 9.2 | 1.02 | HPG357-PPG4000-HPG357a | 15.1 |
| HPGM-PPG4000-HPGM | 4000 | 9.2 | 2.04 | HPG748-PPG4000-HPG748b | 27.2 |
| HPGH-PPG4000-HPGH | 4000 | 9.2 | 4.09 | HPG1580-PPG4000-HPG1580b | 44.1 |
As determined by 1H NMR.
As determined by 13C NMR.
Hydrophilic fraction of the polymers (fphil) = Weight percentage of HPG within the polymer
To further verify the HPG grafting on PPG, the PPG initiator and the synthesized HPGL-PPG4000-HPGL were characterized by MALDI-ToF mass spectrometry. As shown in Fig. 1C, two repeating units (−CH(CH3)−CH2O−, 58.08 g/mol; −CH2−CH2O−CH2O−, 74.08 g/mol) can be assigned to the PPG and HPG components of the HPGL-PPG4000-HPGL, respectively. We noted that, with the same polymer concentration, the HPGL-PPG4000-HPGL has a significantly higher intensity than that of PPG (Fig. 1D). This is because the HPGL-PPG4000-HPGL contains more end hydroxyl groups which are more likely to combine with sodium-ion compared to pure PPG.
2.3. Emulsion-induced polymersomes based on HPG-PPG-HPG
400 mg of HPG-PPG-HPG was weighted in a 20-mL glass vial, 2 mL of DI water was added into the vial, and the mixture was emulsified with a vortex mixer at the speed of 3000 rpm for 20 seconds. All HPG-PPG-HPG polymersomes showed microscopically visible aqueous pockets (Fig. 2A, S5), and the volume of the aqueous pockets (μl per mg of polymer) was proportional to the fphil of the HPG-PPG-HPG polymers (Table 2). These results confirmed our hypothesis that hydrophilic components of the copolymer favor the formation of the aqueous pockets inside the polymersomes. The diameter of polymersomes range from 5 μm to 20 μm, which is inversely proportional to the fphil of the polymers (Table 2).
Figure 2.
Characterization of particles fabricated after emulsifying HPG-PPG-HPG in the water using a vortex mixer at the speed of 3000 rpm for 20 seconds. A) Optical microscope images of HPG-PPG-HPG particles. Red arrow: aqueous pockets. Scale bars: 20 μm. B) A comparison of the percentage of space occupied by the particles in the emulsion between HPG-PPG-HPG and PPG4000. The emulsions were left at room temperature for 192 hours after the vortex procedure. Data are means ± SD, n = 3.
Table 2.
Characterization of HPG-PPG-HPG polymersomes
| Polymersomes | Diametera (μm) | Aqueous pocket volumeb (μl per mg of polymer) | fphil (%) | Physical stablec | |
|---|---|---|---|---|---|
| 0 hour | 48 hours | ||||
| HPGL-PPG1000-HPGL | 18.15 ± 7.34 | / | 4.74 | 8.3 | No |
| HPGH-PPG1000-HPGH | 5.00 ± 1.72 | / | 5.04 | 59.2 | No |
| HPGL-PPG2000-HPGL | 20.78 ± 8.47 | 53.05 ±17.02* | 3.48 | 27.0 | Yes |
| HPGH-PPG2000-HPGH | 4.37 ± 1.57 | 5.20 ± 1.72* | 5.28 | 37.1 | Yes |
| HPGL-PPG4000-HPGL | 18.45 ± 8.11 | 19.08 ± 5.83* | 3.90 | 15.1 | Yes |
| HPGM-PPG4000-HPGM | 13.54 ± 4.54 | 14.24 ± 5.86 | 5.04 | 27.2 | Yes |
| HPGH-PPG4000-HPGH | 5.29 ± 2.28 | 5.75 ± 2.04 | 5.22 | 44.1 | Yes |
Diameter of polymersomes was observed immediately after preparation
Aqueous pocket volume was observed immediately after preparation
Phase separation of polymersomes was observed 192 hours after preparation
P < 0.001
All HPG-PPG-HPG polymersomes based on PPG2000 and PPG4000, such as HPGL-PPG4000-HPGL, HPGH-PPG4000-HPGH, and HPGH-PPG2000-HPGH, have excellent physical stability. These polymersomes maintained their structural integrities (size and hollow structure) after incubating for 48 hours at room temperature (Fig. 2A, Table 2). No oil droplets were observed 192 hours after the vortex procedure (Fig.2B, S6). After centrifuging these polymersomes for 5 min at the speed of 12000 rpm, no change in the particle morphology was observed (Fig. S7). In contrast, HPG-PPG-HPG polymersomes based on PPG1000, such as HPGL-PPG1000-HPGL and HPGH-PPG1000-HPGH polymersomes, are not stable (Fig. S5, Table 2). These results are consistent with the finding that low molecular weight PPG has weak hydrophobic interactions and cannot sufficiently stabilize the polymersome structure.
We also found that the addition of the HPG block not only increases the volume of the aqueous pockets but also stabilizes the polymersomes. For example, compared to the pure PPG4000 particles, the physical stability of HPGL-PPG4000-HPGL polymersomes is much improved (Fig.2B). This phenomenon can be attributed to the existence of aqueous pockets. The presence of aqueous pockets inherently increases the surface area of the polymersome so that the HPG block or hydroxyl groups on the surface can better maintain the polymer/water interface due to their high affinity with water.
2.4. Factors contributing to the stability of HPG-PPG-HPG polymersomes
To investigate the role of the molecular weight of the PPG chain and the molecular weight of the HPG branch in stabilizing HPG-PPG-HPG polymersomes, all-atom MD simulations were carried out for six PPG systems based on PPG1000 and PPG2000. The six PPG-based systems are PPG1000, HPG518-PPG1000-HPG518, PPG740-PPG1000-HPG740, PPG2000, HPG518-PPG2000-HPG518, and PPG740-PPG2000-HPG740, where PPG1000 and PPG2000 are pure PPG chains without HPG branches. Each system was comprised of an array of 256 identical PPG polymers solvated in water, as shown in Fig. S8 (more simulation details are provided in the Supplemental Materials). Representative HPG branches are shown in Fig. 3A. The all-atom MD simulations were used to calculate the average binding energy of each PPG-based polymer. As shown in Fig. 3B, independent of the HPG branch, the binding energy of polymers with a PPG2000 chain is generally lower than that of polymers with a PPG1000 chain. Lower binding energies indicate that PPG2000 polymers form more stable aggregates than PPG1000 polymers. These results agree with the experimental observations (as shown in Table 2) that the formation and stability of polymersomes increase with the increasing molecular weight of PPG chain. Furthermore, comparing the binding energies between PPG1000, HPG518-PPG1000-HPG518 and PPG740-PPG1000-HPG740 as well as between PPG2000, HPG518-PPG2000-HPG518 and PPG740-PPG2000-HPG740, it can be noted that increasing the molecular weight of the HPG branch lowers the average binding energy of the PPG polymers in both the PPG1000 and PPG2000 systems (Fig. 3B). This indicates that increasing the hydrophilicity of the PPG polymer through the addition of HPG branches enables the formation of more stable aggregates. These results suggest that the formation and structural stability of polymersomes can be further improved by increasing the molecular weight of the PPG chain and increasing the hydrophilicity of the polymer through the addition of HPG branches.
Figure 3.
The average binding energy of six unique PPG polymers.
2.5. Drug encapsulation and release from polymersomes
2.5.1. Sodium fluorescein (SF)
We selected sodium fluorescein (SF) (molecular weight 376.27, logP = 2.6) as the model drug to validate the feasibility of EIP in administering small hydrophilic molecules. SF has an absorption maximum at 492 nm in PBS buffer (pH = 7.4). Thus, its concentration can be readily measured by an ultraviolet-visible spectrophotometer. 20 μg of SF was dissolved in 1 mL of phosphate buffered saline (PBS, pH 7.4). 200 mg of polymer was weighted in a 20 mL glass vial. The prepared SF solution was added to the vial, and the mixture was vortexed at the speed of 3000 rpm for 20 seconds. Immediately following the vortex procedure, both the encapsulation efficiency of SF and the in vitro release of SF were measured.
As shown in Fig.4A, the SF encapsulation efficiency is proportional to the molecular weight of HPG and therefore proportional to the volume of the aqueous pocket of HPG-PPG-HPG polymersomes. In particular, the SF encapsulation efficiencies of HPGH-PPG1000-HPGH, HPGH-PPG2000-HPGH, and HPGH-PPG4000-HPGH polymersomes were 84.15 ± 0.60%, 88.65 ± 0.69%, and 86.81 ± 0.20%, respectively (Fig. 4A). In contrast, the SF encapsulation efficiency of pure PPG polymersomes is significantly lower, which indicates that smaller aqueous pockets can provide less storage space for SF. For HPG-PPG-HPG polymersomes that have less volume of aqueous pocket, the SF encapsulation efficiency can be increased by increasing the polymer-to-drug ratio of the formulation. With the SF amount unchanged, when the HPGL-PPG4000-HPGL concentration increased from 200 to 300 mg/ml, the encapsulation efficiency increased substantially from 62.8 ± 5.2% to 89.4 ± 1.3% (Fig. 4B). There were no significant differences in encapsulation efficiency when the concentration increased from 300 to 400 mg/ml. This result indicates that most of the SF has already been encapsulated inside the polymersomes at the polymer concentration of 300 mg/ml.
Figure 4.
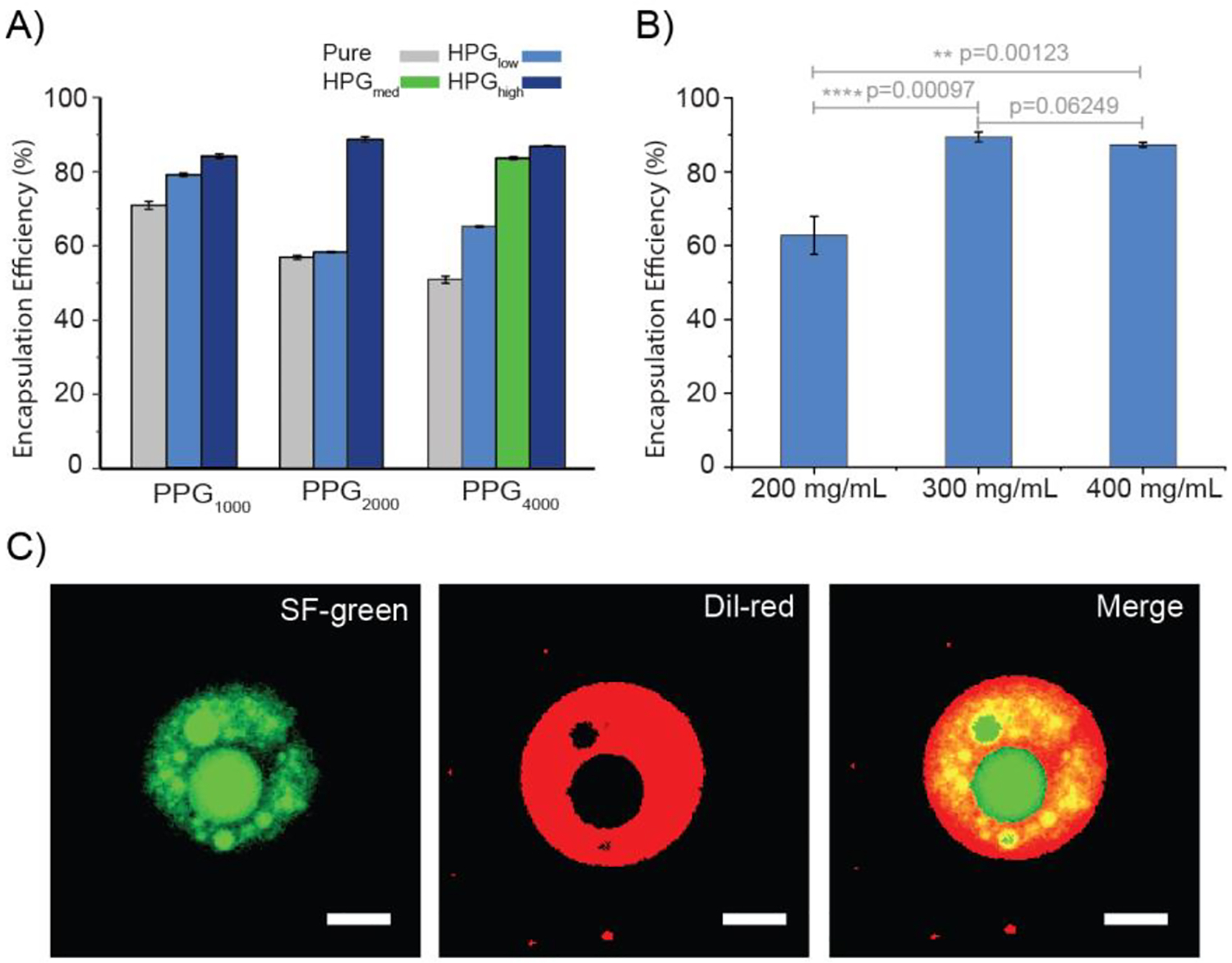
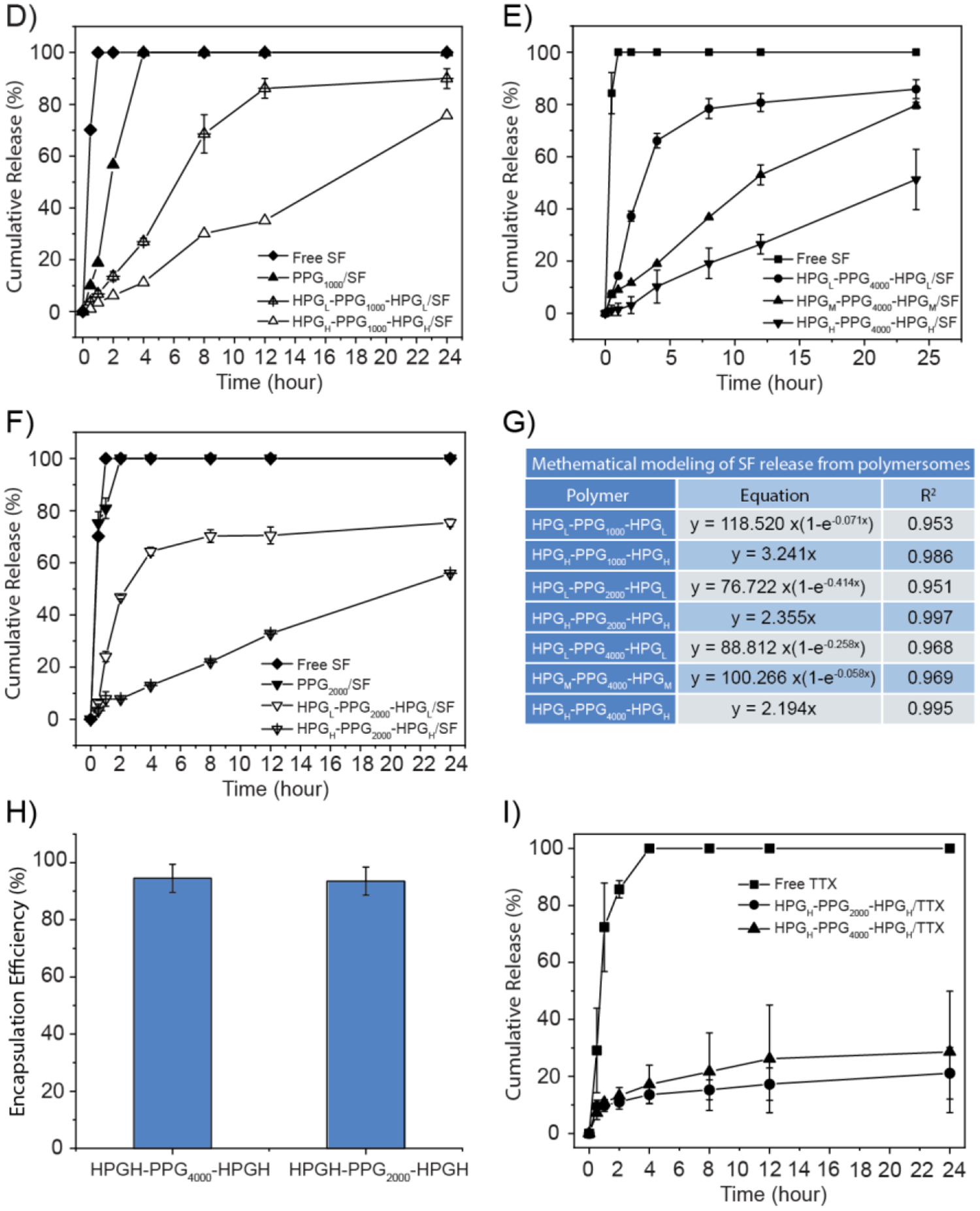
Characterization of drug encapsulation and release characteristics from polymersomes fabricated by the vortex procedure. A) A comparison of the encapsulation efficiency of SF in different polymersomes. B) Effect of the HPGL-PPG4000-HPGL concentration on the encapsulation efficiency of SF. C) Confocal fluorescence microscopy images of polymersomes. Green: SF. Red: Dil. Scale bars: 20 μm. D) A comparison of the cumulative release of SF eluted from different polymersomes in PBS at 37 °C. D-F) A comparison of the cumulative release of SF eluted from different polymersomes in PBS at 37 °C. G) Mathematical modeling of SF release from polymersomes. H) A comparison of the encapsulation efficiency of TTX in polymersomes. I) A comparison of the cumulative release of TTX eluted from different polymersomes in PBS at 37 °C. Data are means ± SD, n = 3.
As shown in the confocal images (Fig. 4C), the aqueous pockets of the HPG-PPG-HPG polymersomes were green (SF stains water as green) and surrounded by the red polymer shell (Dil stains polymer as red). This observation confirmed that the SF molecules were encapsulated inside the HPG-PPG-HPG polymersomes. In contrast, a scarce green color can be found in the PPG4000 and PPG1000 microspheres.
SF release from all polymersomes was slower than observed with SF alone (Fig. 4D), indicating the successful establishment of a sustained release platform by the drug delivery systems. When the PPG block length was kept constant, the release rate decreases as the HPG molecular weight increases. Interestingly, when the HPG molecular weight increases to a certain level, the SF release kinetics turn from a first-order release to a zero-order release. This transition can be attributed to the increase of SF encapsulation efficiency and improvement of the polymersomes stability. Diffusion of SF from the polymersome is the only driving force of the drug release; therefore, it follows the zero-order release. In contrast, for other formulations, the non-encapsulated SF, broken polymersomes, and the typical SF diffusion contribute to drug release. Together, these unique methods of drug release follow the first-order release.
Pure PPG particles have SF encapsulation efficiency above 50%, which may be because of the physical trapping of SF in the PPG phase. The interaction between SF and PPG chains is extremely weak, leading to fast SF release. Fast release may also be attributed to the insufficient polymersome stability. Immediately following the vortex process, pure PPGs form polymersomes and efficiently encapsulated SF inside aqueous pockets. However, the encapsulation is only effective for a limited time due to insufficient structural stability. PPG polymersomes tend to disassemble during the incubation, leading to the fast SF release.
2.5.2. Tetrodotoxin
In addition to SF, the encapsulation and controlled release characteristics of HPGH-PPG4000-HPGH and HPGH-PPG2000-HPGH polymersomes with TTX were also evaluated. 10 μg of TTX was dissolved in 1 mL of PBS. 200 mg of polymer was weighted in a 20 mL glass vial. The prepared TTX solution was added into the vial, and the mixture was vortexed at the speed of 3000 rpm for 20 seconds. Immediately following the vortex procedure, the encapsulation efficiency and in vitro release of TTX were measured. The concentration of non-encapsulated TTX in the filtrate was quantified by ELISA. The results of TTX encapsulation and controlled release are consistent with that of SF. HPGH-PPG4000-HPGH and HPGH-PPG2000-HPGH polymersomes had the TTX encapsulation efficiency of 94.5±4.9%, and 93.5±4.9%, respectively (Fig. 4H). Contrary to almost all free TTX being released within 4 hours, sustained release of TTX from the polymersomes was demonstrated, with less than 40% TTX released from the polymersomes within 24 hours (Fig. 4I).
2.6. Rat sciatic nerve blockade in vivo
Male Sprague-Dawley rats (250–350 g, 4 in each group) were injected at the left sciatic nerve with 0.3 mL of PBS containing free TTX or polymersomal TTX formulations (e.g. an increasing dose of TTX was emulsified with 60 mg of polymers in 0.3 mL of PBS). Next, the rats underwent neurobehavioral testing to determine the duration of functional deficits (i.e. sensory and motor nerve blockade) in both hindpaws. The duration of deficits on the injected (left) side reflected the duration of nerve block. Deficits on the uninjected (right, contralateral) side reflected systemic TTX distribution.
2.6.1. In vivo assessment of tolerance to TTX
Injection with 4 μg of TTX in 0.3 mL of PBS caused contralateral deficits in all rats (e.g. thermal nociceptive test [31], thermal latency going to the 12 seconds cut off in the un-injected limb suggests systemic distribution of TTX) (Table 3) and was uniformly fatal (e.g. rats developed seizures and respiratory distress, and were subsequently euthanized). In contrast, injection of HPGH-PPG2000-HPGH polymersomes containing 11.5 μg TTX did not induce any TTX-related systemic toxicity as evidenced by no rats developing contralateral deficits or seizures and respiratory distress. Two of the four rats injected with HPGH-PPG2000-HPGH polymersomes containing 12 μg TTX developed contralateral deficits, but did not show seizures or respiratory distress. The other two rats did not show any TTX-related systemic toxicity. Of note, 12 μg TTX is three times the lethal dose of TTX alone. Although not as effective as HPGH-PPG2000-HPGH polymersomes, HPGL-PPG4000-HPGL and HPGH-PPG4000-HPGH polymersomes reduced the systemic toxicity of 8 μg TTX, which can be demonstrated by the reduction in contralateral block frequency and mortality (Table 3). These TTX tolerance results were consistent with the results of in vitro TTX encapsulation and controlled release. All tested polymersomes can efficiently encapsulate TTX and achieve sustained TTX release, avoiding burst TTX release that could cause the systemic toxicity of animals after the injection.
Table 3.
Sciatic nerve blockade with free TTX and/or polymersome and/or SOS
| Polymersomea | TTX dose (μg) | SOS (w/v) | Successful block (%) | Duration of blockb (h) | Contralateral blockc (%) | Mortality (%) |
|---|---|---|---|---|---|---|
| TTX alone | 1 | / | 0 | 0 | 0 | 0 |
| 2 | / | 0 | 0 | 0 | 0 | |
| 3 | / | 75 | 1.3±1.3 | 0 | 0 | |
| 4 | / | / | / | 100 | 100 | |
| HPGL-PPG4000-HPGL | 8 | / | 100 | 6.5 | 100 | 75 |
| HPGH-PPG4000-HPGH | 8 | / | 100 | 3.7±1.2 | 75 | 0 |
| HPGH-PPG2000-HPGH | 8 | / | 0 | / | 0 | 0 |
| 9 | / | 50 | 3.0±1.4 | 0 | 0 | |
| 11.5 | / | 75 | 3.7±2.1 | 0 | 0 | |
| 12 | / | 100 | 4.0±2.7 | 50 | 0 | |
| 9 | 2% | 100 | 5.7±2.2 | 0 | 0 | |
| 11.5 | 1% | 100 | 132.0±76.5 | 0 | 0 | |
| 11.5 | 2% | 100 | 540.0±269.0 | 0 | 0 |
Each rat received a single sciatic nerve injection of 0.3 mL PBS containing 60 mg polymer and/or TTX and/or SOS, n=4.
Sensory nerve block. Data are means ± SD, n=4.
Frequency of nerve block in the uninjected (contralateral) leg.
2.6.2. Sciatic nerve block with EIP/TTX
Groups of rats receiving injections of free TTX at the sciatic nerve showed dose-dependent increases in the frequency of successful nerve blocks and median duration of nerve blocks (Table 3). Nerve block obtained from 3 μg of free TTX in 0.3 mL of PBS was successful in 75% of animals and produced a median duration of sensory nerve block of 1.3±1.3 hours. These results are comparable to the effect of 0.5% bupivacaine, an anesthetic commonly used in the clinic. Notably, injection of 1–2 μg of free TTX in 0.3 mL of PBS did not block the nerve, and as described above, injection of 4 μg of free TTX in 0.3 mL of PBS was uniformly fatal.
Groups of rats that received injections of polymersome/TTX formulations at the sciatic nerve showed a TTX dose-dependence on the frequency of successful nerve blocks and the median duration of nerve block (Table 3). In particular, injection of HPGH-PPG2000-HPGH polymersome containing 11.5 μg of TTX successfully induced ipsilateral nerve block in three out of four rats. The median duration of sensory nerve block in the blocked rats was 3.7±2.1 hours, which is a 3-fold longer nerve block duration than achieved by free TTX and compatible to the duration that achieved from a single perineural injection of Exparel (clinically used liposomal bupivacaine; 13.3 mg/mL) in an equal volume.
Even if the loaded TTX is twice or three times the uniformly lethal dose of free TTX (8–11.5 μg), the injection of polymersome/TTX formulation only produced sciatic nerve blockade in a certain percentage of rats. Other rats behaved normally, similar to uninjected rats. These results indicate that most of the loaded TTX was encapsulated in the polymersomes, and the subsequent release is extremely slow, resulting in a local concentration of TTX lower than the therapeutic level required to block the sciatic nerve. In order to make the ineffective TTX concentration effective and ultimately improve the nerve block effect, 1% or 2% w/v SOS, a chemical permeation enhancer (CPE), was added into the HPGH-PPG2000-HPGH/TTX formulation in the process of formulation preparation. The principle of CPE is to reversibly disrupt the perineurium barrier and facilitate the crossing of a polar molecule, such as TTX, through the perineurium. The enhanced flux of TTX to the site of action can improve the nerve block performance of TTX. As expected, nerve block from the SOS containing formulation was successful in 100% of animals. The effect of SOS on the median duration of nerve block showed dose-dependent increases. In particular, the injection of HPGH-PPG2000-HPGH polymersome containing 11.5 μg TTX and 2% SOS produced a median duration of sensory nerve block of 540.0 ± 269.0 hours (e.g., 22.5 ± 11.2 days). The addition of SOS did not induce systemic toxicity. To our knowledge, this is the longest rat sciatic nerve duration reported to date with neurotoxin-based local anesthetics. There was no statistically significant difference between the durations of sensory and motor nerve blockade in any groups (p = 1.000). Rats injected with the any formulations did not show weight loss (Fig. S9).
2.7. Tissue reaction.
A principal obstacle to the development of prolonged duration local anesthesia has been the concern of local tissue toxicity. Inflammation caused by the drug delivery system may have long-lasting effects and the intrinsic myotoxicity and neurotoxicity of compounds could harm the muscles and nerves. As described above, PPG polymer was selected because of its excellent biocompatibility. More importantly, PPG4000 has been proven to not cause local muscle and nerve damage after perineural injection[31]. Here, the emphasis was given to examining the tissue reaction to the HPG-PPG-HPG-based formulations.
Animals injected with formulations were euthanized 4, 14 and 28 days after sciatic nerve injection. At dissection, there were visible residual materials observed at the injection site around the sciatic nerve in all rats in the HPGH-PPG2000-HPGH/TTX group and HPGH-PPG2000-HPGH/TTX/SOS group 4 and 14 days after the injection (Fig. 6). The material residence indicates that the formulation can prevent it from spreading to off-target sciatic nerves after injection, so it can act as a drug reservoir to continuously release TTX acting on the sciatic nerve, which is essential for achieving up to 20 days of sciatic nerve blockade. 28 days after the injection, the visible residues of materials were found only in 2 out of 10 rats injected with the HPGH-PPG2000-HPGH/TTX/SOS formulation, reflecting the clearance of materials. The material clearance is desired after it produces therapeutic effects to avoid tissue toxicity due to long-term material exposure after the completion of drug delivery. At dissection, there were no visible residual materials in all rats in the HPGH-PPG4000-HPGH/TTX group at 4 days and 14 days after injection. This may be due to the higher rigidity of the HPGH-PPG4000-HPGH polymersomes than HPGH-PPG2000-HPGH polymersomes, which increase the fluidity of the formulation. The tissues in all rats did not appear edematous or discolored and had no other signs of tissue injury. Sciatic nerves and surrounding tissues were sectioned and harvested for histologic evaluation. Muscle tissue was processed with hematoxylin-eosin staining, and nerve tissue was processed with toluidine blue staining. At 4, 14, or 28 days after injection, microscopic examination showed no significant myotoxicity and only moderate inflammation in animals (Fig. 6, Table 4). There was no statistically significant difference between the scores in any group injected with HPG-PPG-HPG/TTX or free TTX (p >0.05). Compared with an equal concentration of SOS, HPG-PPG-HPG/TTX/SOS was less toxic to surrounding muscle tissue (e.g. myotoxicity)[44, 45], indicating that compared with immediate release of SOS, slow release of SOS can reduce the risk of chemical injury to muscle tissue.
Figure 6.
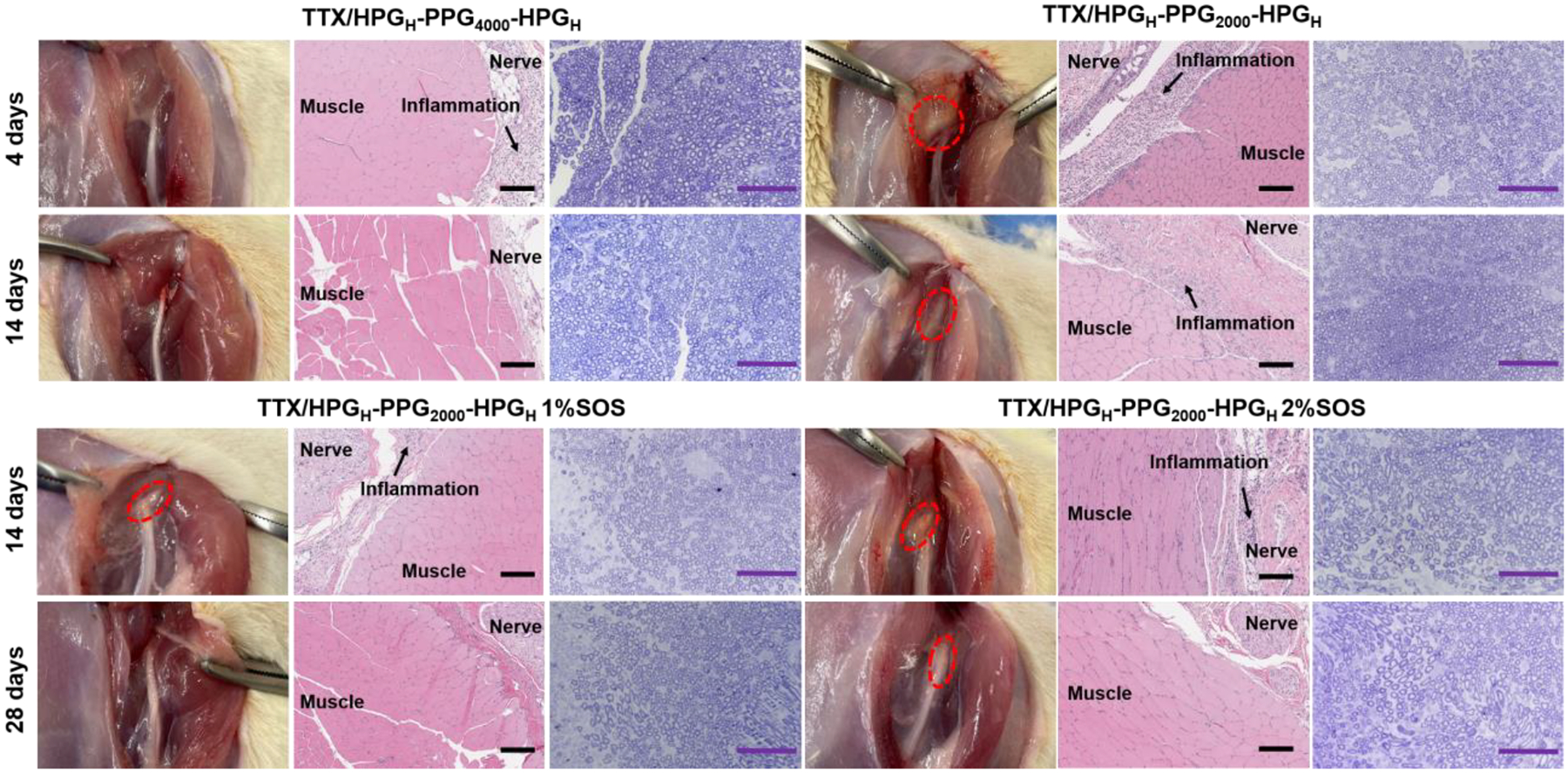
Tissue reaction to formulations 4, 14, and 28 days after injection, including representative photographs of the site of injection upon dissection, representative hematoxylin–eosin stained sections of muscles and adjacent loose connective tissue, and toluidine blue stained sections of nerve. Data are representative of 2–5 animals in each group. Scale bars, 200 μm (H&E), 100 μm (toluidine blue)
Table 4.
Myotoxicity and inflammation
| Myotoxicity Score | Inflammation Score | |||||
|---|---|---|---|---|---|---|
| Day 4 | Day 14 | Day 28 | Day 4 | Day 14 | Day 28 | |
| Free TTX | 0 (0–0) | / | / | 0 (0–0) | / | / |
| HPGH-PPG4000-HPGH | 0 (0–0) | 0 (0–0) | / | 0 (0–1) | 0 (0–0) | / |
| P value vs free TTX | 1 | 1 | 0.505 | 1 | ||
| HPGH-PPG2000-HPGH | 2 (0–3) | 1 (0–1) | / | 1 (0–1) | 1 (0–1) | / |
| P value vs free TTX | 0.197 | 0.188 | 0.188 | 0.188 | ||
| HPGH-PPG2000-HPGH/1% SOS | / | 1 (1–1) | 1 (1–1) | / | 1 (1–1) | 1 (1–1) |
| P value vs free TTX | 0.096 | 0.096 | 0.096 | 0.096 | ||
| P value vs no SOS | 0.683 | 0.683 | 0.683 | 0.683 | ||
| HPGH-PPG2000-HPGH/2% SOS | / | 1 (0–2) | 0 (0–0) | / | 1 (1–2) | 0 (0–2) |
| P value vs free TTX | 0.197 | 1 | 0.059 | 0.505 | ||
| P value vs no SOS | 0.814 | 0.188 | 0.302 | 1 | ||
Data are medians with interquartile ranges. n = 2–5 per experimental group. Inflammation scores range: 0–4; myotoxicity scores range: 0–6
We obtained toluidine blue-stained Epon-embedded sections of the sciatic nerve in animals injected with HPG-PPG-HPG/TTX and HPG-PPG-HPG/TTX/SOS. Neurotoxicity (e.g., nerve fiber density, myelin width) was not detected in any rats injected with HPG-PPG-HPG/TTX. The benign nerve tissue reaction was also evidenced by that HPG-PPG-HPG polymers do not enter the nerves 4 days after sciatic nerve injection (Fig.S10). 5 out of 10 nerves from rats injected with HPG-PPG-HPG/TTX/SOS at 14 and 28 days showed minimal peripheral injury (decreased density of axons) involving < 10% of the nerve (Fig. 6, Fig.S11). That minimal nerve injury was induced by the SOS, which is consistent with the previous report that a high concentration of CPE can cause nerve injury[31]. When reducing the SOS concentration or using a less toxic CPE in the formulation, it is believed that the minimal nerve injury can be eliminated. It is worth noting that in the same animal model, nerve injury was uniformly observed in rats injected with bupivacaine, lidocaine, and bupivacaine liposome and other clinically used local anesthetics[46, 47].
3. Discussion
We present the emulsion approach for the rapid and high-yielding production of micrometer-sized polymersomes. This method differs substantially from existing techniques used to manufacture polymersomes, such as film hydration, solvent evaporation, and microfluidic method[35, 48, 49]. The emulsion method is also rarely used for the fabrication of nanoparticles[50–53] and hydrogels[54–57]. Moreover, the emulsion approach has advantages over these existing techniques, as it is an extremely simple and rapid process that does not require the use of volatile organic solvents, surfactants, and expensive devices.
EIP polymersomes is an ideal drug delivery system because (1) the prepared polymersomes have vesicular structure that can effectively encapsulate and achieve sustained release of water-soluble drugs; (2) the simple, rapid, single-step production allows the formulation preparation to immediately precede injection, reduce the premature release of drugs during storage and transportation, and avoid additional steps that potentially deplete the vehicles’ drug content; (3) the drug encapsulation process is typically proceeded in aqueous solutions, minimizing the risk of drug denaturation when exposed to organic solvents[58]. The emulsification process usually generates minimal heat, which eliminates the possibility of drug thermal denaturation; (4) due to the high drug encapsulation efficiency, the step of removing unencapsulated drug can be omitted, because the small amount of unencapsulated drug will not cause systemic toxicity, but is beneficial to initiate effectiveness rapidly upon in vivo injection. These features enable formulations to be quickly prepared before injection, and the freshly prepared formation can be directly injected, eliminating the problems of drug waste and early drug release during storage and transportation; (5) last but not least, the simple manufacturing procedures serve to reduce costs.
The polymersome/TTX/SOS formulation is a TTX-based local anesthetic that provides longer nerve blocks than can be achieved with the lipophilic anesthetics currently in use. In rats, a single injection of HPG-PPG-HPG/TTX/SOS formulation provided a sensory nerve blockade lasting 22.5 ± 11.2 days; this is much longer than that of liposomal bupivacaine (5.5 ± 0.7 hours)[59] and the Exparel (4 hours)[60]. Of note, Exparel is the FDA-approved bupivacaine liposome injectable suspension for local infiltration in pediatric patients aged 6 years and above. Given that a single injection of Exparel produced local anesthesia in humans lasted for 3 days, the HPG-PPG-HPG/TTX/SOS probably produced weeks to months of local anesthesia in humans, which makes it an attractive candidate medicine for chronic pain treatment.
The polymersome/TTX/SOS formulation is considered safe for use in humans. In a Phase III clinical trial performed in Canada, 30 μg of TTX was administered subcutaneously twice daily for four consecutive days to cancer patients[61]. The results showed that no patients experienced life-threatening side effects, indicating that the human TTX tolerated dose might even be higher than 240 μg. We have demonstrated that the polymersome increases the tolerated dose of TTX in rats by at least 3 times. Therefore, it is estimated that the polymersome carrying 720 μg TTX are safe for humans. On the other hand, as mentioned above, the effectiveness of TTX in a nerve block is approximately 1000 times that of conventional local anesthetics. Whereas the injection of 2.5–5 mg of bupivacaine successfully blocked the human sciatic nerve[62]. Accordingly, 2.5–5 μg of TTX would be sufficient to block human nerves. Therefore, the maximum tolerated TTX dose in the polymer/TTX/SOS formulation is expected to be much higher than the effective dose for nerve blocks in humans.
Given that liposomes as drug carriers have been widely used in pharmaceutical applications[63, 64], and that polymersomes have a core-shell structure similar to liposomes, the EIP technique can be a strategy for encapsulating various hydrophilic and hydrophobic diagnostic or therapeutic agents, and can be applied to many pharmaceutical applications and have a substantial clinical impact. In addition to drug delivery systems, EIP polymersomes can also be used as injectable cellular carriers for tissue regeneration. Firstly, injectable cell carriers from 10 to 100 μm in diameter are mostly used in tissue engineering[65, 66], and the size of EIP polymersomes is in this range. Secondly, cells can be loaded into EIP polymersomes by simply mixing the polymer and cells using a mixer, which is a simple and cell-friendly process.
The presented EIP polymersomes range in diameter from 1–20 μm, which is a good size for local drug administration, as they are injectable. However, drug delivery systems designed for intravenous injection must be within the size range of 10–300 nm[67]. By changing the emulsification conditions, such as stirring time and speed or the addition of a second surfactant, we may use the EIP approach to make smaller polymersomes, and the application of EIP polymersomes can be extended to intravenous injection.
The polymer candidates for the EIP technique are not limited to the presented HPG-PPG-HPG. As described above, liquid, water-insoluble, and amphiphilic polymers meet the criteria for using the EIP technique to make polymersomes. For example, the PPG block of the HPG-PPG-HPG can be replaced by other hydrophobic, liquid polymers such as polycaprolactone (PCL). The HPG block of the HPG-PPG-HPG can be replaced by other hydrophilic polymers such as polyamidoamine (PAMAM) and hyaluronic acid (HA). The substantial structure and composition diversity in EIP polymers allows for rationally designing polymersome properties and customizing polymersome functions for specific biomedical applications. The variety of candidate polymers represents a huge room for optimization of the EIP platform in clinical applications.
4. Conclusion
We developed the EIP polymersomes as a drug delivery system of TTX. TTX was simply emulsified with HPG-PPG-HPG in an aqueous solution, then used directly for sciatic nerve injection in rats. This methodology requires no additional steps that may cause TTX loss. HPG-PPG-HPG/TTX/SOS formulation provided sustained release of TTX and prolonged sensory nerve blockade up to about 20 days with a single injection in rats, without causing any systemic toxicity, and only minimal local tissue toxicity. EIP technique is a significant departure from the existing technologies for polymersome production and drug delivery. More importantly, it achieves a high drug encapsulation efficiency, provides a desired sustained drug release, has a low cost, and is scalable and translatable. We believe that the EIP technique has a high potential for future clinical applications to increase the efficacy of systemic and locally delivered therapeutics in promoting animal and human welfare.
5. Methods
Reagents.
Poly(propylene glycol) (PPG, Mn = 1000, 2000), potassium methoxide (KOCH3, 95%), glycidol (96%), phosphate buffer saline (PBS, pH 7.4, 0.15M, 138mM NaCl, 2.7 mM KCl), chloroform-d (100%, 99.96 atom % D), fluorescein sodium salt, deuterium oxide (99.9 atom % D), dimethyl sulphoxide-[D6] (≥99.8%), fluorescein-5-isothiocyanate (FITC), Tissue-Tek® O.C.T. Compound were purchased from VWR International Ltd (Radnor, PA). Poly(propylene glycol) (PPG, Mn = 4000), and sodium octyl sulfate (≥95%), were purchased from Sigma-Aldrich Inc. (St. Louis, MO). Tetrodotoxin was purchased from Abcam (Cambridge, MA, USA). TTX ELISA kits were purchased from Reagen LLC (Moorestown, NJ, USA).
Synthesis of HPG-PPG-HPG.
Dry PPG (see Table 1 for feeding amount) was added into a nitrogen-protected flask in a 95°C oil bath, and a predetermined amount of KOCH3 was added. The mole ratio of the hydroxyl group of PPG diol to KOCH3 was 1:0.45. The system was kept for 1 hour with stirring (500–600 rpm) under vacuum. Then the flask was left in an 80°C vacuum oven for 24 hours. The flask was refilled with nitrogen and placed in a 95°C oil bath. Glycidol was added by a syringe pump with a speed of 1 mL/h. The system was kept for another 24 hours with stirring (500–600 rpm) after the addition of the glycidol. At the end of reaction time, the reaction mixture was washed with 100–200 mL warm water (80°C) and centrifuged at 12000 rpm for 5 minutes. The supernatant was removed, and the residue was remixed with DI water. The mixture was dialyzed against 1L of DI water for 72 hours (replaced with fresh DI water every 24 hours). The the polymer was lyophilized under 30 mT over 48h to get dry polymer. HPG-PPG-HPG polymers were obtained as viscous liquid. Yields of isolated polymer ranged between 30% and 65%. The polymers were stored in a dry environment before using them.
Synthesis of HPG-PPG-HPG-FITC urethane.
0.2 g of HPG-PPG-HPG polymer was dried in a 50 mL flask in a vacuum oven overnight to eliminate moisture. Then, 5 mL of THF was added to the flask, and 10 mg of FITC dissolved in 5 mL of anhydrous DMSO was added sequentially. The reaction occurred under stirring at 50 °C for 4 hours. At the end of the reaction, the resultant polymer was dialyzed with a semipermeable membrane (molecular weight cutoff, MWCO ~ 12,000) in 1 L water for 48 h, and the water was refreshed every 12 h. The HPG-PPG-HPG-FITC urethane was obtained in 63% yield.
Fabrication of polymersomes.
A predetermined amount (400, 600, 800 mg) of polymer was weighted in a 20-mL glass vial, 2 mL of DI water was added into the vial, and the mixture was vortexed at the speed of 3000 rpm for the predetermined period (10, 20, and 60 s).
Nuclear magnetic resonance (NMR) spectroscopy.
HPG-PPG-HPG polymers were dried in an 80°C vacuum oven for one week. The 1H-NMR and inverse gated 13C-NMR spectra were recorded on a NMR spectrometer (Bruker, Avance 500). DMSO-d6 was used as the solvent. The concentration of the polymer for 1H-NMR and 13C-NMR spectra were 10 mg/ml and 100 mg/ml respectively. The chemical shifts (δ, in ppm) for the peaks corresponding to the hydrogens in italics in HPG-PPG-HPG polymers are provided. s/d/m indicate the shape of a peak (i.e., singlet, doublet, triplet). 1H NMR (HPG-PPG-HPG) (500MHz, DMSO-d6) δ/ppm: 1.14–1.17 (3H, d, -CH3), 3.47–3.63 (1H, d, -CH2O-), 3.75–3.96 (1H, qt, >CH-O-). Based on the 1H-NMR spectra, the Mn of HPG can be calculated with Equation 1.
| (1) |
where r is the weight ratio of HPG to PPG calculated with Equation 2 (I1 and I2 are the integrated information of resonances 3.3 ppm ~3.9 ppm and ~1 ppm, respectively), and x is the given Mn of PPG. The composition of copolymer is HPGrx/2PPGxHPGrx/2.
| (2) |
The Mn of HPG was calculated from inverse gated 13C-NMR spectra based on a previously reported method[68]. The signals of 13C-NMR spectra can be assigned as: (i) linear 1,3-unit (L13): CH2OH carbon at 61.5 ppm, CH2 carbon at 69.9 ppm, and CH carbon at 80.3 ppm; (ii) linear 1,4-unit (L14): both CH2 carbons at 73.3 ppm, CHOH carbon at 69.0 ppm; (iii) terminal unit (T): CH2OH carbon at 63.5 ppm, CHOH carbon at 70.9 ppm, and the CH2 carbon at about 71.2 ppm; (iv) dendritic unit (D): CH carbon at 78.5 ppm, one CH2 carbon at 72.0 ppm, and the other at about 71.2 overlapping with a CH2 carbon of a terminal unit. Based on the 13C-NMR spectra, the Mn of HPG can be calculated with Equation 3.
| (3) |
“n” represents the number of propagation cores. For a linear PPG molecule with two hydroxyl group ends, the value of n is 2. Since the method stated calculates only the average molecule weight of the grafted HPG structure, it should be noted that the prerequisite is only propagation onto the core polymer has occurred.
MALDI-TOF mass spectrometry.
Polymer samples were characterized using a Bruker Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS). 2,5-dihydroxybenzoic acid was used as matrix. Samples were prepared by dissolving the polymer in methanol at a concentration of 5 mg/ml. A 1 μL aliquot was added to 5 μL of matrix solution containing NaCl. A 1 μL aliquot of the resulting mixture was placed onto a target plate to evaporate the methanol and create a thin matrix/analyte film. The ions were accelerated to 21.50 kV and measured in the reflection mode of the spectrometer. Only sodium-cationized ions [M + Na]+ were detected. Red phosphorous was used for an external calibration.
Gel permeation chromatography (GPC).
The GPC was performed using a Visotek system (Malvern Panalytical, PA, USA) equipped with an RI detector, an automatic sampler, a pump, an injector, an inline degasser, a column oven (60 °C), and two in-series Malvern T6000 M SEC columns. DMF was used as the mobile phase at a flow rate of 1 mL/min. Polystyrene was used as standard.
Optical microscope imaging.
Polymers were weighted in a 20-mL glass vial, 1 ml of DI water was added to the vial, and the mixture was vortexed at the speed of 3000 rpm for a predetermined time period (10, 20, and 60 s). Right after the mixture, the solution was dropped onto the glass chip and observed using an optical microscope (Ts2R, Nikon). The sizes of polymersomes were measured and counted by NIS-Elements Imaging Software (Nikon Instruments Inc.). For each sample, ~200 repetitions were observed. The reported sizes were average of the mean size.
Confocal imaging.
1 μg of Dil stain (red, hydrophobic dye to stain the hydrophobic polymer phase) and 10 μg of sodium fluorescein (green, hydrophilic dye to stain the water phase) were dissolved in 0.5 mL of PBS (20 μg/mL), and then formulated with 100 mg polymer under vortex until the polymer was fully emulsified. The mixture was diluted to 10 mg/mL using the PBS solution and the morphology was observed under confocal laser scanning microscopy (Leica TCS SP2 AOBS) at room temperature using a 20× objective.
Measurement of the space occupancy of particles.
100 mg polymer was weighed in a 2-ml centrifuge tube, added with 0.5 mL of DI water, and vortexed at the speed of 3000 rpm. Right after the mixing, the emulsions were transferred to a 1-mL disposable glass tube. The samples were kept still under ambient temperature and photographed at fixed time intervals. The height of the particle phase separated from the emulsion was measured after 192 hours. The percentage of space occupied by the particles in the emulsion was calculated by Equation 4.
| (4) |
Aqueous pocket volume analysis.
Polymersome trapped volume, which is defined as the aqueous pocket volume of polymersomes per unit mass of the polymer (VT, in μl per mg of polymer), was measured as previously reported. Briefly, add a marker (e.g. SF) during the preparation of polymersomes and remove the unencapsulated marker by dialysis or centrifuge. The ratio of the marker enclosed inside polymersomes to the total amount added is equal to the ratio of the volume of polymersomes to the total volume of the mixture. VT was calculated as: (volume of polymersomes - volume of the polymer)/mass of the polymer.
Physical stability of polymersomes.
400 mg of the polymer was weighed in a 2-mL centrifuge tube, added with 2 mL of DI water, and vortexed at the speed of 3000 rpm. Right after the mixing, the emulsions were kept still under ambient temperature and photographed at fixed time intervals. The onset time of the first oil droplet observed was recorded.
The physical stability of HPG-PPG-HPG polymersomes was also assessed by observing the morphology under the optical microscope after incubating the emulsion at ambient temperature for 48 hours and centrifuging the emulsion at the speed of 12000 rpm for 5 minutes.
Determination of encapsulation efficiency.
20 μg of sodium fluorescein or 10 μg of TTX was dissolved in 1 mL of PBS and then formulated with the predetermined amount of polymer under vortex at the speed of 3000 rpm until the polymer was emulsified thoroughly. The measurement was performed by directly injecting the mixture through a 23 G needle into the bottom of a 20-mL sample vial which contained 19 mL of PBS buffer prewarmed at 37°C. The whole mixture was gently stirred to make the unencapsulated drug diffuse into the supernatant. Then, 2 mL of the supernatant was filtered through a 0.22-μm pore size disposable syringe filter. The SF concentration in the solution was measured with an absorbance microplate reader (EMax Plus, Molecular Devices) at a wavelength of 492 nm. Briefly, put 300 μL of the solution into a 96-well plate. Each sample has 3 duplicates. Measure the optical density (OD) value at a wavelength of 492 nm. The TTX concentration in the solution was measured by ELISA.
The encapsulation efficiency was calculated using Equation 5.
| (5) |
Determination of in vitro drug release.
20 μg of sodium fluorescein or 10 μg of TTX was dissolved in 1 mL of PBS and then formulated with 200 mg of the polymer under vortex at the speed of 3000 rpm until the polymer was emulsified thoroughly. Drug release was performed by directly injecting the mixture through a 23 G needle into the bottom of a 20-mL sample vial which contained 19 mL of PBS buffer prewarmed at 37°C. At each time point (30 min, 1h, 2h, 4h, 8h, 12h, 24h), 1 mL of the supernatant was taken for further analysis and another 1 mL of fresh, prewarmed PBS was refilled into the vial. The supernatant was filtered through a 0.22-μm pore size disposable syringe filter. Then, the SF concentration in the solution was measured with an absorbance microplate reader (EMax Plus, Molecular Devices) at a wavelength of 492 nm. The TTX concentration in the solution was measured by ELISA.
The encapsulation efficiency was calculated using Equation 6.
| (6) |
*C(drug) is the concentration of sodium fluorescein or TTX determined at each time point.
Animal studies.
Animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee at University of Alabama (Protocol ID: 19-11-2992). Adult male Sprague-Dawley rats weighing 250–350 g (i.e. adult rats over 70 days old, Charles River Laboratories, Wilmington, MA, USA) were housed in groups under a 12 h/12 h light/dark cycle. Prior to injections, rats were anesthetized briefly with isoflurane by facemask. The injection was conducted by introducing a 23 G × 3/4” needle posteromedial to the greater trochanter, pointing in an anteromedial direction. Once the bone was contacted 0.3 mL of test solution was injected. The left leg was used for blocks; the right served as a control, particularly for systemic toxicity. Neurobehavioral testing was performed at predetermined intervals. Rats were observed as to whether they developed seizures, respiratory distress, and terminal apnea. Any signs of distress generally resulted in euthanasia of the animals.
Sensory nerve blockade was assessed by a modified hotplate test. Briefly, the time that a rat will leave its hindpaw on a hot plate (Model 39D Hot Plate Analgesia Meter, IITC Inc., Woodland Hills, CA) at 56°C was measured by a stopwatch (this time is referred to as thermal latency). The paw was removed from the hotplate after 12 seconds by the investigator to avoid harm to the rat. Measurements were repeated three times at each time point and the median was used for data analysis.
Motor function was tested by measuring Extensor Postural Thrust. The rat was held above a digital balance and allowed to bear weight on one hindpaw at a time. The maximum weight that the rat can bear without its ankle touching the balance was measured. Measurements were repeated three times at each time point and the median was used for data analysis.
The duration of block of sensory nerve blockade is the time required for latency to return to a value of 7 seconds (baseline latency is approximately 2 seconds; thus 7 seconds represents the midpoint between maximal latency, which is set at a cutoff of 12 seconds, and baseline). The duration of motor block was defined as the time until 50% recovery of weight bearing.
Tissue harvesting and histology.
Rats were sacrificed at 4 days (acute inflammation) or 14 days (chronic inflammation) or 28 days (chronic inflammation) after the injection, and the sciatic nerve was harvested together with surrounding tissues. Muscle samples were fixed in 10% neutral buffered formalin and processed for histology (hematoxylin-eosin stained slides) using standard techniques. Muscle samples were stained with Hematoxylin and eosin, and then scored for inflammation (0–4 points) and myotoxicity (0–6 points)[31, 60]. The inflammation score was a subjective assessment of severity (0: no inflammation, 1: peripheral inflammation, 2: deep inflammation, 3: muscular hemifascicular inflammation, 4: muscular holofascicular inflammation). The myotoxicity score reflected two characteristic features of local anesthetic myotoxicity: nuclear internalization and regeneration. Nuclear internalization is characterized by myocytes normal in size and chromaticity, but with nuclei located away from their usual location at the periphery of the cell. Regeneration is characterized by shrunken myocytes with basophilic cytoplasm. Scoring was as follows: 0. normal; 1. perifascicular internalization; 2. deep internalization (>5 cell layers), 3. perifascicular regeneration, 4. deep regeneration, 5. hemifascicular regeneration, 6. holofascicular regeneration.
To evaluate the neurotoxicity, the sciatic nerve samples were processed and fixed in Karnovsky’s KII Solution (2.5 % glutaraldehyde, 2.0 % paraformaldehyde, 0.025 % calcium chloride in 0.1 M cacodylate buffer, pH 7.4). Samples were treated with osmium tetroxide for post-fixation for 2 hours and were subsequently dehydrated in graded acetone solutions for 10 min each (30%, 60%, 90%, 100%, 100%, 100%). Then, the nerves were infiltrated with acetone/Epon resin mixtures (1:1, 1:2, 0:1). After being sectioned by a diamond knife through an ultramicrotome, nerve sections of 1 μm were stained with toluidine blue, followed by light microscopy. Slides were analyzed by an observer blinded to the nature of individual samples.
To evaluate the distribution of PPG-HPG-FITC urethane, the sciatic nerves were harvested and embedded into the OCT compound and frozen sections were prepared. A coverslip was placed, and the slides were imaged by confocal laser scanning microscopy (Leica TCS SP2 AOBS).
Statistical Analysis.
Data are presented as means ± SDs (n = 3 in drug encapsulation and release kinetics, n = 4 in neurobehavioral studies). The statistical differences between groups were tested by one-way analysis of variance (ANOVA) for multiple comparisons. The inflammation scores and myotoxicity scores were reported as medians (n= 2–5 per experimental group) and were compared using the Mann-Whitney U test. This method was selected because the data were ordinal in nature. Statistical analyses were performed with Origin 2021 software (Origin Lab Corp. Northampton, MA). Statistical significance was defined as a p< 0.05.
Supplementary Material
Figure 5.
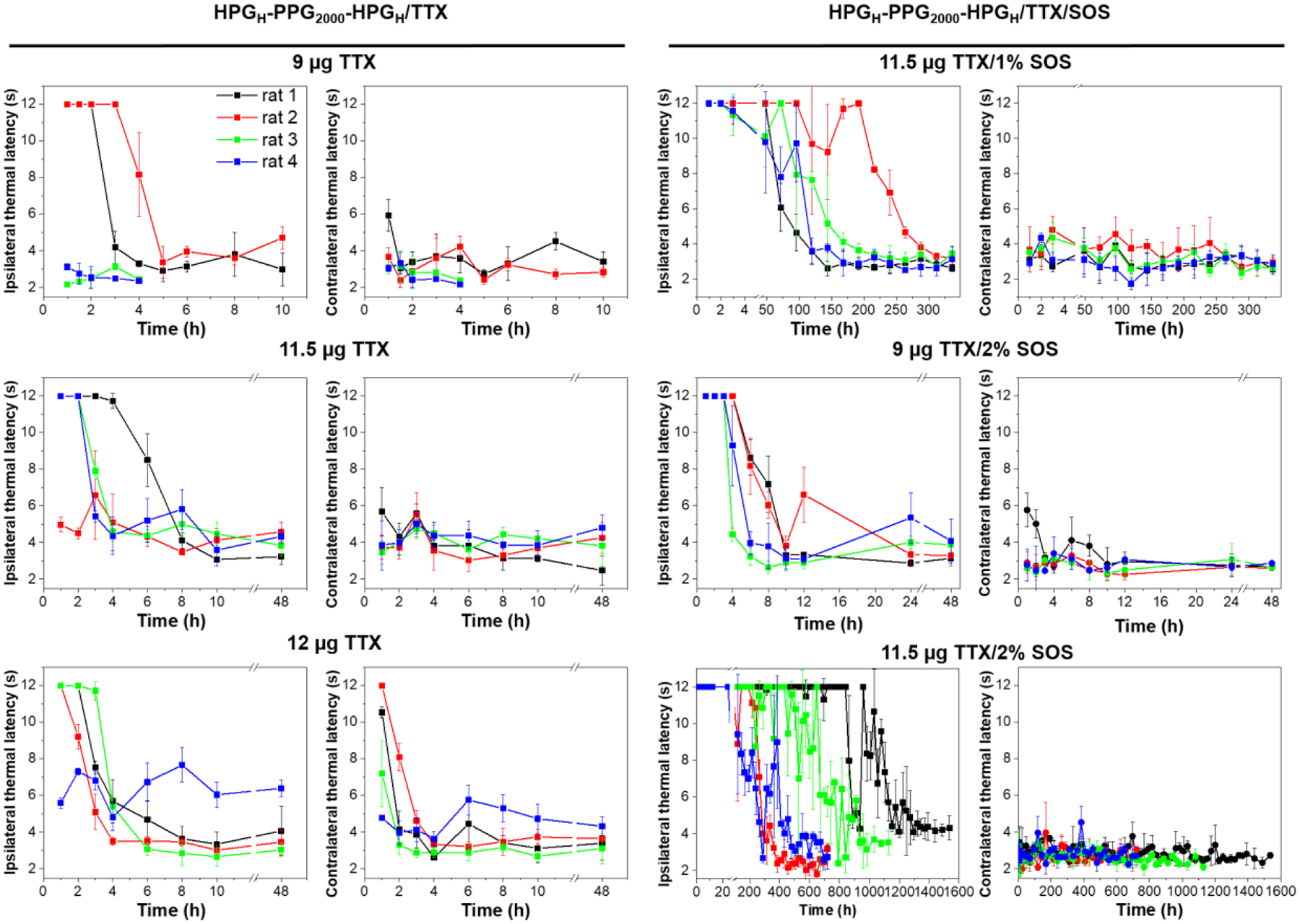
Neurobehavioral assessment of rats that received a single sciatic nerve injection of 0.3 mL PBS containing 60 mg HPG-PPG-HPG polymer and TTX and/or SOS. Ipsilateral and contralateral paw thermal latency to thermal stimulation. n = 4. Data presented are the mean ± SD of the respective treatment groups.
Acknowledgements
Research reported in this publication was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number R15GM139193. This research was partially supported by the University of Alabama’s Small Grant Program Awards (GR14901 and GR14972). U.W. acknowledges support for her pain research program at the University of Alabama at Birmingham through the William A. Lell, M.D. – Paul N. Samuelson, M.D. Endowed Professorship in Anesthesiology.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. U.W. reports research grants from the US National Institutes of Health. She serves on the External Consultant Board for the “NIH Preclinical Screening Platform for Pain”, a novel pre-clinical pain therapy screening platform that has been launched at the National Institute for Neurological Disorders and Stroke in the U.S. In her capacity as a special government employee of the US Food and Drug Administration (FDA), she has served as a voting member of the FDA Anesthetic and Analgesic Drug Products Advisory Committee. She serves as a consultant for Aphrodite Health Inc., Wilmington, DE, Avenue Therapeutics Inc., New York, NY, Bayer Aktiengesellschaft, Leverkusen, Germany, and Biohaven Pharmaceuticals, New Haven, CT, all unrelated to the submitted work
References
- [1].Joshi GP, Bonnet F, Shah R, Wilkinson RC, Camu F, Fischer B, Neugebauer EA, Rawal N, Schug SA, Simanski C, Kehlet H, A systematic review of randomized trials evaluating regional techniques for postthoracotomy analgesia, Anesth Analg 107(3) (2008) 1026–40. [DOI] [PubMed] [Google Scholar]
- [2].Cooper SA, Desjardins PJ, Turk DC, Dworkin RH, Katz NP, Kehlet H, Ballantyne JC, Burke LB, Carragee E, Cowan P, Croll S, Dionne RA, Farrar JT, Gilron I, Gordon DB, Iyengar S, Jay GW, Kalso EA, Kerns RD, McDermott MP, Raja SN, Rappaport BA, Rauschkolb C, Royal MA, Segerdahl M, Stauffer JW, Todd KH, Vanhove GF, Wallace MS, West C, White RE, Wu C, Research design considerations for single-dose analgesic clinical trials in acute pain: IMMPACT recommendations, Pain 157(2) (2016) 288–301. [DOI] [PubMed] [Google Scholar]
- [3].Smith CR, Baharloo R, Nickerson P, Wallace M, Zou B, Fillingim RB, Crispen P, Parvataneni H, Gray C, Prieto H, Machuca T, Hughes S, Murad G, Rashidi P, Tighe PJ, Predicting long-term postsurgical pain by examining the evolution of acute pain, European journal of pain (London, England) 25(3) (2021) 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pak DJ, Yong RJ, Kaye AD, Urman RD, Chronification of Pain: Mechanisms, Current Understanding, and Clinical Implications, Current pain and headache reports 22(2) (2018) 9. [DOI] [PubMed] [Google Scholar]
- [5].Treede R-D, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, Cohen M, Evers S, Finnerup NB, First MB, Giamberardino MA, Kaasa S, Kosek E, Lavand’homme P, Nicholas M, Perrot S, Scholz J, Schug S, Smith BH, Svensson P, Vlaeyen JWS, Wang S-J, A classification of chronic pain for ICD-11, Pain 156(6) (2015) 1003–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Macrae WA, Chronic post-surgical pain: 10 years on, Br J Anaesth 101(1) (2008) 77–86. [DOI] [PubMed] [Google Scholar]
- [7].Glare P, Aubrey KR, Myles PS, Transition from acute to chronic pain after surgery, Lancet (London, England) 393(10180) (2019) 1537–1546. [DOI] [PubMed] [Google Scholar]
- [8].Lavand’homme P, Transition from acute to chronic pain after surgery, Pain 158 Suppl 1 (2017) S50–s54. [DOI] [PubMed] [Google Scholar]
- [9].Weiser TG, Haynes AB, Molina G, Lipsitz SR, Esquivel MM, Uribe-Leitz T, Fu R, Azad T, Chao TE, Berry WR, Gawande AA, Size and distribution of the global volume of surgery in 2012, Bulletin of the World Health Organization 94(3) (2016) 201–209f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rosenblum A, Marsch LA, Joseph H, Portenoy RK, Opioids and the treatment of chronic pain: controversies, current status, and future directions, Exp Clin Psychopharmacol 16(5) (2008) 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baker DW, History of The Joint Commission’s Pain Standards: Lessons for Today’s Prescription Opioid Epidemic, Jama 317(11) (2017) 1117–1118. [DOI] [PubMed] [Google Scholar]
- [12].Hah JM, Bateman BT, Ratliff J, Curtin C, Sun E, Chronic Opioid Use After Surgery: Implications for Perioperative Management in the Face of the Opioid Epidemic, Anesth Analg 125(5) (2017) 1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pathan H, Williams J, Basic opioid pharmacology: an update, Br J Pain 6(1) (2012) 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schaefer CP, Tome ME, Davis TP, The opioid epidemic: a central role for the blood brain barrier in opioid analgesia and abuse, Fluids Barriers CNS 14(1) (2017) 32–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhao S, Chen F, Feng A, Han W, Zhang Y, Risk Factors and Prevention Strategies for Postoperative Opioid Abuse, Pain Research and Management 2019 (2019) 7490801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu CL, King AB, Geiger TM, Grant MC, Grocott MPW, Gupta R, Hah JM, Miller TE, Shaw AD, Gan TJ, Thacker JKM, Mythen MG, McEvoy MD, American Society for Enhanced Recovery and Perioperative Quality Initiative Joint Consensus Statement on Perioperative Opioid Minimization in Opioid-Naïve Patients, Anesth Analg 129(2) (2019) 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kowalski C, Ridenour R, McNutt S, Ba D, Liu G, Bible J, Aynardi M, Garner M, Leslie D, Dhawan A, Risk Factors For Prolonged Opioid Use After Spine Surgery, Global Spine Journal (2021) 21925682211003854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kohane DS, Smith SE, Louis DN, Colombo G, Ghoroghchian P, Hunfeld NGM, Berde CB, Langer R, Prolonged duration local anesthesia from tetrodotoxin-enhanced local anesthetic microspheres, Pain 104(1) (2003) 415–421. [DOI] [PubMed] [Google Scholar]
- [19].Kohane DS, Sankar WN, Shubina M, Hu D, Rifai N, Berde CB, Sciatic nerve blockade in infant, adolescent, and adult rats: a comparison of ropivacaine with bupivacaine, Anesthesiology 89(5) (1998) 1199–208. [DOI] [PubMed] [Google Scholar]
- [20].Kohane DS, Lipp M, Kinney RC, Lotan N, Langer R, Sciatic Nerve Blockade with Lipid-Protein-Sugar Particles Containing Bupivacaine, Pharmaceutical Research 17(10) (2000) 1243–1249. [DOI] [PubMed] [Google Scholar]
- [21].Kohane DS, Berde CB, Strichartz G, Langer RS, Local anesthetic formulations, Google Patents, 2001. [Google Scholar]
- [22].Padera R, Bellas E, Tse JY, Hao D, Kohane DS, Local myotoxicity from sustained release of bupivacaine from microparticles, Anesthesiology 108(5) (2008) 921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].PERE P, WAHLSTRÖM T, WATANABE H, ROSENBERG PH, PITKÄNEN M, Local Myotoxicity of Bupivacaine in Rabbits After Continuous Supraclavicular Brachial Plexus Block, Regional Anesthesia and Pain Medicine 18(5) (1993) 304–307. [PubMed] [Google Scholar]
- [24].Jia X, Colombo G, Padera R, Langer R, Kohane DS, Prolongation of sciatic nerve blockade by in situ cross-linked hyaluronic acid, Biomaterials 25(19) (2004) 4797–4804. [DOI] [PubMed] [Google Scholar]
- [25].Ilfeld BM, Eisenach JC, Gabriel RA, Clinical Effectiveness of Liposomal Bupivacaine Administered by Infiltration or Peripheral Nerve Block to Treat Postoperative Pain, Anesthesiology 134(2) (2021) 283–344. [DOI] [PubMed] [Google Scholar]
- [26].Gabriel RA, Ilfeld BM, Peripheral nerve blocks for postoperative analgesia: From traditional unencapsulated local anesthetic to liposomes, cryoneurolysis and peripheral nerve stimulation, Best practice & research. Clinical anaesthesiology 33(3) (2019) 293–302. [DOI] [PubMed] [Google Scholar]
- [27].Lahaye LA, Butterworth IVJF, Site-1 Sodium Channel Blockers as Local AnestheticsWill Neosaxitoxin Supplant the Need for Continuous Nerve Blocks?, Anesthesiology 123(4) (2015) 741–742. [DOI] [PubMed] [Google Scholar]
- [28].Kohane DS, Lu NT, Gökgöl-Kline AC, Shubina M, Kuang Y, Hall S, Strichartz GR, Berde CB, The local anesthetic properties and toxicity of saxitonin homologues for rat sciatic nerve block in vivo, Regional Anesthesia and Pain Medicine 25(1) (2000) 52–59. [DOI] [PubMed] [Google Scholar]
- [29].Hagen NA, Cantin L, Constant J, Haller T, Blaise G, Ong-Lam M, du Souich P, Korz W, Lapointe B, Tetrodotoxin for Moderate to Severe Cancer-Related Pain: A Multicentre, Randomized, Double-Blind, Placebo-Controlled, Parallel-Design Trial, Pain research & management 2017 (2017) 7212713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kohane Daniel S., Yieh J, Lu Nu T., Langer R, Strichartz Gary R., Berde Charles B., A Re-examination of Tetrodotoxin for Prolonged Duration Local Anesthesia Anesthesiology 89(1) (1998) 119–131. [DOI] [PubMed] [Google Scholar]
- [31].Zhao C, Liu A, Santamaria CM, Shomorony A, Ji T, Wei T, Gordon A, Elofsson H, Mehta M, Yang R, Kohane DS, Polymer-tetrodotoxin conjugates to induce prolonged duration local anesthesia with minimal toxicity, Nat Commun 10(1) (2019) 2566–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li X, Zhao Y, Zhao C, Applications of capillary action in drug delivery, iScience 24(7) (2021) 102810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li X, Li Q, Zhao C, Zero-Order Controlled Release of Water-Soluble Drugs Using a Marker Pen Platform, ACS Omega 6(21) (2021) 13774–13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li Q, Li X, Zhao C, Strategies to Obtain Encapsulation and Controlled Release of Small Hydrophilic Molecules, Front Bioeng Biotechnol 8 (2020) 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhan C, Wang W, Santamaria C, Wang B, Rwei A, Timko BP, Kohane DS, Ultrasensitive Phototriggered Local Anesthesia, Nano Letters 17(2) (2017) 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rwei A, Lee J-J, Zhan C, Liu Q, Ok M, Shankarappa S, Langer R, Kohane D, Repeatable and adjustable on-demand sciatic nerve block with phototriggerable liposomes, Proceedings of the National Academy of Sciences of the United States of America 112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rwei AY, Paris JL, Wang B, Wang W, Axon CD, Vallet-Regí M, Langer R, Kohane DS, Ultrasound-triggered local anaesthesia, Nature biomedical engineering 1 (2017) 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu Q, Santamaria CM, Wei T, Zhao C, Ji T, Yang T, Shomorony A, Wang BY, Kohane DS, Hollow Silica Nanoparticles Penetrate the Peripheral Nerve and Enhance the Nerve Blockade from Tetrodotoxin, Nano Letters 18(1) (2018) 32–37. [DOI] [PubMed] [Google Scholar]
- [39].Joshi G, Gandhi K, Shah N, Gadsden J, Corman SL, Peripheral nerve blocks in the management of postoperative pain: challenges and opportunities, Journal of Clinical Anesthesia 35 (2016) 524–529. [DOI] [PubMed] [Google Scholar]
- [40].Gehman SD, Relationship between Molecular Structure and Physical Properties, Industrial & Engineering Chemistry 44(4) (1952) 730–739. [Google Scholar]
- [41].Nunes RW, Martin JR, Johnson JF, Influence of molecular weight and molecular weight distribution on mechanical properties of polymers, Polymer Engineering & Science 22(4) (1982) 205–228. [Google Scholar]
- [42].Final Report on the Safety Assessment of Propylene Glycol and Polypropylene Glycols, Journal of the American College of Toxicology 13(6) (1994) 437–491. [Google Scholar]
- [43].Deng Y, Saucier-Sawyer JK, Hoimes CJ, Zhang J, Seo Y-E, Andrejecsk JW, Saltzman WM, The effect of hyperbranched polyglycerol coatings on drug delivery using degradable polymer nanoparticles, Biomaterials 35(24) (2014) 6595–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shomorony A, Santamaria CM, Zhao C, Rwei AY, Mehta M, Zurakowski D, Kohane DS, Prolonged Duration Local Anesthesia by Combined Delivery of Capsaicin- and Tetrodotoxin-Loaded Liposomes, Anesthesia & Analgesia 129(3) (2019). [DOI] [PubMed] [Google Scholar]
- [45].Simons EJ, Bellas E, Lawlor MW, Kohane DS, Effect of Chemical Permeation Enhancers on Nerve Blockade, Molecular Pharmaceutics 6(1) (2009) 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Farber SJ, Saheb-Al-Zamani M, Zieske L, Laurido-Soto O, Bery A, Hunter D, Johnson P, Mackinnon SE, Peripheral nerve injury after local anesthetic injection, Anesth Analg 117(3) (2013) 731–739. [DOI] [PubMed] [Google Scholar]
- [47].Selander D, Neurotoxicity of local anesthetics: animal data, Regional anesthesia 18(6 Suppl) (1993) 461–8. [PubMed] [Google Scholar]
- [48].Marsden HR, Gabrielli L, Kros A, Rapid preparation of polymersomes by a water addition/solvent evaporation method, Polymer Chemistry 1(9) (2010) 1512–1518. [Google Scholar]
- [49].Carugo D, Bottaro E, Owen J, Stride E, Nastruzzi C, Liposome production by microfluidics: potential and limiting factors, Sci Rep 6(1) (2016) 25876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Huang X, Liu C, Kong N, Xiao Y, Yurdagul A Jr., Tabas I, Tao W, Synthesis of siRNA nanoparticles to silence plaque-destabilizing gene in atherosclerotic lesional macrophages, Nature protocols 17(3) (2022) 748–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kong N, Zhang R, Wu G, Sui X, Wang J, Kim NY, Blake S, De D, Xie T, Cao Y, Tao W, Intravesical delivery of KDM6A-mRNA via mucoadhesive nanoparticles inhibits the metastasis of bladder cancer, Proceedings of the National Academy of Sciences 119(7) (2022) e2112696119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rane AV, Kanny K, Abitha VK, Thomas S, Chapter 5 - Methods for Synthesis of Nanoparticles and Fabrication of Nanocomposites, in: Mohan Bhagyaraj S, Oluwafemi OS, Kalarikkal N, Thomas S (Eds.), Synthesis of Inorganic Nanomaterials, Woodhead Publishing; 2018, pp. 121–139. [Google Scholar]
- [53].Hayashi Y, Inoue M, Takizawa H, Suganuma K, Nanoparticle Fabrication, 2008, pp. 109–120.
- [54].Ouyang J, Ji X, Zhang X, Feng C, Tang Z, Kong N, Xie A, Wang J, Sui X, Deng L, Liu Y, Kim JS, Cao Y, Tao W, In situ sprayed NIR-responsive, analgesic black phosphorus-based gel for diabetic ulcer treatment, Proceedings of the National Academy of Sciences 117(46) (2020) 28667–28677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Delattre C, Louis F, Akashi M, Matsusaki M, Michaud P, Pierre G, Fabrication Methods of Sustainable Hydrogels, in: Inamuddin, Thomas S, Kumar Mishra R, Asiri AM (Eds.), Sustainable Polymer Composites and Nanocomposites, Springer International Publishing, Cham, 2019, pp. 355–386. [Google Scholar]
- [56].Zhao C, Li X, Li L, Cheng G, Gong X, Zheng J, Dual Functionality of Antimicrobial and Antifouling of Poly(N-hydroxyethylacrylamide)/Salicylate Hydrogels, Langmuir 29(5) (2013) 1517–1524. [DOI] [PubMed] [Google Scholar]
- [57].Campea MA, Majcher MJ, Lofts A, Hoare T, A Review of Design and Fabrication Methods for Nanoparticle Network Hydrogels for Biomedical, Environmental, and Industrial Applications, Advanced Functional Materials 31(33) (2021) 2102355. [Google Scholar]
- [58].Li J, Mooney DJ, Designing hydrogels for controlled drug delivery, Nature Reviews Materials 1(12) (2016) 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rwei AY, Sherburne RT, Zurakowski D, Wang B, Kohane DS, Prolonged Duration Local Anesthesia Using Liposomal Bupivacaine Combined With Liposomal Dexamethasone and Dexmedetomidine, Anesth Analg 126(4) (2018) 1170–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].McAlvin JB, Padera RF, Shankarappa SA, Reznor G, Kwon AH, Chiang HH, Yang J, Kohane DS, Multivesicular liposomal bupivacaine at the sciatic nerve, Biomaterials 35(15) (2014) 4557–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hagen NA, Cantin L, Constant J, Haller T, Blaise G, Ong-Lam M, du Souich P, Korz W, Lapointe B, Tetrodotoxin for Moderate to Severe Cancer-Related Pain: A Multicentre, Randomized, Double-Blind, Placebo-Controlled, Parallel-Design Trial, Pain Research and Management 2017 (2017) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nader A, Kendall MC, De Oliveira GS Jr., Puri L, Tureanu L, Brodskaia A, Asher Y, Parimi V, McCarthy RJ, A dose-ranging study of 0.5% bupivacaine or ropivacaine on the success and duration of the ultrasound-guided, nerve-stimulator-assisted sciatic nerve block: a double-blind, randomized clinical trial, Regional anesthesia and pain medicine 38(6) (2013) 492–502. [DOI] [PubMed] [Google Scholar]
- [63].Liu P, Chen G, Zhang J, A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives, Molecules 27(4) (2022) 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sandhu HK, Miller CC III, Tanaka A, Estrera AL, Charlton-Ouw KM, Effectiveness of Standard Local Anesthetic Bupivacaine and Liposomal Bupivacaine for Postoperative Pain Control in Patients Undergoing Truncal Incisions: A Randomized Clinical Trial, JAMA network open 4(3) (2021) e210753–e210753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hejbøl EK, Sellathurai J, Nair PD, Schrøder HD, Injectable scaffold materials differ in their cell instructive effects on primary human myoblasts, Journal of Tissue Engineering 8 (2017) 2041731417717677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhao C, Tian S, Liu Q, Xiu K, Lei I, Wang Z, Ma PX, Biodegradable Nanofibrous Temperature-Responsive Gelling Microspheres for Heart Regeneration, Advanced Functional Materials 30(21) (2020) 2000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ferrari R, Sponchioni M, Morbidelli M, Moscatelli D, Polymer nanoparticles for the intravenous delivery of anticancer drugs: the checkpoints on the road from the synthesis to clinical translation, Nanoscale 10(48) (2018) 22701–22719. [DOI] [PubMed] [Google Scholar]
- [68].Sunder A, Hanselmann R, Frey H, Mulhaupt R, Controlled synthesis of hyperbranched polyglycerols by ring-opening multibranching polymerization, Macromolecules 32(13) (1999) 4240–4246. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.