

Abstract
Aggregation of otherwise soluble proteins into amyloid structures is a hallmark of many disorders, such as Alzheimer’s and Parkinson’s disease. There is an increasing evidence that the small aggregations, instead of ordered fibrillar aggregates, are the main structures causing toxicity. However, the studies on the small aggregation phase are limited due to the variety of structures and the complexity of the physiological environment. Here, we showed an engineered co-assembling oppositely charged amyloid-like peptide pair ([II]) as a simple tool to establish methodologies to study the mechanism and kinetics of aggregation and relate its aggregation to toxicity. The toxicity mechanism of [II] is through cell membrane damage and stress, shown with YAP and eIF2α, as in the amyloid protein-initiated diseases. Albumin is demonstrated as an extrinsic and physiologically relevant molecule in controlling the aggregation lag time and toxicity of [II]. This study represents a molecular engineering strategy to create simplistic molecular tools for establishing methodologies to study the aggregation process and kinetics of amyloid-like proteins in various conditions. Understanding the nature of protein aggregation kinetics and linking them to their biological functions through engineered peptides paves the way for future designs and drug development applications.
Keywords: aggregation kinetics, membrane damage, peptide co-assembly
1 |. INTRODUCTION
Most proteins and peptides (if not all) aggregate into amyloids, these cross β-sheet fibrillar structures are linked to numerous diseases, such as Alzheimer’s and Parkinson’s.1,2 In Alzheimer’s Disease (AD), although for many decades the accumulation of amyloid plaques was associated with the pathology of the disease, increasing evidence suggests that the misfolded protein aggregates, oligomers, are soluble and form in the process of fibrillization, are the leading cause of the toxicity.3–6 Part of their toxicity process occurs through the interactions of oligomers of amyloid-β or α-synuclein with the lipid membranes of cells, which initiates damage.7,8 The membrane damage often induces immunogenic cell death (ICD) and inflammation.9–11 The aggregation and oligomer formation process is complex because of the convoluted conditions in the body. Dynamic conditions create transient heterogeneous misfolding of oligomers and make the molecular level of understanding, quantifying, and correlating the mechanism of aggregation and toxicity; thus, the development of the target drug can be challenging.6,12 Therefore, a minimalist and simplified synthetic model that mimics the structure, aggregation, and function of amyloids can serve as a powerful tool for studying their mechanism of aggregation.
The molecular events that occur during the formation of small aggregations of proteins and peptides (lag phase) are in a complex environment. Studies with the extracted proteins in an isolated system may not provide an adequate understanding of the pathophysiology of the disease. Yet, studies in biological fluids create huge complexity. Indeed, the amyloidogenic proteins’ kinetics and free energy landscape in the initial aggregation phase are largely unknown and have been studied with variations of new experimental and computational approaches.13–16 A combination of these methodologies establishes strategies for a more accurate understanding of the aggregation process. Such a combination can be possible with molecular tools that have similar behaviors and biological functions in experimental conditions as amyloid-like proteins and are simple enough for computational simulations.17
Co-assembly of oppositely charged peptides (CoOP) is a minimalist strategy enabling the study of intermolecular interactions’ role in peptide aggregation.17 CoOPs are designed to assemble into amyloid fibrils. Among the studied peptide sequences; the oppositely charged pair, KFFIIK and EFFIIE (the pair will be shown hereafter as [II]) led to the most ordered amyloid-like β-sheet assembly. On the cell membrane, aggregation of [II] peptides causes immunogenic rupture (PAIIR), which starts immunogenic cell death (ICD) and results in the release of damage-associated molecular patterns (DAMPs), which act as an immunostimulatory agent.18 PAIIR induces secondary pyroptosis, a programmed ICD, and a common feature of amyloid-inducing cell death as in Alzheimer’s and Parkinson’s diseases.19,20 PAIIR is a simplified peptide-based tool designed to mimic the function instead of the sequence of a protein; thus, there is no natural ortholog of [II].
Albumin is an abundant protein in the human body, flexible, and mostly hydrophobic; thus, it has a high nonspecific binding capacity.21 It is known that albumin impedes the fibrillization of amyloids and increases the soluble misfolded oligomers of higher toxicity.22–24 Akin to amyloid, co-aggregation with albumin creates a soluble oligomeric form of [II] that is more toxic than its fibril form. This study shows control over the aggregation kinetics and mechanism of [II] via albumin. The simplicity of [II] provides details of the characterization of aggregates and their interactions with lipid membranes. The lipid membrane interaction is shown with liposomes and fluorescent tagged peptides. The soluble form of peptides misfolded with albumin integrates into the lipid membrane of liposomes, while the ordered fibril structures do not. The kinetics of the aggregations are studied with spectroscopic methods with fluorescent probes. The morphology of misfolded oligomeric to ordered fibrillar structures is analyzed by dynamic light scattering (DLS) and atomic force microscopy (AFM). The oligomeric form of [II] misfolded with albumin results in membrane damage and cellular cytotoxicity, whereas the fibrillar [II] without albumin does not; a similar trend of amyloid proteins in Alzheimer and Parkinson’s diseases. By delaying the formation of thermodynamically favorable ordered fibrils via albumin or simply introducing the counterparts in different time points, the soluble free peptides can localize into the membrane before they come together. Lastly, mechanical cell stress induced by [II] is identified through phosphorylation of yes-associated protein (YAP) and eukaryotic initiation factor 2 alpha (eIF2α), similar to the mechanism of amyloid-β cytotoxicity. Importantly, we identified these stress markers in cells treated with misfolded oligomers, but not with ordered amyloid fibrillar form of [II]. Overall, [II] offers a simplistic tool for investigating the effects of physical and biological processes in experiments to understand the aggregation mechanism and function of pathological proteins with amyloid-like structures.
2 |. RESULTS AND DISCUSSION
2.1 |. Integration of [II] peptides into liposomal membrane
The membrane interaction of [II] peptides (Figure S1) was analyzed using a liposomal membrane. Liposomes are commonly used for studying interactions between amyloids and lipid membranes.25,26 The fluorescence tagged [II] peptides KFFIIK with FITC (green) and EFFIIE with rhodamine B (red) were used to determine the membrane interactions of the agarose embedded liposomes (blue, approximately 100 μm in diameter, Figure S2) (Figure 1A).27 30 min premixed [II] as in Figure 1A-i initiates the interaction of oppositely charged groups, as the charge neutralization was observed through zeta potential (Figure S3). The high affinity of the peptides for with each other prevented their disassociation and localizing on the lipid membrane of liposomes (Figure 1B).
FIGURE 1.
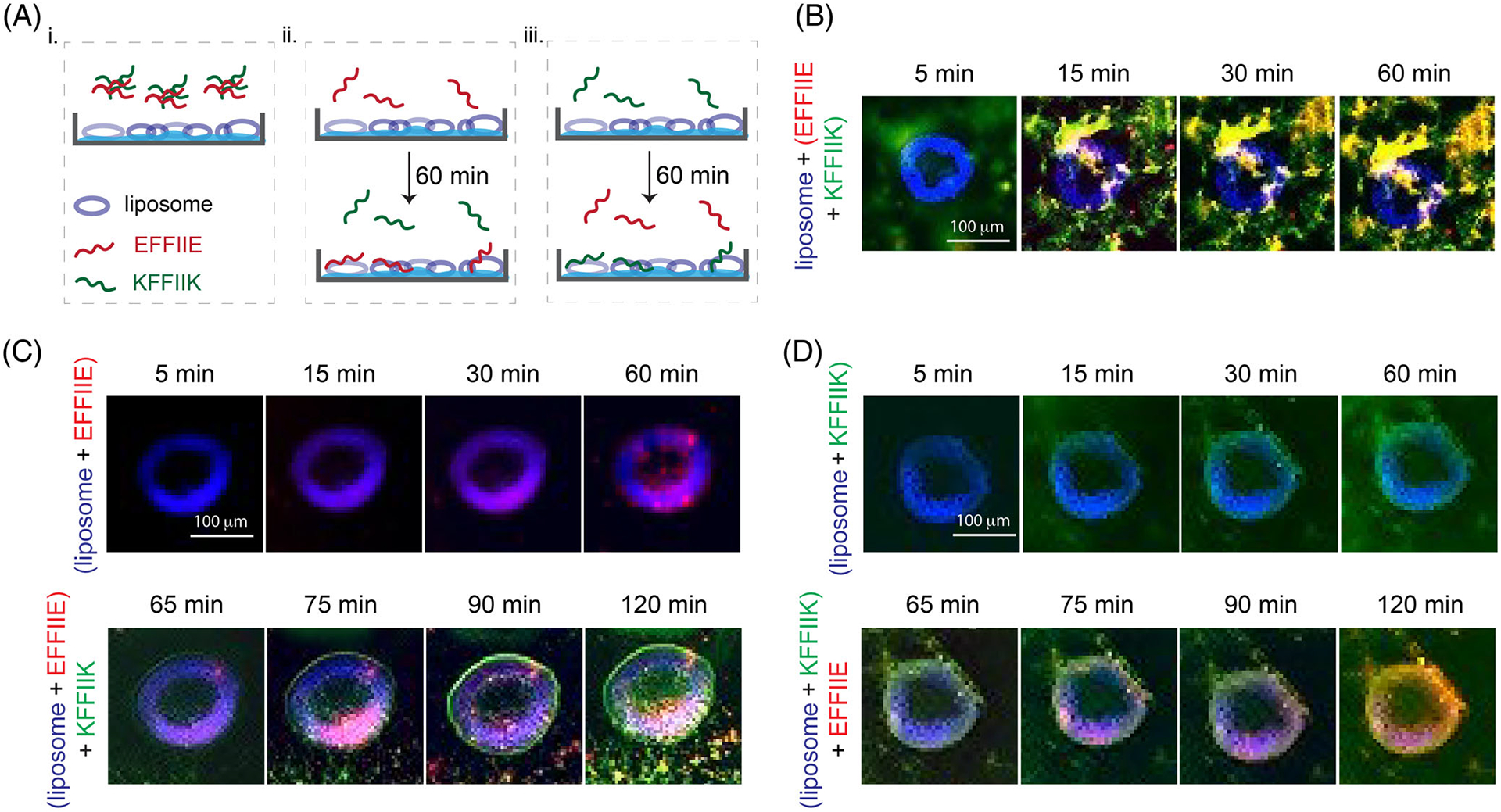
Liposome (blue) membrane integration of [II] and individual peptides; KFFIIK (green), EFFIIE (red). (A) The schematic of membrane localization experiment by using agarose embedded liposomes. (B) Membrane localization of 30 min premixed of EFFIIE + KFFIIK. (C) EFFIIE addition followed by KFFIIK and (D) KFFIIK addition followed by EFFIIE. (Scale bar = 100 μm)
Incubation of EFFIIE alone for 60 min, as in Figure 1A-ii, allowed the integration of the peptide into the cell membrane (red fluorescence on blue liposomes). Despite the charge similarities with the lipid, the hydrophobicity of the FFII group might be the reason for this integration. After introducing KFFIIK on the EFFIIE + liposome, green fluorescent (KFFIIK) increased immediately in 5 min, which shows the localized interaction of [II] on the membrane (Figure 1C). Additionally, amyloid fibril aggregation around liposomes was also observed, indicating that the remaining soluble EFFIIE interacts with its soluble counterpart peptide before integration into lipids. Similarly, incubation of KFFIIK alone, as in Figure 1A-iii, showed integration into the lipids (Figure 1D). The membrane localization of KFFIIK was faster than EFFIIE, likely because of the opposite liposome surface charge.28 Addition of EFFIIE 60 min later also showed membrane integration of both peptides and fibrillar aggregations of [II] outside the membrane.
Overall, these results demonstrated that the soluble forms of individual [II] peptides integrate into a membrane. The premixed [II] did not show peptide integration; they were stable. The membrane-binding capacity of [II] is correlated to the presence of free peptides in the medium. Controlling the aggregation kinetics of [II]—in other words, controlling the time and amount of free peptides in the medium—can control the amount of peptides integrated into the membrane.
2.2 |. Controlling peptide aggregation kinetics
The mechanism of ordered fibrillar amyloid assembly follows sigmoidal growth divided into three steps: (i) lag phase (nucleation), in which the peptides are mostly in misfolded monomeric form; (ii) growth (elongation) phase, in which the larger aggregates (misfolded or ordered) form of peptides; and (iii) final plateau in which grown aggregates (misfolded or ordered) dominate at the final equilibrium.29 Amyloid-forming proteins have hydrophobic domains which can misfold and form nonspecific interactions and disordered aggregates with the other hydrophobic molecules in the body. The stability of these aggregates depends on the affinity of each component. Because the ordered amyloid assembly of peptides has specific interactions that require a specific conformation, they are often more stable than nonspecific hydrophobicity-driven misfolded aggregates. However, the same conformational specificity creates an energy barrier. Therefore, any aggregation in the intermediate energy level, particularly when highly nonspecific, impedes the conformational order for the assembly and delays the equilibrium in the final plateau of stable fibrils. In other words, misfolding of monomers prolongates the initial lag and growth phase, where the monomeric structures are soluble and less stable.
Albumin, as a flexible hydrophobic protein, aggregates with amyloid-forming proteins, prolonging the lag phase.22–24 The unstable and soluble monomers present in the lag phase are able to integrate into the cell membrane and become more toxic.30 Although the prolonged lag time is known to be more toxic; it has been little study on the correlation between the lag time (aggregation kinetics), structures of the aggregates, and cell membrane damage. Since [II] showed amyloid-β characteristics such as highly ordered β-sheet secondary structure as observed from Amide I and II peaks (Figure S8), we hypothesized that albumin could change the aggregation behavior and fibrillar assembly kinetics of [II].17 Figure 2 shows the kinetics and morphology of [II] aggregates in increasing amount of albumin (0, 0.002, 0.02, 0.05, 0.1, 0.2, 0.5, 1, and 2 w%) in PBS. To distinguish the kinetics of ordered amyloid assembly from misfolded oligomer aggregates, we used Thioflavin T (ThT) and 1,6-diphenyl-1,3,5-hexatriene (DPH) assays, respectively. The morphology and size of the aggregates were studied with AFM and DLS.
FIGURE 2.
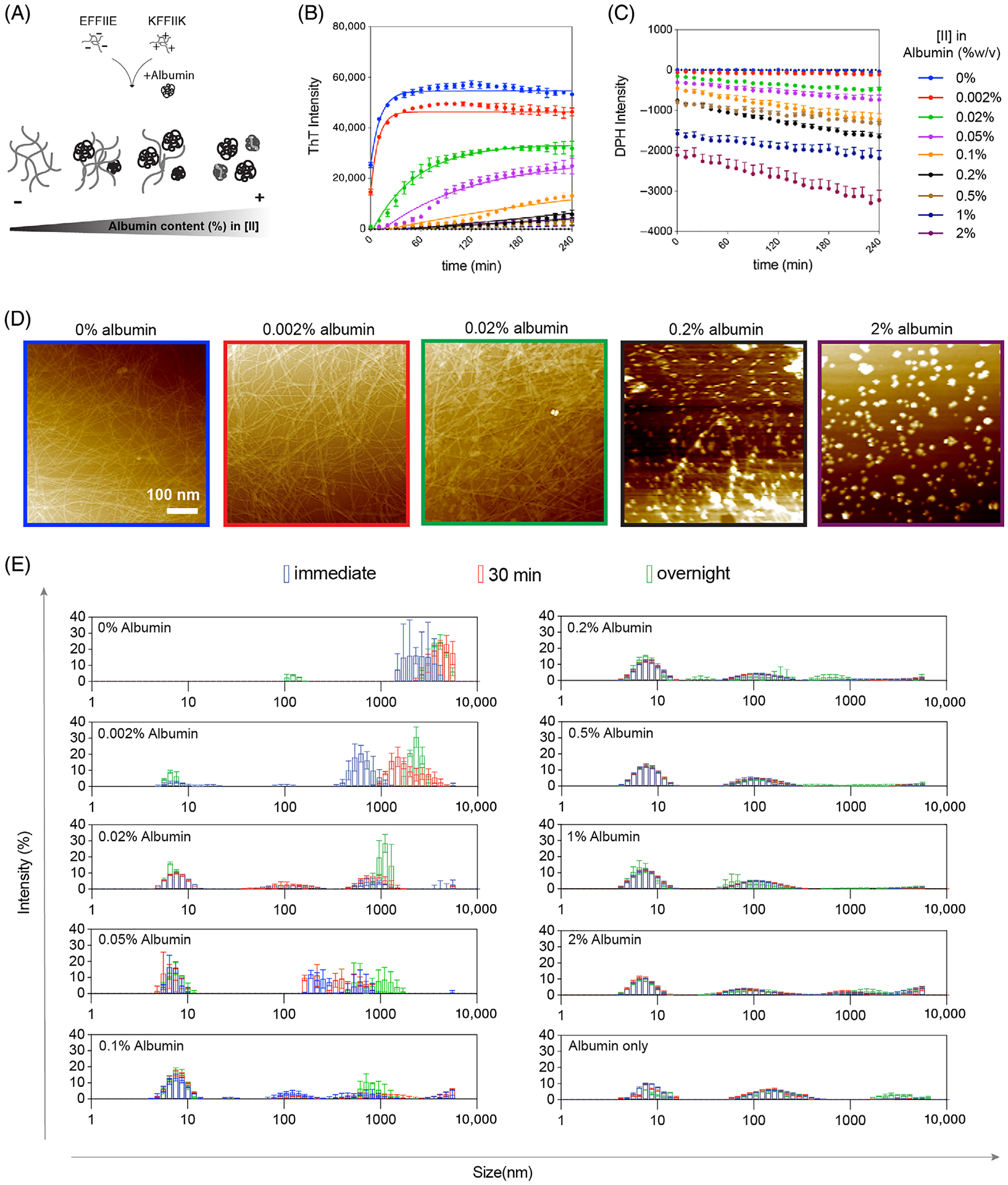
Aggregation kinetics depend on albumin concentration. (A) The schematic of [II] aggregation with albumin. Aggregation kinetics were performed with (B) ThT and (C) DPH assays in increasing albumin content. (D) AFM images of [II]-albumin structures (scale bar = 100 nm). (E) DLS measurements [II]-albumin mixtures
ThT is a fluorescent dye commonly used for β-sheet structure identification.31 The transition from oligomers to fibrils has been characterized by ThT in prion protein32; here, we apply it to [II] individual peptides prepared in PBS with increasing albumin content and constant ThT, and the fluorescence of ThT was tracked for 4 h (Figure 2B). Albumin-only groups with different w% were measured with constant ThT as the background, showing some degree of intensity as the structures have a hydrophobic core but did not increase with time (Figure S4). When mixed, [II] peptides assemble into ordered structures and achieve their final equilibrium of fibrillar form in less than 10 min.17 Fibrillar final equilibrium was only observed in the medium without albumin (0 w%) and 0.002, 0.02, 0.05, and 0.1 w% albumin in 4 h (Figure 2B). Similarly, the fibrillar structures were observed in the overnight incubated samples in these parameters with AFM (Figure 2D). Albumin-only conditions did not show any fibrillization (Figure S5). However, the addition of albumin delayed the formation of ordered amyloid structures by increasing the rate of the lag phase (nucleation) and growth (elongation) phase.33 There was also a reduction in the total amount of fibers generated as indicated by ThT fluorescence signals as shown with AFM, and no fibrillar structure was observed for 0.2 and 2 w% albumin incubated overnight (Figure 2D).
The formation of disordered structures was identified with the DPH assay; DPH is a fluorescent dye with enhanced intensity in hydrophobic environments.34 Because albumin is a hydrophobic protein and forms soluble aggregates in PBS, the initial measurements were performed with only DPH and albumin as the background (Figure S6). Each peptide was prepared in PBS with concentrations increasing in albumin anovd constant in DPH, then mixed and tracked for 4 h (Figure 2C). Decreased intensity of DPH with increasing albumin is interpreted as a decrease in the number and sizes of the aggregates. This might be due to covering the hydrophobic parts of the albumin with the charged peptides creating more soluble and smaller albumin aggregates in PBS. In the PBS with the lowest amount of albumin, 0.002 w%, albumin showed an insignificant intensity. The structures in this solution are mostly fibrillar, as observed through increasing ThT assay, indicating that the amount of disorganized aggregates was negligible. The increased albumin concentrations showed slowly lowered intensities; in other words, more change between the intensity of the [II] + albumin complexes compared to the same amount of albumin alone is because of the formation of smaller aggregates.
While the aggregation of individual peptides did not change with the addition of albumin, several observations are worth noting. When alone, KFFIIK showed slightly higher and more stable ThT intensity, which reduced but remained stable over time with increasing albumin (Figure S4). Similarly, the DPH assay showed increasing intensity over time in KFFIIK alone (Figure S7). This suggests that KFFIIK alone is not forming organized structures but instead leads to disorganized aggregates. These measurements were all performed in PBS, in which the salt ions reduce the charges of the peptides. Therefore, KFFIIK alone shows misfolded aggregation due to the hydrophobic nature of the alkyl tail of Lysine (K).17 Yet, when KFFIIK is mixed with EFFIIK, the ThT intensity changes immediately and dramatically, indicating that the peptides co-assemble in PBS (Figure 2B). The negative counterpart, EFFIIE, in PBS without albumin, does not aggregate in an ordered or disordered manner (Figures S4 and S7). It might be a result of the hydrophobic amino acids’ lower contact probability in the core of EFFIIE compared to KFFIIK and [II].17 Considering that albumin alone also has ThT and DPH intensities due to its hydrophobic nature (Figures S4 and S6), the observed consistent negative intensities of all EFFIIE and KFFIIK solutions with albumin indicated the formation of smaller and more stable aggregates; peptides interact and aggregate with albumin individually. However, when mixed, the increase of ThT intensities, or formation of peptide fibers, indicates that the individual peptide and albumin aggregations are not as favorable as peptide-to-peptide interactions. The addition of albumin to premixed [II] showed stable DPH intensity lower than that of albumin only (Figure S6, no background subtraction), possibly because albumin falls apart and forms nonspecific bindings with the fibrillar structures.
Size measurements of the aggregates were performed with DLS at three different time points: immediately, after 30 min, and after overnight incubation (Figure 2E). Without albumin, [II] aggregated immediately with >1000 nm average size with a shift toward larger sizes after 30 min (Figure 2E, 0 w% albumin). Similarly, the shift toward bigger structures was observed for the conditions between 0.002 and 0.1 w% albumin, which are the conditions where the final fibrillar equilibrium was observed with ThT. After 0.2 w%, size distribution and intensity profiles were similar to the 2 w% albumin-only group. The population of structures around 10 nm was higher in the presence of peptides compared to 2 w% albumin only, indicating smaller aggregates in the presence of peptides, and explaining the reduction in intensities observed with DPH.
Overall, the observed trend of [II] with albumin is similar to that of natural amyloids; the increasing albumin causes the decrease of the maximum ThT fluorescence intensity, or the total amount of fibrils generated, and increases soluble disordered aggregates; oligomers.
2.3 |. Delayed aggregation increased the lipid membrane integration of [II]
Among the studied albumin concentrations, 0.2 w% showed the threshold for diminishing the ordered fibril formation; hence, the rest of the analyses were performed in 0.2 w% albumin. To compare the integration on the lipid membrane of the liposomes, [II] was prepared at 0.2 w%, incubated for 30 min, then added to the liposomes (Figure 3A). Unlike the premixed [II] in 0 w% albumin (Figure 1B), the individual peptides were localized in the liposomes starting from 5 min. Albumin slows down the peptide–peptide interactions by aggregating with the membrane, and the interactions of peptides with the lipid membrane are likely to be more stable than their interactions with albumin. Incubation of individual peptides with 0.2 w% albumin for 60 min showed that KFFIIK integrates to the lipid bilayer even in 5 min, while the negative counterpart, EFFIIE, does not integrate, likely due to the charges of the lipids (Figure S9). The integration of EFFIIE when incubated with albumin and KFFIIK might be because of the initial membrane integration of soluble KFFIIK followed by the aggregation of EFFIIE, even in the lipid interface. These results indicate that without albumin, KFFIIK and EFFIIE co-assemble into fibers, which do not interfere with the integrity of the lipid membrane. In the presence of albumin, the co-assembly process is prolonged due to the aggregation of the individual peptides with albumin. The delay time (lag time or the time required to achieve equilibrium fibril phase) is correlated to the amount of albumin in the solvent.
FIGURE 3.
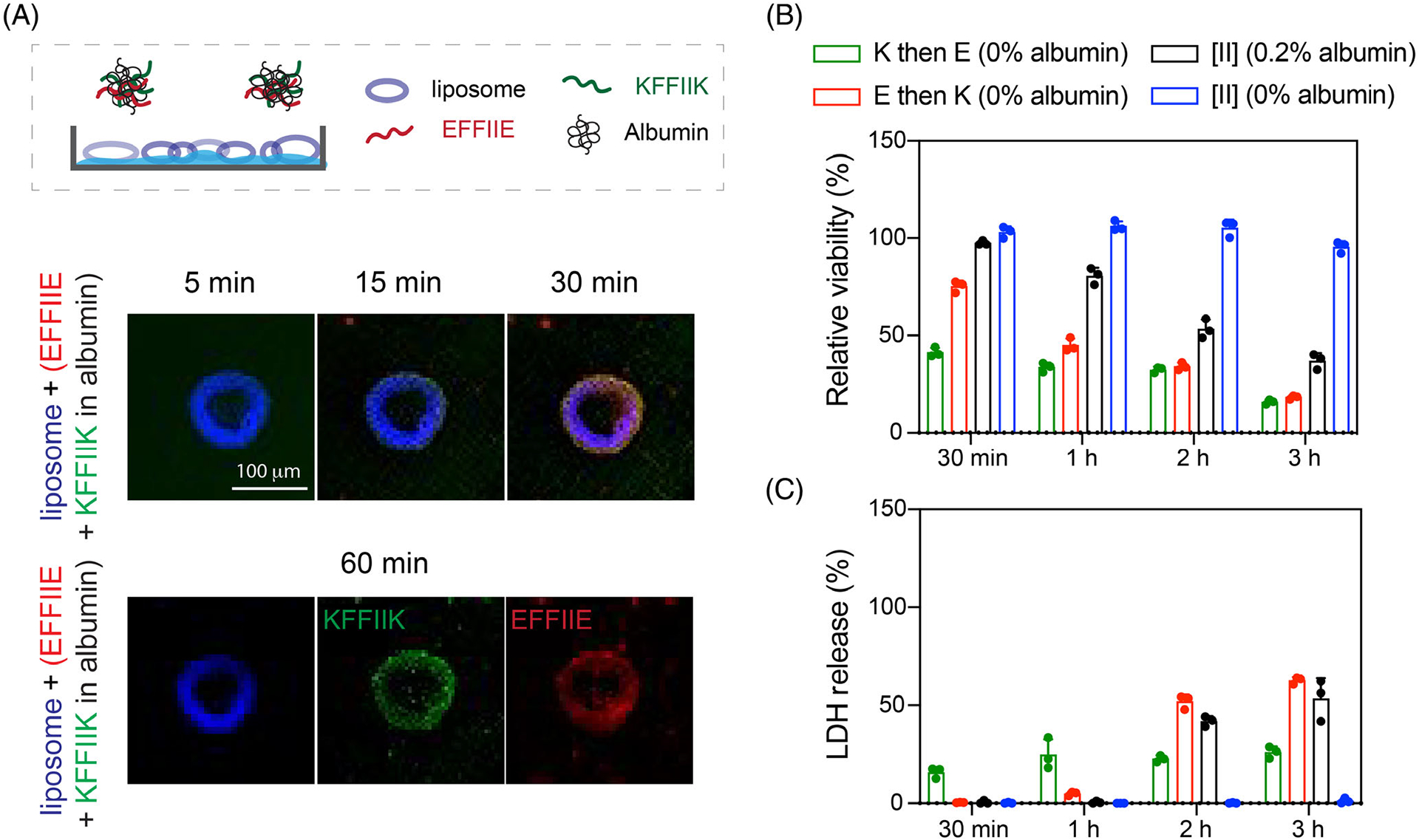
Delayed [II] aggregation increases its integration on the lipid membranes. (A) Liposomal membrane localization of 0.5 mM [II] in 0.2 w% albumin; liposome = blue, EFFIIE = red, KFFIIK = green (scale bar = 100 μm). (B) Relative viability and (C) LDH release of EMT6 cells treated with individual and aggregates with 0.5 mM [II] in 0 and 0.2 w% of albumin in PBS
The effect of enhanced lipid membrane accumulation of [II] altered its toxicity (Figure 3B,C). [II] induces immunogenic rupture of mammalian cell membranes, and initiates immunogenic cell death in 6 h in cell medium supplemented with 10 v% FBS (has app. 0.2 w% albumin) as shown before.18 During 3 h of treatment, [II] in 0 w% (without albumin) did not cause any membrane damage (Figure 3B), as [II] fibrils do not interact with the lipid membrane (Figure 1B). However, the addition of [II] with 0.2 w% albumin showed increasing membrane damage over time which is quantified by the release of lactate dehydrogenase (LDH), a cytoplasmic enzyme released upon membrane damage.35 These results correlate with ThT (high intensity, high ordered fibril assembly) and DPH (low intensity, high disordered aggregation), indicating that delayed fibril formation increases membrane damage. Furthermore, we also tested the effects of consecutively introduced peptides to cells without albumin to correlate the toxicity to the membrane integration. When KFFIIK was given first (green bar) followed by EFFIIE addition, cellular viability decreased to 50% at 30 min and reduced to 25% at 3 h (Figure 3B). The opposite scenario where EFFIIE added first and then KFFIIK (red bar) started from 75% cell viability at 30 min and decreased to 25% at 3 h. As observed from liposomal experiments (Figure 1C,D), KFFIIK accumulated faster on the membrane compared to EFFIIE. Therefore, when peptides were introduced consecutively (30 min), we observed faster cell death in K then in E condition. However, cell death was similar at 3 h regardless of the addition order (Figure 3B). These results show that the toxicity of [II] can be controlled through control over its aggregation kinetics.
2.4 |. FRET analysis for albumin and [II] interactions
FITC and TRITC are fluorescent dyes and FRET couples. When the distance of the two fluorophores is less than 8 nm, the excitation of light with 488 nm wavelength triggers FRET; emission from FITC (donor) at 520 nm excites TRITC (acceptor) that emits light at 578 nm.36 We used albumin conjugated with TRITC and incubated with FITC-KFFIIK for this analysis. Incubation of FITC-KFFIIK with 0.2 w% albumin for 30 min and 24 h, which did not show FRET, indicates that they are not in close proximity (Figure 4A). In the presence of EFFIIE (not fluorescent tagged) also in 0.2 w% albumin showed the FRET intensity, TRITC emission at 580 nm and FITC emission at 488 nm were reduced (Figure 4B), indicating the peptides and albumin are aggregated in closer proximity than 8 nm.
FIGURE 4.
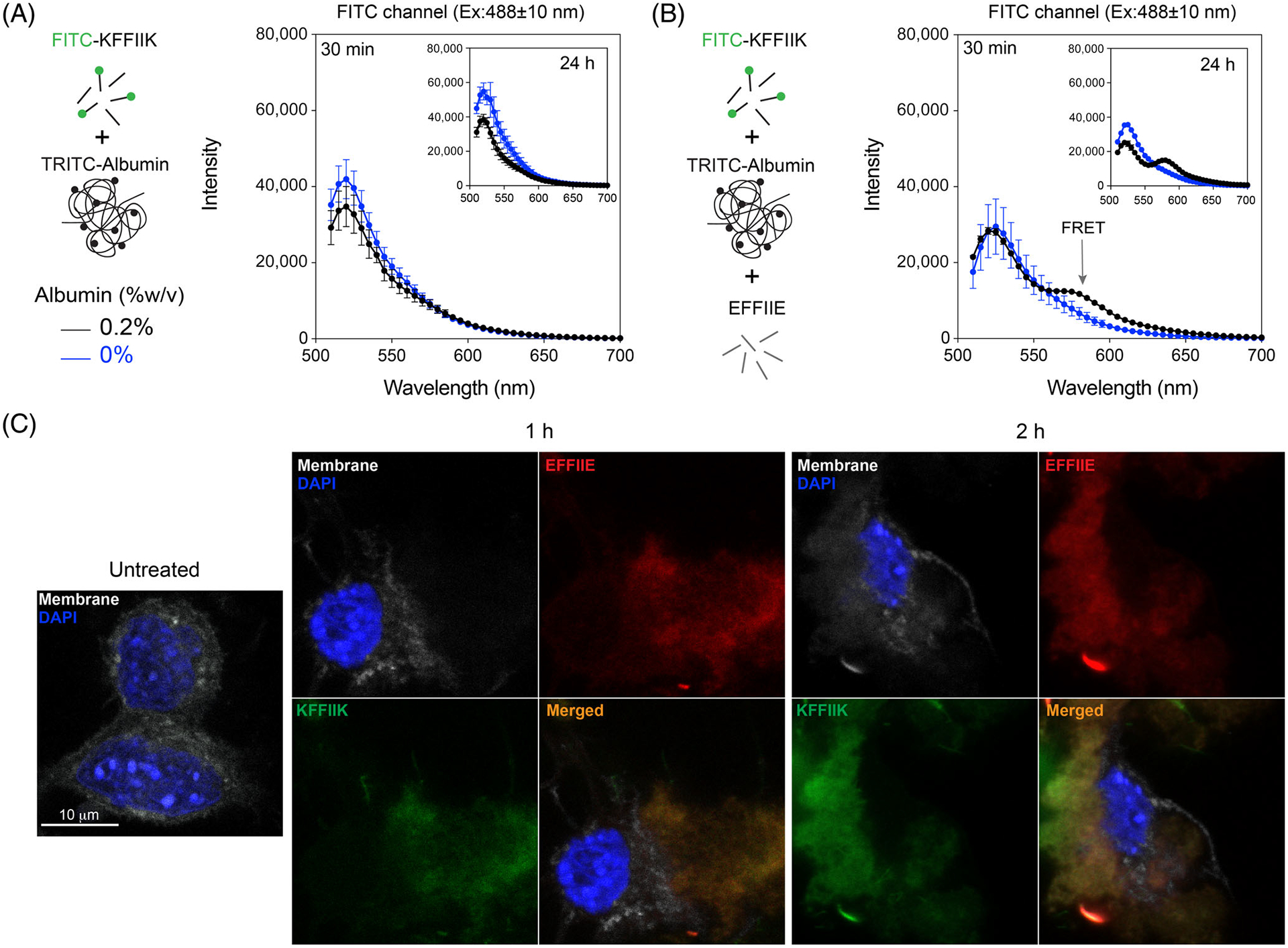
Albumin and [II] interaction. (A) The FRET analysis of the presence and absence of TRITC-albumin with FITC-KFFIIK and (B) with [II]. (C) Confocal analysis of cells treated with 0.5 mM [II] for 30 min, 1 h, and 2 h in the presence of 0.2 w% albumin. Membrane stained with WGA, gray; cells treated with Rhodamine B labeled EFFIIE, red; FITC labeled KFFIIK, green. (Scale bar = 10 μm)
The cell membranes were stained with Wheat Germ Agglutinin (WGA) (gray) and incubated with [II] (FITC-KFFIIK [green] and Rhodamine-EFFIIE [red]) in 0.2 w% albumin for 1 and 2 h to identify the localization of the peptides. Accumulation of each peptide increased from 1 to 2 h with lost integrity of the membrane, which also explained the higher LDH release (Figure 3C). No cell membrane localization was observed when the peptides were preincubated in the absence of albumin for 30 min (Figure S10), which is also in line with viability results. Overall, these results demonstrate that peptides assemble into ordered fibrillar structures without albumin and do not localize to the cell membrane. The presence of the albumin in the solution prevents immediate peptide–peptide interactions and allows time for peptide aggregation to occur within the cell and at the membrane. Therefore, changing albumin concentration changes the peptide aggregation kinetics, which results in controllable membrane accumulation and cell death.
2.5 |. [II] aggregation induces cell stress
The cell plasma membrane senses external stimuli to adapt to changes through intracellular signaling. One pathway responding against various external stimuli such as mechanical stress is the Hippo pathway. Aβ 1–42-mediated neurodegeneration involves the activation of the Hippo pathway.37 One of the central proteins within the Hippo pathway is YAP. YAP is a transcription factor found in the nucleus in active form and gets inactivated when translocated to the cytoplasm. We treated the cells to analyze the effect of [II] aggregation on YAP localization. At the early time points, 30 min, YAP localized in the cytoplasm (Figure 5A). More specifically, cytoplasmic localization of YAP is mediated by its phosphorylation (Serine 127 [S127]), leading to its inactivation38(p1) and degradation.39 Upon administration of [II] with 0.2 w% albumin, YAP phosphorylation in the cytoplasm can be observed green at 30 min and 1 h (Figure 5B, Figure S11). However, after 1 h, phosphorylated YAP in the cytoplasm starts to degrade, which is parallel with YAP localization to the cytoplasm at early time points (30 min). Western blot results also confirmed the phosphorylation of YAP and degradation (Figure 5C). Together these results show that [II] aggregation is inducing cytoplasmic localization of YAP through its phosphorylation, thus related to the mechanical stress on the cell membrane.
FIGURE 5.
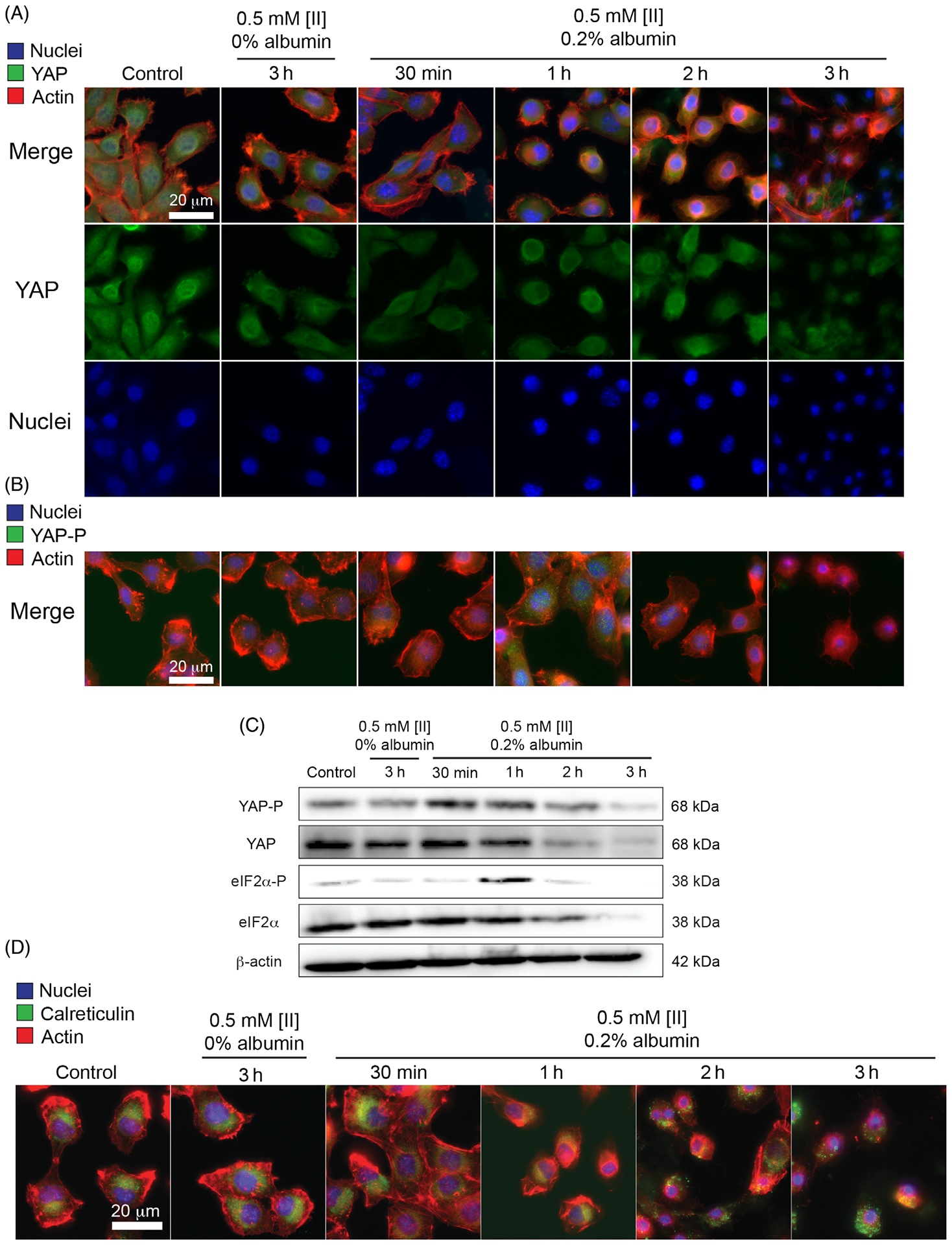
[II] aggregation on the cell membrane induces cell stress. (A) Immunocytochemistry of YAP localization (YAP = green, actin = red, nuclei = blue) (B) and phosphorylation based on aggregation (YAP-P = green). (C) Western blot analysis of YAP, YAP phosphorylation, eIF2α, and eIF2α phosphorylation in different time points, β actin was used as an internal loading control. (D) Immunocytochemistry of calreticulin cell surface localization (calreticulin = green, actin = red, nuclei = blue). (Scale bars = 20 μm)
eIF2α is another factor integrated into the stress response to external stimuli. Under cell stress, eIF2α gets phosphorylated and initiates the activation of genes responsible for stress response. Phosphorylation of eIF2α is a consistent marker in various neurodegenerative diseases,40 including AD.41 Histological analysis on AD patients showed elevated phosphorylation of eIF2α.42 Moreover, in in vitro studies, Aβ 1–42 was shown to induce eIF2α phosphorylation in human neuroblastoma cells.43 We analyzed eIF2α phosphorylation for 0.5 mM [II] in 0.2 w% albumin and 0.5 mM [II] in 0 w% albumin (nonmembrane damaging condition). [II] aggregation-induced eIF2α phosphorylation at 1 h (Figure 5C) followed by rapid degradation due to proteasome activity reported previously.44
Phosphorylation of eIF2α and its downstream effect on calreticulin cell surface localization are identified as hallmarks of ICD.45 Cell membrane-localized calreticulin contributes to the immunogenicity of dying cancer cells, serving as an “eat me” signal to facilitate phagocytosis.46,47 We previously showed that [II] aggregation-induced cell death is ICD through the release of DAMPs in multiple cell lines.18 Given that eIF2α phosphorylation results in calreticulin surface localization, we identified the localization of calreticulin within the cells incubated with [II]. Calreticulin was observed only intracellularly in control cells and cells treated with 0.5 mM [II] in 0 w% albumin. However, calreticulin localized to the cell membrane starting at 2 h in cells treated with 0.5 mM [II] in 0.2 w% albumin (Figure 5D, Figure S12). Moreover, membrane calreticulin fluorescent intensity was increased upon [II] aggregation (Figure S13). These results show that [II] aggregation-induced cell death involves the phosphorylation of eIF2α, followed by calreticulin surface localization, indicating that the observed ICD is due to the stress on the cell membrane with [II] aggregation.
3 |. CONCLUSIONS
Here we show that [II] aggregation induces cell membrane damage and engages cell stress markers as in a variety of neurodegenerative disorders. Furthermore, we showed that this aggregation can be controlled by using albumin. [II] aggregation-induced the phosphorylation of eIF2α and calreticulin surface localization, which highlighted ICD. Overall, the modulation of peptide aggregation presented here allows us to control cell membrane damage and offers us to modulate immunogenic cell death.
Protein and peptide aggregation is a phenomenon seen in both pathogenic conditions and during the developmental stages of amyloid proteins.4,5 Oligomeric species form transiently during the aggregation process and not only act as essential intermediates but also represent major pathogenic agents in disorders.6 However, the transient and complex structures and aggregates of oligomers create confusing results and challenges in drug development.6,12 Indeed, there is a vast difference between the preclinical successes and clinical results.48 The differences mainly arise from the inadequate understanding of the pathophysiological conditions, which require a combination of theoretical and experimental drug development strategies to overcome.49,50 In this study, we showed an engineered peptide complex via the CoOP strategy as a synthetic and simple tool to study the amyloid aggregation mechanism into ordered assemblies (fibrils) and disordered aggregates (oligomers) with the presence of an extrinsic pathophysiological relevant factor, albumin, and correlate it to its toxicity. Importantly, we showed that albumin controls the aggregation kinetics of [II] and enables the integration of the peptides before they aggregate. In the pathological scenario, the shown process might correspond to the misfolding of the amyloid-beta peptides with the albumin in oligomers, integration into the cell, and then folding into fibrillar form. It may also correspond to the part left after the cleavage of amyloid precursor protein (APP), which might be causing stress that induces ICD. The immune response against ICD is potent. Controlling ICD means control over the immune response and progress of amyloidosis-related diseases.
Overall, [II] offers a unique engineered platform to study aggregation kinetics of amyloidosis. The simplicity of [II] as an engineered tool allowed the detailed characterization of disorganized structures to fibril aggregation kinetics and the effects of each step on the cell. Studies with [II] can establish clear and direct results for drug development with amyloidosis.
4 |. EXPERIMENTAL SECTION
4.1 |. Peptide synthesis
KFFIIK and EFFIIE peptides were synthesized via solid phase peptide synthesis with PreludeX automatic peptide synthesizer (Protein Technologies, Inc., Tucson, AZ). They were prepared on a 0.25 scale by repeated amino acid couplings using Fmoc protected amino acid (3 eq. 1 ml), DIC (7.5 eq. 1 ml), and Oxyma (7.5 eq. 1 ml). MBHA Rink Amide resin (High-Loaded, Gyros Protein Technologies) was used as solid support to construct the peptides. Fmoc protected amino acids (Gyros Protein Technologies) were removed through treatment with 20% piperidine/DMF solution for 45 min (three times for 15 min) at 80°C. All the peptide conjugation was performed for 2 h at 80°C and acetylated with treatment with 10% acetonitrile/DMF solution for 15 min twice. Cleavage of the peptides from resin and deprotection of acid labile protected amino acids were carried out with a mixture of TFA/-TIS/water in a ratio of 95:2.5:2.5 for 3 h at room temperature. Excess TFA and organic solvents were removed by evaporation and the remaining peptide was precipitated using diethyl ether at 80°C using a 1:4 volume ratio and stayed overnight. After precipitation, peptides in ether solutions were centrifuged at 8000 rpm for 15 min. Peptide precipitates were collected, and ether was removed. The centrifuged white peptide precipitate was dissolved in either (0.1% formic acid in water for KFFIIK peptide) or (0.1% ammonium hydroxide in water for EFFIIE peptide). Peptides were purified with preparative HPLC (Agilent 1260) with Agilent ZORBAX 300 SB-C18 (9.4 × 250 mm) column with a mobile phase of water/acetonitrile mixture (0.1% ammonium hydroxide) used for negatively charged peptides; water/acetonitrile mixture (0.1% formic acid) was used for positively charged peptides. All peptides were tested with a purity >95%. HPLC run started with 100% water for 3 min, followed by a gradient increase in acetonitrile from 0% to 100% over 25 min, followed by 100% acetonitrile for 3 min, and ended with 100% for 2 min. The flow rate is 2 ml/min, and injection volumes are 10 μl.
4.2 |. Liposome formation and imaging
Giant liposomes were formed based on previously published data with simple modifications.27 First, agarose (UltraPure Agarose, Thermo-Fisher Scientific) was prepared with 1% w/v in water and boiled in a microwave oven until all agarose powder was dissolved. Then, it was poured into a petri dish to cool down at room temperature and form a gel for 2–3 h. Before coating the glass slides, agarose gel was melted in a microwave until all gel turned into a solution. One side of glass slides was dipped into agarose solution, and excess solution was removed. After coating, glass slides were placed on a hotplate (37°C) and waited for 1–2 h until an agarose film was formed on top of the glass slides. For making liposomes, DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids, Inc.) was used with cholesterol (Sigma-Aldrich). At first, DPPC was prepared in 2 mM in chloroform, and cholesterol was prepared in 400 μM in chloroform. Both solutions were mixed with equal volume (1 ml each) to have a 5:1 molar ratio (DPPC: cholesterol). For staining the liposomes with DiR (1,1′-Dioctadecyl-3,3,3′,3′-Tetra methylindotricarbocyanine iodide) dye, 4 μl of DiR (1 mM in chloroform) was mixed with 2 ml DPPC: cholesterol. Then, 50 μl of labeled liposomes were dropped onto agarose-covered glass slides and spread out homogeneously by rolling a glass rod. Then, slides were placed in a vacuum chamber for 30 min. After removing from the vacuum chamber, slides were placed in a clean petri dish, and 1× PBS was added from the side of the petri dish to allow hydration and swelling of liposomes for 3 h. For peptide-membrane localization assay, FITC labeled KFFIIK and Rhodamine-B labeled EFFIIE peptides (Biomatik Corporation) were used. KFFIIK and EFFIIE were first solubilized in 10 mM as a stock solution in water, diluted to 0.5 mM with 1X PBS. The fluorescent portion consisted of a 1:100 molar ratio. For example, FITC-KFFIIK was 50 μM in 0.5 mM KFFIIK peptide as a final concentration. After liposome formation, 0.5 mM of fluorescent labeled one peptide counterpart (200 μl) were incubated with liposomes for 60 min, then oppositely charged 0.5 mM of fluorescent labeled peptide (200 μl) was added and incubated for another 60 min. The imaging was performed with Keyence BZ-X800 with BZ-X filter Cy7 (DiR labeled liposome), BZ-X filter GFP (FITC-KFFIIK), and BZ-X filter mCherry (Rho-EFFIIE).
4.3 |. Aggregation analysis
Thioflavin T (ThT) (Sigma-Aldrich) and 1,6-Diphenyl-1,3,5-hexatriene (DPH) (Sigma-Aldrich) were used to understand the kinetics involved in ordered assembly and aggregation kinetics. Both fluorescence measurements included albumin (bovine serum albumin, Sigma-Aldrich) with different concentrations indicated based on %weight/volume (w%) and without albumin (in 1× PBS). At first, fresh albumin (40 mg/ml, 4 w%) was dissolved in 1× PBS and diluted into 2, 1, 0.5, 0.2, 0.1, 0.05, 0.02, 0.002 w% in 1× PBS. Then, stock peptides (10 mM in water) were diluted (0.5 mM) in different albumin concentrations individually. For ThT measurements, 5 μl from 400 μM ThT in PBS was added to 195 μl of 0.5 mM KFFIIK and EFFIIE (either in different albumin concentrations or in 1× PBS) individually. Then, each peptide in the same albumin concentration was mixed and read with a BioTek Neo2SM microplate reader for 4 h with 10 min intervals (Ex: 440, Em: 480 with gain: 90). For DPH analysis, stock DPH (1 mM in water) was diluted to 80 μM working concentration in 1× PBS. Similar to the ThT assay, 5 μl from 80 μM DPH in 1 × PBS was added to 195 μl of 0.5 mM KFFIIK and EFFIIE (either in different albumin concentrations or in 1× PBS) individually. Then, each peptide in the same albumin concentration was mixed and read with a BioTek Neo2SM microplate reader for 4 h with 10 min intervals (Ex: 360, Em: 450 with gain: 100). Albumin with different concentrations (without any peptide) was also analyzed for both assays and subtracted from [II] + Albumin data. For [II] in 0 w% albumin, 1× PBS (with ThT or DPH) was used as a background for subtraction. For ThT analysis, relative intensity values were calculated within each group.
4.4 |. Atomic Force Microscopy (AFM)
Morphological characterization of [II] was performed with AFM (NX-10 Park Systems Corp) with a noncontact mode by using a cantilever probe (OMCL-AC160TS 5 M). Peptide were prepared by either with albumin (0.002, 0.02, 0.2, and 2 w%) or without albumin (0 w%) in PBS. At first, 0.5 mM KFFIIK (50 μl) was prepared with or without albumin, and 0.5 mM EFFIIE (50 μl) was prepared with or without albumin in PBS. Then, two oppositely charged peptides were mixed and incubated overnight (~20 h). For sample preparation, mixed samples were diluted to 0.1 mM with water, and 20 μl dropped onto a silicon wafer. Prepared samples were dried at room temperature and imaged with a 1 kHz scanning frequency. Data points were taken in a 256 × 256 grid over a 5 μm-by-5 μm area.
4.5 |. Dynamic Light scattering
Hydrodynamic size and ζ potential of single and mixed peptides were measured by DLS. For particle size analysis, the intensity of scattered light was measured under Brownian motion, and hydrodynamic radius, Rh, was calculated by using stokes–Einstein equation (Equation 1)51:
| (1) |
where k is the Boltzmann’s constant, T is the temperature in K (298 K), and η is the solvent viscosity. The diameter measured in DLS is a value that refers to the diffusion of the particle. A ZetaSizer Nano ZS (Malvern, UK) instrument with a detector angle of 173° in a back-scatter mode was used for analysis. Clear disposable cell cuvettes were first washed three times with 0.22-μm filtered deionized water (resistance of 18.2 megaohms cm). Before measurements, peptide solutions in phosphate-buffered saline were also filtered with a 0.22-μm filter to avoid dust that could alter the measurements. For individual peptides, 500 μM (500 μl) positively and negatively charged peptides were used in PBS. For [II], equimolar (500 μM, 250 μl) of KFFIIK and equimolar (500 μM, 250 μl) EFFIIE were mixed and analyzed immediately, at 30 min and overnight (~20 h). For [II] with albumin samples, individual peptides (500 μM each) were prepared in albumin solution (0.002, 0.02, 0.05, 0.1, 0.2, 0.5, 1, or 2 w% albumin in PBS) individually and mixed with equal volume (250 μl each). Then analyzed immediately, at 30 min and overnight (~20 h). ζ potential measurements were performed at 30 min.
4.6 |. FRET analysis
FITC and TRITC could act as the donor and acceptor of a FRET pair, respectively.34 FITC-KFFIIK peptide was prepared as 500 μM (1:50 FITC-KFFIIK: KFFIIK molar ratio), and albumin was prepared as 2% w/v (20 mg/ml, 300 μM) mixed with TRITC-albumin as 1:100 molar ratio. EFFIIE peptide was prepared as 500 μM. For a group consisting of FITC-KFFIIK and TRITC-albumin, 50 μl of FITC-KFFIIK was mixed with 10 μl TRITC-albumin, and 50 μl of PBS was added to have a final volume of 110 μl. For 0 w% albumin, 50 μl of FITC-KFFIIK was mixed with 60 μl of PBS. [II] was prepared as follows. For 0.2 w% albumin. Fifty microliter of FITC-KFFIIK was mixed with 50 μl of EFFIIE, and 10 μl of TRITC-albumin was added to have a final volume of 110 μl. For 0 % albumin, 50 μl of FITC-KFFIIK was mixed with 50 μl of EFFIIE, and 10 μl of PBS was added. These mixtures were incubated for 30 min and 24 h at room temperature and subjected to fluorescence measurements with an excitation of 488 nm for FITC as donor and scan between the emission of 500 and 700 nm for TRITC as acceptor.
4.7 |. FT-IR analysis
Peptides (KFFIIK and EFFIIE) with a concentration of 300 μM were prepared in water, and analysis was performed immediately. For the peptide combination [II], equal volumes of charged peptides were mixed (as a total of 20 μl) and put directly into the FTIR stage. Analysis was performed with Bruker Tensor II with BioATR II unit. For the background spectrum, water was measured first, and then peptide groups were subtracted automatically in the device. Analysis was performed between 1800 and 900 cm−1, with 4 cm−1 resolution and 60 scans.
4.8 |. Cell Culture
EMT6 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium, supplemented with 10% FBS (Hyclone SH30910.03) and 1% antibiotics; penicillin (100 U/ml), and streptomycin (100 μg/ml) (Thermo Fisher 15240062). Cells were cultured in a humidified incubator at 37°C supplied with 5% CO2. T75 flasks (TPP 90076) were used for culturing, and cells were passaged upon 85% confluency using trypsin (Sigma 59418C). Cell culture media was changed every 2 days.
4.9 |. Peptide preparation for in vitro experiments
[II] peptides were prepared in either 0.2 w% albumin or 0 w% albumin for in vitro conditions. First, albumin was prepared as a stock solution in 2 w% (20 mg in 1 ml of the only RPMI medium). Then, it was diluted at 1:10 to reach 0.2 w% in the RPMI medium. The prepared KFFIIK and EFFIIE stock peptides (10 mM in water) were diluted to 0.5 mM separately into 0.2 w% albumin/medium. For 0 w% albumin condition, KFFIIK and EFFIIE stock peptides 10 mM in water were diluted to 0.5 mM separately into albumin/medium. For 96 well plate, each well consisted of 100 μl of the peptide having 50 μl EFFIIE and 50 μl KFFIIK in either only RPMI medium (0 w% albumin) or 0.2 w% albumin in RPMI medium.
4.10 |. Cell Titer Glo2.0 Viability
Cells were seeded onto 96 well plates and left for O/N attachment. The next day, media was removed, and treatments were carried out. Viability was measured with Cell Titer Glo 2.0 solution (Promega G9248). The luminescent signal was measured in accordance with the manufacturer’s instructions. Measurements were carried out with a BioTek Neo2SM microplate reader, and relative viability was calculated by using an untreated control group.
4.11 |. LDH Release
LDH release was measured with Cytoscan-LDH Cytotoxicity Assay (G-BIOSCIENCES 786-210). Briefly, at the end of the treatments, collected supernatants were mixed with the reaction mixture and incubated for 20 min at 37°C. The reaction was stopped with a stop solution, and absorbance was measured at 490 and 680 nm. Triton-X treatment is used as a 100% LDH release control. Absorbance at 680 nm is used as the background signal, and values were subtracted from absorbance values at 490 nm. Measurements were carried out with a BioTek Neo2SM microplate reader, and relative LDH release was quantified based on the positive control for LDH release (maximum LDH release through Triton-X treatment).
4.12 |. Confocal Imaging
EMT6 cells were seeded on glass coverslips in a 24-well plate and incubated O/N for attachment. Before the experiment, cells were labeled with WGA 633 membrane stain (ThermoFisher W21404) according to the manufacturer’s instructions. After membrane staining, cells were washed 2× with PBS and treated in the presence (0.2 w%) and absence (0 w%) of albumin.
4.13 |. Immunoblotting, Reagents
Acrylamide/Bis-acrylamide, 30 w% solution (Sigma A3699), 1.5 M Tris–HCl, pH 8.8 (Teknova T1588), Tris HCl Buffer 0.5 M solution, sterile pH 6.8 (Bio Basic SD8122), Ammonium persulfate (Sigma A3678), UltraPure 10% SDS (Invitrogen 15553-027), TEMED (Thermo Fisher Scientific 17919), Dithiothreitol (DTT) (BIO-RAD 1610610) Tris Base (Fisher Bioreagents BP152), Glycine (Fisher Bioreagents BP381), 4X Laemmli sample buffer (BIO-RAD 1610747), TWEEN 20 (Sigma P9416), Mini Trans-Blot filter paper (BIO-RAD 1703932), Nitrocellulose Membranes 0.45 μm (BIO-RAD 1620115), EveryBlot Blocking Buffer (BIO-RAD 12010020), Clarity Western ECL Substrate (BIO-RAD 170-5060).
4.14 |. Procedure
Samples were prepared in Laemmli buffer and boiled for 8 min at 96°C. Then, proteins were separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (8.5 and 15 w%) and transferred to nitrocellulose membranes (45 μm). After transfer, the membranes were blocked EverBlot Blocking Buffer and washed with Tris Buffered Saline with 0.1% Tween 20 (TBST). Blots were incubated with Phospho-YAP (Ser127) (Cell Signaling 13008), YAP (Cell Signaling 4912), Phospho-eIF2α (S51) (Cell Signaling 9721), and eIF2α (Cell Signaling 9722) overnight. The next day blots were washed and incubated with Goat anti-rabbit secondary antibody (Invitrogen 31460) and imaged with Clarity Western ECL substrate (BIO-RAD 170-5060). β-actin (Santa Cruz sc-47778 HRP) was used as an internal loading control. Blots were imaged by using Azure c600.
4.15 |. Immunocytochemistry
EMT6 cells were seeded on glass coverslips in a 24-well plate and incubated O/N for attachment. After indicated treatments, samples were washed with 1X PBS and fixed with 4% PFA for 20 min. Then, Triton-X (0.5%) was used to permeabilize the cells for 20 min on a shaker. Samples were blocked with 3% BSA for 1 h on a shaker, washed three times with 1X PBS, and incubated with the following antibodies Phospho-YAP (S127) (Cell Signaling 13008), YAP (Cell Signaling 4912), or calreticulin (Cell Signaling D3E6) O/N on shaker at 4°C. The following day samples were washed and incubated for 1 h on a shaker with DyLight™ 488 Donkey anti-rabbit IgG (minimal x-reactivity) antibody (BioLegend 406404). After incubation, samples were washed and incubated with Flash Phalloidin™ Red 594 (BioLegend 424203) for 30 min on a shaker. Lastly, samples were washed and mounted in ProLong™ Glass Antifade Mountant with NucBlue™ Stain (Thermo Fisher P36981) and stored in the dark until imaging.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported in part by a grant from the Research Council of the University of Oklahoma Norman Campus and in part by the Oklahoma Tobacco Settlement Endowment Trust awarded to the University of Oklahoma, Stephenson Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Oklahoma Tobacco Settlement Endowment Trust. Dr. Tingting Gu from Samuel Roberts Noble Microscopy Laboratory at the University of Oklahoma conducted the confocal imaging. EMT6 breast cancer cell line was a gift from Dr. Judy Lieberman, Boston Children’s Hospital, Harvard Medical School. The University of Oklahoma Protein Production provided FT-IR services and a Characterization Core Facility, supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P30GM145423).
Funding information
Oklahoma Tobacco Settlement Endowment Trust; Research Council of the University of Oklahoma Norman Campus; National Institute of General Medical Sciences of the National Institutes of Health, Grant/Award Number: P30GM145423
Footnotes
CONFLICT OF INTEREST The authors have submitted a patent application for the technology described in this study.
SUPPORTING INFORMATION Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1.Chiti F, Webster P, Taddei N, et al. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci. 1999;96(7):3590–3594. doi: 10.1073/pnas.96.7.3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobson CM. Protein misfolding, evolution, and disease. Trends Biochem Sci. 1999;24(9):329–332. doi: 10.1016/S0968-0004(99)01445-0 [DOI] [PubMed] [Google Scholar]
- 3.Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE. A new era for understanding amyloid structures and disease. Nat Rev Mol Cell Biol. 2018;19(12):755–773. doi: 10.1038/s41580-018-0060-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallardo R, Ranson NA, Radford SE. Amyloid structures: much more than just a cross-β fold. Curr Opin Struct Biol. 2020;60:7–16. doi: 10.1016/j.sbi.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia Z, Schmit JD, Chen J. Amyloid assembly is dominated by misregistered kinetic traps on an unbiased energy landscape. Proc Natl Acad Sci. 2020;117(19):10322–10328. doi: 10.1073/pnas.1911153117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulenkampff K, Wolf Perez AM, Sormanni P, Habchi J, Vendruscolo M. Quantifying misfolded protein oligomers as drug targets and biomarkers in Alzheimer and Parkinson diseases. Nat Rev Chem. 2021;5(4):277–294. doi: 10.1038/s41570-021-00254-9 [DOI] [PubMed] [Google Scholar]
- 7.Fusco G, Chen SW, Williamson PTF, et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science. 2017;358(6369):1440–1443. doi: 10.1126/science.aan6160 [DOI] [PubMed] [Google Scholar]
- 8.Yasumoto T, Takamura Y, Tsuji M, et al. High molecular weight amyloid β1–42 oligomers induce neurotoxicity via plasma membrane damage. Alzheimers Dement. 2020;16(S2):e037546. doi: 10.1002/alz.037546 [DOI] [PubMed] [Google Scholar]
- 9.Milner MT, Maddugoda M, Götz J, Burgener SS, Schroder K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr Opin Immunol. 2021;68:116–124. doi: 10.1016/j.coi.2020.10.011 [DOI] [PubMed] [Google Scholar]
- 10.Tanaka H, Homma H, Fujita K, et al. YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat Commun. 2020;11(1):507. doi: 10.1038/s41467-020-14353-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenzie BA, Dixit VM, Power C. Fiery cell death: pyroptosis in the central nervous system. Trends Neurosci. 2020;43(1):55–73. doi: 10.1016/j.tins.2019.11.005 [DOI] [PubMed] [Google Scholar]
- 12.Walsh DM, Selkoe DJ. Amyloid β-protein and beyond: the path forward in Alzheimer’s disease. Curr Opin Neurobiol. 2020;61:116–124. doi: 10.1016/j.conb.2020.02.003 [DOI] [PubMed] [Google Scholar]
- 13.Guo AZ, Fluitt AM, de Pablo JJ. Early-stage human islet amyloid polypeptide aggregation: mechanisms behind dimer formation. J Chem Phys. 2018;149(2):025101. doi: 10.1063/1.5033458 [DOI] [PubMed] [Google Scholar]
- 14.Serrano AL, Lomont JP, Tu LH, Raleigh DP, Zanni MT. A free energy barrier caused by the refolding of an oligomeric intermediate controls the lag time of amyloid formation by hIAPP. J Am Chem Soc. 2017; 139(46):16748–16758. doi: 10.1021/jacs.7b08830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan J, Lan H, Ning W, et al. Modeling amide-I vibrations of alanine dipeptide in solution by using neural network protocol. Spectrochim Acta A Mol Biomol Spectrosc. 2022;268:120675. doi: 10.1016/j.saa.2021.120675 [DOI] [PubMed] [Google Scholar]
- 16.Kazman P, Absmeier RM, Engelhardt H, Buchner J. Dissection of the amyloid formation pathway in AL amyloidosis. Nat Commun. 2021; 12(1):6516. doi: 10.1038/s41467-021-26845-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamsici S, White AD, Acar H. Peptide framework for screening the effects of amino acids on assembly. Sci Adv. 2022;8:eabj0305. doi: 10.1126/sciadv.abj0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunay G, Hamsici S, Lang GA, Lang ML, Kovats S, Acar H. Peptide aggregation induced immunogenic rupture (PAIIR). Adv Sci. 2022;9: 2105868. doi: 10.1002/advs.202105868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021;28(7):2029–2044. doi: 10.1038/s41418-021-00814-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leong YQ, Ng KY, Chye SM, Ling APK, Koh RY. Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death. Metab Brain Dis. 2020;35(1):11–30. doi: 10.1007/s11011-019-00516-y [DOI] [PubMed] [Google Scholar]
- 21.Finn TE, Nunez AC, Sunde M, Easterbrook-Smith SB. Serum albumin prevents protein aggregation and amyloid formation and retains chaperone-like activity in the presence of physiological ligands. J Biol Chem. 2012;287(25):21530–21540. doi: 10.1074/jbc.M112.372961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao M, Guo C. Multipronged regulatory functions of serum albumin in early stages of amyloid-β aggregation. ACS Chem Nerosci. 2021; 12(13):2409–2420. doi: 10.1021/acschemneuro.1c00150 [DOI] [PubMed] [Google Scholar]
- 23.Picón-Pagès P, Bonet J, García-García J, et al. Human albumin impairs amyloid β-peptide fibrillation through its C-terminus: from docking modeling to protection against neurotoxicity in Alzheimer’s disease. Comput Struct Biotechnol J. 2019;17:963–971. doi: 10.1016/j.csbj.2019.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kakinen A, Javed I, Faridi A, Davis TP, Ke PC. Serum albumin impedes the amyloid aggregation and hemolysis of human islet amyloid polypeptide and alpha synuclein. Biochim Biophys Acta Biomembr. 2018;1860(9):1803–1809. doi: 10.1016/j.bbamem.2018.01.015 [DOI] [PubMed] [Google Scholar]
- 25.Savini F, Bobone S, Roversi D, Mangoni ML, Stella L. From liposomes to cells: filling the gap between physicochemical and microbiological studies of the activity and selectivity of host-defense peptides. Pept Sci. 2018;110(5):e24041. doi: 10.1002/pep2.24041 [DOI] [Google Scholar]
- 26.Andrade S, Loureiro JA, Pereira MC. The role of amyloid β-biomembrane interactions in the pathogenesis of Alzheimer’s disease: insights from liposomes as membrane models. ChemPhysChem. 2021;22(15):1547–1565. doi: 10.1002/cphc.202100124 [DOI] [PubMed] [Google Scholar]
- 27.Horger KS, Estes DJ, Capone R, Mayer M. Films of agarose enable rapid formation of giant liposomes in solutions of physiologic ionic strength. J Am Chem Soc. 2009;131(5):1810–1819. doi: 10.1021/ja805625u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chibowski E, Szcześ A. Zeta potential and surface charge of DPPC and DOPC liposomes in the presence of PLC enzyme. Adsorption. 2016;22(4–6):755–765. doi: 10.1007/s10450-016-9767-z [DOI] [Google Scholar]
- 29.Arosio P, Knowles TPJ, Linse S. On the lag phase in amyloid fibril formation. Phys Chem Chem Phys. 2015;17(12):7606–7618. doi: 10.1039/C4CP05563B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stanyon HF, Viles JH. Human serum albumin can regulate amyloid-β peptide fiber growth in the brain interstitium: implications for Alzheimer disease. J Biol Chem. 2012;287(33):28163–28168. doi: 10.1074/jbc.C112.360800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta Proteins Proteom. 2010; 1804(7):1405–1412. doi: 10.1016/j.bbapap.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sang JC, Lee JE, Dear AJ, et al. Direct observation of prion protein oligomer formation reveals an aggregation mechanism with multiple conformationally distinct species. Chem Sci. 2019;10(17):4588–4597. doi: 10.1039/C8SC05627G [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ziaunys M, Sakalauskas A, Sneideris T, Smirnovas V. Lysozyme fibrils alter the mechanism of insulin amyloid aggregation. Int J Mol Sci. 2021;22(4):1775. doi: 10.3390/ijms22041775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makwana PK, Jethva PN, Roy I. Coumarin 6 and 1,6-diphenyl-1,3,5-hexatriene (DPH) as fluorescent probes to monitor protein aggregation. Analyst. 2011;136(10):2161–2167. doi: 10.1039/C0AN00829J [DOI] [PubMed] [Google Scholar]
- 35.Kumar P, Nagarajan A, Uchil PD. Analysis of cell viability by the lactate dehydrogenase assay. Cold Spring Harb Protoc. 2018;2018(6): pdb.prot095497. doi: 10.1101/pdb.prot095497 [DOI] [PubMed] [Google Scholar]
- 36.Lichlyter DJ, Grant SA, Soykan O. Development of a novel FRET immunosensor technique. Biosens Bioelectron. 2003;19(3):219–226. doi: 10.1016/S0956-5663(03)00215-X [DOI] [PubMed] [Google Scholar]
- 37.Irwin M, Tare M, Singh A, et al. A positive feedback loop of hippo- and c-Jun-amino-terminal kinase signaling pathways regulates amyloid-beta-mediated neurodegeneration. Front Cell Dev Biol. 2020; 8:117. doi: 10.3389/fcell.2020.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008; 283(9):5496–5509. doi: 10.1074/jbc.M709037200 [DOI] [PubMed] [Google Scholar]
- 39.Abylkassov R, Xie Y. Role of yes-associated protein in cancer: an update. Oncol Lett. 2016;12(4):2277–2282. doi: 10.3892/ol.2016.4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bond S, Lopez-Lloreda C, Gannon PJ, Akay-Espinoza C, Jordan-Sciutto KL. The integrated stress response and phosphorylated eukaryotic initiation factor 2α in neurodegeneration. J Neuropathol Exp Neurol. 2020;79(2):123–143. doi: 10.1093/jnen/nlz129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohno M Roles of eIF2α kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci. 2014;7:22. doi: 10.3389/fnmol.2014.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang RCC, Wong AKY, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimerʼs disease. Neuroreport. 2002;13(18):2429–2432. doi: 10.1097/00001756-200212200-00011 [DOI] [PubMed] [Google Scholar]
- 43.Lee DY, Lee KS, Lee HJ, et al. Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS One. 2010;5(5):e10489. doi: 10.1371/journal.pone.0010489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yerlikaya A, Kimball SR, Stanley BA. Phosphorylation of eIF2α in response to 26S proteasome inhibition is mediated by the haem-regulated inhibitor (HRI) kinase. Biochem J. 2008;412(3):579–588. doi: 10.1042/BJ20080324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bezu L, Sauvat A, Humeau J, et al. eIF2α phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 2018;25(8): 1375–1393. doi: 10.1038/s41418-017-0044-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obeid M, Panaretakis T, Joza N, et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14(10):1848–1850. doi: 10.1038/sj.cdd.4402201 [DOI] [PubMed] [Google Scholar]
- 47.Fucikova J, Kepp O, Kasikova L, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020; 11(11):1013. doi: 10.1038/s41419-020-03221-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cummings J, Kinney J, Fillit H. Alzheimer’s disease drug development: a research and development ecosystem. In: Fillit H, Kinney J, Cummings J, eds. Alzheimer’s Disease Drug Development: Research and Development Ecosystem. Cambridge University Press; 2022:1–24. doi: 10.1017/9781108975759.002 [DOI] [Google Scholar]
- 49.Cummings J Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin Transl Sci. 2018;11(2):147–152. doi: 10.1111/cts.12491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yiannopoulou KG, Anastasiou AI, Zachariou V, Pelidou SH. Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicine. 2019;7(4):97. doi: 10.3390/biomedicines7040097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saiki M, Shiba K, Okumura M. Structural stability of amyloid fibrils depends on the existence of the peripheral sequence near the core cross-β region. FEBS Lett. 2015;589(23):3541–3547. doi: 10.1016/j.febslet.2015.10.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.