

Abstract
Chemotherapeutic treatment with conventional drug formulations pose numerous challenges, such as poor solubility, high cytotoxicity and serious off-target side effects, low bioavailability, and ultimately subtherapeutic tumoral concentration leading to poor therapeutic outcomes. In the field of Nanomedicine, advances in nanotechnology have been applied with great success to design and develop novel nanoparticle-based formulations for the treatment of various types of cancer. The approval of the first nanomedicine, Doxil® (liposomal doxorubicin) in 1995, paved the path for further development for various types of novel delivery platforms. Several different types of nanoparticles, especially organic (soft) nanoparticles (liposomes, polymeric micelles, and albumin-bound nanoparticles), have been developed and approved for several anticancer drugs. Nanoparticulate drug delivery platform have facilitated to overcome of these challenges and offered key advantages of improved bioavailability, higher intra-tumoral concentration of the drug, reduced toxicity, and improved efficacy. This review introduces various commonly used nanoparticulate systems in biomedical research and their pharmacokinetic (PK) attributes, then focuses on the various physicochemical and physiological factors affecting the in vivo disposition of chemotherapeutic agents encapsulated in nanoparticles in recent years. Further, it provides a review of the current landscape of soft nanoparticulate formulations for the two most widely investigated anticancer drugs, paclitaxel, and doxorubicin, that are either approved or under investigation. Formulation details, PK profiles, and therapeutic outcomes of these novel strategies have been discussed individually and in comparison, to traditional formulations.
This article is categorized under:
Nanotechnology Approaches to Biology > Cells at the Nanoscale
Diagnostic Tools > In Vivo Nanodiagnostics and Imaging
Therapeutic Approaches and Drug Discovery > Nanomedicine for Oncologic Disease
Keywords: chemotherapeutics, doxorubicin, paclitaxel, pharmacokinetics, soft nanoparticles
1 |. INTRODUCTION
Nanoparticles (NPs), defined by the International Organization for Standardization (ISO) as materials with one or more dimensions at the nanoscale, can be classified according to their shape, size, morphology, physical, and chemical properties. They could be spherical, cylindrical, tubular, conical, hollow core, spiral, flat, or irregular and range from 1 nm to 100 nm in size. The surface can be uniform or irregular with or without surface modifications. Some NPs are crystalline or amorphous with single or multi-crystal solids either loose or agglomerated (Ealias & Saravanakumar, 2017). NPs can also be categorized depending on the material of construction, and these include organic or soft, inorganic or hard (Yingchoncharoen et al., 2016), and hybrid NPs. Soft NPs “are made of carbon (organic materials such as lipids and polymers) that are susceptible to change in size and shape when facing different biological conditions such as pH, ionic strength, and pressure” (Estelrich et al., 2014). Soft NPs can be further categorized into polymer-based, and lipid-based NPs. Polymer-based NPs include dendrimers, micelles, nanogels, protein NPs, and drug conjugates. Lipid-based NPs can be divided into liposomes and solid lipid NPs. So far, most of the clinically approved NPs are formulated as polymers or lipids. Each of these systems possesses unique physicochemical and biopharmaceutic features that enable efficient encapsulation and targeting (Table 1). The very first generation of NP-based therapeutics that received FDA approval were lipid systems like liposomes and micelles (Patra et al., 2018). FDA approval of the nanomedicine, Doxil® (liposomal doxorubicin) in 1995, paved the way for more development of various types of NPs for various drugs and indications worldwide. This also encouraged the life cycle management of traditional drugs and repurposing them for better treatment outcomes.
TABLE 1.
Commonly used nanoparticulate systems in biomedical research
| Nano system | Physicochemical attributes | Pharmacokinetic/Pharmacodynamic attributes | Limitations | Examples of approved/marketed/clinical trials |
|---|---|---|---|---|
| Lipid-based nanocarriers | ||||
Liposomes
|
|
|
|
|
Lipid nanoparticles (LNPs)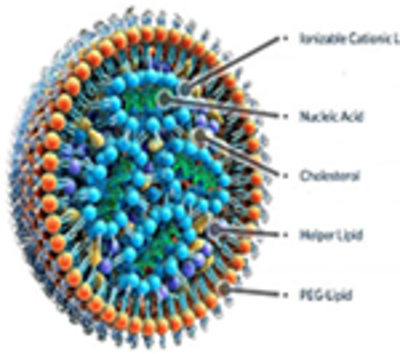
|
|
|
|
|
Solid lipid nanoparticles (SLNs)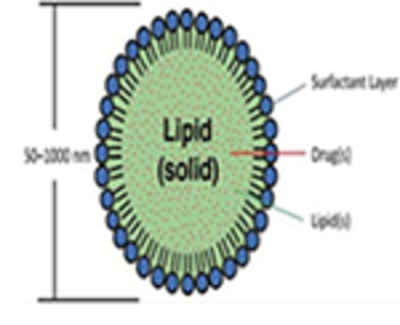
|
|
|
|
|
Nanostructured lipid carriers (NLCs)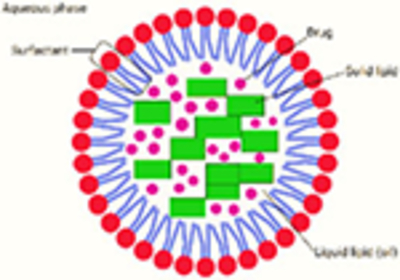
|
|
|
|
|
| Protein/polymer—nanoparticles | ||||
Albumin based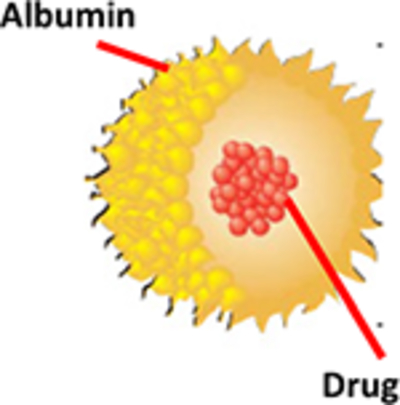
|
|
|
|
|
Polymeric nanoparticles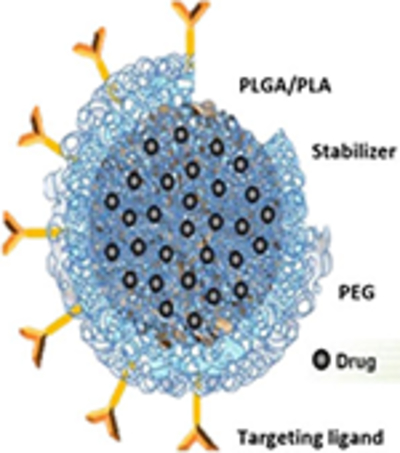
|
|
|
|
BIND-014 (PLGA-based) Phase 2 trial in patients with Metastatic Castration-Resistant Prostate Cancer |
Polymeric micelles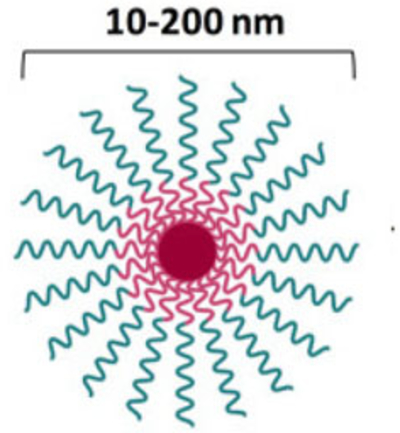
|
|
|
|
Genexol PM® (Paclitaxel) Nanoxel PM® (Docetaxel) NK 105 (Paclitaxel) SP1049C (doxorubicin) |
Dendrimers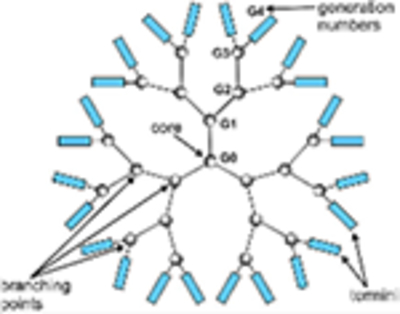
|
|
|
|
|
The pharmacokinetic (PK) disposition of free drugs is governed by its physicochemical properties and consequently by the interaction with various biological components such as cellular membranes, plasma proteins, metabolic enzymes, transporters, receptors, and so on. On the other hand, the PK of drugs encapsulated or immobilized in nanoparticle carrier systems is further impacted by the nature and composition of the carrier, which could be markedly different from the free drug. Since NP drug delivery systems are intended to alter the pharmacokinetics of encapsulated drugs for targeted therapy and improved therapeutic efficacy, it is crucial to assess and completely understand the absorption, distribution, metabolism, and elimination (ADME) of the drug in its encapsulated form. This understanding could be utilized to design and tailor an optimum nanocarrier system. PK parameters that describe the disposition of free and encapsulated drugs can be estimated using mathematical models that simulate the rate and extent of ADME. These PK parameters such as clearance (Cl), volume of distribution (Vd), elimination half-life (t1/2), maximum plasma concentration (Cmax), area under the plasma concentration–time curve (AUC), mean residence time (MRT), and bioavailability (F) can be altered by the encapsulation of free drugs in nanocarrier systems. Modulation of the PK properties of NPs can be achieved by tuning its size, incorporating suitable surface coating to convey stealth, and masking the surface charge on the NPs (Lin et al., 2015).
One of the unique characteristics of nano-formulations is that the encapsulated drug behaves differently from the nonencapsulated free drug after in vivo administration. Pharmacokinetic/pharmacodynamic (PK/PD) models can be used to predict the optimal conditions for a drug carrier to maximize the antitumor effect of the encapsulated drug relative to the free drug (Rodallec et al., 2018). In such PK/PD models, the disposition of the free and encapsulated drugs is often described by compartment models which are linked to the tumor compartment via blood flow rates (Zheng et al., 2019). Using an appropriate cell-kill kinetic PD model, the number of tumor cells can be expressed quantitatively as a function of the free concentration of drug in the tumor compartment. The concentration of the free drug in the tumor compartment can be modulated by optimizing the rate of release of the encapsulated drug from the nanocarrier. The rate of release from the nanocarrier can vary between preclinical species and humans limiting the extrapolation of optimal conditions from experimental animals to humans. Other important considerations in building a suitable PK/PD model for encapsulated chemotherapeutic agents are uptake by tumor cells, tumor heterogeneity, tumor type, size, and growth rate. Since NPs are usually cleared by the reticuloendothelial system (RES), the effect of the efflux of free drugs from the RES on the overall antitumor effect should also be evaluated (Harashima et al., 1999; Zhang et al., 2021).
This review provides an update on the current state of PK of chemotherapeutic drugs encapsulated in organic (soft) nanoparticulate drug delivery systems. It covers the literature published within the past 6 years; however, a few older literature are included to cite breakthrough concepts and classical examples. For clarification, the term “chemotherapeutics” used in this review refers to small molecule anti-cancer drugs that act purposefully as cytotoxic agents, and “nanomedicine” encompasses nano-pharmaceuticals, nanoimaging agents, and theranostics. This article primarily discusses the characteristics of soft NPs such as particle size, morphology, surface modifications, and their impact on the preclinical and clinical PK disposition of encapsulated chemotherapeutic drugs. We have included the example of the two most preclinically and clinically investigated chemotherapeutic drugs, PTX and doxorubicin (DOX), and evaluated their traditional formulations versus the nanoparticulate delivery systems.
2 |. ADVANTAGES OF ENCAPSULATING CHEMOTHERAPEUTICS IN NANOPARTICLES
Chemotherapeutic drugs exert their pharmacological effect by either slowing or stopping the growth of cancer cells. There are some major challenges with conventional chemotherapeutics, such as low bioavailability, low therapeutic index, off-target cytotoxicity, and development of drug resistance. Since these drugs cannot differentiate between healthy and cancerous cells, they indiscriminately kill all vigorously growing cells resulting in significant cytotoxicity. This manifests as serious adverse effects such as hair loss, bone marrow suppression, and gastrointestinal irritation (Navya et al., 2019). These drawbacks can be circumvented by encapsulating the chemotherapeutics in nanoparticulate delivery systems. Chemotherapeutic agents encapsulated in NPs exhibit unique physicochemical and biological properties and serve as a promising approach for their targeted delivery to specific organs in the body (Yetisgin et al., 2020)
NPs drug delivery systems hold immense potential to improve cancer treatment by modifying biodistribution and tumor site targeting and accumulation of traditional chemotherapeutic agents. It can also be used to control the release of chemotherapeutics to improve their biological and pharmacological profiles and enhance their therapeutic efficacy. Targeted therapy allows delivery of therapeutic concentration of chemotherapeutic agent at the desired site of action for a prolonged period. Thus, permitting the use of lower doses. This is particularly important because these agents have a low therapeutic index. Enhancing the drug concentration at the target organ improves the therapeutic index, enhances the efficacy, and increases the overall tolerability of the drug (Sung et al., 2021)
In addition, NPs have also been used as a strategy to mitigate multi-drug resistance (MDR). MDR is a major limitation, where tumors become unresponsive to conventional chemotherapy treatment over a period and long-term treatment is not effective. NPs can be formulated to deliver a combination of drugs sequentially and at specific molar ratios within the tumor microenvironment, allowing for maximal synergy, that is not achieved with conventional drug delivery methods. For example, Vyxeos® (CPX-351), a liposomal formulation that co-delivers cytarabine and daunorubicin at a 5:1 fixed molar ratio has been developed for the treatment of acute myeloid leukemia (AML). Poor treatment outcomes with cytarabine and daunorubicin have been linked to the overexpression of the MDR1 gene and P-gp in certain AML patients (Alfayez et al., 2020). Vyxeos® was developed using a drug design methodology, CombiPlex®, to achieve controlled release of both cytarabine and daunorubicin from nanoscale liposomes. The CombiPlex technology identifies the most synergistic ratio of drugs in vitro and fix this ratio in a nano-scale delivery complex that maintains the synergistic combination after administration. The technology utilizes two proprietary nano-scale delivery platforms: liposomes to control the release and distribution of water-soluble drugs and drugs that are both water- and fat-soluble (amphipathic); and NPs to control the release and distribution of non-water-soluble (hydrophobic) drugs. Vyxeos® demonstrated improved efficacy in two Phase 2 clinical trials, compared with a standard cytarabine and daunorubicin regime, and was approved by the FDA to treat adults with newly diagnosed t-AML or AML-MRC. In a pivotal Phase 3 trial, Vyxeos® improved overall survival compared to the standard of care 7 + 3 (cytarabine and daunorubicin) regimen (Lancet et al., 2018)
3 |. IMPACTS OF PHYSICOCHEMICAL PROPERTIES ON IN VIVO DISPOSITION OF NANOPARTICLES
The in vivo fate of the NP carrier is very important since the PK of the carrier will dictate both PD and PK/PD/toxicokinetics of the encapsulated drug. NPs act as “prodrugs” that are not active until the encapsulated drug is released from the vehicles (Price et al., 2020). Since there are varieties of NPs (liposomes, polymeric micelles, fullerenes, carbon nanotubes, quantum dots, nanoshells, polymers, dendrimers, conjugates, etc.) with distinct composition and physicochemical properties, the biological interactions, and PK characteristics of each type is also unique. Once the NPs enter systemic circulation, in vivo ADME behaviors including clearance, and interaction with barriers inside body such as the tumor microenvironment, and mucus membranes are dependent on factors such as particle size, size distribution, shape, charge, and surface chemistry (Liu et al., 2016). These factors are critical in terms of determining tissue penetration, cellular delivery, and therapeutic efficacy. On the other hand, physiological factors such as excretion, blood flow, coronas, and phagocytic cells can make NPs less stable (Mitchell et al., 2021)
One of the fundamental and critical attributes of NPs is their size which falls in the range between molecules and cell organelles. The unique size range allows them to circulate through the bloodstream, penetrate cells and tissues, elicit minimal immunogenicity and toxicity, and be cleared from the body efficiently (Sangtani et al., 2017). NPs with smaller sizes have larger surface areas, increased mobility, and higher interaction with cell membranes, which can result in enhanced cellular uptake of NPs. A study by Donkor and Tang showed that the cellular and nuclear internalization of 30 nm-sized single-walled carbon nanotubes was higher than 50 nm nanotubes (Donkor & Tang, 2014; Seleci et al., 2016). In addition, the large surface area to volume ratio allows NPs to possess large loading capacity and thus makes them highly attractive candidates for optimized therapy for various human diseases. Particle size influences drug loading and release characteristics, particle stability, toxicity, and in vivo distribution and clearance. Particles greater than 100 nm can be found in blood and highly perfused organs, for example, the spleen, lungs, liver, and kidney (Seleci et al., 2016). The particle size or diameter of NPs can be determined by using different methods to prepare or change certain concentrations of the polymer or the surfactant. Size plays a significant role in determining the mechanism of uptake too. Smaller particles bind to cells and may internalize via vacuolar uptake, whereas particles in the size range of 10–500 nm are internalized into cells via vesicular pathways. Caveolae-mediated pathway may facilitate the entry of NPs of 60–80 nm and in some cases up to 100 nm in diameter (Parhiz et al., 2018). However, below 100 nm, the size of the NPs plays a less important role in possible routes of uptake, as the geometry of each pathway can readily accommodate small NPs (Rennick et al., 2021)
In addition to size, particle shape, and symmetry of NPs is equally important for drug delivery efficiency since it affects the transport and diffusion of NPs. Based on a detailed free energy analysis, the effect of shape was found to be mainly induced by the different membrane bending energies during endocytosis. Spherical particles demonstrated smooth movements with minimal membrane bending energy attributed to their inherent symmetry while irregularly shaped particles tumbled with the flow. The effect of the shape is more pronounced in filtering organs like kidney, spleen, and liver (Figure 1). The spherical NPs thus demonstrated the fastest internalization rate, followed by cubic-, then rod- and disk-shaped NPs (Seleci et al., 2016). Although size and shape are essential features, surface charge and functionality are also significant for the interaction of NPs with the biological system
FIGURE 1.
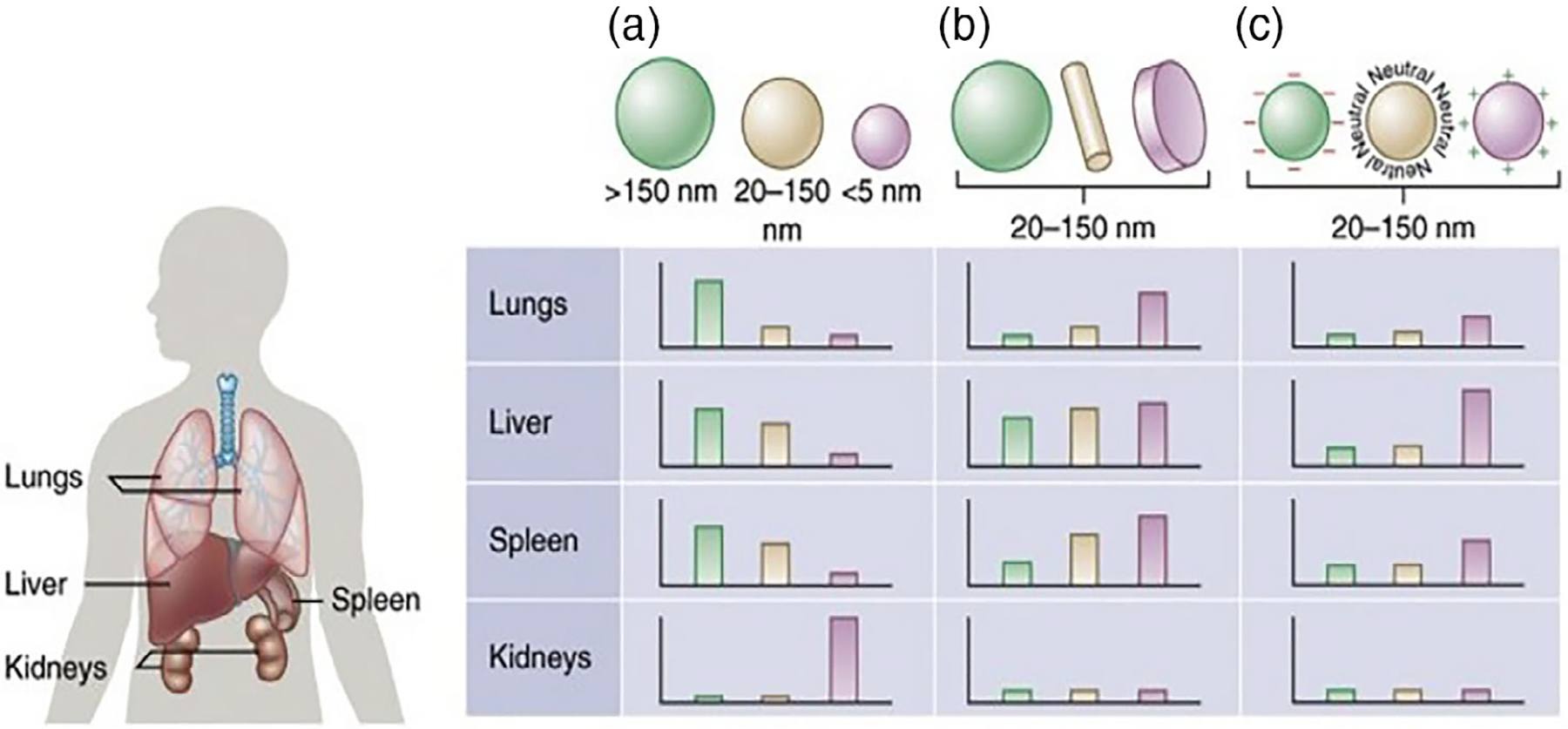
Effect of nanoparticle (a) size, (b) shape, and (c) surface charge on biodistribution among the different organs including the lungs, liver, spleen, and kidneys (adapted from Blanco et al., 2015)
Enhanced permeability and retention (EPR) have been utilized as guiding strategies for the targeted delivery of anti-cancer NPs. On the other hand, RES plays a critical role in the clearance of NPs from the body. Both RES system and EPR effect play a significant role in the pharmacokinetics of NPs and consequently, the PD effect of encapsulated drugs.
Overcoming clearance by RES has long been a major challenge in using NPs as carriers for drug delivery. RES is a network of phagocytic cells (monocytes, macrophages, dendritic cells, and neutrophils) whose primary function is to sequester and clear foreign materials found in blood and lymph nodes. The RES predominantly exists in the liver and spleen (Bourquin et al., 2018). NPs larger than 200 nm are cleared by the phagocytic cells in the RES while particles smaller than 10 nm can be easily cleared by other physiological systems such as filtration through the kidney. It is well documented that therapeutic NPs in 20–200 nm size range accumulate better in tumors because they are not easily recognized by the RES, therefore, possessing longer circulation. PEGylation is the most common strategy to escape RES (impart stealth) and prolong systemic circulation time. While modification of the surface of NPs with polyethylene glycol allows the particles to avoid clearance by macrophages in the RES, it also suppresses internalization by the target cells (Salvioni et al., 2019; Tang et al., 2019).
Surface chemistry is another important factor that affects the interaction of NPs with biological environments. The surface of NPs is usually hydrophilic to enable them to escape macrophage capture. This is typically achieved by coating the surface of NPs with a hydrophilic polymer such as PEG. Surface modification of NPs with PEG was found to reduce NPs accumulation in off-target organs such as liver and spleen, offering stealth properties. PEG-coated NPs have a high solubility in several solvents and exhibit reduced adsorption of blood proteins, leading to a prolonged circulation half-life compared to non-PEGylated NPs. Mechanistically, the conjugation of PEG to the surface of liposomes leads to steric hindrance that reduces the interaction of liposomes with plasma proteins. As a result, this prevents plasma proteins, such as opsonin, from adsorbing to the surface of liposomes reducing opsonization and uptake of liposomes by RES. The success of stealth NPs for tumor therapy is highly dependent on reduced RES uptake and prolonged circulation time in the blood. While PEGylation is important for liposomal stealth, it has also been demonstrated that it causes hindrance to cellular uptake of encapsulated drugs. A study by Soininen et al. demonstrated lower cellular uptake of Caelyx® in rat glioma cell line; approximately 3-fold higher extracellular concentrations compared to DOX in free form (Soininen et al., 2016). Cellular concentrations of free DOX increased linearly with the extracellular concentrations (range of 0.011 to 8.7 μg/ml), while the fraction of free DOX found in nuclei decreased from approximately 100% to 20% as the extracellular and cellular concentrations increased. Nuclear accumulation of DOX was shown to be dependent on the cellular drug concentration. Soininen et al. concluded that PEGylation (with 5 mol% of lipids) forms a “steric” barrier on the surface of liposome reducing interactions with the cell surface and suppressing endocytosis. Poor cellular internalization of other PEGylated liposomal DOX like LipoDox® have also been shown in murine mammary carcinoma cells using fluorescence imaging. Lesser degree of PEGylation (2.5 mol% of lipids) did not prevent interactions with the cell surface (Soininen et al., 2016).
In addition to conferring stealth, the PEG chains on the surface of NPs can be functionalized with alcohols, carboxylic acids amines, and thiols to conjugate small molecules or target ligands. Coating with Polysorbate 80 has demonstrated increased delivery of DOX using NPs, since polysorbate 80 is an inhibitor of P-glycoprotein (P-gp), which obstructs chemotherapeutic agents like DOX (a P-gp substrate) from entering the cytosol. Also, the surface charge of NPs also impacts its distribution and interaction with cell membranes. Surface charge is commonly characterized as zeta potential, which reflects the electrostatic potential of particles. Particles with a zeta potential more positive than +30 mV or more negative than −30 mV are normally considered to be physically stable. The negative membrane of cells interacts differently with positively/negatively charged NPs. Positively charged NPs are generally known to be more easily internalized than neutral and negatively charged NPs (Soininen et al., 2016).
EPR serves a unique mechanism that allows high-molecular-weight molecules to accumulate in tissues conferring increased vascular permeability at sites of inflammation or cancer. In comparison to normal cells, EPR effect is prolonged in cancerous cells and facilitates the supply of nutrients and oxygen for rapid growth. The blood vessels in tumor area are disrupted therefore having higher vascular permeability, with poor lymphatic drainage. This physiological anomaly has been widely exploited for passive drug delivery (convection or passive diffusion) of NPs (micelles, liposomes, protein–polymer conjugates, molecules bigger than 40 kDa) and antibody therapy: NPs leak from the tumor vessel and accumulate in tumor tissues (Chenthamara et al., 2019). Also, NPs can extravasate from leaky tumor vessels if their hydrodynamic diameter size is larger than the renal clearance threshold (Shi et al., 2020). Lipid NPs exhibit preferential extravasation because tumor neo-vasculature contains apertures with diameters up to 200 nm, leading to penetration of small particulate systems into the tumor tissue.
The EPR effect has been effectively demonstrated in a variety of small animal xenograft models, but the prominence and extent of this in humans is strongly debated. For instance, PEGylated liposomal DOX (Doxil®, 85 nm) has higher tumor accumulations and better efficacy in mouse breast cancer xenograft models, but showed no efficacy difference in treating human metastatic breast cancer in comparison to DOX (Sun et al., 2020). Researchers have demonstrated that mechanisms of entry of NPs into solid tumors are very complex (Lammers, 2019; Sindhwani et al., 2020) and immune cells in the tumor microenvironment play a significant role in the accumulation, retention, and intra-tumoral distribution of NPs (Miller et al., 2015). Although EPR provides a strategy to deliver drugs to the targeted tumor, alternative strategies are needed to improve targeting for larger payloads. One such strategy is active targeting. With active targeting, the surface of NPs is decorated with certain ligands to specifically bind to the target cells. This method has been proven to improve the efficiency of drug delivery to tumors.
4 |. SOFT NANOFORMULATIONS OF COMMON CHEMOTHERAPEUTICS
At present, almost 250 nanotechnology-based formulations of chemotherapeutic drugs have been approved worldwide (Ahmad et al., 2021). Since the introduction of Doxil® in 1995, several nanoparticulate delivery systems of chemotherapeutic drugs have received regulatory approval and become available on market. A few popular products include Abraxane® (albumin-bound Paclitaxel), DaunoXome® (non-PEGylated Daunorubicin liposome), Onivyde® (PEGylated Irinotecan liposome), DepoCyt® (Cytarabine liposome), Marqibo® (non-PEGylated Vincristine liposome), Lupron Depot® (PLGA microspheres).
Of all anti-cancer drugs, paclitaxel (PTX) and doxorubicin (DOX) are the most extensively researched for nanomedicine development. The most plausible reason is both of the drugs have broad-spectrum efficacy against numerous types of cancers and offer widespread clinical utility. In addition, these two drugs have been used as the most innovative nanotechnology platforms for drug delivery. Various kinds of nanocarrier systems have been successfully developed and marketed for these two drugs. Thus, in this section, we will discuss PTX and DOX nanoformulations that have been approved, marketed, and are being further investigated in the preclinical/clinical stages of development.
4.1 |. Paclitaxel
PTX belongs to the taxane class of compounds, one of the most important groups of oncology drugs. It is currently approved by FDA for ovarian, breast, and lung cancer, as well as Kaposi’s sarcoma. Conventional taxanes (PTX, docetaxel) are associated with significant toxicity, including myelosuppression, peripheral neuropathy, myalgia/arthralgia, cardiovascular events, alopecia, and gastrointestinal toxicity. Neutropenia is dose-dependent and has dose-limiting toxicity (Slingerland et al., 2013).
One of the major challenges of using PTX as an anticancer agent is its extremely low water solubility (approximately 0.4 μg/ml). For parenteral use, it is formulated at 6 mg/ml in a mixture of polyoxyethylated castor oil (Cremophor EL, CrEL, Kolliphor® EL) and dehydrated ethanol (50:50, v/v) to overcome poor solubilization of PTX. This formulation is known as Taxol® and is the first PTX formulation to be approved by FDA (Ma & Mumper, 2013).
Prior to administration, Taxol® is diluted to a concentration of 0.3–1.2 mg/ml. Despite dilution, the concentration of Cremophor EL necessary to solubilize the required dose of PTX, is significantly higher and causes serious hypersensitivity reactions in humans (Li, Zhang, He, et al., 2018). In addition, it also carries risk of adverse effects such as peripheral neurotoxicity. In the initial clinical use of PTX, the incidence of severe infusion-related hypersensitivity reactions was approximately 41%, and nearly 2% of patients experience potentially life-threatening reactions (Berger et al., 2015). Consequently, premedication and prolonged infusion time are required (175 mg/m2 over a period of 3 h). In addition to the adverse effects mentioned above, the alteration of PK profile of PTX to an unpredictable nonlinear plasma pharmacokinetics, has been well established and attributed to the presence of Cremophor EL (Stage et al., 2018). The nonlinearity of PTX pharmacokinetics is exhibited by both Cmax and CL with disproportionate increases with dose. Moreover, PTX is a known substrate for several ATP-binding cassettes (ABC) efflux transporters, including P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance-associated proteins2 (MRP2) which actively pushes PTX out of the cells and is accountable for very low oral bioavailability and induces drug resistance. In addition, PTX is also a substrate for the influx transporters, organic anion transporter (OAT) polypeptide 1B3 (OATP1B3) and renal OAT2, responsible for hepatic and renal uptake, respectively, and play an important role in the distribution and elimination of PTX (Nieuweboer et al., 2014; Stage et al., 2021).
4.1.1 |. Nanoformulations of PTX
Considerable efforts have been put forward to formulate novel nano-delivery systems of PTX to overcome the inherent disadvantages of toxicity associated with the drug and Cremophor EL in Taxol®. To overcome clinical challenges presented by Taxol®, different and innovative drug formulations have been employed (Slingerland et al., 2013). The incorporation of functional and active ligands to the surface of PTX lipid nano-formulation allows specific targeting to tumor cells, increasing the therapeutic concentration of the drug in the tumor and significantly reducing off-target effects. Several of these approaches have resulted in non-Cremophor formulations and drug delivery systems, such as emulsion, micelles, water-soluble prodrugs, and conjugates which either are approved/marketed or currently under clinical investigation (Table 2). The major advantage exhibited by most of these new products is reduced adverse effects, more favorable tolerability profiles, and superior antitumor efficacy. PTX is marketed in three different nanoplatforms for its parenteral delivery: polymeric micelles (Genexol®, Nanoxel®, and Paclical®, Apealea®), polymeric NPs (Abraxane®), and liposomes (Lipusu®). Several other products are under investigation, including polymeric conjugates (CT-2103, PNU166945), polymeric micelles (NK105, Paxceed®), liposomes (LEP-ETU), and many more.
TABLE 2.
Paclitaxel (PTX) formulations
| Formulations | Manufacturer | Composition details | Regulatory status |
|---|---|---|---|
| Approved/marketed | |||
| Taxol® | Bristol-Myers Squibb | Each ml of sterile nonpyrogenic nonaqueous solution contains 6 mg paclitaxel, 527 mg of purified Cremophor®
EL* (polyoxyethylated castor oil) and 49.7% (v/v) dehydrated alcohol, USP. Drug Loading: approximately 1% wt PTX |
Approved in 1992 (Weaver, 2014) |
| Abraxane® (nab-paclitaxel; ABI-007) | Abraxis BioScience, Inc. (now Celgene) | Each single-dose vial contains 100 mg of paclitaxel (bound to human albumin) and approximately 900 mg of human albumin (containing sodium caprylate and sodium acetyl tryptophanate). Each milliliter (ml) of reconstituted suspension contains 5 mg paclitaxel formulated as albumin-bound NPs Drug loading: approximately 10% wt PTX |
Approved by FDA in 2005 for metastatic breast. In 2012, approved for lung (NSCLC) and pancreatic cancer [FDA label] |
| Lipusu® | Luye Pharma Group Ltd. | Liposomes lecithin and cholesterol in a ratio of 87:13 wt% (Steffes et al., 2017; Ye et al., 2013) | Approved in China in 2003 for ovarian cancer, breast cancer, and nonsmall cell lung cancer (Ye et al., 2013) |
| Apealea® Paclical® |
Oasmia Pharmaceutical AB | Micellar paclitaxel containing XR-17 (N-(all-trans-retinoyl)-l-cysteic acid methyl ester sodium salt and N-(13-cis-retinoyl)-l-cysteic acid methyl ester sodium salt t; Vergote et al., 2020) One vial of powder (Greenish yellow to yellow powder) contains 60 mg of paclitaxel. After reconstitution, each ml of solution contains 1 mg of paclitaxel (micellar) |
Authorized for marketing by the European Commission in 2018. Apealea® in combination with Carboplatin is approved for the treatment of adult patients with the first relapse of platinum-sensitive epithelial ovarian cancer, primary peritoneal cancer, and fallopian tube cancer. Elevar Therapeutics licensed the global rights for Apealea® except for Nordics, Russia, and select CIS countries. |
| Genexol PM | Samyang Biopharmaceuticals Corporation | Lyophilized mPEG-PDLLA polymeric micellar formulation of paclitaxel. mPEG-PDLLA [monomethoxy-poly (ethylene glycol)-block-poly (d,l-lactide)] as solubilizer. Prepared with Samyang’s plant cell culture technology and proprietary polymeric micelle technology. Drug loading: approximately 16% wt PTX |
Approved in South Korea in 2007 |
| Clinical Trials | |||
| EndoTAG-1® (SB05PC) | Medigene SynCore Biotechnology Co., Ltd., Taipei, Taiwan | Paclitaxel [Liposomes containing cationic lipid dioleoyloxypropyltrimethyl ammonium (DOTAP) and neutral lipid (DOPC)] [DOTAP:DOPC:PTX in 50:47:3] | FDA grants orphan drug designation in 2009 Phase 3 clinical trial (NCT03126435 and NCT03002103) for locally advanced and/or metastatic adenocarcinoma of the pancreas |
| LEP-ETU (Liposome Entrapped Paclitaxel Easy to Use) | Neopharm Labs Sponsored by Insys Therapeutics | Paclitaxel (liposome composed of a 90:5:5 molar ratio of DOPC, cholesterol, and cardiolipin and a final total lipid to drug molar ratio of 33:1). | FDA granted Orphan Drug Designation in 2015 for Ovarian cancer |
| NK-105 | NanoCarrier, Chiba, Japan | Self-assembled micellar formulation from PEG-poly (aspartic acid) (PEG-P[Asp]) modified with 4-phenyl-1-butanol to increase the hydrophobicity NK-105 was formulated as a freeze-dried formulation and administered as an infusion Drug loading: approximately 23% w/w. Average particle size of 85 nm (20–430 nm) |
Phase 3 (NCT01644890) for treatment of metastatic breast cancer Phase 3 for recurrent gastric cancer. |
| Taxoprexin® (docosahexaenoic acid-paclitaxel conjugate) | Luitpold Pharmaceuticals, Inc. | Prodrug of PTX A prodrug composed of the naturally occurring omega-3 fatty acid (docosahexaenoic acid, DHA) covalently conjugated to paclitaxel. Contains 73% parent drug |
Phase 3 study (in combination with Carboplatin in patients with Non-Small Cell Lung Cancer; NCT00243867) |
| Paclical® poliglumex (XYOTAX, CT-2103) | CTI Life Sciences, Ltd | Ester conjugate of α-poly (l)-glutamic acid (PGA) and paclitaxel Prodrug of PTX (consist of 37% parent drug) Reconstitution with 10 ml sterile water for injections yields a solution containing 9 mg/ml paclitaxel, in conjugated form with 5 mg/ml poloxamer 188, 20 mg/ml disodium phosphate, and 16 mg/ml sodium dihydrogen phosphate. |
Phase 2 study (in combination with Capecitabine in patients with breast cancer; NCT00265733) |
| Paclitaxel-hyaluronic acid | Fidia Farmaceutici S.p.A. | Hyaluronic acid-Paclitaxel conjugate | Orphan drug designation in 2021 for treatment of Malignant Mesothelioma Phase I trial (NCT04798703) |
| ANG1005 | Angiochem Inc | Paclitaxel is linked to angiopep-2 (brain peptide vector). | Open-label Phase 3 study n HER2-negative breast cancer patients with the newly diagnosed leptomeningeal disease and previously treated brain metastases. Phase 3 trial (NCT03613181) |
Micellar formulations of PTX
Several polymeric micellar formulations of PTX have been approved and marketed. For example, Genexol-PM® is a polymeric micellar formulation, composed of hundreds of amphiphilic PLA-b-PEG diblock co-polymers with a diameter of 20–50 nm and 16 wt% of PTX, as compared to 1% wt in Taxol® (He et al., 2016). PEG and PLA in Genexol-PM® have molecular weights of 2000 and 1750 g/mol, respectively. These micelles interact with cell membrane and release the encapsulated PTX. Genexol-PM® does not increase the circulation time of PTX, consistent with the rapid release of PTX from PEG-b-PLA micelles (Cho et al., 2016). After release, PTX enters the cells via the lipid/raft/caveolae-mediated endocytosis pathway. Genexol-PM® showed 2- to 3-fold higher drug concentration in tumors of nude mice, as compared to Taxol® indicating improved In vivo antitumor efficacy. In a phase I trial, Genexol-PM® demonstrated a demonstrated 3-fold higher MTD (maximum tolerated dose) than Taxol®. Also, plasma AUC and Cmax increased by 3 and 4 folds, respectively, when the dose increased from 80 to 200 mg/m2 suggesting dose-proportional pharmacokinetics of PTX from Genexol-PM® across the studied dose range. Phase II clinical studies found that Genexol-PM® is safe and effective in patients with metastatic breast or advanced pancreatic cancer. Genexol-PM® plus carboplatin, as a first-line treatment, was evaluated in a Phase II trial in epithelial ovarian cancer patients and have demonstrated non-inferior efficacy and well-tolerated toxicities compared with the standard PTX regimen (Lee et al., 2018). Paxceed® is another polymeric micelle in which PTX is encapsulated in PLA-b-mPEG diblock co-polymers. This micelle was investigated for the treatment of severe psoriasis and rheumatoid arthritis.
The aqueous solubility of PTX is significantly increased when it is coupled with water-soluble polymers or encapsulated by lipid-based NPs and use of novel excipients composed of a surfactant-based derivative of retinoic acid (XR-17). This eliminates the need for organic solvents like CrEL and ethanol to solubilize PTX, thus reducing the side effects caused by these excipients. XR-17 is Oasmia’s proprietary nontoxic surfactant-based derivative of retinoic acid excipient. In the aqueous solution for infusion, these constituents are soluble and form micellar NPs with a size of approximately 20–40 nm and eliminate the need for solvents like CrEL. Moreover, XR-17 facilitates ease of administration and allows for higher doses for drugs including PTX. Once the encapsulated molecule is released, XR-17 is metabolized naturally. It received market authorization by the European Commission in November of 2018 for use in epithelial ovarian cancer (Borga et al., 2019; Vergote et al., 2020). Apealea® is an Cremophor®-free, water-soluble, intravenously injectable micellar formulation of PTX, developed by Oasmia Pharmaceutical AB in Sweden utilizing their proprietary XR17 micelle platform technology. The formulation comprises two novel micelle-forming excipients, notably N-(all-trans-retinoyl)-l-cysteic acid methyl ester sodium salt and N-(13-cis-retinoyl)-l-cysteic acid methyl ester sodium salt. The drug:surfactant ratio was optimized to 1:1.33 (w/w). Using the same technology, a veterinary product of PTX has been developed, known as Paccal Vet-CA1 (Reckelhoff et al., 2019).
Liposomal formulations of PTX
Liposomal formulations have been deployed for the delivery of chemotherapeutic agents to increase the solubility of drugs, enhance blood circulation, and improve accumulation in tissues. Lipusu® is the only commercialized liposomal formulation of PTX available in China. It is composed of lecithin and cholesterol, with a particle size of 400 nm. The formulation is approved by the State Food and Drug Administration of China in 2006 (Zhou et al., 2021).
A new promising approach has been the use of electrically charged lipids to achieve an electrostatic attraction between the charged lipid and oppositely charged drug to create a stable liposome drug formulation, as developed in LEP-ETU (Liposome Entrapped PTX Easy to Use). The use of synthetic electrostatic cardiolipin has enabled the liposome encapsulation of a variety of chemotherapeutic agents, including PTX (Slingerland et al., 2013). The mean particle size of the liposomes is about 150 nm before and after lyophilization, and the drug-entrapment efficiency is in the range of 94%–100% (Zhang, 2005) Stability data indicated that the lyophilized LEP-ETU was physically stable for at least 12 months at 2–8°C and chemically stable for at least 12 months at 25°C. The LEP-ETU formulation allows for the possible administration of PTX to patients without the need for premedication with corticosteroids because the well-characterized, synthetic phospholipids, and cholesterol appear to be better tolerated than polyethoxylated castor oil.
EndoTAG-1 is another novel liposomal formulation of PTX. These cationic liposomes displayed superior anti-vascular and antiangiogenic activities by binding and internalizing tumor endothelial cells after intravenous administration. With this formulation, the cytostatic activity of PTX is targeted and limited to the activated tumor endothelial cells (Chen & Su, 2020). An active, randomized, controlled, open Label, Adaptive Phase-3 Trial is ongoing to evaluate the safety and efficacy of EndoTAG-1 plus gemcitabine versus gemcitabine alone in patients with measurable locally advanced and/or metastatic adenocarcinoma of the pancreas (NCT03126435).
Conjugation with biomolecules
Another strategy for maximizing the anticancer effects of PTX and specifically targeting tumor delivery is by generating novel conjugates with biomolecules. Some examples of this strategy include Abraxane® (serum albumin), Taxoprexin® (Docosahexaenoic acid [DHA]–PTX), Paclical® poliglumex (poly-[l-glutamic acid]-PTX), hyaluronic acid–PTX conjugate, and PTX–lipoate.
Abraxane®, albumin-bound PTX was the first PTX nano-formulation approved by the FDA in 2005 for metastatic breast cancer. PTX is conjugated to human serum albumin (HSA) and is actively moved into tumors via a selective 60 kDa glycoprotein (gp60) receptor (albondin). This formulation showed reduced toxicity compared to Taxol®, and the total dose can be administered within 30 min without pretreatment.
Taxoprexin® is a 2′-O-acyl conjugate of PTX covalently bound to the essential natural fatty acid docosahexaenoic acid (DHA) via an ester bond. DHA (22 carbon atoms) is a prevalent fatty acid and is approved for exogenous administration by the European regulatory authorities and the World Health Organization. The DHA–PTX conjugate is nontoxic and does not have microtubule assembly activity. The inert prodrug accumulates preferentially in tumor tissue, where it is cleaved to PTX by esterases (Payne et al., 2006). Equitoxic or equimolar doses of DHA-PTX resulted in significantly greater intratumor concentrations of PTX and a larger cytotoxic effect than PTX alone, which allowed the administration of 4.4-fold higher doses. The unique pharmacokinetics profile of this PTX conjugate may partly be explained by extensive binding to plasma proteins. The clinical preparation of DHA–PTX is formulated in a vehicle containing 80% less CrEL and ethanol on a molar basis than the standard formulation PTX. This agent can be reconstituted in dextrose 5% to a maximum concentration of 8 mg/ml and administered intravenously over 2 h every 21 days (Bradley et al., 2001; Sparreboom et al., 2003). At the recommended Phase 2 dose of 1100 mg/m2, DHA–PTX exhibited a small volume of distribution, a long terminal half-life, and a slow system clearance. The clinical dose of DHA–PTX during Phase 1 study was set to 900 mg/m2 (molar equivalent of 657 mg/m2 of PTX) based on toxicity among the first 13 patients treated 1100 mg/m2 (molar equivalent of 803 mg/m2 of PTX) for every 3 weeks. At the recommended Phase 2 dose of 1100 mg/m2, DHA–PTX exhibited a small volume of distribution, a long terminal half-life, and a slow system clearance. A Phase 3 trial was conducted to compare the efficacy and toxicity profiles of DHA–PTX with those of dacarbazine for the treatment of metastatic malignant melanoma. There were no significant differences in response rate, duration of response, time to progression, and time to treatment failure between the two drugs. Myelosuppression was more common with DHA–PTX (Bedikian et al., 2011). DHA–PTX has little activity in patients with advanced non-small cell lung cancer (900 mg/m2), as a single agent (Payne et al., 2006).
ANG1005 is a PTX-Angiopep-2 peptide conjugate with 3:1 molar ratio. Angiopep-2 is a 19 amino-acid peptide targeting low-density lipoprotein receptor-related protein 1 (LRP1) receptor to aid PTX to across blood-brain barrier to treat brain tumor. In preclinical studies, ANG1005 showed improved antitumor efficacy and increased survival time in several mouse models. In the clinic, the brain penetrating peptide–drug conjugate showed activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases (Kumthekar et al., 2020).
PTX-lipoate (IDD-1040) is a conjugate formed by adding a lipoic acid moiety to the C2′ of PTX. IDD-1040 was studied in a nonsmall-cell lung cancer (NSCLC) xenograft mouse model (administered intravenously in a vehicle containing 14% CrEL, 3.5% ethanol, and 82.5% saline) and demonstrated a significantly increased MTD (250 mg/kg) compared to PTX alone (20 mg/kg). Furthermore, IDD-1040 showed a higher anti-tumor efficiency, which was dose-dependent. More interestingly, the anti-tumor activity lasted for 2 weeks after stopping IDD-1040 treatment, while for PTX, after stopping the treatment the tumor relapsed. This proved that IDD-1040 has excellent tumor inhibition efficiency with controlled release (Falah et al., 2019).
4.1.2 |. Pharmacokinetics of PTX in nanoformulations
Compared to its original co-solvent formulation, the PK disposition of PTX is significantly altered by encapsulating it in NPs. In this section, we have discussed in detail the preclinical and clinical pharmacokinetics and biodistribution pattern of PTX from co-solvent formulation, Taxol®, and other nanoformulations. A comparison of protein-bound PTX (Abraxane®) and Taxol® has been discussed in detail to show the impact of nanoparticle formulation on the PK of PTX.
Cremophor EL formulation
Taxol® consists of 30 mg PTX dissolved in 5 ml of 1:1 v/v mixture of polyoxyethylated castor oil derivative Cremophor® EL (CrEL) and dehydrated ethanol. CrEL is a formulation vehicle that is used to dissolve very poorly water-soluble drugs including anesthetics, vitamins, and so forth (cyclosporin A, tacrolimus), and some anticancer drugs (teniposide, Didemnin B, Aplidine; Gupta & Middleton, 2011; Nan, 2015). Contrary to the requirement of excipient inertness, CrEL is known to be pharmacologically active and exerts a range of biological, some of which have significant clinical implications. The average amount of CrEL administered to a patient in a single dose is 5 ml (range, 1.5–10.3 ml), but PTX is an exception since the amount of CrEL is approximately 26 ml per administration. A patient receiving a regular dose of 175 mg/m2 of PTX, is also receiving a high dose of about 14 ml/m2 of Cremophor (Aronson, 2016). Clinically, the most widely used dosage regimen for Taxol® is a 3-h infusion of 175 mg/m2. Following IV administration, plasma concentrations of PTX declined in a biphasic manner, with the initial rapid decline representing distribution from the central to the peripheral compartment (blood to various organs and tissues) and the slower second phase representing drug elimination from the central compartment. The terminal half-life was about 27 h.
CrEL, a surfactant used to increase the solubility of PTX in Taxol®, is known to alter PK of several intravenously administered drugs (Aronson, 2016). One of the important improvements is significant increase in drug circulation with concomitantly reduced systemic clearance. A 1.7-fold increase in PTX dose given as 3-h infusion the Cmax increases 3-fold. The nonlinear PK disposition of PTX is observed in both preclinical and clinical studies with Taxol®. Several studies have described the PK behavior of PTX from Taxol® as nonlinear disposition. Earlier speculations thought it to be a result of saturable distribution and elimination processes (van Zuylen et al., 2001). However, the currently accepted fact based on numerous experimental data is that CrEL causes micellar encapsulation of PTX which leads to change in free drug concentration (Borga et al., 2019). A study by Sparreboom et al., investigated the pharmacokinetics of PTX at different dosages and drug formulations (Cremophor EL-ethanol, Diethyl acetamide, and Tween 80-ethanol) to determine the effects of Cremophor EL on the disposition of PTX (2–10 mg/kg in mice) showed nonlinear PK behavior with decreasing clearance with increasing doses, whereas formulations with DMA and Tween 80 showed linear distribution and elimination processes (Sparreboom et al., 1996). Further, in vitro studies have demonstrated that CrEL alters blood distribution by entrapment of PTX in the hydrophobic core of CrEL micelles, reducing free drug fraction for cellular partitioning. It has been shown that the encapsulation amount of total PTX in micelles increases disproportionately with a higher amount of CrEL, thereby reducing the unbound (free) PTX concentration in tumor, tissues, metabolism, and biliary and intestinal secretion (Chiang & Yang, 2021). The binding affinity of PTX is highest for CrEL (when concentration is above the critical micellar concentration, i.e., 0.01%). followed by plasma proteins and human serum albumin (Scripture et al., 2005).
A comprehensive PK analysis carried out by van Zuylen et al. (2001), compared whole blood and plasma levels of PTX to determine the root cause of its nonlinear disposition and identify the saturable kinetic process accountable for this phenomenon. Seven patients with solid tumors were treated with PTX infused over 3 h, each at consecutive 3-weekly dose levels of 225, 175, and 135 mg/m2 (CrEL dose levels, 18.8, 14.6, and 11.3 ml/m2, respectively). Results indicated that PTX Cmax and AUCs in whole blood increased linearly with dose, whereas plasma levels showed substantial deviation from linearity. In addition, dose-dependent plasma clearance of PTX was observed, which decreased significantly with an increase in drug dose (16.7 ± 2.53 L/h/m2 at 135 mg/m2 to 9.75 ± 2.78 at 225 mg/m2, p = 0.03). However, whole blood clearance is independent of dose. Blood to plasma concentration ratio changed significantly from 0.83 ± 0.11 (135 mg/m2) to 0.68 ± 0.07 (225 mg/m2; van Zuylen et al., 2001). At Cmax, Cb/Cp ratios decreased in a dose-dependent manner from 0.83 ± 0.11 (135 mg/m2) to 0.80 ± 0.18 (175 mg/m2) and 0.68 ± 0.07 (225 mg/m2). The terminal half-life was consistent across the different dose levels.
These observations confirmed that CrEL is responsible for the nonlinear disposition of PTX by disproportionate drug accumulation in the plasma fraction (preferential affinity of PTX for Cremophor) thus leading to concentration-dependent decrease in PTX uptake in red blood cells. With this data, a three-compartment PK model was developed that could precisely describe the blood and plasma concentration of PTX and validated the theory of micellar encapsulation of PTX in CrEL (Figure 2). In contrast to standard linear compartmental, this model assumes the existence of PTX in bound, unbound, and micellar forms which are available for distribution (van Zuylen et al., 2001).
FIGURE 2.
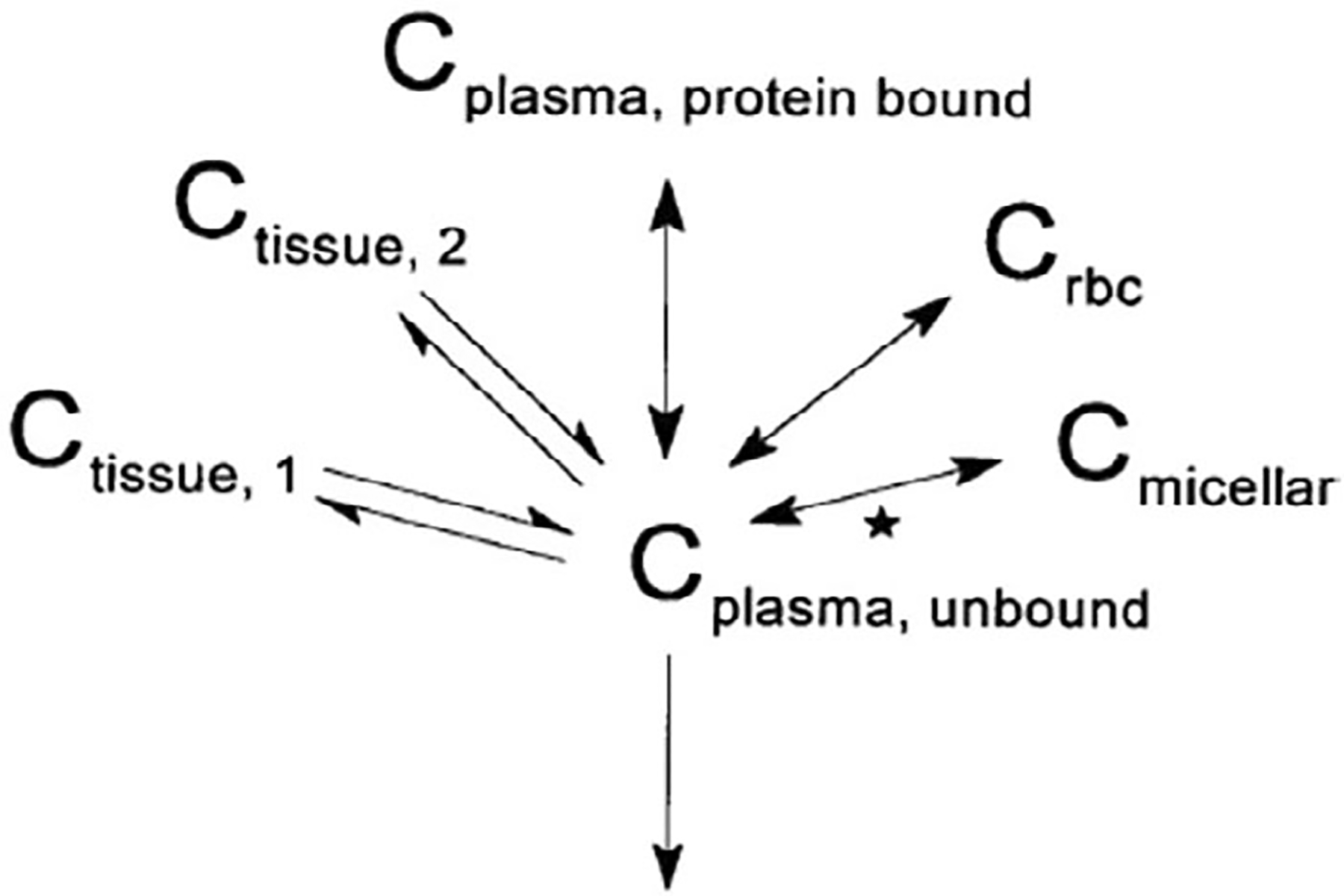
Proposed model of paclitaxel pharmacokinetics, assuming pharmacokinetically distinguishable forms of paclitaxel in the blood compartment; Pharmacokinetically distinguishable forms: Unbound (CUnbound), bound to plasma protein (CBound), in micellar form together with CrEL (CMicellar), and bound to or distributed into red blood cells (Crbc). Further, C was assumed to be in equilibrium with tissues (Double-headed arrows denote processes with assumed instantaneous equilibrium. The star indicates a nonlinear process; van Zuylen et al., 2001)
Another interesting aspect is that PTX pharmacokinetics are nonlinear for short infusion times (approximately 6 h) but not for longer infusions (>24 h), especially for CrEL-containing PTX formulations (Stage et al., 2018). The plasma concentration of PTX appeared to exceed the metabolic capacity of its elimination pathways when administered at higher doses with shorter infusion rates (3 h vs. 24 h at dose levels of 135 and 175 mg/m2). This resulted in a significant increase in circulation to PTX with a concomitantly reduced systemic clearance, leading to altered PD characteristics of the solubilized drug and patients at risk for severe systemic toxicities (Hennenfent & Govindan, 2006). A 30% increase in dose with the 24-h infusion, resulted in a disproportional increase in Cmax by 87% (195 vs. 365 ng/ml), whereas the AUC0–∞ remained proportional. However, with a 3-h infusion, a 30% increase in dose, resulted in 68% and 89% increase in Cmax (2170 vs. 3650 ng/ml) and AUC0–∞ (7952 vs. 15,007 ng h/ml), respectively. Extensive systematic data mining was performed on more than 50 clinical studies (30 different doses and six different infusion times) to explore the effect of dose and infusion time on CL, Cmax, and T > 0.05 μM (the time above a threshold PTX plasma concentration [0.05 μM] is important for the efficacy and toxicity of the drug). A strong correlation was illustrated between PTX dose and Cmax; dose-dependent PTX clearance, with CL decreasing with increasing dose. A quadratic equation best described the Cmax–dose relationship for PTX given as a short infusion. In contrast, a linear relationship provides the best fit for data from long infusion times (Stage et al., 2018). Increasing infusion time from 3 to 24 h did not significantly increase T > 0.05 μM. Nonlinear PK of PTX for shorter infusions was attributed to dilution of PTX in Cremophor EL (Stage et al., 2018).
In contrast to PTX, CrEL shows a linear and dose-independent, but schedule-dependent PK behavior. CrEL clearance increased from approximately 160 to 300 to 400 ml/h/m2, when the length of infusion was increased from 1 to 3 to 24 h. Saturation of serum esterase-mediated metabolic degradation is suggested to cause this schedule-dependent behavior, leading to increased systemic exposure and associated side effects of Cremophor (Gelderblom et al., 2001).
Several nanoformulations for PTX have been developed which have demonstrated safety, tolerability, and efficacy outcomes, linear PK, in comparison to Taxol®. More diverse types of cancer can be treated with these new PTX formulations.
Albumin bound formulation
Abraxane® (ABI-007) is a human albumin-bound PTX form. In this formulation, 6–7 PTX molecules are noncovalently bound to albumin forming a primary PTX-albumin aggregate of 4–14 nm. The primary aggregates further join to form bigger PTX-albumin particles (approximately 130 nm), which eventually dissociate rapidly in the bloodstream (Sofias et al., 2017). Abraxane® is approved for pancreatic cancer, in which Taxol® had failed to demonstrate efficacy. The observed differences in tolerability and efficacy between different PTX formulations are speculated to be a result of altered PK characteristics compared to that of Taxol®.
Abraxane® has a mean particle size of approximately 130 nm and is administered as an IV injectable suspension. It is used in the dose range of 135–375 mg/m2, depending on the cancer to be treated. Human serum albumin reversibly binds hydrophobic chemical substances, carrying them through the body to the cell surface to unload. The absence of solvents makes Abraxane® superior to Taxol®, allowing the use of higher doses (without premedication with corticosteroid) with a shorter duration of infusions with comparable side effects and lack of solvent-related hypersensitivity reactions. The Phase III clinical trial of Abraxane® compared its antitumor activity and tolerability in women with measurable metastatic breast cancer, to those of Taxol®. Despite the increased dose of PTX in Abraxane®, the incidence of grade 4 neutropenia was significantly lower than Taxol®. The overall response rate (ORR) for Abraxane® was almost twice that of Taxol® (Wang et al., 2017).
PTX displays linear PK from Abraxane® over the clinically relevant dose range of 135–300 mg/m2, and independent of the duration of administration (based on the available clinical data). The 2.2-fold dose increase results in a 2.2-fold increase in peak concentration (Cmax) and a 2.7-fold increase in AUC and independent of the duration of administration. The mean maximum concentration at the end of the infusion was 18,741 ng/ml (dose of 260 mg/m2). The total volume of distribution is 1741 L, indicating extensive extravascular distribution and/or tissue binding of PTX. The mean total clearance and terminal half-life range from 13 to 30 L/h/m2, and 13 to 27 hours, respectively, across the dose range of 80–300 mg/m2. At the clinical dose of 260 mg/m2 over 30 min infusion, mean values for cumulative urinary recovery of unchanged drug (4%) indicated extensive nonrenal clearance (Abraxane® label, FDA).
The MTD of ABI-007 was ~70%–80% higher than that reported for Taxol® for both an every 3 weeks regimen (300 vs. 175 mg/m2) and a weekly regimen (150 vs. 80 mg/m2). Sparreboom et al. conducted a comparative preclinical and clinical PK study with two clinically relevant PTX formulations: albumin-bound PTX nanoformulation (Abraxane®) and CrEL-containing solvent formulation (Taxol®). The results from the study demonstrated that the volume of distribution at a steady state and clearance for PTX from ABI-007 nanoparticles were significantly greater than those for PTX formulated with CrEL in rats (refer to Table 3). Results showed that Cmax occurred at the end of infusion for both the formulations and it was 6.5-fold greater for Abraxane® than for Taxol® (22,968.6 vs. 3543.3 ng/ml; p approximately 0.001); Tmax was significantly less for Abraxane® as compared to Taxol® (0.36 vs. 2.65 h; p < 0.001), reflecting the shorter infusion duration. However, mean PTX AUC∞ from ABI-007 260 mg/m2 (14,788.6 ng h/ml) was similar to Taxol® 175 mg/m2 (12,602.7 ng·h/ml) despite the difference in dose. PTX clearance and volume of distribution were significantly higher for ABI-007 than for Taxol® in humans [21.13 vs. 14.76 L/h/m2 (p = 0.048) and 663.8 versus 433.4 L/m2 (p = 0.040)], respectively. Terminal half-life of PTX was similar for both formulations which suggested that the primary impact of CrEL on PTX clearance is associated with drug distribution rather than drug elimination. Thus, PK assessment of clinical data showed ~50% greater CL and Vz with ABI-007 than with Taxol®, which is consistent with the results of the preclinical studies (Sparreboom et al., 2005).
TABLE 3.
Comparison of preclinical and clinical data of paclitaxel nanoformulations
| ABI-007(Abraxane®) | Taxol® | |
|---|---|---|
| Preclinical data in rats | ||
| Dose (mg/kg) | 5 | 5 |
| t1/2(h) | 11.42 | 7.24 |
| Tmax (h) | 0.033 | 0.033 |
| Cmax (μg/ml) | 4.0 | 11.8 |
| AUC∞ (μg·h/ml) | 4.59 | 5.85 |
| Vz (L/kg) | 18.33 | 8.75 |
| CL (L/h/kg) | 1.112 | 0.837 |
| Clinical data | ||
| Dose | 260 mg/m2, 30 min (n = 14) | 175 mg/m2, 3 h (n = 12) |
| MTD | 300 mg/m2 | 240 mg/m2 |
| t1/2(h) | 21.6 (17.2) | 20.5 (14.6) |
| Tmax (h) | 0.36 (45.2) | 2.65 (27.6) |
| Cmax (ng/ml) | 22,968.6 (112.5) | 3543.3 (57.2) |
| AUC∞ (ng·h/ml) | 14,778.6 (45.3) | 12,602.7 (21.0) |
| Vz (L/m2) | 663.8 (48.1) | 433.4 (31.1) |
| Cl (L/h/m2) | 21.13 (43.8) | 14.76 (31.8) |
Note: The data presented here has been compiled from the article published by Sparreboom et al. (2005). Preclinical: ABI-007 and Taxol were given i.v. to Harlan Sprague–Dawley male rats (n = 10). An i.v. bolus of either [3H] ABI-007 or [3H] Taxol at a PTX dose of 5 mg/kg was administered. Blood PTX concentration was determined from extracted blood pooled from 10 rats, plotted versus time, and fitted using noncompartmental analysis. Two methods were used for blood PTX quantitation: radioactivity and HPLC. The data from HPLC is presented here. Clinical: ABI-007 (Abraxane®) 260 mg/m2 over 0.5 h or Taxol® 175 mg/m2 over 3 h were randomized to give patients with advanced solid tumors. Data are presented as mean values (% coefficient of variation).
Moreover, two studies with clinical data have shown that the fraction of unbound PTX is inversely correlated to the circulating concentration of Cremophor (Henningsson et al., 2003).
The higher Vz of PTX formulated as Abraxane® (albumin NPs) confirmed that Cremophor in Taxol® prohibited the distribution of PTX out of the circulation and into the tissues. Moreover, the volume of distribution of CrEL is extremely low, implying that the tissue and tumor delivery of CrEL is insignificant and thus tissue accumulation of PTX is low.
Li et al. carried out an extensive investigation on the PK and tissue distribution of different types of nanoparticulate formulations of PTX which represented the approved and marketed formulations. This included albumin-bound PTX, micellar, and polymeric nanoparticle PTX. The plasma PK profile and tissue distribution were characterized and compared to Taxol®, in female CD-1 mice (6–8 weeks old). Each of the formulations with different nanocarrier delivery systems resulted in similar plasma PTX profile but distinctly different tissue concentration-time profiles (Figure 3). All nanoformulations were associated with increased penetration of PTX in all tissues, and increased tissue: plasma ratios compared with pac-T. The nanoformulations of PTX resulted in different penetrations in tissues, which may link to distinct anticancer efficacy and safety profiles in animals and humans. For example, albumin-bound PTX (nab-P; Abraxane®) resulted in higher PTX delivery efficiency to the pancreas compared to pac-P (Paclical®), pac-G (Genexol-PM®), and pac-T (Taxol®). Moreover, higher penetrations in lung and fat pad tissues associated with all the tested nanoformulations (nab-P, Pac-G, and pac-P) might be reflective of improved efficacy in lung and breast cancer treatments. The study established that differences in observed efficacy and toxicity profiles can be explained by the different pharmacokinetics and tissue distribution. These results rationalize the clinical observation of improved survival benefit seen in the MPACT study combining Abraxane® with gemcitabine as compared to the current standard of care, gemcitabine monotherapy in this highly aggressive tumor with limited treatment options.
FIGURE 3.
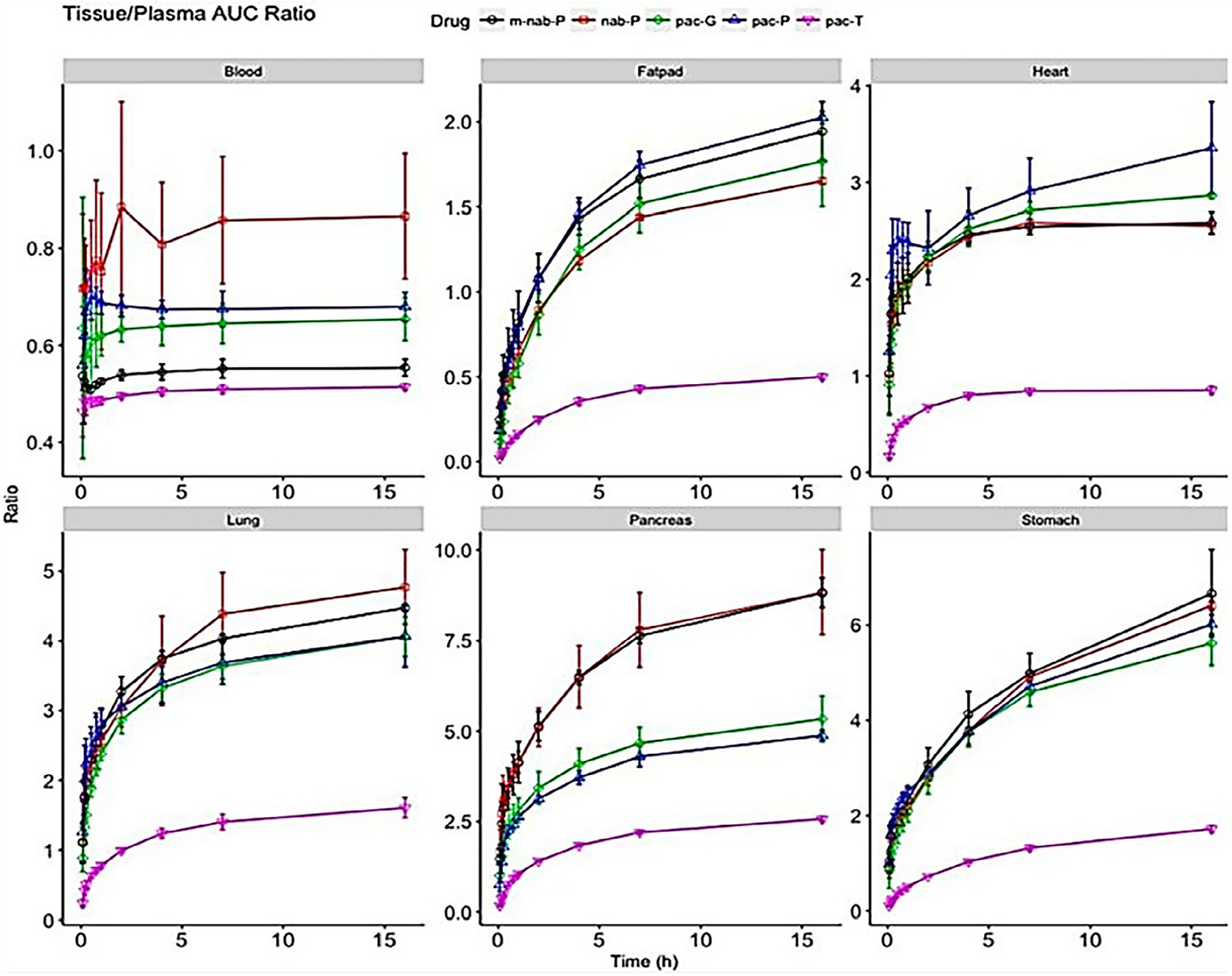
The ratio of area under the curve (AUC0–t) between select tissues (blood, fat pad, heart, lung, pancreas, and stomach) and plasma. Ratio (y-axis) of accumulated AUC in tissues scaled to its corresponding accumulated AUC of plasma for solvent-based PTX (pac-T), nab-PTX (nab-P), mouse albumin nab-PTX (m-nab-P), micellar PTX (pac-P), and polymeric nanoparticle PTX (pac-G; Li, Zhang, Zhu, et al., 2018)
Further, in a recent study, semi-mechanistic model was developed to describe the PK behavior of PTX from different formulations (nab–PTX and CrEL–PTX). Clinical data from patients were analyzed using a semi-mechanistic model in NONMEM, with the simultaneous disposition of PTX–carrier complexes and the total PTX released from the complexes. Sensitivity analysis was performed to identify the key factors driving PTX exposure in circulation and peripheral tissues. nab–PTX demonstrated four times faster and 10 times more extensive distribution to peripheral tissues than that of CrEL–PTX micelles, resulting in distinct tissue PTX profiles. Moreover, sensitivity analyses showed that the plasma concentration–time profile was insensitive to rapid rates of tissue distribution and decomposition of PTX–carrier complexes but the tissue distribution profile of PTX was highly sensitive to it. Tissue distribution of PTX is carrier complex system dependent. Thus, both preclinical studies by Li et al., and the modeling study emphasized that different delivery systems result in distinct tissue PTX profiles but similar concentration–time profiles in plasma or blood (Li et al., 2015).
Similar investigation on the liposomal formulation of PTX, LEP-ETU, also demonstrated the difference in tissue distribution in preclinical studies (Fetterly & Straubinger, 2003) when compared to Taxol®. These studies demonstrated that the clinical PK properties of PTX are markedly affected by the composition characteristics of the drug formulation.
A clinical study by Hertz et al. identified a PTX exposure target associated with an acceptable risk of treatment-limiting peripheral neuropathy (25%). Among several toxic effects of PTX, peripheral neuropathy is the dose-limiting toxicity of weekly PTX administered to patients with breast cancer. Previous work has focused on time above threshold (Tc>0.05) as the exposure parameter of interest. This study suggested that measurement of PTX plasma concentration during the first dose of treatment could be useful to predict peripheral neuropathy that necessitates PTX treatment disruption. As per the model developed, a patient with no prior neuropathy receiving 12 weekly doses of PTX 80 mg/m2 with an exposure of Cmax = 2885 ng/ml or Tc > 0.05 = 14.06 h would have an approximately 25% risk of peripheral neuropathy-induced treatment disruption during treatment. The chosen threshold of 25% risk is like the rates of grade 2+ peripheral neuropathy in clinical trials of weekly PTX which is typically the threshold at which treatment disruption is mandated by the trial protocol (Hertz et al., 2018).
4.2 |. Doxorubicin
DOX is a commonly used chemotherapeutic drug since its discovery in the late 1950s and is indicated for the treatment of numerous cancers. It is an antineoplastic anthracycline antibiotic, originally isolated from cultures of Streptomyces peucetius var. caesius (soil fungus) and semi-synthetically produced. DOX consists of a naphthacene quinone nucleus linked through a glycosidic bond at ring atom 7 to an amino sugar, daunosamine. DOX was approved for medical use in the United States in 1974. Despite its high potency, its utility in clinical oncology is restricted by dose-dependent toxicity (myelosuppression and cardiotoxicity), the emergence of multidrug resistance, and its low specificity against cancer cells.
Plasma pharmacokinetics of DOX (Adriamycin) and its metabolite were first described in 1983 by Greene et al., in metastatic breast cancer patients (n = 10) undergoing single-agent therapy with DOX at the dose of 75 mg/m2 over 15 min i.v. infusion (Greene et al., 1983). The PK disposition of Adriamycin could best be fit to a biexponential equation of the form:
where Cp(t) is Adriamycin concentration (nM), and time, t (h). A Cmax of 3880 ± 1078 nM was observed. A rapid initial distribution phase (half-life of 8 min) was followed by a slower decrease in concentration which was log-linear beyond 6 h (terminal half-life of 30 h). The main drug elimination occurred during the terminal phase where drug concentrations were generally less than 10−7 M (0.05 μg/ml). The volume of distribution and clearance was estimated to be 28 ± 3.3 L/kg and 514 ± 56 ml/min/m2, respectively.
Plasma PK of the major active metabolite, Adriamycinol, was also characterized. An initial rapid increase of Adriamycinol concentration was observed in plasma, followed by a slow declining elimination phase, parallel to Adriamycin, indicating a formation rate limited metabolite elimination kinetics. The relative drug exposure of Adriamycinol to Adriamycin was approximately 50% at steady state (AUCAdriamycin = 4427 ± 418 vs. AUCAdriamycinol = 2137 ± 472 nM-h).
4.2.1 |. Nanoformulations of DOX
Nanoformulations of DOX have offered the obvious benefits of reduced off-target systemic toxicity, controlled drug release, increased tumor penetration and accumulation, altered biodistribution, and thus enhanced the therapeutic effectiveness of DOX (Prados et al., 2012). In addition, encapsulation of the drug into NPs has been shown to be a successful strategy to bypass efflux-mediated drug resistance (Pieper et al., 2019; Onafuye et al., 2019). The first DOX-loaded liposomes were developed to effectively and efficiently treat aggressive tumors, like breast and ovary metastatic cancers and Kaposi’s sarcoma (Lao et al., 2013). The first ever nanoparticulate formulation of DOX was approved in the US in 1995 (Doxil®) and in Europe in 1996 (Caelyx®). These were PEGylated liposomal formulations, also called as “Stealth Liposomes”. Table 4 summarizes the various novel formulations of approved/marketed and investigational formulations of DOX.
TABLE 4.
Doxorubicin formulations
| Formulations | Manufacturer | Composition/Strength | Regulatory status |
|---|---|---|---|
| Adriamycin PFS™ (Doxorubicin HCl Injection, USP; 2 mg/ml sterile, isotonic solution) Adriamycin RDF™ (Powder for Injection; doxorubicin hydrochloride for injection, USP; Sterile red-orange lyophilized powder) |
Pharmacia & Upjohn S.p.A (Now Pfizer) | Each ml contains doxorubicin HCI 2 mg, USP, and the following inactive ingredients: sodium chloride 0.9% and water for injection q.s. Hydrochloric acid is used to adjust the pH to a target pH of 3.0. | Received FDA approval in July 1993 Discontinued in the US |
| Nanoformulations | |||
| Doxil® Caelyx® (PEGylated liposomes) |
Janssen Pharmaceuticals Janssen-Cilag International NV |
2 mg/ml of doxorubicin, 3.19 mg/ml of N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt (DSPE-PEG 2000), 9.58 mg/ml of fully hydrogenated soy phosphatidylcholine (HSPC), and 3.19 mg/ml of cholesterol. Mean particle size: 87.3 ± 8.5 nm |
FDA approval of Doxil® in 1995 EMEA approval of Caelyx® in 1996 |
| MYOCET™ (non-PEGylated liposomes) | Zeneus Teva BV | Single lamellar liposomes (approximately 150 nm) composed of egg phosphatidylcholine: cholesterol (55:45 mole percent) containing 300 mM citrate at pH 4.5. Encapsulation efficiency >95% After encapsulation of doxorubicin, the drug-to-lipid ratio of Myocet is approximately 0.25:1 (wt:wt) and the pH is 6.5–8.5. Encapsulation is achieved via an active loading process, which utilizes an acidic (pH 4.5) proton concentration gradient. The internal complex is a flexible assembly of doxorubicin monomers stacked into fibers that are cross-linked by citrate into a hexagonal array with a 35 Å lattice repeat. Supplied as a three-vial system: doxorubicin HCl, liposomes and buffer, 0.9% (w/v) sodium chloride solution for injection |
Myocet was approved in Europe and Canada in 2000 and is currently undergoing a Phase 3 global trial. |
| LIPODOX (SPIL DXR HCl liposome injection) generic version of Doxil®/Caelyx®) | Sun Pharmaceutical Industries Ltd. | Available as 2 mg/ml (20 mg/10 ml and 50 mg/25 ml) Doxorubicin hydrochloride liposome injection. The drug product appears as a translucent, red liposomal dispersion. | Due to shortage of Doxil® in 2011, FDA allowed temporary import of Lipo-DOX for the treatment of epithelial ovarian cancer (EOC) FDA approved the generic version of Doxil® in 2013 |
| Lipodox® (second-generation PEGylated liposomal doxorubicin) | TTY Biopharm Company Ltd, Taipei, Taiwan | Distearoylphosphatidylcholine (DSPC)/PEG-DSPE liposome with a slightly higher phase transition temperature than HSPC, distearoylphosphatidylcholine (DSPC), and cholesterol with a surface coating of PEG. | Commercially available in Taiwan since 1998 |
| Thermodox® (thermally sensitive liposomal doxorubicin) | Celsion Corporation | DPPC, MSPC, and PEG 2000-DSPE (90:10:4 molar ratio) | Phase III trial (OPTIMA study) for treatment of nonresectable hepatocellular carcinoma (HCC), in conjunction with standardized radiofrequency ablation (sRFA ≥ 45). |
| TLD1/TaliDox® (glycan targeted liposomal Xdoxorubicin) | InnoMedica | Comprises of 1,2-distearoyl-snglycero-3-phosphocholine (DSPC), cholesterol, and DSPE-MPEG 2000 Particle size: approximately 60 nm with a PDI < 0.01 |
Phase I/IIa in Switzerland (NCT03387917) in patients with advanced solid tumors. InnoMedica has decided to submit the dossier for marketing authorization to Swissmedic in the first half of 2021. |
| EGFR(V)-EDV-Dox™ | EnGeneIC Ltd. | EGFR (vectibix sequence)-targeted EnGeneIC delivery vehicle (EDV) containing doxorubicin. Bacterially derived nanocell packages Doxorubicin, into a 400 nm particle which targets specific cancer cells using bispecific antibodies (BsAb) | Phase I ACTRN12609000672257 Phase I NCT02766699 |
In cancer patients with higher risk of cardiac disease (i.e., patients with cardiomyopathy or myocardial infarction), PEGylated liposomal DOX have increased their survival, compared to conventional DOX. Patients treated with liposomal DOX (i.e., Doxil®) exhibited better safety profiles and less adverse effects (cardiotoxicity, nausea, vomiting, and myelosuppression), in comparison to conventional DOX-treated patients. The fewer side effects of the liposome preparations have been attributed to greater accumulation in tumor tissue (lesser myocardial drug concentration) through (EPR) effect. Gyöngyösi et al. performed preclinical studies in pigs to investigate molecular mechanisms and the impact of DOX on gene expression profiles that could explain reduced cardiotoxicity of liposomal DOX (Myocet®) in comparison to free drug. The animals were treated with doses equivalent to human treatment regimens (60 mg/m2 body surface area) as a single 1 h i.v. infusion every 21 days (at Days 1, 22, and 43). Although the plasma concentration of DOX was several folds higher in the liposomal group than the free DOX group (due to faster clearance of the free drug), a lower myocardial concentration was observed in the liposomal DOX group. Also, they observed that interferon-stimulated genes, linked to DNA damage repair and cell survival, were downregulated by DOX, but upregulated by liposomal DOX and the expression of cardioprotective translocator protein was inhibited by free DOX, but not the liposomal formulation. Clearly, liposomal packaging of DOX is an important strategy to lower cardiotoxicity caused by DOX and this is of high clinical importance in anticancer treatment (Gyongyosi et al., 2020).
Talidox® (also known as TLD-1) is a new liposomal DOX formulation with a particle size twice as small as conventional liposomal formulations like Caelyx®; which enables deeper penetration into tumors. A safety assessment study (Phase I/IIa) was conducted on a late-stage cancer patient (advanced solid tumors) by the Swiss Group for Clinical Cancer Research (SAKK) in five Swiss hospitals in November 2018 (NCT03387917). The study aimed to determine an optimal dose and investigate the PK and toxicity profile of Talidox®. The dose of 40 mg/m2 was considered optimal in terms of benefits and side effects. The report on the first clinical study concluded that treatment with Talidox® leads to substantially less pronounced side effects (heart problems, hair loss, nausea, and vomiting) as compared to conventional chemotherapies, Adriblastin, Taxol®, and Doxil®. This includes side effects such as Talidox® reduced tumor growth in 60% of patients’ tumors were substantially reduced in size or almost eliminated in one in seven patients. InnoMedica submitted the dossier for market authorization to Swissmedic in the fall of 2021 (InnoMedica, 2022).
4.2.2 |. Development of novel targeted DOX formulations
Although PEGylated liposomal DOX formulations have a reduced side effects profile compared to free DOX, they do not offer superior efficacy over nonliposomal DOX. Though PEG increases the accumulation of liposomes in tumor by EPR effect, it also creates a steric barrier that could lead to reduced interaction with the target cells, and less uptake of the entrapped drugs. In recent years, several modifications have been attempted on stealth liposomes, such as conjugation with proteins, peptides, antibodies, aptamers, and ligands (transferrin, folate) which are actively taken up by target cells via endocytosis, finally improving overall efficacy.
Numerous molecular-targeting strategies have been explored for liposomal delivery of DOX. Several stimulus-controlled systems using heat, light, and pH to trigger drug release have been developed to address the slow release of DOX from its carrier systems (Carter et al., 2019).
Ligands present in tumor micro-environment have for a long time been exploited for the targeted delivery of chemotherapeutic agents to tumors. Rapidly growing tumor cells require various nutrients and vitamins in large quantities for development, and hence, overexpress many tumor-specific receptors (e.g., folate, biotin, transferrin, EGF). For example, folate receptor is overexpressed in more than 40% of tumors and thus, folic acid has been broadly applied to conjugate to liposomes for higher specific cancer cell uptake. Folate-tagged liposomal DOX have been prepared via the conjugation of folic acid to cholesterol. Biodistribution study showed that folate-tagged liposomes reduced uptake of liposomes in liver and spleen and increased the uptake to a significant level in tumor (Lohade et al., 2016). Long circulating pH-sensitive DOX-liposomes were assembled via the conjugation of folic acid to DSPE-PEG2000. Biodistribution studies using radio-labeled liposomes showed that the uptake of the folate-coated liposomes was significantly higher than that of noncoated liposomes (de Oliveira Silva et al., 2019).
Luo et al. synthesized a robust sterically stabilized, long-circulating stealth porphyrin-phospholipid (PoP) liposome formulation to extend the blood circulation time of encapsulated DOX. The encapsulated drugs were released when triggered by near-infrared (NIR) light. Their DOX-loaded stealth PoP liposomes exhibited a circulating half-life of 21.9 h with an area under the curve (AUC) of 4837 μg/(ml h), compared with the half-life of PEGylated DOX of 16.9 h with an AUC of 5695 μg/(ml h). Following an intravenous injection of DOX-loaded stealth PoP liposomes and NIR irradiation, DOX disposition increased by approximately 7-fold in subcutaneously treated human pancreatic xenografts (Luo et al., 2016).
Xie et al. loaded DOX into selenium-plated liposomes (DOX-SeLPs). Selenium is an essential micronutrient for human beings that functions as cofactor for reduction of antioxidant enzymes. Selenium NPs can be internalized into cancer cells through endocytosis, thus inducing cell death by triggering mitochondria-mediated apoptosis. PK studies in SD rats, did not show any significant difference between free DOX (AUC = 26.09 ± 6.12 μg min/ml, β t1/2 = 166.41 ± 8.48 min, CL = 115.48 ± 46.35 μl/min/kg) and conventional DOX-loaded liposomes (AUC = 26.92 ± 6.09 μg min/ml, β t1/2 = 177.64 ± 23.46 min, CL = 110.15 ± 25.83 μl/min/kg). However, DOX-SeLPs exhibited a strikingly enhanced AUC (87.03 ± 20.18 μg·min/ml), an extended elimination half-life of 227.72 ± 19.64 min, reduced clearance (25.14 ± 1.50 μl/min/kg) compared with DOX-SeLPs and free DOX. The half-life and mean residence time (MRT) of DOX-SeLPs were significantly longer than that of free DOX and conventional DOX-loaded liposomes. Reversely, the clearance (CL) of DOX-SeLPs was significantly smaller, demonstrating a long-circulation effect of DOX-SeLPs in the blood (Xie et al., 2018). Thus, selenium-coated liposomes with excellent plasma stability, greatly improved the PK property of DOX. Moreover, antitumor efficacy of free and liposomal DOX was measured in A549 cells xenograft model after treatment for 14 days. Free DOX and DOX-LPs inhibited the tumor growth to some extent, and DOX-SeLPs showed the strongest tumor inhibitory effect. The tumor volume of free DOX, DOX-LPs, and DOX-SeLPs group was 63.2%, 69.7%, and 41.7% of the saline group on the 24th day of inoculation, respectively. This study concluded that Dox-SeLPs provide higher AUC value, longer t1/2, and MRT in vivo compared to free DOX and DOX-LPs. Moreover, DOX-SeLPs possess improved pharmacokinetics that results in high distribution of DOX into the tumor, better antitumor efficacy, and lower systemic toxicity in vivo.
Li et al. synthesized a dual-targeting, PEGylated carrier co-modified with d-mannose and l-fucose to improve the tumor-targeting ability and enhance the therapeutic effect of DOX. Mannose receptors (CD206) are selectively overexpressed on M2-like tumor-associated macrophages. On the other hand, l-fucose at the terminal of CD15 acts as a calcium chelator mediating recognition with E-selectin expressed by the tumor. The expression of E-selectin is inversely related to the distance between vascular endothelial cells and tumor tissues; its expression is stimulated by cytokines like interleukin-1 (IL-1) and tumor necrosis factor (TNF-α) originating from tumor cells. Targeting mannose receptors and E-selectin allowed Li et al. to precisely deliver DOX into tumor tissues. In PK studies, they compared the Cmax, AUC, and t1/2 between free DOX, non-PEGylated DOX (DOX-CL), PEGylated DOX (DOX-PL), mannose modified PEGylated DOX (DOX-MPL), fucose modified PEGylated DOX (DOX-FPL), and co-modified PEGylated DOX (DOX-MFPL) after an intravenous injection of 5 mg/kg. Compared to free DOX, DOX-CL showed significantly higher Cmax and AUC, and extended half-life; and DOX-PL further enhanced the difference (Figure 4). There was no significant difference in PK parameters between DOX-PL DOX and all other modified PEGylated DOX formulations. However, DOX-MFPL presented a distinctly higher DOX accumulation in the tumor. The cumulative concentration of DOX in tumor (AUCtumor) of DOX-MFPL was 28.24, 3.33, 1.84, 1.25, and 1.22 times higher than that of free DOX, DOX-CL non-PegL DOX, PegL DOX, FPegL DOX, and MPegL DOX, respectively (p < 0.01 with all groups). Their study suggested that dual anchored (Man-PEG and Fuc-PEG) DOX liposomes improve the tumor-targeting effect (Li et al., 2019). This study demonstrated that dual targeting of liposomes significantly impacted the accumulation of DOX at tumor site, but had a limited effect on its PK.
FIGURE 4.

(a) Schematic that shows how DOX-MFPL target S180 tumor in mice B. Pharmacokinetic behavior of different DOX preparations in S180 tumor-bearing mice. The data are expressed as mean ± SD (n = 3). (c) Quantification of the DOX distribution in tumor after i.v. administration of different DOX formulations. (d) AUC of DOX in tumors after intravenous administration of different DOX formulations into the S180-bearing mice at a DOX dosage of 5 mg/kg. **p < 0.01. (e) The S180 tumor stroma volume versus time curve of tumor-bearing mice i.v. injected with various DOX preparation (Li et al., 2019)
4.2.3 |. ThermoDox®
Lysolipid temperature-sensitive liposomes or low-temperature-sensitive liposomes (LTSLs) are specifically designed to provide targeted tumor therapy in combination with mild localized hyperthermia (Lyon et al., 2021). The integration of hyperthermia and liposomes have resulted in improved efficacy of liposomal drug delivery (Kong & Dewhirst, 1999). Comparable temperatures have been shown to trigger drug release from specially designed thermosensitive liposomes (TSL; Magin & Niesman, 1984). This clinically advanced approach relies on heating the target tissue within the range of ablative temperatures in combination with the administration of chemotherapy using thermosensitive liposomes.
ThermoDox® is a 100 nm DOX containing LTSL. It is composed of three synthetic phospholipids: DPPC (1,2-dipalmitoyl-sn-glycero-3 phosphocholine), MSPC (1-Stearoyl-2-hydroxy-sn-glycero-3-phosphocholine), and DSPE-mPEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxypolyethyleneglycol 2000) in optimal molar ratio of 86:10:4 (DPPC: MSPC:DSPE-PEG2000). Localized heat at mild hyperthermia temperatures (greater than 39.5°C) augments liposome accumulation through increased perfusion and vascular permeability and releases the encapsulated drug from the liposome enabling high concentrations of DOX to be deposited in and around the tumor. A comprehensive study was conducted to understand the effect of different temperature-sensitive liposome carriers (low non-traditional, traditional thermosensitive liposomes; LTSL, NTSL, and TTSL) on drug release properties and systemic PK and biodistribution profiles of DOX in vivo. Each of them displayed distinct serum stability and temperature sensitivity.
LTSL showed a lower transition temperature compared to TTSL with the main phase transition peaks at 41.5 and 44.2°C, respectively. No phase transition peak was observed with NTSL. The lower transition temperature of LTSL is due to MSPC lysolipid existing in the lipid bilayer, which leads to a slightly less ordered phospholipid molecule arrangement in the gel. The lack of NTSL transition temperature is due to the high cholesterol content in the liposome formulation. Serum stability was determined by the release of encapsulated DOX from LTSL, NTSL, and TTSL, incubated in 50% mouse serum at 37°C. LTSL-DOX released 30% of encapsulated DOX after 30 min at 37°C. At 42°C (mimicking hypodermic conditions), LTSL released 100% of their DOX content within 1 min while TTSL released 70% in the first 5 min of incubation and reached 100% after 15 min of incubation. NTSL was stable showing no DOX release up to 24 h. Stability of liposome carriers in blood was determined by release of DOX from dual-labeled (3H/14C) LTSL-DOX, TTSL-DOX, and NTSL-DOX liposomes in B16F10 tumor-bearing C57BL6 mice. Results showed that LTSL had the shortest blood circulation time with 20% DOX remaining after 6 h of injection while TTSL-DOX showed longer blood circulation, like NTSL-DOX, with almost 40% ID and 15% ID of the injected liposome dose detected in the blood after 6 and 24 h, respectively. Further, mean particle size of liposomes has an impact on blood circulation for the robust and circulating NTSL systems but is of less importance for LTSL. Smaller NTSL particles (90 nm) showed longer blood circulation, as compared to bigger particles (175 nm) up to 24 h post-injection in B16F10 tumor-bearing C57BL6 mice. However, no difference in the blood level was observed with LTSL-DOX of different sizes between 2 and 24 h. Organ uptake studies of 3H-labeled liposomes and 14C-DOX after 1 and 24 h post-injection, showed no marked difference in the tissue concentration of liposomes between different liposome carrier groups (Al-Jamal et al., 2012).
Radiofrequency ablation (RFA) is a minimally invasive thermal therapy that is clinically applied to shrink the size of tumors of the liver, kidney, lungs, and bone. Preclinical studies demonstrated that at temperatures ≥39.5°C, ThermoDox® yielded DOX tumor concentrations up to 15-fold greater than the same dose of free DOX. ThermoDox delivered 25 ng DOX/mg tissue to the tumor whereas non-TSL and traditional TSL formulations delivered a substantially lower amount of 7–8 ng DOX/mg tissue at the same temperature. Increasing the duration of ablation increases local drug concentration and the tissue volume exposed to the drug (Swenson et al., 2015). In a Phase I study of ThermoDox® in combination with RFA, the concentration of DOX peaked at 30 min and then decreased as DOX was cleared (initial half-life 0.92 h) from the plasma. More than 92% of entire plasma AUC(0–∞) for DOX occurred in the 3.5 h post-infusion. Consistent with liposomal therapies, the total body clearance of DOX from TSL was 1.1 L/h/m2 which was much slower than DOX clearance 30.2 L/h/m2. Similarly, Vss of DOX from TSL was 5.1 L/ m2, approximately 1.75 times standard blood volume, and was also much smaller than free DOX (approximately 947 L/m2). Thus, data suggested that DOX associated with ThermoDox® therapy is largely contained in the blood compartment (Wood et al., 2012).
Although Phase I studies of ThermoDox® were very promising, phase III HEAT trial had disappointing results with a lack of sufficient evidence of clinical effectiveness, and improvement in progression-free survival in patients with intermediate, unresectable hepatocarcinoma (Ho et al., 2018). The duration of RFA was inadequate and thus resulted in subtherapeutic DOX concentrations in local tumor tissue (Tak et al., 2018). Several other clinical trials with Thermodox® are ongoing. ThermoDox® in combination with other means of heat such as Magnetic Resonance Guided High Intensity Focused Ultrasound (MR-HIFU) for metastatic breast cancer (NCT02536183) is undergoing Phase I study. A preclinical study with MR-HIFU showed a 23× increase in intratumoral DOX concentration with ThermoDox® + HIFU, compared with a 2× increase in intratumoral DOX concentration with free DOX plus HIFU (Figure 5; Farr et al., 2018). A Phase I study in nonresectable pancreatic cancer with ThermoDox® (PanDox) in combination with HIFU is ongoing with active patient recruitment (NCT04852367).
FIGURE 5.
(a) Box-and-Whisker plot of doxorubicin (DOX) concentration in the tumors of KPC mice treated with 15 mg DOX/kg low-temperature-sensitive liposomal doxorubicin (LTSL-DOX) and nonliposomal doxorubicin (DOX), with and without the application of MR-HIFU. *Denotes significance at the p < 0.05 level. (b) Median doxorubicin concentration ([interquartile range] measured in pancreatic tumors following different treatment regimens (n = 4 in each group; Farr et al., 2018)
4.2.4 |. Doxorubicin-loaded EGFR-targeting nanocell
DOX-loaded EGFR-targeting Nanocell (EGFR(V)-EDV-Dox™) uses EnGeneIC delivery vehicle (EDV™) which is a novel compound that can package theoretically effective concentrations of chemotherapeutic drugs into minicells (400 nm NPs derived from Salmonella typhimurium). EnGeneIC’s EDV™ is a unique cyto-immunotherapy that can directly kill cancer cells as well as provoke a potent anti-tumor immune response with little to no toxicity, even in late-stage drug-resistant cancers (Taylor et al., 2015).
This proprietary EDV™ nanocellular technology can be used for the targeted delivery of cancer therapeutics via minicell-surface attached bispecific antibodies. Minicells can be packaged with chemotherapeutic drugs via incubation with the chemotherapeutic drugs; hydrophilic (irinotecan), hydrophobic (PTX, cisplatin, carboplatin), or amphipathic (DOX, vinblastine, 5-fluorouracil). Epidermal growth factor receptor (EGFR) is overexpressed in 40%–50% of patients with glioblastoma (GBM) and is a promising target for new therapeutics. EGFR(V)-EDV-Dox™ targets EGFR through Vectibix. In a study of, by Whittle et al., VEDVDox, contains a mean of 500 ± 100 ng of DOX and less than 5 mg of anti-human EGFR (Vectibix, panitumumab sequence) per 109 EDV and targets the epidermal growth factor receptor (EGFR) protein on the tumor cell. The nanocells are stable in the bloodstream and the anti-EGFR bsAb moiety targets and binds to EGFR-expressing tumor cells. Thereafter, the nanocell is internalized and DOX is released intracellularly.
In a preclinical study of EDV-DOX by MacDiarmid et al., they packaged DOX in minicells and targeted EGFR using an anti-EGFR BsAb. They compared the in vivo biodistribution of DOX following i.v. administration of free DOX, nontargeted minicell DOX, EGFR nanocells DOX to MDA-MB-468 xenografts bearing nude mice. At 6 h, EGFR EDV-DOX delivered approximately 30% of the total DOX dose administered to the tumor compared to approximately 1% from free DOX and about 0.34% from nontargeted minicells DOX (MacDiarmid et al., 2007). In subsequent studies in dogs with late-stage brain cancers, they demonstrated that EGFR(V)-EDV-Dox/EGFR (vectibix sequence)-targeted EDV containing DOX demonstrated superior efficacy (MacDiarmid et al., 2016).
EGFR(V)-EDV-Dox™ has also been studied in the clinic by MacDiarmid et al. In a Phase I open-label clinical trial (NCT02766699), they evaluated the safety, tolerability, and immunogenicity of EGFR(V)-EDV-DOX in subjects with recurrent GBM (astrocytoma, grade IV; Sagnella et al., 2020). Eligible subjects enrolled in the study received EGFR(V)-EDV-Dox™ administered weekly for 7 weeks via a 20-min IV infusion, followed by radiological evaluation at week 8. This trial demonstrated that EGFR(V)-EDV-DOX can be administered safely to patients with recurrent GBM, however, the efficacy readout suggested that it has minimal efficacy as a single agent. There have been other clinical trials using the EnGeneIC delivery vehicle loaded with either DOX or PTX. In another open-label, multicenter, Phase I/II clinical study, the single agent EGFR(V)-EDV-Dox™ was administered intravenously over 20 min once weekly to patients with recurrent GBM expressing EGFR. This study demonstrated that EGFR(V)-EDV-DOX can be administered safely to patients with recurrent GBM (Goncalves et al., 2020; Kaitu’u-Lino et al., 2013; Whittle et al., 2015).
4.2.5 |. Red blood cells compatible with DOX-loaded liposomes
Surface property modification of liposomes by the addition of polyethylene-glycol (PEG) to escape RES, prolong circulation time and reduce plasma clearance is a common technique, but studies have found that PEG induces an immune response and thus alternatives to PEG have been extensively researched. RBCs have been widely explored as an alternative bio-compatible, drug delivery carrier system (Sun et al., 2017; Xu et al., 2016). The biological origin of red blood cells offers inherent protection against immune reactions. The long circulation times of red blood cells, superior biocompatibility, absence of toxic by-products during degradation, large space available to encapsulate drugs and sustained release ability (Sun et al., 2017) make them an attractive carrier for drug delivery. In addition to the above advantages, the manufacturing process is very simple. Several methods of drug encapsulation onto erythrocytes have been employed such as cell-penetrating peptide mediation method, Dimethyl sulfoxide osmotic pulse, electroporation, and osmotic-based methods (hypnotic preswelling, hypnotic dialysis; Bourgeaux et al., 2016).
Two studies have demonstrated the successful use of RBCs as delivery carriers for DOX, one in humans for the treatment of lymphoma (Skorokhod et al., 2007) and one in dogs for the treatment of lymphosarcoma. A more recent study by Lucas et al. investigated the antitumorigenic effects of RBC-DOX in human colon cancer cells (HT-29), and in colon cancer xenograft models. Electroporation was used to entrap DOX into RBCs. Micropores were created using high pulse generator that allowed slow diffusion of DOX from saline into the RBCs. The potential difference was created across the erythrocyte membrane that created pores in the membrane. Rapid distribution of ions between the intracellular and extracellular compartments caused erythrocyte swelling and drug entry. RBCs (at 50% hematocrit) were suspended in saline solution with DOX at 5 mg/ml. RBCs were resealed by incubation at 4°C followed by 37°C for 1 h. Finally, RBC-DOX were washed with PBS with 0.5% HSA. RBC-DOX was characterized using fluorescent microscopy. RBC-DOX showed an increased tumor cytotoxicity as compared to DOX (IC50 was 1.77 μg/ml for the free DOX group, and 1.45 μg/ml for the RBC-DOX group).
The plasma concentration versus time plots for free DOX and DOX-RBC after a single 5 mg/kg IV dose is shown in Figure 6. The area under the concentration–time curve (0–24 h) was statistically higher for RBC-DOX (3024 ± 89 μg h/L) compared to DOX (1289 ± 48 μg h/L). The total body clearance was statistically greater and almost double for RBC-DOX as compared to DOX (2.79 ± 0.31 vs. 1.49 ± 0.09 L/h/kg). These results indicated a longer-lasting presence and enhanced bioavailability of DOX in the body when loaded into RBCs as compared to free DOX. Tumor volume for the RBC-DOX group was smaller than the free DOX groups in the HT-29 xenografts models. RBC-DOX group showed statistically higher concentrations of DOX in the liver, spleen, and lungs as compared to the free DOX group, with no significant differences in tumor biodistribution. The authors explained that RBC-DOX resulted in repeated exposures at equivalent concentrations of DOX over longer periods of time. The repeated exposure resulted from a recurring flow of RBC-DOX through the tumor vasculature, which eventually led to a significant decrease in tumor size without a significant increase in DOX tumor concentration for the RBC-DOX group (Figure 6). The lungs also had a significantly larger concentration of DOX for the RBC-DOX group. The concentration of large liposomes (5–10 μm) in the lungs has been observed previously, and it is attributed to the effect of bloodborne macrophages, which tend to engulf the liposomes and then migrate to the alveoli where they become resident alveolar macrophages The results of this study suggest that RBC-DOX can be a potential delivery carrier for DOX, with superior antitumorigenic effects, decreased myelosuppression, and limited cardiac toxicity compared to equivalent doses of free DOX (Lucas et al., 2019).
FIGURE 6.
(a) Plasma concentration–time plot over a 24 h period of DOX after a 5 mg/kg intravenous injection in DOX and RBC-DOX groups. (b) Tumor growth over a 24-day period for control (gray), DOX (blue), and RBC-DOX (red) in treated mice. (c) Biodistribution of DOX in liver, spleen, lungs, and kidney. (d) Biodistribution of DOX in heart, skin, and tumor-treated mice after the 24-day treatment period (Lucas et al., 2019)
Like erythrocytes, several other cell-based drug delivery systems, utilizing leukocytes, platelets, and stem cells, are being extensively studied to achieve ideal drug delivery platform technology. These cells are enveloped in NPs because of innate properties of biocompatibility, biodegradability, and immune evasion.
5 |. CONCLUSION
The state of nanotechnology in cancer therapy is evolving at an exponential rate as evidenced by the enormous number of scientific publications and increasing number of regulatory approvals of different types of NPs. The growing range of novel nanotechnology platforms have led to the metamorphosis of several traditional chemotherapeutic drugs, such as PTX, DOX, daunorubicin, and cytarabine, and have widened the application of NPs in the treatment of the different type of cancers. The most striking advantage of NPs is their potential to minimize serious side effects of cytotoxic drugs by targeting to specific cancer cells. In addition, the nano delivery systems can be engineered to impart stability to unstable drug substances from degradation, improve the solubility of poorly water-soluble drugs, and provide sustained and controlled release at site of action.
The goal to achieve an optimum therapeutic PK/PD response is significantly influenced by several factors such as the core composition, structure, size, surface ligands, and presence of surface coatings of the nanoparticles. The unique size/shape/surface chemistry of NPs allows them to navigate and overcome biological barriers and release a therapeutic drug quantity in the optimal dosage range. However, it is important to remember that the NP size and distribution is critical since it must strike a balance between maximizing uptake by tumor site and minimizing capture by the RES system, and thus ensure the efficacy of treatment.
The nanocarrier system governs the in vivo distribution pattern of the NPs and thus the overall therapeutic efficacy and treatment outcome. Numerous processes mediate the PK of NPs, with the ADME being poorly understood and is widely different from the traditional formulation. NPs can be tailored to modify the tissue distribution, PK profile, and targeting characteristics to prolong their systemic circulation.
NPs exhibit an exceptional challenge for investigations pertaining to disposition and PK due to their extended circulation times, nonlinear PK profiles, and broad distribution patterns. A comprehensive investigation of PK and PD behavior is a critical step in the translation of NPs studies from preclinical animal studies to humans.
Several novel platforms are being investigated and many have successfully translated from preclinical studies to human trials, but very few have received regulatory approval for market authorization and commercialization. Despite the relentless research efforts to develop novel, improved, and sophisticated nanomedicines, there is a huge gap in translation from research to market. A thorough understanding of challenges imposed by physicochemical characterization, biocompatibility, PK, and PD assessment, scale-up manufacturing, and stability of nanomedicines are needed to bridge this gap, especially with the recent emergence of highly sophisticated and complex nano delivery systems. Moreover, translation from preclinical to clinical also requires a meticulous understanding of the similarity and differences of biological phenomena of animal models and human cancers. For example, the existence and prominence of EPR phenomenon in animals is demonstrated but in the case of humans, it is strongly debated. Moreover, tumor heterogeneity observed in humans is not fully replicated in animal models which can lead to erroneous conclusions.
The concern of regulatory agencies over the quality, safety, toxicity, and efficacy of complex nanostructured delivery systems imposes additional challenges in advancing the nanomedicines to market.
Alternatively, the establishment of a guidance document outlining critical material attributes, process parameters, manufacturing control, identification, and characterization techniques, and other quality attributes is required to guide the development and approval of nanomedicines.
Hence, there is a growing need in enhancing this knowledge base and enhance nanomedicine research, also providing resources to bridge the knowledge gap for successful translation of innovative nanomedicines to the clinic.
5.1 |. Key points
PK (in vivo disposition) of drugs encapsulated in NPs is dependent on the nanocarrier system until the drug is released.
NPs alter the plasma PK and the biodistribution pattern of the encapsulated drug to achieve targeted delivery and sustained effect.
Particle size and particle size distribution are crucial in determining the overall in vivo performance of nanoformulations. Generally, NPs in 20–200 nm size range with unformal size accumulate better in tumors.
More and more novel and sophisticated nanoparticulate delivery approaches are being investigated and advancing from preclinical to clinical trials
FUNDING INFORMATION
This study was supported by the National Institute on Minority Health and Health Disparities (NIMHD) of the National Institute of Health (NIH) under Award Number U54MD007605 and Cancer Prevention & Research Institute of Texas (CPRIT) Core Facilities Support Awards RP180748.
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Ahmad S, Idris RAM, Wan Hanaffi WN, Perumal K, Boer JC, Plebanski M, Jaafar J, Lim JK, & Mohamud R (2021). Cancer nanomedicine and immune system—Interactions and challenges. Frontiers in Nanotechnology, 3. 10.3389/fnano.2021.681305 [DOI] [Google Scholar]
- Ait-Oudhia S, Mager DE, & Straubinger RM (2014). Application of pharmacokinetic and pharmacodynamic analysis to the development of liposomal formulations for oncology. Pharmaceutics, 6(1), 137–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfayez M, Kantarjian H, Kadia T, Ravandi-Kashani F, & Daver N (2020). CPX-351 (vyxeos) in AML. Leukemia & Lymphoma, 61(2), 288–297. [DOI] [PubMed] [Google Scholar]
- Al-Jamal WT, Al-Ahmady ZS, & Kostarelos K (2012). Pharmacokinetics & tissue distribution of temperature-sensitive liposomal doxorubicin in tumor-bearing mice triggered with mild hyperthermia. Biomaterials, 33(18), 4608–4617. [DOI] [PubMed] [Google Scholar]
- Aronson JK (2016). Cremophor. In Aronson JK (Ed.), Meyler’s side effects of drugs (16th ed., p. 763). Elsevier. [Google Scholar]
- Bedikian AY, DeConti RC, Conry R, Agarwala S, Papadopoulos N, Kim KB, & Ernstoff M (2011). Phase 3 study of docosahexaenoic acid-paclitaxel versus dacarbazine in patients with metastatic malignant melanoma. Annals of Oncology, 22(4), 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MJ, Vargo C, Vincent M, Shaver K, Phillips G, Layman R, Macrae E, Mrozek E, Ramaswamy B, Wesolowski R, Shapiro CL, & Lustberg MB (2015). Stopping paclitaxel premedication after two doses in patients not experiencing a previous infusion hypersensitivity reaction. Support Care Cancer, 23(7), 2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco E, Shen H, & Ferrari M (2015). Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature Biotechnology, 33(9), 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borga O, Lilienberg E, Bjermo H, Hansson F, Heldring N, & Dediu R (2019). Pharmacokinetics of Total and unbound paclitaxel after Administration of Paclitaxel Micellar or nab-paclitaxel: An open, randomized, cross-over, explorative study in breast cancer patients. Advances in Therapy, 36(10), 2825–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeaux V, Lanao JM, Bax BE, & Godfrin Y (2016). Drug-loaded erythrocytes: On the road toward marketing approval. Drug Design, Development and Therapy, 10, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourquin J, Milosevic A, Hauser D, Lehner R, Blank F, Petri-Fink A, & Rothen-Rutishauser B (2018). Biodistribution, clearance, and long-term fate of clinically relevant nanomaterials. Advanced Materials, 30(19), e1704307. [DOI] [PubMed] [Google Scholar]
- Bradley MO, Swindell CS, Anthony FH, Witman PA, Devanesan P, Webb NL, Baker SD, Wolff AC, & Donehower RC (2001). Tumor targeting by conjugation of DHA to paclitaxel. Journal of Controlled Release, 74(1–3), 233–236. [DOI] [PubMed] [Google Scholar]
- Bulbake U, Doppalapudi S, Kommineni N, & Khan W (2017). Liposomal formulations in clinical use: An updated review. Pharmaceutics, 9(2), 12. 10.3390/pharmaceutics9020012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter KA, Luo D, Geng J, Stern ST, & Lovell JF (2019). Blood interactions, pharmacokinetics, and depth-dependent ablation of rat mammary tumors with photoactivatable, liposomal doxorubicin. Molecular Cancer Therapeutics, 18(3), 592–601. [DOI] [PubMed] [Google Scholar]
- Chen L-T, & Su M-H (2020). EndoTAG-1 plus gemcitabine versus gemcitabine alone in patients with measurable locally advanced and/or metastatic adenocarcinoma of the pancreas failed on FOLFIRINOX treatment (NCT03126435). Journal of Clinical Oncology, 38-(15_suppl), TPS4669. [Google Scholar]
- Chenthamara D, Subramaniam S, Ramakrishnan SG, Krishnaswamy S, Essa MM, Lin FH, & Qoronfleh MW (2019). Therapeutic efficacy of nanoparticles and routes of administration. Biomaterials Research, 23, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JL, & Yang YW (2021). Modulation of the anticancer activities of paclitaxel by Cremophor micelles. International Journal of Pharmaceutics, 603, 120699. [DOI] [PubMed] [Google Scholar]
- Chis AA, Dobrea C, Morgovan C, Arseniu AM, Rus LL, Butuca A, Juncan AM, Totan M, Vonica-Tincu AL, Cormos G, Muntean AC, Muresan ML, Gligor FG, & Frum A (2020). Applications and limitations of dendrimers in biomedicine. Molecules, 25(17), 3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Gao J, & Kwon GS (2016). PEG-b-PLA micelles and PLGA-b-PEG-b-PLGA sol-gels for drug delivery. Journal of Controlled Release, 240, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daraee H, Etemadi A, Kouhi M, Alimirzalu S, & Akbarzadeh A (2016). Application of liposomes in medicine and drug delivery. Artificial Cells, Nanomedicine, and Biotechnology, 44(1), 381–391. [DOI] [PubMed] [Google Scholar]
- de Oliveira Silva J, Fernandes RS, Ramos Oda CM, Ferreira TH, Machado Botelho AF, Martins Melo M, de Miranda MC, Assis Gomes D, Dantas Cassali G, Townsend DM, Rubello D, Oliveira MC, & de Barros ALB (2019). Folate-coated, long-circulating and pH-sensitive liposomes enhance doxorubicin antitumor effect in a breast cancer animal model. Biomedicine & Pharmacotherapy, 118, 109323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkor DA, & Tang XS (2014). Tube length and cell type-dependent cellular responses to ultra-short single-walled carbon nanotube. Biomaterials, 35(9), 3121–3131. [DOI] [PubMed] [Google Scholar]
- Ealias AM, & Saravanakumar MP (2017). A review on the classification, characterisation, synthesis of nanoparticles and their application. IOP Conference Series: Materials Science and Engineering, 263, 032019. [Google Scholar]
- Estelrich J, Quesada-Pérez M, Forcada J, & Callejas-Fernández J (2014). Introductory aspects of soft nanoparticles. Royal Society of Chemistry. [Google Scholar]
- Falah M, Rayan M, & Rayan A (2019). A novel paclitaxel conjugate with higher efficiency and lower toxicity: A new drug candidate for cancer treatment. International Journal of Molecular Sciences, 20(19), 4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr N, Wang Y-N, D’Andrea S, Starr F, Partanen A, Gravelle KM, McCune JS, Risler LJ, Whang SG, & Chang A (2018). Hyperthermia-enhanced targeted drug delivery using magnetic resonance-guided focussed ultrasound: A pre-clinical study in a genetic model of pancreatic cancer. International Journal of Hyperthermia, 34(3), 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetterly GJ, & Straubinger RM (2003). Pharmacokinetics of paclitaxel-containing liposomes in rats. AAPS PharmSci, 5(4), E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom H, Verweij J, Nooter K, & Sparreboom A (2001). Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. European Journal of Cancer, 37(13), 1590–1598. [DOI] [PubMed] [Google Scholar]
- Goncalves M, Mignani S, Rodrigues J, & Tomas H (2020). A glance over doxorubicin based-nanotherapeutics: From proof-of-concept studies to solutions in the market. Journal of Controlled Release, 317, 347–374. [DOI] [PubMed] [Google Scholar]
- Greene RF, Collins JM, Jenkins JF, Speyer JL, & Myers CE (1983). Plasma pharmacokinetics of adriamycin and adriamycinol: Implications for the design of in vitro experiments and treatment protocols. Cancer Research, 43(7), 3417–3421. [PubMed] [Google Scholar]
- Gupta A, & Middleton M (2011). Chapter 45—Cytostatic and cytotoxic drugs. In Aronson JK (Ed.), Side effects of drugs annual (Vol. 33, pp. 935–962). Elsevier. [Google Scholar]
- Gyongyosi M, Lukovic D, Zlabinger K, Spannbauer A, Gugerell A, Pavo N, Traxler D, Pils D, Maurer G, Jakab A, Riesenhuber M, Pircher A, Winkler J, & Bergler-Klein J (2020). Liposomal doxorubicin attenuates cardiotoxicity via induction of interferon-related DNA damage resistance. Cardiovascular Research, 116(5), 970–982. [DOI] [PubMed] [Google Scholar]
- Harashima H, Tsuchihashi M, Iida S, Doi H, & Kiwada H (1999). Pharmacokinetic/pharmacodynamic modeling of antitumor agents encapsulated into liposomes. Advanced Drug Delivery Reviews, 40(1–2), 39–61. [DOI] [PubMed] [Google Scholar]
- He Z, Wan X, Schulz A, Bludau H, Dobrovolskaia MA, Stern ST, Montgomery SA, Yuan H, Li Z, Alakhova D, Sokolsky M, Darr DB, Perou CM, Jordan R, Luxenhofer R, & Kabanov AV (2016). A high capacity polymeric micelle of paclitaxel: Implication of high dose drug therapy to safety and in vivo anti-cancer activity. Biomaterials, 101, 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennenfent KL, & Govindan R (2006). Novel formulations of taxanes: A review. Old wine in a new bottle? Annals of Oncology, 17(5), 735–749. [DOI] [PubMed] [Google Scholar]
- Henningsson A, Sparreboom A, Sandstrom M, Freijs A, Larsson R, Bergh J, Nygren P, & Karlsson MO (2003). Population pharmacokinetic modelling of unbound and total plasma concentrations of paclitaxel in cancer patients. European Journal of Cancer, 39(8), 1105–1114. [DOI] [PubMed] [Google Scholar]
- Hertz DL, Kidwell KM, Vangipuram K, Li F, Pai MP, Burness M, Griggs JJ, Schott AF, Van Poznak C, Hayes DF, Lavoie Smith EM, & Henry NL (2018). Paclitaxel plasma concentration after the first infusion predicts treatment-limiting peripheral neuropathy. Clinical Cancer Research, 24(15), 3602–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Bokharaei M, & Li SD (2018). Current update of a thermosensitive liposomes composed of DPPC and Brij78. Journal of Drug Targeting, 26(5–6), 407–419. [DOI] [PubMed] [Google Scholar]
- InnoMedica. (2022). Talidox. Retrieved from https://innomedica.com/en/products/talidox/.
- Kaitu’u-Lino TJ, Pattison S, Ye L, Tuohey L, Sluka P, MacDiarmid J, Brahmbhatt H, Johns T, Horne AW, Brown J, & Tong S (2013). Targeted nanoparticle delivery of doxorubicin into placental tissues to treat ectopic pregnancies. Endocrinology, 154(2), 911–919. [DOI] [PubMed] [Google Scholar]
- Karimi M, Bahrami S, Ravari SB, Zangabad PS, Mirshekari H, Bozorgomid M, Shahreza S, Sori M, & Hamblin MR (2016). Albumin nanostructures as advanced drug delivery systems. Expert Opinion in Drug Delivery, 13(11), 1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong G, & Dewhirst MW (1999). Hyperthermia and liposomes. International Journal of Hyperthermia, 15(5), 345–370. [DOI] [PubMed] [Google Scholar]
- Kumthekar P, Tang SC, Brenner AJ, Kesari S, Piccioni DE, Anders C, Carrillo J, Chalasani P, Kabos P, Puhalla S, Tkaczuk K, Garcia AA, Ahluwalia MS, Wefel JS, Lakhani N, & Ibrahim N (2020). ANG1005, a brain-penetrating peptide-drug conjugate, shows activity in patients with breast cancer with leptomeningeal Carcinomatosis and recurrent brain metastases. Clinical Cancer Research, 26(12), 2789–2799. [DOI] [PubMed] [Google Scholar]
- Lammers T (2019). Macro-nanomedicine: Targeting the big picture. Journal of Controlled Release, 294, 372–375. [DOI] [PubMed] [Google Scholar]
- Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, Stuart RK, Strickland SA, Hogge D, Solomon SR, Stone RM, Bixby DL, Kolitz JE, Schiller GJ, Wieduwilt MJ, Ryan DH, Hoering A, Banerjee K, Chiarella M, … Medeiros BC (2018). CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. Journal of Clinical Oncology, 36(26), 2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao J, Madani J, Puertolas T, Alvarez M, Hernandez A, Pazo-Cid R, Artal A, & Anton Torres A (2013). Liposomal doxorubicin in the treatment of breast cancer patients: A review. Journal of Drug Delivery, 2013, 456409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Kim YM, Cho CH, Kim YT, Kim SM, Hur SY, Kim JH, Kim BG, Kim SC, Ryu HS, & Kang SB (2018). An open-label, randomized, parallel, phase II trial to evaluate the efficacy and safety of a Cremophor-free polymeric micelle formulation of paclitaxel as first-line treatment for ovarian cancer: A Korean gynecologic oncology group study (KGOG-3021). Cancer Research and Treatment, 50(1), 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Lai C, Qiu Q, Luo X, Hu L, Zheng H, Lu Y, Liu M, Zhang H, Liu X, Deng Y, & Song Y (2019). Dual-ligand modification of PEGylated liposomes used for targeted doxorubicin delivery to enhance anticancer efficacy. AAPS PharmSciTech, 20(5), 188. [DOI] [PubMed] [Google Scholar]
- Li F, Zhang H, He M, Liao J, Chen N, Li Y, Zhou S, Palmisano M, Yu A, Pai MP, Yuan H, & Sun D (2018). Different Nanoformulations Alter the tissue distribution of paclitaxel, which aligns with reported distinct efficacy and safety profiles. Molecular Pharmaceutics, 15(10), 4505–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhang H, Zhu X, Liu C, Wu M, Li C, Li X, Gao L, & Ding Y (2018). Tolerance, variability and pharmacokinetics of albumin-bound paclitaxel in Chinese breast cancer patients. Frontiers in Pharmacology, 9, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen N, Palmisano M, & Zhou S (2015). Pharmacologic sensitivity of paclitaxel to its delivery vehicles drives distinct clinical outcomes of paclitaxel formulations. Molecular Pharmaceutics, 12(4), 1308–1317. [DOI] [PubMed] [Google Scholar]
- Lin Z, Monteiro-Riviere NA, & Riviere JE (2015). Pharmacokinetics of metallic nanoparticles. WIREs Nanomedicine and Nanobiotechnology, 7(2), 189–217. [DOI] [PubMed] [Google Scholar]
- Liu E, Zhang M, & Huang Y (2016). Pharmacokinetics and pharmacodynamics (PK/PD) of bionanomaterials. Biomedical Nanomaterials, 1–60. [Google Scholar]
- Lohade AA, Jain RR, Iyer K, Roy SK, Shimpi HH, Pawar Y, Rajan MG, & Menon MD (2016). A novel folate-targeted Nanoliposomal system of doxorubicin for cancer targeting. AAPS PharmSciTech, 17(6), 1298–1311. [DOI] [PubMed] [Google Scholar]
- Lucas A, Lam D, & Cabrales P (2019). Doxorubicin-loaded red blood cells reduced cardiac toxicity and preserved anticancer activity. Drug Delivery, 26(1), 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Carter KA, Razi A, Geng J, Shao S, Giraldo D, Sunar U, Ortega J, & Lovell JF (2016). Doxorubicin encapsulated in stealth liposomes conferred with light-triggered drug release. Biomaterials, 75, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon PC, Mannaris C, Gray M, Carlisle R, Gleeson FV, Cranston D, Wu F, & Coussios CC (2021). Large-volume hyperthermia for safe and cost-effective targeted drug delivery using a clinical ultrasound-guided focused ultrasound device. Ultrasound in Medicine & Biology, 47(4), 982–997. [DOI] [PubMed] [Google Scholar]
- Ma P, & Mumper RJ (2013). Paclitaxel nano-delivery systems: A comprehensive review. Journal of Nanomedicine & Nanotechnology, 4(2), 1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDiarmid JA, Langova V, Bailey D, Pattison ST, Pattison SL, Christensen N, Armstrong LR, Brahmbhatt VN, Smolarczyk K, Harrison MT, Costa M, Mugridge NB, Sedliarou I, Grimes NA, Kiss DL, Stillman B, Hann CL, Gallia GL, Graham RM, & Brahmbhatt H (2016). Targeted doxorubicin delivery to brain tumors via Minicells: Proof of principle using dogs with spontaneously occurring tumors as a model. PLoS One, 11(4), e0151832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, Haasdyk JE, Dickson KA, Brahmbhatt VN, Pattison ST, James AC, Al Bakri G, Straw RC, Stillman B, Graham RM, & Brahmbhatt H (2007). Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell, 11(5), 431–445. [DOI] [PubMed] [Google Scholar]
- Magin RL, & Niesman MR (1984). Temperature-dependent drug release from large unilamellar liposomes. Cancer Drug Delivery, 1(2), 109–117. [DOI] [PubMed] [Google Scholar]
- Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC, & Weissleder R (2015). Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nature Communications, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, & Langer R (2021). Engineering precision nanoparticles for drug delivery. Nature Reviews. Drug Discovery, 20(2), 101–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan A (2015). Chapter 47—Miscellaneous drugs, materials, medical devices and techniques. In Ray SD (Ed.), Side effects of drugs annual (Vol. 37, pp. 603–619). Elsevier. [Google Scholar]
- Nardecchia S, Sanchez-Moreno P, Vicente J, Marchal JA, & Boulaiz H (2019). Clinical trials of Thermosensitive nanomaterials: An overview. Nanomaterials (Basel), 9(2), 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navya PN, Kaphle A, Srinivas SP, Bhargava SK, Rotello VM, & Daima HK (2019). Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Convergence, 6(1), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuweboer AJ, Hu S, Gui C, Hagenbuch B, Ghobadi Moghaddam-Helmantel IM, Gibson AA, de Bruijn P, Mathijssen RH, & Sparreboom A (2014). Influence of drug formulation on OATP1B-mediated transport of paclitaxel. Cancer Research, 74(11), 3137–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onafuye H, Pieper S, Mulac D, Cinatl J Jr., Wass MN, Langer K, & Michaelis M (2019). Doxorubicin-loaded human serum albumin nanoparticles overcome transporter-mediated drug resistance in drug-adapted cancer cells. Beilstein Journal of Nanotechnology, 10, 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhiz H, Khoshnejad M, Myerson JW, Hood E, Patel PN, Brenner JS, & Muzykantov VR (2018). Unintended effects of drug carriers: Big issues of small particles. Advanced Drug Delivery Reviews, 130, 90–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra JK, Das G, Fraceto LF, Campos EVR, Rodriguez-Torres MDP, Acosta-Torres LS, Diaz-Torres LA, Grillo R, Swamy MK, Sharma S, Habtemariam S, & Shin HS (2018). Nano based drug delivery systems: Recent developments and future prospects. Journal of Nanobiotechnology, 16(1), 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne M, Ellis P, Dunlop D, Ranson M, Danson S, Schacter L, & Talbot D (2006). DHA-paclitaxel (Taxoprexin) as first-line treatment in patients with stage IIIB or IV non-small cell lung cancer: Report of a phase II open-label multicenter trial. Journal of Thoracic Oncology, 1(9), 984–990. [PubMed] [Google Scholar]
- Pieper S, Onafuye H, Mulac D, Cinatl J Jr., Wass MN, Michaelis M, & Langer K (2019). Incorporation of doxorubicin in different polymer nanoparticles and their anticancer activity. Beilstein Journal of Nanotechnology, 10, 2062–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prados J, Melguizo C, Ortiz R, Velez C, Alvarez PJ, Arias JL, Ruiz MA, Gallardo V, & Aranega A (2012). Doxorubicin-loaded nanoparticles: New advances in breast cancer therapy. Anti-Cancer Agents in Medicinal Chemistry, 12(9), 1058–1070. [DOI] [PubMed] [Google Scholar]
- Price LSL, Stern ST, Deal AM, Kabanov AV, & Zamboni WC (2020). A reanalysis of nanoparticle tumor delivery using classical pharmacokinetic metrics. Science Advances, 6(29), eaay9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reckelhoff CR, Lejeune A, Thompson PM, & Shiomitsu K (2019). In vitro effects of the chemotherapy agent water-soluble micellar paclitaxel (Paccal vet) on canine hemangiosarcoma cell lines. Veterinary and Comparative Oncology, 17(1), 32–41. [DOI] [PubMed] [Google Scholar]
- Rennick JJ, Johnston APR, & Parton RG (2021). Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nature Nanotechnology, 16(3), 266–276. [DOI] [PubMed] [Google Scholar]
- Rodallec A, Fanciullino R, Lacarelle B, & Ciccolini J (2018). Seek and destroy: Improving PK/PD profiles of anticancer agents with nanoparticles. Expert Review of Clinical Pharmacology, 11(6), 599–610. [DOI] [PubMed] [Google Scholar]
- Sagnella SM, Yang L, Stubbs GE, Boslem E, Martino-Echarri E, Smolarczyk K, Pattison SL, Vanegas N, St Clair E, Clarke S, Boockvar J, MacDiarmid JA, & Brahmbhatt H (2020). Cyto-immuno-therapy for cancer: A pathway elicited by tumor-targeted, cytotoxic drug-packaged bacterially derived nanocells. Cancer Cell, 37(3), 354–370 e357. [DOI] [PubMed] [Google Scholar]
- Salvioni L, Rizzuto MA, Bertolini JA, Pandolfi L, Colombo M, & Prosperi D (2019). Thirty years of cancer nanomedicine: Success, frustration, and Hope. Cancers (Basel), 11(12), 1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangtani A, Nag OK, Field LD, Breger JC, & Delehanty JB (2017). Multifunctional nanoparticle composites: Progress in the use of soft and hard nanoparticles for drug delivery and imaging. WIREs Nanomedicine and Nanobiotechnology, 9(6), e1466. [DOI] [PubMed] [Google Scholar]
- Scripture CD, Figg WD, & Sparreboom A (2005). Paclitaxel chemotherapy: From empiricism to a mechanism-based formulation strategy. Therapeutics and Clinical Risk Management, 1(2), 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seleci M, Seleci DA, Joncyzk R, Stahl F, Blume C, & Scheper T (2016). Smart multifunctional nanoparticles in nanomedicine. BioNanoMaterials, 17(1–2), 33–41. [Google Scholar]
- Shi Y, van der Meel R, Chen X, & Lammers T (2020). The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics, 10(17), 7921–7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J, MacMillan P, Zhang Y, Rajesh NU, Hoang T, Wu JLY, Wilhelm S, Zilman A, Gadde S, Sulaiman A, Ouyang B, Lin Z, Wang L, Egeblad M, & Chan WCW (2020). The entry of nanoparticles into solid tumours. Nature Materials, 19(5), 566–575. [DOI] [PubMed] [Google Scholar]
- Skorokhod O, Kulikova EV, Galkina NM, Medvedev PV, Zybunova EE, Vitvitsky VM, Pivnik AV, & Ataullakhanov FI (2007). Doxorubicin pharmacokinetics in lymphoma patients treated with doxorubicin-loaded erythrocytes. Haematologica, 92(4), 570–571. [DOI] [PubMed] [Google Scholar]
- Slingerland M, Guchelaar HJ, Rosing H, Scheulen ME, van Warmerdam LJ, Beijnen JH, & Gelderblom H (2013). Bioequivalence of liposome-entrapped paclitaxel easy-to-use (LEP-ETU) formulation and paclitaxel in polyethoxylated castor oil: A randomized, two-period crossover study in patients with advanced cancer. Clinical Therapeutics, 35(12), 1946–1954. [DOI] [PubMed] [Google Scholar]
- Sofias AM, Dunne M, Storm G, & Allen C (2017). The battle of “nano” paclitaxel. Advanced Drug Delivery Reviews, 122, 20–30. [DOI] [PubMed] [Google Scholar]
- Soininen SK, Vellonen KS, Heikkinen AT, Auriola S, Ranta VP, Urtti A, & Ruponen M (2016). Intracellular PK/PD relationships of free and liposomal doxorubicin: Quantitative analyses and PK/PD modeling. Molecular Pharmaceutics, 13(4), 1358–1365. [DOI] [PubMed] [Google Scholar]
- Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, & Desai N (2005). Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clinical Cancer Research, 11(11), 4136–4143. [DOI] [PubMed] [Google Scholar]
- Sparreboom A, van Tellingen O, Nooijen WJ, & Beijnen JH (1996). Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Research, 56(9), 2112–2115. [PubMed] [Google Scholar]
- Sparreboom A, Wolff AC, Verweij J, Zabelina Y, van Zomeren DM, McIntire GL, Swindell CS, Donehower RC, & Baker SD (2003). Disposition of docosahexaenoic acid-paclitaxel, a novel taxane, in blood: In vitro and clinical pharmacokinetic studies. Clinical Cancer Research, 9(1), 151–159. [PubMed] [Google Scholar]
- Stage TB, Bergmann TK, & Kroetz DL (2018). Clinical pharmacokinetics of paclitaxel monotherapy: An updated literature review. Clinical Pharmacokinetics, 57(1), 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stage TB, Hu S, Sparreboom A, & Kroetz DL (2021). Role for drug transporters in chemotherapy-induced peripheral neuropathy. Clinical and Translational Science, 14(2), 460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffes VM, Murali MM, Park Y, Fletcher BJ, Ewert KK, & Safinya CR (2017). Distinct solubility and cytotoxicity regimes of paclitaxel-loaded cationic liposomes at low and high drug content revealed by kinetic phase behavior and cancer cell viability studies. Biomaterials, 145, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Zhou S, & Gao W (2020). What went wrong with anticancer Nanomedicine design and how to make it right. ACS Nano, 14(10), 12281–12290. [DOI] [PubMed] [Google Scholar]
- Sun Y, Su J, Liu G, Chen J, Zhang X, Zhang R, Jiang M, & Qiu M (2017). Advances of blood cell-based drug delivery systems. European Journal of Pharmaceutical Sciences, 96, 115–128. [DOI] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, & Bray F (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249. [DOI] [PubMed] [Google Scholar]
- Swenson CE, Haemmerich D, Maul DH, Knox B, Ehrhart N, & Reed RA (2015). Increased duration of heating boosts local drug deposition during radiofrequency ablation in combination with thermally sensitive liposomes (ThermoDox) in a porcine model. PLoS One, 10(10), e0139752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak WY, Lin SM, Wang Y, Zheng J, Vecchione A, Park SY, Chen MH, Wong S, Xu R, Peng CY, Chiou YY, Huang GT, Cai J, Abdullah BJJ, Lee JS, Lee JY, Choi JY, Gopez-Cervantes J, Sherman M, … Lencioni R (2018). Phase III HEAT study adding lyso-thermosensitive liposomal doxorubicin to radiofrequency ablation in patients with unresectable hepatocellular carcinoma lesions. Clinical Cancer Research, 24(1), 73–83. [DOI] [PubMed] [Google Scholar]
- Tang Y, Wang X, Li J, Nie Y, Liao G, Yu Y, & Li C (2019). Overcoming the reticuloendothelial system barrier to drug delivery with a “Don’t-eat-us” strategy. ACS Nano, 13(11), 13015–13026. [DOI] [PubMed] [Google Scholar]
- Taylor K, Howard CB, Jones ML, Sedliarou I, MacDiarmid J, Brahmbhatt H, Munro TP, & Mahler SM (2015). Nanocell targeting using engineered bispecific antibodies. MAbs, 7(1), 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zuylen L, Karlsson MO, Verweij J, Brouwer E, de Bruijn P, Nooter K, Stoter G, & Sparreboom A (2001). Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemotherapy and Pharmacology, 47(4), 309–318. [DOI] [PubMed] [Google Scholar]
- Vergote I, Bergfeldt K, Franquet A, Lisyanskaya AS, Bjermo H, Heldring N, Buyse M, & Brize A (2020). A randomized phase III trial in patients with recurrent platinum sensitive ovarian cancer comparing efficacy and safety of paclitaxel micellar and cremophor EL-paclitaxel. Gynecologic Oncology, 156(2), 293–300. [DOI] [PubMed] [Google Scholar]
- Wang F, Porter M, Konstantopoulos A, Zhang P, & Cui H (2017). Preclinical development of drug delivery systems for paclitaxel-based cancer chemotherapy. Journal of Controlled Release, 267, 100–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA (2014). How taxol/paclitaxel kills cancer cells. Molecular Biology of the Cell, 25(18), 2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle JR, Lickliter JD, Gan HK, Scott AM, Simes J, Solomon BJ, MacDiarmid JA, Brahmbhatt H, & Rosenthal MA (2015). First in human nanotechnology doxorubicin delivery system to target epidermal growth factor receptors in recurrent glioblastoma. Journal of Clinical Neuroscience, 22(12), 1889–1894. [DOI] [PubMed] [Google Scholar]
- Wood BJ, Poon RT, Locklin JK, Dreher MR, Ng KK, Eugeni M, Seidel G, Dromi S, Neeman Z, Kolf M, Black CD, Prabhakar R, & Libutti SK (2012). Phase I study of heat-deployed liposomal doxorubicin during radiofrequency ablation for hepatic malignancies. Journal of Vascular and Interventional Radiology, 23(2), 248–255.e247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Deng W, Yuan X, Wang H, Ma Z, Wu B, & Zhang X (2018). Selenium-functionalized liposomes for systemic delivery of doxorubicin with enhanced pharmacokinetics and anticancer effect. European Journal of Pharmaceutics and Biopharmaceutics, 122, 87–95. [DOI] [PubMed] [Google Scholar]
- Xu P, Wang R, Wang X, & Ouyang J (2016). Recent advancements in erythrocytes, platelets, and albumin as delivery systems. Oncotargets and Therapy, 9, 2873–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, He J, Hu Z, Dong Q, Wang H, Fu F, & Tian J (2013). Antitumor effect and toxicity of Lipusu in rat ovarian cancer xenografts. Food and Chemical Toxicology, 52, 200–206. [DOI] [PubMed] [Google Scholar]
- Yetisgin AA, Cetinel S, Zuvin M, Kosar A, & Kutlu O (2020). Therapeutic nanoparticles and their targeted delivery applications. Molecules, 25(9), 2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingchoncharoen P, Kalinowski DS, & Richardson DR (2016). Lipid-based drug delivery systems in cancer therapy: What is available and what is yet to come. Pharmacological Reviews, 68(3), 701–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JA, Anyarambhatla G, Ma L, Ugwu S, Xuan T, Sardone T, & Ahmad I (2005). Development and characterization of a novel Cremophor® EL free liposome-based paclitaxel (LEP-ETU) formulation. European Journal of Pharmaceutics and Biopharmaceutics, 59 (1), 177–187. 10.1016/j.ejpb.2004.06.009 [DOI] [PubMed] [Google Scholar]
- Zhang M, Gao S, Yang D, Fang Y, Lin X, Jin X, Liu Y, Liu X, Su K, & Shi K (2021). Influencing factors and strategies of enhancing nanoparticles into tumors in vivo. Acta Pharmaceutica Sinica B, 11(8), 2265–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Lian B, Xu G, Liu X, Li X, & Ji J (2019). Development of a subcellular semimechanism-based pharmacokinetic/pharmacodynamic model to characterize paclitaxel effects delivered by polymeric micelles. Journal of Pharmaceutical Sciences, 108(1), 725–731. [DOI] [PubMed] [Google Scholar]
- Zhou H, Yan J, Chen W, Yang J, Liu M, Zhang Y, Shen X, Ma Y, Hu X, Wang Y, Du K, & Li G (2021). Population pharmacokinetics and exposure–safety relationship of paclitaxel liposome in patients with non-small cell lung cancer. Frontiers in Oncology, 10. 10.3389/fonc.2020.01731 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.