

Abstract
Excess dopamine release in the dorsal striatum (DS) is linked to psychosis. Antipsychotics are thought to work by blocking striatal D2 dopamine receptors, but they lack efficacy for the negative and cognitive symptoms of schizophrenia. These observations and the fact that increasing brain-wide dopamine improves cognition have fueled the dogma that excess dopamine is not involved in negative and cognitive symptoms. However, this idea has never been explicitly tested with DS-pathway specificity. To determine if excess DS dopamine is involved in cognitive and negative symptoms, we selectively re-expressed excitatory TRPV1 receptors in DS-projecting dopamine neurons of Trpv1 knockout mice. We treated these mice with capsaicin (TRPV1 agonist) to selectively activate these neurons, validated this approach with fiber photometry, and assessed its effects on social interaction and working memory, behavioral constructs related to negative and cognitive symptoms. We combined this manipulation with antipsychotic treatment (haloperidol) and compared it to brain-wide dopamine release via amphetamine treatment. We found that selectively activating DS-projecting dopamine neurons increased DS (but not cortical) dopamine release and increased locomotor activity. Surprisingly, this manipulation also impaired social interaction and working memory. Haloperidol normalized locomotion, but only partially rescued working memory and had no effect on social interaction. By contrast, amphetamine increased locomotion but did not impair social interaction or working memory. These results suggest that excess dopamine release, when restricted to the DS, causes behavioral deficits linked to negative and cognitive symptoms. Future therapies should address this disregarded role for excess striatal dopamine in the treatment-resistant symptoms of psychosis.
Subject terms: Psychosis, Schizophrenia
Introduction
Current antipsychotic treatments are largely ineffective for the cognitive and negative symptoms of schizophrenia [1]. Negative symptoms such as the loss of desire for social engagement and cognitive symptoms like deficits in working memory are just as debilitating to quality of life as the hallmark positive symptoms of psychosis (e.g., hallucinations and delusions) [2]. Therefore, there is an immediate need to address these treatment-resistant symptoms. A major barrier to this has been an imprecise understanding of the underlying neural substrates of psychosis and how its neuropathology maps onto different symptoms.
For decades, we have known that the neurotransmitter dopamine plays an important role in psychosis, but the mechanisms underlying this role remain unclear. Nearly all antipsychotic drugs block D2 dopamine receptors (D2Rs), and functional imaging studies show that schizophrenia patients have increased dopamine in the dorsal striatum (DS), where D2Rs are abundant [3–6]. These observations fueled the prominent hypothesis that excess striatal dopamine is responsible for the D2R antagonist-responsive, positive symptoms of schizophrenia but not its D2R antagonist-resistant, negative and cognitive symptoms [7]. This idea is further supported by the fact that amphetamine (a dopamine releasing drug) can acutely induce positive symptoms [8] but actually improves cognitive function in schizophrenia patients [9].
However, D2Rs are not the only dopamine receptors in the striatum, which equally expresses D1 dopamine receptors (D1Rs) [10]. Because D1Rs are not a principal target of existing antipsychotics, their signaling may contribute to symptoms that are resistant to treatment with D2R antagonist-based antipsychotics. Moreover, the pro-cognitive effects of amphetamine in schizophrenia patients could stem from the fact that it increases dopamine release throughout the brain (not only in the DS, as observed in schizophrenia), including in the prefrontal cortex (PFC) where dopamine release is actually decreased in these patients [10–12]. Moreover, the dorsal striatum is anatomically interconnected with the PFC and other brain structures implicated in cognitive and social function [13–15]. These circuit-level interactions and the specificity of antipsychotics for D2Rs suggest that excess dopamine signaling, when restricted to the DS, could contribute to the negative and cognitive symptoms. However, this idea that has never been directly tested.
To explore this, we developed an experimental approach using the capsaicin-gated ion channel, TRPV1, to accurately recapitulate the pathway-specific increase in dopamine transmission observed in schizophrenia and examined its effects on antipsychotic treatment-responsive and resistant behavioral processes in mice. In contrast to the prevailing view, selectively driving DS dopamine transmission impaired behavioral processes related to the negative and cognitive symptoms of schizophrenia (social interaction and working memory). These behavioral deficits were largely unresponsive to the D2R-antagonist/antipsychotic drug haloperidol and did not occur following treatment with the non-selective dopamine releaser amphetamine. Our findings suggest that excess striatal dopamine plays a broader role in the symptomatology of psychosis than previously thought. This insight exposes a gap in our understanding of dopamine’s role in psychosis and the tools established here provide a path forward to close this gap and develop more comprehensive antipsychotic treatments.
Materials and methods
Animals
We housed and handled all mice according to guidelines approved by the Northwestern University Animal Care and Use Committee. We used both male and female mice housed on a reverse light cycle for all experiments. For capsaicin experiments, we crossed homozygous DATIREScre mice (Jax #0006660) with Trpv1 knockout (KO) mice (Jax #003770) to generate double heterozygous mice that we then crossed to generate DATcre/+; TRPV1 KO mice [16, 17]. We maintained all founder mouse lines through backcrossing to C57BL/6 J mice (Jax #000664), the same mouse strain we used for behavioral experiments with amphetamine. All mice were 12–24 weeks at the start of experimental testing. Detailed materials and methods are provided in the Supplementary Information.
Drugs
We injected all drugs subcutaneously at volume of 10 mL·kg−1. We dissolved capsaicin (3.5–10 mg·kg−1; Alomone Labs) in 3.33% Tween 80 in PBS, D-Amphetamine hemisulfate (0.5–10 mg·kg−1; Sigma) in saline, and haloperidol (0.032–0.1 mg·kg−1; Sigma) in 0.3% tartaric acid.
Surgical procedures
We stereotaxically injected either AAV2/5-hSyn-FLEX-TRPV1-mCherry or AAV2/5-hSyn-DIO-mCherry bilaterally in the substantia nigra pars compacta (SNc) of DATcre/+; TRPV1 KO mice. For animals used for fiber photometry, we also unilaterally injected AAV2/9-CAG-dLight1.3b into the dorsomedial striatum (DMS) and medial prefrontal cortex (mPFC), followed by fiberoptic cannula implants into the same regions (Figs. 1A, 2A).
Fig. 1. Histological validation of the selective expression of functional TRPV1 in SNc dopamine neurons.
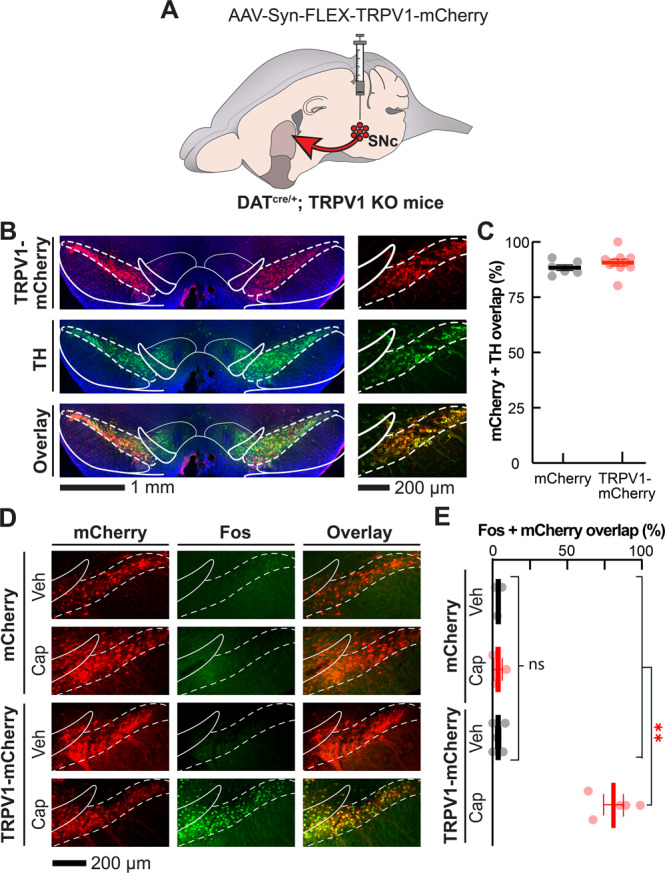
A We virally expressed Cre-dependent mCherry (control) or TRPV1-mCherry (experimental) in DS-projecting SNc dopamine neurons of DATcre/+; TRPV1 KO mice. B Coronal brain sections containing SNc and VTA from a representative experimental (TRPV1) mouse shown at 10× (left) and 16× resolution (right; blue: DAPI nuclear stain; red: α-RFP; green: α-TH). C Mean ± s.e.m. percentage of mCherry-expressing neurons that are TH positive in control and TRPV1 mice (N = 6 and 10 respectively; average of 3 brain slices per mouse). D Coronal brain sections containing SNc from representative control (top) and TRPV1 (bottom) mice immunostained for Fos (green) and RFP (red) 75 min after systemic vehicle or capsaicin treatment (10 mg·kg−1). E Mean ± s.e.m. percentage of mCherry-expressing neurons that are Fos positive as a function of treatment and experimental group (N = 3–5 mice per group; average of 3 brain slices per mouse). In all images, white dashed lines indicate boundaries of SNc and solid lines indicate boundaries of adjacent brain structures. **P < 0.01 comparing treatments and groups; Kruskal-Wallis unpaired one-way ANOVA in (E). Details for these and all other statistical comparisons are presented in the Supplementary Table.
Fig. 2. Recordings of dLight1.3b fluorescence in DMS and mPFC following systemic capsaicin or amphetamine treatment.

A We virally expressed mCherry (control) or TRPV1-mCherry (experimental) in DS-projecting SNc dopamine neurons of DATcre/+; TRPV1 KO mice and the fluorescent dopamine sensor dLight1.3b in DMS and mPFC. We then implanted fiber-optic cannulas and used fiber photometry to simultaneously record dopamine transmission in these regions. B Representative coronal brain sections from an experimental, TRPV1 mouse expressing dLight1.3b in mPFC and DMS (green: α -GFP; blue: DAPI nuclear stain; scale bar: 1 mm). White dashed lines indicate the position of the implanted fiber-optic probe. C Schematic representation of the time course of dLight1.3b recordings and drug treatments. We administered vehicle at t = 0 min, and capsaicin (10 mg·kg−1) or amphetamine (10 mg·kg−1) at t = 15 min. We recorded dLight1.3b fluorescence for 15-, 30-, or 60-min following vehicle, capsaicin, or amphetamine treatment, respectively. D Example trace (% ΔF/F) of dLight1.3b fluorescence in DMS and mPFC in response to capsaicin treatment in a representative TRPV1 mouse. E, F Mean ± s.e.m. dLight1.3b fluorescence in DMS, (E), and mPFC, (F), calculated using area-under-the-curve (AUC) in 1-min time bins and normalized to values in the final 10 min following vehicle treatment in control and TRPV1 mice. Capsaicin responses are truncated to the first 10 min when changes in dopamine were most evident. G, H Mean ± s.e.m. dLight1.3b fluorescence in DMS, (G), and mPFC, (H), calculated using AUC in 1-min time bins and normalized to values in the final 10-min following vehicle treatment in control and TRPV1 mice. Amphetamine responses are truncated to the first 40 min when changes in dopamine were most evident. I, J dLight1.3b fluorescence in DMS, (I), and mPFC, (J), calculated using AUC in 1-min time bins, normalized to values in the final 10-min following vehicle treatment, and averaged across the 10, 1-min time bins surrounding the peak response following systemic capsaicin or amphetamine treatment in control and TRPV1 mice. Both control and TRPV1 groups were combined in the analysis of amphetamine treatment effects (G–J). In these and all other box-and-whisker plots, the horizontal lines denote median values, boxes cover the middle two quartiles and whiskers span 1.5× the interquartile range. N = 5–7 mCherry, N = 8–11 TRPV1, and N = 13–18 combined amphetamine mice; ***P < 0.001 and **P < 0.01; Holm–Sidak’s multiple comparison test.
Fiber photometry
We used a commercial fiber photometry system and software (TDT) to record each mouse’s baseline dLight1.3b activity in the DMS and mPFC for 15 min, subcutaneously injected vehicle, recorded for an additional 15 min, and then subcutaneously injected capsaicin or amphetamine (10 mg·kg−1 each) and recorded dLight1.3b activity for 30 or 60 min, respectively (Fig. 2C). We used custom MATLAB scripts to compute normalized dLight1.3b fluorescent traces (% ΔF/F) and area-under-the-curve (AUC) measurements for traces in 1-min time bins.
Open field locomotor activity
We habituated mice to the open field for 20 min, injected vehicle, recorded locomotion for 30 min or 1 h, injected capsaicin or amphetamine at varying doses (one dose per day) and recorded locomotion for 30 min or 1 h, respectively (Figs. 3B, 5A). For haloperidol experiments, we pretreated mice with either vehicle or varying doses of haloperidol (one dose per day), recorded locomotion for 30 min, injected capsaicin (3.5 mg·kg−1) and again recorded locomotion for 30 min (Fig. 5H). To analyze locomotor speed, we used a video camera and custom software written in ImageJ to track each mouse’s position over time in the open field [18]. The open field locomotor experiments for mice treated with vehicle followed by capsaicin (3.5 mg·kg−1) were experimentally equivalent and the data statistically indistinguishable between the first cohort of mice (Fig. 3B, C) and the cohort used for the haloperidol experiments (Fig. 5H, I), so we combined all mice in those treatment groups for statistical comparisons.
Fig. 3. Selectively driving nigrostriatal dopamine transmission dose-dependently increases locomotion in TRPV1, but not control mice.

A We recorded locomotor activity in an open field arena for 30 min following vehicle then capsaicin treatment (left). Example trajectories (10-min duration) for an experimental TRPV1 mouse following systemic treatment with vehicle (middle) or capsaicin (right; 10 mg·kg−1). B, C Systemic capsaicin treatment dose-dependently increased locomotor activity in TRPV1 but not control mice. Data in (B) are mean ± s.e.m. normalized to values following vehicle treatment and plotted in 3-min time bins; the control data are averaged across all capsaicin doses. Data in (C) are averaged during the first 15-min following vehicle or capsaicin treatment (N = 8 mCherry and N = 14–29 TRPV1 mice). D, E Locomotor response to repeated capsaicin injections (3.5 mg·kg−1 every 15 min) in experimental and control mice. Data in (D) are mean ± s.e.m. normalized to values following vehicle treatment (t = 0–15 min) and plotted in 3-min time bins. Data in (E) are 15-min averages following each injection, normalized to associated values following vehicle treatment (N = 5 mCherry and N = 11 TRPV1 mice). ****P < 0.0001, ***P < 0.001, **P < 0.01 and *P < 0.05 comparing capsaicin and vehicle treatments; two-way ANOVA in (B); Holm–Sidak’s multiple comparison test in (C–E).
Fig. 5. Amphetamine treatment fails to disrupt social exploration and working memory, while haloperidol incompletely normalizes these deficits caused by selective nigrostriatal dopamine excess.

A, B Systemically treating C57BL/6 J mice with amphetamine dose-dependently increased locomotor speed in the open field. Data in (A) are mean ± s.e.m. normalized to vehicle treatment and plotted in 5-min time bins. Data in (B) are 15-min averages following each injection, normalized to values following vehicle treatment. C Amphetamine treatment (1 mg·kg−1) in C57BL/6 J mice induced locomotion equivalently to capsaicin treatment (3.5 mg·kg−1) in TRPV1 mice in the 15-min following injection, normalized to values following vehicle treatment. D, E Amphetamine treatment (1 mg·kg−1) did not significantly affect the time C57BL/6 J mice spend interacting with a novel juvenile conspecific mouse, (D), and there was no difference, (E), in social interaction time when normalized, within each mouse, to values following vehicle treatment (mean ± s.e.m.). F, G Amphetamine treatment (1 mg·kg−1) did not significantly affect the percent of correct choices C57BL/6 J mice made in the T-maze, DNMS task, (F), and there was no difference, (G), in percent correct choice following amphetamine treatment normalized to vehicle (mean ± s.e.m.; N = 9–10 C57BL/6 J mice and N = 29 TRPV1 mice; ****P < 0.0001, ***P < 0.001, and **P < 0.01 comparing amphetamine, capsaicin, and vehicle treatments; Holm–Sidak’s multiple comparison test in (A–C, F, G); Wilcoxon signed-rank test in (D, E). H, I Pre-treating TRPV1 mice with the first-generation antipsychotic haloperidol suppressed the increase in locomotion caused by systemic capsaicin treatment (3.5 mg·kg−1). Data in (H) are mean ± s.e.m. normalized to values following vehicle treatment and plotted in 3-min time bins. Data in (I) are averaged during the first 15-min following capsaicin treatment, normalized to values following vehicle treatment (N = 12–29 TRPV1 mice). J, K Haloperidol pre-treatment (0.1 mg·kg−1) failed to normalize the decrease in juvenile social interaction time in TRPV1 mice caused by capsaicin treatment (3.5 mg·kg−1), (J), or normalized social interaction time within each mouse, (K), between capsaicin and vehicle treatment (mean ± s.e.m; N = 12 TRPV1 mice). L, M Mean ± s.e.m. percent correct choice, (L), and change in percent choice from vehicle treatment following capsaicin treatment (3.5 mg·kg−1), (M), in the DNMS T-maze task was reduced in TRPV1 mice and only partially rescued by haloperidol pre-treatment (N = 11 TRPV1 mice). N Summary of behavioral results in the different experimental groups included in this study. Arrows indicate directionality and magnitude of effects. Holm–Sidak’s multiple comparison test compared to treatment with vehicle only (****P < 0.0001, ***P < 0.001) and vehicle + capsaicin (####P < 0.0001, ##P < 0.01 and #P < 0.05) in (H, I), or to vehicle–vehicle (***P < 0.001, **P < 0.01 and *P < 0.05) treatment in (J–N).
Juvenile social exploration
We habituated individual mice in their home cage for 10 min, injected vehicle, capsaicin (3.5 mg·kg−1), or amphetamine (1 mg·kg−1), waited 2 min (capsaicin) or 4 min (amphetamine), and placed a novel juvenile (postnatal day 21–35) conspecific mouse in the adult’s home cage (Fig. 4A). For haloperidol experiments, we pretreated mice with either vehicle or haloperidol (0.1 mg·kg−1), waited 10 min, then injected capsaicin (3.5 mg·kg−1), waited 2 min, and placed the novel juvenile in the adult’s home cage (Fig. 5J, K). On each day, we measured the amount of time the adult experimental mouse spent interacting with the juvenile test subject for 5 min, which included the sum duration of sniffing, grooming, approaching, or pawing initiated by the adult mouse towards the juvenile [19].
Fig. 4. Selective nigrostriatal dopamine excess disrupts social and cognitive function.

A We recorded behavior during an assay of juvenile social exploration in which control (mCherry) and experimental (TRPV1) adult mice interacted with a novel juvenile, conspecific mouse following vehicle or capsaicin (3.5 mg·kg−1) treatment for 5 min. B, C Mean ± s.e.m. interaction time, quantified as the sum duration of sniffing, grooming, approaching, or pawing initiated by the adult mouse towards the juvenile, (B), or normalized to values following vehicle treatment, (C), decreased following capsaicin treatment in TRPV1, but not control mice (N = 10 mCherry and N = 9 TRPV1 mice). D We used an automated T-maze to evaluate spatial working memory with a delayed non-match to sample (DNMS) protocol, where mice alternated between forced and choice trials with varying delay periods (2, 10 or 60 s) in a center holding area between trials. E, F Mean ± s.e.m. percent correct choice in control, (E), and TRPV1, (F), mice following vehicle or capsaicin treatment (3.5 mg·kg−1) in the DNMS task at different delay periods. G Mean ± s.e.m. difference in percent correct choice between capsaicin and vehicle treatment conditions in control and TRPV1 mice (N = 5 mCherry and N = 8 TRPV1 mice; **P < 0.01 and *P < 0.05 comparing capsaicin and vehicle treatments; Wilcoxon signed-rank test in (B, C); two-way ANOVA in (E, F); Holm–Sidak’s multiple comparison test in (G).
Spatial working memory
We individually housed and gradually food restricted mice to 85% of their ad libitum body weight. We then habituated mice to an automated T-maze (Maze Engineers) for 2 days. Following habituation, we pre-trained mice in sessions consisting of 10 alternating (left vs. right) forced trials from the center holding area to a baited goal arm and then back to the center holding area. The day after pre-training, we trained mice in a delayed non-match to sample (DNMS) task in which we interleaved forced and choice trials [20]. During the choice trial, mice were required to choose the arm opposite to the immediately preceding forced trial to obtain a food reward. Once mice had at least three consecutive days with ≥70% correct choice trials in DNMS training, we proceeded with pharmacological testing. In each session, we injected mice with vehicle, capsaicin (3.5 mg·kg−1), or amphetamine (1 mg·kg−1), waited 2 min (capsaicin) or 4 min (amphetamine), and tested their DNMS performance with a delay period of 2, 10, or 60 s between forced and choice trials on sequential days (Figs. 4E–G, 5F–G). For sessions that exceeded 15 min, we administered additional capsaicin injections every 15 min to sustain the drug’s behavioral effect (Fig. 3B). For haloperidol experiments, we pretreated mice with either vehicle or haloperidol (0.1 mg·kg−1), waited 10 min, then injected capsaicin (3.5 mg·kg−1), waited 2 min, and tested their performance with the same 2, 10, and 60 s delays while administering capsaicin every 15 min until task completion (Fig. 5L, M).
Histology
Following all photometry and behavioral experiments, we euthanized and intracardially perfused mice, then removed and sliced their brains for immunostaining. We used α-TH or α-Fos with α-RFP antibodies to quantify TH or Fos colocalization with mCherry in SNc. For dlight1.3b analysis, we used an α-GFP antibody in DMS and mPFC. We imaged fluorescence using a multiphoton or wide-field fluorescence microscope to verify the accuracy of injection and/or implantation (Figs. 1, 2B).
Statistical analysis
We used Prism (GraphPad) to perform all statistical tests. For comparisons between data that did not conform to a normal distribution, we used non-parametric statistical comparisons. For comparisons of more than two groups, we used one-way ANOVA. For comparisons of two or more groups across conditions or time, we used two-way repeated measures ANOVA. For all post-hoc analyses, we used Holm–Sidak’s correction for multiple comparisons. All statistical comparisons are presented in the Supplementary Table.
Results
Selective expression and activation of TRPV1 in SNc dopamine neurons
To selectively drive dopamine release in the DS, we re-expressed the capsaicin-sensitive, excitatory cation channel TRPV1 in DS-projecting, substantia nigra pars compacta (SNc) dopamine neurons. Specifically, we bilaterally injected a Cre-dependent virus expressing TRPV1-mCherry (or mCherry control) into the SNc of DATcre/+ mice that lack the endogenous Trpv1 gene (DATcre/+; TRPV1 KO mice) (Fig. 1A). This approach resulted in a high degree of overlap between neurons that express mCherry and neurons that were immunopositive for the dopamine biosynthesis enzyme tyrosine hydroxylase (TH), specifically in the SNc (Fig. 1B, C). Considering these mice lack the Trpv1 gene, this allowed us to inject them systemically with the TRPV1 agonist capsaicin and selectively activate neurons at the site of virus injection without engaging endogenous TRPV1 receptors in the periphery [21, 22]. To confirm that virally expressed TRPV1 was functional in SNc dopamine neurons, we systemically administered vehicle or capsaicin (10 mg·kg−1) to control and experimental (TRPV1) mice. 75-min following injection, there was a high degree of overlap between neurons expressing mCherry and the immediate early gene product Fos in TRPV1 mice after capsaicin treatment. By contrast, there was little to no overlap of Fos with mCherry in control mice injected with capsaicin, or in TRPV1 mice injected with vehicle (Fig. 1D, E). These results indicate that TRPV1 expression was restricted to SNc dopamine neurons in these mice and that systemic capsaicin treatment selectively activated these neurons.
Selective activation of nigrostriatal dopamine release in vivo
To confirm that capsaicin treatment increases dopamine release in a pathway-specific manner in these mice, we virally expressed the fluorescent dopamine sensor dLight (AAV2/9-CAG-dLight1.3b) and implanted fiber-optic probes unilaterally into the dorsomedial striatum (DMS) and medial prefrontal cortex (mPFC) for dual-site, fiber photometry recordings (Fig. 2A, B). We then recorded dLight fluorescence in the DMS and mPFC following systemic treatment with vehicle, capsaicin, or the non-selective dopamine releaser amphetamine (Fig. 2C). Capsaicin treatment (10 mg·kg−1) increased dopamine transmission (ΔF/F) in the DMS of TRPV1, but not control mice (Fig. 2D, E). Importantly, capsaicin treatment did not increase dopamine release in the mPFC of either TRPV1 or control mice (Fig. 2D, F).
In contrast to capsaicin, amphetamine treatment (10 mg·kg−1) increased dopamine release in both the DMS and mPFC of these mice (Fig. 2G, H), indicating that dLight1.3b is suitable for measuring dopamine release even in the mPFC, where dopamine levels are relatively lower. Taken together, these findings demonstrate that our approach selectively drives activity in DS-projecting, SNc dopamine neurons and mimics the pathway-specific excess of dopamine observed in schizophrenia patients [12] (Fig. 2I, J).
Selective nigrostriatal dopamine excess increases locomotor activity
Locomotor hyperactivity is associated with the positive symptoms of schizophrenia in that both are reversed by effective antipsychotic treatments [23]. We recorded the locomotor activity of control and TRPV1 mice in an open field arena following treatment with a range of capsaicin doses (Fig. 3A). Capsaicin treatment dose-dependently induced locomotor hyperactivity in TRPV1, but not control mice (Fig. 3B, C). The effect of capsaicin on locomotion started rapidly (<~3 min) and lasted about 15 min after injection for the lowest dosage tested (3.5 mg·kg−1; Fig. 3B). To determine whether we could repeatedly inject capsaicin without de-sensitizing its behavioral effects, we administered three successive injections separated by 15 min. These repeated capsaicin treatments consistently induced hyperlocomotion in TRPV1 mice, but not control mice, with no evidence of desensitization (Fig. 3D, E). These results show that selectively driving DS dopamine release with capsaicin increases locomotor activity and establishes the time-course and dose-dependence of its behavioral effect. Given that the lowest dose tested here (3.5 mg·kg−1) only modestly increased locomotion and reliably elicited a behavioral response with no obvious desensitization, we used this dose for all subsequent behavioral assessments.
Selective nigrostriatal dopamine excess disrupts social and cognitive function
The negative and cognitive symptoms of schizophrenia include social withdrawal and deficits in working memory. To determine if selectively driving DS dopamine transmission affects behaviors associated with these symptoms, we evaluated the effects of capsaicin treatment on juvenile social exploration (JSE) and spatial working memory (WM). Surprisingly, capsaicin treatment reduced social interaction with a juvenile conspecific mouse in TRPV1, but not control mice (Fig. 4A–C). Likewise, capsaicin treatment disrupted working memory in TRPV1 but not control mice in a T-maze, delayed non-match to sample (DNMS) task requiring mice to choose the arm opposite from the one visited on the preceding forced-choice trial (Fig. 4D–G). Notably, this DNMS performance deficit was pronounced at all delay durations, even when the delay duration was only 2 s (the minimum possible in this T-Maze), suggesting the possible disruption of processes other than working memory. However, when factoring the time required to return to the start arm (Fig. 4D), the true delay duration was much longer for each condition (e.g., 8 s for the 2-s condition; Supplementary Fig. 1A–C). Treating control or TRPV1 mice with capsaicin had no effects on these longer, true delay durations, the directional bias of choices, or the latency to choose in either correct or incorrect trials in the DNMS working memory task (Supplementary Fig. 1A–H). Finally, capsaicin treatment had no effect on performance in a subset of these TRPV1 mice trained in a version of the T-Maze task requiring mice to repeatedly choose the same reward arm where working memory was not required (Supplementary Fig. 1I). These findings indicate that capsaicin treatment did not grossly alter the performance strategy of TRPV1 mice or diminish their engagement in the working memory task. Altogether, our results show that excess dopamine, when restricted to the DS, diminishes cognitive and social function.
Non-selective dopamine excess alters locomotor activity but not social or cognitive function
Amphetamine is a psychostimulant that is often used to approximate dopamine dysregulation in psychosis in rodents [23]. In contrast to the viral-genetic approach used here, amphetamine increases dopamine release throughout the brain (not selectively in DS), including in the PFC [24]. Consistent with previous studies, treating C57BL/6 J mice with amphetamine dose-dependently increased locomotor activity in the open field (Fig. 5A, B). A 1 mg·kg−1 amphetamine dose increased locomotor speed most comparably to the 3.5 mg·kg−1 dose of capsaicin we used for the WM and JSE experiments in TRPV1 mice (Fig. 5C), so we used this dose for subsequent behavioral experiments. Despite inducing similar levels of locomotion, amphetamine treatment had no effect on social interaction or working memory performance compared to vehicle treatment (Fig. 5D–G). These results suggest that the behavioral deficits caused by capsaicin treatment in TRPV1 mice were not solely due to changes in their locomotor activity. Moreover, these findings highlight the differences between the effects of DS-restricted and brain wide excess in dopamine, in that only the former produces deficits in behaviors related to negative and cognitive symptoms (Figs. 2, 4, 5).
Haloperidol normalizes locomotor hyperactivity and partially normalizes cognitive but not social deficits caused by selective nigrostriatal dopamine excess
Classical antipsychotic drugs are largely ineffective for treating the negative and cognitive symptoms of schizophrenia [1]. To determine whether the common antipsychotic drug haloperidol normalizes the behavioral deficits of a selective excess in DS dopamine, we tested its effects in TRPV1 mice. Pretreating these mice with a range of haloperidol doses blocked their increase in locomotor activity following systemic capsaicin treatment (Fig. 5H, I). For subsequent behavioral tests in TRPV1 mice, we used the highest dose of haloperidol tested in the open field (0.1 mg·kg−1). Despite normalizing capsaicin-induced locomotion, haloperidol pretreatment failed to normalize the capsaicin-induced decrease in social interaction in TRPV1 mice (Fig. 5J, K). Furthermore, haloperidol only partially rescued the disruption of working memory caused by capsaicin treatment in these mice (Fig. 5L, M).
Taken together, these data show that haloperidol normalized behavioral processes that are typically normalized by antipsychotic drugs in rodents (i.e., hyperlocomotion). However, haloperidol failed to normalize, or only partially normalized behavioral processes associated with symptoms of schisophrenia that are resistant to treatment in patients (social interaction and working memory, respectively) (Fig. 5N). Despite blocking striatally enriched D2Rs, haloperidol was only partially effective for the behavioral changes driven by a selective excess in DS dopamine.
Discussion
The adverse effects of cognitive and negative symptoms are widely recognized, yet their underlying etiology is poorly understood. The prevailing view is that excess dopamine is not involved in these symptoms, since antipsychotic drugs that block dopamine receptors do not alleviate these symptoms, and drugs that increase dopamine do not reliably induce them. In the current study, we directly tested this idea in a manner that explicitly models the pathway-specific increase in dopamine observed in schizophrenia patients.
To do this, we adapted an approach that was previously used to activate all dopamine neurons [21]. Specifically, we generated a virus to selectively express excitatory TRPV1 receptors in DS-projecting, SNc dopamine neurons of DATcre/+ mice that lack the endogenous Trpv1 gene. In contrast to the transgenic approach previous studies used to selectively express TRPV1 [21, 22], our method allowed us to specifically activate genetically defined neurons at the site of virus injection. This virus-mediated approach is widely applicable to manipulating circuits throughout the brain. We leveraged this regional and genetic selectivity to transiently activate DS-projecting SNc neurons (but not other dopamine neurons) using the TRPV1 agonist capsaicin and selectively induce dopamine release in the DS, but not mPFC. Capsaicin’s effects on behavior and dopamine release lasted approximately 15–20 min, consistent with earlier studies using capsaicin to activate all dopamine neurons [21]. This transient duration is an advantage for experimental designs requiring a modest duration (minutes) of reversible activation, but a limitation for experiments requiring shorter or longer stimulations (sub-seconds or hours). Other advantages include the compatibility with traditional chemogenetic approaches (since capsaicin and DREADD ligands are distinct) and in vivo imaging approaches that are constrained by limited cranial space for implanting optogenetic fibers [18, 25, 26]. A notable limitation of this approach is the requirement that the mice must be on a Trpv1 knockout background, which requires at least two generations of crosses to generate experimental animals. The lack of endogenous TRPV1 expression also has the potential to confound behavioral phenotypes. Although here we used control mice that also lacked the Trpv1 gene, this could particularly confound studies of behavioral processes such as sensation or pain that are influenced by TRPV1 [17]. Another potential caveat of the virus-mediated approach used here is that, although they are a minority, subpopulations of molecularly defined SNc dopamine neurons are known to project to areas other than the dorsal striatum. For instance, Vglut2 + /Calb1+ SNc dopamine neurons mostly project to tail of the dorsal striatum, but also extend ventrally to central amygdala, while Sox6 + /Ndnf+ dorsal SNc dopamine neurons send modest projections to the nucleus accumbens [27]. Although these extra-DS projections are minimal, their existence warrants consideration when interpreting the effects of activating all SNc dopamine neurons on behavior. As we better understand the diversity of dopamine cell sub-types, it will be important to take advantage of intersectional genetic approaches, and potentially the approaches used here, to understand the roles of these subpopulations in specific behavioral processes [28, 29].
Treating TRPV1 mice with capsaicin and C57BL6/J mice with amphetamine both induced hyperlocomotion. A drug’s ability to suppress amphetamine-driven locomotion in mice is commonly used to assess its antipsychotic potential in humans [23]. Albeit circular, there is logic to this idea. Most antipsychotic drugs attenuate dopamine receptor signaling, and hyperlocomotion results from amphetamine-induced increases in striatal dopamine release [30]. Because both amphetamine-driven locomotion in rodents and the positive symptoms of schizophrenia in patients respond to antipsychotic treatment, they are speculated to engage partly overlapping neural substrates. Although further experiments are necessary to confirm that capsaicin treatment induces behavioral changes associated with positive symptoms in TRPV1 mice, the fact that it induces haloperidol-responsive hyperlocomotion suggests it is engaging dopamine pathways related to psychosis.
Although capsaicin (3.5 mg·kg−1 in TRPV1 mice) and amphetamine (1 mg·kg−1 in C57BL6/J mice) treatments equivalently induced hyperlocomotion, only the former additionally perturbed social interaction and working memory. Social exploration and working memory are complex processes that rely on distributed brain areas [20, 31–33]. The mPFC, in particular, is crucially involved in both processes [34–36], and the brains of schizophrenia patients exhibit an array of pathological changes in the prefrontal cortex that are thought to underlie cognitive and negative symptoms [37]. In fact, the onset of these symptoms can precede that of the positive symptoms, further supporting distinct underlying etiologies [38, 39]. However, patients with prodromal schizophrenia symptoms also have elevated dopamine signaling in the DS that correlates with neurocognitive dysfunction [40]. Furthermore, the DS is both directly and indirectly connected to brain structures implicated in cognitive and social function. For instance, inhibiting DMS-projecting mPFC neurons during the delay period of a T-maze working memory task impairs performance, and DMS neurons are sequentially active during this delay period [13, 41]. The DS is also connected through basal ganglia outputs to the thalamocortical connections thought to sustain persistent activity in the PFC during delay periods of working memory [20, 42]. Striatal dysfunction has also been repeatedly implicated in social behavior deficits, particularly in the context of autism-spectrum disorders [43–45]. For example, optogenetically activating nigrostriatal dopamine neurons was recently shown to reduce social preference and decrease social interaction in mice [43]. Therefore, our own findings are consistent with previous studies implicating striatal function in social interaction and working memory. Our findings add to this understanding by distinguishing nigrostriatal from brain-wide dopamine excess (i.e., following amphetamine treatment), which did not disrupt social interaction or working memory. This distinction could result from several differences between treatment with amphetamine and capsaicin in our model. One possibility is that the amphetamine-driven dopamine release outside of the striatum (e.g., in the PFC) has pro-cognitive and pro-social effects that compensate for the deficits caused by selective nigrostriatal dopamine excess [9]. Intriguingly, it has been postulated that multiple memory systems compete for cognitive resources, and dopamine released asymmetrically across these systems could sub-optimally promote this competition [15]. Related to this idea, there is an inverse relationship between striatal and cortical dopamine signaling in human and in animal studies of schizophrenia, suggesting a circuit-level interaction between the two structures that could drive a diversity of symptoms [46–52]. Another possible explanation for the behavioral differences between amphetamine and capsaicin treatment are the different temporal profiles with which they drive dopamine release in the striatum (Fig. 2E, G). We attempted to control for these differences by dosing amphetamine and capsaicin according to the duration of the behaviors we studied and the time-course and magnitude of each drug’s effects on locomotor activity (Fig. 5C). Because both drugs increase dopamine release at the minutes-timescale, it is reasonable to assume that both preferentially increase “tonic” dopamine levels in the striatum. Nonetheless, different timescales of dopamine transmission have been linked to specific brain processes, and we cannot rule out that such differences did not occur here [53, 54]. Finally, it is notable that amphetamine promotes the release of neurotransmitters other than dopamine (notably serotonin and norepinephrine [55]), and SNc dopamine neurons can also co-release glutamate or GABA [56]. Therefore, differences between the additional neurotransmitters released by amphetamine (i.e., serotonin and norepinephrine) and capsaicin (i.e., glutamate and GABA) treatment could potentially explain their differential effects on the behaviors tested here. Nonetheless, our findings underscore important differences between selectively activating SNc dopamine neurons and systemic amphetamine treatment, which continues to be widely used in preclinical psychosis research.
If nigrostriatal dopamine excess contributes to cognitive and negative symptoms, then why are antipsychotics that block D2 dopamine receptors largely ineffective for these symptoms? The simplest explanation is that these drugs fail to properly engage the neural substrates responsible for these symptoms (e.g., decreased dopamine, glutamate, or neuropathology in the PFC). While a lack of effects outside of the striatum undoubtedly contributes to their limited efficacy, even within the striatum, antipsychotics do not fully address the consequences of dopamine excess. Approximately half of the neurons in the striatum express D1Rs rather than D2Rs. Because antipsychotics do not principally target them, striatal D1Rs could contribute to the treatment-resistant symptoms. Consistent with this idea, ongoing work in our laboratory found that clozapine, a highly efficacious antipsychotic drug with some pro-cognitive effects, preferentially normalizes activity in D1R-expressing spiny projection neurons (SPNs) in the DMS under hyperdopaminergic conditions [57, 58]. By contrast, haloperidol affected both D1- and D2-SPN activity following amphetamine treatment [57]. Although these findings have yet to complete peer review, they could potentially explain why haloperidol partially rescued working memory in capsaicin-treated TRPV1 mice, and suggest that drugs that preferentially normalize D1-SPN activity may have greater therapeutic advantages over drugs like haloperidol that principally block D2Rs [57]. However, it remains to be determined how capsaicin treatment alters D1- and D2-SPN activity in this mouse model and how it changes their encoding of the behaviors studied here. After establishing these parameters, combining the approaches used here with those underway in our lab will provide a powerful approach to determine the mechanistic basis of the efficacy or lack thereof of antipsychotic drugs.
In summary, our results show that excess dopamine, when restricted to the dorsal striatum, alters behavioral processes linked to the positive, negative, and cognitive symptom subclasses of schizophrenia. Non-selectively driving dopamine release did not disrupt the processes associated with negative and cognitive symptoms, and haloperidol treatment only partially normalized the deficits in these behaviors in our model. Our results implicate nigrostriatal hyperdopaminegia in the etiology of schizophrenia symptoms that do not respond to current treatments, but other brain areas are also undoubtedly involved, particularly the PFC. Within the context of our earlier studies, treatments targeted to striatal D1-, rather than D2-SPNs may have therapeutic benefits over classical antipsychotic drugs. Further studies are necessary to unravel the intricacies of nigrostriatal dopamine excess in the behavioral processes examined here. In particular, the DS is a large, heterogeneous brain structure encompassing several subregions that contain multiple cell-types innervated by multiple dopamine cell sub-types. A better understanding of these intricacies holds the promise to develop more targeted therapies that better address the diverse symptoms of psychosis.
Supplementary information
Acknowledgements
We thank Dr. Ben Arenkiel for providing the cDNA plasmid for Trpv1. The authors have no financial disclosures to declare.
Author contributions
Conceptualization and methodology: NAM, LRB, LSZ, JGP; Investigation and analysis: NAM, SY, SWF, MMM, JAN, JGP; Writing and editing: NAM and JGP; All authors edited and approved draft prior to submission.
Funding
This work was supported by the Whitehall Foundation (JGP) and National Institutes of Mental Health: K01 MH11313201 (JGP) and 2T32 MH067564 (NAM), Neurological Disorders and Stroke R01 NS122840 (JGP), Drug Abuse P30DA048736 (LSZ) and Diabetes and Digestive and Kidney Diseases R01 DK128477 (LRB).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41386-022-01492-1.
References
- 1.Huhn M, Nikolakopoulou A, Schneider-Thoma J, Krause M, Samara M, Peter N, et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: a systematic review and network meta-analysis. Lancet. 2019;394:939–51. doi: 10.1016/S0140-6736(19)31135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harvey PD, Strassnig M. Predicting the severity of everyday functional disability in people with schizophrenia: cognitive deficits, functional capacity, symptoms, and health status. World Psychiatry. 2012;11:73–9. doi: 10.1016/j.wpsyc.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261:717–9. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- 4.Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–3. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 5.Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA. 2000;97:8104–9. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurd YL, Suzuki M, Sedvall GC. D1 and D2 dopamine receptor mRNA expression in whole hemisphere sections of the human brain. J Chem Neuroanat. 2001;22:127–37. doi: 10.1016/S0891-0618(01)00122-3. [DOI] [PubMed] [Google Scholar]
- 7.Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–86. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 8.Bell DS. The experimental reproduction of amphetamine psychosis. Arch Gen Psychiatry. 1973;29:35–40. doi: 10.1001/archpsyc.1973.04200010020003. [DOI] [PubMed] [Google Scholar]
- 9.Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophr Res. 2005;77:43–58. doi: 10.1016/j.schres.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 10.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 11.Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry. 2015;72:316–24. doi: 10.1001/jamapsychiatry.2014.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci. 2019;42:205–20. doi: 10.1016/j.tins.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akhlaghpour H, Wiskerke J, Choi JY, Taliaferro JP, Au J, Witten IB. Dissociated sequential activity and stimulus encoding in the dorsomedial striatum during spatial working memory. Elife. 2016;55:e19507. [DOI] [PMC free article] [PubMed]
- 14.Graybiel AM. The basal ganglia and cognitive pattern generators. Schizophr Bull. 1997;23:459–69. doi: 10.1093/schbul/23.3.459. [DOI] [PubMed] [Google Scholar]
- 15.Poldrack RA, Packard MG. Competition among multiple memory systems: converging evidence from animal and human brain studies. Neuropsychologia. 2003;41:245–51. doi: 10.1016/S0028-3932(02)00157-4. [DOI] [PubMed] [Google Scholar]
- 16.Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, et al. Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–90. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- 17.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–13. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 18.Parker JG, Marshall JD, Ahanonu B, Wu YW, Kim TH, Grewe BF, et al. Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature. 2018;557:177–82. doi: 10.1038/s41586-018-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez Espejo E. Prefrontocortical dopamine loss in rats delays long-term extinction of contextual conditioned fear, and reduces social interaction without affecting short-term social interaction memory. Neuropsychopharmacology. 2003;28:490–8. doi: 10.1038/sj.npp.1300066. [DOI] [PubMed] [Google Scholar]
- 20.Bolkan SS, Stujenske JM, Parnaudeau S, Spellman TJ, Rauffenbart C, Abbas AI, et al. Thalamic projections sustain prefrontal activity during working memory maintenance. Nat Neurosci. 2017;20:987–96. doi: 10.1038/nn.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guler AD, Rainwater A, Parker JG, Jones GL, Argilli E, Arenkiel BR, et al. Transient activation of specific neurons in mice by selective expression of the capsaicin receptor. Nat Commun. 2012;3:746. doi: 10.1038/ncomms1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dietrich MO, Zimmer MR, Bober J, Horvath TL. Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell. 2015;160:1222–32. doi: 10.1016/j.cell.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van den Buuse M. Modeling the positive symptoms of schizophrenia in genetically modified mice: pharmacology and methodology aspects. Schizophr Bull. 2010;36:246–70. doi: 10.1093/schbul/sbp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moghaddam B, Bunney BS. Differential effect of cocaine on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens: comparison to amphetamine. Synapse. 1989;4:156–61. doi: 10.1002/syn.890040209. [DOI] [PubMed] [Google Scholar]
- 25.Roth BL. DREADDs for neuroscientists. Neuron. 2016;89:683–94. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beutler LR, Chen Y, Ahn JS, Lin YC, Essner RA, Knight ZA. Dynamics of gut-brain communication underlying hunger. Neuron. 2017;96:461–75.e5. doi: 10.1016/j.neuron.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, et al. Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci. 2018;21:1260–71. doi: 10.1038/s41593-018-0203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulin JF, Gaertner Z, Moreno-Ramos OA, Awatramani R. Classification of midbrain dopamine neurons using single-cell gene expression profiling approaches. Trends Neurosci. 2020;43:155–69. doi: 10.1016/j.tins.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azcorra M, Gaertner Z, Davidson C, Ramakrishnan C, Fenno L, Kim YS, et al. Dopaminergic axons track somatic signaling in behaving mice. bioRxiv. 2022:2022.06.20.496872.
- 30.Kelley AE, Gauthier AM, Lang CG. Amphetamine microinjections into distinct striatal subregions cause dissociable effects on motor and ingestive behavior. Behav Brain Res. 1989;35:27–39. doi: 10.1016/S0166-4328(89)80005-1. [DOI] [PubMed] [Google Scholar]
- 31.Kim Y, Venkataraju KU, Pradhan K, Mende C, Taranda J, Turaga SC, et al. Mapping social behavior-induced brain activation at cellular resolution in the mouse. Cell Rep. 2015;10:292–305. doi: 10.1016/j.celrep.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo ZV, Inagaki HK, Daie K, Druckmann S, Gerfen CR, Svoboda K. Maintenance of persistent activity in a frontal thalamocortical loop. Nature. 2017;545:181–86. doi: 10.1038/nature22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNab F, Klingberg T. Prefrontal cortex and basal ganglia control access to working memory. Nat Neurosci. 2008;11:103–7. doi: 10.1038/nn2024. [DOI] [PubMed] [Google Scholar]
- 34.Yizhar O, Levy DR. The social dilemma: prefrontal control of mammalian sociability. Curr Opin Neurobiol. 2021;68:67–75. doi: 10.1016/j.conb.2021.01.007. [DOI] [PubMed] [Google Scholar]
- 35.Funahashi S, Chafee MV, Goldman-Rakic PS. Prefrontal neuronal activity in rhesus monkeys performing a delayed anti-saccade task. Nature. 1993;365:753–6. doi: 10.1038/365753a0. [DOI] [PubMed] [Google Scholar]
- 36.Kim IH, Kim N, Kim S, Toda K, Catavero CM, Courtland JL, et al. Dysregulation of the synaptic cytoskeleton in the PFC drives neural circuit pathology, leading to social dysfunction. Cell Rep. 2020;32:107965. doi: 10.1016/j.celrep.2020.107965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis DA. Cortical circuit dysfunction and cognitive deficits in schizophrenia–implications for preemptive interventions. Eur J Neurosci. 2012;35:1871–8. doi: 10.1111/j.1460-9568.2012.08156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piskulic D, Addington J, Cadenhead KS, Cannon TD, Cornblatt BA, Heinssen R, et al. Negative symptoms in individuals at clinical high risk of psychosis. Psychiatry Res. 2012;196:220–4. doi: 10.1016/j.psychres.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bora E, Lin A, Wood SJ, Yung AR, McGorry PD, Pantelis C. Cognitive deficits in youth with familial and clinical high risk to psychosis: a systematic review and meta-analysis. Acta Psychiatr Scand. 2014;130:1–15. doi: 10.1111/acps.12261. [DOI] [PubMed] [Google Scholar]
- 40.Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66:13–20. doi: 10.1001/archgenpsychiatry.2008.514. [DOI] [PubMed] [Google Scholar]
- 41.Chernysheva M, Sych Y, Fomins A, Alatorre Warren JL, Lewis C, Capdevila LS, et al. Striatum-projecting prefrontal cortex neurons support working memory maintenance. bioRxiv. 2021:2021.12.03.471159. [DOI] [PMC free article] [PubMed]
- 42.McElvain LE, Chen Y, Moore JD, Brigidi GS, Bloodgood BL, Lim BK, et al. Specific populations of basal ganglia output neurons target distinct brain stem areas while collateralizing throughout the diencephalon. Neuron. 2021;109:1721–38.e4. doi: 10.1016/j.neuron.2021.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee Y, Kim H, Kim JE, Park JY, Choi J, Lee JE, et al. Excessive D1 dopamine receptor activation in the dorsal striatum promotes autistic-like behaviors. Mol Neurobiol. 2018;55:5658–71. doi: 10.1007/s12035-017-0770-5. [DOI] [PubMed] [Google Scholar]
- 44.Kim H, Lee Y, Park JY, Kim JE, Kim TK, Choi J, et al. Loss of adenylyl cyclase type-5 in the dorsal striatum produces autistic-like behaviors. Mol Neurobiol. 2017;54:7994–8008. doi: 10.1007/s12035-016-0256-x. [DOI] [PubMed] [Google Scholar]
- 45.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–42. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krabbe S, Duda J, Schiemann J, Poetschke C, Schneider G, Kandel ER, et al. Increased dopamine D2 receptor activity in the striatum alters the firing pattern of dopamine neurons in the ventral tegmental area. Proc Natl Acad Sci. 2015;112:E1498. doi: 10.1073/pnas.1500450112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, et al. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron. 2006;49:603–15. doi: 10.1016/j.neuron.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 48.Wilkinson LS. The nature of interactions involving prefrontal and striatal dopamine systems. J Psychopharmacol (Oxf, Engl) 1997;11:143–50. doi: 10.1177/026988119701100207. [DOI] [PubMed] [Google Scholar]
- 49.Pycock CJ, Kerwin RW, Carter CJ. Effect of lesion of cortical dopamine terminals on subcortical dopamine receptors in rats. Nature. 1980;286:74–77. doi: 10.1038/286074a0. [DOI] [PubMed] [Google Scholar]
- 50.Li YC, Kellendonk C, Simpson EH, Kandel ER, Gao WJ. D2 receptor overexpression in the striatum leads to a deficit in inhibitory transmission and dopamine sensitivity in mouse prefrontal cortex. Proc Natl Acad Sci USA. 2011;108:12107–12. doi: 10.1073/pnas.1109718108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim IH, Rossi MA, Aryal DK, Racz B, Kim N, Uezu A, et al. Spine pruning drives antipsychotic-sensitive locomotion via circuit control of striatal dopamine. Nat Neurosci. 2015;18:883–91. doi: 10.1038/nn.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casado-Sainz A, Gudmundsen F, Baerentzen SL, Lange D, Ringsted A, Martinez-Tejada I, et al. Dorsal striatal dopamine induces fronto-cortical hypoactivity and attenuates anxiety and compulsive behaviors in rats. Neuropsychopharmacology. 2022;47:454–64. doi: 10.1038/s41386-021-01207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berke JD. What does dopamine mean? Nat Neurosci. 2018;21:787–93. doi: 10.1038/s41593-018-0152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. 2016;17:524–32. doi: 10.1038/nrn.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elliott JM, Beveridge TJ. Psychostimulants and monoamine transporters: upsetting the balance. Curr Opin Pharm. 2005;5:94–100. doi: 10.1016/j.coph.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature. 2012;490:262–6. doi: 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yun S, Yang B, Martin M, Yeh N-H, Contractor A, Parker J. Modulating D1 rather than D2 receptor-expressing spiny-projection neurons corresponds to optimal antipsychotic effect. bioRxiv; 2021.
- 58.Lee MA, Thompson PA, Meltzer HY. Effects of clozapine on cognitive function in schizophrenia. J Clin Psychiatry. 1994;55:82–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.