

Abstract
In this perspective, the authors summarise some properties of the solid tumour micro-environment that have been explored during the last 55 years. It is well established that the concentrations of nutrients, including oxygen, decrease with increasing distance from tumour blood vessels, and that low extracellular pH is found in nutrient-poor regions. Cell proliferation is dependent on nutrient metabolites and decreases in regions distal from patent blood vessels. Proliferating cells cause migration of neighbouring cells further from blood vessels where they may die, and their breakdown products pass into regions of necrosis. Anticancer drugs reach solid tumours via the vascular system and establish concentration gradients such that drug concentration within tumours may be quite variable. Treatment with chemotherapy such as doxorubicin or docetaxel can kill well-nourished proliferating cells close to blood vessels, thereby interrupting migration toward necrotic regions and lead to re-oxygenation and renewed proliferation of distal cells, as can occur with radiotherapy. This effect leads to the paradox that cancer treatment can rescue cells that were destined to die in the untreated tumour. Renewed and sometimes accelerated repopulation of surviving tumour cells can counter the effects of cell killing from repeated treatments, leading to tumour shrinkage and regrowth without changes in the intrinsic sensitivity of cells to the administered treatment. Strategies to prevent these effects include the combined use of chemotherapy with agents that selectively kill hypoxic tumour cells, including inhibitors of autophagy, since this is a process that may allow recycling of cellular macromolecules from dying cells and improve their survival.
Subject terms: Cancer microenvironment, Cancer therapeutic resistance
Cell proliferation in relation to tumour blood vessels
Dr Tannock joined the Institute of Cancer Research as a PhD student in the Department of Biophysics in 1965 at a time when cell proliferation in tumours and normal tissues was being studied by using thymidine autoradiography. Seminal work had been undertaken by his mentor, Dr Gordon Steel, whose contributions had established that tumours did not grow because of the uniform proliferation of cancer cells within them: rather there were non-dividing tumour cells and often a high rate of cell loss [1, 2]. It was known that tumours developed a limited vascular supply [3] although factors that induced angiogenesis were characterised later, and that they often contained regions of necrosis. It seemed probable that the proliferation of cancer cells might relate to nutrient concentration, although the Warburg hypothesis had posited that anaerobic glycolysis was a key driving event in tumour formation and progression [4]. From this background knowledge, a study of cell proliferation in relation to the vascular system seemed important and was facilitated by the availability of an experimental tumour that had cords of viable cells around blood vessels, as described in human tumours by Thomlinson and Gray [5]. This led to the study published in BJC in 1968, where tumour cell proliferation was characterised in relation to the central blood vessel of cords in a murine tumour using thymidine autoradiography [6]. These experiments established that there was a gradient of cell proliferation in the tumour, and that there was a dynamic process of proliferation in perivascular tumour regions that caused migration of cells away from blood vessels, with death occurring distally and movement of cell residua into necrotic regions. There is a clear analogy with patterns of proliferation and death in normal renewing tissues such as the bone marrow and gut, although there is no evidence of differentiation in the tumour that was studied.
It is not surprising that there is a gradient from high to low proliferation with increasing distance from patent blood vessels, because other studies have demonstrated gradients in oxygen and other nutrients within solid tumours [7, 8]. If cells have less nutrients available, they are less likely to be able to produce the energy (i.e., ATP) necessary for the synthesis of macromolecules that are necessary for cell division. However, an amusing anecdote stems from a meeting in Lindau, Germany in 1966 at which graduate students were invited to listen to presentations by, and to meet with, Nobel prize winners in physiology and medicine [https://www.mediatheque.lindau-nobel.org/meetings/1966]. A very nervous Tannock summarised his early results at a round table hosted by Otto Warburg and asked why, if hypoxic cells were responsible for tumour formation and progression, the well-oxygenated tumour cells close to blood vessels had the highest rate of cell proliferation. This was met with curt disbelief by the Nobel laureate.
The laborious methods for studying cell proliferation using thymidine autoradiography and microscopy have long been replaced by methods amenable to automation, such as separation by flow cytometry of tumour cells at different distances from patent tumour blood vessels using gradients of injected markers such as Hoechst 33342 and studying markers of cell proliferation such as Ki67 that can be recognised by fluorescence-labelled antibodies [9]. These methods have confirmed the general property that tumour cells are less likely to proliferate in regions that have low concentrations of nutrient metabolites (such as oxygen and glucose) and high concentrations of breakdown products of dying cells that may diffuse from necrotic regions, and consequent low extracellular pH (Fig. 1) [10].
Fig. 1. Schematic representation of a functional blood vessel and its surrounding cells.

Cells that are closest to the blood vessel are richly nourished with oxygen and other nutrients and therefore proliferate rapidly. Cells that are farther away are nutrient-deprived and may become hypoxic and have an acidic microenvironment; they tend to proliferate slowly. Modified from Tan et al. [10]. ECM extracellular matrix.
Tumour microenvironment and cancer treatment
It was known that solid tumours contained hypoxic cells that were resistant to radiation, and that following radiation treatment hypoxic cells could reoxygenate and repopulate tumours [11–13]. This effect presumably occurred because radiation killed the more sensitive aerobic cells close to tumour blood vessels, leading to improved diffusion of oxygen and other nutrients to more distant cells. Indeed, subsequent experiments showed that one effect of radiation was the narrowing of the cords of apparently viable cells within corded tumours, as surviving tumour cells migrated closer to the vascular source of nutrition [14].
The tumour microanatomy and resulting nutritional microenvironment is equally important for understanding the effects of drug treatment. Almost all chemotherapy drugs, and many molecular targeted agents, are more active against rapidly proliferating cancer cells, and will therefore tend to kill cells closer to tumour blood vessels. In addition, drugs reach tumours via the bloodstream, and several studies have shown that following injection of most anticancer drugs, gradients of drug concentration are established such that tumour cells distant from blood vessels are exposed to low drug concentrations (Fig. 2) [15–17]. Hence, poorly nourished cells in solid tumours are resistant to many anticancer drugs, both because they are slowly proliferating, and because they are exposed to lower drug concentrations. By injecting into mice two markers of hypoxic cells (EF5 and pimonidazole) that could be recognised by fluorescence-tagged monoclonal antibodies, with an interval between them that allowed changes in oxygenation to be studied, the following events were shown to occur following treatment of tumours in mice with the anticancer drugs doxorubicin or docetaxel [18]:
In the absence of treatment, tumour cells die, and their breakdown products pass into regions of necrosis; this is due to the proliferation of cells proximal to blood vessels forcing the migration of more distant cells into regions of hypoxia and poor nutrition.
Following treatment with the anticancer drugs doxorubicin or docetaxel (and presumably many others), the process of natural cell death is interrupted as cells proximal to blood vessels are killed or inhibited from cycling. This leads to the paradoxical finding that chemotherapy can rescue tumour cells that were destined to die in untreated tumours.
Some of the previously hypoxic cells are no longer hypoxic following drug treatment (i.e., there is reoxygenation, as occurs after radiation treatment)
Injection of Ki67 also showed that the “rescued” surviving cells increase their rate of proliferation.
Fig. 2. Distribution of doxorubicin in solid tumours.

a Distribution of fluorescent doxorubicin (false-colour blue) around blood vessels (recognised by CD31 on endothelial cells: false-colour red) in a mouse mammary tumour. Hypoxic areas are recognised by binding to EF5 (false-colour green). b Doxorubicin fluorescence as a function of distance from the nearest blood vessel in sections from three mouse mammary tumours. Modified from Primeau et al. [16].
These events are shown diagrammatically in Fig. 3. Responses to fractionated radiotherapy have been characterised by “five R’s”: radiosensitivity (or its opposite, resistance), repair, reoxygenation, repopulation and redistribution around the cell cycle [19]. The five R’s apply also to the response of tumours after drug treatment [20].
Fig. 3. Time-dependent changes in hypoxic cells, and in their proliferation, in doxorubicin-treated and control mice bearing MCF-7 xenografts.

Mice were injected with 2 hypoxic cell markers (pimonidazole and EF5) with a variable interval between. a Percent of tumour cells that were originally hypoxic (pimo + ) as a function of time. b Percent of hypoxic tumour cells that are newly hypoxic (pimo−/EF5 + ) as a function of time. There is dynamic flux of cells through the hypoxic compartment. c Percent of originally hypoxic cells that are no longer hypoxic (pimo + /EF5−) at 24 h after injection of saline or doxorubicin. d Percent of reoxygenated cells that are Ki67 positive at 24 h after injection of saline or doxorubicin. Reoxygenation and repopulation of hypoxic cells occur after treatment with doxorubicin. Similar results were obtained after treatment of prostate cancer PC3 xenografts with docetaxel. Adapted from Saggar and Tannock [18].
Drug resistance
A review of publications relating to the resistance of anticancer drugs would reveal many thousands of papers that investigate molecular causes of resistance. This is important, but not the only cause of resistance. Far less appreciated is the effect of limited drug distribution in tumours (as shown in Fig. 2): if drugs do not reach some of the tumour cells in a lethal concentration, then there will be resistance to therapy—regardless of the sensitivity of constituent cells when exposed to the drug at a uniform concentration in tissue culture. Pharmacokinetic studies may report drug concentration in tumour, blood, and normal tissues as a function of time after drug administration, and investigators may talk about the “tumour drug concentration”. However, the average drug concentration in a tumour has very little meaning if the concentration varies from high levels to close to zero in microanatomical regions—for example, from regions bordering patent blood vessels to regions that are nutrient-deprived.
A second neglected cause of drug resistance relates to repopulation. Cells surviving initial drug treatment may increase their proliferation after chemotherapy, so that if drug treatments are given at intervals (typically 3 weeks for chemotherapy), the increasing rate of repopulation between times of drug administration may eventually overcome the killing effect of the applied therapy (Fig. 4) [21, 22]. A similar effect has been described for radiotherapy with greater repopulation towards the end of a fractionated course leading to decreasing benefit [23]. The priming of repopulation by chemotherapy might also explain the absence of benefit for some types of human tumour when neoadjuvant chemotherapy is given prior to initiating radiotherapy [24].
Fig. 4. Schematic diagram to illustrate how accelerating repopulation of tumours (that may occur with improving nutrition) can overcome the killing effects of repeated doses of drugs or radiation.

It is assumed that 70% of tumour cells are killed after each administration of chemotherapy, which is given at 3-week intervals. a Assumes a constant rate of repopulation of surviving tumour cells between treatments, characterised by a doubling time of either 10 days or 2 months. b Assumes accelerating repopulation of surviving tumour cells between successive courses of chemotherapy, characterised by the indicated doubling times. c Assumes a delay in onset of repopulation after each cycle of chemotherapy, followed by accelerating repopulation of surviving tumour cells with the indicated doubling times. Note that accelerated repopulation can lead to the remission and regrowth of tumours during chemotherapy, as is commonly observed in clinical practice, without any change in the intrinsic chemosensitivity of the tumour cells. TD cell doubling time. Adapted from Kim and Tannock [22].
Combating microenvironmental drug resistance
There are potential strategies to overcome drug resistance due to microenvironmental and/or proliferative factors, whereas increasing the sensitivity of cells that are intrinsically drug-resistant is difficult. Strategies to combat resistance of cancer cells that are situated in a nutrient-derived microenvironment include:
Improving the distribution of anticancer drugs within solid tumours.
Combining conventional drug therapy with agents that have selective toxicity for hypoxic or nutrient-deprived cells.
Inhibiting cell survival mechanisms for nutrient-deprived cells.
Improving the tumour distribution of drugs that have a short half-life in blood is difficult because better distribution to cells distal from blood vessels is only likely to be achieved by inhibiting uptake in proximal cells, which may decrease the effectiveness of such treatment. Administering drugs by chronic infusion may establish a more uniform distribution, although multiple factors influence the efficacy of variant schedules. Drugs with a long half-life in the blood (e.g., monoclonal antibodies) are likely to reach equilibrium and establish a more uniform concentration in tumours.
Pro-drugs that require bio-reduction to a toxic form may have selective activity for hypoxic cells, and the toxic products may also diffuse to kill cells that are not themselves hypoxic [25]. Agents of this type include tirapazamine and evofosfamide (TH-302). These agents are effective in augmenting the effects of chemotherapy drugs against tumours that contain hypoxic cells in experimental animals [26–28]. Both agents have been evaluated in large Phase 3 clinical trials together with standard chemotherapy but did not significantly improve the survival of treated patients [29–32]. These trials have been criticised because no effort was made to select patients with tumours that contained a substantial proportion of hypoxic cells [33]; this is unfortunate because the negative results of the trials have discouraged further research into a strategy that retains the potential for improving clinical outcomes.
Autophagy (or self-eating) is a well-characterised mechanism in which macromolecular fragments from damaged cells are processed into autophagosomes that fuse with acidic lysosomes, where there is further breakdown under the action of cathepsins and other enzymes that are active under acidic conditions. In some circumstances, this leads to cell death, but autophagy can also promote the survival of poorly nourished or damaged tumour cells by acting as a recycling mechanism for the breakdown products of macromolecules. Experiments in animal models have shown that markers of autophagy (such as LC3B) co-localise with hypoxia in tumours [34], and knockout cells that are autophagy-deficient have poor survival under hypoxic conditions [34, 35]. Treatment of cultured cells with different chemotherapy drugs induces autophagy [36], and inhibition of autophagy by drugs that inhibit the acidification of endosomes, such as pantoprazole, have been shown to augment the activity of chemotherapy in experimental models by increasing cell death in regions distal from tumour blood vessels (Fig. 5) [37]. Agents used to inhibit autophagy have this far been repurposed from other uses: chloroquine and hydroxychloroquine used to treat malaria and autoimmune diseases, and drugs that inhibit acidification of cells lining the stomach such as omeprazole and pantoprazole that are used to treat or prevent stomach ulcers. The development of more specific inhibitors of autophagy via mechanisms other than inhibition of endosomal acidification deserves high priority for clinical evaluation.
Fig. 5. Inhibition of Autophagy can increase the effectiveness of anticancer drugs.
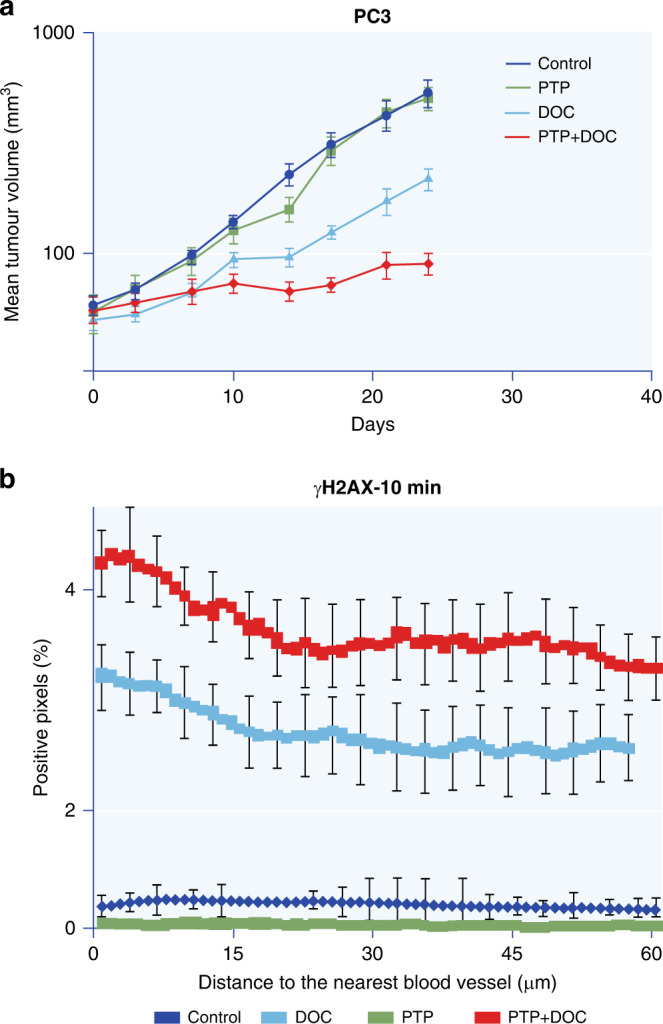
a Growth curves of PC3 prostate cancer xenografts following treatment with docetaxel (DOC), pantoprazole (PTP), or both drugs. b Staining of γH2AX (a marker of DNA damage) as a function of distance from the nearest tumour blood vessel at 10 min after treatment of PC3 xenografts with saline, docetaxel, pantoprazole or both drugs. Note that PTP, an inhibitor of autophagy, has no effect alone but augments the activity of docetaxel. Adapted from Tan et al. [37].
Conclusion
In this brief perspective, we have outlined research which relates to the broad theme of nutritional heterogeneity in the microenvironment of solid tumours that has taken place in the 50+ years since the publication of the original article relating cell proliferation to the vascular system in an experimental solid tumour [6]. During that period, various strategies have led to profound improvements in cancer treatment, but there remains considerable potential for further improvements that are directed to overcoming resistance to both radiation and drug therapy that require a thorough understanding of the microanatomy of solid tumours.
Author contributions
Both authors contributed to the writing of the MS.
Funding
None.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Steel GG, Lamerton LF. The growth rate of human tumours. Br J Cancer. 1966;20:74–86. doi: 10.1038/bjc.1966.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steel GG. Cell loss as a factor in the growth rate of human tumours. Eur J Cancer. 1967;3:381–7. doi: 10.1016/0014-2964(67)90022-9. [DOI] [PubMed] [Google Scholar]
- 3.Goldacre RJ, Sylven B. On the access of blood-borne dyes to various tumour regions. Br J Cancer. 1962;16:306–22. doi: 10.1038/bjc.1962.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9:539–49. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tannock IF. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer. 1968;22:258–73. doi: 10.1038/bjc.1968.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–65. [PubMed] [Google Scholar]
- 8.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–82. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 9.Chaplin DJ, Durand RE, Olive PL. Cell selection from a murine tumour using the fluorescent probe Hoechst 33342. Br J Cancer. 1985;51:569–72. doi: 10.1038/bjc.1985.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan Q, Saggar JK, Yu M, Wang M, Tannock IF. Mechanisms of drug resistance related to the microenvironment of solid tumors and possible strategies to inhibit them. Cancer J. 2015;21:254–62. doi: 10.1097/PPO.0000000000000131. [DOI] [PubMed] [Google Scholar]
- 11.Begg AC, Denekamp J, Howes AE. Re-oxygenation of tumours after irradiation and cell population kinetics. J Physiol. 1969;202:16P–17P. [PubMed] [Google Scholar]
- 12.Kallman RF. Reoxygenation and repopulation in irradiated tumors. Front Radiat Ther Oncol. 1988;22:30–49. doi: 10.1159/000415094. [DOI] [PubMed] [Google Scholar]
- 13.Speke AK, Hill RP. The effects of clamping and reoxygenation on repopulation during fractionated irradiation. Int J Radiat Oncol Biol Phys. 1995;31:857–63. doi: 10.1016/0360-3016(94)00501-X. [DOI] [PubMed] [Google Scholar]
- 14.Tannock I, Howes A. The response of viable tumor cords to a single dose of radiation. Radiat Res. 1973;55:477–86. doi: 10.2307/3573853. [DOI] [PubMed] [Google Scholar]
- 15.Lankelma J, Dekker H, Luque FR, Luykx S, Hoekman K, van der Valk P, et al. Doxorubicin gradients in human breast cancer. Clin Cancer Res. 1999;5:1703–7. [PubMed] [Google Scholar]
- 16.Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;15:8782–8. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]
- 17.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 18.Saggar JK, Tannock IF. Chemotherapy rescues hypoxic tumor cells and induces their reoxygenation and repopulation—an effect that is inhibited by the hypoxia-activated prodrug TH-302. Clin Cancer Res. 2015;21:2107–14. doi: 10.1158/1078-0432.CCR-14-2298. [DOI] [PubMed] [Google Scholar]
- 19.Klement RJ, Champ CE. Calories, carbohydrates, and cancer therapy with radiation: exploiting the five R’s through dietary manipulation. Cancer Metastasis Rev. 2014;33:217–29. doi: 10.1007/s10555-014-9495-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tannock IF. The five Rs of chemotherapy. Lancet Oncol. 2016;17:703–5. doi: 10.1016/S1470-2045(16)00103-0. [DOI] [PubMed] [Google Scholar]
- 21.Davis AJ, Tannock JF. Repopulation of tumour cells between cycles of chemotherapy: a neglected factor. Lancet Oncol. 2000;1:86–93. doi: 10.1016/S1470-2045(00)00019-X. [DOI] [PubMed] [Google Scholar]
- 22.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer. 2005;5:516–25. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 23.Withers HR, Taylor JM, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988;27:131–46. doi: 10.3109/02841868809090333. [DOI] [PubMed] [Google Scholar]
- 24.Brade AM, Tannock IF. Scheduling of radiation and chemotherapy for limited-stage small-cell lung cancer: repopulation as a cause of treatment failure? J Clin Oncol. 2006;24:1020–2. doi: 10.1200/JCO.2005.04.9676. [DOI] [PubMed] [Google Scholar]
- 25.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 26.Dorie MJ, Brown JM. Modification of the antitumor activity of chemotherapeutic drugs by the hypoxic cytotoxic agent tirapazamine. Cancer Chemother Pharm. 1997;39:361–6. doi: 10.1007/s002800050584. [DOI] [PubMed] [Google Scholar]
- 27.Saggar JK, Tannock IF. Activity of the hypoxia-activated pro-drug TH-302 in hypoxic and perivascular regions of solid tumors and its potential to enhance therapeutic effects of chemotherapy. Int J Cancer. 2014;134:2726–34. doi: 10.1002/ijc.28595. [DOI] [PubMed] [Google Scholar]
- 28.Sun JD, Liu Q, Ahluwalia D, Li W, Meng F, Wang Y, et al. Efficacy and safety of the hypoxia-activated prodrug TH-302 in combination with gemcitabine and nab-paclitaxel in human tumor xenograft models of pancreatic cancer. Cancer Biol Ther. 2015;16:438–49. doi: 10.1080/15384047.2014.1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williamson SK, Crowley JJ, Lara PN, Jr, McCoy J, Lau DHM, Tucker RW, et al. Phase III trial of paclitaxel plus carboplatin with or without tirapazamine in advanced non-small-cell lung cancer: Southwest Oncology Group Trial S0003. J Clin Oncol. 2005;23:9097–104. doi: 10.1200/JCO.2005.01.3771. [DOI] [PubMed] [Google Scholar]
- 30.DiSilvestro PA, Ali S, Craighead PS, Lucci JA, Lee Y-C, Cohn DE, et al. Phase III randomized trial of weekly cisplatin and irradiation versus cisplatin and tirapazamine and irradiation in stages IB2, IIA, IIB, IIIB, and IVA cervical carcinoma limited to the pelvis: a Gynecologic Oncology Group study. J Clin Oncol. 2014;32:458–64. doi: 10.1200/JCO.2013.51.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Cutsem E, Lenz H-J, Furuse J, Tabernero J, Heinemann V, Ioka, T, et al. MAESTRO: a randomized, double-blind phase III study of evofosfamide in combination with gemcitabine in previously untreated patients with metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma. J Clin Oncol. 2016;34(Suppl):Abstract 4007.
- 32.Tap WD, Papai Z, Van Tine BA, Attia S, Ganjoo KN, Jones RL, et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2017;18:1089–103. doi: 10.1016/S1470-2045(17)30381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter FW, Wouters BG, Wilson WR. Hypoxia-activated prodrugs: paths forward in the era of personalised medicine. Br J Cancer. 2016;114:1071–7. doi: 10.1038/bjc.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan Q, Wang M, Yu M, Zhang J, Bristow RG, Hill RP, et al. Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia. 2016;18:347–55. doi: 10.1016/j.neo.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Tan Q, Joshua AM, Wang M, Bristow RG, Wouters BG, Allen CJ, et al. Up-regulation of autophagy is a mechanism of resistance to chemotherapy and can be inhibited by pantoprazole to increase drug sensitivity. Cancer Chemother Pharm. 2017;79:959–69. doi: 10.1007/s00280-017-3298-5. [DOI] [PubMed] [Google Scholar]
- 37.Tan Q, Joshua AM, Saggar JK, Yu M, Wang M, Kanga N, et al. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. Br J Cancer. 2015;112:832–40. doi: 10.1038/bjc.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]