

Abstract
Background
Germline variants in the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) cause Lynch syndrome, an autosomal dominant hereditary cancer susceptibility syndrome. The risk for endometrial cancer is significantly higher in women with MSH6 pathogenic/likely pathogenic (P/LP) variants compared with that for MLH1 or MSH2 variants.
Methods
The proband was tested via a clinical testing, Memorial Sloan Kettering‐Integrated Mutation Profiling of Actionable Cancer Targets (MSK‐IMPACT). RT‐PCR was performed using patient's blood DNA and cDNA was analyzed by DNA sequencing and a cloning approach.
Results
We report a 56‐year‐old female with endometrial cancer who carries a germline variant, MSH6 c.4001G > C, located at the last nucleotide of exon 9. While the pathogenicity of this variant was previously unknown, functional studies demonstrated that this variant completely abolished normal splicing and caused exon 9 skipping, which is expected to lead to a prematurely truncated or abnormal protein.
Conclusion
Our results indicate that this variant likely contributes to cancer predisposition through disruption of normal splicing, and is classified as likely pathogenic.
Keywords: c.4001G > C, germline, Lynch syndrome, MSH6, splice site variant
We report a 56‐year‐old female with endometrial cancer who carries a germline variant, MSH6 c.4001G > C, located in the last nucleotide of exon 9. While the pathogenicity of this variant was previously unknown, functional studies demonstrated that this variant completely abolished normal splicing and caused exon 9 skipping, which is expected to lead to a prematurely truncated or abnormal protein. Our results indicate that this variant likely contributes to cancer predisposition through disruption of normal splicing, and is classified as likely pathogenic.

1. INTRODUCTION
Lynch syndrome (LS), also known as hereditary non‐polyposis colorectal cancer (HNPCC), is an autosomal dominant cancer predisposition syndrome accounting for approximately 1%–5% of all diagnosed colorectal cancers (CRC) (Hampel et al., 2005; Rubenstein et al., 2015; Vasen, 2005). LS is characterized by a significantly increased risk of developing endometrial cancer (EC) and CRC, as well as ovarian, small bowel, stomach, hepatobiliary, urinary, brain or central nervous system cancer, and sebaceous tumors (Cohen & Leininger, 2014). LS is caused by a defect in the DNA mismatch repair (MMR) pathway, (Kunkel & Erie, 2005; Tamura et al., 2019), or by a deletion in the epithelial cell adhesion molecule (EPCAM) (OMIM#: 185535) gene, leading to a transcriptional read‐through which silences the downstream mutS homolog 2 (MSH2) (OMIM#: 60309) gene (Goel et al., 2011; Kuiper et al., 2011; Niessen et al., 2009).
The MMR proteins MLH1, MSH2, MSH6, and PMS2 are encoded by MutL homolog1 (MLH1) (OMIM#: 120436), MSH2, mutS homolog 6 (MSH6) (OMIM#: 600678) and post‐meiotic segregation increased 2 (PMS2) (OMIM#: 600259) genes, respectively. MSH2 couples with either MSH6 or MSH3, and MLH1 interacts with PMS2 or MLH3 to form heterodimeric complexes (Jiricny, 2006). These complexes are responsible for surveillance and correction of errors made during DNA replication, repair, and recombination (Jiricny, 2006). Germline P/LP variants in the MMR gene generally lead to tumors with characteristic mutational signature, microsatellite instability (MSI), and loss of expression of one or more MMR proteins detected by immunohistochemistry (IHC) (Boland et al., 2008). Although EPCAM is not an MMR gene, germline deletions of 3′ end of the EPCAM gene leads to epigenetic silencing of the neighboring MMR gene MSH2 by hypermethylation (Kuiper et al., 2011; Ligtenberg et al., 2009).
Germline variants in MMR genes result in a cumulative risk of up to 60% to develop CRC in men, and up to 50% in women; and a risk of up to 50% to develop EC at 75 years of age (Dominguez‐Valentin et al., 2020). MLH1 and MSH2 P/LP variants account for more than 50% of all LS colorectal cancer in many studies (Hampel et al., 2008; Moller et al., 2017; Sjursen et al., 2016; Yurgelun et al., 2015). MSH6 and PMS2 germline P/LP variants are less common, accounting for about 6%–17% and less than 15% of all MMR gene deleterious variants in LS patients respectively (Bonadona et al., 2011; Hampel et al., 2008; Moller et al., 2017; Sjursen et al., 2016). However, more recent studies indicate MSH6 and PMS2 P/LP variants account for 24%–29% and 22%–24%, respectively, of all germline P/LP variants associated with Lynch syndrome which is much more prevalent than the previous studies (Espenschied et al., 2017; Latham et al., 2019). Germline EPCAM deletions occur in at least 1%–3% of the LS families (Tutlewska et al., 2013). Individuals with MSH6 P/LP variants tend to develop CRC at an older age than those who carry MLH1 or MSH2 P/LP variants and have reduced penetrance (Baglietto et al., 2010; Berends et al., 2002; Hendriks et al., 2004; Wijnen et al., 1998). In women harboring MSH6 P/LP variants, the risk for colorectal cancer is significantly lower than that in individuals harboring MLH1 and MSH2 P/LP variants, while the risk for endometrial cancer is significantly higher by age 70 (Hendriks et al., 2004). The cumulative risk for diagnosis of endometrial cancer through lifetime is 16%–49% for individuals who contains MSH6 P/LP variants (Baglietto et al., 2010; Bonadona et al., 2011; Moller et al., 2018). The incidence of EC is 26‐fold higher in women who carry MSH6 pathogenic variants, compared with incidence for the general population (Baglietto et al., 2010). Therefore, determining MSH6 variant pathogenicity is of significant clinical relevance, particularly for predicting cancer risks.
The clinical interpretation of variants involving the last nucleotide of an exon is difficult due to uncertain molecular effects of such alterations. Variants at this position may result in missense substitutions (Kanai et al., 1999), and/or disruptions of normal splicing leading to skipping of one or more exons (Barreiros et al., 2018; Vettore et al., 2010; Yamada et al., 2007). Typically, variants in the last nucleotide of an exon are classified as a variant of uncertain significance (VUS) according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines (Richards et al., 2015) in the absence of additional functional, segregation, and splicing studies. A variant in the last nucleotide of exon 9 of MSH6 gene (c.4001G > A) has been reported to segregate with disease in multiple LS families (Hendriks et al., 2004; Klarskov et al., 2011; Wijnen et al., 1999), and has been classified as pathogenic. However, there are no functional data supporting the pathogenicity of the other variants affecting the same nucleotide, including c.4001G > C. In this report, we demonstrated that the MSH6 c.4001G > C variant, identified in 56‐year‐old woman diagnosed with uterine endometrioid carcinoma with microsatellite instability high (MSI‐H) and loss of MSH6 protein expression, disrupts normal splicing and results in complete loss of exon 9, which presumably leads to premature protein truncation or abnormal protein. Our results indicate that this variant likely contributes to cancer predisposition through disruption of normal splicing, and can be classified as likely pathogenic based on ACMG/AMP guidelines.
2. MATERIAL AND METHODS
2.1. Subject
Our proband is a 58‐year‐old woman who was diagnosed with uterine endometrioid carcinoma at age 56 with MSI‐H and loss of MSH6 protein expression. A four‐generation pedigree (Figure 1) indicated that at least three family members were affected with LS‐related cancers. The proband's maternal grandfather was affected with prostate cancer. The proband's mother and brothers were diagnosed with LS. The proband was tested via a clinical‐grade testing using NYSDOH‐ and CLIA‐approved Memorial Sloan Kettering‐Integrated Mutation Profiling of Actionable Cancer Targets (MSK‐IMPACT) and was identified to carry the MSH6 (NM_000179.2) c.4001G > C. This variant was classified as VUS and likely pathogenic by different laboratories in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/233214/). Given the conflicting interpretations of pathogenicity of this variant, the patient was assigned onto an IRB‐approved protocol and agreed to provide additional blood samples for further characterization of this variant at Memorial Sloan Kettering Cancer Center (MSKCC). Peripheral blood samples were collected and submitted to the Diagnostics Molecular Genetics Laboratory at MSKCC. Control RNAs were from unrelated cancer patients who do not carry the MSH6 variant.
FIGURE 1.
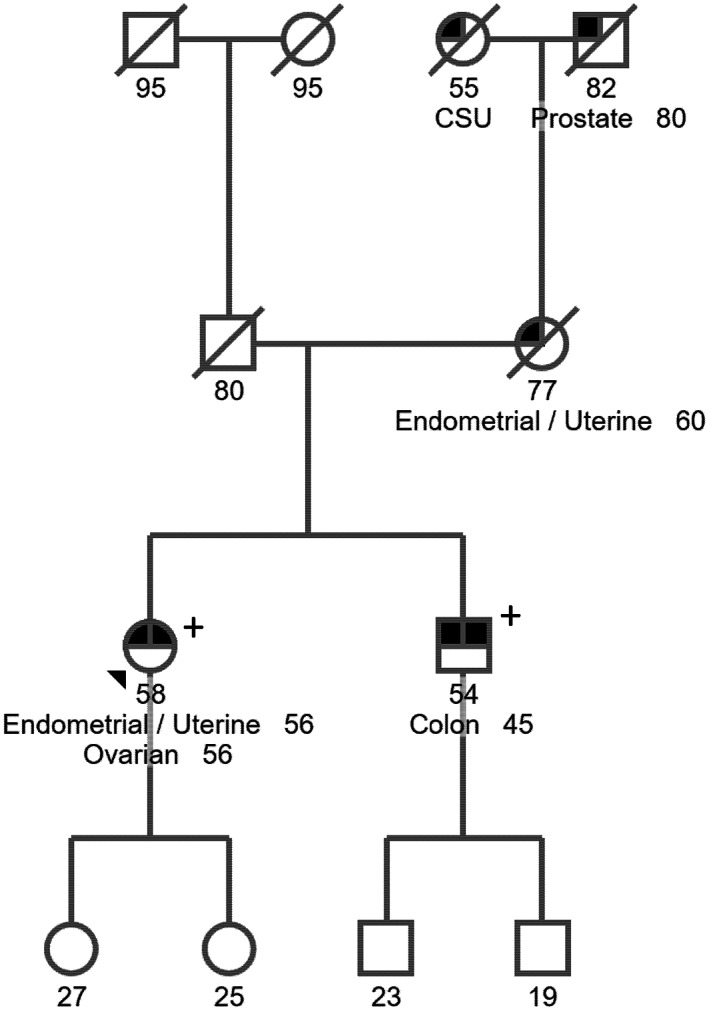
Patient pedigree. The patient described here is a 58‐year‐old female who was diagnosed with endometrial and ovarian cancers at age 56. Her brother who also carries the variant MSH6 NM_000179.2 c.4001G > C was affected with colon cancer at age 45 and her mother was diagnosed with endometrial cancer at her 60s.
2.2. In silico analysis
Sequence data spanning the MSH6 locus for Homo sapiens [Chromosome 2: 47,783,082.0.47,810,101] was obtained from the Ensembl Genome Browser (http://www.ensembl.org/index.html). Primers were designed using the Primer 3 software (http://bioinfo.ut.ee/primer3‐0.4.0/). In silico evaluation of the variants was performed through Alamut (Interactive Biosoftwar), which include SSF, MaxEnt, NNSPLICE, and GeneSplicer tools.
2.3. cDNA analysis
Total RNA from the patient was extracted using the PAXgene BloodRNA Kit (PreAnalytiX, Qiagen, Valencia, CA) and was subsequently used for cDNA synthesis (Superscript III First‐Strand Synthesis SuperMix, Invitrogen Life Technologies, Carlsbad, CA). Control RNA was extracted from another unrelated cancer individual who did not carry the MSH6 variant. RT‐PCR was performed through SuperScript™ III First‐Strand Synthesis SuperMix (Invitrogen) for RT and then the JumpStart REDTaq Ready Mix (Sigma) for PCR, with control cDNA or the patient's cDNA in the presence of M13‐tagged forward and reverse primers (Forward, E7F: 5′‐GTA AAA CGA CGG CCA GT TGAAACTGCCAGCATACTCAT‐3′; Reverse, E10R: 5′‐CAG GAA ACA GCT ATG AC TCAACTCAAAGCTTCCAATG‐3′). Each PCR reaction contains 12.5 μl 2× JumpStart REDTaq Ready Mix, 2 μl 10 μM primers (1 μl for each primer), 2 μl cDNA, and water to make a final volume of 25 μl. PCR reactions were performed under the following conditions: 96°C for 5 min, 94°C for 30 s (35×), 64°C for 45 s (35×), and 72°C for 60 s (35×) with a final extension at 72°C for 5 min (1×).
2.4. Cloning
To test whether the mutant allele is able to generate any normal transcript, RT‐PCR products were cloned into pCR4 TOPO vectors (Invitrogen, Carlsbad, CA), following procedures of the pCR4 TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA). DNA from colonies was amplified using the forward E7F and the reverse primer E10R, and subjected to direct DNA sequencing analysis using the forward PCR primer (BigDye Terminator v3.1 Cycle Sequencing kit and 3730 DNA Analyzer, Applied Biosystems, Foster City, CA).
3. RESULTS
3.1. The MSH6 c.4001G > C variant disrupts normal splicing and presumably leads to premature protein truncation
This variant c.4001G > C affects the last nucleotide of exon 9 of the MSH6 coding sequence, which is part of the consensus splice site for this exon. To evaluate the potential effects of the variant on splicing, we used Alamut software, which incorporates four tools to predict the potential effect of MSH6 c.4001G > C on mRNA splicing. Three out of the four tools predicted that the variant significantly weakens the 3′ splice acceptor site with two of them predicting a complete loss of the canonical acceptor site and another one predicting a score reduction of 57%. The last tool (GeneSplicer) predicted that this variant may not affect splicing (Figure 2a). Another learning‐based splicing tool, SpliceAI (https://spliceailookup.broadinstitute.org/), predicts a loss of the canonical donor site at c.4001, with the high prediction score (0.88) (Figure 2b).
FIGURE 2.
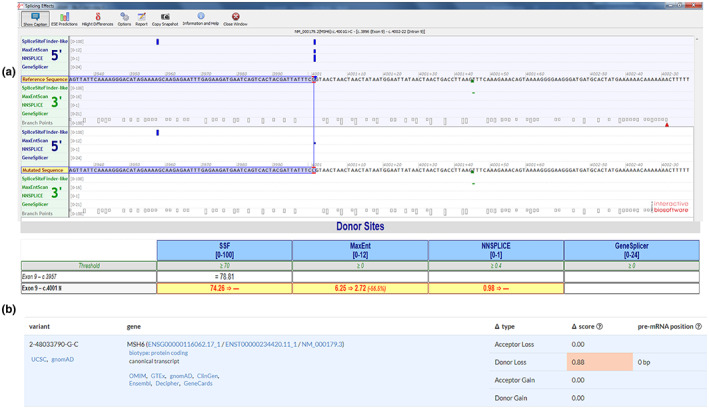
In silico predictions of the c.4001G > C variant. (a) The Alamut software was used to evaluate the potential effects of the variant on splicing. Three out of the four tools predicted that the variant significantly weakens the 5′ donor splice site with two of them predicted complete loss of the canonical donor site and another one predicted a score reduction of 57%. The other tool predicted that this variant does not significantly affect splicing. (b) SpliceAI predicts a loss of the donor site with a high prediction score.
The effect of MSH6 c.4001G > C variant on RNA splicing was evaluated by amplifying relevant regions of MSH6 from cDNA derived from the patient. PCR was designed to generate a fragment that spanned exons likely to be affected by this variant, including a part of exon 7, and the entire coding regions of exons 8, 9, and 10. Identification of an additional PCR product suggested the presence of an aberrantly spliced MSH6 transcript in the patient sample (Figure 3a). Further sequencing analysis revealed that the entire coding region of MSH6 exon 9 is deleted from the aberrantly spliced MSH6 transcript (Figure 3b). The deletion of MSH6 exon 9 is predicted to result in an absent or truncated protein product NP_000170.1: (p.?). The truncation disrupts a significant C‐terminal portion of the MSH2 interaction domain of the MSH6 protein (residues Ala1302‐Leu1360) (Guerrette et al., 1998; Kariola et al., 2002).
FIGURE 3.
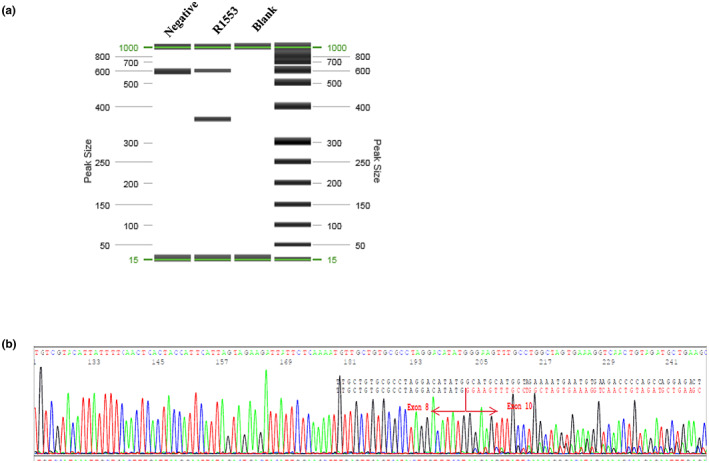
RT‐PCR analysis demonstrates c.4001G > C leads to exon 9 skipping. (a) RT‐PCR products run on QIAxcel. Two extra bands were observed in the patient, but not in the control. (b) Electropherogram showing that the variant causes exon skipping. The boundary of exons is marked by red arrow.
3.2. The variant MSH6 c.4001G > C completely disrupts normal splicing in the mutant allele
We used a cloning approach to determine whether the c.4001G > C variant completely abolishes normal splicing. The RT‐PCR products were cloned into the TOPO sequencing vector, and 135 colonies were sequenced to assess the effect of this variant on splicing. One hundred and nine out of 135 colonies (109/135; 80.7%) contained the aberrantly spliced transcript lacking exon 9 (Figure 4b). The remaining 26 clones (26/135; 19.3%) contained the full‐length transcript and all had the wild‐type nucleotide at the c.4001 position (i.e., G), indicating that the mutant allele was unable to generate any full‐length transcript (Figure 4a).
FIGURE 4.
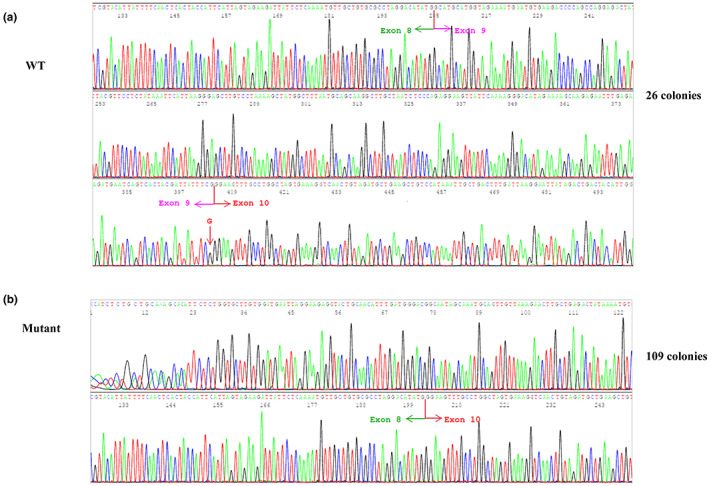
cDNA cloning demonstrates that the mutant allele does not produce any wild‐type transcript. (a) All clones (n = 26) with the full‐length transcript containing the normal G at the c.4001 position; (b) All clones (n = 109) with the exon 9 deletion are generated from the mutant C allele.
4. DISCUSSION
Missense changes at the last nucleotide of an exon have been reported to cause missense substitutions, as well as aberrant splicing leading to exon skipping (Kanai et al., 1999; Vettore et al., 2010; Yamada et al., 2007). Therefore, the clinical significance of such variants remains uncertain without further functional, and/or segregation analysis. In this study, we report that a missense substitution c.4001G > C located in the last nucleotide of exon 9 of MSH6 gene completely abolishes normal splicing of the MSH6 transcript, and is predicted to lead to a prematurely truncated or absent protein.
The c.4001G > C variant has not, to our knowledge, been previously reported in the literature, and is absent from large reference population databases (e.g., The Genome Aggregation Database) (PM2_supporting). Although the arginine residue is only moderately conserved, this substitution was classified as likely pathogenic in 2019 by Ambry and as a VUS by Invitae in 2019 (https://www.ncbi.nlm.nih.gov/clinvar/variation/233214/). Of note, another variant affecting the same nucleotide (c.4001G > A; p.Arg1334Gln) is a well‐known pathogenic change based on segregation data from two LS families (Hendriks et al., 2004; Klarskov et al., 2011; Wijnen et al., 1999) and observations in other unrelated individuals affected with LS‐associated cancers (Overbeek et al., 2007; Susswein et al., 2016; You et al., 2010). The pathogenicity of the c.4001G > C variant was uncertain based on the current version of ACMG/AMP variant interpretation guidelines due to the lack of more definitive functional or segregation data.
A variety of software tools have been developed to predict the effect of an alteration on creation of novel, or changes to existing splice sites. Although these tools cannot be used to definitively classify variants in a clinical laboratory setting, they can help prioritize variants of uncertain significance for further investigation, including in vitro splicing studies. Splicing prediction tools, including SplinceSiteFinder‐like, MaxEntScan, NNSPLICE, and SpliceAI, suggested that the MSH6 c.4001G > C variant may affect RNA splicing, which inspired us to pursue further analysis of patient‐derived RNA. The c.4001G > C variant completely disrupted normal splicing leading to skipping of exon 9 according to in vitro RT‐PCR results. Since exon 9 is the penultimate exon of MSH6, the c.4001G > C variant is likely to escape nonsense‐mediated decay and result in a truncated protein (p.Ala1268Glyfs*6) (Karousis & Muhlemann, 2019; Kurosaki et al., 2019).
The MSH6 protein contains two interaction regions which help in forming a heterodimer with the MSH2 protein (Guerrette et al., 1998; Kariola et al., 2002): the amino‐terminal interaction region (residues 326 to 575), and the carboxy‐terminal interaction region (residues 1302 to 1360) (Guerrette et al., 1998; Kariola et al., 2002). Our RT‐PCR and cDNA sequencing analysis demonstrated that the MSH6 c.4001G > C variant, which may lead to a truncated protein (p.Ala1268Glyfs*6), is expected to disrupt a significant portion of the C‐terminal MSH2 interaction domain (PS3). Notably, multiple truncating variants downstream of Ala1268 have been reported in individuals with LS (Baglietto et al., 2010; Barnetson et al., 2006; Devlin et al., 2008; Raskin et al., 2011). Furthermore, MSH6 c.3984_3987dup (p.Leu1330Valfs*12), a well‐characterized LS founder mutation in the Ashkenazi Jewish population (Goldberg et al., 2010; Raskin et al., 2011), has been shown to segregate with disease in a family affected with colorectal cancer (Peterlongo et al., 2003), and result in lack of MSH6 staining and tumor microsatellite instability (Goldberg et al., 2010). Taken together, these reports strongly indicate that the last 31 amino acid residues are critical for MSH6 protein function (PM1), and the c.4001G > C variant likely results in a loss‐of‐function. It is worth noting that tumor from the proband exhibits MSI‐H and loss of MSH2 and MSH6 protein expression, and harbors a second hit with the variant c.3646+1G>A located in intron 7 of MSH6, but not in MSH2 as tested by MSK‐IMPACT (Data not shown), indicating that the loss of MSH2 is likely due to loss of MSH6. The proband's brother who was diagnosed with colon cancer at age 45 also had the same germline variant (PP1).Taken together, the strong LS family history (PP4) and our data indicate that the MSH6 c.4001G > C variant results in a likely loss‐of‐function, and should be classified as likely pathogenic according to ACMG/AMP guidelines.
The improved understanding of this variant has significant impact on the patient's medical management and for counseling of the patient and family members regarding disease risk and reproductive planning.
AUTHOR CONTRIBUTIONS
Ciyu Yang and Liying Zhang designed this study. Ciyu Yang performed the experiments and drafted the manuscript. Maksym Misyura and Vikas Rai assisted with generating laboratory data. Sarah Kane and Alicia Latham provided clinical data. All authors reviewed the manuscript.
FUNDING INFORMATION
This study was funded by Department of Pathology, Memorial Sloan Kettering Cancer Center (Grant: P30 CA008748).
CONFLICT OF INTEREST
Dr. Zhang reports honoraria (Future Technology Research LLC, BGI, Illumina); honoraria and Travel and accommodation expenses (Roche Diagnostics Asia Pacific). Family members hold leadership position and ownership interests of Decipher Medicine.
ETHICAL STANDARDS
The patient signed the IRB‐approved protocol (#12–245) approved by Memorial Sloan Kettering Cancer Center.
ACKNOWLEDGEMENTS
We thank members in the Diagnostic Molecular Genetics (DMG) Laboratory, MSK‐IMPACT team and Clinical Genetics Service for providing the laboratory and clinical data that allowed us to further study this variant. We also thank Department of Pathology and MSKCC for the funding.
Yang, C. , Misyura, M. , Kane, S. , Rai, V. , Latham, A. , & Zhang, L. (2023). Characterization of a germline variant MSH6 c.4001G > C in a Lynch syndrome family. Molecular Genetics & Genomic Medicine, 11, e2104. 10.1002/mgg3.2104
DATA AVAILABILITY STATEMENT
The data was submitted in Clinvar. The submission ID is SCV002060005.
REFERENCES
- Baglietto, L. , Lindor, N. M. , Dowty, J. G. , White, D. M. , Wagner, A. , Gomez Garcia, E. B. , Vriends, A. H. , Dutch Lynch Syndrome Study Group , Cartwright, N. R. , Barnetson, R. A. , Farrington, S. M. , Tenesa, A. , Hampel, H. , Buchanan, D. , Arnold, S. , Young, J. , Walsh, M. D. , Jass, J. , Macrae, F. , … Jenkins, M. A. (2010). Risks of lynch syndrome cancers for MSH6 mutation carriers. Journal of the National Cancer Institute, 102(3), 193–201. 10.1093/jnci/djp473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnetson, R. A. , Tenesa, A. , Farrington, S. M. , Nicholl, I. D. , Cetnarskyj, R. , Porteous, M. E. , Campbell, H. , & Dunlop, M. G. (2006). Identification and survival of carriers of mutations in DNA mismatch‐repair genes in colon cancer. The New England Journal of Medicine, 354(26), 2751–2763. 10.1056/NEJMoa053493 [DOI] [PubMed] [Google Scholar]
- Barreiros, L. A. , Segundo, G. R. S. , Grumach, A. S. , Roxo‐Junior, P. , Torgerson, T. R. , Ochs, H. D. , & Condino‐Neto, A. (2018). A novel homozygous JAK3 mutation leading to T‐B+NK‐ SCID in two Brazilian patients. Frontiers in Pediatrics, 6, 230. 10.3389/fped.2018.00230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berends, M. J. , Wu, Y. , Sijmons, R. H. , Mensink, R. G. , van der Sluis, T. , Hordijk‐Hos, J. M. , de Vries, E. G. , Hollema, H. , Karrenbeld, A. , Buys, C. H. , van der Zee, A. , Hofstra, R. M. , & Kleibeuker, J. H. (2002). Molecular and clinical characteristics of MSH6 variants: An analysis of 25 index carriers of a germline variant. American Journal of Human Genetics, 70(1), 26–37. 10.1086/337944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland, C. R. , Koi, M. , Chang, D. K. , & Carethers, J. M. (2008). The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in lynch syndrome: From bench to bedside. Familial Cancer, 7(1), 41–52. 10.1007/s10689-007-9145-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonadona, V. , Bonaiti, B. , Olschwang, S. , Grandjouan, S. , Huiart, L. , Longy, M. , Guimbaud, R. , Buecher, B. , Bignon, Y. J. , Caron, O. , Colas, C. , Noguès, C. , Lejeune‐Dumoulin, S. , Olivier‐Faivre, L. , Polycarpe‐Osaer, F. , Nguyen, T. D. , Desseigne, F. , Saurin, J.‐C. , Berthet, P. , … for the French Cancer Genetics Network . (2011). Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in lynch syndrome. JAMA, 305(22), 2304–2310. 10.1001/jama.2011.743 [DOI] [PubMed] [Google Scholar]
- Cohen, S. A. , & Leininger, A. (2014). The genetic basis of lynch syndrome and its implications for clinical practice and risk management. The Application of Clinical Genetics, 7, 147–158. 10.2147/TACG.S51483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin, L. A. , Graham, C. A. , Price, J. H. , & Morrison, P. J. (2008). Germline MSH6 mutations are more prevalent in endometrial cancer patient cohorts than hereditary non polyposis colorectal cancer cohorts. The Ulster Medical Journal, 77(1), 25–30. [PMC free article] [PubMed] [Google Scholar]
- Dominguez‐Valentin, M. , Sampson, J. R. , Seppälä, T. T. , ten Broeke, S. , Plazzer, J. P. , Nakken, S. , Engel, C. , Aretz, S. , Jenkins, M. A. , Sunde, L. , Bernstein, I. , Capella, G. , Balaguer, F. , Thomas, H. , Evans, D. G. , Burn, J. , Greenblatt, M. , Hovig, E. , de Vos tot Nederveen Cappel, W. , … Møller, P. (2020). Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the prospective lynch syndrome database. Genetics in Medicine, 22(1), 15–25. 10.1038/s41436-019-0596-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenschied, C. R. , LaDuca, H. , Li, S. , McFarland, R. , Gau, C. L. , & Hampel, H. (2017). Multigene panel testing provides a new perspective on lynch syndrome. Journal of Clinical Oncology, 35(22), 2568–2575. 10.1200/JCO.2016.71.9260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel, A. , Nguyen, T. P. , Leung, H. C. , Nagasaka, T. , Rhees, J. , Hotchkiss, E. , Arnold, M. , Banerji, P. , Koi, M. , Kwok, C. T. , Packham, D. , Lipton, L. , Boland, C. R. , Ward, R. L. , & Hitchins, M. P. (2011). De novo constitutional MLH1 epimutations confer early‐onset colorectal cancer in two new sporadic lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. International Journal of Cancer, 128(4), 869–878. 10.1002/ijc.25422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, Y. , Porat, R. M. , Kedar, I. , Shochat, C. , Galinsky, D. , Hamburger, T. , Hubert, A. , Strul, H. , Kariiv, R. , Ben‐Avi, L. , Savion, M. , Pikarsky, E. , Abeliovich, D. , Bercovich, D. , Lerer, I. , & Peretz, T. (2010). An Ashkenazi founder mutation in the MSH6 gene leading to HNPCC. Familial Cancer, 9(2), 141–150. 10.1007/s10689-009-9298-9 [DOI] [PubMed] [Google Scholar]
- Guerrette, S. , Wilson, T. , Gradia, S. , & Fishel, R. (1998). Interactions of human hMSH2 with hMSH3 and hMSH2 with hMSH6: Examination of mutations found in hereditary nonpolyposis colorectal cancer. Molecular and Cellular Biology, 18(11), 6616–6623. 10.1128/mcb.18.11.6616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Frankel, W. L. , Martin, E. , Arnold, M. , Khanduja, K. , Kuebler, P. , Clendenning, M. , Sotamaa, K. , Prior, T. , Westman, J. A. , Panescu, J. , Fix, D. , Lockman, J. , LaJeunesse, J. , Comeras, I. , & de la Chapelle, A. (2008). Feasibility of screening for lynch syndrome among patients with colorectal cancer. Journal of Clinical Oncology, 26(35), 5783–5788. 10.1200/JCO.2008.17.5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Frankel, W. L. , Martin, E. , Arnold, M. , Khanduja, K. , Kuebler, P. , Nakagawa, H. , Sotamaa, K. , Prior, T. W. , Westman, J. , Panescu, J. , Dan Fix, B. S. , Janet Lockman, B. S. , Ilene Comeras, R. N. , & de la Chapelle, A. (2005). Screening for the lynch syndrome (hereditary nonpolyposis colorectal cancer). The New England Journal of Medicine, 352(18), 1851–1860. 10.1056/NEJMoa043146 [DOI] [PubMed] [Google Scholar]
- Hendriks, Y. M. , Wagner, A. , Morreau, H. , Menko, F. , Stormorken, A. , Quehenberger, F. , Sandkuijl, L. , Møller, P. , Genuardi, M. , van Houwelingen, H. , Tops, C. , van Puijenbroek, M. , Verkuijlen, P. , Kenter, G. , van Mil, A. , Meijers‐Heijboer, H. , Tan, G. B. , Breuning, M. H. , Fodde, R. , … Vasen, H. (2004). Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: Impact on counseling and surveillance. Gastroenterology, 127(1), 17–25. 10.1053/j.gastro.2004.03.068 [DOI] [PubMed] [Google Scholar]
- Jiricny, J. (2006). The multifaceted mismatch‐repair system. Nature Reviews. Molecular Cell Biology, 7(5), 335–346. 10.1038/nrm1907 [DOI] [PubMed] [Google Scholar]
- Kanai, N. , Yanai, F. , Hirose, S. , Nibu, K. , Izuhara, K. , Tani, T. , Kubota, T. , & Mitsudome, A. (1999). A G to A transition at the last nucleotide of exon 6 of the gamma c gene (868G‐‐>A) may result in either a splice or missense mutation in patients with X‐linked severe combined immunodeficiency. Human Genetics, 104(1), 36–42. 10.1007/s004390050907 [DOI] [PubMed] [Google Scholar]
- Kariola, R. , Raevaara, T. E. , Lonnqvist, K. E. , & Nystrom‐Lahti, M. (2002). Functional analysis of MSH6 mutations linked to kindreds with putative hereditary non‐polyposis colorectal cancer syndrome. Human Molecular Genetics, 11(11), 1303–1310. 10.1093/hmg/11.11.1303 [DOI] [PubMed] [Google Scholar]
- Karousis, E. D. , & Muhlemann, O. (2019). Nonsense‐mediated mRNA decay begins where translation ends. Cold Spring Harbor Perspectives in Biology, 11(2), a032862. 10.1101/cshperspect.a032862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarskov, L. , Holck, S. , Bernstein, I. , Okkels, H. , Rambech, E. , Baldetorp, B. , & Nilbert, M. (2011). Challenges in the identification of MSH6‐associated colorectal cancer: Rectal location, less typical histology, and a subset with retained mismatch repair function. The American Journal of Surgical Pathology, 35(9), 1391–1399. 10.1097/PAS.0b013e318225c3f0 [DOI] [PubMed] [Google Scholar]
- Kuiper, R. P. , Vissers, L. E. , Venkatachalam, R. , Bodmer, D. , Hoenselaar, E. , Goossens, M. , Haufe, A. , Kamping, E. , Niessen, R. C. , Hogervorst, F. B. , Gille, J. J. , Redeker, B. , Tops, C. M. , van Gijn, M. , van den Ouweland, A. , Rahner, N. , Steinke, V. , Kahl, P. , Holinski‐Feder, E. , … Ligtenberg, M. J. (2011). Recurrence and variability of germline EPCAM deletions in lynch syndrome. Human Mutation, 32(4), 407–414. 10.1002/humu.21446 [DOI] [PubMed] [Google Scholar]
- Kunkel, T. A. , & Erie, D. A. (2005). DNA mismatch repair. Annual Review of Biochemistry, 74, 681–710. 10.1146/annurev.biochem.74.082803.133243 [DOI] [PubMed] [Google Scholar]
- Kurosaki, T. , Popp, M. W. , & Maquat, L. E. (2019). Quality and quantity control of gene expression by nonsense‐mediated mRNA decay. Nature Reviews. Molecular Cell Biology, 20(7), 406–420. 10.1038/s41580-019-0126-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham, A. , Srinivasan, P. , Kemel, Y. , Shia, J. , Bandlamudi, C. , Mandelker, D. , Middha, S. , Hechtman, J. , Zehir, A. , Dubard‐Gault, M. , Tran, C. , Stewart, C. , Sheehan, M. , Penson, A. , DeLair, D. , Yaeger, R. , Vijai, J. , Mukherjee, S. , Galle, J. , … Stadler, Z. K. (2019). Microsatellite instability is associated with the presence of lynch syndrome pan‐cancer. Journal of Clinical Oncology, 37(4), 286–295. 10.1200/JCO.18.00283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligtenberg, M. J. , Kuiper, R. P. , Chan, T. L. , Goossens, M. , Hebeda, K. M. , Voorendt, M. , Lee, T. Y. , Bodmer, D. , Hoenselaar, E. , Hendriks‐Cornelissen, S. J. , Tsui, W. Y. , Kong, C. K. , Brunner, H. G. , van Kessel, A. , Yuen, S. T. , van Krieken, J. , Leung, S. Y. , & Hoogerbrugge, N. (2009). Heritable somatic methylation and inactivation of MSH2 in families with lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nature Genetics, 41(1), 112–117. 10.1038/ng.283 [DOI] [PubMed] [Google Scholar]
- Møller, P. , Seppälä, T. , Bernstein, I. , Holinski‐Feder, E. , Sala, P. , Evans, D. G. , Lindblom, A. , Macrae, F. , Blanco, I. , Sijmons, R. , Jeffries, J. , Vasen, H. , Burn, J. , Nakken, S. , Hovig, E. , Rødland, E. A. , Tharmaratnam, K. , de Vos tot Nederveen Cappel, W. , Hill, J. , … Mallorca Group (http://mallorca‐group.eu). (2017). Cancer incidence and survival in lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective lynch syndrome database. Gut, 66(3), 464–472. 10.1136/gutjnl-2015-309675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller, P. , Seppälä, T. T. , Bernstein, I. , Holinski‐Feder, E. , Sala, P. , Gareth Evans, D. , Lindblom, A. , Macrae, F. , Blanco, I. , Sijmons, R. H. , Jeffries, J. , Vasen, H. F. A. , Burn, J. , Nakken, S. , Hovig, E. , Rødland, E. A. , Tharmaratnam, K. , de Vos tot Nederveen Cappel, W. , Hill, J. , … Mallorca Group . (2018). Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the prospective lynch syndrome database. Gut, 67(7), 1306–1316. 10.1136/gutjnl-2017-314057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen, R. C. , Hofstra, R. M. , Westers, H. , Ligtenberg, M. J. , Kooi, K. , Jager, P. O. , de Groote, M. L. , Dijkhuizen, T. , Olderode‐Berends, M. J. , Hollema, H. , Kleibeuker, J. H. , & Sijmons, R. H. (2009). Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of lynch syndrome. Genes, Chromosomes & Cancer, 48(8), 737–744. 10.1002/gcc.20678 [DOI] [PubMed] [Google Scholar]
- Overbeek, L. I. , Kets, C. M. , Hebeda, K. M. , Bodmer, D. , van der Looij, E. , Willems, R. , Goossens, M. , Arts, N. , Brunner, H. G. , van Krieken, J. , Hoogerbrugge, N. , & Ligtenberg, M. J. (2007). Patients with an unexplained microsatellite instable tumour have a low risk of familial cancer. British Journal of Cancer, 96(10), 1605–1612. 10.1038/sj.bjc.6603754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlongo, P. , Nafa, K. , Lerman, G. S. , Glogowski, E. , Shia, J. , Ye, T. Z. , Markowitz, A. J. , Guillem, J. G. , Kolachana, P. , Boyd, J. A. , Offit, K. , & Ellis, N. A. (2003). MSH6 germline mutations are rare in colorectal cancer families. International Journal of Cancer, 107(4), 571–579. 10.1002/ijc.11415 [DOI] [PubMed] [Google Scholar]
- Raskin, L. , Schwenter, F. , Freytsis, M. , Tischkowitz, M. , Wong, N. , Chong, G. , Narod, S. A. , Levine, D. A. , Bogomolniy, F. , Aronson, M. , Thibodeau, S. N. , Hunt, K. S. , Rennert, G. , Gallinger, S. , Gruber, S. B. , & Foulkes, W. D. (2011). Characterization of two Ashkenazi Jewish founder mutations in MSH6 gene causing lynch syndrome. Clinical Genetics, 79(6), 512–522. 10.1111/j.1399-0004.2010.01594.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein, J. H. , Enns, R. , Heidelbaugh, J. , Barkun, A. , & Clinical Guidelines Committee . (2015). American Gastroenterological Association Institute guideline on the diagnosis and management of lynch syndrome. Gastroenterology, 149(3), 777–782; quiz e716‐777. 10.1053/j.gastro.2015.07.036 [DOI] [PubMed] [Google Scholar]
- Sjursen, W. , McPhillips, M. , Scott, R. J. , & Talseth‐Palmer, B. A. (2016). Lynch syndrome mutation spectrum in New South Wales, Australia, including 55 novel mutations. Molecular Genetics & Genomic Medicine, 4(2), 223–231. 10.1002/mgg3.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susswein, L. R. , Marshall, M. L. , Nusbaum, R. , Vogel Postula, K. J. , Weissman, S. M. , Yackowski, L. , Vaccari, E. M. , Bissonnette, J. , Booker, J. K. , Cremona, M. L. , Gibellini, F. , Murphy, P. D. , Pineda‐Alvarez, D. E. , Pollevick, G. D. , Xu, Z. , Richard, G. , Bale, S. , Klein, R. T. , Hruska, K. S. , & Chung, W. K. (2016). Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next‐generation cancer panel testing. Genetics in Medicine, 18(8), 823–832. 10.1038/gim.2015.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Kaneda, M. , Futagawa, M. , Takeshita, M. , Kim, S. , Nakama, M. , Kawashita, N. , & Tatsumi‐Miyajima, J. (2019). Genetic and genomic basis of the mismatch repair system involved in lynch syndrome. International Journal of Clinical Oncology, 24(9), 999–1011. 10.1007/s10147-019-01494-y [DOI] [PubMed] [Google Scholar]
- Tutlewska, K. , Lubinski, J. , & Kurzawski, G. (2013). Germline deletions in the EPCAM gene as a cause of lynch syndrome ‐ literature review. Hereditary Cancer in Clinical Practice, 11(1), 9. 10.1186/1897-4287-11-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasen, H. F. (2005). Clinical description of the lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Familial Cancer, 4(3), 219–225. 10.1007/s10689-004-3906-5 [DOI] [PubMed] [Google Scholar]
- Vettore, S. , de Rocco, D. , Gerber, B. , Scandellari, R. , Bianco, A. M. , Balduini, C. L. , Pecci, A. , Fabris, F. , & Savoia, A. (2010). A G to C transversion at the last nucleotide of exon 25 of the MYH9 gene results in a missense mutation rather than in a splicing defect. European Journal of Medical Genetics, 53(5), 256–260. 10.1016/j.ejmg.2010.06.010 [DOI] [PubMed] [Google Scholar]
- Wijnen, J. , de Leeuw, W. , Vasen, H. , van der Klift, H. , Moller, P. , Stormorken, A. , Meijers‐Heijboer, H. , Lindhout, D. , Menko, F. , Vossen, S. , Möslein, G. , Tops, C. , Bröcker‐Vriends, A. , Wu, Y. , Hofstra, R. , Sijmons, R. , Cornelisse, C. , Morreau, H. , Fodde, R. , & Fodde, R. (1999). Familial endometrial cancer in female carriers of MSH6 germline mutations. Nature Genetics, 23(2), 142–144. 10.1038/13773 [DOI] [PubMed] [Google Scholar]
- Wijnen, J. T. , Vasen, H. F. , Khan, P. M. , Zwinderman, A. H. , van der Klift, H. , Mulder, A. , Tops, C. , Møller, P. , & Fodde, R. (1998). Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. The New England Journal of Medicine, 339(8), 511–518. 10.1056/NEJM199808203390804 [DOI] [PubMed] [Google Scholar]
- Yamada, K. , Fukao, T. , Zhang, G. , Sakurai, S. , Ruiter, J. P. , Wanders, R. J. , & Kondo, N. (2007). Single‐base substitution at the last nucleotide of exon 6 (c.671G>a), resulting in the skipping of exon 6, and exons 6 and 7 in human succinyl‐CoA:3‐ketoacid CoA transferase (SCOT) gene. Molecular Genetics and Metabolism, 90(3), 291–297. 10.1016/j.ymgme.2006.10.010 [DOI] [PubMed] [Google Scholar]
- You, J. F. , Buhard, O. , Ligtenberg, M. J. , Kets, C. M. , Niessen, R. C. , Hofstra, R. M. , Wagner, A. , Dinjens, W. N. , Colas, C. , Lascols, O. , Collura, A. , Flejou, J. F. , Duval, A. , & Hamelin, R. (2010). Tumours with loss of MSH6 expression are MSI‐H when screened with a pentaplex of five mononucleotide repeats. British Journal of Cancer, 103(12), 1840–1845. 10.1038/sj.bjc.6605988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurgelun, M. B. , Allen, B. , Kaldate, R. R. , Bowles, K. R. , Judkins, T. , Kaushik, P. , Roa, B. B. , Wenstrup, R. J. , Hartman, A. R. , & Syngal, S. (2015). Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology, 149(3), 604–613 e620. 10.1053/j.gastro.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data was submitted in Clinvar. The submission ID is SCV002060005.