

Abstract
JAK2 is a non-receptor tyrosine kinase that regulates hematopoiesis through the JAK-STAT pathway. The pseudokinase domain (JH2) is an important regulator of the activity of the kinase domain (JH1). V617F mutation in JH2 has been associated with the pathogenesis of various myeloproliferative neoplasms, but JAK2 JH2 has been poorly explored as a pharmacological target. In light of this, we aimed to develop JAK2 JH2 binders that could selectively target JH2 over JH1 and test their capacity to modulate JAK2 activity in cells. Toward this goal, we optimized a diaminotriazole lead compound into potent, selective, and cell-permeable JH2 binders leveraging computational design, synthesis, binding affinity measurements for the JH1, JH2 WT, and JH2 V617F domains, permeability measurements, crystallography, and cell assays. Optimized diaminotriazoles are capable of inhibiting STAT5 phosphorylation by both WT and V617F JAK2 in cells.
Keywords: Janus Kinase 2, JAK2, JAK2 JH2, Pseudokinase, V617F, Structure-Based Drug Design, Fluorescence Polarization, PAMPA, Protein Crystallography
INTRODUCTION
Janus Kinase 2 (JAK2) is one of four members of the JAK family of nonreceptor tyrosine kinases [JAK1, JAK2, JAK3 and Tyrosine Kinase 2 (TYK2).1 JAKs are associated with the cytoplasmic tails of cytokine receptors, and have an important role in signal transduction for the regulation of hematopoiesis and immune response. Cytokine binding to the corresponding receptor leads to a cascade of phosphorylation events that results in JAK activation, and subsequent binding of Signal Transducer and Activator of Transcription (STAT). STATs are thereby phosphorylated, dimerize, and the dimers translocate to the nucleus to bind DNA and regulate gene expression (JAK-STAT pathway).2,3
Mutations in JAKs have been linked to hematological diseases.4 V617F, the most frequently-occurring mutation in JAK2, has been associated with the pathogenesis of myeloproliferative neoplasms (MPNs), like polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (MF).5,6 Structurally, JAK2 is comprised of seven Janus Homology (JH) domains, including a C-terminal kinase domain (JH1) responsible for the catalytic activity and an adjacent pseudokinase domain (JH2). The pseudokinase domain adopts a prototypical protein-kinase fold and can bind ATP, but it lacks critical residues for significant phosphorylation catalysis.7 Its primary role is to regulate the function of the JH1 domain.8,9 Recent insights shed light into the mechanism of cytokine receptor activation, revealing a critical role of the pseudokinase domain in receptor dimerization. The insights suggest that activating mutations like V617F drive stronger interactions between the pseudokinase domains, stabilizing the JAK2-receptor dimer and resulting in cytokine independent activation.10
Despite the fact that occurrence of V617F mutation in the JH2 domain results in hyperactivation of the kinase activity, so far only the kinase domain of JAK2 has been utilized for therapeutic targeting by type-I ATP-competitive inhibitors.11 Current therapies for MPNs, like the FDA approved drug ruxolitinib (JAK1/JAK2 inhibitor) for the treatment of myelofibrosis, are non-selective and cause hematopoietic toxicities.12 Evidence from mutagenesis studies suggest that the hyperactivation caused by the V617F mutant could be attenuated by displacing ATP from the binding site of the JAK2 JH2 domain.13 Moreover, the studies indicate that the displacement event itself could attenuate the hyperactive V617F variant without affecting wild-type JAK2.13
Despite these auspicious insights, the potential value of the pseudokinase domain of JAK2 as a pharmacological target has yet to be demonstrated by small molecules. Previously reported potent JAK2 JH2 ligands were devoid of selectivity for the pseudokinase over the kinase domain, which prohibited accurate evaluation of the JH2 domain targetability.14 Along these lines, we endeavored to identify small molecules that could selectively bind the JH2 over the JH1 domain and aim to use them as chemical probes to test this hypothesis. These efforts have led to the development of several series of novel JAK2 JH2 binders: indoloxytriazines,15 diaminotriazoles,16 and pyrrolopyrimidines (Figure 1).17,18
Figure 1.

Representative structures of JAK2 JH2 binders discovered previously: (A) indoloxytriazines with single-digit μM affinity for JH2 and up to 14-fold selectivity over JH1,15 (B) diaminotriazoles with low triple-digit nM affinity for JH2 and up to 19-fold selectivity over JH1,16 and (C) pyrrolopyrimidines with double-digit nM affinity for JH2 and up to 360-fold selectivity over JH1.18
An initial step in these studies was the identification of JNJ7706621, a known Aurora A/B kinase and pan-CDK inhibitor,19,20 as a non-selective JAK2 JH2 ligand (Kd=0.67 ± 0.18 μM for JH1 and 0.46 ± 0.12 μM for JH2) through a high-throughput fluorescence polarization (FP) screen (Figure 2, compound 1).21 We then developed a FP assay with a fluorescein-conjugate of JNJ7706621 as the tracer, which facilitated rapid binding affinity measurements with a lower detection limit.22 Leveraging structural data and computer-aided design, we previously optimized JNJ7706621 into a series of novel diaminotriazole ureas, which provided potent and selective JAK2 JH2 ligands.16 However, the affinity and properties of the ligands were not sufficient for testing in cellular assays. Newly discovered insights on essential pharmacophore elements of JH2 leads18 prompted us to further optimize the diaminotriazole urea series, aiming to achieve higher potency, selectivity, and enable cell testing. Toward this goal, we report here affinity and permeability optimizations toward the development of potent, selective, and cell-permeable lead compounds (Figure 2).
Figure 2.

Lead optimization studies on diaminotriazole ligands. Compound 1 is the known pan-CDK and pan-JAK kinase inhibitor JNJ7706621. Compound 2 is a previously reported nM and selective JAK2 JH2 binder from optimization studies on 1.16 Compounds 3 – 14 are new JAK2 JH2-binding molecules reported in this work. Lead optimization stages: Ring and substituent optimization with eventual appending of the OBn moiety (phenyl-benzyloxy analogs), and permeability optimization.
RESULTS AND DISCUSSION
Structure-Based Design
BOMB (Biochemical and Organic Model Builder)23 was utilized to model in silico various 6-membered ring substituents for the optimization of 2. The generated protein-ligand complexes were energy-minimized with MCPRO24 using the OPLS-AA/M force field for proteins25 and OPLS/CM1A for ligands.26 Docking methods were also used starting from the crystal structures of 1 in complex with JH2 WT (PDB ID 5USZ) and JH1 (PDB ID: 5USY) (Table S3, Figure S6, Figure S8 B).21 The crystal structure of 11 in complex with JH2 WT (PDB ID: 7T0P) was used in retrospective analyses (Table S3, Figure S7). The desired ligands for docking were generated with Maestro27,28 and LigPrep29 using the OPLS3 force field30–33 and Epik.34–36 They were docked with Glide SP.37,38 Details about the docking are reported in the Supporting Information.
Assays
Competitive Fluorescence Polarization (FP) Assay
Binding affinities (Kd) with the JAK2 JH2 domain were evaluated using the previously developed FP assay22 and compound 1 as a control. An initial screening is conducted at 50 μM, and binding affinities (Kd’s) are measured for those exhibiting >50% binding at 50 μM (Table 1). Replacement of the oxazole in 2 with 6-membered aromatic rings produced compounds with a binding range of 0.129 to 1.4 μM. Conversion of the carboxylate of compound 7 to a methyl ester (8) or just hydrogen (9) resulted in large loss of binding affinity, and the addition of a benzyloxy group gave compounds (11–14) with an affinity range of 0.033 to 0.075 μM. Selectivity measurements were conducted subsequently for the most potent JAK2 JH2 ligands.
Table 1.
Measured Binding Affinity (Kd, μM) with JAK2 JH2a
| Compound | Kd (μM) |
|---|---|
| 1 | 0.456 ± 0.124 |
| 2 | 0.346 ± 0.034 |
| 3 | 0.934 ± 0.256 |
| 4 | 1.4 ± 0.1 |
| 5 | 0.652 ± 0.045 |
| 6 | 0.394 ± 0.044 |
| 7 | 0.129 ± 0.002 |
| 8 | 27 % @ 50 μM |
| 9 | 30.5 % @ 50 μM |
| 10 | 0.155 ± 0.013 |
| 11 | 0.03710 ± 0.0005 |
| 12 | 0.075 ± 0.015 |
| 13 | 0.0334 ± 0.0033 |
| 14 | 0.044 ± 0.003 |
For each Kd measurement, 2-3 independent experiments were carried out in quadruplicate using the competitive fluorescence polarization (FP) assay.
Parallel Artificial Membrane Permeation Assay (PAMPA)
To gauge the permeability of selected compounds, the Parallel Artificial Membrane Permeation Assay (PAMPA) was used. It simulates a cell membrane with a mix of dodecane and 4% lecithin applied to a filter plate. Theophylline, diclofenac, and chloramphenicol were used as low, medium, and high permeability controls, respectively.39,40 Further details are reported in the Experimental Section.
Cell Assays41
The major objective of the cell experiments was to assess the effects of the tested compounds in JAK2 downstream signaling by measuring STAT phosphorylation (PSTAT5). Human erythroleukemia cell lines HEL (containing the JAK2 V617F mutant, cytokine independent) and TF-1 (containing wild-type JAK2, cytokine (GM-CSF) dependent) were selected.42 The TF-1 cells and HEL cells were treated with various concentrations of JAK2 pseudokinase domain binders for 1-3 hours, and inactivation of JAK2 was monitored by the depletion of PSTAT5 levels in a dose-dependent manner. Further details are reported in the Experimental Section.
Synthesis of New Compounds
The new compounds reported here are numbered 3 – 14 in Figure 2; complete synthetic details and characterization data are provided in the Experimental Section. The general approach can be illustrated with the retrosynthesis shown in Scheme 1. We had previously established the conditions for carrying out the key urea formation reaction,16 through a 1H-[1,2,4]triazole-3,5-diamine (Fragment A – Synthesis is reported in Scheme 3, Experimental Section) and a phenyl carbamate (Fragment B). Although not highly regioselective for the desired 1H-triazole, the reaction has the advantage of great tolerance to functional groups (polar amines and sulfonamide moieties of the triazole fragment, and free carboxylic acid/carboxylate salts of the carbamate fragment), which enabled the synthesis of various substrates, by coupling either ester precursors or directly the free carboxylates to the triazolyl fragments.
Scheme 1.
Retrosynthetic Design for the Target Compounds
Scheme 3. Synthesis of Substituted Triazolesa.

aReagents and conditions: (a) A2i: THF, reflux, 28 h, 55 %; (b) A2: THF, reflux, 3 h, 76 %. Note: Methods for the synthesis of Fragments A1i and A1 (Scheme 3) were previously reported.16
One of the key elements accompanying the synthesis of this series was the preparation of the corresponding aniline precursors. Suzuki coupling of an appropriate p-amino-phenyl boronic acid and an aryl/heteroaryl bromide ester was convenient for preparing these substrates, due to the ease of the method and the availability of various aryl and heteroaryl bromides and boronic acids. To this end, we screened various conditions, both benchtop and microwave reactions, and palladium sources to establish an efficient method for the C-C (sp2-sp2) bond formation for the desired substrates (Table S1). Use of [1,1’-bis(diphenylphosphino)ferrocene] dichloro palladium in N,N-Dimethylformamide provided optimal results among the conditions screened. Applying this protocol, the synthesis of aryl-heteroaryl carboxylate analogs 3-6 proceeded through Method A (Scheme 2), where Boc-protected amines were prepared via Suzuki coupling and were subsequently deprotected to form the corresponding biaryl carbamate esters. Upon urea formation, the esters were converted to carboxylic acids via hydrolysis mediated by Li-salts (detailed synthesis of analogs 3-6 is reported in Scheme 4, Experimental Section).16,43 Synthesis of simple analogs 7-9 required only two-steps; carbamate and urea formation (Method B, Scheme 2), since the diphenyl aniline precursors were commercially available (detailed synthesis of analogs 7-9 is reported in Scheme 5, Experimental Section).
Scheme 2.

Approaches for the Synthesis of the Series Based on the Chemical Nature and Availability of the Corresponding Aniline Precursor Scaffolds
Scheme 4. Synthesis of 6-membered bi-aryl N-Heterocyclic analogs 3-6 a.

aReagents and conditions: (a) Cs2CO3, 10 mol % Pd(dppf)Cl2, DMF, 60 °C. B1ii: 6.5 h, 79 %; B2ii: 4 h, 77 %; B3ii: 6 h, 69 %; B4ii: 1 h 25 min, 68 % (b) DCM, rt. B1i: 1 h*; B2i: 1.5 h*; B3i: 1 h 10 min*; B4i: 1 h 10 min*; (c) NaHCO3, THF/H2O, 0 °C. B1: 55 min, 98 %; B2: 2 h*; B3: 73 min, 98 %; B4: 45 min, 93 %; (d) Et3N, Dioxane. 3i: 100 °C, 2 h, 28 %; 4i: 110 °C, 2 h, 13 %; 5i: 100 °C, 2 h 45 min, 67 %; 6i: 110 °C, 2 h 45 min, 66 %; (e) DBN, LiBr, MeCN, 2 vol % H2O, rt. 3: 52 h 10 min, 41 %; 4: 32 h, 27 %; 5: 98 h, 21 %; 6: 29 h 20min, 34 %.
*Yield not available; material was carried to the next step without further purification.
Scheme 5. Synthesis of biphenyl analogs 7-9a.

aReagents and conditions: (a) NaHCO3, THF/H2O, 0 °C; B5: 2 h, 32 %; B6: 110 min, 85%, B7: 1 h, Quant. (b) Et3N, Dioxane, 100 °C; 7: 6 h 20 min, 3%, 8: 4 h, 8.2 %, 9: 3h, 25 %.
Compound 10 was a special case. Although 4-(p-aminophenyl)benzoic acid is commercially available, efforts to proceed with Method B proved fruitless, since the corresponding carbamate was unstable. We therefore had to adopt a slightly different strategy. Starting with 4-bromobenzoic acid, we installed a t-Butyl ester and then followed the steps of Method A. Reaching the last step, the t-Bu group appeared to be quite sturdy and required strong acidic conditions to afford the hydrolysis. The urea core remained unaffected under stirring with neat trifluoroacetic acid, providing carboxylate analog 10 (detailed synthesis of analog 10 is reported in Scheme 6, Experimental Section).
Scheme 6. Synthesis of biphenyl analog 10a.

aReagents and conditions: a. DMAP, THF, rt, 28 h, 33 %; b. Cs2CO3, 10 mol % Pd(dppf)Cl2, DMF, 60 °C, 24.5 h, 57 %; c. NaHCO3, THF: H2O, 0 °C, 1.5 h, 78 %; d. Et3N, dioxane, 100 °C, 3.5 h, 58 %; e. TFA, rt, 2.5 h, 57 %.
The synthesis of diphenyl benzyloxy analogs 11-14 proceeded through Method C (Scheme 2), since none of the previous routes (Methods A and B, Scheme 2) was viable. Firstly, it was observed that the benzyloxy moiety was acid-sensitive and thus would be incompatible with the current Boc-protection strategy followed in Method A (Scheme 2). Furthermore, core functionalization with the OBn moiety late in the synthesis would require a very complex protection strategy and add burdensome steps to the synthesis. To overcome this challenge, we opted to carry-out the Suzuki reaction directly with the unprotected aniline (Method C, Scheme 2). This proved to be viable, though much longer reaction times were required.
An additional challenge was the hydrolysis of the ester. Hydrolysis mediated with Li salts, followed in Method A (Scheme 2), was dependent on the presence of an α- or β-heteroatom adjacent to the carbonyl group.43 In addition, hydrolysis with Li salts became less efficient as substrates became more complex. The most convenient solution was to carry out hydrolysis earlier in the synthesis. The aniline methyl ester precursors seemed ideal for hydrolysis using strong bases, since carbamates and ureas are unstable to hydroxide bases. Proceeding with this strategy followed by carbamate and urea formation of the free carboxylates of the benzyloxy-phenyl analogs (Method C, Scheme 2), the desired compounds 11-14 were prepared (detailed synthesis of analogs 11-14 is reported in Scheme 7, Experimental Section).
Scheme 7. Synthesis of benzyloxy-biphenyl analogs 11-14a.

aReagents and conditions: (a) K2CO3, DMF, 60 °C. B9iii: 24 h, 92 %, B10iii: 24 h, Quant. (b) Cs2CO3, 10 mol % Pd(dppf)Cl2, DMF, 60 °C. B9ii: 28 h, 53 %, B10ii: 27 h, 73 % (c) 2N NaOH, dioxane, rt. B9i: 55 h 20 min, 98 %, B10i: 4 d, 2 h, 77 %. (d) NaHCO3, THF/H2O, 0 °C. B9-Na: 1 h 50 min, 75 %; B9-H: 2 h 30 min; B10: 2 h 35 min, 98 %; (e) Et3N, Dioxane, 100 °C. 11: 6 h, 2 %; 12: 6 h, 2 %, 13: 3 h, 46 %, 14: 8.5 h, 1.8%.
Lead Optimization
Compound 2 was previously identified as a strong JAK2 JH2 binder, with a binding affinity of 0.346 ± 0.034 μM and 19-fold selectivity over JH1. With this starting point, we aimed to further optimize the affinity and selectivity to a level that would be propitious for testing in cells. The first step was to find alternatives for the oxazole ring that could lead to improved interactions of the attached carboxylic acid with T555, T557, and R715. Guided by the JH2 structure in complex with 2 (PDB: 6OCC), some goals were envisioned: (A) improve the distance and the directionality of the hydrogen bond between the carboxylate moiety and R715 (Figure 3A) and (B) project a substituent into the groove terminating in W718. This feature seemed readily accessible to our series, wherein W718 is 10.6 Å from the center of the oxazole ring of compound 2 (Figure 3B).
Figure 3.

Crystal structure of 2 in complex with WT JAK2 JH2. Shown is the biaryl-end of the ligand, bound to the polar ATP-binding region in the JH2 site (PDB ID: 6OCC). Lead optimization strategies were A) to improve the hydrogen-bonding network and interactions with T555, T557 and R715, and B) grow towards W718.
Ring Optimization
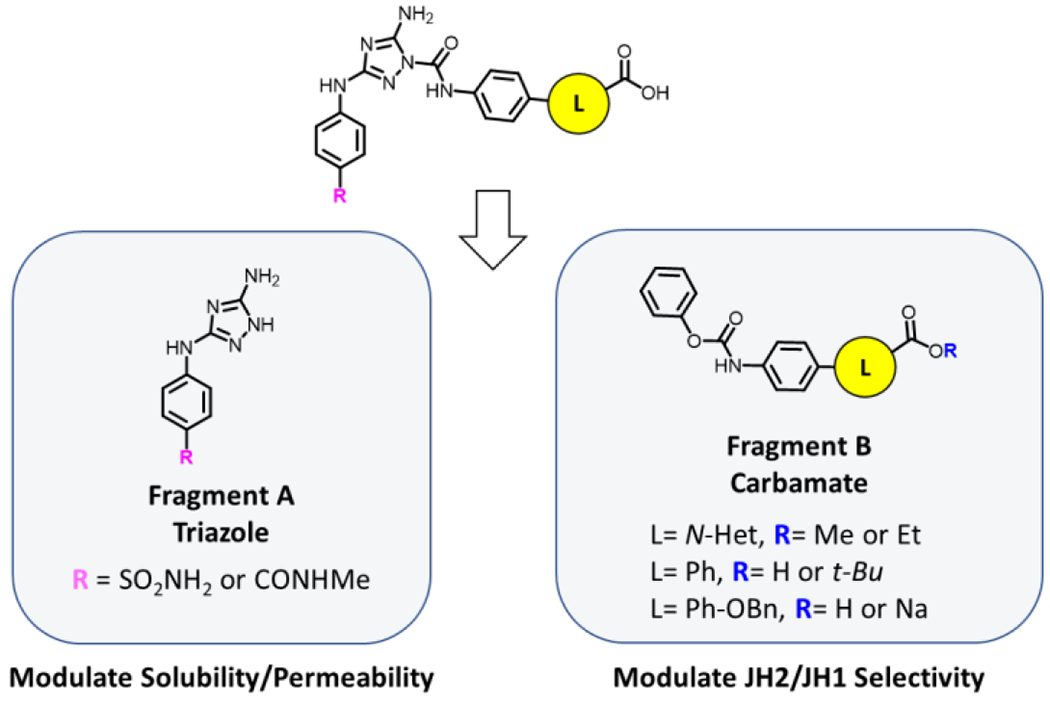
Seeking alternatives to the oxazole ring, we decided to explore 6-membered N-heterocycles. Modeling with BOMB revealed promising pyridine and pyrazine analogs. As seen in Figure 4, pyridine heterocycles presented the opportunity to project the carboxylic acid moiety closer to the T555-T557-R715 cluster (Figure 4, distances r) and at different directionality (Figure 4, dihedrals θ). Utilizing these heterocycles, we could also alter the ring electronics without inducing substantial changes to the core (Figure 4, alignments).
Figure 4.
Comparison of 2 (crystal-structure pose in JAK2 JH2, PDB ID: 6OCC) with BOMB-generated poses for 6-membered heterocycle analogs 3, 5, and 6.
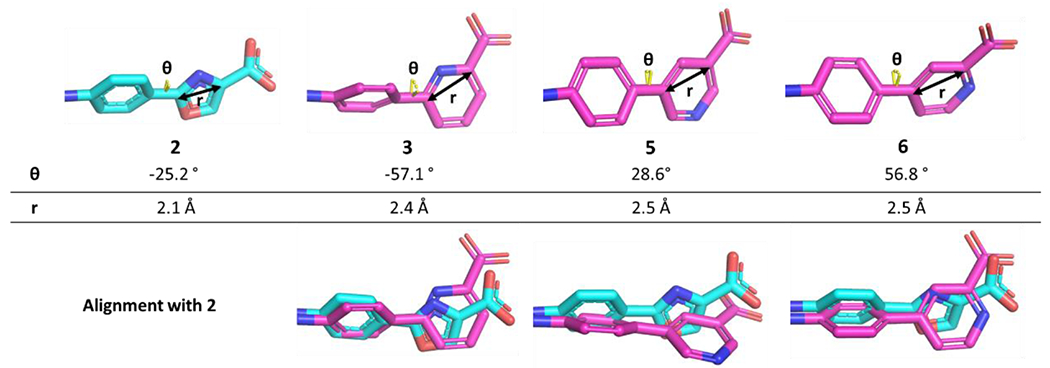
The 2-pyridyl analog, 3, which positions the N-atom similarly to the oxazole ring (Figure 4, alignment) exhibited a 3-fold loss in binding affinity compared to 2 (Table 1), indicating possible disruption of the elaborate hydrogen-bonding network that the oxazolic-N was facilitating. The pyrazine analog 4 could participate in a similar hydrogen-bonding network and provide an additional hydrogen-bond acceptor; however, it turned out to be an even weaker binder. Moving the N around the ring, the 3-pyridyl analog 5 appeared promising bringing the carboxylate closer to R715 at 2.8 Å, and it yielded a Kd of 0.652 ± 0.045 μM. The 4-pyridyl analog 6 optimized the N-position in the ring with a Kd of 0.394 ± 0.044 μM, similar to that for 2. A crystal structure of 6 bound to JAK2 JH2 was obtained (PDB ID: 7SZW - Crystallographic details are reported in Table S2 and Figure S1) and showed an extensive hydrogen-bonding network including a hydrogen bond between the pyridyl-N and a water molecule that bridges with R715 and N673 (Figure 5).
Figure 5.
Crystal structure of 6 bound to WT JAK2 JH2 (PDB ID: 7SZW), showing an extensive hydrogen-bonding network in the east side of the JH2 binding site. The pyridine N is hydrogen-bonded to a water molecule that also contacts the carboxylate group, N673, and R715.
The biphenyl-3-carboxylate analog 7 was also prepared as a reference for the SAR on the 6-membered rings. Curiously, 7 exhibited enhanced binding with a Kd of 0.129 ± 0.002. An explanation may be that the rigid, electron-rich biphenyl carboxylate improves the cation-π interaction with K581, which is discussed further below. We then sought to quantify the importance of the carboxylate group by preparing the methyl ester 8 and the unsubstituted biphenyl analog 9. Both compounds were found to lose about 3 orders of magnitude in binding affinity compared to 7 (Table 1). Then, in order to test placement of the carboxylate group closer to R715, the para isomer of 7, compound 10, was prepared. The binding affinity of 10, 0.155 ± 0.013 μM, turned out to be very similar to that of 7; however, its synthesis required 3 additional steps owing to the instability of the carbamate of the unprotected carboxylic acid. Therefore, 7 was selected for further optimization.
Growing Towards W718
In the pyrrolopyrimidine series,18 we identified a phenethyl group as a viable substituent to project into the W718 pocket as exemplified by the ligand JAK292 (Figure 1C, X=CH2). To consider if the addition of a similar group into the diaminotriazole series is feasible, the crystal structures of 6 (Figure 6, cyan) and the pyrrolopyrimidine ligand JAK292 (Figure 6, magenta) bound to WT JAK2 JH2 were overlaid. The protein backbones align with an RMSD of 0.178 Å and upon closer inspection, the best position for a phenethyl or isosteric group to be placed should be ortho to the carboxylic acid.
Figure 6.

Superposition of the crystal structures of compound 6 (cyan, PDB ID: 7SZW) and a pyrrolopyrimidine analog JAK292 (also shown in Figure 1C, where X=CH2), bound to WT JAK2 JH2 (magenta, PDB ID: 7T1T). The phenethyl group projects from the 4-position of the pyridine ring of 6.
In the crystal structure of JAK2 JH2 with the pyrrolopyrimidine ligand JAK292, N673 was resolved in two different conformations, either “inward” close to the ligand, or pointing “outward”, to accommodate the phenethyl group (Figure 6). The inwards conformation was in close vicinity to the phenethyl moiety, 3.5 Å and 4.1 Å away from the β and γ homophenylalanine carbons respectively. As such, it seemed that a benzyloxy group might form a hydrogen bond with the terminal amino group of N673 and extend the phenyl group to W718. Thus, the benzyloxy derivative 11 was prepared. It gave a Kd of 0.03710 ± 0.0005 μM with WT JAK2 JH2, almost 3.5x more potent than 7.
Crystallographic characterization of 11 in complex with JH2 WT (PDB ID: 7T0P -Crystallographic details are reported in Table S2 and Figure S1) confirmed the design. The benzyloxy moiety is projected to the bottom of the W718 pocket (Figure 7). It is engaged in an edge-to-face aryl-aryl interaction with W718, while R715 lies parallel to the benzyloxy substituent and forms a cation-π interaction with the terminal phenyl ring. These features justify the boost in binding affinity (Figure 7). The usual cation-π interaction with K581 is also present, and there is an extensive hydrogen bonding network for the carboxylate group with four water molecules, T555, and T557. In this structure, N673 is solely in the “outward” conformation forming hydrogen bonds to neighboring P700, I702, and R715 (Figure S8 A).
Figure 7.

Crystal structure of 11 bound to WT JAK2 JH2 (PDB ID: 7T0P). There are hydrogen bonds for the carboxylate group with T557, T555, and four water molecules. The benzyloxy phenyl ring makes an aryl-aryl interaction with W718 and a cation-π interaction with R715.
Presumably due its relative rigidity and size, 11 also distorts the structure of JAK2 JH2 upon binding, most prominently in the β3-αC and β4-β5 loops. As illustrated in Figure 8, binding of 11 causes a domino effect, shifting F595 and F594, and the β3-αC and β4-β5 loops into a conformation previously observed in the crystal structure of the V617F mutant of JAK2 JH2 with ATP (Figure 8A).7 In addition, the carboxylate moiety of 11 pushes the backbone of residues Q553-G554-T555 (β1 region) upwards (Figure 8B). Interestingly, soaking of compound 11 into apo JAK2 JH2 crystals did not yield in a complex structure; co-crystallization was required to generate the protein-ligand complex in a new space group. Given that the structures of JAK2 JH2 complexes with compounds of similar size have been previously determined by crystal soaking,16,18 this observation may suggest that the structural changes observed in the JAK2 JH2-11 complex are induced by compound binding and not from crystal packing effects.
Figure 8.

Crystal structure alignment showing the distortion caused in the αC helix, the β3-αC and β4-β5 loops of JAK2 JH2 upon binding of 11. WT JH2 bound to 11 (green, PDB ID: 7T0P), WT JAK2 JH2 bound to ATP (magenta, PDB ID: 4FVQ), and V617F JAK2 JH2 bound to ATP (purple, PDB ID: 4FVR). Binding of 11 causes F594 and F595 to adopt a mutant-like conformation (A), as observed in the conformation of β3-αC and β4-β5 loops (B). In addition, the carboxylic acid of 11 pushes the backbone of residues Q553-G554-T555 (β1 region) upwards.
Finally, given the success with 11, we wanted to explore its meta-benzyloxy isomer, 12. Though a better binder than 7, 12 turned out to be a 2-fold weaker binder than 11 with a Kd of 0.075 ± 0.015 μM for WT JAK2 JH2 (Table 1). It is expected that the contacts with R715 and W718 are less optimal in this case, though crystallographic characterization was not pursued.
Permeability
As the size of the ligands expanded during lead optimization combined with the presence of four aromatic rings, and the high total polar surface area (contributed by various polar groups, the free amines, the sulfonamide, and the ionic carboxylate moiety) potential problems with cell permeability could be envisioned. Thus, PAMPA was used for selected compounds to assess permeability prior to cell testing. Initially, the permeability of compound 7 and its derivatives 8 and 9 were examined to determine the effect of the carboxylic acid. As shown in Table 2, all three compounds were measured to be essentially impermeable, with PAMPA values close or equal to 0. For 7, this problem was thought to be associated with the anionic and polar groups of the ligand, however, the result for the methyl ester 8 indicates that the carboxylate is not the only source of poor permeability. For the biphenyl analog 9, poor aqueous solubility may have contributed to the observed poor permeability.
Table 2.
Permeability Measured with PAMPA for Selected Compoundsa
| Compound | 7 | 8 | 9 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|
|
Permeability
(x 10−6 cm/s) |
0.015 | 0.09 | 0.000 | 0.488 | 1.15 | 0.24 | 0.971 |
| TPSA (Å2) | 192.6 | 181.6 | 155.3 | 201.8 | 201.8 | 170.7 | 170.7 |
Experiments were performed in duplicate for each compound. Controls: theophylline (low control) 0.27-0.33 x 10−6 cm/s, diclofenac (medium control) 2.91-3.13 x 10−6 cm/s, and chloramphenicol (high control) 4.36-4.99 x 10−6 cm/s.
Nevertheless, compound 11 yielded a permeability value above the low control (theophylline) despite the presence of the carboxylic acid, possibly due to increased lipophilicity contributed by the benzyloxy group. The m-benzyloxy analog 12 was tested as well, and it exhibited a 3-fold increase in permeability compared to its isomer 11. From this result, it became apparent that different substitution patterns at the right side of the ligand were significantly affecting permeability, and the presence of the carboxylic acid can be tolerated. At this point we wanted to explore the effects of other polar moieties on permeability. The sulfonamide moiety in 1 - 12 is solvent exposed in their complexes so its modification was not expected to have large effects on binding, while its polarity can contribute to aqueous solubility but slow the rate of membrane penetration. As an initial study, a carboxylic amide arose as a synthetically tractable alternative with less polar surface area and molecular weight. Consequently, compounds 13 and 14 were synthesized. Compound 13, the N-methyl amide analog of 11, exhibited similar binding affinity for WT JAK2 JH2 (Table 1), but surprisingly almost 2-fold reduced permeability (Table 2). Compound 14, the N-methyl amide analog of 12, exhibited 2-fold improved binding affinity (0.044 μM) without significantly different permeability compared to 12.
Selectivity
An essential next step was to evaluate the selectivity of compound 6 and the potent compounds 11-14 with regard to WT JH1, WT JH2, and the V617F JH2 domains. Kd results (Table 3) were obtained via the competitive fluorescence polarization (FP) assay (Kd graphs are shown in Figure S2). As expected, the compounds did not show any significant selectivity for the V617F JH2 variant compared to WT JH2, since the two domains are identical in the vicinity of the ATP-binding site. An exception to this was compound 6, which exhibited 3-fold selectivity toward the mutant JH2. On the other hand, the binding affinities of the tested compounds for the JH1 domain showed only minimal variation throughout lead optimization, ranging from 3-8 μM (Table 3). In combination with the increased affinity for the WT JAK2 JH2, high selectivity was achieved. Compound 6 showed ca. 20-fold selectivity for JAK2 JH2, similar to the parent 2 (ca. 19-fold). Appending the benzyloxy moiety had a profound impact on the selectivity, with the sulfonamide 11 as ca. 155-fold selective and its N-methyl amide analog 13 as ca. 200-fold selective. The corresponding m-benzyloxy analogs 12 and 14 are less selective, ca. 45-fold for 12 and 134-fold for 14.
Table 3.
Binding Affinity (Kd, μM) for Different JAK2 Domains, and Selectivity for JH2 over JH1a
| WT JH1 | WT JH2 | V617F JH2 | JH2/JH1 Selectivity | |
|---|---|---|---|---|
| 2 | 6.6 ± 0.90 | 0.346 ± 0.034 | 0.478 ± 0.135 | 19x |
| 6 | 8.1 ± 1.2 | 0.394 ± 0.044 | 0.131 ± 0.020 | 20x |
| 11 | 5.75 ± 0.23 | 0.03710 ± 0.0005 | 0.033 ± 0.001 | 155x |
| 12 | 3.36 ± 0.54 | 0.075 ± 0.015 | 0.062 ± 0.001 | 45x |
| 13 | 6.81 ± 0.93 | 0.0334 ± 0.0033 | 0.043 ± 0.005 | 200x |
| 14 | 5.91 ± 0.47 | 0.044 ± 0.003 | 0.050 ± 0.002 | 134x |
For each Kd measurement, 2-3 independent experiments were carried out in quadruplicate and measured with the competitive fluorescence polarization (FP) assay.
A hypothesis for the strong selectivity of the benzyloxy analogs could be deduced from the crystal structures as well as the conformation of 11 bound to JH1 obtained by docking, in comparison to its JH2 binding (Figure S8). Owing to the different accessibility of the Trp pocket (W718 in JH2 and W1020 in JH1), the benzyloxy moiety is capable of contacting the Trp only for the JH2 domain. Upon ligand binding in WT JAK2 JH2, N673 flips outward forming hydrogen-bonds to neighboring residues P700, I702, and R715 (Figure S8 A1 -A2). This opens the space for the benzyloxy moiety to access W718. In JH1 (Figure S8 B) the rigidly bound N981-R980-D976 cluster limits accessibility to the W1020 pocket. To bind into the active site, the ligand is forced to flip the carboxylate moiety downwards, facilitating the formation of a salt bridge with R980. The benzyloxy moiety is accommodated upwards in an edge-to-face aryl interaction with F860, which is unfavored considering the polarity of the region.
Cell Assay Results
The improved affinity for the WT JAK21 JH2, the large selectivity window for binding to the JH2 domain in preference to the JH1 domain, and the promising permeability results for compounds 11-14 encouraged us to proceed with initial studies in cells. Our first goal was to test if 11 and 13 were active in HEL cells, which express only the V617F JAK2 variant. Compound 13 showed complete inhibition of STAT-5 phosphorylation at 20 μM, and proved to be more active than 11, which did not show any inhibition up to 40 μM with 1-hour incubation (Figure 9A). The increased potency of compound 13 was surprising given the PAMPA results and the similarity in Kd’s with the JH1 and JH2 domains. The PAMPA model, of course, provides a highly simplified model of a cell membrane. For reference, some cell experiments were also carried out with ruxolitinib, an ATP-competitive inhibitor of the JAK2 kinase domain, and 1, as a non-selective JAK2 kinase/pseudokinase binder. Ruxolitinib was tested at only one concentration (2 μM), and it showed complete inhibition of STAT-5 phosphorylation for both HEL and TF-1 cells (Figure S3 A). These results indicated that it has an IC50 below 2 μM, which is in accord with literature reports of IC50 values of 274 nM44 and 30-100 nM.45,46 Compound 1 exhibited the same inhibition patterns for HEL and TF-1 cells, with an estimated IC50 between 1-5 μM for HEL cells (Figure S3 B), which is also in accord with the literature.47
Figure 9.

(A) Western blot analysis measuring the levels of STAT5 and phosphorylated-STAT5 (P-STAT5) in lysates from HEL cells after treatment with compounds 11 and 13 for 3 hours at the indicated concentrations. (B) Western blot analysis measuring the levels of STAT5 and phosphorylated-STAT5 (P-STAT5) in lysates from HEL and TF-1 cells after incubation with compound 13 at the indicated concentrations for 1 hour. The compound has similar activity in both cell lines, exhibiting complete inhibition at 20 μM. Original blots have been cropped for clarity.
Given 13’s cellular activity, its effects in HEL cells and against TF1 cells (expressing only WT JAK2) were examined in more detail. The compound was tested at a range of concentrations 2-30 μM and the results indicated that inhibition was observed between 10-20 μM for both HEL and TF-1 cells (Figure 9B). The time-dependence of the activity of 13 was also investigated with experiments covering 3-48 h of incubation. At 3-24 h no difference was observed in inhibition compared to the 1-h incubation, suggesting that equilibrium of 13 within the cells was achieved. Given these results, it is unclear whether the effect of 13 is due to JH2 and/or JH1-binding. For the m-benzyloxy analogs 12 (sulfonamide) and 14 (N-Me amide), no inhibition was observed in HEL cells up to 25 μM for 12 and up to 30 μM for 14 (Figure S4). Thus, the cell assay results are sensitive to the structural details with only 13 among 11-14 showing inhibition of phosphorylation in HEL cells.
Finally, we were interested in exploring whether the V617F-like conformational change observed upon binding of 11 in the WT JH2 domain (Figure 8) translated to an activating effect in cells containing WT JAK2. Utilizing compound 13, a window of tested concentrations lower than the Kd of 13 for JH1 binding (6.81±0.93 μM) was chosen in order to eliminate any possibility of inhibition that could result from JH1-binding. After incubation of TF-1 cells starved of cytokine with 13 at 0.1, 0.5, and 2 μM concentrations, no stimulation of STAT5 phosphorylation was observed (Figure S5).
CONCLUSION
In the studies reported here, using compound 2 as a lead, low-nanomolar, selective, and cell-permeable JAK2 JH2 binding molecules were discovered. Compound 13 represents the most potent WT JAK2 JH2 binder that has been reported, exhibiting 10-fold stronger binding for WT JH2, and 10-fold greater selectivity over JH1 binding than 2. Optimization of the series caused little change in the affinity for the JH1 domain, and high selectivity was obtained through optimization of the JH2 binding. Additionally, detailed structural insights were reported, especially, the accommodation of the benzyloxy group in the crystal structure of the complex of WT JAK2 JH2 with 11 (Figure 7).
Initial assays in cells expressing JAK2 were then undertaken. Specifically, the desire was to test the hypothesis from previous mutagenesis studies that a selective, ATP-competitive JH2 binder could revert the hyperactivation of V617F JAK2 without affecting wild-type JAK2 activity.13 Though 11-14 are such compounds, only 13 was found to show inhibition of JAK2 phosphorylation in HEL or TF1 cells, and the inactivation was at the same level in both cell lines. It was also noted that there is structural distortion in WT JAK2 JH2 caused by binding 11 that makes the protein adopt a more mutant-like conformation. Though this might promote activation by 11 for WT JAK2, no measurable effect on JAK2 activity (neither inhibition nor activation) was observed that could be attributed JH2-related JAK2 modulation.
It may be noted that McNally et al. reported small molecule JH2 ligands with nanomolar affinity that could modulate the structure of V617F JAK2 JH2 to a conformation that is characteristic of the wild-type JH2. When tested in cells, however, these ligands only partially inhibited STAT5 phosphorylation at 20 μM concentrations in the JAK mutant-containing SET-2 cell line despite having sub micromolar affinities for JAK2 JH2.14 This, combined with the structural alterations observed upon binding of 11, suggests that small molecules are capable of affecting the structure of both the WT and V617F JAK2 JH2 allosteric sites. However, it remains to be established if a ligand which selectively binds to and distorts JAK JH2 can alter the kinase activity of WT or variant JAK2.
The outcomes of this study illustrate the complicated nature of JAK2 regulation. Attention has been focused on the role of JH2 which may function either in an intramolecular or intermolecular manner. Based on mechanistic studies of JAK2 activation,10 it is possible that molecules which can disrupt JH2-JH2 dimerization may provide alternative means to regulate JAK2 activity in addition to or instead of molecules which solely displace ATP from JH2 domains.13 It is clear that further studies are needed to unravel the details and possibilities for small-molecule modulation of JAK2 activity. Increased variety of strong, JH2-selective binding molecules is needed along with more extensive cell testing.
EXPERIMENTAL SECTION
General Synthetic Methods
Reagents and solvents were obtained from commercial supplies and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) using Merck pre-coated silica gel plates (analytical, SiO2-60, F254). TLC plates were visualized under UV light (254 nm/365 nm). Organic solutions were concentrated under reduced pressure on a Büchi rotary evaporator.
Low resolution mass analysis of intermediates was performed with an Agilent 6120 Quadrupole LC/MS instrument via electrospray ionization. Chromatographic purification with flash column chromatography was performed with a Teledyne ISCO Combiflash® Rf+ automated system employing RediSep Normal Phase Silica (particle size: 35-70 μm; pore size: 60 Å) or RediSep Gold Normal Phase Silica (particle size: 20-40 μm; pore size: 60 Å) disposable cartridge columns. RediSep Gold C18 reusable columns (particle size: 20 – 40 μm spherical; pore size: 100 Å) were employed for reverse phase chromatography. Preparative Reverse Phase HPLC Systems
System A:
a Shimadzu Prominence system equipped with LC-20AP pumps, CBM-20A Communications BUS module, SPD-20A UV/vis detector, SIL-10AP autosampler, FRC-10A fractions collector and a Waters SymmetryPrepTM C8, 19 x 300 mm column (particle size: 7 μm; pore size: 100 Å) with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase. (Purification of Compounds 3-6).
System B:
an Agilent 1260 Infinity II system equipped with G7161A Preparative Binary Pump., G7115A Diode Array Detector WR., G7157A Preparative Autosampler, G7159B Agilent Preparative Open-Bed Fraction Collector and an Agilent 100Å C18, 21.2 x 100 mm, 5 μm particle size preparative column with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase. Purity assessment was conducted with the same system, using an Agilent C18 Scalar column, 100Å, 4.6 x 100 mm, 5μM particle size, with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase. (Purification of Compounds 3i, 4i, 7, 12-14).
Nuclear magnetic resonance (NMR) spectra were recorded on an Agilent DD2 400 (1H NMR, 13C NMR recorded at 400, and 101 MHz, respectively), an Agilent DD2 500 (1H NMR, 13C NMR recorded at 500, and 126 MHz, respectively), or an Agilent DD2 600 (1H NMR, 13C NMR recorded at 600, and 151 MHz, respectively). All spectra were recorded at room temperature. Chemical shifts are reported in ppm relative to deuterated solvent as an internal standard (δH DMSO-d6 2.50 ppm, δC DMSO-d6 39.52 ppm; δH Methanol-d6 3.31 ppm, δC Methanol-d6 49.00 ppm δH Acetone-d6 2.05 ppm, δC Acetone-d6 29.84 ppm, 206.26 ppm; δH Chloroform-d 7.26 ppm, δC Chloroform-d 77.16 ppm) with the following convention for describing multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, br = broad signal, dd = doublet of doublets, etc).
High resolution mass spectroscopy (HRMS) Instruments
A) a Waters Xevo QTOF with a Z-spray electrospray ionization source. Purity was determined on a Shimadzu Prominence HPLC equipped with an Agilent Poroshell 120 SB-C18 2.7 μm column, using 0.1% TFA in water and 0.1% TFA in acetonitrile as the mobile phase. (Compounds 3-6)
B) a Shimadzu Scientific Instruments QToF 9030 LC-MS system, equipped with a Nexera LC-40D xs UHPLC, consisting of a CBM-40 Lite system controller, a DGU-405 Degasser Unit, two LC-40D XS UHPLC pumps, a SIL-40C XS autosampler and a Column Oven CTO-40S. UV data was collected with a Shimadzu Nexera HPLC/UHPLC Photodiode Array Detector SPD M-40 in the range of 190 – 800 nm. Mass spectra were subsequently recorded with the quadrupole time-of-flight (QToF) 9030 mass spectrometer. The samples were held at 4 °C in the autosampler compartment. 0.3 μL of each spiked solution were injected into a sample loop and separated on a Shim-pack Scepter C18-120, 1.9 μm, 2.1x100 mm Column, equilibrated at 40 °C in a column oven. A binary gradient was used: Solvent A: Water, HPLC grade Chromasolv, with 0.1 % Formic Acid, Solvent B: Acetonitrile, HPLC grade Chromasolv, with 0.1 % Formic Acid. The ionization source was run in “ESI” mode, with the electrospray needle held at +4.5 kV. Nebulizer Gas was at 2 L/min, Heating Gas Flow at 10 L/min and the Interface at 300 °C. Dry Gas was at 10 L/min, the Desolvation Line at 250 °C and the heating block at 400 °C. Mass spectra were recorded in the range of 50 to 2000 m/z in the positive ion mode. Measurements and data post-processing were performed with LabSolutions 5.97 Realtime Analysis and PostRun. (Compounds 7-14).
The purity of all the compounds was determined to be ≥ 95 % by integration of the UV traces. The samples showed no minor peaks above 3 %.
Phenyl N’-cyano-N-(4-(methylcarbamoyl)phenyl)carbamimidate (Fragment A2i)
4-amino-N-methylbenzamide (1.00 g, 6.66 mmol, 1.0 eq.) was mixed with diphenyl N-cyanocarbonimidate (2.38 g, 9.99 mmol, 1.5 eq.) in anhydrous tetrahydrofuran (42 mL) and the reaction was heated to reflux for 28 h. Subsequently, solvent was evaporated under reduced pressure and the residue was first washed with hexanes (5 to 8 times) and then with a mixture of hexanes and ethyl acetate (5 to 10 times). The product was dried under high vacuum overnight. Product: Off-white Solid. Yield: 1.07 g, 55 %. HRMS (ESI) m/z calculated for C16H14N4O2 [M+H]+= 295.1190, found: 295.1190.
1H NMR (600 MHz, DMSO-d6) δ 11.02 (s, 1H), 8.42 (q, J = 4.4 Hz, 1H), 7.87 – 7.83 (m, 2H), 7.58 – 7.53 (m, 2H), 7.48 – 7.43 (m, 2H), 7.35 – 7.28 (m, 3H), 2.78 (d, J = 4.5 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.9, 151.6, 138.5, 131.7, 129.9, 127.8, 126.3, 122.8, 120.7, 117.2, 113.0, 26.2.
4-((5-amino-1H-1,2,4-triazol-3-yl)amino)-N-methylbenzamide (Fragment A2)
Phenyl N’-cyano-N-(4-(methylcarbamoyl)phenyl)carbamimidate (A2i) (588.6 mg, 2.0 mmol, 1.0 eq.) was mixed with anhydrous tetrahydrofuran (12 mL). The mixture was brought to 0 °C and hydrazine (120 μL, 3.8 mmol, 1.9 eq.) was added slowly. The reaction was heated to reflux for 3 h, and solvent was evaporated. The reaction residue was washed with tetrahydrofuran and dried under high vacuum overnight. Product: Off-white Solid. Yield: 355.3 mg, 76 %. HRMS (ESI) m/z calculated for C10H12N6O [M+H]+= 233.1145, found: 233.1148.
1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.00 (s, 1H), 8.11 (q, J = 4.2 Hz, 1H), 7.67 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 8.8 Hz, 2H), 5.93 (s, 2H), 2.74 (d, J = 4.5 Hz, 3H). 13C NMR (151 MHz DMSO-d6) δ 166.5, 157.5, 155.4, 145.0, 127.9, 124.1, 114.5, 26.1.
General Method A: Suzuki coupling of bromides with phenylboronic acids
In a flame-dried vial equipped with a rubber septum was added appropriate bromide (1.61-1.94 mmol, 1.0 eq.), appropriate boronic acid (1.0 eq.) and cesium carbonate (2.0-3.0 eq.) in 8-9.4 mL anhydrous dimethylformamide (~4.7-5.0 mL/mmol). The mixture was degassed under vigorous stirring for 20-30 min, and subsequently Pd(dppf)Cl2 (0.10 eq.) was added. The mixture was flushed with nitrogen and was heated to 60 °C for the indicated time. Unless otherwise stated, solvent was evaporated under reduced pressure, and the residue was diluted with 40 mL ethyl acetate. The organic phase was washed with water (3 times x 10 mL) and with brine (1 time x 10 mL). The product was additionally back-extracted from the aqueous phase with ethyl acetate (3 times x 20 mL). The combined organic phase was dried under sodium sulfate, and after solvent evaporation the mixture was chromatographed using a gradient of ethyl acetate in hexanes.
General Method B: Deprotection of Boc-anilines
Appropriate Boc-aniline (1.0 eq) was dissolved in anhydrous dichloromethane (2.6 mL/mmol). The reaction mixture was cooled to 0 °C and trifluoroacetic acid (34.0 eq. unless otherwise stated) was added dropwise. At the end of the addition, the reaction was warmed to rt and was allowed to run for the indicated time. The pH was then adjusted to ~ 8 with a sat. solution of sodium bicarbonate (addition at 0 °C). The aqueous phase was extracted with ethyl acetate (5 times x 20 mL). The combined organic phase was washed with brine (2 times x 20 mL), dried over sodium sulfate, and solvent was evaporated to afford the title product.
General Method C: Carbamate formation48
Appropriate aniline (1.0 eq.) was suspended in a mixture of tetrahydrofuran/water (~2:1, as indicated). The mixture was cooled at 0 °C and sodium bicarbonate (1.2-2.2 eq.) was added. A solution of phenyl chloroformate (1.05 eq.) in tetrahydrofuran (1 mL/mmol) was slowly added to the mixture. Upon addition, the reaction was allowed to run at 0 °C for the indicated time, at which point TLC indicated completion. Unless otherwise stated, the mixture was diluted with 30-40 mL ethyl acetate, and was washed with water (3 times x 10 mL) and brine (2 times x 10 mL). The aqueous phase was back-extracted with ethyl acetate 2 times, and the combined organic phase was washed with brine and dried over sodium sulfate. Solvent was evaporated to afford the title products.
General Method D: Urea formation
Mixture A: Unless otherwise stated, in a flame-dried vial, fragment A1 or A2 (1.0 eq) was suspended in anhydrous dioxane (1.4-6.8 mL/mmol) and the mixture was heated for 5-10 min at 100 °C, followed by the addition of triethylamine (1.0-2.1 eq). The mixture was allowed to stir for another 10 min and subsequently was added dropwise to Mixture B. The vial was rinsed with anhydrous dioxane (3 mL/mmol substrate) which was also added to Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, appropriate carbamate (1.0 eq.) in anhydrous dioxane (1.9-6.8 mL/mmol) was heated at 100 °C for 10 min, under nitrogen. 1.0-2.1 eq. triethylamine was added, followed by the addition of Mixture A.
The reaction ran at 100-110 °C for the time indicated in each case. Workup details are reported in the individual procedures.
General Method E: Hydrolysis of esters mediated by Li salts16,43
Appropriate ester (1.0 eq.) was suspended in a mixture of acetonitrile (43 mL per mmol of ester) and 2 vol % water. 1,5-Diazabicyclo[4.3.0]non-5-ene [DBN] (3.0 eq.) and lithium bromide (10.0 eq.) were added to the reaction, and then the mixture was allowed to stir at rt for the indicated time, when MS indicated completion.
Workup: Solvent was evaporated under a stream of nitrogen and an amount of water (29 mL/mmol of substrate) was added to the residue. The pH was adjusted to ~4 (using 1N HCl and sat. sodium bicarbonate solution), and the mixture was kept at low temperature overnight (−20 °C). The mixture was then centrifuged, and the precipitate was collected, dried, and purified with reverse phase HPLC (System A), using batches of 10 mg from the crude mixture. HPLC fractions were lyophilized to afford the title product.
Ethyl 6-(4-((tert-butoxycarbonyl)amino)phenyl)picolinate (B1ii)
According to General Method A:
Ethyl 6-bromopicolinate (425.7 mg, 1.85 mmol) was mixed with (4-((tert-butoxycarbonyl)amino)phenyl)boronic acid (438.6 mg, 1.85 mmol), and cesium carbonate (1206 mg, 3.70 mmol, 2.0 eq.) in 9 mL anhydrous dimethylformamide. The mixture was degassed under vigorous stirring for 20 min, and subsequently Pd(dppf)Cl2 (139.0 mg, 0.19 mmol) was added. The reaction ran for 6.5 h, when TLC and LCMS indicated completion. Product: White Solid. Yield: 500.2 mg, 79 %. HRMS (ESI) m/z calculated for C19H22N2O4 [M+H]+= 343.1652, found: 343.1659. 1H NMR (600 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.13 (d, J = 7.9 Hz, 1H), 8.07 – 8.03 (m, 2H), 8.02 (t, J = 7.8 Hz, 1H), 7.93 (d, J = 7.6 Hz, 1H), 7.60 (d, J = 8.6 Hz, 2H), 4.38 (q, J = 7.1 Hz, 2H), 1.49 (s, 9H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 156.0, 152.7, 147.6, 141.0, 138.4, 131.3, 127.3, 122.8, 122.7, 118.0, 79.4, 61.2, 28.1, 14.2.
Methyl 6-(4-((tert-butoxycarbonyl)amino)phenyl)pyrazine-2-carboxylate (B2ii)
According to General Method A:
Methyl 6-bromopyrazine-2-carboxylate (401.5 mg, 1.85 mmol) was mixed with (4-((tert-butoxycarbonyl)amino)phenyl)boronic acid (438.6 mg, 1.85 mmol), and cesium carbonate (1206 mg, 3.70 mmol, 2.0 eq.) in 9 mL anhydrous dimethylformamide. The mixture was degassed under vigorous stirring for 20 min, and subsequently Pd(dppf)Cl2 (139.0 mg, 0.19 mmol) was added. The reaction ran for 4 h, when TLC and LCMS indicated completion. Slightly-modified workup: Solvent was evaporated under reduced pressure, and the residue was diluted with 70 mL ethyl acetate. The organic phase was washed with water (3 times x 30 mL) and with brine (1 time x 30 mL). The product was additionally back-extracted from the aqueous phase with ethyl acetate (3 times x 20 mL). The combined organic phase was dried under sodium sulfate, and after solvent evaporation the mixture was chromatographed using a gradient of ethyl acetate in hexanes. Product: Off-white Solid. Yield: 467.4 mg, 77 %. MS (ESI) m/z = 330.1 [M+ H]+.
1H NMR (600 MHz, DMSO-d6) δ 9.67 (s, 1H), 9.42 (s, 1H), 9.07 (s, 1H), 8.14 – 8.10 (m, 2H), 7.65 (d, J = 8.7 Hz, 2H), 3.96 (s, 3H), 1.50 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 164.3, 152.6, 150.9, 144.5, 142.8, 142.0, 141.9, 128.4, 127.7, 118.2, 79.5, 52.8, 28.1.
Ethyl 5-(4-((tert-butoxycarbonyl)amino)phenyl)nicotinate (B3ii)
According to General Method A:
Ethyl 5-bromonicotinate (425.7 mg, 1.85 mmol) was mixed with (4-((tert-butoxycarbonyl)amino)phenyl)boronic acid (438.6 mg, 1.85 mmol), and cesium carbonate (1206 mg, 3.70 mmol, 2.0 eq.) in 9 mL anhydrous dimethylformamide. The mixture was degassed under vigorous stirring for 20 min, and subsequently Pd(dppf)Cl2 (139.0 mg, 0.19 mmol) was added. The reaction ran for 6 h, when TLC and LCMS indicated completion. Slightly-modified workup: Solvent was evaporated under reduced pressure, and the residue was diluted with 80 mL ethyl acetate. The organic phase was washed with water (3 times x 20 mL) and with brine (1 time x 20 mL). The product was additionally back-extracted from the aqueous phase with ethyl acetate (3 times x 20 mL). The combined organic phase was dried under sodium sulfate, and after solvent evaporation the mixture was chromatographed using a gradient of ethyl acetate in hexanes to afford the title product. Product: White-pink solid. Yield: 439.2 mg, 69 %. HRMS (ESI) m/z calculated for C19H22N2O4 [M+H]+= 343.1652, found: 343.1662.
1H NMR (600 MHz, DMSO-d6) δ 9.56 (s, 1H), 9.10 (d, J = 2.2 Hz, 1H), 9.01 (d, J = 1.9 Hz, 1H), 8.41 (t, J = 2.2 Hz, 1H), 7.73 – 7.70 (m, 2H), 7.61 (d, J = 8.5 Hz, 2H), 4.38 (q, J = 7.1 Hz, 2H), 1.49 (s, 9H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 152.7, 151.2, 148.0, 140.2, 135.2, 133.5, 129.2, 127.4, 125.9, 118.5, 79.3, 61.3, 28.1, 14.1.
Methyl 4-(4-((tert-butoxycarbonyl)amino)phenyl)picolinate (B4ii)
According to General Method A:
Methyl 4-bromopicolinate (399.7 mg, 1.85 mmol) was mixed with (4-((tert-butoxycarbonyl)amino)phenyl)boronic acid (438.6 mg, 1.85 mmol), and cesium carbonate (1206 mg, 3.70 mmol, 2.0 eq.) in 9 mL anhydrous dimethylformamide. The mixture was degassed under vigorous stirring for 20 min, and subsequently Pd(dppf)Cl2 (139.0 mg, 0.19 mmol) was added. The reaction ran for 1 h 25 min, when TLC indicated completion. Product: White Solid. Yield: 416.0 mg, 68 %. HRMS (ESI) m/z calculated for C18H20N2O4 [M+H]+=329.1496, found: 329.1489.
1H NMR (600 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.70 (d, J = 5.1 Hz, 1H), 8.26 (d, J = 1.5 Hz, 1H), 7.92 (dd, J = 5.1, 1.8 Hz, 1H), 7.82 – 7.78 (m, 2H), 7.63 (d, J = 8.6 Hz, 2H), 3.91 (s, 3H), 1.49 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 165.4, 152.7 150.4, 148.3, 147.9, 141.3, 129.4, 127.4, 123.7, 121.2, 118.4, 79.5, 52.5, 28.1.
Ethyl 6-(4-aminophenyl)picolinate (B1i)
According to General Method B:
Ethyl 6-(4-((tert-butoxycarbonyl)amino)phenyl)picolinate (B1ii) (500.0 mg, 1.46 mmol) reacted with trifluoroacetic acid (3.8 mL, 49.6 mmol) in 3.8 mL anhydrous dichloromethane for 1 h. Product: Light Brown Solid. 402.5 mg of material were isolated and carried to the next step without further purification. HRMS (ESI) m/z calculated for C14H14N2O2 [M+H]+= 243.1128, found: 243.1124.
1H NMR (600 MHz, DMSO-d6) δ 7.98 (dd, J = 8.1, 0.7 Hz, 1H), 7.91 (t, J = 7.8 Hz, 1H), 7.88 – 7.84 (m, 2H), 7.80 (dd, J = 7.6, 0.7 Hz, 1H), 6.68 – 6.64 (m, 2H), 5.53 (s, 2H), 4.36 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.1 Hz, 3H).13C NMR (151 MHz, DMSO-d6) δ 165.0, 156.9, 150.5, 147.4, 137.9, 127.8, 124.9, 121.5, 121.4, 113.7, 61.1, 14.2.
Methyl 6-(4-aminophenyl)pyrazine-2-carboxylate (B2i)
According to General Method B:
Methyl 6-(4-((tert-butoxycarbonyl)amino)phenyl)pyrazine-2-carboxylate (B2ii) (465.2 mg, 1.41 mmol) reacted with trifluoroacetic acid (3.7 mL, 48.3 mmol) in 3.7 mL anhydrous dichloromethane for 1.5 h. Product: Light Brown Solid. 397 mg of material were isolated and carried to the next step without further purification. HRMS (ESI) m/z calculated for C12H11N3O2 [M+H]+= 230.0924, found: 230.0909.
1H NMR (600 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.91 (s, 1H), 7.94 – 7.90 (m, 2H), 6.70 – 6.67 (m, 2H), 5.74 (s, 2H), 3.93 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.6, 151.8, 151.4, 143.5, 141.8, 141.2, 128.2, 121.6, 113.8, 52.7.
Ethyl 5-(4-aminophenyl)nicotinate (B3i)
According to General Method B:
Ethyl 5-(4-((tert-butoxycarbonyl)amino)phenyl)nicotinate (B3ii) (437.0 mg, 1.28 mmol) reacted with trifluoroacetic acid (3.3 mL, 43.1 mmol) in 3.3 mL anhydrous dichloromethane for 1 h 10 min. Product: Light Yellow Solid. 325.4 mg of material were isolated and carried to the next step without further purification. HRMS (ESI) m/z calculated for C14H14N2O2 [M+H]+= 243.1128, found: 243.1120.
1H NMR (600 MHz, DMSO-d6) δ 9.01 (d, J = 2.2 Hz, 1H), 8.91 (d, J = 1.7 Hz, 1H), 8.31 (t, J = 2.1 Hz, 1H), 7.50 – 7.46 (m, 2H), 6.72 – 6.65 (m, 2H), 5.45 (s, 2H), 4.37 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.9, 150.4, 149.6, 146.8, 136.0, 132.4, 127.6, 125.8, 122.4, 114.3, 61.2, 14.1.
Methyl 4-(4-aminophenyl)picolinate (B4i)
According to General Method B:
Methyl 4-(4-((tert-butoxycarbonyl)amino)phenyl)picolinate (B4ii) (413.0 mg, 1.26 mmol) reacted with trifluoroacetic acid (3.2 mL, 41.8 mmol) in 3.2 mL anhydrous dichloromethane for 1 h 10 min. Product: Light Brown Solid. 481.5 mg of material were isolated and carried to the next step without further purification. MS (ESI) m/z = 229.1 [M+ H]+.
1H NMR (600 MHz, DMSO-d6) δ 8.59 (d, J = 5.2 Hz, 1H), 8.17 (d, J = 1.5 Hz, 1H), 7.81 (dd, J = 5.2, 1.9 Hz, 1H), 7.62 – 7.56 (m, 2H), 6.71 – 6.65 (m, 2H), 5.64 (s, 2H), 3.89 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.6, 150.8, 150.1, 148.5, 148.1, 127.7, 122.4, 122.3, 120.1, 114.1, 52.4.
Ethyl 6-(4-((phenoxycarbonyl)amino)phenyl)picolinate (B1)
According to General Method C:
Ethyl 6-(4-aminophenyl)picolinate (B1i) (353.7 mg, 1.46 mmol) was suspended in 3.0 mL tetrahydrofuran and 1.5 mL water. Upon mixing with the other reagents, the reaction was allowed to run at 0 °C for 55 min. Product: Pale Yellow Solid. Yield: 517.8 mg, 98 %. MS (ESI) m/z = 363.1 [M+H]+.
1H NMR (600 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.14 – 8.11 (m, 2H), 8.04 (t, J = 7.8 Hz, 1H), 7.95 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 8.6 Hz, 2H), 7.46 – 7.43 (m, 2H), 7.30 – 7.24 (m, 3H), 4.39 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 155.8, 151.7, 150.4, 147.6, 140.1, 138.5, 132.3, 129.5, 127.5, 125.6, 123.0, 122.9, 122.0, 118.4, 61.2, 14.2.
Methyl 6-(4-((phenoxycarbonyl)amino)phenyl)pyrazine-2-carboxylate (B2)
According to General Method C:
Methyl 6-(4-aminophenyl)pyrazine-2-carboxylate (B2i) (323.0 mg, 1.41 mmol) was suspended in 3.0 mL tetrahydrofuran and 1.5 mL water. Upon mixing with the other reagents, the reaction was allowed to run at 0 °C for 2 h. Product: Dark Yellow Solid. 515.8 mg of material were isolated and carried to the next step without further purification. MS (ESI) m/z = 350.1 [M+H]+.
1H NMR (600 MHz, DMSO-d6) δ 10.55 (s, 1H), 9.45 (s, 1H), 9.09 (s, 1H), 8.20 – 8.18 (m, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.47 – 7.43 (m, 2H), 7.30 – 7.24 (m, 3H), 3.96 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.3, 151.7, 150.7, 150.4, 144.6, 143.1, 142.1, 140.9, 129.5, 129.3, 127.9, 125.6, 122.0, 118.6, 52.8.
Ethyl 5-(4-((phenoxycarbonyl)amino)phenyl)nicotinate (B3)
According to General Method C:
Ethyl 5-(4-aminophenyl)nicotinate (B3i) (310.1 mg, 1.28 mmol) was suspended in 2.6 mL tetrahydrofuran and 1.3 mL water. Upon mixing with the other reagents, the reaction was allowed to run at 0 °C for 73 min. The back-extraction step was excluded. Product: Pale Yellow Solid. Yield: 452.9 mg, 98 %. MS (ESI) m/z = 363.1 [M+H]+.
1H NMR (600 MHz, DMSO-d6) δ 10.45 (s, 1H), 9.13 (d, J = 2.2 Hz, 1H), 9.03 (d, J = 1.8 Hz, 1H), 8.45 (t, J = 2.1 Hz, 1H), 7.82 – 7.79 (m, 2H), 7.67 (d, J = 8.5 Hz, 2H), 7.48 – 7.42 (m, 2H), 7.31 – 7.23 (m, 3H), 4.39 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 151.7, 151.2, 150.4, 148.2, 139.3, 135.1, 133.7, 130.2, 129.5, 127.7, 126.0, 125.6, 122.0, 118.9, 61.3, 14.1.
Methyl 4-(4-((phenoxycarbonyl)amino)phenyl)picolinate (B4)
According to General Method C:
Methyl 4-(4-aminophenyl)picolinate (B4i) (287.6 mg, 1.26 mmol) was suspended in 3.1 mL tetrahydrofuran and 1.3 mL water. Upon mixing with the other reagents, the reaction was allowed to run at 0 °C for 45 min. Product: Pale Yellow Solid. Yield: 408.0 mg, 93 %. MS (ESI) m/z = 349.1 [M+H]+.
1H NMR (600 MHz, DMSO-d6) δ 10.51 (s, 1H), 8.73 (d, J = 5.1 Hz, 1H), 8.31 – 8.28 (m, 1H), 7.95 (dd, J = 5.1, 1.7 Hz, 1H), 7.91 – 7.86 (m, 2H), 7.69 (d, J = 8.6 Hz, 2H), 7.48 – 7.41 (m, 2H), 7.30 – 7.24 (m, 3H), 3.91 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.3, 151.7, 150.41, 150.38, 148.3, 147.8, 140.3, 130.4, 129.5, 127.7, 125.6, 123.9, 122.0, 121.4, 118.8, 52.5.
Ethyl 6-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)picolinate (3i)
According to General Method D:
Ethyl 6-(4-((phenoxycarbonyl)amino)phenyl)picolinate (B1) (116.0 mg, 0.32 mmol) reacted at 100 °C for 2 h (1.9 mL/mmol anhydrous dioxane, 1 eq. triethylamine). Solvent was evaporated and 20-30 mL of water were added. The aqueous layer was washed with ethyl acetate 3-4 times. The two phases were separated and the aqueous layer was collected and concentrated in vacuo. The residue was further purified with preparative reverse phase HPLC, using an Agilent 5 Prep-C18 21.2 × 100 mm column with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase (System B). Product: Off-white solid. Yield: 46.7 mg, 28 %. HRMS (ESI) m/z calculated for C23H22N8O5S [M+H]+= 523.1507, found: 523.1520.
1H NMR (600 MHz, DMSO-d6) δ 9.76 (s, 1H), 9.71 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 8.21 – 8.17 (m, 2H), 8.07 (t, J = 7.8 Hz, 1H), 7.98 (d, J = 7.7 Hz, 1H), 7.88 – 7.85 (m, 2H), 7.85 – 7.81 (m, 2H), 7.72 (d, J = 8.9 Hz, 2H), 7.47 (br s, 2H), 7.15 (s, 2H), 4.40 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 156.7, 155.9, 155.7, 149.0, 147.7, 143.8, 138.63, 138.57, 134.9, 133.5, 127.2, 126.8, 123.2, 123.1, 121.5, 116.2, 61.3, 14.2.
Methyl 6-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)pyrazine-2-carboxylate (4i)
According to General Method D:
Methyl 6-(4-((phenoxycarbonyl)amino)phenyl)pyrazine-2-carboxylate (B2) (111.8 mg, 0.32 mmol), reacted at 110 °C for 2 h (1.9 mL/mmol anhydrous dioxane, 1 eq. triethylamine). The reaction was monitored with TLC in long wave and with LCMS. The reaction mixture was dried and was chromatographed using normal phase chromatography and a gradient of acetonitrile in dichloromethane. Additional purification was necessary with preparative reverse phase HPLC, using an Agilent 5 Prep-C18 21.2 × 100 mm column with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase (System B). Product: Light Yellow Solid. 21.5 mg, 13 %. MS (ESI) m/z = 510.2 [M+H]+.
1H NMR (600 MHz, DMSO-d6) δ 9.83 (s, 1H), 9.72 (s, 1H), 9.51 (s, 1H), 9.13 (s, 1H), 8.26 (d, J = 8.2 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.49 (br s, 2H), 7.16 (s, 2H), 3.97 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.3, 156.8, 156.0, 150.6, 148.9, 144.8, 143.8, 143.3, 142.1, 139.5, 134.9, 130.5, 127.5, 126.8, 121.5, 116.2, 52.8.
Ethyl 5-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)nicotinate (5i)
According to General Method D:
Ethyl 5-(4-((phenoxycarbonyl)amino)phenyl)nicotinate (B3) (116.0 mg, 0.32 mmol) reacted at 100 °C for 2 h 45 min (1.9 mL/mmol anhydrous dioxane, 1 eq. triethylamine). Solvent was evaporated and 20-30 mL of water were added. The aqueous layer was washed with ethyl acetate 3-4 times. The two phases were separated and the aqueous layer was collected and concentrated in vacuo to afford the title product. Product: White Solid. Yield: 111.6 mg, 67 %. HRMS (ESI) m/z calculated for C23H22N8O5S [M+H]+= 523.1507, found: 523.1506.
1H NMR (500 MHz, DMSO-d6) δ 9.75 (s, 1H), 9.70 (s, 1H), 9.18 (d, J = 2.1 Hz, 1H), 9.06 (d, J = 1.7 Hz, 1H), 8.50 (t, J = 2.0 Hz, 1H), 7.87 (s, 4H), 7.83 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.8 Hz, 2H), 7.46 (br s, 2H), 7.15 (s, 2H), 4.40 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 156.8, 156.0, 151.4, 149.1, 148.4, 143.9, 137.9, 135.0, 134.9, 133.9, 131.5, 127.4, 126.8, 126.0, 122.0, 116.3, 61.4, 14.1.
Methyl 4-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)picolinate (6i)
According to General Method D:
Methyl 4-(4-((phenoxycarbonyl)amino)phenyl)picolinate (B4) (81.4 mg, 0.32 mmol) reacted at 110 °C for 2 h 45 min (1.9 mL/mmol anhydrous dioxane, 1 eq. triethylamine). Solvent was evaporated and 20-30 mL of water were added. The aqueous layer was washed with ethyl acetate 3-4 times. The two phases were separated and the aqueous layer was collected and concentrated in vacuo to afford the title product. Product: Pale Yellow Solid. Yield: 108.2 mg, 66 %. HRMS (ESI) m/z calculated for C22H20N8O5S [M+H]+= 509.1350, found: 509.1354.
1H NMR (600 MHz, DMSO-d6) δ 9.80 (s, 1H), 9.72 (s, 1H), 8.76 (d, J = 5.1 Hz, 1H), 8.34 (d, J = 1.4 Hz, 1H), 8.01 (dd, J = 5.1, 1.7 Hz, 1H), 7.95 (d, J = 8.8 Hz, 2H), 7.90 (d, J = 8.7 Hz, 2H), 7.86 – 7.81 (m, 2H), 7.72 (d, J = 8.8 Hz, 2H), 7.48 (br s, 2H), 7.16 (s, 2H), 3.93 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.4, 156.8, 156.0, 150.5, 149.0, 148.4, 147.7, 143.8, 138.9, 134.9, 131.7, 127.4, 126.8, 124.1, 121.8, 121.5, 116.2, 52.5.
6-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)picolinic acid (3)
According to General Method E:
Ethyl6-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)picolinate (3i) (40.8 mg, 0.078 mmol) was hydrolyzed according to the conditions reported. Reaction time: 52 h 10 min. Product: White Solid. Yield: 16.0 mg, 41 % (Purity: 95 %). HRMS (ESI) m/z calculated for C21H18N8O5S [M+H]+= 495.1194, found: 495.1178.
1H NMR (600 MHz, DMSO-d6) δ 9.76 (s, 1H), 9.71 (s, 1H), 8.24 (d, J = 8.1 Hz, 2H), 8.19 (d, J = 7.8 Hz, 1H), 8.04 (t, J = 7.7 Hz, 1H), 7.96 (d, J = 7.4 Hz, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.84 (d, J = 9.0 Hz, 2H), 7.72 (d, J = 8.3 Hz, 2H), 7.47 (s, 2H), 7.15 (s, 2H) [Proton of the COOH moiety was not observed]. 13C NMR (151 MHz, DMSO-d6) δ 166.4, 156.7, 155.9, 155.4, 149.0, 143.8, 138.5, 138.4, 134.9, 133.6, 127.2, 126.8, 122.8, 122.7, 121.4, 116.2 (one carbon peak overlaps with the rest in the aromatic region).
6-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)pyrazine-2-carboxylic acid (4)
According to General Method E:
Methyl6-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)pyrazine-2-carboxylate (4i) (17.3 mg, 0.034 mmol) was hydrolyzed according to the conditions reported. Reaction time: 32 h. Product: Pale White Solid. Yield: 4.6 mg, 27 % (Purity: 96 %). HRMS (ESI) m/z calculated for C20H17N9O5S [M+H]+= 496.1146, found: 496.1130.
1H NMR (600 MHz, DMSO-d6) δ 9.80 (s, 1H), 9.71 (s, 1H), 9.42 (s, 1H), 9.07 (s, 1H), 8.27 (d, J = 8.1 Hz, 2H), 7.91 (d, J = 7.8 Hz, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 7.48 (br s, 2H), 7.15 (s, 2H) [Proton of the COOH moiety was not observed]. 13C NMR (151 MHz, DMSO-d6) δ 165.5, 156.8, 156.0, 150.3, 149.0, 144.6, 143.8, 143.6, 143.3, 139.3, 134.9, 131.0, 127.5, 126.8, 121.5, 116.3.
5-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)nicotinic acid (5)
According to General Method E:
Ethyl5-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)nicotinate (5i) (40.8 mg, 0.078 mmol) was hydrolyzed according to the conditions reported. Reaction time: 98 h. Product: Yellow Solid. Yield: 8.0 mg, 21 % (Purity: 95 %). HRMS (ESI) m/z calculated for C21H18N8O5S [M+H]+= 495.1194, found: 495.1193.
1H NMR (600 MHz, DMSO-d6) δ 9.72 (s, 1H), 9.70 (s, 1H), 8.96 (s, 1H), 8.91 (s, 1H), 8.40 (s, 1H), 7.84 (d, J = 8.0 Hz, 4H), 7.79 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.46 (br s, 2H), 7.15 (s, 2H) [Proton of the COOH moiety was not observed]. 13C NMR (151 MHz, DMSO-d6) δ 166.9, 164.1, 156.7, 155.9, 149.2, 149.0, 148.0, 143.9, 137.3, 134.9, 134.1, 133.8, 132.9, 127.1, 126.8, 122.0, 116.2.
4-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)picolinic acid (6)
According to General Method E:
Methyl4-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)picolinate (6i) (40.0 mg, 0.079 mmol) was hydrolyzed according to the conditions reported. Reaction time: 29 h 20 min. Product: Yellow Solid. Yield: 13.4 mg, 34 % (Purity: 97 %). HRMS (ESI) m/z calculated for C21H18N8O5S [M+H]+= 495.1194, found: 495.1185.
1H NMR (600 MHz, DMSO-d6) δ 9.78 (s, 1H), 9.71 (s, 1H), 8.69 (s, 1H), 8.30 (s, 1H), 7.94 – 7.87 (m, 5H), 7.84 (d, J = 8.1 Hz, 2H), 7.72 (d, J = 8.3 Hz, 2H), 7.47 (br s, 2H), 7.15 (s, 2H) [Proton of the COOH moiety was not observed].13C NMR (151 MHz, DMSO-d6) δ 166.8, 163.6, 156.7, 155.9, 149.7, 149.0, 147.3, 143.8, 138.6, 134.9, 132.2, 127.2, 126.8, 122.8, 121.8, 121.0, 116.2.
4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylic acid (B5)
According to General Method C:
4’-amino-[1,1’-biphenyl]-3-carboxylic acid (705.8 mg, 3.31 mmol) was suspended in 6.6 mL tetrahydrofuran containing 3.3 mL water. The mixture was brought to 0 °C followed by the addition of sodium bicarbonate (611.6 mg, 7.28 mmol). The mixture was allowed to stir for 5-10 min and then a solution of phenyl chloroformate (0.44 mL, 3.48 mmol) in 3.3 mL tetrahydrofuran was added dropwise. The reaction was allowed to stir for 2 h at 0 °C, at which point TLC indicated completion. Water (10 mL) was added to the reaction, followed by methanol until cloudy. The solid was filtered out and the filtrate was collected and further processed as follows: 25 mL water was added and the pH was adjusted to ~4 with 1N HCl. The aqueous phase was extracted with ethyl acetate (3 times x 40 mL). The combined organic layer was washed with brine (2 times x 30mL) and was dried over sodium sulfate. Solvent was evaporated under reduced pressure to afford the title product. Product: Pale Pink Solid. Yield: 348.8 mg, 32 %. HRMS (ESI) m/z calculated for C20H15NO4 [M+H]+= 334.1074, found: 334.1100.
1H NMR (600 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.17 (s, 1H), 7.90 (dt, J = 7.9, 1.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H), 7.57 (t, J = 7.7 Hz, 1H), 7.44 (t, J = 7.8 Hz, 2H), 7.34 – 7.19 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.3, 151.7, 150.5, 140.0, 138.5, 133.7, 131.6, 130.6, 129.5, 129.3, 127.9, 127.3, 126.9, 125.5, 122.0, 118.9.
Methyl 4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylate (B6)
According to General Method C:
Methyl 4’-amino-[1,1’-biphenyl]-3-carboxylate (752.4 mg, 3.31 mmol) was suspended in a mixture of tetrahydrofuran/water 2:1 mixture (2 mL THF/mmol of substrate). The mixture was cooled to 0 °C and sodium bicarbonate (333.5 mg, 3.97 mmol) was added. A solution of phenyl chloroformate (0.44 mL, 3.48 mmol) in tetrahydrofuran (3.3 ml) was added dropwise to the mixture. Upon addition, the reaction was allowed to run at 0 °C for 110 min, at which point TLC indicated completion. The mixture was diluted with 65 mL ethyl acetate and was washed with water (3 times x 20 mL) and brine (2 times x 20 mL). The combined organic phase was then dried over sodium sulfate. After filtration, solvent was evaporated under reduced pressure and the solid was further dried under high vacuum overnight to afford the title product. Product: White Solid. Yield: 982.1 mg, 85 %. HRMS (ESI) m/z calculated for C21H17NO4 [M+H]+= 348.1230, found: 348.1228.
1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.18 (t, J = 1.7 Hz, 1H), 7.98 – 7.88 (m, 2H), 7.72 – 7.68 (m, 2H), 7.64 (d, J = 8.7 Hz, 2H), 7.61 (t, J = 7.8 Hz, 1H), 7.49 – 7.40 (m, 2H), 7.30 – 7.22 (m, 3H), 3.89 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.2, 151.7, 150.5, 140.2, 138.6, 133.5, 131.1, 130.3, 129.48, 129.45, 127.7, 127.3, 126.6, 125.5, 122.0, 118.9, 52.3.
Phenyl [1,1’-biphenyl]-4-ylcarbamate (B7)
According to General Method C:
[1,1’-biphenyl]-4-amine (560.2 mg, 3.31 mmol) was suspended in a mixture of tetrahydrofuran/water 2:1 mixture (2 mL THF/mmol of substrate). The mixture was cooled to 0 °C and sodium bicarbonate (333.5 mg, 3.97 mmol) was added. A solution of phenyl chloroformate (0.44 mL, 3.48 mmol) in tetrahydrofuran (3.3 mL) was added dropwise to the mixture. Upon addition, the reaction was allowed to run at 0 °C for 1 h, at which point TLC indicated completion. The mixture was diluted with 65 mL ethyl acetate, and was washed with water (3 times x 20 mL) and brine (2 times x 20 mL). The combined organic phase was then dried over sodium sulfate. After filtration, solvent was evaporated to afford the title product. Product: Brown Solid. Yield: 954.2 mg, Quant. HRMS (ESI) m/z calculated for C19H15NO2 [M+H]+= 290.1176, found: 290.1173.
1H NMR (600 MHz, DMSO-d6) δ 10.35 (s, 1H), 7.67 – 7.64 (m, 4H), 7.61 (d, J = 8.6 Hz, 2H), 7.46 – 7.42 (m, 4H), 7.35 – 7.31 (m, 1H), 7.29 – 7.26 (m, 1H), 7.26 – 7.23 (m, 2H).13C NMR (151 MHz, DMSO-d6) δ 151.7, 150.5, 139.6, 138.1, 134.7, 129.5, 128.9, 127.1, 127.0, 126.3, 125.5, 122.0, 118.8.
4’-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-[1,1’-biphenyl]-3-carboxylic acid (7)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A1 (89.8 mg, 0.35 mmol) was suspended in 0.5 mL anhydrous dioxane and the mixture was heated for a 10-15 min at 80-100 °C, followed by the addition of triethylamine (50 μL, 0.35 mmol). The mixture was allowed to stir for another 10 min and subsequently was added dropwise to Mixture B. The vial was rinsed with 2.0 mL anhydrous dioxane, which was also added to Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, 4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylic acid (B5) (117.7 mg, 0.35 mmol) in 0.8 mL anhydrous dioxane was heated at 100 °C for 10 min, under nitrogen. Triethylamine (100 μL, 0.72 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 6 h 20 min. Solvent was evaporated under a stream of nitrogen and water was added. The crude mixture was acidified to pH ~4 with 1N HCl and the precipitate was collected by centrifugation and dried. The crude mixture was purified in batches of 20 mg using preparative reverse phase HPLC, an Agilent 5 Prep-C18 21.2 × 100 mm column with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase (System B). Product: White Solid. Yield: 43.7 mg, 25 % (Purity: 90-95 %)/ 5.6 mg, 3% (Purity: 97.6 %). HRMS (ESI) m/z calculated for C22H19N7O5S [M+H]+= 494.1241, found: 494.1260.
1H NMR (500 MHz, DMSO-d6) δ 9.71 (s, 1H), 9.70 (s, 1H), 8.21 (s, 1H), 7.95 – 7.90 (m, 2H), 7.86 – 7.80 (m, 4H), 7.75 (d, J = 8.6 Hz, 2H), 7.72 (d, J = 8.8 Hz, 2H), 7.58 (t, J = 7.7 Hz, 1H), 7.46 (br s, 2H), 7.15 (s, 2H) [Proton of the COOH moiety was not observed]. 13C NMR (126 MHz, DMSO-d6) δ 167.5, 156.7, 155.9, 149.0, 143.9, 139.7, 137.0, 135.2, 134.9, 132.7, 130.3, 129.2, 128.0, 126.98, 126.95, 126.8, 122.0, 116.2.
Methyl 4’-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-[1,1’-biphenyl]-3-carboxylate (8)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A1 (63.6 mg, 0.25 mmol) was suspended in 1.0 mL anhydrous dioxane and the mixture was heated for 5-10 min at 100 °C, followed by the addition of triethylamine (35 μL, 0.25 mmol). The mixture was allowed to stir for another 5-10 min and subsequently was added dropwise to Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, methyl 4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylate (B6) (86.8 mg, 0.25 mmol) in 0.5 mL anhydrous dioxane was heated at 100 °C for 10 min, under nitrogen. Triethylamine (35 μL, 0.25 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 4 h. Solvent was evaporated and the crude mixture was chromatographed using a gradient of methanol in dichloromethane, to afford the title product. Product: White Solid. Yield: 10.4 mg, 8.2% (Purity: 98.3%). HRMS (ESI) m/z calculated for C23H21N7O5S [M+H]+= 508.1398, found: 508.1391.
1H NMR (600 MHz, DMSO-d6) δ 9.72 (s, 1H), 9.72 (s, 1H), 8.22 (t, J = 1.8 Hz, 1H), 7.99 (ddd, J = 7.7, 1.8, 1.0 Hz, 1H), 7.95 (dt, J = 7.8, 1.3 Hz, 1H), 7.86 – 7.81 (m, 4H), 7.79 – 7.74 (m, 2H), 7.74 – 7.69 (m, 2H), 7.63 (t, J = 7.7 Hz, 1H), 7.46 (br s, 2H), 7.16 (s, 2H), 3.90 (s, 3H).13C NMR (151 MHz, DMSO-d6) δ 166.2, 156.7, 155.9, 149.0, 143.8, 140.1, 137.2, 134.9, 134.8, 131.2, 130.4, 129.5, 127.9, 127.0, 126.79, 126.76, 122.0, 116.2, 52.3.
N-([1,1’-biphenyl]-4-yl)-5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamide (9)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A1 (89.0 mg, 0.35 mmol) was suspended in 0.9 mL anhydrous dioxane and the mixture was heated for a 10-15 min at 100 °C, followed by the addition of triethylamine (49 μL, 0.35 mmol). The mixture was allowed to stir for another 10 min and subsequently was added dropwise to Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, phenyl [1,1’-biphenyl]-4-ylcarbamate (B7) (101.0 mg, 0.35 mmol) in 0.3 mL anhydrous dioxane was heated at 100 °C for 10 min, under nitrogen. Triethylamine (49 μL, 0.35 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 3 h. Solvent was evaporated and the crude mixture was chromatographed using a gradient of methanol in dichloromethane, to afford the title product. Product: White Solid. Yield: 39.6 mg, 25 % (Purity: 96 %). HRMS (ESI) m/z calculated for C21H19N7O3S [M+H]+= 450.1343, found: 450.1340.
1H NMR (500 MHz, DMSO-d6) δ 9.69 (s, 1H), 9.68 (s, 1H), 7.85 – 7.81 (m, 2H), 7.80 – 7.76 (m, 2H), 7.75 – 7.67 (m, 6H), 7.51 – 7.41 (m, 4H), 7.38 – 7.34 (m, 1H), 7.15 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 156.7, 155.9, 149.1, 143.9, 139.5, 136.6, 136.1, 134.9, 129.0, 127.3, 126.8, 126.4, 121.9, 116.2 (one carbon peak overlaps with the rest in the aromatic region).
Tert-butyl 4-bromobenzoate (B8ii)
In a round-bottom flask equipped with a rubber septum, a catalytic amount of 4-dimethylaminopyridine (DMAP) (0.7 g, 5.7 mmol) was added to a mixture of 4-bromobenzoic acid (5.0 g, 24.9 mmol) in tetrahydrofuran (47 mL). The mixture was allowed to stir at room temperature for 10 min, and subsequently di-tert-butyl dicarbonate (16.6 g, 76.1 mmol) was added portion wise, while vortexing the mixture from time to time. Once the addition was complete, the reaction was allowed to run at room temperature for 28 h. The solvent was evaporated and the crude mixture was chromatographed using a gradient of ethyl acetate in hexanes to afford the title product. Product: Light Yellow Oil. Yield: 2.13 g, 33 %. Product could not be detected in the MS.49
1H NMR (600 MHz, DMSO-d6) δ 7.83 – 7.80 (m, 2H), 7.72 – 7.69 (m, 2H), 1.53 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 164.2, 131.7, 131.0, 130.4, 126.9, 81.2, 27.7.
tert-butyl 4’-amino-[1,1’-biphenyl]-4-carboxylate (B8i)
According to General Method A:
In a flame-dried vial equipped with a rubber septum was added tert-butyl 4-bromobenzoate (B8ii) (498.7 mg, 1.94 mmol), 4-aminophenylboronic acid hydrochloride (336.4 mg, 1.94 mmol) and cesium carbonate (1896.3 mg, 5.82 mmol, 3.0 eq.) in 9.4 mL anhydrous dimethylformamide. The mixture was degassed under vigorous stirring for 30 min, and subsequently Pd(dppf)Cl2 (141.9 mg, 0.194 mmol) was added. The mixture was flushed with nitrogen and was heated to 60 °C for 24.5 h, when TLC indicated completion. Slightly-modified workup: Solvent was evaporated under reduced pressure, and the crude mixture was directly purified with chromatography using a gradient of ethyl acetate in hexanes, to afford the title product. Product: Pale Yellow solid. Yield: 298.9 mg, 57 %. HRMS (ESI) m/z calculated for C17H19NO2 [M+H]+= 270.1489, found: 270.1489.
1H NMR (600 MHz, DMSO-d6) δ 7.90 – 7.85 (m, 2H), 7.67 – 7.64 (m, 2H), 7.46 – 7.43 (m, 2H), 6.67 – 6.63 (m, 2H), 5.40 (s, 2H), 1.55 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 165.0, 149.4, 144.8, 129.6, 128.2, 127.6, 125.6, 124.9, 114.1, 80.3, 27.9.
Tert-butyl 4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-4-carboxylate (B8)
According to General Method C:
Tert-butyl 4’-amino-[1,1’-biphenyl]-4-carboxylate (B8i) (293.0 mg, 1.09 mmol) in tetrahydrofuran:water (2.2:1.1 mL) reacted with phenyl chloroformate for 1.5 h. The mixture was processed without the back-extraction step. Product: Pale Yellow Solid. Yield: 332.8 mg, 78 %. HRMS (ESI) m/z calculated for C24H23NO4 [M+H]+= 390.1700, found: 390.1697.
1H NMR (600 MHz, DMSO-d6) δ 10.42 (s, 1H), 7.97 – 7.94 (m, 2H), 7.80 – 7.77 (m, 2H), 7.75 – 7.72 (m, 2H), 7.64 (d, J = 8.7 Hz, 2H), 7.46 – 7.42 (m, 2H), 7.29 – 7.23 (m, 3H), 1.56 (s, 9H).
13C NMR (151 MHz, DMSO-d6) δ 164.8, 151.7, 150.5, 143.8, 139.0, 133.3, 129.7, 129.5, 129.4, 127.5, 126.2, 125.5, 122.0, 118.8, 80.7, 27.8.
Tert-butyl 4’-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-[1,1’-biphenyl]-4-carboxylate (10i)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A1 (63.6 mg, 0.25 mmol) was suspended in 1.0 mL anhydrous dioxane and the mixture was heated for 5 min at 100 °C, followed by the addition of triethylamine (35 μL, 0.25 mmol). The mixture was allowed to stir for another 5 min and subsequently it was added dropwise to Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, tert-butyl 4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-4-carboxylate (B8) (97.4 mg, 0.25 mmol) in 1.0 mL anhydrous dioxane was heated at 100 °C for 5 min, under nitrogen. Triethylamine (35 μL, 0.25 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 3.5 h, at which point solvent was evaporated. The crude mixture was purified with normal-phase chromatography using a gradient of methanol in dichloromethane. Product: White Solid. Yield: 79.7 mg, 58 %. HRMS (ESI) m/z calculated for C26H27N7O5S [M+H]+= 550.1867, found: 550.1867.
1H NMR (600 MHz, DMSO-d6) δ 9.73 (s, 1H), 9.70 (s, 1H), 8.00 – 7.95 (m, 2H), 7.86 – 7.82 (m, 6H), 7.80 – 7.77 (m, 2H), 7.74 – 7.70 (m, 2H), 7.46 (br s, 2H), 7.15 (s, 2H), 1.57 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 156.7, 155.9, 149.0, 143.9, 143.7, 137.6, 134.9, 134.6, 129.9, 129.7, 127.2, 126.8, 126.4, 121.8, 116.2, 80.7, 27.8.
4’-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-[1,1’-biphenyl]-4-carboxylic acid (10)
Tert-butyl 4’-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-[1,1’-biphenyl]-4-carboxylate (10i) (44.0 mg, 0.08 mmol) was suspended in trifluoroacetic acid (1.84 mL, 24.0 mmol). The mixture was allowed to stir at room temperature for 2.5 h, at which point it was dried under a stream of nitrogen. Subsequently water (2.0 mL) was added and the pH was adjusted to ~4 using acetic acid and triethylamine. The residue was collected after centrifugation, and it was dried. Finally, the residue was triturated, aided by sonication and vortexing, with ethyl ether (2 times) and with a mixture of ethyl ether and acetonitrile (5 times) to afford the title product. Product: White Solid. Yield: 22.6 mg, 57 % Purity: 96.6 %. HRMS (ESI) m/z calculated for C22H19N7O3S [M+H]+= 494.1241, found: 494.1238.
1H NMR (600 MHz, DMSO-d6) δ 12.95 (s, 1H), 9.72 (s, 1H), 9.70 (s, 1H), 8.06 – 7.98 (m, 2H), 7.86 – 7.82 (m, 6H), 7.81 – 7.78 (m, 2H), 7.74 – 7.70 (m, 2H), 7.46 (br s, 2H), 7.15 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.2, 156.7, 155.9, 149.0, 143.9, 143.6, 137.5, 134.9, 134.7, 130.0, 129.4, 127.2, 126.8, 126.4, 121.8, 116.2.
Methyl 2-(benzyloxy)-5-bromobenzoate (B9iii)
In an oven-dried vial, methyl 5-bromo-2-hydroxybenzoate (462.1 mg, 2.00 mmol) was combined with potassium carbonate (346.9 mg, 2.51 mmol) in 1.4 mL anhydrous dimethylformamide. The mixture was allowed to stir until homogenized and subsequently benzyl bromide (0.2 mL, 1.67 mmol) was added dropwise. When the addition was complete, the reaction was heated at 60 °C for 24 h, at which point TLC indicated consumption of the starting material. Then solvent was evaporated and the mixture was diluted with 30 mL ethyl acetate. The organic phase was washed with water (3 times x 10 mL), 2N NaOH (2 times x 10 mL), brine (2 times x 10 mL) and was dried over sodium sulfate. The mixture was filtered and solvent was evaporated under reduced pressure to afford the title product. Product: Light Yellow Oil. Yield: 591.9 mg, 92 %. HRMS (ESI) m/z calculated for C15H13BrO3 [M+H]+=321.0121, found: 321.0113.
1H NMR (500 MHz, DMSO-d6) δ 7.79 (d, J = 2.1 Hz, 1H), 7.70 (dd, J = 8.9, 2.1 Hz, 1H), 7.47 (d, J = 7.4 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.22 (d, J = 9.0 Hz, 1H), 5.22 (s, 2H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.7, 156.4, 136.5, 135.8, 132.8, 128.4, 127.7, 127.0, 122.4, 116.5, 111.6, 69.9, 52.2.
Methyl 3-(benzyloxy)-5-bromobenzoate (B10iii)
In an oven-dried vial, methyl 3-bromo-5-hydroxybenzoate (1.71 g, 7.40 mmol) was combined with potassium carbonate (1.28 g, 9.26 mmol) in 5 mL anhydrous dimethylformamide. The mixture was allowed to stir until it was homogenized and subsequently benzyl bromide (0.73 mL, 6.17 mmol) was added dropwise. When the addition was complete, the reaction was heated at 60 °C and stirred for 24 h, at which point TLC indicated consumption of the starting material. Solvent was evaporated and the mixture was diluted with 100 mL ethyl acetate. The organic phase was washed with water (4 times x 30-40 mL), 2N NaOH (5 times x 30-40 mL), brine (4 times x 30 mL) and was dried over sodium sulfate. The mixture was filtered, solvent was evaporated under reduced pressure, and the residue was further dried under high vacuum overnight to afford the title product. Product: Gold-yellow Oil. Yield: 2.00 g, Quant. HRMS (ESI) m/z calculated for C15H13BrO3 [M+H]+=321.0121, found: 321.0108.
1H NMR (600 MHz, DMSO-d6) δ 7.64 (t, J = 1.5 Hz, 1H), 7.57 – 7.54 (m, 1H), 7.52 (dd, J = 2.4, 1.3 Hz, 1H), 7.47 – 7.44 (m, 2H), 7.42 – 7.38 (m, 2H), 7.37 – 7.32 (m, 1H), 5.20 (s, 2H), 3.85 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 159.3, 136.2, 132.6, 128.5, 128.1, 127.8, 124.0, 122.5, 122.3, 114.7, 69.9, 52.6.
Methyl 4’-amino-4-(benzyloxy)-[1,1’-biphenyl]-3-carboxylate (B9ii)
According to General Method A:
In a flame-dried vial equipped with a rubber septum was added methyl 2-(benzyloxy)-5-bromobenzoate (B9iii) (515.7 mg, 1.61 mmol), 4-aminophenylboronic acid hydrochloride (279.2 mg, 1.61 mmol) and cesium carbonate (1573.7 mg, 4.83 mmol, 3.0 eq) in 8 mL anhydrous dimethylformamide. The mixture was degassed under vigorous stirring for 20 min, and subsequently Pd(dppf)Cl2 (117.8 mg, 0.16 mmol) was added. The mixture was purged with nitrogen and was heated at 60 °C for 28 h when TLC indicated completion. Solvent was evaporated under reduced pressure, and the residue was diluted with ethyl acetate. The organic phase was washed with water (2 times x 30 mL) and with brine (2 times x 30 mL). The organic phase was then concentrated under reduced pressure, and the mixture was chromatographed using a gradient of ethyl acetate in hexanes to afford the title product. Product: White Solid. Yield: 285.4 mg, 53 %. HRMS (ESI) m/z calculated for C21H19NO3 [M+H]+=334.1438, found: 334.1431.
1H NMR (500 MHz, DMSO-d6) δ 7.80 (d, J = 2.0 Hz, 1H), 7.68 (dd, J = 8.7, 2.2 Hz, 1H), 7.50 (d, J = 7.6 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.35 – 7.27 (m, 3H), 7.23 (d, J = 8.7 Hz, 1H), 6.63 (d, J = 8.2 Hz, 2H), 5.22 (s, 2H), 5.20 (s, 2H), 3.83 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.2, 155.4, 148.2, 137.1, 133.2, 130.0, 128.4, 127.6, 127.3, 127.0, 126.8, 126.0, 120.8, 114.7, 114.3, 69.7, 51.9.
Methyl 4’-amino-5-(benzyloxy)-[1,1’-biphenyl]-3-carboxylate (B10ii)
According to General Method A:
In a flame-dried vial equipped with a rubber septum was added methyl 3-(benzyloxy)-5-bromobenzoate (B10iii) (610.0 mg, 1.9 mmol), 4-aminophenylboronic acid hydrochloride (329.5 mg, 1.9 mmol) and cesium carbonate (1857.2 mg, 5.7 mmol, 3.0 eq) in 9 mL anhydrous dimethylformamide. The mixture was degassed under vigorous stirring for 20 min, and subsequently Pd(dppf)Cl2 (139.0 mg, 0.19 mmol) was added. The mixture was flushed with nitrogen and was heated to 60 °C for 27 h, when TLC indicated completion. Solvent was evaporated under reduced pressure, and the residue was diluted with ethyl acetate. The organic phase was washed with water (2 times x 30 mL), brine (2 times x 30 mL) and the organic phase was collected and concentrated under reduced pressure. The crude mixture was further purified with chromatography, using a gradient of ethyl acetate in hexanes, to afford the title product. Product: Pale Yellow Solid. Yield: 463.1 mg, 73 %. HRMS (ESI) m/z calculated for C21H19NO3 [M+H]+=334.1438, found: 334.1435.
1H NMR (500 MHz, DMSO-d6) δ 7.69 (t, J = 1.4 Hz, 1H), 7.52 – 7.46 (m, 2H), 7.45 – 7.37 (m, 6H), 7.37 – 7.31 (m, 1H), 6.67 – 6.62 (m, 2H), 5.33 (s, 2H), 5.23 (s, 2H), 3.86 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.2, 158.9, 149.1, 142.6, 136.9, 131.3, 128.4, 127.9, 127.7, 127.4, 125.8, 118.5, 116.6, 114.1, 112.1, 69.5, 52.2.
Sodium 4’-amino-4-(benzyloxy)-[1,1’-biphenyl]-3-carboxylate (B9i)
Methyl 4’-amino-4-(benzyloxy)-[1,1’-biphenyl]-3-carboxylate (B9ii) (71.3 mg, 0.21 mmol) was mixed with dioxane (5 mL) and 2N NaOH (1.06 mL, 2.12 mmol) was added dropwise. The reaction was allowed to stir at rt for 55 h 20 min. Then the mixture was neutralized (pH ~7) with 2N HCl (at 0 °C) and solvent was evaporated under reduced pressure. The residue was then chromatographed using a gradient of methanol in dichloromethane to afford the title product. Product: Pale White Solid. Yield: 70.1 mg, 98 %. HRMS (ESI) m/z calculated for C20H17NO3 [M+Na]+=342.1106, found: 342.1094.
1H NMR (500 MHz, DMSO-d6) δ 7.76 (d, J = 2.5 Hz, 1H), 7.62 (dd, J = 8.7, 2.5 Hz, 1H), 7.50 (d, J = 7.3 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.35 – 7.27 (m, 3H), 7.18 (d, J = 8.7 Hz, 1H), 6.65 – 6.60 (m, 2H), 5.21 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.6, 155.2, 148.1, 137.2, 133.1, 129.3, 128.3, 127.6, 127.2, 127.1, 126.7, 126.3, 122.6, 114.5, 114.3, 69.7.
4’-amino-5-(benzyloxy)-[1,1’-biphenyl]-3-carboxylic acid (B10i)
Methyl 4’-amino-5-(benzyloxy)-[1,1’-biphenyl]-3-carboxylate (B10ii) (449.0 mg, 1.35 mmol) was mixed with dioxane (32 mL) and 2N NaOH (6.75 mL, 13.5 mmol) was added dropwise. The reaction was allowed to stir at rt for 4 d 2 h. Then the mixture was neutralized with 2N HCl (at 0 °C) and solvent was evaporated under reduced pressure. The residue was chromatographed using a gradient of methanol in dichloromethane. The collected material was dried and then it was suspended in water (20 mL). The mixture was acidified to pH~4 with 1N HCl and subsequently was extracted with ethyl acetate (80 mL). The organic layer was washed with brine and was dried over sodium sulfate. The mixture was filtered, the solvent was evaporated, and the residue was dried under high vacuum overnight to afford the title product. Product: Light Yellow Solid. Yield: 330.2 mg, 77 %. HRMS (ESI) m/z calculated for C20H17NO3 [M+H]+= 320.1281, found: 320.1282.
1H NMR (500 MHz, DMSO-d6) δ 7.69 (s, 1H), 7.49 (d, J = 7.3 Hz, 2H), 7.43 – 7.36 (m, 6H), 7.34 (t, J = 7.3 Hz, 1H), 6.64 (d, J = 8.5 Hz, 2H), 5.22 (s, 2H) [(protons of the NH2 and COOH were not observed)]. 13C NMR (126 MHz, DMSO-d6) δ 167.2, 158.8, 149.0, 142.4, 137.0, 132.5, 128.5, 127.8, 127.6, 127.3, 126.1, 118.8, 116.2, 114.2, 112.3, 69.4.
Sodium 4-(benzyloxy)-4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylate (B9-Na)
According to General Method C:
Sodium 4’-amino-4-(benzyloxy)-[1,1’-biphenyl]-3-carboxylate (B9i) (67.9 mg, 0.20 mmol) was suspended in a mixture of tetrahydrofuran (1.0 mL) and water (0.5 mL). The mixture was cooled to 0 °C and sodium bicarbonate (20.2 mg, 0.24 mmol) was added, followed by the slow addition of a solution of phenyl chloroformate (26 μL, 0.21 mmol) in 0.5 mL tetrahydrofuran. The reaction was allowed to stir for 1 h 50 min at 0 °C, at which point TLC indicated consumption of the starting material. The reaction mixture was then diluted with ethyl acetate (20 mL), and was washed with brine (3 times x 2-5 mL). The organic phase was collected, solvent was evaporated under reduced pressure, and then the residue was dried in high vacuum overnight to afford the title product. Product: Pale-Yellow Solid. Yield: 69.3 mg, 75 %. HRMS (ESI) m/z calculated for C27H21NO5 [M+Na]+= 462.1312 found: 462.1299.
1H NMR (600 MHz, DMSO-d6) δ 10.34 (s, 1H), 7.69 (d, J = 2.3 Hz, 1H), 7.62 – 7.56 (m, 4H), 7.56 – 7.53 (m, 1H), 7.51 (d, J = 7.1 Hz, 2H), 7.46 – 7.40 (m, 2H), 7.39 – 7.35 (m, 2H), 7.32 – 7.28 (m, 1H), 7.26 (tt, J = 7.5, 1.0 Hz, 1H), 7.25 – 7.21 (m, 2H), 7.10 (d, J = 8.7 Hz, 1H), 5.19 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 169.6, 155.0, 151.7, 150.5, 137.6, 137.5, 134.3, 131.7, 129.4, 128.3, 127.4, 127.14, 127.05, 126.5, 125.4, 122.0, 118.7, 114.31, 114.25, 69.6.
4-(benzyloxy)-4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylic acid (B9-H)
According to General Method C:
Sodium 4’-amino-4-(benzyloxy)-[1,1’-biphenyl]-3-carboxylate (B9i) (173.5 mg, 0.51 mmol) was suspended in a mixture of tetrahydrofuran (4.0 mL) and water (2.0 mL). The mixture was cooled to 0 °C and sodium bicarbonate (51.4 mg, 0.61 mmol) was added, followed by the slow addition of a solution of phenyl chloroformate (68 μL, 0.54 mmol) in 2.0 mL tetrahydrofuran. The reaction was allowed to stir for 2 h 30 min at 0 °C, at which point TLC indicated consumption of the starting material. The reaction mixture was then acidified to pH~4 with 1N HCl and then it was extracted with ethyl acetate (50 mL). The aqueous layer was removed, and the organic phase was washed with water (3 times x 5 mL) and brine (2 times x 5 mL). The organic phase was collected, solvent was evaporated under reduced pressure and then the residue was dried in high vacuum overnight to afford the title product. Product: Pale Yellow Solid. Yield: 362.4 mg of material were isolated and carried to the next step without further purification. HRMS (ESI) m/z calculated for C27H21NO5 [M+H]+=440.1493, found: 440.1482.
1H NMR (600 MHz, DMSO-d6) δ 12.79 (s, 1H), 10.36 (s, 1H), 7.89 (d, J = 2.6 Hz, 1H), 7.76 (dd, J = 8.7, 2.6 Hz, 1H), 7.65 – 7.57 (m, 4H), 7.53 – 7.49 (m, 2H), 7.46 – 7.42 (m, 2H), 7.42 – 7.37 (m, 2H), 7.34 – 7.30 (m, 1H), 7.29 – 7.25 (m, 2H), 7.25 – 7.22 (m, 2H), 5.25 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.4, 156.1, 151.7, 150.5, 137.9, 137.0, 133.5, 131.9, 130.4, 129.4, 128.4, 128.1, 127.7, 127.1, 126.7, 125.5, 122.5, 122.0, 118.7, 114.5, 69.7.
Sodium 5-(benzyloxy)-4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylate (B10)
According to General Method C:
4’-amino-5-(benzyloxy)-[1,1’-biphenyl]-3-carboxylic acid (B10i) (325.0 mg, 1.02 mmol) was suspended in a mixture of tetrahydrofuran (5 mL) and water (2.5 mL). The mixture was cooled to 0 °C and sodium bicarbonate (188.2 mg, 2.24 mmol) was added, followed by the slow addition of a solution of phenyl chloroformate (0.13 mL, 1.07 mmol) in 2.5 mL tetrahydrofuran. The reaction ran for 2 h 35 min at 0 °C, at which point TLC indicated consumption of the starting material. Ethyl acetate (80 mL) was added, the organic phase was washed with brine (3 times x 30 mL), solvent was evaporated under reduced pressure and the residue was dried in high vacuum overnight. The residue was washed with water, it was dried over the Buchner, and then it was suspended in ethyl acetate. The mixture was filtered and the filtrate was dried again in high vacuum to afford the title product. Product: Beige Solid. Yield: 463.0 mg, 98%. HRMS (ESI) m/z calculated for C27H21NO5 [M+H]+= 440.1493, found: 440.1487.
1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1H), 7.78 (s, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.54 – 7.47 (m, 4H), 7.43 (dt, J = 14.8, 7.8 Hz, 4H), 7.34 (t, J = 7.3 Hz, 1H), 7.30 – 7.22 (m, 3H), 5.25 (s, 2H).
13C NMR (126 MHz, DMSO-d6) δ 167.0, 158.9, 151.7, 150.5, 141.4, 138.7, 136.8, 133.5, 132.7, 129.4, 128.5, 127.9, 127.7, 127.3, 125.5, 121.9, 119.6, 118.8, 117.3, 113.6, 69.5.
4’-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-4-(benzyloxy)-[1,1’-biphenyl]-3-carboxylic acid (11)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A1 (35.8 mg, 0.14 mmol) was suspended in 0.8 mL anhydrous dioxane and the mixture was heated for a 10-15 min at 100 °C, followed by the addition of triethylamine (20 μL, 0.14 mmol). The mixture was allowed to stir for another 10 min and subsequently it was added dropwise to Mixture B. The vial was rinsed with 0.5 mL anhydrous dioxane, which was also added to the Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, sodium 4-(benzyloxy)-4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylate (B9-Na) (65.0 mg, 0.14 mmol) in 2.0 mL anhydrous dioxane was heated at 80 °C for 10 min, under nitrogen. Triethylamine (20 μL, 0.14 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 6 h. The mixture was then allowed to settle and the supernatant was removed. The residue was triturated with methanol and was dried. Water (1.0 mL) was added and the mixture was acidified to pH~4 with 1N HCl. The precipitate was collected by centrifugation and was subsequently purified with reversed-phase chromatography using a RediSep Gold C18 reusable column (particle size: 20 – 40 μm spherical; pore size: 100 Å) with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase. Product: White Solid. Yield: 1.7 mg, 2 % (Purity: 95.9 %). HRMS (ESI) m/z calculated for C29H25N7O6S [M+H]+=600.1660, found: 600.1670.
1H NMR (600 MHz, DMSO-d6) δ 9.69 (s, 1H), 9.66 (s, 1H), 7.89 (s, 1H), 7.83 (d, J = 8.7 Hz, 2H), 7.79 – 7.74 (m, 3H), 7.72 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 7.4 Hz, 2H), 7.44 (br s, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.25 (d, J = 8.6 Hz, 1H), 7.14 (s, 2H), 5.25 (s, 2H) [Proton of the COOH moiety was not observed]. 13C NMR (151 MHz, DMSO-d6) δ 167.7, 156.7, 156.1, 155.9, 149.0, 143.9, 137.1, 136.3, 135.0, 134.9, 131.7, 129.8, 128.4, 128.0, 127.6, 127.1, 126.8, 126.4, 122.0, 116.2, 114.5, 69.7 (one carbon peak overlaps with the rest in the aromatic region).
4’-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-5-(benzyloxy)-[1,1’-biphenyl]-3-carboxylic acid (12)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A1 (63.6 mg, 0.25 mmol) was suspended in 1.0 mL anhydrous dioxane and the mixture was heated for a 5-10 min at 100 °C, followed by the addition of triethylamine (35 μL, 0.25 mmol). The mixture was allowed to stir for another 5 min and subsequently it was added dropwise to Mixture B. The vial was rinsed with 1.0 mL anhydrous dioxane, which was also added to the Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, sodium 5-(benzyloxy)-4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylate (B10) (115.4 mg, 0.25 mmol) in 1.0 mL anhydrous dioxane was heated at 100 °C for 5-10 min, under nitrogen. Triethylamine (35 μL, 0.25 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 6 h. Solvent was evaporated, water was added to the residue and the mixture was acidified to pH~4 with 1N HCl. The precipitate was collected by centrifugation and was subsequently purified with preparative reverse-phase HPLC, using an Agilent 5 Prep-C18 21.2 × 100 mm column with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase (System B). Product: White Solid. Yield: 3.1 mg, 2 % (Purity: 96.7 %). HRMS (ESI) m/z calculated for C29H25N7O6S [M+H]+= 600.1660, found: 600.1671.
1H NMR (600 MHz, DMSO-d6) δ 9.70 (s, 1H), 9.69 (s, 1H), 7.84 (d, J = 8.6 Hz, 2H), 7.82 – 7.76 (m, 3H), 7.76 – 7.69 (m, 4H), 7.54 – 7.48 (m, 3H), 7.46 (br s, 2H), 7.44 – 7.38 (m, 3H), 7.34 (t, J = 7.3 Hz, 1H), 7.15 (s, 2H), 5.23 (s, 2H) [Proton of the COOH moiety was not observed]. 13C NMR (126 MHz, DMSO-d6) δ 167.9, 158.6, 156.7, 155.9, 149.0, 143.9, 140.4, 137.2, 136.9, 135.6, 134.9, 128.5, 127.8, 127.7, 126.9, 126.8, 121.9, 119.8, 116.2, 115.4, 114.5, 113.8, 69.4.
4’-(5-amino-3-((4-(methylcarbamoyl)phenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-4-(benzyloxy)-[1,1’-biphenyl]-3-carboxylic acid (13)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A2 (58.1 mg, 0.25 mmol) was suspended in 1.7 mL anhydrous dioxane and the mixture was heated for a 10-15 min at 100 °C, followed by the addition of triethylamine (35 μL, 0.25 mmol). The mixture was allowed to stir for another 10 min and subsequently was added dropwise to Mixture B. The vial was rinsed with 1.0 mL anhydrous dioxane, which was also added to Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, 4-(benzyloxy)-4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylic acid (B9-H) (109.9 mg, 0.25 mmol) in 1.0 mL anhydrous dioxane was heated at 100 °C for 5 min, under nitrogen. Triethylamine (70 μL, 0.50 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 3 h. The reaction mixture was then dried, the residue was triturated with ethyl acetate and it was dried again. Water (1.0 mL) was added and the mixture was acidified to pH~4 with 1N HCl. The precipitate was collected by centrifugation and it was subsequently purified using preparative reverse-phase HPLC, with an Agilent 5 Prep-C18 21.2 × 100 mm column with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase (System B). Product: White Solid. Yield: 66.1 mg, 46 % (Purity: 95.6 %). HRMS (ESI) m/z calculated for C31H27N7O5 [M+H]+=578.2146, found: 578.2144.
1H NMR (500 MHz, DMSO-d6) δ 9.67 (s, 1H), 9.51 (s, 1H), 8.20 (q, J = 4.7 Hz, 1H), 7.89 (s, 1H), 7.83 – 7.75 (m, 5H), 7.73 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 7.3 Hz, 2H), 7.44 – 7.37 (m, 4H), 7.32 (t, J = 7.1 Hz, 1H), 7.26 (d, J = 8.7 Hz, 1H), 5.25 (s, 2H), 2.76 (d, J = 4.4 Hz, 3H) [Proton of the COOH moiety was not observed]. 13C NMR (126 MHz, DMSO-d6) δ 168.1, 166.4, 156.9, 155.9, 149.1, 143.4, 137.2, 136.3, 135.0, 131.7, 129.4, 128.4, 128.0, 127.9, 127.6, 127.1, 126.7, 126.3, 125.7, 121.8, 116.0, 114.5, 114.3, 69.7, 26.2.
4’-(5-amino-3-((4-(methylcarbamoyl)phenyl)amino)-1H-1,2,4-triazole-1-carboxamido)-5-(benzyloxy)-[1,1’-biphenyl]-3-carboxylic acid (14)
According to General Method D:
Mixture A: In a flame-dried vial, Fragment A2 (41.5 mg, 0.18 mmol) was suspended in 1.0 mL anhydrous dioxane and the mixture was heated for a 5-10 min at 100 °C, followed by the addition of triethylamine (25 μL, 0.18 mmol). The mixture was allowed to stir for another 5 min and subsequently it was added dropwise to Mixture B. The vial was rinsed with 1.0 mL anhydrous dioxane, which was also added to the Mixture B.
Mixture B: In a flame-dried vial equipped with a rubber septum, sodium 5-(benzyloxy)-4’-((phenoxycarbonyl)amino)-[1,1’-biphenyl]-3-carboxylate (B10) (83.1 mg, 0.18 mmol) in 1.0 mL anhydrous dioxane was heated at 100 °C for 5-10 min, under nitrogen. Triethylamine (25 μL, 0.18 mmol) was added, followed by the addition of Mixture A. The reaction ran at 100 °C for 8.5 h. Solvent was evaporated, and the residue was triturated with ethyl acetate and it was dried. Then water was added and the mixture was acidified to pH~4 with 1N HCl. The precipitate was collected by centrifugation and was subsequently purified with preparative reverse-phase HPLC, using an Agilent 5 Prep-C18 21.2 × 100 mm column with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase (System B). Product: Off-white Solid. Yield: 1.9 mg, 1.8 % (Purity: 98.6 %). HRMS (ESI) m/z calculated for C31H27N7O5 [M+H]+=578.2146, found: 578.2145. 1H NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H), 9.51 (s, 1H), 8.22 (q, J = 4.2 Hz, 1H), 7.83 – 7.77 (m, 5H), 7.76 – 7.71 (m, 4H), 7.54 – 7.49 (m, 3H), 7.47 – 7.39 (m, 5H), 7.34 (t, J = 7.1 Hz, 1H), 5.24 (s, 2H), 2.77 (d, J = 4.2 Hz, 3H) [Proton of the COOH moiety was not observed]. 13C NMR (126 MHz, DMSO-d6) δ 167.9, 166.4, 158.7, 156.9, 155.9, 149.0, 143.4, 140.6, 137.1, 137.0, 135.3, 128.4, 128.0, 127.8, 127.7, 126.9, 125.7, 121.6, 119.8, 116.0, 115.9, 113.8, 69.4, 26.2 (one carbon peak overlaps with the rest in the aromatic region).
JAK2 Protein Expression and Purification
The JH1 and JH2 domains of JAK2 were expressed in Sf9 cells transfected with baculovirus generated from recombinant bacmid as previously described.16 JH2 was purified as previously described.16 JH1 used for this study was purified either as described previously16 or as following. Transfected Sf9 cells were resuspended in lysis buffer containing 20 mM Tris pH 8.0, 500 mM NaCl, 20% glycerol, 0.25 mM TCEP, and cOmplete EDTA-free protease inhibitor (Roche) and were lysed by pressure homogenization using an Emulsiflex cell disruptor (Avestin). Lysate was cleared by centrifugation at 16,500 rpm in an SS-34 rotor for 45 minutes. Ni-NTA agarose beads (Qiagen) were added to the supernatant and incubated for 2 h at 4 °C. Beads were washed with lysis buffer containing 10 mM imidazole, and JH1 was eluted with lysis buffer containing 200 mM imidazole. The eluent was concentrated and exchanged into a buffer composed of 20 mM Tris pH 8.0, 250 mM NaCl, 5% glycerol, and 1.0 mM TCEP using an Amicon Ultra-15 centrifugal filter unit (10 kDa MWCO) and applied to a Superdex 75 10/300 (GE Healthcare) mounted on an ÄKTA pure protein purification system (GE Healthcare) pre-equilibrated with the same buffer. Resulting JH1 fractions were pooled, concentrated to a final concentration of 4 mg/ml, aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C.
Crystal structure determination and refinement
Crystals of JH2 in complex with compounds 6 and 11 were obtained using co-crystallization methods described previously.16 X-ray data on frozen crystals were collected on a Rigaku MicroMax-007HF+ Xray generator (Cu rotating anode; λ = 1.54 Å) equipped with a Rigaku Saturn 944+ CCD detector. Data sets were processed with XDS.50 Phases were determined by molecular replacement with the program Phaser51 using a protein chain from the previously solved structure PDB 6OCC16 as the initial search model. All model building into electron density was performed with COOT52 and the structures were refined using Phenix Refine.53 For each refinement, 5% of all reflections were omitted and used for the calculation of Rfree. Successive rounds of simulated annealing, XYZ coordinate, and individual B-factor refinement were performed until acceptable R factors, geometry statistics, and Ramachandran statistics were achieved. Data collection, diffraction, and refinement statistics can be found in Table S2. All atomic coordinates and structure factors and have been deposited in the Protein Data Bank with PDB ID codes 7SZW and 7T0P. All figures were prepared by PyMOL.54
Competitive Fluorescence Polarization (FP) Assay21,22
In a black, flat-bottom, 96-well plate (BRANDplates® pureGrade™ 96-Well x 350μL Assay Microplate, Sterile, Black, with F-Bottom), FP-buffer (pH 8.0, with 20 mM Tris-HCl, 150 mM NaCl, 20% Glycerol, 0.5 mM TCEP, 0.01% Tween 20) was added using a multichannel pipette as follows: 200 μL to column 1 (blank), 150 μL to column 2 (low-FP control), 140 μL to column 3 (high-FP control) and also 140 μL to columns 4-12 (measured compounds). 10 μL of protein (at concentrations: 2.96 μM for JAK2 JH2 WT, 3.52 μM for JAK2 JH2 V617F, 6.93 μM for JAK2 JH1, all prepared in FP-buffer) was added to columns 3-12. Tested compounds were prepared in 9 concentrations with serial dilutions in DMSO, and 2 μl of each solution was added to columns 4-12, in quadruplicate. For consistency, 2 μL of DMSO were added to columns 1-3. Finally, 50 μl of tracer (24.0 nM in FP-buffer) were added to columns 2-12, and after mixing, fluorescence polarization was measured at λexc=485±20 nm, λem=535±25 nm using a TECAN Spark microplate reader, recording FP measurements at t=0, 15 min, and 30 min. Data at t =30 min was analyzed with a least-squares, non-linear fit, using Prism 9 in order to determine the compounds’ IC50. IC50 values were then converted to Kd’s using the following equation based on the IC50, Kd of the tracer (Kdt), total (Lt) and bound (Lb) tracer, as well as total protein concentration (Pt):
Two to three independent experiments were carried out to determine the Kd of each compound.
Parallel Artificial Membrane Permeation Assay (PAMPA)
PAMPA experiments were performed in a 96-well 0.45 μm filter plate containing hydrophobic PVDF membranes (Millipore) as described previously.39–40 The artificial membrane was formed by adding 5 μL of a solution containing 4 % lecithin (Sigma P5638) in dodecane to each filter. 200 μL of donor solutions containing saturated or up to 500 μM concentrations of compound in PBS pH 7.4 and 5 % DMSO were added to the top donor wells. 300 μL of PBS pH 7.4 buffer were added to the receiving wells of the acceptor plate. The stacked donor and acceptor plates were incubated at room temperature for 18 hours, and the concentration of compound in the acceptor solutions was then measured by a multi-wavelength UV plate reader. Experiments were performed in duplicate for each compound. An equilibrium standard was made for each compound containing 40 % concentration of the donor solution which represents the concentration of the acceptor solution if full permeability was achieved. The apparent permeability of each compound was calculated by the following equation:
VD and VA are the volumes of the donor and acceptor solutions, S is the membrane surface area, t is the time of incubation, and ODA and ODE are the absorbances of the acceptor solution and equilibrium standards, respectively.
It should be noted that these permeability measurements rely on a balance of both aqueous solubility and lipophilicity, therefore poorly soluble molecules could present challenges in PAMPA measurements due to their precipitation in the wells over the equilibration time and also due difficulties in accurate UV quantitation (The latter could also be the case for compounds with poor UV-absorption).
Cell culture and Western blot analysis41
Human cell lines HEL (containing the JAK2 V617F mutant) and TF-1 (containing wild-type JAK2) were obtained from the ATCC (Philadelphia, PA) and were cultured in RPMI media (ATCC modification; containing L-glutamine, HEPES, Sodium Pyruvate, high glucose and low sodium bicarbonate) in 100 mm culture dishes. RPMI media for TF-1 cell culture was supplemented with an additional 2 ng/ml GM-CSF. To measure inhibition of STAT5 phosphorylation, approximately 1 X 106 cells per well in a 6-well plate were incubated for 1 hour with different concentrations of inhibitor added from a concentrated DMSO stock. Cells were lysed and equal amounts of protein as determined by a BCA assay from each experiment were run by 12 % SDS-PAGE. For Western blots, membranes were incubated for 1 hour at room temperature with anti-STAT5 (Cell Signaling Technology, #25656S) and anti-phospho-STAT5 (Y694) (Abcam, ab32364) followed by incubation with Anti-rabbit IgG HRP-linked antibody (Cell Signaling Technology, #7074) for 1 hour. Signal was detected on an iBright FL1000 Imaging System after addition of BioRad Clarity Max Western ECL substrate onto the membrane.
Supplementary Material
Acknowledgements
Gratitude is expressed to the U. S. National Institutes of Health (GM032136) for research support. This research used resources of the Yale Macromolecular X-ray Core Facility (NIH Grant 1S10OD018007-01) and the Chemical and Biophysical Instrumentation Center (CBIC) at Yale University (RRID:SCR_021738). The authors thank Dr. Brandon Mercado at the CBIC at Yale University for assistance with X-ray data collection and Dr. Fabian Menges at the CBIC at Yale University for assistance with Mass Spectrometry and purity (HPLC) measurements.
Abbreviations
- ATP
adenosine triphosphate
- FP
fluorescence polarization
- JAKs
Janus kinases
- JH1
kinase domain
- JH2
pseudokinase domain
- PAMPA
parallel artificial membrane permeability assay
- STAT
signal transducer and activator of transcription
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Molecular formula strings for compounds 3-14; details for the Suzuki reaction; 1H and 13CNMR spectra for final compounds 3-14 and HPLC traces for compounds 11-14; results from docking; additional results from cell culture experiments; details for the FP measurements of compounds 11-14; crystallographic data for complexes of 6 and 11 with JAK2 JH2, which have been deposited in the RCSB Protein Data Bank with the PDB ID codes 7SZW and 7T0P. Authors will release the atomic coordinates upon article publication.
Yale University has filed patent applications for JAK2 binding agents.
References
- (1).Yamaoka K; Saharinen P; Pesu M; Holt VET; Silvennoinen O; O’Shea JJ The janus kinases (JAKs). Genome Biol. 2004, 5, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).O’Shea JJ; Holland SM; Staudt LM JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med 2013, 368, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Leonard WJ; O’Shea JJ; JAKs and STATs: biological implications. Annu. Rev. Immunol 1998, 16, 293–322. [DOI] [PubMed] [Google Scholar]
- (4).Raivola J; Haikarainen T; Abraham BG; Silvennoinen O Janus kinases in leukemia. Cancers. 2021, 13, 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Levine RL; Wadleigh M; Cools J; Ebert BL; Wernig G; Huntly BJ; Boggon TJ; Wlodarska I; Clark JJ; Moore S; Adelsperger J Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer cell. 2005, 7, 387–397. [DOI] [PubMed] [Google Scholar]
- (6).Baxter EJ; Scott LM; Campbell PJ; East C; Fourouclas N; Swanton S; Vassiliou GS; Bench AJ; Boyd EM; Curtin N; Scott MA Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005, 365, 1054–1061. [DOI] [PubMed] [Google Scholar]
- (7).Bandaranayake RM; Ungureanu D; Shan Y; Shaw DE; Silvennoinen O; Hubbard SR Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat. Struct. Mol. Biol 2012, 19, 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Saharinen P; Takaluoma K; Silvennoinen O Regulation of the JAK2 tyrosine kinase by its pseudokinase domain. Mol. Cell. Biol 2000, 20, 3387–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ungureanu D; Wu J; Pekkala T; Niranjan Y; Young C; Jensen ON; Xu CF; Neubert TA; Skoda RC; Hubbard SR; Silvennoinen O The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat. Struct. Mol. Biol 2011, 18, 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wilmes S; Hafer M; Vuorio J; Tucker JA; Winkelmann H; Löchte S; Stanly TA; Prieto KDP; Poojari C; Sharma V; Richter CP Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. Science. 2020, 367, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Leroy E; Constantinescu SN Rethinking JAK2 inhibition: towards novel strategies of more specific and versatile janus kinase inhibition. Leukemia. 2017, 31, 1023–1038. [DOI] [PubMed] [Google Scholar]
- (12).Tefferi A JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012, 119, 2721–2730. [DOI] [PubMed] [Google Scholar]
- (13).Hammaren HM; Ungureanu D; Grisouard J; Skoda RC; Hubbard SR; Silvennoinen O ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 4642–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McNally R; Li Q; Li K; Dekker C; Vangrevelinghe E; Jones M; Chène P; Machauer R; Radimerski T; Eck MJ. Discovery and structural characterization of ATP-site ligands for the wild-type and V617F mutant JAK2 pseudokinase domain. ACS Chem. Biol 2019, 14, 587–593. [DOI] [PubMed] [Google Scholar]
- (15).Newton AS; Liosi M-E; Henry SP; Deiana L; Faver JC; Krimmer SG; Puleo DE; Schlessinger J; Jorgensen WL Indoloxytriazines as binding molecules for the JAK2 JH2 pseudokinase domain and its V617F variant. Tetrahedron Lett. 2021, 77, 153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liosi M-E; Krimmer SG; Newton AS; Dawson TK; Puleo DE; Cutrona KJ; Suzuki Y; Schlessinger J; Jorgensen WL Selective janus kinase 2 (JAK2) pseudokinase ligands with a diaminotriazole core. J. Med. Chem 2020, 63, 5324–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cutrona KJ; Newton AS; Krimmer SG; Tirado-Rives J; Jorgensen WL Metadynamics as a postprocessing method for virtual screening with application to the pseudokinase domain of JAK2. J. Chem. Inf. Model 2020, 60, 4403–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Henry SP; Liosi M-E; Cutrona KJ; Ippolito JA; Krimmer SG; Newton AS; Schlessinger J; Jorgensen WL Conversion of a false-positive into selective binders for the JAK2 JH2 domain using convergent design strategies. 2022. Submitted to ACS. Med. Chem. Lett [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lin RH; Connolly PJ; Huang SL; Wetter SK; Lu YH; Murray WV; Emanuel SL; Gruninger RH; Fuentes-Pesquera AR; Rugg CA; Middleton SA; Jolliffe LK 1-acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: synthesis and evaluation of biological activities. J. Med. Chem 2005, 48, 4208–4211. [DOI] [PubMed] [Google Scholar]
- (20).Karaman MW; Herrgard S; Treiber DK; Gallant P; Atteridge CE; Campbell BT; Chan KW; Ciceri P; Davis MI; Edeen PT; Faraoni R; Floyd M; Hunt JP; Lockhart DJ; Milanov ZV; Morrison MJ; Pallares G; Patel HK; Pritchard S; Wodicka LM; Zarrinkar PP A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- (21).Puleo DE; Kucera K; Hammarén H; Ungureanu D; Newton AS; Silvennoinen O; Jorgensen WL; Schlessinger J. Identification and characterization of JAK2 pseudokinase domain small molecule binders. ACS Med. Chem. Lett 2017, 8, 618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Newton AS; Deiana L; Puleo DE; Cisneros JA; Cutrona KJ; Schlessinger J; Jorgensen WL. JAK2 JH2 fluorescence polarization assay and crystal structures for complexes with three small molecules. ACS Med. Chem. Lett 2017, 8, 614–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jorgensen WL Efficient drug lead discovery and optimization. Acc. Chem. Res 2009, 42, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jorgensen WL; Tirado-Rives J Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem 2005, 26, 1689–1700. [DOI] [PubMed] [Google Scholar]
- (25).Robertson MJ; Tirado-Rives J; Jorgensen WL Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput 2015, 11, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jorgensen WL; Tirado-Rives J Potential energy functions for atomic-level simulations of organic and biomolecular systems. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 6665–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Schrödinger release 2015-4: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2015; Impact, Schrödinger, LLC, New York, NY, 2015; Prime, Schrödinger, LLC, New York, NY, 2015. [Google Scholar]
- (28).Schrödinger release 2015-4: Maestro, Schrödinger, LLC, New York, NY, 2015. [Google Scholar]
- (29).Schrödinger release 2015-4: LigPrep, Schrödinger, LLC, New York, NY, 2015. [Google Scholar]
- (30).Jorgensen WL; Tirado-Rives J The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- (31).Jorgensen WL; Maxwell DS; Tirado-Rives J Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- (32).Shivakumar D; Williams J; Wu Y; Damm W; Shelley J; Sherman W Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput 2010, 6, 1509–1519. [DOI] [PubMed] [Google Scholar]
- (33).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (34).Shelley JC; Cholleti A; Frye LL; Greenwood JR; Timlin MR; Uchimaya M Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des 2007, 21, 681–691. [DOI] [PubMed] [Google Scholar]
- (35).Greenwood JR; Calkins D; Sullivan AP; Shelley JC Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des 2010, 24, 591–604. [DOI] [PubMed] [Google Scholar]
- (36).Schrödinger release 2015-4: Epik, Schrödinger, LLC, New York, NY, 2015. [Google Scholar]
- (37).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (38).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (39).Kansy M; Senner F; Gubernator K Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- (40).Berben P; Bauer-Brandl A; Brandl M; Faller B; Flaten GE; Jacobsen AC; Brouwers J; Augustijns P Drug permeability profiling using cell-free permeation tools: overview and applications. Eur. J. Pharm. Sci 2018, 119, 219–233. [DOI] [PubMed] [Google Scholar]
- (41).Andraos R; Qian Z; Bonenfant D; Rubert J; Vangrevelinghe E; Scheufler C; Marque F; Régnier CH; De Pover A; Ryckelynck H; Bhagwat N; Koppikar P; Aviva Goel A; Wyder L; Tavares G; Baffert F; Pissot-Soldermann C; Manley PW; Gaul C; Voshol H; Levine RL; Sellers WR; Hofmann F, Radimerski T Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012, 2, 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Quentmeier H; MacLeod R; Zaborski M; Drexler HG JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia. 2006, 20, 471–476. [DOI] [PubMed] [Google Scholar]
- (43).Mattsson S; Dahlström M; Karlsson S A mild hydrolysis of esters mediated by lithium salts. Tetrahedron Lett. 2007, 48, 2497–2499. [Google Scholar]
- (44).Barrio S; Gallardo M; Arenas A; Ayala R; Rapado I; Rueda D; Jimenez A; Albizua E; Burgaleta C Gilsanz F; Martinez-Lopez J. Inhibition of related JAK/STAT pathways with molecular targeted drugs shows strong synergy with ruxolitinib in chronic myeloproliferative neoplasm. Br. J. Haematol 2013, 161, 667–676. [DOI] [PubMed] [Google Scholar]
- (45).de Melo Campos P; Machado-Neto JA; Eide CA; Savage SL; Scopim-Ribeiro R; Duarte ADSS; Favaro P; Lorand-Metze I; Costa FF; Tognon CE ; Druker BJ. IRS2 silencing increases apoptosis and potentiates the effects of ruxolitinib in JAK2V617F-positive myeloproliferative neoplasms. Oncotarget. 2016, 7, 6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lima K; Lopes LR; Machado-Neto JA Exploring redox vulnerabilities in JAK2 V617F-positive cellular models. Hematol. Transfus. Cell Ther 2021, 43, 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Gäbler K; Rolvering C; Kaczor J; Eulenfeld R; Méndez SÁ; Berchem G; Palissot V; Behrmann I; Haan C Cooperative effects of janus and aurora kinase inhibition by CEP 701 in cells expressing JAK2V617F. J. Cell. Mol. Med 2013, 17, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hron R; Jursic BS Preparation of substituted semicarbazides from corresponding amines and hydrazines via phenyl carbamates. Tetrahedron Lett. 2014, 55, 1540–1543. [Google Scholar]
- (49).Richards JE; Philp D A reactive nitrone-based organogel that self-assembles from its constituents in chloroform. Chem. Commun 2016, 52, 4995–4998. [DOI] [PubMed] [Google Scholar]
- (50).Kabsch W Xds. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of COOT. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).DeLano WL The PyMOL molecular graphics system. 2002. http://www.pymol.org
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.