

Abstract
Despite lipid nanoparticles’ (LNP) success in the effective and safe delivery of mRNA vaccines, an inhalation-based mRNA therapy for lung diseases remains challenging. LNP tend to disintegrate due to shear stress during aerosolization, leading to ineffective delivery. Therefore, LNP need to remain stable through the process of nebulization and mucus penetration, yet labile enough for endosomal escape. To meet these opposing needs, we utilized PEG lipid to enhance the surficial stability of LNP with the inclusion of cholesterol analogs, β-sitosterol, to improve endosomal escape. Increased PEG concentrations in LNP enhanced the shear resistance and mucus penetration, while β-sitosterol provided LNP with a polyhedral shape facilitating endosomal escape. The optimized LNP exhibited a uniform particle distribution, a polyhedral morphology, and a rapid mucosal diffusion with enhanced gene transfection. Inhaled LNPs led to localized protein production in the mouse lung without pulmonary or systemic toxicity. Repeated administration of these LNP led to sustained protein production in the lungs. Lastly, mRNA encoding the cystic fibrosis transmembrane conductance regulator (CFTR) was delivered after nebulization to a CFTR deficient animal model, resulting in the pulmonary expression of this therapeutic protein. This study demonstrated the rational design approach for clinical translation of inhalable LNP-based mRNA therapies.
Keywords: Inhalation, Nebulization, Pulmonary delivery, Cystic fibrosis, Lung delivery, mRNA therapy, β-sitosterol
Graphical Abstract
INTRODUCTION
Pulmonary gene therapy has gained significant interest as a modality to cure lung diseases, including inherited diseases and cancers. Especially, genetic disorders, such as cystic fibrosis (CF) and α1-antitrypsin deficiency, could be treated by gene therapy that repairs mutations or provides normal protein expression.1,2 Among various gene therapies, messenger RNA (mRNA) therapy aims to produce proteins to restore the proteins’ functions and alleviate symptoms. Previous proof-of-principle studies demonstrated the feasibility of mRNA therapy for treating lung diseases.3–5 For instance, CFTR mRNA treatment restored the chloride ion efflux in the nasal epithelium of CFTR-deficient mice.6 However, despite encouraging results in preclinical studies, effective mRNA treatment for pulmonary diseases remains challenging primarily due to insufficient delivery of mRNA therapeutics.7
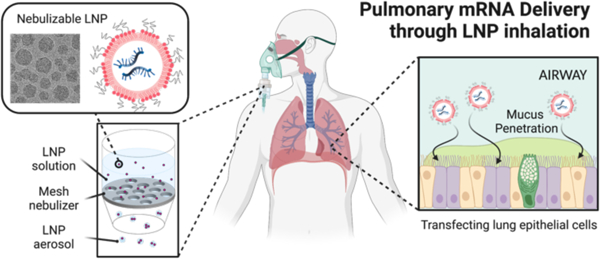
Recent clinical successes of mRNA-based vaccines proved that lipid nanoparticles (LNP; Figure 1a) can deliver mRNA to humans.8 LNP’s ability to deliver mRNA to the inside of cells is not only limited to vaccination but has versatile applications such as treating genetic disorders. In fact, LNP has been extensively investigated to deliver mRNA therapies targeting various genetic diseases.9,10 Recently, LNP was employed in the clinical trial to deliver Cas9 mRNA and guide RNA for editing the gene causing transthyretin amyloidosis.11 However, therapeutic application of the LNP platform is restricted mostly for hepatic diseases because LNP innately accumulates in the liver when administered systemically, which significantly limits access to other organs.12 Despite recent studies showing that modulating nanoparticle surface charge permits the systemically administered LNP to reach the lungs,13–15 focused delivery of mRNA therapy to the pulmonary system via inhalation still represents a promising route to achieve lung transfection. Inhaled LNP encapsulating mRNA can access the pulmonary system, particularly lung epithelium, avoiding potential off-target effects in other organs. For LNP inhalation, a vibrating mesh nebulizer, which turns a liquid into a mist, could be useful due to its ability to generate a uniform aerosol and its compatibility with aqueous biopharmaceutical (Figure 1b).16,17 However, LNP undergo (1) considerable shear force in the nebulizer, leading to the destruction of the nanoparticle structure and genetic payloads (Figure 1c).18 Nebulized LNP then encounter biological barriers that limit entry of all gene therapy vectors into the lungs: (2) the airway mucus that impedes the nanoparticles from reaching epithelial cells, and (3) inadequate cytosolic availability of mRNA after uptake by cells due to endosomal entrapment. Hence, there is a critical need for LNP formulations capable of overcoming aforementioned barriers and delivering mRNA to the lungs.
Figure 1.
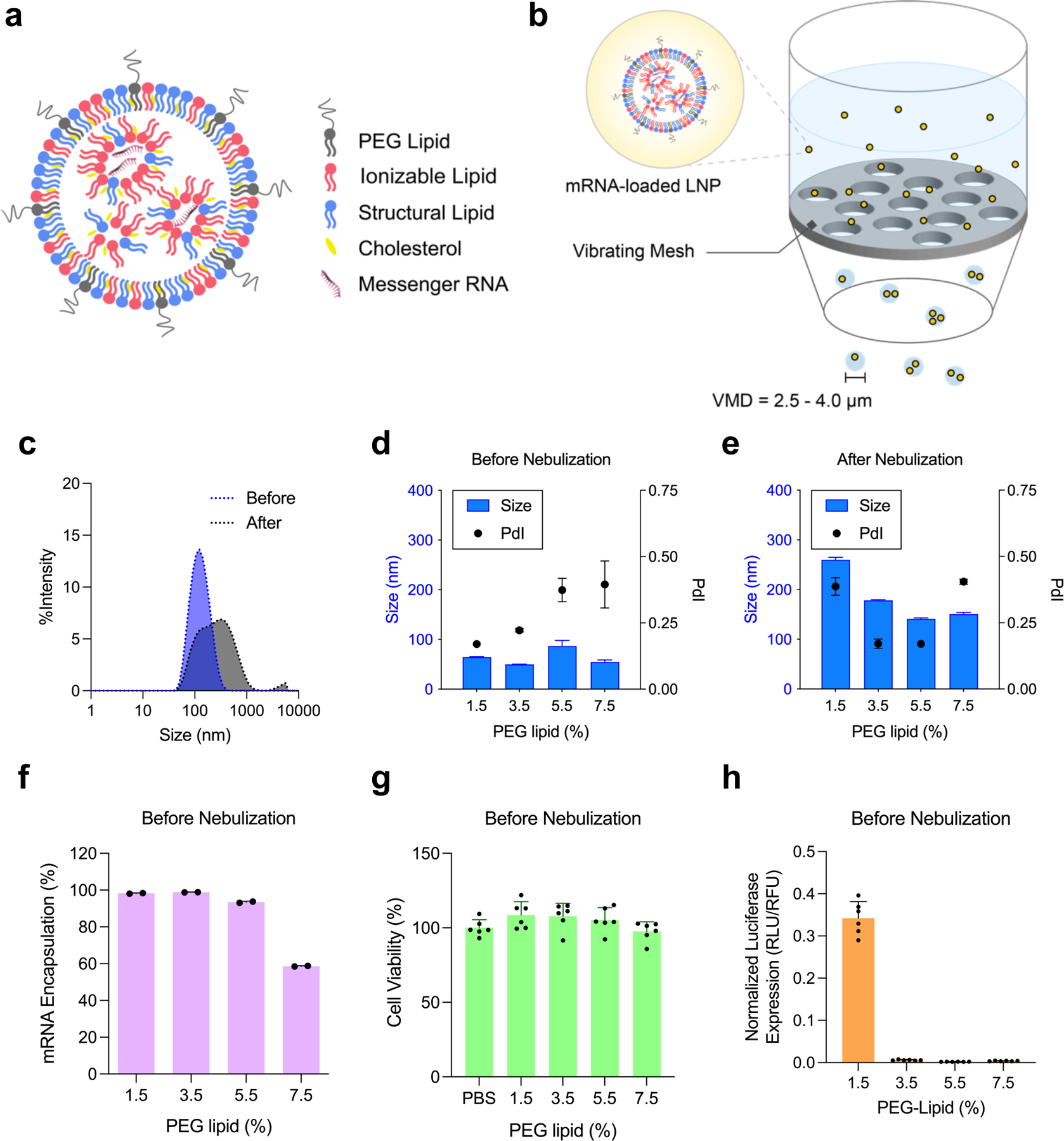
Nebulization process influence physicochemical properties of LNP. (a) A schematic representation of mRNA-loaded LNP. Ionizable lipid (red), cholesterol (yellow), structural lipid (blue), and PEG lipid (gray) form a spherical nanoparticle, encapsulating mRNA inside. (b) A schematic of nebulization process of mRNA-loaded LNP. VMD; volume mean diameter. (c) Size change of Onpattro® LNP formulation during nebulization. Blue; before nebulization, Gray; after nebulization. (d-h) Characterization of LNP-Chol with various PEG lipid contents. (d,e) Size and PdI of LNP-Chol (d) before and (e) after nebulization. (f) mRNA encapsulation of LNP-Chol with various PEG lipid contents. (g) Cell viability and (h) normalized luciferase expression of HeLa cells treated with various LNP-Chol at 50 ng mRNA per well for 24 h. Data were presented in mean ± standard deviation.
Typical LNP contains four functional lipids that play distinctive roles: (1) ionizable lipid, (2) PEG lipid, (3) cholesterol, and (4) structural lipid (Figure 1a). The chemistry of ionizable lipids has been of primary interest due to its significance in transfection efficiency; however, the robustness of the LNP membrane is primarily associated with the other lipids.8,19 Specifically, cholesterol impacts the biophysical properties of unsaturated lipid membranes by increasing the rigidity of the bilayer.20 Besides, substituting cholesterol with its C-24 alkyl phytosterols or its oxidized derivatives in LNP enhances intracellular delivery of mRNA, suggesting the important roles of sterols in endosomal escape.21–24 PEG lipid prevents LNP from aggregation and improves particle stability by providing steric hindrance.25 Amount of PEG lipid in LNP is also related to the resulting particle size.25,26 As shown in recent studies, the presence of a dense PEG layer helps LNP to achieve pulmonary delivery of mRNA following nebulization.27,28 Furthermore, PEG density affects the Brownian motion of nanoparticles, thereby changing the particle mobility in mucus.29,30 Yet, the PEG layer inhibits the receptor-mediated endocytosis by reducing adsorption of serum protein and interferes with the endosomal escape of LNP, significantly restricting the intracellular delivery of mRNA.8,19,25 Therefore, successful pulmonary delivery of mRNA via LNP nebulization will require opposing particle characteristics: particle stabilization and mucus penetration are achievable by a dense PEG layer, whereas subcellular delivery of mRNA relies on a light PEG layer. To meet such criteria, we exploited β-sitosterol, a phytosterol facilitating LNP’s endosomal escape. We hypothesized that combinations of β-sitosterol and high PEG contents would permit nebulization, mucus penetration, and endosomal escape of LNP. In this study, we identified a LNP formulation compliant with nebulization by screening a small library and named nebulizable LNP (nLNP). This nLNP retained its physicochemical properties and efficiently delivered mRNA after nebulization. We consider nLNP delivering mRNA to the lung epithelia with high efficiency in a non-invasive and direct manner.
RESULTS/DISCUSSION
First, to test the influence of nebulization on LNP, we used the lipid composition of Onpattro® consisting of DLin-MC3-DMA, 1,2-dimyristoyl-rac-glycerol-methoxy(poly(ethylene glycol)-2000 (DMG-PEG2K), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) at 50/1.5/38.5/10 molar ratios, respectively (LNP-Chol/1.5). The dynamic light scattering (DLS) analysis showed that LNP-Chol/1.5 encapsulating Fluc mRNA became larger, aggregated, and unstable in nebulization (Figure 1c). To make LNP stabler and smaller for nebulization, we varied the amount of PEG lipid, ranging from 1.5% to 7.5% (LNP-Chol/1.5–7.5; Table S1, Supporting Information). Increasing PEG lipid from 1.5% to 3.5% (LNP-Chol/1.5 vs. LNP-Chol/3.5) led to a decrease in LNP size as shown in the previous study.31 However, LNP-Chol/5.5 and LNP-Chol/7.5 exhibited a relatively larger size and polydispersity index (PdI), suggestive of the destabilization of the LNP containing PEG lipid more than 5.5% (Figure 1d). After nebulization, LNP-Chol/1.5 exhibited the larger size and PdI than the LNP with more PEG lipid (Figure 1e), which corroborates that having high PEG contents helps LNP be more resistant to shear stress. mRNA encapsulation appeared to decrease in LNP-Chol/5.5, suggesting that having excessive PEG lipid might be detrimental to the RNA encapsulation (Figure 1f). Increasing PEG lipid in LNP did not appear cytotoxic to HeLa cells (Figure 1g) but it significantly deprived LNP of the ability to transfect cells (Figure 1h), which is likely due to the excessive PEG inhibiting the endocytosis.12,32
To overcome the loss of transfection derived from high PEG contents, we focused on cholesterol derivatives. Previously, we demonstrated that a simple substitution of cholesterol (Figure 2a) with β-sitosterol (Figure 2b) in LNP led to up to a 200-fold increase in mRNA transfection.21,22 Cholesterol derivatives carrying C24 alkyl variations were further shown to change LNP morphology and improve the LNP endosomal escape, resulting in greater mRNA transfection.22,23 Based on these findings, we hypothesized that a combination of β-sitosterol and high PEG contents could enable LNP to be stable enough for nebulization and penetrate mucosal barriers while effectively delivering mRNA to cells. To verify the effects of sterol substitution, LNP containing cholesterol (LNP-Chol/1.5) and β-sitosterol (LNP-Sito/1.5) were prepared and tested in an array of human cell lines, including several lung epithelial cell lines: A549, 16HBE14o-, and CFBE. As with the previous results, LNP-Sito/1.5 delivered Fluc mRNA more efficiently than LNP-Chol/1.5 in all cell lines tested (Figure S1, Supporting Information). To evaluate if the substitution of β-sitosterol in place of cholesterol in LNP could offset the loss of transfection received from high PEG contents, we tested the effects of the PEG contents in LNP-Chol and LNP-Sito, ranging from 1.5% to 4.5% of PEG lipid (Table S1, Supporting Information). It was shown that LNP size decreased in response to the increasing PEG contents (Figure 2c), and all LNP formulations displayed high mRNA encapsulations (> 90%) prior to nebulization (Figure 2d). After nebulization, LNP-Chol/1.5–4.5 showed modest increases in size and PdI. Nebulized LNP-Sito/1.5 and LNP-Sito/2.5 exhibited notable increases in size and PdI; however, LNP-Sito/3.5 and LNP-Sito/4.5 displayed similar values in size and PdI as compared to LNP-Chol/3.5 and LNP-Chol/4.5 (Figure 2e). It might indicate that the level of PEG lipid to inhibit LNP-Sito from aggregation during nebulization is around 3.5%. mRNA encapsulation within LNP decreased after nebulization (Figure 2f). It likely represents the leakage of the encapsulated mRNA to the outside of LNP caused by the rearrangement of the LNP structure during nebulization. Although all nebulized LNP exhibited lower than 50% RNA encapsulation, having relatively high PEG contents appeared to retain mRNA better within nebulized LNP during nebulization (Figure 2f). In vitro transfection assay in HeLa cells revealed that LNP-Chol and LNP-Sito both caused negligible cytotoxicity regardless of nebulization (Figure S2a,b, Supporting Information). In the following luciferase assay performed in HeLa cells, LNP-Sito showed significantly greater Fluc mRNA transfection than LNP-Chol in all various PEG lipid contents (Figure 2g). Both LNP-Chol and LNP-Sito exhibited decreasing luciferase expression when PEG lipid percentages increased, and LNP-Sito/1.5 led to the highest luciferase expression among all tested formulation (Figure 2g). Notably, mRNA transfection by nebulized LNP was different by showing that LNP-Sito containing relatively high PEG contents outperformed the one containing low PEG contents (Figure 2h). Luciferase expressions of nebulized LNP-Sito were constantly higher than those of nebulized LNP-Chol. Especially, LNP-Sito with 4.5% PEG resulted in a higher luciferase expression than all other LNP, 12-fold greater than LNP-Chol/1.5 (Onpattro® formulation; Figure 2h). We further tested the LNP library in two lung epithelial cell lines: 16HBE14o- and A549. Likewise, LNP-Sito showed higher transfection than LNP-Chol in both cells with decreasing potencies in response to increasing PEG lipid contents (Figure S3a,b, Supporting Information). After nebulization, LNP-Sito similarly produced higher luciferase expressions than LNP-Chol (Figure S4a,b, Supporting Information). Unlike HeLa cells, the effects of PEG lipids in the mRNA transfection of LNP-Sito were less noticeable in A549 and 16HBE14o- cells (Figure S4a,b, Supporting Information). Summarizing in vitro results, LNP-Sito containing high PEG lipid contents are capable of delivering mRNA to the cells effectively while preventing particles from aggregation.
Figure 2.
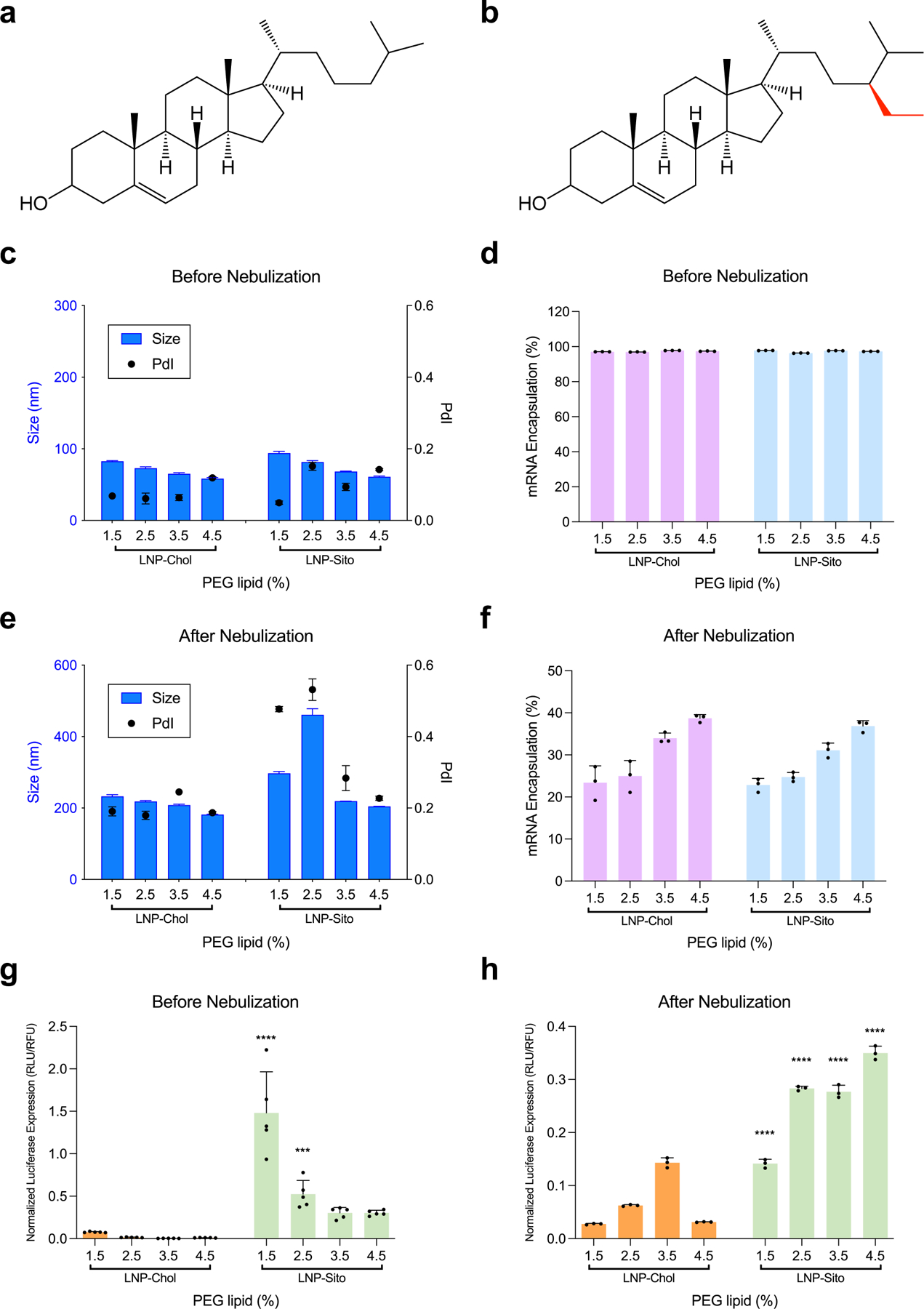
Enhanced mRNA transfection by substitution of cholesterol to β-sitosterol in LNP. (a,b) Chemical structures of (a) cholesterol and (b) β-sitosterol. β-sitosterol has an additional alkyl group (red) in comparison to cholesterol. (c-f) Physicochemical characterization of LNP-Chol and LNP-Sito containing various amounts of PEG lipid. (c,e) DLS analysis and (d,f) mRNA encapsulation of LNP (c,d) before and (e,f) after nebulization. (g,h) Normalized luciferase expression in HeLa cells transfected with (g) LNP solution and (h) LNP aerosol at 50 ng and 1 μg mRNA per well, respectively. Data were presented in mean ± standard deviation. ***p<0.001, ****p<0.0001; significant analysis by two-way ANOVA with Sidak’s multiple comparison test.
Next, we investigated whether varying PEG lipid contents affects LNP morphology using cryogenic transmission electron microscopy (cryoTEM). As with our previous study, 22 LNP-Chol produced uni-lamellar shapes, and LNP-Sito showed polymorphic, polyhedral, multi-lamellar shapes (Figure 3). Moreover, altering PEG lipid contents changed the particle size. Size comparison of LNP by cryoTEM and DLS showed a similar trend: increasing PEG lipid contents decreased the particle size (Table S2, Supporting Information). Both methods consistently revealed that, at the same concentration of PEG lipid incorporated, LNP-Chol were smaller than LNP-Sito. While the particle size varied depending on PEG lipid contents, polyhedral shapes of LNP-Sito remained. This size difference was more pronounced in cryoTEM results due to the low contrast of PEG layers in the micrographs (Table S2, Supporting Information).22 When PEG lipid was included at greater than or equal to 3.5%, LNP size became comparable between LNP-Chol and LNP-Sito (Table S2, Supporting Information). It was shown that anisotropy of LNP-Chol was higher than that of LNP-Sito, indicative of influence of sterol substitution on lipid membrane rigidity.22 Considering the different morphology, size, and membrane rigidity of LNP-Chol and LNP-Sito, these structural and morphological features may have led to the rearrangement of the nanoparticles at different degrees when exposed to nebulization, which could have contributed to the transfection.
Figure 3.

Cryogenic transmission electron microscopy (cryoTEM) imaging of LNP-Chol (left) and LNP-Sito (right) containing varying amounts of PEG lipids. Scale bars indicate 50 nm.
Having confirmed the potential advantages of combining β-sitosterol and a high PEG content for LNP nebulization, we continued evaluations in vivo. We used the aerosol exposure system connecting a rodent restraint to a vibrating mesh nebulizer via a spacer so that nebulized LNP can reach the mouse nose by laminar flow of medical-grade compressed air (Figure S5, Supporting Information). This system enables the effective administration of LNP at a relatively low dose compared to a whole-body nebulization chamber.33 Further, we used nanoluciferase (Nluc) mRNA instead of Fluc mRNA because Nluc emits brighter signals than Fluc, which increases the signal-to-noise ratio of bioluminescence upon mRNA transfection. The synthesized Nluc mRNA showed approximately 800 nucleotides in length under the agarose gel electrophoresis (Figure S6, Supporting Information). We nebulized LNP-Chol and LNP-Sito containing Nluc mRNA and various PEG lipid contents at a dose of 100 μg Nluc mRNA per mouse and measured luminescence in collected mouse lungs 24 h after nebulization (Figure S5, Supporting Information). Ex vivo imaging results proved that nebulized LNP transfected the lungs (Figure 4a). Of note, no other organs (including the liver) showed any luciferase expression after inhalation of nebulized LNP, indicating that LNP inhalation offers the selective transfection of the lungs (Figure 4a). Additionally, luciferase expression was detected in all five lung lobes, suggesting that nebulized LNP travel through airways evenly (Figure 4b,c). The difference in luciferase expression detected in each lung lobe was insignificant (p>0.5; Figure 4c). We further measured luminescent signals in the lung homogenates using a luminometer to assay luciferase transfection, followed by normalization against total protein concentrations (Figure S5, Supporting Information). As a result, inhalation of LNP-Sito led to higher luciferase expression than LNP-Chol at 1.5%, 2.5%, and 3.5% of the PEG lipid included (Figure 4d). Particularly, LNP-Sito produced significantly higher luciferase expressions than LNP-Chol when 1.5 and 3.5% PEG lipid were used (Figure 4d). LNP-Chol/4.5 and LNP-Sito/4.5 led to the comparable luciferase expressions. LNP-Chol exhibited relatively consistent despite the various amounts of PEG lipid (Figure 4d). Notably, inhalation of nebulized LNP-Sito/1.5 still produced a substantial protein expression in mouse lungs despite its destabilized and aggregated structure (Figure 2e and 4d). It is likely because the lack of mucus glands and a relatively large airway in murine lungs allowed the inhaled LNP with a large size and less PEG contents to avoid mucosal barriers and to reach the epithelial tissues.29,34
Figure 4.
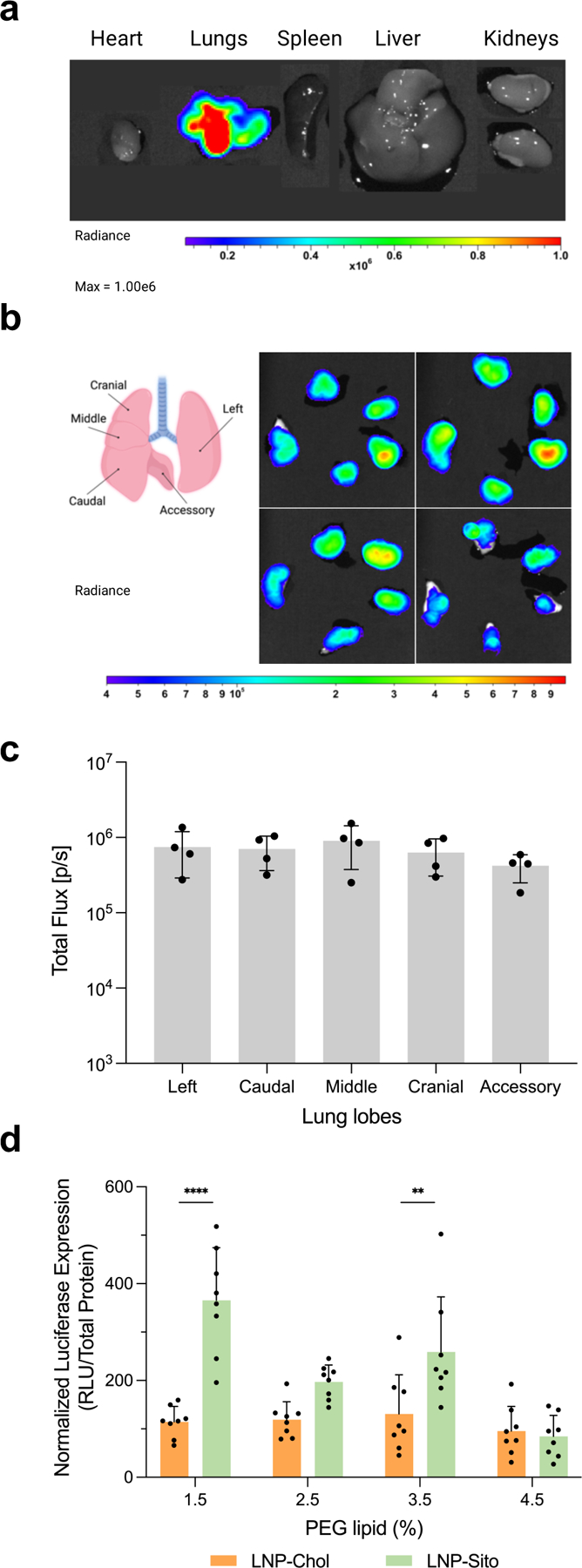
LNP inhalation produces selective lung transfection. (a) A representative bioluminescent image of isolated organs showing a localized luciferase expression in the lungs. (b) A diagram of mouse lung anatomy showing five lung lobes (left), and ex vivo bioluminescent images of mouse lung lobes 24 h after LNP inhalation (right, n=4). (c) Quantification of total flux in individual lung lobes harvested from BALB/c mouse at 24 h after LNP-Sito/1.5 inhalation at a dose of 100 μg mRNA (n=4). (d) Normalized Nluc expression in mouse lung homogenates harvested from BALB/c mice at 24 h after inhalation of LNP-Chol and LNP-Sito containing Nluc mRNA and various amount of PEG lipid. Data were presented in mean ± standard deviation. ****p<0.001, **p<0.01; significant analysis by two-way ANOVA with Sidak’s multiple comparison test (n=8).
To further optimize LNP for inhalation, we evaluated the nanoparticle mobility in a glycoprotein-rich condition. Airway mucus, a viscoelastic matrix in which water and mucin are the primary components, is secreted by secretory cells and submucosal glands.35 We used a porcine mucin suspension to mimic mucosal barriers against inhaled nanoparticles.29 To compare the diffusivity of LNP-Sito with different PEG-lipid contents in mucin suspension, we employed three-dimensional (3D) Single-Molecule Active Real-time Tracking (3D-SMART) to measure the diffusion of individual nanoparticles.36,37 Two formulations, LNP-Sito/1.5 and LNP-Sito/3.5, encapsulating Cy5-labelled EGFP-mRNA were added to a 10 mg/mL mucin suspension. 3D-SMART was used to capture the three-dimensional motion of individual nanoparticles within these mixtures for about 4 hours. Representative trajectories of LNP-Sito/1.5 and LNP-Sito/3.5 in 10 mg/mL mucin suspension were visualized (Figure 5a; Video S1, Supporting Information). Mean square displacement (MSD) analysis was performed on each trajectory and the diffusion coefficient and alpha were calculated for both formulations (LNP-Sito/1.5: n=323; LNP-Sito/3.5, n=353). The alpha values, which indicates the linearity of the MSD curve, were less than 1 for both formulations, but there was no significant difference between the two test groups (p = 0.34, Figure S7, Supporting Information). This indicates that both formulations showed a small amount of sub-diffusive character in mucin suspension, but that the degree of sub-diffusions is not affected by the concentration of PEG lipid. Despite the similar alpha values, analysis of the diffusion coefficients of individual particles in mucin suspensions revealed that a relatively small addition of PEG lipid contents to LNP dramatically expands the travel ranges of the nanoparticles in a mucin suspension (Figure 5b). Precisely, LNP-Sito/1.5 (1.69 ± 0.81 μm2/s) was significantly less diffusive than LNP-Sito/3.5 (3.11 ± 1.66 μm2/s, p<0.0001) in a mucin suspension (Figure 5c). This enhancement in diffusivity could be explained by the fact that the PEG reduces the affinity of the LNP for mucins,29 which could facilitate the delivery process across mucosal barriers. Next, we studied the impact of nebulization on LNP morphology (Figure S8 and Table S3, Supporting Information). CryoTEM imaging of nebulized LNP revealed that the numbers of particles on the grid were very low, indicating the disintegration of LNP during nebulization. LNP-Sito/1.5 and LNP-Sito/3.5 retained their polyhedral shapes after nebulization (Figure S8a,b, Supporting Information). Interestingly, some large particles displayed round shapes rather than polyhedral shapes, indicating the impact of nebulization on the particle shape of LNP (Figure S8a,b, Supporting Information). It might be due to the rearrangement of lipid membrane or the loss of β-sitosterol during nebulization. In addition, it was shown that nebulization increased the particle size of both LNP formulations (Figure S8a-c and Table S3, Supporting Information). As with the DLS data (Figure 2e), LNP-Sito/3.5 had a significantly smaller average particle size than LNP-Sito/1.5 (Figure S8c and Table S3, Supporting Information, p<0.0001), supporting that LNP-Sito/3.5 retained the original particle size better than LNP-Sito/1.5. Consequently, we selected LNP-Sito/3.5 as the optimized formulation for further inhalation studies based on its physicochemical properties, mucosal mobility, and in vivo efficacy, and named it nebulizable LNP (nLNP).
Figure 5.
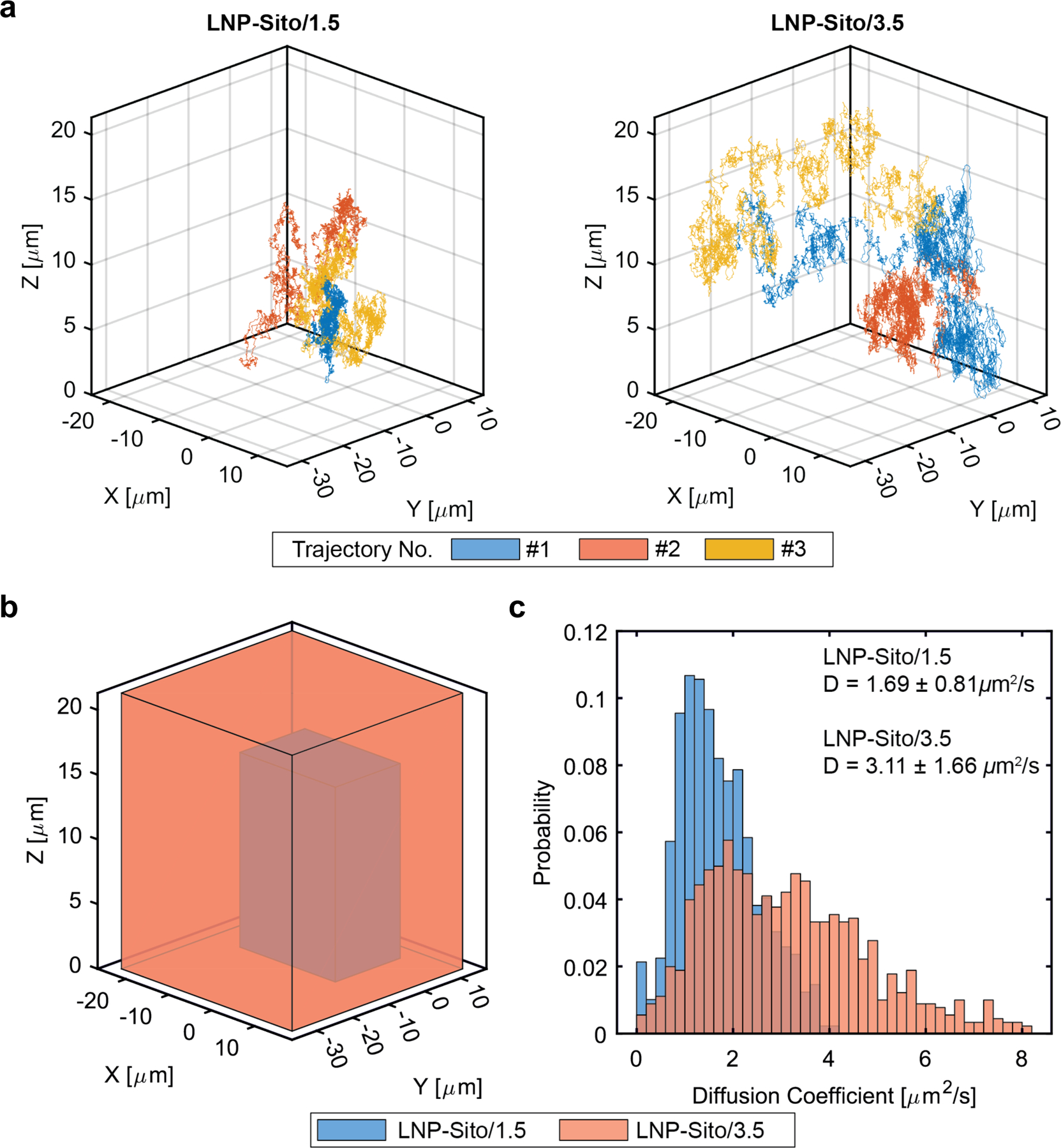
3D-SMART was performed for high-speed 3D particle tracking to capture nanoparticle diffusion. (a) Three 40 second-long representative trajectories of LNP-Sito/1.5 and LNP-Sito/3.5. The measured 3D position is plotted at 1 ms temporal resolution. (b) The boundary of the representative trajectories drawn as boxes, demonstrating the dramatically increased travel range of the LNP-Sito/3.5 (orange) compared to LNP-Sito/1.5 (blue). (c) Analysis of trajectories for LNP-Sito/1.5 (blue, n=323) and LNP-Sito/3.5 (orange, n=353), mean ± standard deviation. The two formulations show significant differences in diffusion coefficient (p<0.0001).
To determine the extent of mRNA delivery throughout the lungs using nLNP, we studied the tissue-specific gene expression in the lungs using Ai9 tdTomato reporter mouse that exhibits tdTomato protein expression after Cre-Lox recombination (Figure 6a). nLNP was prepared with encapsulating Cre mRNA and administered to Ai9 mice via inhalation, followed by five-day incubation for the site-specific recombination. In the lung sections of the Ai9 mice exposed to LNP inhalation, tdTomato expression was detected in alveoli and respiratory bronchioles, suggesting that inhaled LNP accumulate to a wide range of the bronchial tree. (Figure 6b,c). In the lung sections of untreated Ai9 mouse, tdTomato expression was insignificant (Figure S9a,b, Supporting Information).
Figure 6.
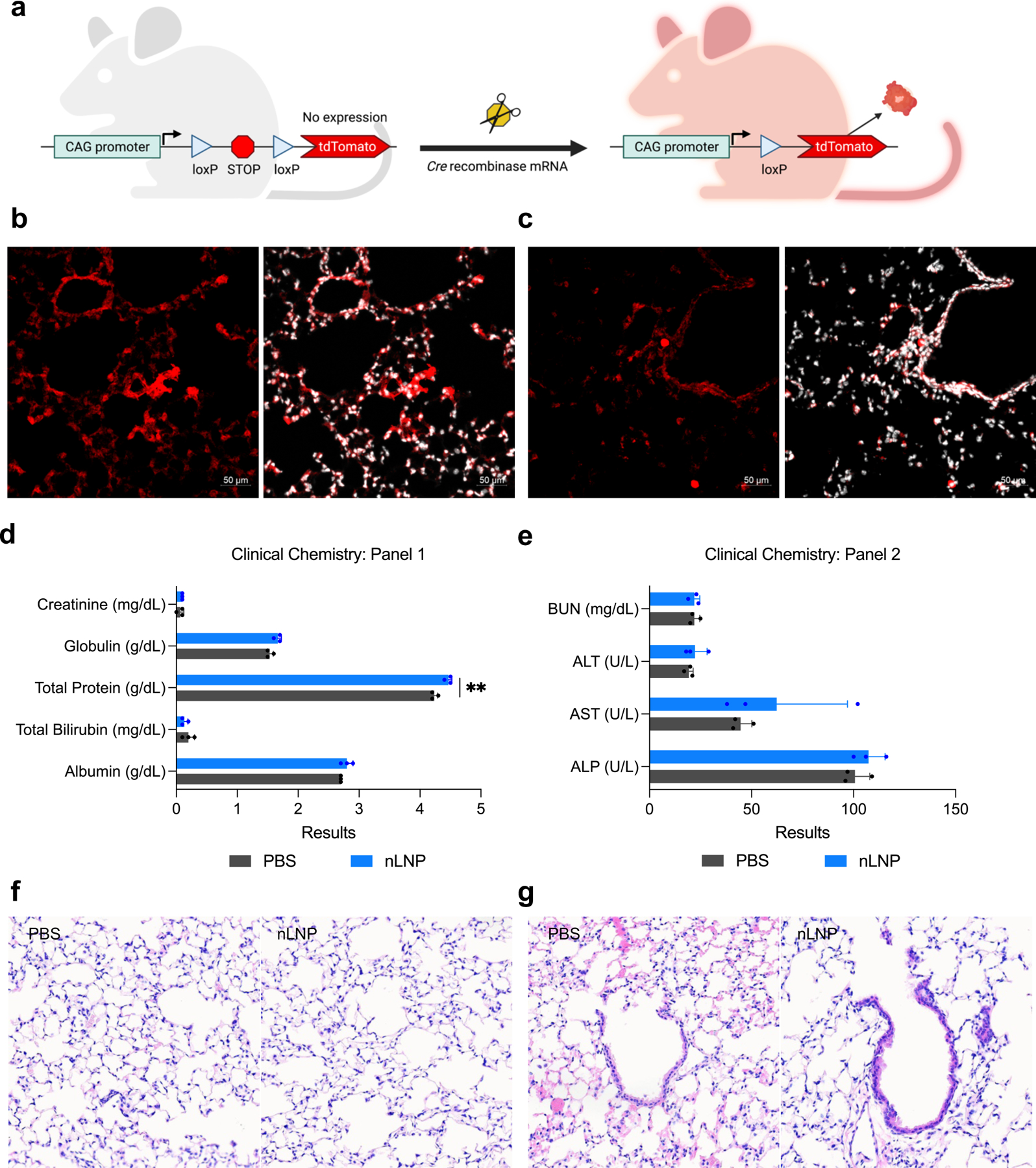
Inhaled nLNP transfects airway epithelium without causing acute toxicity. (a) A diagram of Cre-lox recombination in Ai9 transgenic Cre reporter mouse that expresses tdTomato when loxP recombination has occurred. (b-c) Representative immunohistochemistry of Ai9 mouse lung sections displaying tdTomato expression after inhalation of nLNP encapsulating Cre mRNA. tdTomato (red) and nuclei (white) were detected in (b) alveolar spaces and (e) airway bronchioles. 20x magnification. Scale bars refer to 50 μm. (d,e) Clinical chemistry test of mouse sera harvested at 24 h after PBS or nLNP inhalation. Data were presented in mean ± standard deviation. **p<0.01, significant analysis by multiple unpaired t-test (n=3). (f,g) Histopathological analysis of (f) alveolar spaces and (g) bronchial epithelium of murine lungs harvested at 24 h after PBS (left) or nLNP (right) inhalation. Hematoxylin and eosin; 20x magnification. Arrows indicate the bronchial epithelium.
Next, we tested acute adverse effects of nLNP inhalation to BALB/c mice using clinical chemistry and histopathology. In the clinical chemistry test, the overall results of the LNP-treated group were similar to those of the PBS-treated group, suggesting the safety of inhalation nLNP (Figure 6d,e and Table S4, Supporting Information). Total protein concentration was significantly higher in nLNP-treated group than PBS-treated group; however, the mean value was still within the normal range of BALB/c mouse (Figure 6d and Table S4, Supporting Information).38 The elevation of blood aspartate aminotransferase (AST) of nLNP-treated group was insignificant when compared to the level of PBS-treated group (p = 0.4; Figure 6e and Table S4, Supporting Information). We further evaluated the potential acute lung damage after nLNP inhalation by histopathology (Figure 6f,g). In the histopathological analysis of the lung sections exposed to PBS or nLNP, the lungs from both groups were mildly atelectatic (Figure S10 and Table S5, Supporting Information). However, no significant difference was observed between the two groups, indicating that the abnormalities are artefactual findings and not treatment-related (Figure 6f,g and Table S5, Supporting Information).
The transient nature of mRNA therapeutic allows for the controllable expression of the target protein; however, persistent administration is necessary to maintain the protein expression within the therapeutic window. Accordingly, we tested whether the optimized LNP can be given repeatedly (Figure 7a). Throughout three measurements of luciferase expression post-inhalation, the Nluc expression levels were maintained (Figure 7b and Figure S11a, b, Supporting Information). The body weight changes of the animals were also insignificant during the study (Figure 7c). Based on these results, we determined that single- or repeated-inhalation of nLNP did not pose any risk of acute toxicity in vivo.
Figure 7.
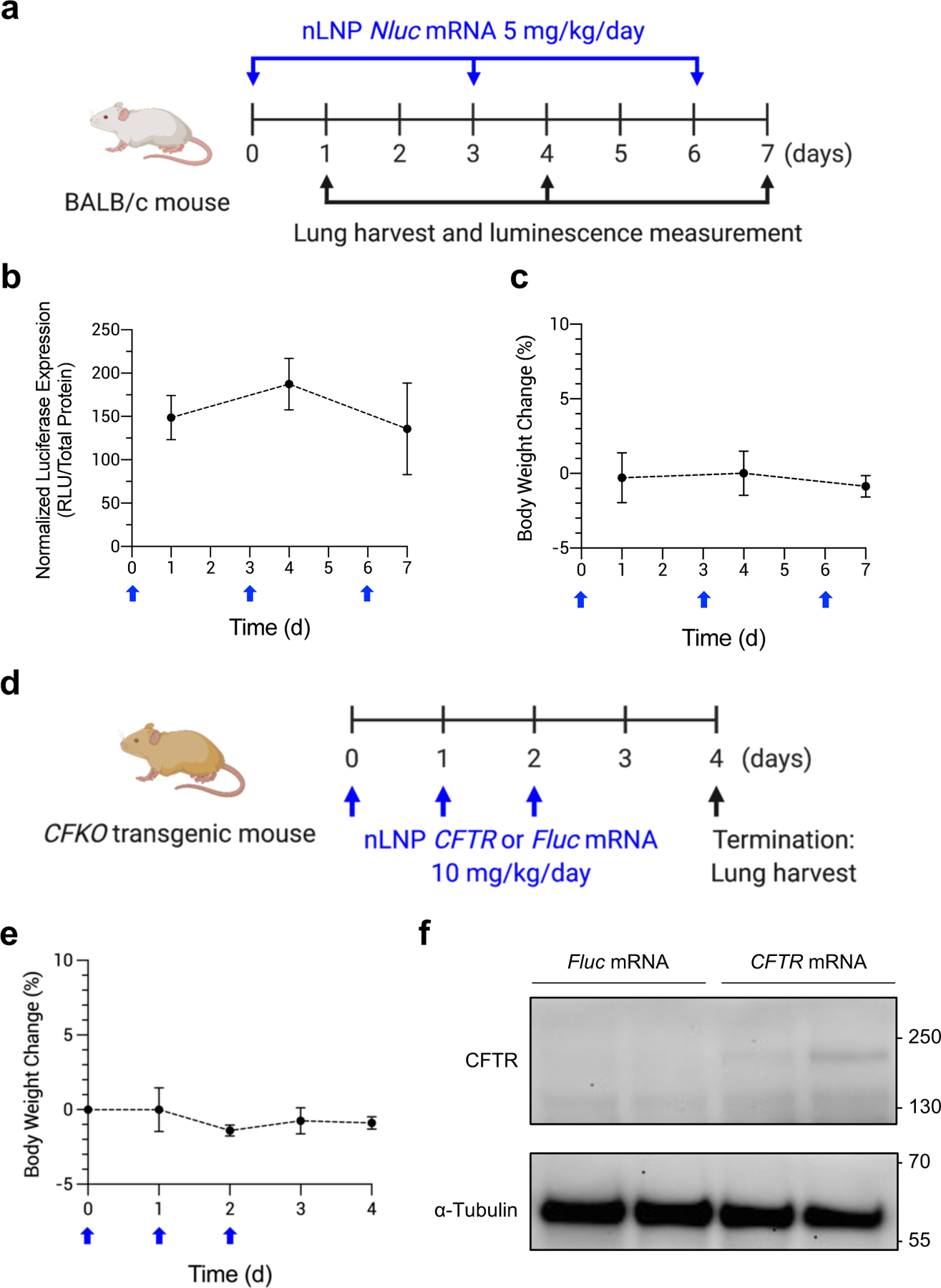
Repeated inhalation of nLNP to sustain production of therapeutic protein. (a) A dosing regimen for persistent inhalation of nLNP encapsulating Nluc mRNA. nLNP was administered via inhalation every 3 days at a dose of 5 mg/kg/day mRNA (blue arrows). At 24 h after each dose, mouse lungs were collected to measure luciferase expression (black arrows). (b) Normalized luciferase expression in mouse lungs and (c) body weight change of BALB/c after repeat dosing (blue arrow). Data were presented in mean ± standard deviation (n=4–12). (d) A dosing regimen for CFTR mRNA delivery via inhalation. nLNP encapsulating CFTR or Fluc mRNA was administered to CFKO mice daily for 3 d (blue arrows), followed by harvesting lungs to detect CFTR proteins (black arrow). (e) Body weight change of CFKO transgenic mice after repeat dosing (blue arrow). (f) Western blot images after immunoprecipitation using an anti-CFTR antibody. mRNA delivered by nLNP is noted above the images. Upper and lower blots were probed using anti-CFTR and anti-α-Tubulin antibodies, respectively. Approximate molecular weights are marked on the left sides of the images.
Lastly, we delivered human CFTR (hCFTR) mRNA using nLNP to test whether nLNP can deliver therapeutic mRNA (Figure 7d). To study the expression of hCFTR protein, we utilized a well-characterized bitransgenic CFKO mouse model. This mouse model is fully knocked out for the endogenous mouse CFTR. Three doses of mRNA were given to the CFKO mouse model via inhalation, followed by collecting lungs to detect hCFTR. During the treatment, we did not observe any significant change in the body weight of CFKO mice (Figure 7e). An immunoprecipitation (IP) approach was performed to examine the presence of hCFTR protein in the lungs of CFKO mice after mRNA inhalation. The IP revealed the presence of hCFTR protein at approximately 170 kDa in the precipitated samples of the CFKO mice treated with CFTR mRNA inhalation (Figure 7f). In the precipitated samples of the CFKO mice treated with Fluc mRNA inhalation, the expression of hCFTR protein was not detected (Figure 7f). α-Tubulin bands were detected in the flow-through samples of both groups (Figure 7f). These data indicated that hCFTR mRNA delivered by nLNP produced hCFTR protein in the mouse lungs. Non-specific bands were also found in the blots of the precipitated samples (Figure S12a,b, Supporting Information). Considering that the same bands were observed when we conducted the IP with PBS, they seemed to be the anti-CFTR antibody and its heavy and light chains (Figure S12b, Supporting Information). Taken together, our results support that CFTR mRNA could be delivered using inhalation of nLNP to the lungs for the potential treatment of CF.
CONCLUSIONS
Extrahepatic delivery of therapeutic mRNA via LNP enables the treatment of diseases unrelated to the liver. The lungs are associated with various diseases, including inherited genetic disorders, cancers, and infectious diseases. Nevertheless, pulmonary delivery of mRNA is a difficult task since many nanoparticles preferentially accumulate to the liver when administered systemically. Furthermore, lipid-based vectors are particularly challenging to move beyond the liver because of strong interactions with apolipoprotein E (ApoE), leading to the low-density-lipoprotein (LDL) receptor-mediated cellular uptake to the hepatocytes.12 Therefore, inhalation route has been of interest for pulmonary delivery of mRNA. A recent study demonstrated that mRNA could be inhaled by complexing with cationic polymers for transfecting lung epithelium using a nebulizer.33 Polymeric nanoparticles have the advantage of self-assembling with mRNA, which could be advantageous to recover nanostructures after being nebulized. However, it is challenging to synthesize monodispersed polymers on an industrial scale, and the positive charges in cationic polymers often lead to cytotoxicity.8,39 In contrast, LNP are considered more potent and tolerant than polymeric nanoparticles.8 Additionally, the manufacturing capabilities of mRNA vaccines will also enable the large-scale production of LNP-based mRNA therapeutics. To date, LNP chemistry has been optimized mainly for intravenous or intramuscular administrations. However, it is not readily translatable for inhalation due to the different requirements for compatibility with inhalation devices and the unique characteristics of the respiratory system, such as its anatomical structure and the existence of mucosal barriers. Therefore, the formulation criteria for LNP inhalation are likely different to those for other administration routes.
This study has explored the design criteria for LNP nebulization with the clinically relevant ionizable lipid, DLin-MC3-DMA. Consistent with the recent studies,27,28 we found that the molar percentage of PEG lipid among lipid components is critical to control the LNP size after nebulization. However, we found that excess PEG lipid is disadvantageous for mRNA transfection. We reasoned that dense PEG would inhibit the formation of biomolecular corona on the nanoparticles, thus negatively affecting the cellular entry of nanoparticles. As ApoE assists the LNP uptake in the systemic circulation, potential facilitators could exist in the airway surface liquid for LNP uptake. In the pulmonary system, alveolar macrophages abundantly produce ApoE, and lung epithelial cells express LDL receptors.40,41 Therefore, if ApoE assists LNP uptake in the lungs in the same way it does in the systemic circulation, the restricted interactions between heavily PEGylated LNP and ApoE would diminish the cellular uptake of LNP and the resulting mRNA transfection. To overcome this double-edged effect of PEG lipid in LNP nebulization, we have explored a small library of LNP formulations and identified the optimized LNP (nLNP) for inhalation-mediated mRNA delivery to the lungs. nLNP contains β-sitosterol and high PEG lipid contents, and it was resilient to shear stresses during nebulization. In addition, 3D-SMART revealed that nLNP displayed the superior diffusivity in mucin suspension at single-nanoparticle resolution. This feature could be to treat the lung diseases characterized with hypersecretion and impaired clearance of sputum; for example, asthma, chronic obstructive pulmonary disease, and cystic fibrosis.35,42 Considering that airway mucus is a crucial factor stopping LNP from transfecting epithelial cells, LNP having high diffusivity would enhance the chance of traversing mucosal barriers and reaching airway epithelium. Inhaled nLNP also provided effective mRNA transfection in mouse epithelial cells without off-target transfection in the liver. Furthermore, it did not induce any significant change in serum toxicology tests as well as histopathology, supporting its safety.
In summary, our work proposes the LNP design for nebulization and inhalation-mediated mRNA transfection; yet there is room for improvement. First, given that mucus penetration of nanoparticle is important for successful mRNA delivery to the lung epithelium, utilization of animal models that have thick mucus will be helpful to better estimate the trafficking of LNP across mucosal barriers. For example, the β-epithelial sodium channel overexpressing mouse and the ovalbumin-sensitized mouse exhibit the mucus plugging in their lungs.43 CF pigs and ferrets not only exhibit mucus plugging in the airways but have pulmonary anatomies closer to humans.44 Alternatively, nonhuman primates have similar pulmonary anatomies to humans, such as overall lung masses and branching of conducting airways.45 The lack of these anatomical and physiological features in murine models that were used in this study might have contributed to the dilute benefits of LNP optimization for mRNA inhalation. Second, the immune systems are an integral factor for successful mRNA delivery to the pulmonary system. Diseased lungs generally accompany the pulmonary infiltration of immune cells due to inflammations.46–48 Considering that immune cells are responsible for removing nanoparticles, the stimulated immune system in the diseased lungs is likely to alter the nanoparticle clearance. Additionally, potential advantages of our LNP optimization might be explained better where RNase secretion is stimulated by pulmonary inflammation.49 Last, to demonstrate functional rescue of CF, physiological changes including but not limited to chloride ion efflux, sodium ion absorption, and airway surface liquid are necessary after CFTR mRNA inhalation.3,6 With the functional correction of CFTR, CFTR mRNA would be considered translatable as a CF treatment. Despite these questions to be addressed, our results show the importance of PEG lipid concentration on nebulizing LNP and delivering mRNA to the lungs and the cholesterol substitution to enhance mRNA transfection in vivo. Further, with increasing interests in pulmonary vaccination, this study may help to develop intranasal mRNA vaccines. We hope that our strategy helps to shed light on designing the next generation inhalable LNP for pulmonary gene delivery.
METHODS/EXPERIMENTAL
Materials:
Fluc mRNA, Cre mRNA, EGFP mRNA, Cy5-labelled EGFP, and hCFTR mRNA were purchased from TriLink Biotechnologies (CA, USA). Uridine of Fluc, Cre, EGFP, and Cy5-labelled EGFP mRNA was fully substituted with 5-methoxyuridine. Uridine and cytidine of hCFTR mRNA was fully substituted with pseudouridine and 5-methylcytidine, respectively. Cholesterol and β-sitosterol were purchased from Sigma-Aldrich (MO, USA). DMG-PEG2K was bought from NOF America. DLin-MC3-DMA and DSPC were obtained from BioFine International Inc. and Avanti Polar Lipids, Inc., respectively.
LNP formulation and characterization:
LNP composed of DLin-MC3-DMA, Cholesterol or β-sitosterol, DMG-PEG2K, DSPC, and mRNA were prepared using microfluidic mixing as described.50 Briefly, mRNA was diluted in sterile 50 mM citrate buffer, and lipid components were prepared at 5.5 mM in 100% ethanol at a 50:38.5:1.5:10 molar ratio. The lipid and mRNA solutions were mixed using the NanoAssemblr Benchtop at a 1:3 ratio, followed by overnight dialysis against sterile PBS using a Slide-A-Lyzer G2 cassette with 10,000 Da molecular-weight-cut-off (Thermo Fisher, MA). Dialyzed LNP solutions were concentrated using Amicon® Ultra centrifugal filter units with 10,000 Da molecular-weight cut-off (Millipore). Hydrodynamic size and PDI of the LNP were measured in dynamic light scattering using the Zetasizer Nano ZSP (Malvern Instruments). mRNA encapsulation was assayed using a Quant-iT™ RiboGreen® RNA Assay kit (Thermo Fisher, MA) and a multimode microplate reader (Tecan Trading AG, Switzerland).
Cell culture
HeLa and A549 were kindly gifted from Prof. Robert Langer at MIT and Prof. Adam Alani at OSU. 16HBE14o- and CFBE41o- cells were kindly provided by Prof. Kelvin MacDonald at OHSU. HEK293T/17 was purchased from ATCC. SW480 and HT-29 were kindly provided by Prof. Melissa Wong at OHSU. HeLa and A549 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin (Thermo Fisher, MA). SW480 and HT-29 cells were cultured according to the ATCC culture protocols. 16HBE14o- and CFBE41o- cells were cultured in MEM supplemented with 10% heat-inactivated FBS, and 1% penicillin/streptomycin/glutamine (Thermo Fisher, MA).
In vitro Fluc mRNA transfection assay
For in vitro Fluc mRNA transfection assays, cells were seeded on a white 96 well plate at 4×103 cells/well, followed by overnight incubation for cell attachment. Cells were incubated with LNP encapsulating Fluc mRNA for 24 h and analyzed for cell viability and luciferase activity with the ONE-Glo™+Tox luciferase reporter and cell viability assay kit (Promega) using a multimode microplate reader (Tecan Trading AG, Switzerland).
LNP nebulization
Nebulization of LNP was performed as described.50 In brief, an Aeroneb® Lab Control Module equipped with a lab nebulizer unit (Aerogen, Ireland) was used to nebulize LNP. LNP solutions were added to the nebulizer unit, and the nebulized LNP were collected for analysis. For in vitro mRNA transfection, cells were seeded onto a 12 well plate at 5 × 104 cells/well and allowed to attach for overnight incubation. LNP were diluted with serum-free media and nebulized with a small nebulizer unit directly onto the seeded cells (1 μg/well), followed by 24-hour incubation. Cell viability and in vitro luciferase expression was assayed using ONE-Glo™+Tox luciferase reporter and cell viability assay kit (Promega) and a multimode microplate reader (Tecan Trading AG, Switzerland).
In vitro transcription of mRNA
A linearized plasmid containing nanoluciferase (Nluc) under the T7 promoter was used as a template for in vitro transcription. Nluc mRNA was synthesized using the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs Inc., MA) and CleanCap® Reagent AG (TriLink Biotechnologies, CA) according to the manufacturer’s instructions. Synthesized mRNA was purified using the Monarch® RNA Cleanup Kit (New England Biolabs) and stored at -80°C. Concentration of Nluc mRNA was measured using a multimode microplate reader (Tecan Trading AG, Switzerland). For agarose gel electrophoresis, 1 μg of IVT mRNA or RiboRuler high range RNA ladder (Thermo) were denatured and loaded on 1.5 % agarose-formaldehyde gel prestained with GelRed (Biotium, CA). The gel was run at 85V for 2 h, followed by UV visualization.
Animals
All animal studies were conducted at Oregon Health and Sciences University and approved by the Institutional Animal Care and Use Committee (IACUC, IP00001707). Female BALB/c mice were purchased from Charles River Laboratories (MA, USA). B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Ai9) mice were purchased from the Jackson Laboratory (JAX, 007909). Cftr−/−tm1Unc Tg(FABPCFTR)1Jaw/J bitransgenic CFKO mice were purchased from The Jackson Laboratory (JAX 002364). Mice exhibited full knockout of the endogenous mouse Cftr. To avoid intestinal complications, hCFTR is locally expressed under the rat fatty acid binding protein 2, intestinal gene promoter.
Pulmonary Nluc mRNA transfection by LNP inhalation
To assay pulmonary Nluc mRNA transfection in mice, a mouse nebulizer delivery system (Kent Scientific) was used. In brief, BALB/c mice (5–10 week old) were restrained in a tube rodent holder. The tube holder is attached to a plastic spacer connecting a nebulizer unit and airflow at 1 LPM (liter per minute). LNP solution was prepared at 0.5 mg mRNA per ml and added into the nebulizer unit at a rate of 25 μl per minute using a syringe pump. After 24 hours post-inhalation, mouse lungs were harvested, briefly washed with sterile PBS, and incubated in Nano-Glo® substrate (Promega) diluted 40-fold in PBS for 5 min at room temperature, followed by ex vivo bioluminescence imaging in IVIS® Lumina XRMS (PerkinElmer). After imaging, lungs were homogenized with sterile PBS and centrifuged at 17,000 g for 30 min at 4°C to collect supernatants. 30 μl of the supernatants was incubated with 60 μl of Nano-Glo® substrate diluted 80-fold in PBS for 5 min at room temperature in a white 96 well plate for luminescent detection using a multimode microplate reader (Tecan Trading AG). Total protein concentration in the samples was measured using Micro BCA™ protein assay kit (Thermo Fisher, MA). Luminescence values were normalized by the total protein concentration of the supernatants.
Cryogenic transmission electron microscopy (cryoTEM)
CryoTEM acquisition was performed on Glacios cryoelectron microscope equipped with Gatan K3 camera at 200 kV. 2 μl of the sample was dispensed on a plasma cleaned grid in the FEI Vitrobot chamber at 100% relative humidity and allowed to rest for 10 seconds. Then, the grid was blotted for 1 seconds with filter paper and plunged into liquid ethane cooled by liquid nitrogen. The frozen grids were then checked for visible defects and assembled into cassettes. The collected images were then processed and analyzed manually using Fiji. Each LNP population contained at least 35 particles. Only unobstructed LNP with clearly defined edges were included in the statistical analysis to ensure accurate representation of LNP populations and to avoid issues associated with ice thickness. The polydispersity index (PdI) was calculated as PdI =(σ/μ)2, where σ - standard deviation, μ -mean of particle size.
High-temporal resolution single particle tracking
Three-dimensional trajectories of LNPs were collected using an active feedback tracking microscopy, 3D single-molecule active real-time tracking (3D-SMART). A 640nm excitation laser (OBIS 640LX, Coherent, ~76 nW at the focus) is focused exclusively onto the sample through an objective lens (Zeiss Plan Apo, 100×, NA=1.49) and is deflected in a 1μm×1μm×2μm 3D pattern by a pair of electro-optic deflectors (EODs; M310A, ConOptics) and a tunable acoustic gradient lens (TAG Lens 2.5, TAG Optics). Fluorescence photons from the moving probe are collected by the objective lens and are focused through a bandpass filter (ET706/95m, Chroma) onto a single-photon counting avalanche photodiode (Excelitas SPCM-ARQH-15). The photon arrival time is used to calculate the real-time 3D position of the moving probe within the laser scan using a field programmable gate array (NI-7852R) with a Kalman filter. The real-time position is then used to drive a galvo mirror (SG7220-A, Sino Galvo) and a piezoelectric nanopositioner (Z-Nano-OP65HS, MadCity Labs) to center the target in focal volume. All trajectories shown are sampled every 1 millisecond. To collect more trajectories and avoid uninterested stuck particles, the maximum trajectory time was set to 120 seconds, i.e., tracking will be automatically disengaged after collecting data for 120 seconds followed with a fresh round of searching process. Mucin suspensions were prepared as described.29 Briefly, native mucin from bovine submaxillary gland (Sigma-Aldrich, MO) was dissolved in distilled water at 10 mg/ml, followed by centrifugation at 10,000 g for 30 min to separate undissolved parts. The mucin-containing supernatant was lyophilized and resuspended in distilled water at 10 mg/ml. To quantify the diffusion, mean square displacement (MSD) analysis was applied to every trajectory longer than 5 seconds in duration. To keep consistent analysis, trajectories longer than 5 seconds in duration were broken up into 5 second segments, with a maximum of three segments (15 seconds) used for all trajectories. Diffusion coefficients extracted from different segments on the same particle were average together. The MSD was calculated as follows:
Here, τ is the lag time. x(n), y(n), and z(n) are the coordinates of the trajectory at timepoint n. N is the total number of data points, 5000 for all segments analyzed in the present work. When available, the 95% confidence interval of the diffusion coefficient was calculated from the linear fit of MSD(τ) using the MATLAB (MathWorks) function confint. The alpha was calculated from the power law fit of MSD(τ).
To clean up the data, outliers were removed using the MATLAB function rmoutliers, in which an outlier is defined as a value more than three scaled median absolute deviations (MAD) away from the median. This acted to remove immobilized particles that were stuck to the coverslip during sample preparation.
Immunohistochemistry (IHC)
Ai9 mice (8–12 week old) were exposed to nebulized LNP encapsulating Cre recombinase mRNA for two days at a dose of 100 μg mRNA per day (3 mice per group). After 5 days, mice sedated by isoflurane were euthanized by cardiac puncture, and their hearts were perfused with sterile PBS through the right ventricle. Through the catheter inserted to the trachea, 4% paraformaldehyde (PFA) in PBS solution were instilled at 25 cm above the level of mouse to inflate the mouse lungs. After overnight incubation in 4% PFA, the inflated lungs were stored at 30% sucrose/PBS solution at 4°C until the lungs sunk to the bottom of tubes. The lungs were embedded in the optimal cutting temperature (O.C.T) compound (Sakura Finetek USA, Inc., CA) and frozen using liquid nitrogen. The frozen sections were cut with 10 μm thickness on a cryostat. The sectioned tissues were permeabilized using 5% Triton X-200 in PBS for 10 min and blocked with 5% donkey serum in PBS for 1 h. A primary antibody consisting of an anti-RFP antibody (1:100, rabbit, ab62341) in PBS containing 1% donkey serum covered the sections overnight at 4°C. The next day, the sections were washed with PBS and incubated in the secondary antibody (Donkey anti-rabbit Alexa Fluor Plus 647, 1:200, A32795, Thermo) in PBS containing 1% donkey serum for 1 h at RT. The sections were washed with PBS, followed by nucleus staining and mounting with coverslips. Negative controls include the Cre mRNA treated lung sections stained with secondary antibody only and the untreated lung sections fully stained. Confocal images were obtained with the ZEISS LSM 880 (Carl Zeiss AG). Z-stacks (spanned 10 μm with 1.08 μm interval) were collected using a 20X objective, and maximum intensity projections were reported.
Histopathology and serum chemistry
BALB/c mice were exposed to nebulized PBS or LNP. After 24 h, mice were euthanized, and the whole blood was collected by cardiac puncture. Serum was extracted from the whole blood samples using serum-separating tubes (BD). Afterward, mouse lungs were perfused with sterile PBS from the right ventricle. 20G catheter was inserted into the trachea to inflate lungs with 10% neutral buffered formalin (Fisher Scientific) at a pressure of 25 cm from the surgical plane. The inflated lungs were harvested and kept in formalin 24 h. In the following days, the lungs were dissected, placed in tissue embedding cassettes, and submerged in 70% ethanol for dehydration. Tissues were paraffin embedded, sectioned, mounted on slides, routinely stained with hematoxylin and eosin, and cover-slipped allowing histopathologic evaluation by IDEXX BioAnalytics. Microscopic changes were graded as to severity utilizing a standard grading system whereby 0 = no significant change, 1 = minimal, 2 = mild, 3 = moderate, and 4 = severe. International Harmonization of Nomenclature and Diagnostic (INHAND) Criteria standards are used as the basis of evaluation (https://www.toxpath.org/inhand.asp). Use of numerical grades allows a mechanism to calculate a total score lesion score which can be used to assess prevalence and severity of tissue changes within and between groups. Mouse sera were proceeded to clinical chemistry test by IDEXX BioAnalytics.
Pulmonary CFTR mRNA transfection by LNP inhalation
CFKO mice were exposed to nebulized LNP encapsulating Fluc or CFTR mRNA at a dose of 10 mg/kg/day for three days. At 48 hours after the last administration, mice were properly euthanized, and lungs were collected. The lungs were homogenized with sterile PBS and centrifuged at 17,000 g for 30 min at 4°C to collect supernatants, followed by supplementing protease and phosphatase inhibitor cocktail (Thermo Fisher, MA). Total protein concentration in the samples was measured using BCA™ protein assay kit (Thermo Fisher, MA). To detect CFTR protein, immunoprecipitation was performed. CFTR antibodies for pull-down and the following Western blot were an anti-CFTR mouse monoclonal antibody (596, Cystic Fibrosis Antibodies Distribution Program). Briefly, Dynabeads™ Protein G (Thermo Fisher, MA) were incubated with anti-CFTR antibodies at room temperature with rotation. The collected lung lysates containing 200 μg of total protein were incubated with the beads having anti-CFTR antibody with rotation. Following three washes with wash buffer, samples were eluted using elution buffer, denatured using LDS sample buffer and reducing agent at 37°C, and run on 8% Bis-Tris gels, followed by wet transfer to nitrocellulose membrane. The blots were blocked using 5% skim milk for 1 h at room temperature. The primary antibodies used were anti-CFTR rabbit polyclonal antibody (HPA021939, Sigma-Aldrich, MO) at 1:1,000 and anti-α-Tubulin at 1:1,000 (Cell Signaling Technology, #2125). The secondary antibody was goat polyclonal anti-rabbit HRP (Jackson ImmunoResearch, 111-035-003) at 1:2,000 and 1:5,000 for CFTR and α-Tubulin, respectively. For detection and documentation, SuperSignal™ West Pico Plus Chemiluminescent Substrate and myECL imager were used (Thermo Fisher, MA). The Western blot images were analyzed using ImageJ (Version: 2.0.0).
Statistical Analysis
Statistical analysis was performed using Prism 9 software (GraphPad, CA, USA).
Supplementary Material
ACKNOWLEDGMENT
We thank the Advanced Light Microscopy Core in OHSU for the help with slide imaging. Some schematics were created with BioRender.com.
Funding Sources
This project was supported through funding from the National Heart Lung and Blood Institute (NHLBI) 1R01HL146736-01 (G.S) and Cystic Fibrosis Foundation - SAHAY 19XX0.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supporting figures, lipid nanoparticle composition and characterization, and additional data demonstrating the efficacy of lipid nanoparticles in vitro and in vivo.
Supporting video, representative trajectories of LNP-Sito with 1.5% and 3.5% PEG lipid.
The authors declare the following competing financial interest(s): G.S. is an inventor in patent application US20200129445A1 that details LNP-Sito. G.S and J.K have submitted a US provisional patent application (No. 63/225,766) related to this work. G.S. is a co-founder of EnterX Bio and RNAvax Bio, and has an advisory role to Saliogen Therapeutics Inc., Rare Air Inc., Circ Bio Inc., and Sanofi.
REFERENCES
- (1).O’Sullivan BP; Freedman SD Cystic Fibrosis. The Lancet 2009, 373, 1891–1904. 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- (2).De Boeck K; Amaral MD Progress in Therapies for Cystic Fibrosis. Lancet Respir. Med 2016, 4, 662–674. 10.1016/S2213-2600(16)00023-0. [DOI] [PubMed] [Google Scholar]
- (3).Mukherjee A; MacDonald KD; Kim J; Henderson MI; Eygeris Y; Sahay G Engineered Mutant α-ENaC Subunit MRNA Delivered by Lipid Nanoparticles Reduces Amiloride Currents in Cystic Fibrosis–Based Cell and Mice Models. Sci. Adv 2020, 6, eabc5911. 10.1126/sciadv.abc5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Haque AKMA; Dewerth A; Antony JS; Riethmüller J; Schweizer GR; Weinmann P; Latifi N; Yasar H; Pedemonte N; Sondo E; Weidensee B; Ralhan A; Laval J; Schlegel P; Seitz C; Loretz B; Lehr C-M; Handgretinger R; Kormann MSD Chemically Modified HCFTR MRNAs Recuperate Lung Function in a Mouse Model of Cystic Fibrosis. Sci. Rep 2018, 8, 16776. 10.1038/s41598-018-34960-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Suzuki S; Crane AM; Anirudhan V; Barillà C; Matthias N; Randell SH; Rab A; Sorscher EJ; Kerschner JL; Yin S; Harris A; Mendel M; Kim K; Zhang L; Conway A; Davis BR Highly Efficient Gene Editing of Cystic Fibrosis Patient-Derived Airway Basal Cells Results in Functional CFTR Correction. Mol. Ther 2020, 28, 1684–1695. 10.1016/j.ymthe.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Robinson E; MacDonald KD; Slaughter K; McKinney M; Patel S; Sun C; Sahay G Lipid Nanoparticle-Delivered Chemically Modified MRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther 2018, 26, 2034–2046. 10.1016/j.ymthe.2018.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yin H; Kanasty RL; Eltoukhy AA; Vegas AJ; Dorkin JR; Anderson DG Non-Viral Vectors for Gene-Based Therapy. Nat. Rev. Genet 2014, 15, 541–555. 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- (8).Kim J; Eygeris Y; Gupta M; Sahay G Self-Assembled MRNA Vaccines. Adv. Drug Deliv. Rev 2021, 170, 83–112. 10.1016/j.addr.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Karikó K; Muramatsu H; Keller JM; Weissman D Increased Erythropoiesis in Mice Injected With Submicrogram Quantities of Pseudouridine-Containing MRNA Encoding Erythropoietin. Mol. Ther 2012, 20, 948–953. 10.1038/mt.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jiang L; Berraondo P; Jericó D; Guey LT; Sampedro A; Frassetto A; Benenato KE; Burke K; Santamaría E; Alegre M; Pejenaute Á; Kalariya M; Butcher W; Park J-S; Zhu X; Sabnis S; Kumarasinghe ES; Salerno T; Kenney M; Lukacs CM; Ávila MA; Martini PGV; Fontanellas A Systemic Messenger RNA as an Etiological Treatment for Acute Intermittent Porphyria. Nat. Med 2018, 24, 1899–1909. 10.1038/s41591-018-0199-z. [DOI] [PubMed] [Google Scholar]
- (11).Gillmore JD; Gane E; Taubel J; Kao J; Fontana M; Maitland ML; Seitzer J; O’Connell D; Walsh KR; Wood K; Phillips J; Xu Y; Amaral A; Boyd AP; Cehelsky JE; McKee MD; Schiermeier A; Harari O; Murphy A; Kyratsous CA; Zambrowicz B; Soltys R; Gutstein DE; Leonard J; Sepp-Lorenzino L; Lebwohl D CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med 2021, 385, 493–502. 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- (12).Akinc A; Querbes W; De S; Qin J; Frank-Kamenetsky M; Jayaprakash KN; Jayaraman M; Rajeev KG; Cantley WL; Dorkin JR; Butler JS; Qin L; Racie T; Sprague A; Fava E; Zeigerer A; Hope MJ; Zerial M; Sah DW; Fitzgerald K; Tracy MA; Manoharan M; Koteliansky V; Fougerolles A. de; Maier MA Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther 2010, 18, 1357–1364. 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cheng Q; Wei T; Farbiak L; Johnson LT; Dilliard SA; Siegwart DJ Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific MRNA Delivery and CRISPR–Cas Gene Editing. Nat. Nanotechnol 2020, 15, 313–320. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu S; Cheng Q; Wei T; Yu X; Johnson LT; Farbiak L; Siegwart DJ Membrane-Destabilizing Ionizable Phospholipids for Organ-Selective MRNA Delivery and CRISPR–Cas Gene Editing. Nat. Mater 2021. 10.1038/s41563-020-00886-0. [DOI] [PMC free article] [PubMed]
- (15).Dilliard SA; Cheng Q; Siegwart DJ On the Mechanism of Tissue-Specific MRNA Delivery by Selective Organ Targeting Nanoparticles. Proc. Natl. Acad. Sci 2021, 118, e2109256118. 10.1073/pnas.2109256118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou Q. (Tony); Tang P; Leung SSY; Chan JGY; Chan H-K Emerging Inhalation Aerosol Devices and Strategies: Where Are We Headed? Improv. Effic. Inhaled Drugs Sev. Lung Dis. Emerg. Pulm. Deliv. Strateg 2014, 75, 3–17. 10.1016/j.addr.2014.03.006. [DOI] [PubMed] [Google Scholar]
- (17).Collins N Nebulizer Therapy in Cystic Fibrosis: An Overview. J. R. Soc. Med 2009, 102, 11–17. 10.1258/jrsm.2009.s19003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cortez-Jugo C; Qi A; Rajapaksa A; Friend JR; Yeo LY Pulmonary Monoclonal Antibody Delivery via a Portable Microfluidic Nebulization Platform. Biomicrofluidics 2015, 9, 052603. 10.1063/1.4917181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Eygeris Y; Gupta M; Kim J; Sahay G Chemistry of Lipid Nanoparticles for RNA Delivery. Acc. Chem. Res 2022, 55, 2–12. 10.1021/acs.accounts.1c00544. [DOI] [PubMed] [Google Scholar]
- (20).Chakraborty S; Doktorova M; Molugu TR; Heberle FA; Scott HL; Dzikovski B; Nagao M; Stingaciu L-R; Standaert RF; Barrera FN; Katsaras J; Khelashvili G; Brown MF; Ashkar R How Cholesterol Stiffens Unsaturated Lipid Membranes. Proc. Natl. Acad. Sci 2020, 117, 21896–21905. 10.1073/pnas.2004807117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Patel S; Ashwanikumar N; Robinson E; Xia Y; Mihai C; Griffith JP; Hou S; Esposito AA; Ketova T; Welsher K; Joyal JL; Almarsson Ö; Sahay G Naturally-Occurring Cholesterol Analogues in Lipid Nanoparticles Induce Polymorphic Shape and Enhance Intracellular Delivery of MRNA. Nat. Commun 2020, 11, 983. 10.1038/s41467-020-14527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Eygeris Y; Patel S; Jozic A; Sahay G Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett 2020, 20, 4543–4549. 10.1021/acs.nanolett.0c01386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Herrera M; Kim J; Eygeris Y; Jozic A; Sahay G Illuminating Endosomal Escape of Polymorphic Lipid Nanoparticles That Boost MRNA Delivery. Biomater. Sci 2021, 9, 4289–4300. 10.1039/D0BM01947J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Paunovska K; Da Silva Sanchez AJ; Sago CD; Gan Z; Lokugamage MP; Islam FZ; Kalathoor S; Krupczak BR; Dahlman JE Nanoparticles Containing Oxidized Cholesterol Deliver MRNA to the Liver Microenvironment at Clinically Relevant Doses. Adv. Mater 2019, 31, 1807748. 10.1002/adma.201807748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mui BL; Tam YK; Jayaraman M; Ansell SM; Du X; Tam YYC; Lin PJ; Chen S; Narayanannair JK; Rajeev KG; Manoharan M; Akinc A; Maier MA; Cullis P; Madden TD; Hope MJ Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of SiRNA Lipid Nanoparticles. Mol. Ther. - Nucleic Acids 2013, 2, e139. 10.1038/mtna.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yanez Arteta M; Kjellman T; Bartesaghi S; Wallin S; Wu X; Kvist AJ; Dabkowska A; Székely N; Radulescu A; Bergenholtz J; Lindfors L Successful Reprogramming of Cellular Protein Production through MRNA Delivered by Functionalized Lipid Nanoparticles. Proc. Natl. Acad. Sci 2018, 115, E3351–E3360. 10.1073/pnas.1720542115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lokugamage MP; Vanover D; Beyersdorf J; Hatit MZC; Rotolo L; Echeverri ES; Peck HE; Ni H; Yoon J-K; Kim Y; Santangelo PJ; Dahlman JE Optimization of Lipid Nanoparticles for the Delivery of Nebulized Therapeutic MRNA to the Lungs. Nat. Biomed. Eng 2021, 5, 1059–1068. 10.1038/s41551-021-00786-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhang H; Leal J; Soto MR; Smyth HDC; Ghosh D Aerosolizable Lipid Nanoparticles for Pulmonary Delivery of MRNA through Design of Experiments. Pharmaceutics 2020, 12, 1042. 10.3390/pharmaceutics12111042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xu Q; Ensign LM; Boylan NJ; Schön A; Gong X; Yang J-C; Lamb NW; Cai S; Yu T; Freire E; Hanes J Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus Ex Vivo and Distribution in Vivo. ACS Nano 2015, 9, 9217–9227. 10.1021/acsnano.5b03876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Suk JS; Xu Q; Kim N; Hanes J; Ensign LM PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Deliv. Rev 2016, 99, 28–51. 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chen S; Tam YYC; Lin PJC; Sung MMH; Tam YK; Cullis PR Influence of Particle Size on the in Vivo Potency of Lipid Nanoparticle Formulations of SiRNA. J. Controlled Release 2016, 235, 236–244. 10.1016/j.jconrel.2016.05.059. [DOI] [PubMed] [Google Scholar]
- (32).Perry JL; Reuter KG; Kai MP; Herlihy KP; Jones SW; Luft JC; Napier M; Bear JE; DeSimone JM PEGylated PRINT Nanoparticles: The Impact of PEG Density on Protein Binding, Macrophage Association, Biodistribution, and Pharmacokinetics. Nano Lett 2012, 12, 5304–5310. 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Patel AK; Kaczmarek JC; Bose S; Kauffman KJ; Mir F; Heartlein MW; DeRosa F; Langer R; Anderson DG Inhaled Nanoformulated MRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater 2019, 31, 1805116. 10.1002/adma.201805116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Irvin CG; Bates JH Measuring the Lung Function in the Mouse: The Challenge of Size. Respir. Res 2003, 4, 1. 10.1186/rr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fahy JV; Dickey BF Airway Mucus Function and Dysfunction. N. Engl. J. Med 2010, 363, 2233–2247. 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hou S; Exell J; Welsher K Real-Time 3D Single Molecule Tracking. Nat. Commun 2020, 11, 3607. 10.1038/s41467-020-17444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hou S; Lang X; Welsher K Robust Real-Time 3D Single-Particle Tracking Using a Dynamically Moving Laser Spot. Opt. Lett 2017, 42, 2390. 10.1364/OL.42.002390. [DOI] [PubMed] [Google Scholar]
- (38).The Clinical Chemistry of Laboratory Animals, 3rd ed.; Kurtz DM, Travlos GS, Eds.; CRC Press: Third edition. | Boca Raton : Taylor & Francis, 2017., 2017. 10.1201/9781315155807. [DOI] [Google Scholar]
- (39).Samal SK; Dash M; Van Vlierberghe S; Kaplan DL; Chiellini E; van Blitterswijk C; Moroni L; Dubruel P Cationic Polymers and Their Therapeutic Potential. Chem. Soc. Rev 2012, 41, 7147. 10.1039/c2cs35094g. [DOI] [PubMed] [Google Scholar]
- (40).Cui H; Jiang D; Banerjee S; Xie N; Kulkarni T; Liu R-M; Duncan SR; Liu G Monocyte-Derived Alveolar Macrophage Apolipoprotein E Participates in Pulmonary Fibrosis Resolution. JCI Insight 2020, 5, e134539. 10.1172/jci.insight.134539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yao X; Gordon EM; Figueroa DM; Barochia AV; Levine SJ Emerging Roles of Apolipoprotein E and Apolipoprotein A-I in the Pathogenesis and Treatment of Lung Disease. Am. J. Respir. Cell Mol. Biol 2016, 55, 159–169. 10.1165/rcmb.2016-0060TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Melton L Does Mucus Hypersecretion Matter in Airway Disease? The Lancet 2002, 359, 1924. 10.1016/S0140-6736(02)08788-3. [DOI] [PubMed] [Google Scholar]
- (43).Ehre C; Worthington EN; Liesman RM; Grubb BR; Barbier D; O’Neal WK; Sallenave J-M; Pickles RJ; Boucher RC Overexpressing Mouse Model Demonstrates the Protective Role of Muc5ac in the Lungs. Proc. Natl. Acad. Sci 2012, 109, 16528–16533. 10.1073/pnas.1206552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).McCarron A; Donnelley M; Parsons D Airway Disease Phenotypes in Animal Models of Cystic Fibrosis. Respir. Res 2018, 19, 54. 10.1186/s12931-018-0750-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Miller LA; Royer CM; Pinkerton KE; Schelegle ES Nonhuman Primate Models of Respiratory Disease: Past, Present, and Future. ILAR J 2017, 58, 269–280. 10.1093/ilar/ilx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Moldoveanu B; Otmishi P; Jani P; Walker J; Sarmiento X; Guardiola J; Saad M; Yu J Inflammatory Mechanisms in the Lung. J. Inflamm. Res 2009, 2, 1–11. [PMC free article] [PubMed] [Google Scholar]
- (47).Myerson JW; Patel PN; Rubey KM; Zamora ME; Zaleski MH; Habibi N; Walsh LR; Lee Y-W; Luther DC; Ferguson LT; Marcos-Contreras OA; Glassman PM; Mazaleuskaya LL; Johnston I; Hood ED; Shuvaeva T; Wu J; Zhang H-Y; Gregory JV; Kiseleva RY; Nong J; Grosser T; Greineder CF; Mitragotri S; Worthen GS; Rotello VM; Lahann J; Muzykantov VR; Brenner JS Supramolecular Arrangement of Protein in Nanoparticle Structures Predicts Nanoparticle Tropism for Neutrophils in Acute Lung Inflammation. Nat. Nanotechnol 2022, 17, 86–97. 10.1038/s41565-021-00997-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kim J; Sahay G Nanomedicine Hitchhikes on Neutrophils to the Inflamed Lung. Nat. Nanotechnol 2021, 17, 1–2. 10.1038/s41565-021-00981-6. [DOI] [PubMed] [Google Scholar]
- (49).Lu L; Li J; Moussaoui M; Boix E Immune Modulation by Human Secreted RNases at the Extracellular Space. Front. Immunol 2018, 9, 1012. 10.3389/fimmu.2018.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kim J; Jozic A; Sahay G Naturally Derived Membrane Lipids Impact Nanoparticle-Based Messenger RNA Delivery. Cell. Mol. Bioeng 2020, 13, 463–474. 10.1007/s12195-020-00619-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.