

Summary:
Before antigen-specific immunity arises, the complement system responds by activation through the classical and/or alternative pathways leading to the covalent deposition of complement fragments. Three models, not mutually exclusive, have been proposed to explain how these complement fragments interact with their receptors, CD21/CD35, to enhance humoral immune responses: i) CD21/CD35 retain and focus antigens for optimal presentation; ii) CD21/CD35 on B cells serve as enhancing co-receptors for B-cell antigen receptor (BCR) signaling; iii) CD21/CD35 regulate B-cell responses, by CD19 aggregation. The co-receptor model led us to predict that CD21/CD35 may lower the threshold of BCR affinity to diversify the repertoire of humoral immune responses, but surprisingly, CD21/CD35-deficient mice expressing a transgenic BCR with very low affinity (Ka ≈1.2×105 M−1) for the (4-hydroxy-3-nitrophenyl)acetyl hapten generated significant antibody and germinal center responses to even low doses of antigens in alum. The magnitudes of these responses were much below those of normal controls but higher doses of antigens substantially rectified these deficits. Thus, while CD21/CD35 play important roles in humoral immunity, our observations provide little support to the hypothesis that CD21/CD35 promote clonal diversity in immune responses by helping recruit low-affinity B cells.
Introduction
Vertebrates possess two branches of immunity: innate (or natural) and acquired responses against microbial invaders. One prominent component of the innate immune system is the complement system consisting of about 30 soluble and membrane proteins. Many of the components are inactive proenzymes circulating in serum; these are activated through two highly regulated enzymatic cascades, the classical and alternative pathways. The binding of antibody to antigens initiates the classical pathway: C1q binds to Fc regions of Ig and induces the enzyme activity of C1r leading to a sequential activation of C1s, C4 and C2 to form a C3 convertase, C4b2a. C3 convertases can also be generated through the alternative pathway. For example, spontaneous C3 hydrolysis can lead to the covalent attachment of C3b to microbial surfaces. C3b then interacts with factor B and factor D to become another C3 convertase, C3bBb. Both the classical and alternative pathways converge at C3 and lead to sequential activation of C5 through C9, generating a membrane-attack complex (MAC), C5b-9, that directly lyses bacteria. Complement activation cascades also generate component fragments covalently deposited on microbial surfaces that mediate the phagocytosis of microbes, even before the generation of antigen-specific acquired immune responses (1).
Three decades ago, Nussenzweig and colleagues discovered that B lymphocytes expressed complement receptors (2–4). This finding indicated that complement might have a then-unexpected role – regulation of acquired humoral immunity. To test this idea, Pepys (5) studied antibody responses in mice transiently depleted of C3 with cobra venom factor (CoF) and found that C3 depletion shortly before or after immunization inhibited antibody responses to the T-dependent antigen sheep red blood cells (SRBC) (5). Later, defective antibody production was also observed in individuals with genetic deficiencies in C2, C3 or C4 (6–10). More recently, targeted gene disruption has been used to generate mice deficient in specific complement components or receptors. Mice deficient in C3 or C4 failed to generate antibody responses to doses of bacteriophage ϕX174 that are fully immunogenic in wild-type mice (11). Increasing the dose of bacteriophage 10-fold elicited modest IgG responses in knockout animals but these responses remained 10-fold below that of normal controls. Deficiency in the complement receptors CD21 and CD35 gave the same pattern of impaired antibody responses in mice (12), suggesting that complement may enhance humoral immune responses through interaction with CD21/CD35.
CD21/CD35 and their roles in enhancing humoral immunity
Two principal complement receptors have been described, CR1 (CD35) and CR2 (CD21). In humans, the two receptors are encoded by different genes but in mice, CD21 and CD35 are alternative splice products of the same gene, Cr2 (13). These two receptors are co-expressed on follicular dendritic cells (FDCs) and B cells. CD21 and CD35 are assembled from tandem repeat motifs called short consensus repeats (SCRs). SCRs are conserved units of 60–70 amino acids and some contain the binding site for complement fragments. Human CD21 (hCD21) binds iC3b, C3dg, C3d and C4d, and human CD35 (hCD35) binds C3b and C4b. Mouse CD21 (mCD21) binds the same ligands as hCD21; mCD35 binds not only C3b and C4b but also recognizes the mCD21 ligands. Unlike their human homologs, mCD35 and mCD21 share 15 SCRs and mCD35 includes six additional SCRs that comprise the C3b/C4b binding site. Both hCD35 and mCD35 generate ligands for CD21 by functioning as co-factors to catalyze the conversion of C3b to iC3b and C3dg (13–15).
On the surface of human and murine B cells, CD21 associates with CD19 and CD81 to form a co-receptor complex (16–19). Human CD35 may associate with CD21 but does not become a part of the co-receptor complex (16). In contrast, on mouse B cells, both CD35 and CD21 can associate with CD19 (19). Participation in the co-receptor complex presumably reflects the shared genetic origins of mCD35 and mCD21 rather than species differences in CD19 (15). CD19 is a membrane anchor protein for Src-family tyrosine kinases and a potent regulator of signaling in B cells (20); thus its association with CD21 suggests a role for CD21 in B-cell activation. Indeed, abundant evidence supports the ability of CD21/CD35 to enhance B-cell responses. For example, cross-linking of CD21 and membrane IgM (mIgM) on human B cells synergistically enhances calcium mobilization in vitro (21, 22). To test the function of CD21 in vivo, Hebell et al. (23) made a chimeric Ig/CD21 protein by fusing with an IgG1 the two N-terminal SCRs of CD21 that specify the C3 binding site. This fusion protein suppressed antibody responses to the T-dependent antigens SRBC and keyhole limpet hemocyanin (KLH) when injected at the time of immunization and was hypothesized to compete with endogenous CD21 for C3d ligands, leading to the impaired antibody responses. Interestingly, the extent of immune suppression by this Ig/CD21 fusion protein and treatment with CoF was similar, suggesting that CD21 mediated immune enhancement by C3 (23). In vivo function for CD21/CD35 has also been demonstrated by Heyman et al. (24). Passive administration of the monoclonal anti-mouse CD21/CD35 antibody 7G6 suppressed serum antibody responses when injected into mice 24 h before immunization (24). Suppression was most obvious at suboptimal concentrations of immunogens; a 600-fold increase in antigen dose rectified humoral responses in 7G6-treated mice from 3–4% to about 50% of control antibody levels (24). Treating mice with the 7G6 antibody also inhibited antibody responses to type II T-independent antigens, including haptenated Salmonella and Ficoll (25). This impairment was also ameliorated by higher doses of antigens (25). A hapten-carrier system and adoptive transfer experiments demonstrated that while 7G6 antibody severely inhibited primary antibody responses, it did not reduce priming of T cells; adoptively transferred T cells from 7G6-treated animals were as capable as normally primed T cells in providing B-cell help (26).
The 7G6 antibody binds both mCD21 and mCD35; therefore, its inhibitory effect on antibody responses could indicate regulatory roles for either molecule, or both. The role of CD35 in enhancing B-cell responses is unclear, with some studies observing effects and others not (21, 22, 27, 28). In vitro, polyclonal anti-hCD35 F(ab’)2 enhanced pokeweed mitogen-stimulated IgM, IgG and IgA production from human B cells 10–20- fold (27). Another in vitro study showed that monoclonal anti-hCD35 antibodies increased antigen-specific antibody responses about 4-fold (28). However, in vitro signaling studies on normal and leukemic human B cells showed that co-ligation of mIgM and CD21, but not CD35, enhanced calcium influx. Consistent with the fact that hCD21 but not CD35 complexes with CD19, these observations suggest that CD21 rather than CD35 is important in the early events of B-cell signaling (21, 22). Nonetheless, these observations do not exclude a later role for CD35 in B-cell activation and differentiation. CD35 function has been studied in vivo as well. Injection of the monoclonal antibody, 8C12, which recognizes mCD35 but not mCD21, suppressed antibody responses less well than an equal amount of antibody 7G6 (2-fold suppression for 8C12 vs 30-fold for 7G6) (24). Although consistent with the observation that co-cross-linking of hCD35 with mIgM did not enhance calcium influx, this experiment does not rule out a role for CD35 in B-cell activation. Unfortunately, 8C12, unlike 7G6, downregulates CD35 weakly and differences in the suppressive capacity of these two antibodies may be trivial (24). Indeed, another monoclonal antibody, which binds both CD21 and CD35 but downregulates these receptors less efficiently than 7G6, had an even more modest effect on antibody responses in vivo than did 8C12 (24). Thus, although the current view of CD35 emphasizes its role as an auxiliary molecule for the generation of ligands for CD21 (15), little evidence is available to clarify the function of CD35 in vivo.
To study the function of CD21/CD35 better, two independent lines of mice deficient in CD21/CD35 were generated by targeted disruption of the Cr2 locus (12, 29). B-cell development is overtly normal in Cr2−/− mice except for a 2-fold reduction in the B1-a cell population (12, 29). In one line of Cr2−/− mice, generated by Molina et al. (29), IgG responses against SRBC were impaired 3- to 10-fold whereas IgM responses were only modestly affected. In contrast to earlier suppression experiments (24, 25), a 200-fold increase in antigen dose did not significantly improve antibody responses (29). The other line of Cr2−/− mice, generated by Carroll and colleagues (12), mounted little serum IgG responses against a low dose of bacteriophage ϕX174. A 10-fold increase in antigen dose elicited antibody responses but the response is 10-fold lower than that of wild-type controls (12). Impaired antibody production was not due to a defect in B-cell antigen receptor (BCR) signaling because B cells from Cr2−/− mice responded normally to IgM and/or CD40L cross-linking (12). These genetic studies provide definitive evidence for the critical role of CD21/CD35 in enhancing antibody responses.
CD21/CD35 are also implicated in another pathway of B-cell response – the germinal center (GC) reaction. B cells undergo somatic mutation and clonal selection in GCs, leading to the generation of memory B cells (30, 31). In vitro, treating GC B cells with an anti-CD21/CD35 antibody (7G6) or anti-C3 antibody suppressed antigen-specific antibody production by GC B cells (32). Whether and how much CD21/CD35 deficiency impairs the GC reaction in vivo is controversial. An Ig/CD21 fusion protein suppressed GC reactions (23). However, Molina and colleagues reported that Cr2−/− mice exhibited normal GC morphology and only a mild quantitative reduction in GC responses to SRBC (29, 33). Nonetheless, Carroll and colleagues showed that GC reactions to bacteriophage were reduced about 10-fold in the absence of CD21/CD35 (12), consistent with their observation that C3−/− and C4−/− mice also exhibited impaired GC reactions (11). This discrepancy could result from the different nature of antigens and/or dosage of antigenic stimulation.
CD21 has been hypothesized to provide a survival signal for GC B cells (34). This hypothesis was intuitively reasonable because GC B cells are thought to die by default unless rescued by antigenic signals (30, 31). The antigen resource in GCs is in the form of immune complexes decorated with complement fragments (13, 35–38). To test experimentally the role of CD21 on the survival of GC B cells, GC B cells isolated from human tonsils were cultured with anti-CD21 monoclonal antibodies (34). It was found that a subset of anti-CD21 antibodies rescued GC B cells from apoptosis and the rescued GC B cells increased expression of the Bcl-2 proto-oncogene (34). In another in vitro study (39), co-cross-linking CD21/CD35 with mIgM rescued mouse resting B cells from apoptosis induced by ligation of mIgM alone (39). Recently, Roberts & Snow (40) showed that stimulation of resting hen egg lysozyme (HEL)-transgenic B cells with HEL alone induced Bcl-xL but not Bcl-2 expression in vitro; cross-linking of CD19 was necessary for Bcl-2 upregulation (40). The physiological relevance of these in vitro studies is unclear. Whereas naïve and resting memory B cells from human tonsils express Bcl-2, GC B cells downregulate Bcl-2 expression (41–43), even though these cells should have been stimulated through both BCRs and CD21/CD35.
CD21/CD35 were also tested in vivo for their role in the survival of GC B cells (44). Cr2+/+ or Cr2−/− HEL-specific transgenic B cells, the BCRs of which bind to duck (DEL) and turkey (TEL) lysozyme with Ka 1×107 and 2×1010 M−1, respectively, were adoptively transferred into antigen-primed wild-type mice (44). Unlike Cr2+/+ B cells, Cr2−/− cells transferred along with DEL into DEL-immunized mice were not retained in splenic B-cell follicles. When Cr2−/− B cells were transferred along with TEL into TEL-primed mice, they responded with follicle retention, but the retention was impaired about 2-fold. Even with the extraordinarily high affinity of transgenic BCRs to TEL, Cr2−/− B cells occupied GCs 10-fold less effectively than did Cr2+/+ controls. For the few Cr2−/− cells that did enter wild-type GCs, they failed to proliferate (44). Therefore, Cr2−/− B cells cannot compete with normal cells for follicular retention and survival in GCs.
Models for the function of CD21/CD35
Three models, not mutually exclusive, have been proposed to explain how CD21 and CD35 might interact with complement ligands to enhance humoral immunity (Fig. 1). First, CD21 and CD35 may enhance and prolong humoral responses by focusing and retaining antigen/antibody/complement complexes (35–38); second, complement-decorated antigens may bridge the BCR and CD21/CD19 co-receptor to strengthen BCR/CD19 signals (13–15, 45); third, complement-decorated antigens may aggregate CD21/CD19 complexes independently of the BCR and cause the activation CD19-associated tyrosine kinases (20). Here we discuss the experimental evidence for and against each of these models.
Fig. 1. Three different models, not mutually exclusive, are proposed to explain how CD21/CD35 enhance humoral immunity.
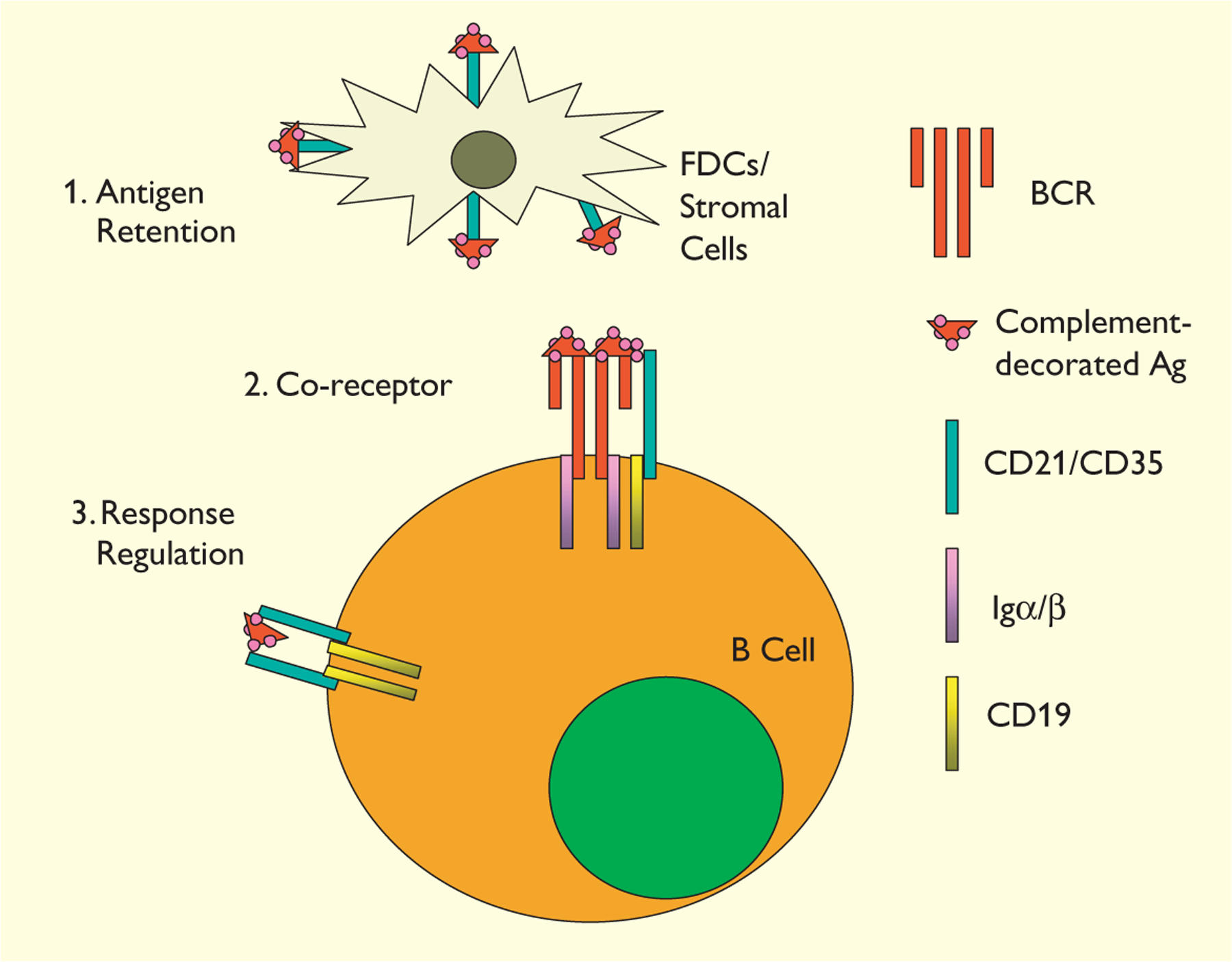
1. CD21/CD35 on FDCs/stromal cells enhance antigen retention. 2. CD21/CD35 on B cells serve as a co- receptor recruiting CD19 to the BCR signaling complexes. 3. Independently of the BCR–antigen interactions, aggregation of CD21/CD19 complexes activates CD19-associated tyrosine kinases to upregulate of B-cell responsiveness.
CD21/CD35 mediate antigen-retention on FDCs/stromal cells
The role of complement in antigen retention was tested first by Papamichail and colleagues in 1975 (46). They found that decomplementation of mice by CoF inhibited the localization of aggregated human IgG in GCs. Injection of anti-CoF antibodies into decomplemented animals restored serum C3 levels and returned aggregated IgG to GCs (46). Antigens that lack the intrinsic abilities to activate complement become localized in splenic follicles only in the presence of complement-fixing antibody (47). In contrast, antigens that activate the alternative pathway become concentrated in lymphoid follicles in the absence of antibody (47). C3-depletion by CoF treatment abrogates the localization of antigens or antigen–antibody complexes into splenic follicles (48), suggesting that C3 and/or its derivatives may mediate this antigen localization. In B-cell follicles, immune complexes are retained on FDCs (35–38). Localization of immune complexes to FDCs depends on Ig isotype; in contrast to IgG, IgM has little (36) or no (35) effect on enhancing antigen retention by FDCs even though IgM effectively activates complement through the classical pathway (49). One possible explanation for the inability of IgM to direct antigens to the FDC surface could be the rapid clearance of IgM. Indeed, decay rates for the passively injected IgM (T1/2≈2 days) are 2 to 4 times that of passive IgG (4–8 days) (50). Another possible explanation for the inability of IgM to localize antigens to FDCs is that FcγR might direct localization of immune complexes into follicles. FcγR does not bind IgM (51). Although some in vitro evidence suggests that FDC may bind immune complexes through FcγR (37), common FcR γ-chain knockout mice, in which the expression of FcγRI and FcγRIII is abolished, exhibit increased, rather than decreased, antigen retention on FDCs. This increase may reflect impaired clearance of immune complexes (52). FcγRIIb-deficient mice have elevated antibody levels (53), although antigen localization on FDCs in these mice has not been investigated. On the other hand, antigen retention in B-cell follicles is dramatically reduced (13) or is undetectable (33) in Cr2−/− mice. Therefore, CD21/CD35 are the major, if not sole, receptors mediating antigen retention on FDCs. This antigen resource may sustain the humoral immune responses.
Indeed, the role of CD21/CD35 on FDCs in antibody responses was shown both in vitro and in vivo (33, 54). In an in vitro study (54), FDCs from wild-type mice enhanced antigen- or lipopolysaccharide (LPS)-stimulated antibody production; treating FDCs with soluble CD21 diminished their immune-enhancing effect (54). Consistently, FDCs from C3−/− mice were not effective in promoting antibody production in vitro. A recent study by Molina and colleagues (33) provided perhaps the strongest evidence in vivo for the role of CD21/CD35 expressed on FDC/stromal cells. In this study, chimeric mice were generated using bone marrow reconstitution of lethally irradiated host mice. In such chimeric mice, many or all FDCs remain to be of host origin (55), whereas B cells can only be regenerated by donor bone marrow. Cr2−/− hosts were reconstituted with Cr2+/+ bone marrow (Cr2−/−BM+/+), or Cr2+/+ hosts with Cr2−/− bone marrow (Cr2+/+BM−/−). These mice were compared to Cr2+/+BM+/+ and Cr2−/−BM−/− mock chimeras for IgG responses to T-dependent antigens. Both Cr2−/−BM+/+ and Cr2+/+BM−/− chimeras exhibited impaired primary and secondary antibody responses. Surprisingly, the suppression of antibody responses in Cr2−/−BM+/+ mice was more severe than that present in Cr2+/+BM−/− chimeras (33). This observation suggests that CD21/CD35 on FDCs/stromal cells contribute more to the enhancement of antibody responses than does CD21/CD35 on B cells.
However, this observation contradicts a previous report from Carroll’s group (12) that Cr2−/− BM+/+ chimeric mice generated antibody and GC reactions comparable to wild-type animals. This group concluded that CD21/CD35 on B cells was the prime, if not sole, mediator of immune enhancement. It is difficult to reconcile these contradicting reports. Molina (33) and Carroll’s (12) groups used different experimental systems to test humoral responses. Fang and colleagues (33) tested primary antibody responses and GC reactions to different doses of SRBC. Ahearn et al. (12) used bacteriophage as antigens and significant antibody responses were not detected until 1 week after secondary challenge (12). The difference in the nature of antigens and methods of immunizations might account for the discrepancy. Nonetheless, CD21/CD35 on FDCs/stromal cells may indeed play an important role in enhancing humoral immunity.
CD21/CD35 on B cells: co-receptor vs response regulator
The importance of CD21 and CD35 on B cells in antibody responses is well established. In one study, Ahearn et al. (12) reconstituted lethally irradiated Cr2−/− mice with Cr2+/+ bone marrow to achieve normal CD21/CD35 expression on B cells whereas most FDCs remained deficient in CD21/CD35. These chimeric mice generated normal antibody responses and GC reactions, indicating that impaired immune response in Cr2−/− mice is caused by the absence of CD21/CD35 on B cells rather than FDCs. The same group of researchers tested this idea by another approach. RAG-2-deficient blastocysts were complemented with Cr2−/− stem cells to generate chimeric mice in which B cells could be generated solely by Cr2−/− stem cells, whereas FDC/stromal cells could be derived from both Cr2+/+ and Cr2−/− progenitors. Like Cr2−/− mice, these chimeric mice generated no detectable antibody response against (4-hydroxy-3-nitrophenyl)acetyl (NP)–KLH (56). Independently, Fang et al. (33) used bone marrow reconstitution to generate chimeric mice comprised of Cr2−/− B lymphocytes and Cr2+/+ FDCs/stromal cells; antibody responses to T-dependent antigens in these chimeric anomals were also impaired in comparison to Cr2+/+ controls, although suppression was milder than that in Cr2−/− mice (33). In vitro, normal FDCs dramatically enhance LPS-stimulated antibody production by B cells from wild-type mice. However, this enhancement is much limited if B cells are from Cr2−/− mice (54). These observations indicate that the presence of CD21and CD35 on B cells is critical for the generation of normal antibody responses.
How do CD21/CD35 on B cells regulate humoral responses? In an in vitro study, co-ligation of mIgM and CD21 or CD35 was achieved by antibody-mediated cross-linking (21). In this study, anti-CD21 and anti-IgM antibodies synergized to increase cytoplasmic free calcium and proliferation. In contrast, no synergism was observed between CD35 and mIgM cross-linking, although the anti-CD21 and anti-CD35 antibodies bound to B cells equally (21). Similar approaches were used to study interaction of CD21 and CD35 with mIgM on leukemic B cells; again, co-cross-linking of CD21 but not CD35 with mIgM induced rise in intracellular calcium (22). CD21 may function through CD19, as co-ligation of CD19 with mIgM on human B cells also decreased the minimum concentration of anti-IgM required to induce B-cell proliferation (57). To test directly the effect of bridging CD21/CD35 with the antigen-specific BCR, Fearon’s group (58) designed a recombinant antigen comprised of a model antigen HEL fused to murine C3d. In vitro, HEL-specific transgenic B cells require minimum of 7 nM unmodified HEL to elicit increases of free calcium in the cytoplasm (58). Multiple copies (1, 2 or 3) of C3d fused to HEL (HEL-C3d1, HEL-C3d2 etc.) decreased this threshold of antigen requirement 10-, 100- and 1,000-fold, respectively, although HEL-C3d3 did not bind more efficiently to the transgenic B cells (58). The enhancing effect of HEL-C3d was obviated by treating the responding B cells with a monoclonal anti-CD21/CD35 antibody (7G6) (58). To explain these observations, a “co-receptor” model was proposed (13–15, 45). In this model, complement-decorated antigens, like the recombinant HEL-C3d proteins, cross-link CD21/CD19 and BCR complexes. This bridging recruits CD19-activating signals to antigen-engaged BCR aggregates and lowers the threshold of B-cell signaling (Fig. 1).
The co-receptor model is fascinating, but cannot explain observations that co-ligating CD21/CD35 with the BCR is not the only means to reveal the function of CD21/CD35 in enhancing B-cell responses. For example, CD21/CD35 promote B-cell responses even if they are triggered independently of BCR stimulation (59, 60). Polymerized, but not soluble, human C3 or C3d enhances the proliferation of LPS-activated mouse B cells (59, 60). Polyclonal anti-CD21 F(ab’)2 promotes cytokine-induced human B-cell proliferation (61), and human B cells, in the presence of irradiated autologous T cells, proliferate in response to anti-CD21 antibodies or F(ab’)2 fragments in a dosage-dependent fashion (62). One monoclonal anti-CD21 antibody, OKB7, blocks both C3d and Epstein–Barr virus binding to B cells. This antibody not only stimulated 50- to 200-fold DNA synthesis by B cells in cultures of unseparated T and B lymphocytes from normal donors, but also induced polyclonal Ig secretion in such cultures (63). However, CD21 stimulation alone does not induce proliferation or differentiation of purified resting B cells (64). These findings indicate that signals from CD21 enhance the responses of B cells that are activated by means other than stimulating the BCR. Since only aggregated but not soluble C3d enhance proliferation of activated B cells (59, 60), CD21 may have to be cross-linked by polymeric ligands to achieve this function. Indeed, preincubation of human tonsillar B cells with polymerized, but not monomeric, C3dg enhanced subsequent anti-IgM-induced B-cell proliferation (65). In another study, Tsokos et al. (66) designed monovalent polypeptide ligands containing the CD21-binding motif of C3d, and polyvalent conjugates of the same polypeptide, to test the role of CD21 cross-linking in BCR signaling. They found that monovalent CD21 ligands inhibited anti-mIgM-stimulated calcium influx but polyvalent CD21 ligands synergized with anti-IgM antibodies to increase cytoplasmic free calcium in human B cells (66). That CD21/CD19 complexes can regulate B-cell activation independently of the BCR is also suggested in studies of transgenic B cells that over-express CD19. B cells from mice that express transgenic human CD19 (hCD19) in addition to endogenous mouse CD19 exhibit enhanced B-cell survival and spontaneous proliferation in vitro (67). Furthermore, stimulation with LPS, cross-linking of mIgM or ligation of CD40 induced stronger proliferation in these transgenic B cells than in wild-type controls (67–69). Therefore, even without bridging with the BCR, cross-linking of CD21 may enhance B-cell proliferation and differentiation.
In explanation of the observations that cross-linking of CD21 can enhance B-cell responses in the absence of co-ligation to the BCR, a model of “response regulation” has been proposed (20). In this model, polymeric C3d on antigens cross-link CD21 in a BCR-independent manner. The aggregation of CD21/CD19 complexes heightens the signaling thresholds of B cells (Fig. 1) and serves to adjust the responsiveness of B cells rather than the CD21/CD19 complex acting as co-receptors (20).
Technically, it is very difficult to discriminate between these two models. HEL-C3d recombinant antigens (58) were designed to test the co-receptor model. However, since HEL tends to form aggregates, recombinant HEL-C3d may cross-link CD21/CD19 independently of BCR ligation, i.e. as in the response regulator model (Fig. 1) (20). Although HEL-C3d3 is reported not to enhance anti-mIgM-induced calcium influx (58), this failure does not exclude roles for CD21/CD35 cross-linking on other aspects of B-cell responses. For example, Carter et al. (65) have shown that whereas pretreatment of B cells with polymeric C3dg did not lower the threshold of anti-mIgM stimulation necessary for proliferation, it did increase the magnitude of induced proliferation (65). Discrimination between these two models is important for conceptual and quite practical reasons. The co-receptor model predicts that complement only enhances antigen-specific B-cell responses, whereas the response regulator model dictates that activation of complement may upregulate B-cell responsiveness in general and antigen-specific immunity in particular. In the event of systemic microbial infection, the response regulator model predicts that large amount of complement fragments deposited on microbial surfaces may cause extensive partial-activation of bystander B cells.
Perhaps there is truth in all three models. In vivo, the interaction of CD21/CD35 with their ligands may promote B-cell proliferation and differentiation through a combination of mechanisms proposed in all the three models. Humoral polymeric C3d, which could signal a systemic microbial infection, may alert all circulating B cells by cross-linking CD21/CD19 complexes, regardless of antigen specificity. Antigen retention and co-receptor functions of CD21/CD35 would lower the minimum amount of antigens required to generate B-cell responses. Together, these effects of CD21/CD35 would help B cells generate robust humoral immune responses to physiological (low) concentrations of antigens.
Antibody responses and GC reactions in mice deficient in CD21/CD35 and expressing low-affinity transgenic BCRs
To characterize the role of CD21/CD35 in antibody responses and GC reactions further, we bred the Cr2−/− mutation onto homozygous Ighb background of C57BL/6 mice and studied B-cell responses to the NP hapten conjugated to chicken y-globulin (NP-CG). All strains of mice carrying the Ighb allotype and Igλ1 loci generate clonally restricted primary antibody responses and GC reactions to immunogenic conjugates of NP (70). Most NP-specific antibodies in these mice carry λ1 light (L−) chains and heavy (H−) chains encoded by VH186.2–DFL16.1–JH2 rearrangements (71, 72). In GCs, the initial repertoire of NP-responding B cells is diverse, but by day 8 of the response, ≥80% of NP-reactive B cells carry VH186.2–DFL16.1 VDJ rearrangements (73). We took advantage of this well-defined model system to study the B-cell responses in the absence of CD21/CD35. Cr2−/− Ighb/b mice generated antibody responses and GC reactions to 5 μg of NP-CG in alum, but the magnitude of the responses was greatly reduced in comparison to Cr2+/− controls; as the antigen dose was increased to 50 μg, differences between Cr2−/− and control mice diminished (Fig. 2) (74). When immunized with 50 μg of NP-CG in alum, Cr2−/− mice generated GC reactions kinetically similar to that of normal controls. Somatic mutation and clonal selection was not reduced in the GCs of Cr2−/− animals. Interestingly, persistence of IgM and IgG1 responses was impaired in Cr2−/− mice but a more rapid loss of serum antibody came with enhanced affinity maturation. The increased rates of IgG1 decay and affinity maturation in Cr2−/− mice were observed at times well after GC reactions were prominent, suggesting that CD21/CD35 may play a role in maintenance and selection of long term memory effector B cells – the antibody-forming cells (AFCs) in bone marrow (75–78). Indeed, Cr2−/− mice exhibited preferential loss of bone marrow AFCs secreting lower-affinity IgG1 antibody (74).
Fig. 2. Antibody responses and GC reactions by Cr2−/− B cells expressing a very low affinity BCR.
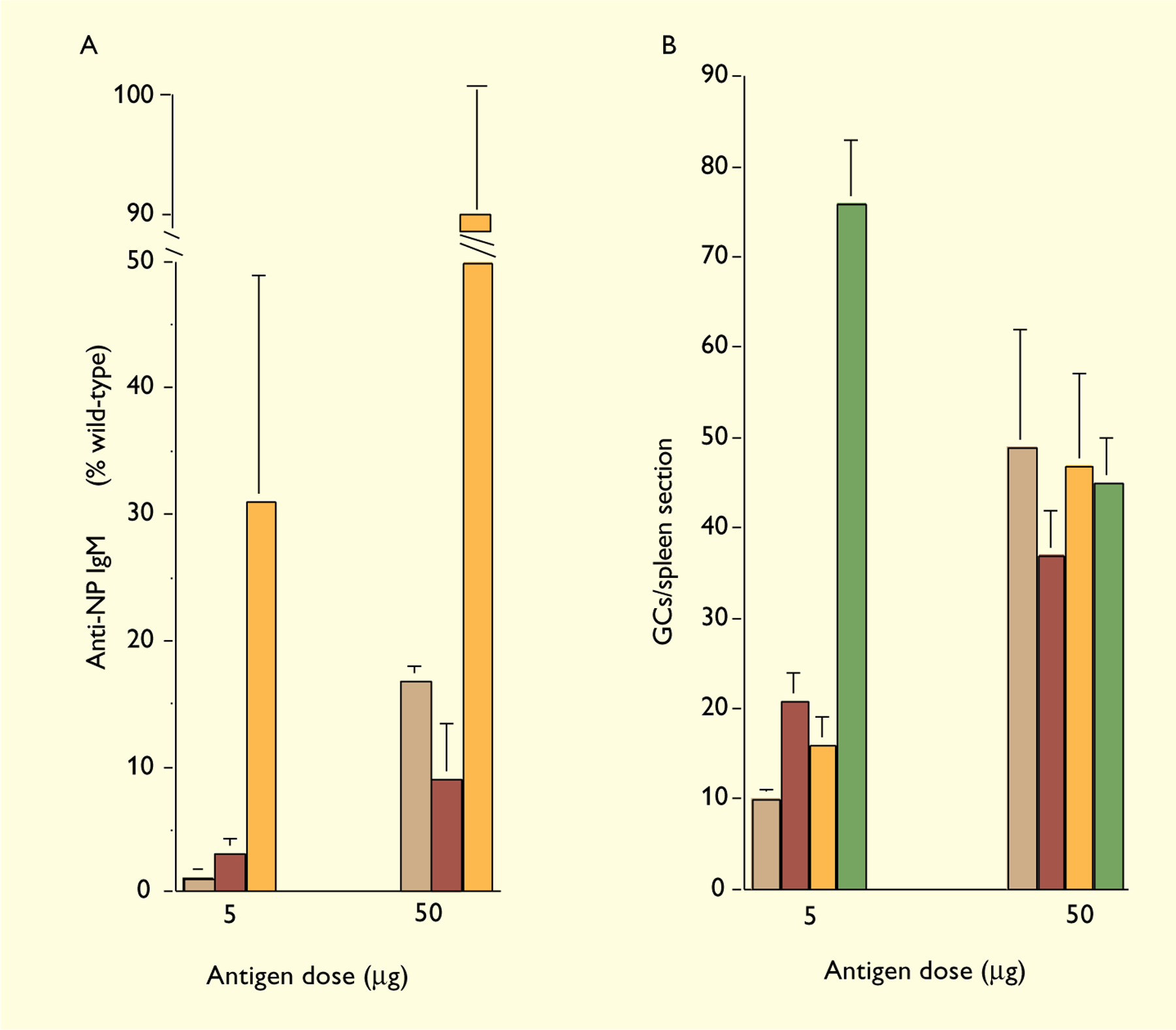
C57BL/6 (green), Cr2−/− (orange), H50G+/−Cr2+/+ (red) and H50G+/−Cr2−/− (brown) mice were immunized with 5 or 50 µg of NP12-CG precipitated in alum and sacrificed at day 10 post-immunization. NP-specific serum IgM was quantified by ELISA. The GC reaction was evaluated in individual mice by immunohistochemical staining with peanut agglutinin. GCs were enumerated under a microscope in two distant sections for each spleen. Each bar represents the mean (±SEM) numbers of GCs per section from 3 to 9 mice.
However, the affinity of early serum antibody (by day 16 post-immunization) in Cr2−/− mice was comparable to that in heterozygous and C57BL/6 controls, and early GC B cells populations in Cr2−/− mice are even less restricted to VH186.2. These observations indicate that the lack of CD21/CD35 does not limit the clonal diversity of B-cell responses; B cells expressing a broad range of BCR affinities can be activated and recruited into humoral immune responses (74).
In the presence of CD21 and CD35, NP-specific B cells with a wide range of affinities for NP, from 5×104 to 1.3×106 M−1, can be activated by antigens to become plasmacytes or participate in GC reactions (79). B1–8, a canonical NP-reactive antibody, which carries an H-chain encoded by a germline VH186.2–DFL16.1–JH2 rearrangement and a λ1 L-chain, binds NP with affinity of 9.6×105 M−1 (79). A point mutation at codon 50 (H50G) of this VDJ rearrangement lowers the affinity of the mutant antibody to 1.2×105 M−1. Despite its remarkably low affinity, B cells carrying this receptor survived and generated progeny in NP-specific GCs (79). To follow antigen-driven activation of low affinity B cells in vivo, we made transgenic mice, called H50G, carrying a transgene H-chain encoded by this mutant VDJ rearrangement and a μa constant region (79) on homozygous Ighb background. The response of transgenic B cells is followed with IgMa allotypic marker (J.M. Dal Porto, G. Kelsoe, unpublished data). After immunization with NP-CG, these transgenic mice generated NP-specific serum antibody that is λ1+ and IgMa+. The magnitude of the responses is reduced in comparison to wild-type mice (Fig. 2) (J. M. Dal Porto, G. Kelsoe, unpublished data).
To test the role of CD21/CD35 in antigen-driven activation of low affinity B cells in vivo, we bred the H50G transgene onto Cr2−/− background. H50G+/−Cr2−/− mice and H50G+/− Cr2+/+ controls were immunized intraperitoneally with 5 or 50 μg of NP-CG in alum. Serum antibody responses and GC reactions were estimated at day 10 post-immunization. NP-specific serum IgMa was determined by ELISA (79) and presented as a percentage of antibody levels in wild-type mice (Fig. 2A). At the antigen dose of 5 μg, IgM responses in Cr2−/− mice are about 3-fold lower than that in wild-type controls (Fig. 2A); surprisingly, despite of the very low affinity of the BCR, B cells in H50G+/−Cr2−/− mice generated IgMa responses that are still just about 3-fold lower than that in H50G+/−Cr2+/+ controls (1.1±0.7 vs 3.2±1.9 (±SEM)). When the antigen dose is increased 10-fold to 50 μg, there is no longer a significant difference between H50G+/−Cr2−/− mice and H50G+/−Cr2+/+ controls or Cr2−/− mice and wild-type controls (Fig. 2A). To estimate GC reactions, spleen sections were stained in tandem with horse-radish peroxidase conjugated peanut agglutinin and a biotinylated monoclonal anti–λ1 antibody, Ls136, followed by streptavidin–alkaline phosphatase as described elsewhere (80). As shown in Fig. 2B, 5 μg of NP-CG in alum elicits 5-fold less GCs in Cr2−/− mice (16±3) than in wild-type controls (76±6) and 2-fold less GCs in H50G+/−Cr2−/− mice (10±3) than in H50G+/−Cr2+/+ controls (21±5). Increasing antigen dose to 50 μg augmented GC reactions in Cr2−/− (47±10), H50G+/−Cr2+/+ (37±5), and H50G+/−Cr2−/− mice (49±13) to such an extent that GC reactions in these groups are comparable to wild-type mice (45±5) (Fig. 2B). Interestingly, GC reactions in wild-type mice were reduced when the antigen dose was increased from 5 to 50 μg (Fig. 2B), whereas IgM titers are not changed, and IgG1 antibody levels increased (74). Reduction in GC reactions to 50 μg of NP-CG was not observed in mice expressing low-affinity transgenic BCR and/or deficient in CD21/CD35, suggesting that excessive B-cell stimulation may limit the GC reactions, perhaps by apoptosis (81–83). This excessive stimulation may be contributed by interaction of high affinity BCR with abundant antigens retained by CD21 and CD35 on FDCs and/or synergism between BCR engagement and CD21/CD35 stimulation on B cells.
It was unexpected that the combination of a low-affinity BCR with deficiency in CD21/CD35 did not exacerbate the immune impairment by the loss of CD21/CD35 function. The co-receptor model of CD21/CD35 led us to hypothesize that presence of CD21/CD35 may lower the threshold of BCR affinity required for B-cell activation and thus diversify the repertoire of B-cell responses. However, in the absence of CD21/CD35, B cells with affinities as low as 1.2×105 M−1 to antigens were activated and generated antibody responses and GC reactions in vivo. Our observations suggest that CD21/CD35 may only have a limited, if any, role in promoting clonal diversity of humoral immunity. In most studies regarding the roles of complement and complement receptors in humoral immunity, effects of complement and complement receptors were more observable when antigens were delivered without adjuvant. For example, depletion of C3 with CoF in mice dramatically inhibited antibody responses to SRBC in the absence adjuvant (5); however, this treatment only modestly reduced antibody titers to protein antigens delivered with alum or incomplete Freund’s adjuvant (84). In our studies, alum was used as adjuvant to elicit NP-specific antibody responses and GC reactions. It is noteworthy that alum serves as an antigen depot, a function that may be simulated by CD21/CD35.
Acknowledgements
This work was supported in part by U. S. Public Health Service grants AI-24335, AG-13789, AG10207.
References
- 1.Kuby J. Immunology, 3rd ed. New York: Freeman; 1997. [Google Scholar]
- 2.Lay WH, Nussenzweig V. Receptors for complement of leukocytes. J Exp Med 1968;128:991–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianco C, Patrick R, Nussenzweig V. A population of lymphocytes bearing a membrane receptor for antigen-antibody-complement complexes. I. Separation and characterization. J Exp Med 1970;132:702–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nussenzweig V Receptors for immune complexes on lymphocytes. Adv Immunol 1974;19:217–258. [DOI] [PubMed] [Google Scholar]
- 5.Pepys MB. Role of complement in induction of the allergic response. Nat New Biol 1972;237:157–159. [DOI] [PubMed] [Google Scholar]
- 6.Ellman L, Green I, Judge F, Frank MM. In vivo studies in C4-deficient guinea pigs. J Exp Med 1971;134:162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson CG, Ochs HD, Wedgwood RJ. Immune response of a patient with deficiency of the fourth component of complement and systemic lupus erythematosus. N Engl J Med 1979;300:1124–1129. [DOI] [PubMed] [Google Scholar]
- 8.Ochs HD, Wedgwood RJ, Frank MM, Heller SR, Hosea SW. The role of complement in the induction of antibody responses. Clin Exp Immunol 1983;53:208–216. [PMC free article] [PubMed] [Google Scholar]
- 9.Bottger EC, Metzger S, Bitter-Suermann D, Stevenson G, Kleindienst S, Burger R. Impaired humoral immune response in complement C3-deficient guinea pigs: absence of secondary antibody response. Eur J Immunol 1986;16:1231–1235. [DOI] [PubMed] [Google Scholar]
- 10.Bitter-Suermann D, Burger R. Guinea pigs deficient in C2, C4, C3 or the C3a receptor. Prog Allergy 1986;39:134–158. [PubMed] [Google Scholar]
- 11.Fischer MB, et al. Regulation of the B cell response to T-dependent antigens by classical pathway complement. J Immunol 1996;157:549–556. [PubMed] [Google Scholar]
- 12.Ahearn JM, et al. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity 1996;4:251–262. [DOI] [PubMed] [Google Scholar]
- 13.Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol 1998;16:545–568. [DOI] [PubMed] [Google Scholar]
- 14.Carroll MC. CD21/CD35 in B cell activation. Semin Immunol 1998;10:279–286. [DOI] [PubMed] [Google Scholar]
- 15.Fearon DT, Carter RH. The CD19/CR2/TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu Rev Immunol 1995;13:127–149. [DOI] [PubMed] [Google Scholar]
- 16.Tuveson DA, Ahearn JM, Matsumoto AK, Fearon DT. Molecular interactions of complement receptors on B lymphocytes: a CR1/CR2 complex distinct from the CR2/CD19 complex. J Exp Med 1991;173:1083–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto AK, Kopicky-Burd J, Carter RH, Tuveson DA, Tedder TF, Fearon DT. Intersection of the complement and immune systems: a signal transduction complex of the B lymphocyte-containing complement receptor type 2 and CD19. J Exp Med 1991;173:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradbury LE, Kansas GS, Levy S, Evans RL, Tedder TF. The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody-1 and Leu-13 molecules. J Immunol 1992;149:2841–2850. [PubMed] [Google Scholar]
- 19.Krop I, Shaffer AL, Fearon DT, Schlissel MS. The signaling activity of murine CD19 is regulated during cell development. J Immunol 1996;157:48–56. [PubMed] [Google Scholar]
- 20.Tedder TF, Inaoki M, Sato S. The CD19-CD21 complex regulates signal transduction thresholds governing humoral immunity and autoimmunity. Immunity 1997;6:107–118. [DOI] [PubMed] [Google Scholar]
- 21.Carter RH, Spycher MO, Ng YC, Hoffman R, Fearon DT. Synergistic interaction between complement receptor type 2 and membrane IgM on B lymphocytes. J Immunol 1988;141:457–463. [PubMed] [Google Scholar]
- 22.Hivroz C, Fischer E, Kazatchkine MD, Grillot-Courvalin C. Differential effects of the stimulation of complement receptors CR1 (CD35) and CR2 (CD21) on cell proliferation and intracellular Ca2+ mobilization of chronic lymphocytic leukemia B cells. J Immunol 1991;146:1766–1772. [PubMed] [Google Scholar]
- 23.Hebell T, Ahearn JM, Fearon DT. Suppression of the immune response by a soluble complement receptor of B lymphocytes. Science 1991;254:102–105. [DOI] [PubMed] [Google Scholar]
- 24.Heyman B, Wiersma EJ, Kinoshita T. In vivo inhibition of the antibody response by a complement receptor-specific monoclonal antibody. J Exp Med 1990;172:665–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thyphronitis G, et al. Modulation of mouse complement receptors 1 and 2 suppresses antibody responses in vivo. J Immunol 1991;147:224–230. [PubMed] [Google Scholar]
- 26.Gustavsson S, Kinoshita T, Heyman B. Antibodies to murine complement receptor 1 and 2 can inhibit the antibody response in vivo without inhibiting T helper cell induction. J Immunol 1995;154:6524–6528. [PubMed] [Google Scholar]
- 27.Daha MR, Bloem AC, Ballieux RE. Immunoglobulin production by human peripheral lymphocytes induced by anti-C3 receptor antibodies. J Immunol 1984;132:1197–1201. [PubMed] [Google Scholar]
- 28.Weiss L, Delfraissy JF, Vazquez A, Wallon C, Galanaud P, Kazatchkine MD. Monoclonal antibodies to the human C3b/C4b receptor (CR1) enhance specific B cell differentiation. J Immunol 1987;138:2988–2993. [PubMed] [Google Scholar]
- 29.Molina H, et al. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci USA 1996;93:3357–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelsoe G. Life and death in germinal centers (redux). Immunity 1996;4:107–111. [DOI] [PubMed] [Google Scholar]
- 31.MacLennan IC. Germinal centers. Annu Rev Immunol 1994;12:117–139. [DOI] [PubMed] [Google Scholar]
- 32.Kopf M, Herren S, Wiles MV, Pepys MB, Kosco-Vilbois MH. Interleukin 6 influences germinal center development and antibody production via a contribution of C3 complement component. J Exp Med 1998;188:1895–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol 1998;160:5273–5279. [PubMed] [Google Scholar]
- 34.Bonnefoy JY, Henchoz S, Hardie D, Holder MJ, Gordon J. A subset of anti-CD21 antibodies promote the rescue of germinal center B cells from apoptosis. Eur J Immunol 1993;23:969–972. [DOI] [PubMed] [Google Scholar]
- 35.Klaus GG, Humphrey JH, Kunkl A, Dongworth DW. The follicular dendritic cell: its role in antigen presentation in the generation of immunological memory. Immunol Rev 1980;53:3–28. [DOI] [PubMed] [Google Scholar]
- 36.Mandel TE, Phipps RP, Abbot A, Tew JG. The follicular dendritic cell: long term antigen retention during immunity. Immunol Rev 1980;53:29–59. [DOI] [PubMed] [Google Scholar]
- 37.Schriever F, Nadler LM. The central role of follicular dendritic cells in lymphoid tissues. Adv Immunol 1992;51:243–284. [DOI] [PubMed] [Google Scholar]
- 38.Tew JG, Wu J, Qin D, Helm S, Burton GF, Szakal AK. Follicular dendritic cells and presentation of antigen and costimulatory signals to B cells. Immunol Rev 1997;156:39–52. [DOI] [PubMed] [Google Scholar]
- 39.Kozono Y, Duke RC, Schleicher MS, Holers VM. Co-ligation of mouse complement receptors 1 and 2 with surface IgM rescues splenic B cells and WEHI-231 cells from anti-surface IgM-induced apoptosis. Eur J Immunol 1995;25:1013–1017. [DOI] [PubMed] [Google Scholar]
- 40.Roberts T, Snow EC. Recruitment of the CD19/CD21 coreceptor to B cell antigen receptor is required for antigen-mediated expression of Bcl-2 by resting and cycling hen egg lysozyme transgenic B cells. J Immunol 1999;162:4377–4380. [PubMed] [Google Scholar]
- 41.Martinez-Valdez H, Guret C, de Bouteiller O, Fugier I, Banchereau J, Liu YJ. Human germinal center B cells express the apoptosis-inducing genes Fas, c-myc, P53, and Bax but not the survival gene bcl-2. J Exp Med 1996;183:971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuscano JM, Druey KM, Riva A, Pena J, Thompson CB, Kehrl JH. Bcl-x rather than Bcl-2 mediates CD40-dependent centrocyte survival in the germinal center. Blood 1996;88:1359–1364. [PubMed] [Google Scholar]
- 43.Takahashi Y, et al. Relaxed negative selection in germinal centers and impaired affinity maturation in bcl-xL transgenic mice. J Exp Med 1999;190:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer MB, et al. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science 1998;280:582–585. [DOI] [PubMed] [Google Scholar]
- 45.Van Noesel CJ, Lankester AC, van Lier RA. Dual antigen recognition by B cells. Immunol Today 1993;14:8–11. [DOI] [PubMed] [Google Scholar]
- 46.Papamichail M, Gutierrez C, Embling P, Johnson P, Holborow EJ, Pepys MB. Complement dependence of localisation of aggregated IgG in germinal centres. Scand J Immunol 1975;4:343–347. [DOI] [PubMed] [Google Scholar]
- 47.White RG, Henderson DC, Eslami MB, Neilsen KH. Localization of a protein antigen in the chicken spleen. Effect of various manipulative procedures on the morphogenesis of the germinal centre. Immunology 1975;28:1–21. [PMC free article] [PubMed] [Google Scholar]
- 48.Klaus GG, Humphrey JH. The generation of memory cells. I. The role of C3 in the generation of B memory cells. Immunology 1977;33:31–40. [PMC free article] [PubMed] [Google Scholar]
- 49.Neuberger MS, Rajewsky K. Activation of mouse complement by monoclonal mouse antibodies. Eur J Immunol 1981;11:1012–1016. [DOI] [PubMed] [Google Scholar]
- 50.Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol 1988;18:313–316. [DOI] [PubMed] [Google Scholar]
- 51.Unkeless JC, Scigliano E, Freedman VH. Structure and function of human and murine receptors for IgG. Annu Rev Immunol 1988;6:251–281. [DOI] [PubMed] [Google Scholar]
- 52.Vora KA, Ravetch JV, Manser T. Amplified follicular immune complex deposition in mice lacking the Fc receptor y-chain does not alter maturation of the B cell response. J Immunol 1997;159:2116–2124. [PubMed] [Google Scholar]
- 53.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc y RII-deficient mice. Nature 1996;379:346–349. [DOI] [PubMed] [Google Scholar]
- 54.Qin D, Wu J, Carroll MC, Burton GF, Szakal AK, Tew JG. Evidence for an important interaction between a complement-derived CD21 ligand on follicular dendritic cells and CD21 on B cells in the initiation of IgG responses. J Immunol 1998;161:4549–4554. [PubMed] [Google Scholar]
- 55.Humphrey JH, Grennan D, Sundaram V. The origin of follicular dendritic cells in the mouse and the mechanism of trapping of immune complexes on them. Eur J Immunol 1984;14:859–864. [DOI] [PubMed] [Google Scholar]
- 56.Croix DA, et al. Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J Exp Med 1996;183:1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science 1992;256:105–107. [PubMed] [Google Scholar]
- 58.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 1996;271:348–350. [DOI] [PubMed] [Google Scholar]
- 59.Erdei A, Melchers F, Schulz T, Dierich M. The action of human C3 in soluble or cross-linked form with resting and activated murine B lymphocytes. Eur J Immunol 1985;15:184–188. [DOI] [PubMed] [Google Scholar]
- 60.Melchers F, Erdei A, Schulz T, Dierich MP. Growth control of activated, synchronized murine B cells by the C3d fragment of human complement. Nature 1985;317:264–267. [DOI] [PubMed] [Google Scholar]
- 61.Frade R, et al. Enhancement of human B cell proliferation by an antibody to the C3d receptor, the gp 140 molecule. Eur J Immunol 1985;15:73–76. [DOI] [PubMed] [Google Scholar]
- 62.Wilson BS, Platt JL, Kay NE. Monoclonal antibodies to the 140,000 mol wt glycoprotein of B lymphocyte membranes (CR2 receptor) initiates proliferation of B cells in vitro. Blood 1985;66:824–829. [PubMed] [Google Scholar]
- 63.Nemerow GR, McNaughton ME, Cooper NR. Binding of monoclonal antibody to the Epstein Barr virus (EBV)/CR2 receptor induces activation and differentiation of human B lymphocytes. J Immunol 1985;135:3068–3073. [PubMed] [Google Scholar]
- 64.Tedder TF, Weis JJ, Clement LT, Fearon DT, Cooper MD. The role of receptors for complement in the induction of polyclonal B-cell proliferation and differentiation. J Clin Immunol 1986;6:65–73. [DOI] [PubMed] [Google Scholar]
- 65.Carter RH, Fearon DT. Polymeric C3dg primes human B lymphocytes for proliferation induced by anti-IgM. J Immunol 1989;143:1755–1760. [PubMed] [Google Scholar]
- 66.Tsokos GC, Lambris JD, Finkelman FD, Anastassiou ED, June CH. Monovalent ligands of complement receptor 2 inhibit whereas polyvalent ligands enhance anti-Ig-induced human B cell intracytoplasmic free calcium concentration. J Immunol 1990;144:1640–1645. [PubMed] [Google Scholar]
- 67.Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity 1995;3:39–50. [DOI] [PubMed] [Google Scholar]
- 68.Zhou LJ, Smith HM, Waldschmidt TJ, Schwarting R, Daley J, Tedder TF. Tissue-specific expression of the human CD19 gene in transgenic mice inhibits antigen-independent B-lymphocyte development. Mol Cell Biol 1994;14:3884–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato S, Steeber DA, Jansen PJ, Tedder TF. CD19 expression levels regulate B lymphocyte development: human CD19 restores normal function in mice lacking endogenous CD19. J Immunol 1997;158:4662–4669. [PubMed] [Google Scholar]
- 70.Kelsoe G In situ studies of the germinal center reaction. Adv Immunol 1995;60:267–288. [DOI] [PubMed] [Google Scholar]
- 71.Makela O, Karjalainen K. Inherited immunoglobulin idiotypes of the mouse. Immunol Rev 1977;34:119–138. [DOI] [PubMed] [Google Scholar]
- 72.Reth M, Hämmerling GJ, Rajewsky K. Analysis of the repertoire of anti-NP antibodies in C57BL/6 mice by cell fusion. I. Characterization of antibody families in the primary and hyperimmune response. Eur J Immunol 1978;8:393–400. [DOI] [PubMed] [Google Scholar]
- 73.Jacob J, Przylepa J, Miller C, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. J Exp Med 1993;178:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Z, Koralov SB, Gendelman M, Carroll MC, Kelsoe G. Humoral immune responses in Cr2−/− mice: enhanced affinity maturation but impaired antibody persistence. J Immunol 2000;164:4522–4532. [DOI] [PubMed] [Google Scholar]
- 75.Slifka MK, Matloubian M, Ahmed R. Bone marrow is a major site of long-term antibody production after acute viral infection. J Virol 1995;69:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature 1997;388:133–134. [DOI] [PubMed] [Google Scholar]
- 77.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J Exp Med 1998;187:885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity 1998;8:363–372. [DOI] [PubMed] [Google Scholar]
- 79.Dal Porto JM, Haberman AM, Shlomchik MJ, Kelsoe G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J Immunol 1998;161:5373–5381. [PubMed] [Google Scholar]
- 80.Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med 1991;173:1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pulendran B, Smith KG, Nossal GJ. Soluble antigen can impede affinity maturation and the germinal center reaction but enhance extrafollicular immunoglobulin production. J Immunol 1995;155:1141–1150. [PubMed] [Google Scholar]
- 82.Shokat KM, Goodnow CC. Antigen-induced B-cell death and elimination during germinal-centre immune responses. Nature 1995;375:334–338. [DOI] [PubMed] [Google Scholar]
- 83.Han S, Zheng B, Dal Porto J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. IV. Affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J Exp Med 1995;182:1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pepys MB. Role of complement in induction of antibody production in vivo. Effect of cobra factor and other C3-reactive agents on thymus-dependent and thymus-independent antibody responses. J Exp Med 1974;140:126–145. [DOI] [PMC free article] [PubMed] [Google Scholar]