

Abstract
A major component of the anticarcinogenic activity of the dietary chemopreventive agent sulforaphane (SFN) is attributed to its ability to induce expression of phase II detoxification genes containing the antioxidant response element (ARE) within their promoters. Because SFN is a reactive electrophile––readily forming conjugates with glutathione (GSH)––we asked whether expression of glutathione S-transferase (GST) P1–1 and the GSH conjugate efflux pump, multidrug resistance or resistance-associated protein (MRP) 1, would significantly modify the cellular response to SFN exposure. This was investigated using GST- and MRP1-poor parental MCF7 cells and transgenic derivatives expressing GSTP1–1 and/or MRP1. Compared with parental cells, expression of GSTP1–1 alone enhanced the rate of intracellular accumulation of SFN and its glutathione conjugate, SFN-SG––an effect that was associated with increased ARE-containing reporter gene induction. Expression of MRP1 greatly reduced SFN/SFN-SG accumulation and resulted in significant attenuation of SFN-mediated induction of ARE-containing reporter and endogenous gene expression. Coexpression of GSTP1–1 with MRP1 further reduced the level of induction. Depletion of GSH prior to SFN treatment or the substitution of tert-butylhydroquinone for SFN abolished the effects of MRP1/GSTP1–1 on ARE-containing gene induction—indicating that these effects are GSH dependent. Lastly, analysis of NF-E2-related factor 2 (Nrf2)––a transcription factor operating via binding to the ARE––showed that the increased levels of Nrf2 following SFN treatment were considerably less sustained in MRP1-expressing, especially those coexpressing GSTP1–1, than in MRP1-poor cells. These results suggest that the regulating effects of MRP1 and GSTP1–1 expression on SFN-dependent induction of phase II genes are ultimately mediated by altering nuclear Nrf2 levels.
Introduction
The isothiocyanates (ITCs), such as sulforaphane (SFN), are important dietary cancer chemopreventive agents found abundantly in cruciferous vegetables (1,2). Epidemiological and animal data reveal an inverse correlation between consumption of dietary chemopreventive agents, such as ITCs, and the risk of developing cancer (2–4). While there is evidence that ITCs may have direct anticancer activities through inhibition of cell growth and stimulation of apoptosis in susceptible cancer cells (5–7), an extensive literature suggests that a major component of the chemopreventive activity of ITCs occurs via induction of genes encoding phase II detoxification enzymes (8–10)––a diverse family of enzymes that can metabolize a variety of reactive carcinogens, mutagens and other toxins (11). The phase II genes that are induced contain antioxidant (or electrophile) response elements (AREs or EpRE) in their promoters and include genes encoding NAD(P)H:quinone oxidoreductase (NQO1), glutathione S-transferases (GSTs), uridine 5-diphosphate-glucuronosyl transferase, epoxide hydrolase, ferritin, γ-glutamate-cysteine ligase and catalase (11).
The ITCs can enhance phase II gene expression by increasing the levels and activation of the NF-E2-related factor 2 (Nrf2) transcription factor. Nuclear Nrf2 can heterodimerize with partner transcription factors, bind AREs and thereby activate transcription of target phase II genes (12–14). Under basal conditions, Nrf2 interacts with the Kelch ECH-associating protein 1 (Keap1) (13). Recent evidence indicates that Keap1 controls the stability of Nrf2 protein (15)––serving as an adapter protein which links Nrf2 to E3 ubiquintin ligase thus targeting Nrf2 for proteosomal degradation (9,16). It is widely believed that electrophiles such as ITCs disrupt Nrf2–Keap1 interactions by reacting with key free thiols within the cysteine-rich Keap1 proteins (17–19). Nrf2 so freed from Keap1 and proteosomal degradation is stabilized, accumulates and is translocated to the nucleus where it can enhance phase II gene transcription. In addition, ARE-containing gene expression can be regulated by activation of mitogen-activated protein kinase pathways, direct phosphorylation of Nrf2 by protein kinase C or PKR-like ER kinase or by the actions of other transcription factors and transcription factor modulators (20–24).
The goal of this study is to understand some of the cellular factors that may differentially govern the chemopreventive response to ITC exposure of various cells and tissues. In particular, we examined the role of glutathione (GSH)-dependent metabolism and efflux of the ITC, SFN, in modulating SFN bioactivity––especially, its ability to induce ARE-dependent gene expression. Prior studies have shown that ITCs readily undergo reversible Michael addition reactions with cellular nucleophiles including protein thiols and GSH. The reaction with GSH occurs both enzymatically (GST catalyzed) and non-enzymatically at physiological pH (25,26). Additionally, there is evidence that ITCs and/or their GSH conjugates, dithiocarbamates, are transported from cells and the role of multidrug resistance or resistance-associated protein (MRP) is implicated in this transport (27,28). Here, we examine the influence of GSTP1–1 and the GSH-conjugate efflux pump, MRP1, on cellular response to SFN using model transgenic MCF7 cell lines expressing GSTP1–1 and MRP1 alone or in combination. Specifically, we test the hypothesis that––by affecting the dynamics of SFN conjugation and conjugate [glutathione conjugate of sulforaphane (SFN-SG)] efflux––the levels of GSTP1–1 and MRP1 expressed at the time of SFN exposure will modulate the acute cellular response to SFN treatment. The studies described support the ideas that (i) MRP1 and GSTP1–1 influence ARE-dependent gene expression by a mechanism that involves altered intracellular SFN/SFN-SG accumulation and (ii) modulation of ARE-containing gene expression by MRP1/GSTP1–1 expression is ultimately mediated by changes in the nuclear Nrf2 levels.
Materials and methods
Cell lines and culture
We have described previously the construction and analysis of stably transduced derivatives of parental MCF7 cells (MCF7/wild type (WT) or WT): these include transduced MCF7 cells expressing the I104, A113 allele of human GSTP1–1, GSTP1a-1a (MCF7/WTπ or WTπ); MRP1 (MCF7/MRP1 or MRP1) and GSTP1a-1a plus MRP1 (MCF7/MRP1π or MRP1π) (29,30). All cell lines were grown in Dulbecco’s modified Eagle medium plus 10% fetal calf serum and penicillin–streptomycin at 37°C, 5% CO2. Transgenic derivatives were maintained in selecting drugs (1.5 mg/ml G418, MCF7/MRP1 and MCF7/MRP1π cells; 0.2 mg/ml hygromycin, MCF7/WTπ and MCF7/MRP1π cells) until several days prior to planned experiments at which time drugs were removed from the medium. GST activities (31) and MRP1 expression (32) were measured periodically and remained stable in the four cell lines throughout the studies: GST activity was <5 nmol/min/mg in MCF7/WT and MCF7/MRP1 cells and was 250–350 nmol/min/mg in MCF7/WTπ and MCF7/MRP1π cells. MRP1 protein (western blot), undetectable in MCF7/WT and MCF7/WTπ cells, was consistently and uniformly high in both MCF7/MRP1 and MCF7/MRP1π cells. Previously, we have examined in MCF7 cells the levels of other MRP family proteins, MRP2–6 (32–35): the studies showed that the expression levels of these MRP proteins are low to absent in parental MCF7/WT cells––in particular, MRP2 (protein and messenger RNA) is undetectable. SFN cytotoxicity experiments were accomplished using the sulforhodamine B assay as described previously (36). In these experiments, cells were exposed to varying concentrations of D,L-SFN (LKT Laboratories, St Paul, MN)––freshly diluted in dimethylsulfoxide––for 3 h, medium was replaced with fresh SFN-free medium and incubations were continued for 6 days.
Synthesis and MRP1-mediated transport of the GSH conjugate of SFN and SFN-SG
Synthesis of radiolabeled SFN-SG was adapted from the method described by Kolm et al. (25) and reaction progress was monitored by the Δ absorbance at 274 nm. For 100 μl reactions, solutions containing 100 μM GSH (1 μCi/nmol [glycine-2-3H]-glutathione, PerkinElmer, Waltham, MA) in 40 mM sodium phosphate (pH 8.4) were extracted three times with 250 μl ethyl acetate. The reactions were initiated in the aqueous phase by the addition of SFN to a final concentration of 500 μM. The reactions were complete within 10 min at 25°C. The SFN-SG was separated by reverse phase high-performance liquid chromatography (C18, Beckman, Ultrasphere, Beckman Coulter, Fullerton, CA ODS, 5 μm, 4.6 × 250 mm). Chromatography was accomplished at 1 ml/min using linear gradients of 100% solvent A to 89% solvent A/11% solvent B over 14 min and then 89% A/11% B to 21% A/79% B over the next ~14 min (where solvent A is 5% acetonitrile, 0.05% trifluoroacetic acid and solvent B is 100% acetonitrile). Chromatograms were monitored at 250 nm and the conjugate, eluting as a single peak at ~ 14 min, was collected and dried under N2.
MRP1-mediated, adenosine triphosphate (ATP)-dependent transport of [3H]-SFN-SG was determined using inside-out membrane vesicles derived from MRP1-expressing MCF7 cells (MCF7/MRP1) as described previously (37). ATP dependence was determined by subtracting isotope uptake by membrane vesicles incubated with 4 mM β,γ-methyleneadenosine 5′-triphosphate (non-hydrolyzable ATP analog) from uptake by vesicles incubated with 4 mM ATP. In control experiments, [3H]-SFN-SG uptake was determined using inside-out vesicles derived from MRP1-poor, MCF7/WT cells. Initial velocities of uptake were fitted to the Michaelis–Menten equation with Synergy Kaleida-Graph software and used to calculate kinetic parameters.
Analysis of intracellular SFN/SFN-SG and GSH levels in SFN-treated cells
To determine the effect of MRP1 and GSTP1–1 expression on intracellular accumulation of SFN and SFN-SG, cells (3 × 105 per well) were plated in replicate 12-well tissue culture dishes. Twenty-four hours later, cells were treated for1h at 37°C, 5% CO2 in medium containing 25 μM SFN. Cells were harvested at periodic intervals throughout the incubations; plates were chilled on ice and cells were rinsed 4× with 1 ml of ice-cold phosphate-buffered saline. Washed cells were lysed directly on tissue culture plates in 100 μl β-mercaptoethanol/N-lauryl sarcosine solution (38) (1 h 37°C with periodic shaking) and processed for intracellular SFN plus SFN-SG levels by the cyclocondensation reaction developed by Zhang et al. (38). Briefly, lysates were transferred to microfuge tubes, centrifuged at 12 000g for 5 min at 25°C and the supernatants saved for the determination of protein (39) and for SFN/SFN-SG levels. SFN/SFN-SG levels were determined by their quantitative reaction with 1,2-benzenedithiol to generate the chromophore, 1,3-benzodithiole-2-thione (BTT), as outlined (38). The BTT was separated by reverse phase high-performance liquid chromatography (C18, Beckman, Ultrasphere ODS, 5 μm, 4.6 × 250 mm) using an isocratic 80% methanol elution at 1.3 ml/min. Chromatograms were monitored at 365 nm with BTT peaks eluting at ~6 min. SFN/SFN-SG levels were calculated for unknown samples by comparing the integrated BTT peak areas of unknown samples with integrated peak areas of BTT derived from cyclocondensation reactions with known quantities of phenyl ITC standards. Intracellular GSH levels were determined in control and SFN-treated cells using the method described by Tietze (40). Cells (3 × 106 per plate) were seeded on replicate 150 mm plates. Twenty-four hours later, cells were treated with 25 μM SFN for 1 h. Cells were harvested at intervals during the 1 h incubation for determination of total intracellular GSH. The basal GSH levels in unstressed, untreated (no SFN) cells were similar among the four cell lines used: these levels ranged from 7 to 12 mM and were comparable with those reported previously for MCF7 derivatives by our laboratory (41).
Induction of ARE-dependent reporter and endogenous gene expression
For reporter gene analysis of ARE-dependent transcription activation, cells were plated in replicate six-well tissue culture dishes at a density of 1 × 105 cells per well. Twenty-four hours later, cells were cotransfected with 0.5 μg of the ARE-containing firefly luciferase reporter gene, pARE-TI-LUC (42), and 5 pg of the control CMV-Renilla luciferase reporter gene, pGL4.75 (Promega, Madison, WI) using Superfect reagent (Qiagen, Valencia, CA) according to the manufacturer’s recommendations. Medium was replaced after a 3 h exposure to the transfected reporter DNA. After overnight incubation, cells were treated with medium containing inducing agent [25 μM SFN or 50 μM tert-butylhydroquinone (tBHQ)] or vehicle control (dimethysulfoxide) for 3 h. Medium was replaced with inducing agent-free medium and cells were harvested for luciferase assays 21 h later. Firefly and Renilla luciferase activities were determined using the Dual Luciferase Assay System (Promega) and a Turner TD 20/20 luminometer. Renilla luciferase activities were used to correct firefly pARE-TI-LUC reporter gene activities for variation in transfection efficiencies and non-specific induction. The pARE-TI-LUC vector was constructed with the 41 bp ARE-containing insert derived from the murine gsta1 gene linked to a TATA-Inr minimal promoter as described by Wasserman et al. (42).
For analysis of endogenous ARE-dependent gene induction, 2 × 106 cells were seeded on 100 mm dishes and 24 h later treated with medium containing 25 μM SFN or vehicle control for 3 h. Medium was replaced with SFN-free medium and cells were harvested for total RNA preparation and northern blot analysis as described previously (43). The northern blot was probed with a cDNA insert, containing the entire coding region of human NQO1, which was labeled with [α−32P]-dcytidine 5-triphosphate by random primer labeling (43). The cDNAwas derived from MCF7 cells by real-time polymerase chain reaction of total cellular RNA using the following oligonucleotides: 5′-CACGAGCCCAGCCAATCA-3′ and 5′-CCAGGATAAGGAATCTCA-3′. The ARE located within the human NQO1 ARE contains an embedded AP-1 site, whereas the murine gsta1 ARE, found in the pARE-TI-LUC reporter, does not (44).
Analysis of cytoplasmic and nuclear Nrf2
Cells were plated at a density of 2 × 106 per 100 mm dish. Twenty-four hours later, cells were treated for 3 h in medium containing 25 μM SFN or vehicle control. After the 3 h exposure, medium was replaced with SFN-free medium and incubations continued for 2–18 h before harvesting. Cells were rinsed and pelleted in phosphate-buffered saline. Cell pellets were suspended in ~3 volumes of ice-cold hypotonic buffer [20 mM potassium 4-(2-hydroxyethyl)-1-piperazineethanesulfonate (pH 7.9), 10 mM KCl, 1 mM ethylenediaminetetraacetic acid, 10% glycerol, 1 mM dithiothreitol, 0.2% NP40, 1 mM phenylmethylsulfonyl fluoride, 50 μg/ml aprotin, 0.5 μg/ml leupeptin and 1 μg/ml pepstatin]. Hypotonic buffer was added slowly with continuous gentle vortexing, which was continued for 10 s after the addition of buffer. This lysate was incubated on ice for 5 min and was then pelleted by microcentrifugation at full speed for 10 s. The cytosolic supernatant was removed and saved while the nuclear pellet was suspended in ~3 volumes of ice-cold extraction buffer (hypotonic buffer supplemented with glycerol and 5 M NaCl to give final concentrations of 20% and 420 mM, respectively) added slowly with continuous gentle vortexing. The sample was held on ice for 30 min with periodic gentle vortexing every 5–10 min. The sample was centrifuged at 12 000g, 5 min, 4°C and the nuclear extract (supernatant) was collected. The nuclear and cytoplasmic fractions were processed for protein assay (39) and western blotting.
Cytoplasmic and nuclear extracts (50 μg protein per sample) were separated by 8% acrylamide sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to Hybond-ECL nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). Western analysis was accomplished using rabbit anti-Nrf2 primary antibody (H300, Santa Cruz Biotechnology, Santa Cruz, CA), horseradish peroxidase-conjugated goat anti-rabbit secondary antibody and enhanced chemiluminescence (PerkinElmer) using standard methods (29). To verify similar loading and transfer efficiencies, blots were stripped and reprobed with anti-human actin (C2, Santa Cruz Biotechnology) or anti-human topoisomerase I (23B11, Alexis, San Diego, CA) primary antibodies and the appropriate horseradish peroxidase-conjugated secondary antibodies.
Results
MRP1 and GSTP1–1 influence the dynamics of SFN and SFN-SG accumulation and efflux
Previous work has indicated that GSH conjugates of ITCs are actively transported out of cells (28). It was expected that the GSH-conjugate efflux pump, MRP1, could support this active efflux of SFN-SG. To verify that this is true and to estimate the efficiency of efflux, the kinetics of ATP-dependent, MRP1-mediated transport of SFN-SG was measured using inside-out plasma membrane vesicles. As shown in Figure 1, vesicles derived from MRP1-expressing MCF7 cells support robust ATP-dependent transport of SFN-SG, whereas vesicles derived from MRP1-poor MCF7 cells do not. From these data, the calculated kinetic parameters of SFN-SG transport are KM = 18 μM, Vmax = 160 pmol/min/mg protein (Figure 1) and for the transport of the GSH conjugate of another ITC, phenethyl ITC, are KM = 9 μM, Vmax = 250 pmol/min/mg. These kinetic values are comparable with the values obtained, using similar vesicle preparations, for other GSH conjugates that are efficiently transported by MRP1 (45).
Fig. 1.
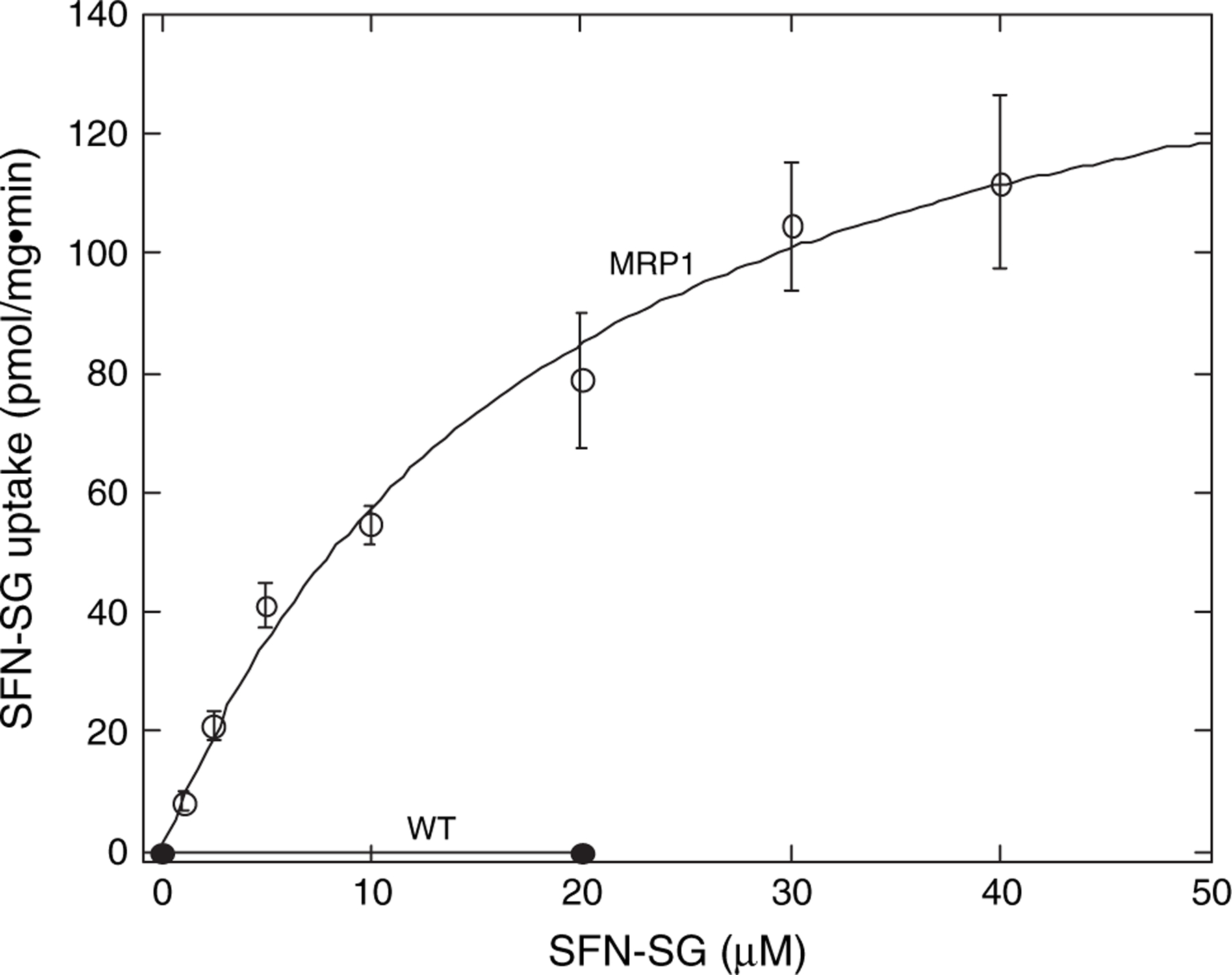
MRP1-mediates efficient ATP-dependent transport of SFN-SG. Shown are the initial velocities of ATP-dependent uptake of radiolabeled SFN-SG by inside-out membrane vesicles derived from MRP1-rich MCF7/MRP1 (MRP1) or MRP1-poor MCF7/WT (WT) cells. Points represent the means of triplicate determinations and the error bars indicate ±1 SD.
While GSH conjugation with SFN occurs readily and non-enzymatically at physiological pH, cytosolic GSTs can greatly accelerate these reactions with a reported catalytic rate enhancement factor for GSTP1–1 of 4.7 × 105 (25). Having shown that MRP1 can support active efflux of these conjugates, we asked whether and by how much expression of GSTP1–1 and MRP1 could influence the intracellular accumulation of SFN/SFN-SG and depletion of GSH in cells exposed to SFN. In MRP1-poor cells [MCF7/WT (WT) and MCF7/WTπ (WTπ)] treated with 25 μM SFN, SFN and SFN-SG accumulation was rapid and sustained throughout the 1 h exposure period. The expression of GSTP1–1 alone [MCF7/WTπ (WTπ)] supported the greatest initial rate of accumulation following exposure to SFN––a result attributable to GST catalysis of SFN-SG formation (Figure 2A). In contrast, expression of MRP1 [MCF7/MRP1 (MRP1) and MCF7/MRP1π (MRP1π) cells] resulted in considerably lower levels of SFN/SFN-SG accumulation throughout the 1 h exposure period. It should be noted that the cyclocondensation reaction used does not distinguish between conjugated and unconjugated ITCs. However, as the lipophilic SFN is expected to readily equilibrate across the plasma membrane whereas its amphiphilic conjugate, SFN-SG, is not, the large accumulation in MRP1-poor cells most likely represents predominantly the membrane-impermeable SFN-SG.
Fig. 2.
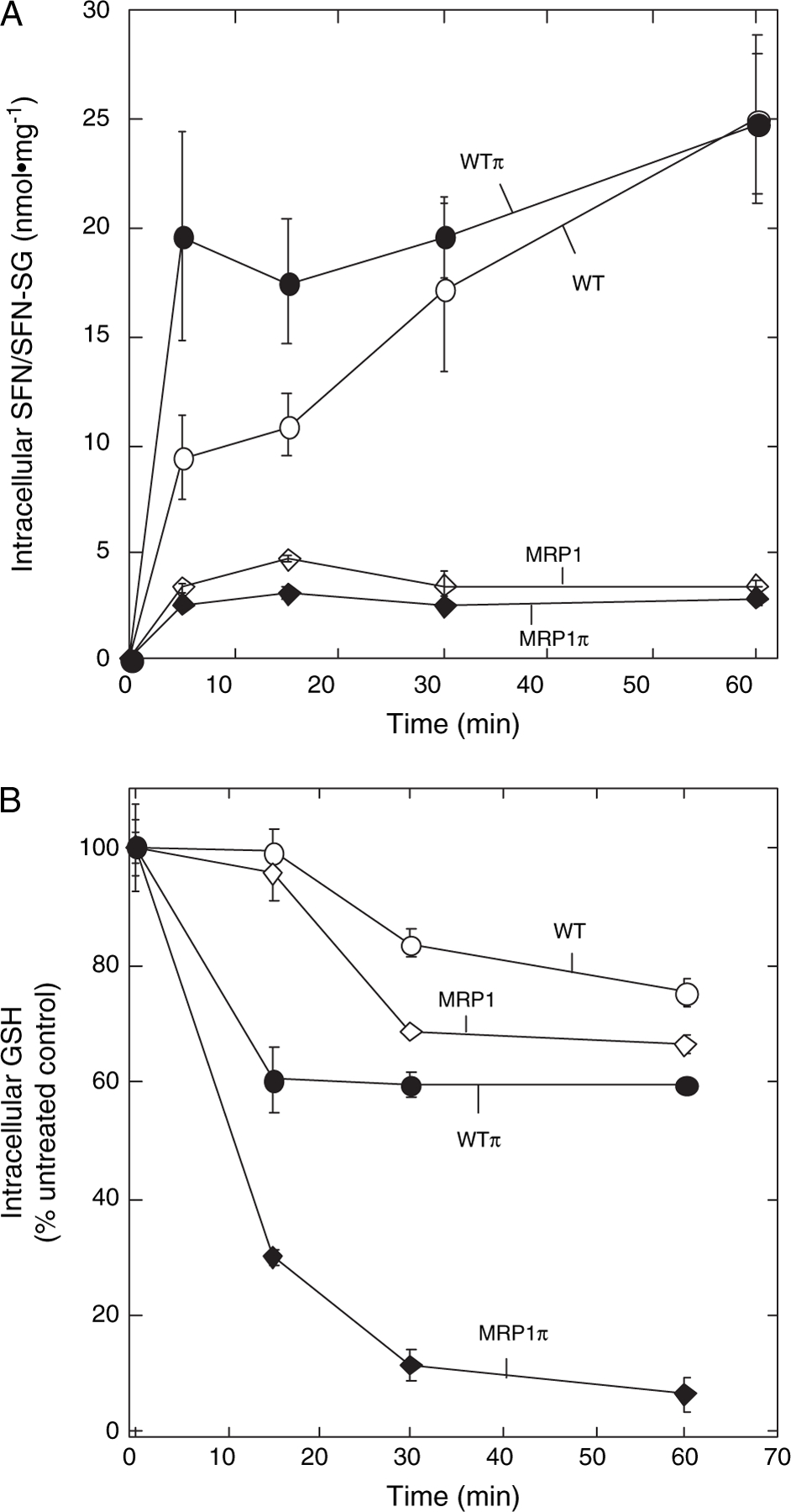
Effect of MRP1 and GSTP1–1 expression on intracellular accumulation of SFN, its GSH conjugate (SFN-SG) and intracellular GSH depletion following treatment with 25 μM SFN. (A) Cells were treated with 25 μM SFN for 1 h and harvested at the indicated times during the 1 h incubation for determination of total SFN (free, SFN, plus conjugated, SFN-SG) using the cyclocondensation assay as described in Materials and Methods. Indicated are parental MCF7 cells (MCF7/WT, WT; open circles) lacking MRP1 and GSTP1–1 and transgenic derivatives expressing GSTP1–1 alone (MCF7/WTπ, WTπ; closed circles), MRP1 alone (MCF7/MRP1, MRP1; open diamonds) or GSTP1–1 and MRP1 in combination (MCF7/MRP1π, MRP1π; closed diamonds). SFN/SFN-SG levels are expressed as nmol/mg total cellular protein (means of triplicate determinations ±1 SE). (B) Parental and transgenic MCF7 derivatives expressing GSTP1–1 and MRP1, alone or in combination, were treated with 25 μM SFN and harvested at the indicated times for total GSH determinations as described in Materials and Methods. Intracellular GSH levels (means of triplicate determinations ±1 SD are expressed as the percentage of untreated control cells.
GSTP1–1 can catalyze SFN conjugation with GSH and may, therefore, significantly alter the rate of intracellular GSH consumption in cells treated with SFN. Since changes in intracellular GSH can influence sensitivity to redox/electrophile stress (including stress due to SFN exposure), we investigated whether the increased rate of SFN-SG formation in GSTP1–1-expressing cells would be associated with measurable differences in the rates of GSH depletion. As shown in Figure 2B, GSTP1–1 expression ± MRP1 (WTπ and MRP1π cells) resulted in higher initial rates of GSH depletion than in cells that lacked GSTP1–1 (WT and MRP1). Although expression of GSTP1–1 alone (WTπ) was associated with increased initial rates of GSH depletion, the extent of GSH depletion at later times was similar to GSTP1–1-minus cells (WT and MRP1). In contrast, coexpression of MRP1 with GSTP1–1 (MRP1π) resulted in marked increases in both the rate and extent of GSH depletion.
Consequences on SFN bioactivities of altered SFN/SFN-SG and GSH dynamics mediated by MRP1 and GSTP1–1 expression
At higher exposure levels, ITCs are known cellular toxins. To determine whether the altered SFN and GSH metabolism associated with MRP1 and GSTP1–1 expression would affect sensitivity to SFN cytotoxicity, the four cell lines were exposed to varying concentration of SFN for 3 h. Very little toxicity was observed in any of the cell lines at SFN concentrations ≤ 25 μM SFN (Figure 3). Moreover, despite significant differences in SFN and GSH metabolism, there were no consistent differences in sensitivity to SFN cytotoxicity at higher exposure levels among the four cell lines. Additionally, no differences in relative sensitivities to SFN cytotoxicity were observed among the four cell lines even when the duration of SFN exposure was varied between 1 and 24 h (not shown).
Fig. 3.
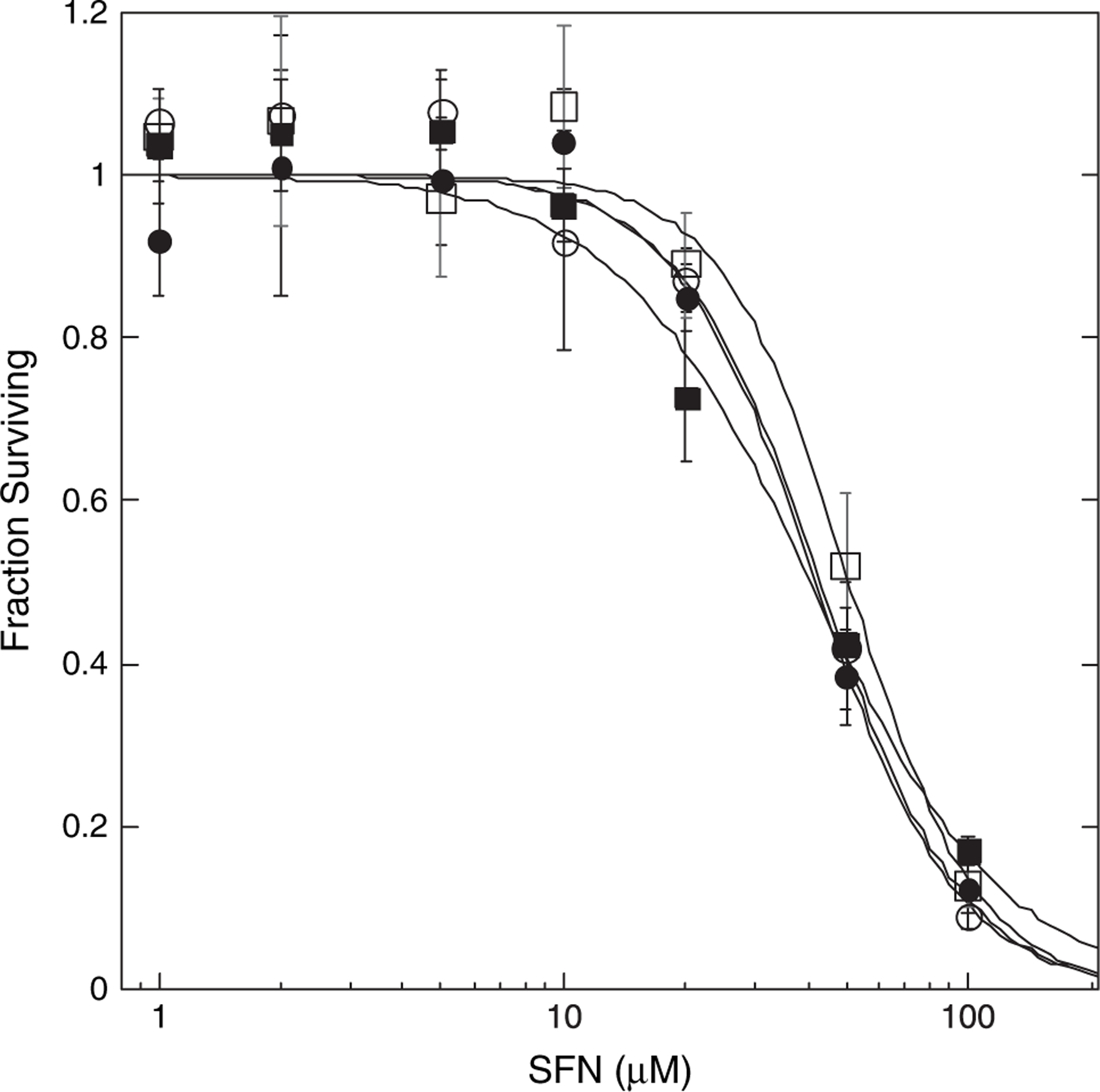
Cytotoxicity of SFN in parental and transgenic MCF7 cells. Cells were exposed to varying concentrations of SFN or vehicle control for 3 h after which medium was replaced with SFN-free medium. Cells were processed 6 days later for cytotoxicity assay using the sulforhodamine B method. Data are expressed as fractions of control (−SFN) surviving. Shown are the means of eight replicate determinations ±1 SD for MCF7/WT (open circles), MCF7/WTπ (closed circles), MCF7/MRP1 (open squares) and MCF7/MRP1π (closed squares) cells.
We next examined the effect of MRP1 and GSTP1–1 expression on SFN-mediated induction of ARE-containing gene expression. While ARE-containing reporter gene induction was observed in MCF7/WT cells treated with a range of SFN concentrations (5–50 μM, not shown), 25 μM was chosen for these and subsequent experiments as a dosage that supported robust induction with minimal cytotoxicity. Results using the ARE-containing reporter gene transiently transfected into the four cell lines are summarized in Figure 4A. The most striking finding was that expression of MRP1 (MCF7/MRP1 cells) resulted in significant attenuation of the SFN-mediated induction observed in MRP1-poor MCF7/WT cells. Coexpression of GSTP1–1 with MRP1 (MCF7/MRP1π cells) further reduced SFN-mediated induction. Lastly, in comparison with MCF7/WT cells, expression of GSTP1–1 alone (MCF7/WTπ cells) produced a modest but reproducible increase in reporter gene induction. Overall, these results demonstrate that MRP1 and GSTP1–1 influence the magnitude of SFN-mediated ARE-containing gene induction and that the level of reporter gene induction correlates positively with the initial rates of SFN/SFN-SG accumulation (Figure 2A) in the four cell lines (i.e. WTπ > WT > MRP1 and MRP1π) or the maximum levels of SFN/SFN-SG accumulated (i.e. WT and WTπ > MRP1 and MRP1π). Additionally, induction of an ARE-containing, SFN-responsive endogenous gene, NQO1, was examined in the four cell lines treated with 25 μM SFN. Northern blot analysis (Figure 4B) demonstrated that, as with the reporter gene, induced levels of endogenous NQO1 messenger RNA were significantly reduced in cells expressing MRP1 in comparison with MRP1-poor MCF7/WT (WT) and MCF7/WTπ (WTπ) cells. Coexpression of GSTP1–1 with MRP1 (MCF7/MRP1π (MRP1π) cells) further reduced SFN-induced levels of NQO1 messenger RNA.
Fig. 4.
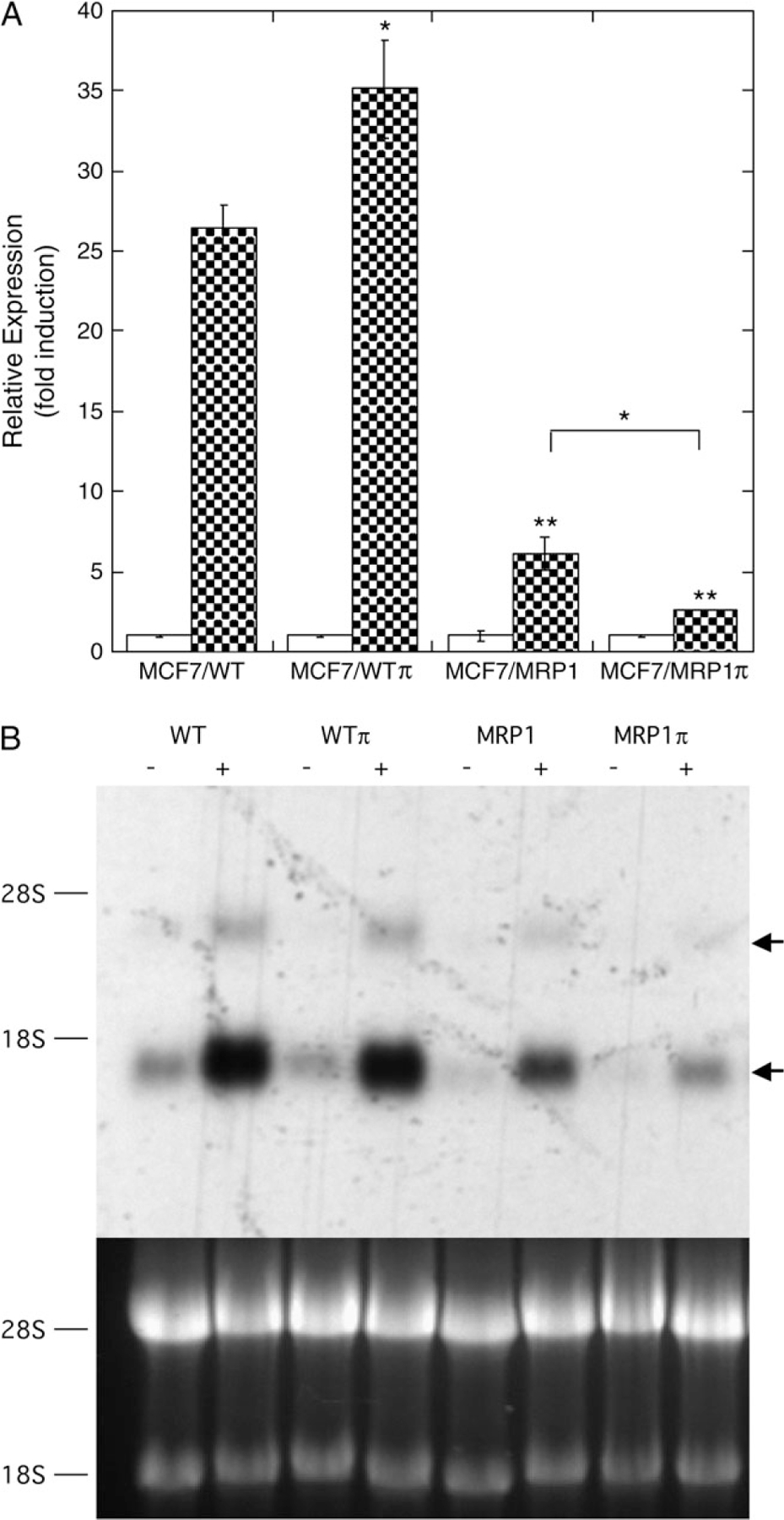
Effect of MRP1 and GSTP1–1 on SFN-mediated induction of ARE-containing gene expression. (A) Reporter gene expression. Indicated cell lines were cotransfected with the ARE-containing firefly luciferase reporter gene (0.5 μg pARE-TI-LUC) and control CMV-Renilla luciferase reporter gene (5 pg pGL4.75) as described in Materials and Methods. Twenty-four hours later, cells were treated with 25 μM SFN (closed bars) or vehicle control (open bars) for 3 h, medium was replaced with SFN-free medium and cells were processed for luciferase assays 21 h later. Renilla luciferase activity was used to correct ARE-driven reporter gene activity for differences in transfection efficiency. Data so corrected was normalized to control (−SFN) activity and expressed as relative expression (fold induction). Each bar represents the mean values ±1 SD of three to six independent transfections. Statistical significance of differences between the induced values (+SFN, closed bars) of parental (MCF7/WT) and the transgenic (MCF7/WTπ, MCF7/MRP1, and MCF7/MRP1π) cells (asterisks directly above bars) and differences between MCF7/MRP1 and MCF7/MRP1π cells (asterisk above bracket) were determined by the Student’s t-test: *P < 0.03, **P < 0.001. (B) Endogenous gene expression. Shown are a representative northern blot, probed for NQO1 expression, of total cellular RNA derived from the four cell lines ± SFN treatment (upper panel) and the corresponding ethidium bromide stained gel (lower panel). Indicated are the positions of 18S and 28S RNA (left) and NQO1 messenger RNA and its incompletely spliced precursor (arrows, right).
The results described above are consistent with the interpretation that MRP1 and GSTP1–1 modulate SFN-mediated ARE-containing gene induction: (i) by influencing the rate of SFN-SG conjugate efflux and, hence, the level of SFN/SFN-SG accumulated intracellularly (MRP1 effect) and (ii) by influencing the rate of SFN-SG formation (GSTP1–1 effect). Clearly, central to both processes are the capacity for the inducing electrophile, SFN, to form GSH conjugates and the presence of ample intracellular GSH to support conjugation. If this view is true then MRP1 and GSTP1–1 should have negligible effects on the activation of ARE-containing genes by other phase II gene inducers, such as tBHQ, that do not readily form GSH conjugates and are, therefore, unlikely to be transported by MRP1. To test this idea, induction of the ARE-containing reporter gene was examined following treatment with 50 μM tBHQ (below). The validity of using tBHQ in this manner requires some explanation. First, we have conducted in vitro experiments in which 50 μM tBHQ was incubated with GSH for >1 h under physiological conditions (pH 7.5, 37°C, 5 mM GSH). Analysis of this mixture by ultraviolet/visible spectroscopy and high-performance liquid chromatography revealed no evidence of tBHQ depletion, conjugation with GSH or any other reaction of tBHQ with GSH (not shown). In contrast, incubation of 50 μM SFN with GSH under similar conditions resulted in >90% conversion of SFN to SFN-SG within 1 min. While tBHQ can be oxidized to a quinone within the cell and, as shown by Nakamura et al. (46), the quinone has the potential to form GSH conjugates, two considerations suggest that GSH conjugation is not a major pathway for tBHQ disposition in the cell: (i) The quinone can be reduced back to the hydroquinone by cellular reductases limiting that amount of substrate vulnerable to Michael addition with GSH. (ii) In the Nakamura paper, the rate of tBHQ-SG formation was extremely slow and the extent of conjugation was very low. Indeed, in cells exposed to 10 μM tBHQ for several hours, tBHQ-SG accumulated to concentrations of only 2–3 nM. In contrast, treatment of MCF7/WT cells with 25 μM SFN resulted in accumulation of SFN-SG to levels exceeding 1 mM within 5 min (Figure 2A) (estimation based upon total intracellular protein concentration ~150 mg/ml).
As shown in Figure 5A, there were no significant differences in tBHQ-mediated induction of ARE-reporter gene activity among the four cell lines. To further investigate the importance of SFN/GSH conjugation in the modulation by MRP1 and GSTP1–1 of SFN-mediated induction, experiments were conducted in which GSH was depleted by 24 h exposure to 50 μM L-buthionine-(S,R)-sulfoximine prior to SFN treatment. Such L-buthionine-(S,R)-sulfoximine treatment results in ≥90% reduction of intracellular GSH (33). Following GSH depletion, SFN-mediated ARE-reporter gene activation was similar among the four cell lines (Figure 5B). In aggregate, these results indicate that the modulatory effects of MRP1 and GSTP1–1 expression on SFN bioactivity are indeed GSH dependent.
Fig. 5.
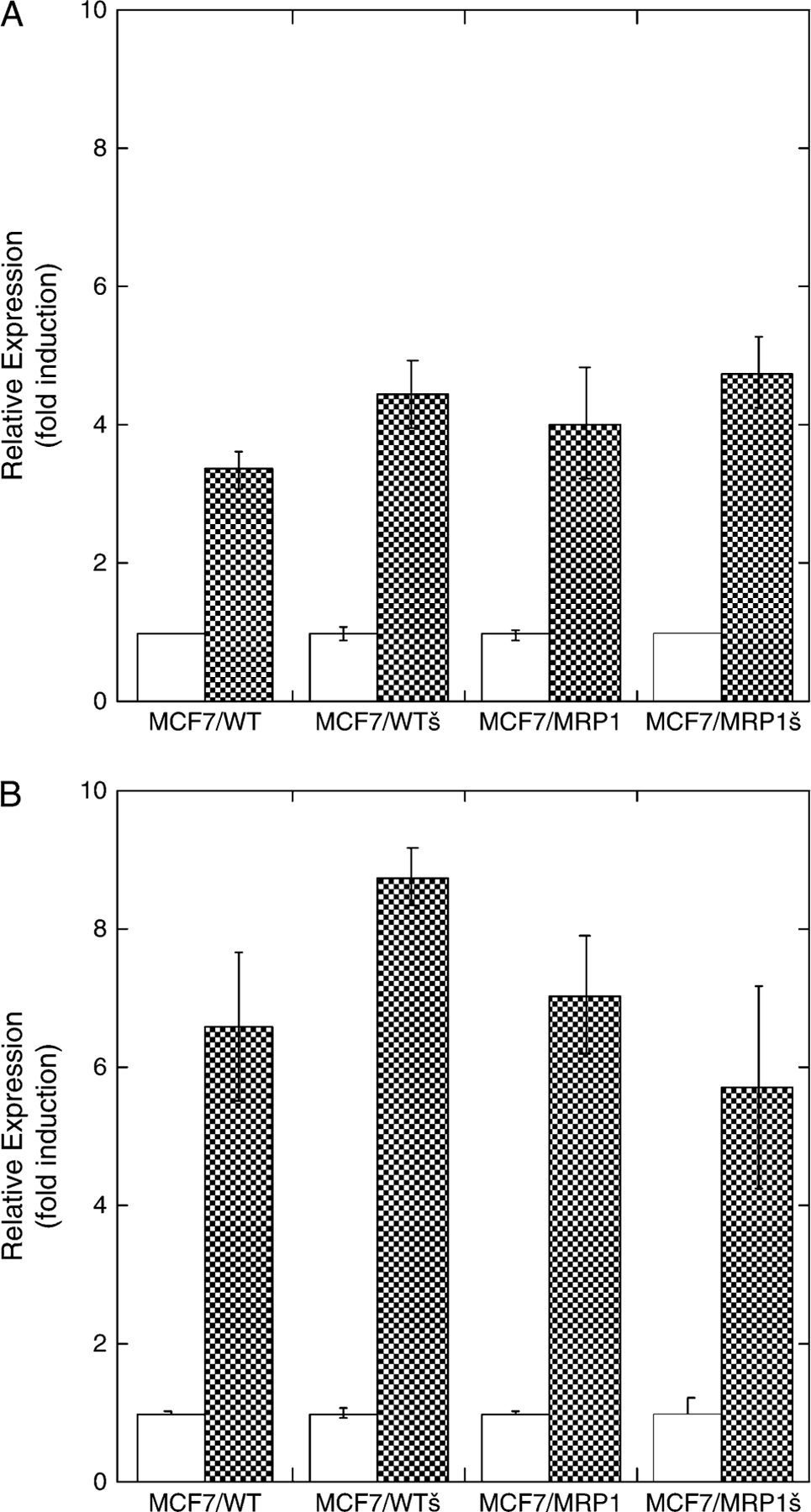
The effects of MRP1 and GSTP1–1 on ARE-containing gene induction are dependent upon GSH and GSH conjugation. (A) Induction with tBHQ. Reporter gene experiments were conducted, analyzed and displayed exactly as described in Figure 4A except that treatment with tBHQ (50 μM)––a compound that in its native form does not form a conjugate with GSH––replaces treatment with SFN. Differences in reporter gene induction between parental (MCF7/WT) and the transgenic (MCF7/WTπ, MCF7/MRP1, and MCF7/MRP1π) cells were not statistically significant (n = 5 6; Student’s t-test, P > 0.1). (B) Induction with SFN after prior depletion of intracellular GSH. Reporter gene experiments were conducted with modifications as described in Materials and Methods. Briefly, immediately following the 3 h transient transfection, cells were treated in medium containing 50 μM L-buthionine-(S,R)-sulfoximine to deplete intracellular GSH. Twenty-four hours later, cells were treated with 25 μM SFN or vehicle control and processed for reporter gene activity as described (Figure 4A) except that L-buthionine-(S,R)-sulfoximine treatment was continued throughout the remaining incubations. Differences in reporter gene induction between parental (MCF7/WT) and the transgenic (MCF7/WTπ, MCF7/MRP1, and MCF7/MRP1π) cells were not statistically significant (n = 5 3; Student’s t-test, P > 0.1).
MRP1 expression attenuates SFN-mediated increases in Nrf2 levels
The transcription factor, Nrf2, binds as a heterodimer to the ARE motifs and is thereby a principle mediator of ARE-containing promoter activation (12). To gain insights into the mechanisms by which MRP1 and GSTP1–1 modulate SFN-induced gene activation, we compared changes among the four cell lines in the levels and subcellular localization of Nrf2 following SFN treatment. Experiments shown in Figure 6 demonstrated a robust increase in cytosolic Nrf2 levels in all the four cell lines within 2 h following treatment with 25 μM SFN (Figure 6A). However, with continued incubation, the increased levels of Nrf2 fell––most rapidly in MRP1 expressing cells. This differentially rapid decline in Nrf2 resulted in substantially lower levels of Nrf2 in MRP1π cells than in WT or WTπ cells at 4–18 h post-SFN treatment and lower levels in MRP1 cells apparent at 10–18 h post-SFN treatment (Figure 6A). Similarly, SFN-mediated increases in nuclear Nrf2 levels were considerably less sustained in MRP1-expressing cells––especially those coexpressing GSTP1–1 (MRPπ)––than in WT or WTπ cells at 4–18 h post-SFN treatment (Figure 6B). Thus, the increased SFN-SG efflux and reduced intracellular SFN/SFN-SG accumulation in MRP1-expressing cells is associated with markedly less sustained increases in Nrf2 levels than observed in MRP1-poor cells. These data indicate that the effects of MRP1 and GSTP1–1 on SFN-induction of ARE-containing gene transcription are ultimately mediated by altered Nrf2 levels, especially in the nucleus.
Fig. 6.
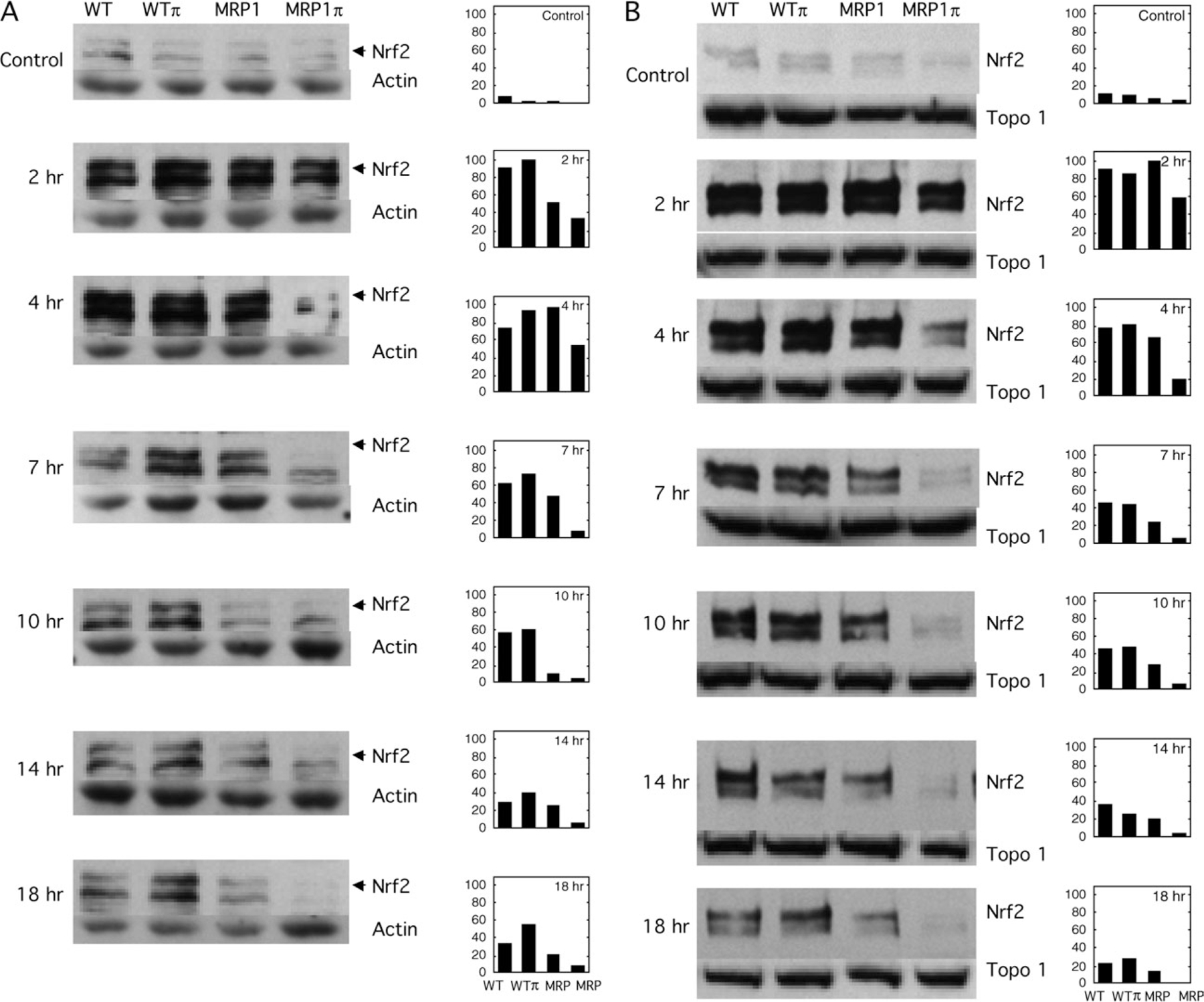
Effect of MRP1 and GSTP1–1 expression on SFN-mediated changes in cytoplasmic and nuclear Nrf2 levels. Parental (MCF7/WT, WT) and transgenic cells (MCF7/WTπ, WTπ; MCF7/MRP1, MRP and MCF7/MRP1π, MRPπ) were treated for 3 h with 25 μM SFN or vehicle (control) and then processed at the indicated times post-SFN treatment for determinations of cytoplasmic (A) or nuclear (B) Nrf2 levels as described in Materials and Methods. Experiments shown in A and B are derived from samples obtained in a single representative experiment. On the left are shown western blots for Nrf2 expression (uppermost panels of A and B), actin expression (lower panels of A), topoisomerase 1 (lower panels of B) and, on the right, densitometry data of relative Nrf2 expression (expressed as a percentage of the highest Nrf2 level achieved in the experiment).
Discussion
Induction of ARE-containing phase II gene transcription is a key component of the chemopreventive response to ITC exposure in vivo and in vitro (8–10). Since ITCs readily form GSH conjugates––both enzymatically and non-enzymatically––and are good substrates of cytosolic GSTs, we reasoned that expression of GST and the GSH-conjugate efflux pump, MRP1, at the time of ITC exposure would have a profound effect on the metabolism, clearance and bioactivity of ITCs in particular cells and tissues. Indeed, the studies described herein demonstrate that the GSH conjugate of SFN is efficiently transported by MRP1 (Figure 1). When compared with MRP1-poor cells, the increased rate of SFN-SG efflux in MRP1-rich MCF7 cells results in a dramatic reduction in the levels of SFN plus SFN-SG accumulated during a 1 h SFN exposure (Figure 2A). Expression of MRP1 and reduced SFN/SFN-SG accumulation are associated with considerably reduced levels of induced ARE-containing reporter and endogenous gene expression (Figure 4A and B). Coexpression of GSTP1–1 with MRP1 resulted in a further decrease in the level of ARE-dependent gene expression upon SFN treatment, whereas expression of GSTP1–1 alone resulted in an increase, relative to GST- and MRP-minus cells, in the initial rate of SFN/SFN-SG accumulation and was associated with a significant increase in reporter gene expression. The observed correlation between intracellular SFN plus SFN-SG accumulation and ARE-dependent gene induction are consistent with the previous work of Ye et al. (47). Moreover, these findings suggest that MRP1 and GSTP1–1 modulate SFN/SFN-SG levels and, hence, gene expression at least in part by governing the rates of conjugate (SFN-SG) efflux and formation, respectively. Supporting this idea and the essential role of GSH (GSH-conjugation) in MRP1/GSTP1–1 regulation of cellular response are the following. First, reporter gene induction by tBHQ––an activator of ARE-dependent transcription that, in its native form, does not readily form GSH conjugates nor is it likely to be transported by GSH-conjugate efflux transporters––is unaffected by expression of MRP1 or GSTP1–1 (Figure 5A). Moreover, prior depletion of intracellular GSH essentially eliminates the effects of GSTP1–1 and MRP1 expression on SFN-mediated ARE-reporter induction (Figure 5B).
A variety of transcription factors, regulatory proteins and signaling pathways have been implicated in the regulation of ARE-dependent, phase II gene transcription (8,21–23,48,49). The best studied is Nrf2, a transcription factor that in combination with its partners forms a heterodimer which binds to the ARE motif and activates transcription. Under basal conditions, in most cells Nrf2 is negatively regulated by Keap1, a cysteine-rich cytosolic protein with which Nrf2 interacts. It has been found that thiol-reactive electrophiles, such as ITCs, interact with key cysteine residues on Keap1 either via adduct formation and/or inducing intra- or intermolecular disulfide bond formation (17,19). Such ITC-dependent reactions are believed to disrupt Keap1–Nrf2 interactions. Initially, it was believed that this simply resulted in the release of Nrf2 sequestered in the cytosol, allowing its translocation to the nucleus where target genes are activated (13). Recent findings suggest a more active role for Keap1 in the repression of Nrf2: Keap1 serves as an adapter protein that links Nrf2 to Cul3-based E3 ubiquitin ligase, thus targeting Nrf2 for proteosomal degradation (16). Hence, disrupting the Keap1–Nrf2 interactions results in increased Nrf2 levels via stabilization of Nrf2. Our results are consistent with the view that SFN-induced phase II gene activation involves accumulation of SFN/SFN-SG which in turn mediates Nrf2 stabilization and accumulation within the nucleus and that expression of MRP1 ± GSTP1–1 interferes with Nrf2 accumulation by reducing SFN/SFN-SG accumulation. First, the increase in Nrf2 levels is very fast: within 2 h following SFN treatment, cytoplasmic Nrf2 rises from nearly undetectable to quite high levels in all the cell lines (Figure 6A). The high rate with which this increase occurs is compatible with a mechanism that involves stabilization of a Nrf2 protein whose synthesis is constitutive and rapid––although we cannot rule out the possible contribution of increased synthesis as suggested in some models (50). At later times––beyond 2 h following SFN treatment––the effects of MRP1 and GSTP1–1 on increased Nrf2 expression become apparent with consistently lower levels of Nrf2 persisting in the cytosolic and nuclear fractions of MRP1-rich (MCF7/MRP1 and MCF7/MRP1p) than in MRP1-poor (MCF7/WT and MCF7/π) cells (Figure 6).
In aggregate, our results indicate that, by regulating intracellular levels of SFN/SFN-SG after SFN treatment, MRP1 and GSTP1–1 modulate ARE-dependent gene expression via altering nuclear Nrf2 levels. As discussed in Results, the bulk of SFN-derived material accumulated within MRP1-poor cells probably represents SFN-SG. The mechanism by which SFN-SG, as opposed to unconjugated SFN, may increase Nrf2 expression is not entirely clear. However, it is plausible that SFN-SG may react with the crucial thiols of Keap1 following a retro-Michael regeneration of free SFN or via a thiol exchange reaction resulting in release and stabilization of Nrf2.
In addition, there are other mechanisms proposed for the induction of ARE-dependent gene expression that need be considered. Redox or electrophilic stress may, independently of direct Keap1-SFN or Keap1-SFN-SG interactions, activate mitogen-activated protein kinase pathways which in turn directly or indirectly modulate ARE-containing gene expression (24,51) or apoptosis (7). In addition, direct phosphorylation of Nrf2 by protein kinase C and PKR-like ER kinase have been implicated in Nrf2 activation (21,52). Such considerations of redox or electrophilic stress are certainly germane to our model system. Indeed, we find that coexpression of GST and MRP1 significantly increases the rate and extent of intracellular GSH depletion (Figure 2B). This is not unexpected in view of the catalytic and conjugate transport roles of GST and MRP1, which together can greatly enhance the net flux of GSH (as conjugate) from cells exposed to high levels of electrophiles. It is not unreasonable to suggest, then, that the extreme GSH depletion associated with SFN treatment of cells expressing both MRP1 and GSTP1 (MCF7/MRP1π) would render these cells particularly vulnerable to further redox stress caused by SFN as well as endogenous prooxidants. Based upon the literature of redox signaling, if variable GSH depletion was an important component of the differential response to SFN observed in our model cell lines, then the increased redox stress in MCF7/MRP1π cells might result in enhanced activation of ERK/JNK signaling pathways, which are reported to enhance ARE-dependent transactivation (51). In addition, or alternatively, the increased redox signaling could be manifested as enhanced apoptosis (6,7). However, among the four cell lines, MRPπ cells, despite showing the greatest depletion of intracellular GSH, are the least responsive to SFN-mediated induction of ARE-gene expression. Furthermore, there were no measurable differences in SFN cytotoxicity among the four cell lines (Figure 3). From these considerations, it is most probable that the accumulation of intracellular SFN/SFN-SG––not global redox stress that may result from GSH depletion––is the key determinant of MRP1/GST modulation of SFN bioactivities. In particular, the consequence of reduced SFN/SFN-SG levels in MRP1-expressing cells is a significantly less sustained increase in Nrf2 levels, resulting in attenuated SFN-dependent ARE-gene induction.
The studies described suggest that the GST and MRP phenotype of a particular cell type or tissue, at the time of exposure, may have a profound effect on the ultimate response to chemopreventive ITCs such as SFN. This has particular relevance in vivo as the levels of expression of MRP and GST family proteins vary widely in the different tissues that are the desired targets of chemoprevention. For example, MRP1, while expressed in most tissues, is nearly undetectable in liver but present at particularly high levels in lung, testis, kidney, skeletal muscle and heart (53). Moreover, MRP1 is differentially expressed within various cell types of particular tissues. The tissue distributions of the alternative GSH-conjugate efflux pumps, MRP2–4, are even more restricted than MRP1 (54,55). The cytosolic isozymes of GST known to catalyze ITC conjugation also have unique tissue distribution and expression level patterns (56,57). For example, GSTP1–1, while widely expressed but at variable levels in many tissues and tumors, is absent in others such as adult liver. The ITC-inducible GSTs of the alpha class are expressed at high levels in liver but differentially and at lower basal levels in other tissues including kidney, adrenal gland and gut. It is likely that the variable MRP/GST phenotypes observed among different tissues and cell types could have a significant influence on the magnitude and efficacy of the chemopreventive response to SFN or other ITC exposure.
It should also be noted that ITCs can induce enzymes that influence their own metabolism including alpha class GSTs (58) and, in the mouse at least, MRP family proteins (59,60). In MCF7/WT cells, GSTA1/2 expression, while undetectable at the protein level, is apparent, in very low copy number, at the RNA level (~20–50 copies/μg RNA, quantitative real-time polymerase chain reaction, C. Morrow unpublished data). Indeed, GSTA1/2 RNA was induced 2–6-fold in MCF7/WT cells treated with 25 μM SFN but the protein remained undetectable and GST activity was unchanged (<5 nmol/min/mg) throughout the 3 h induction and 24 h postinduction period (not shown). However, in other cell types and in vivo with prolonged or repeated SFN exposure, GST levels may change sufficiently to autoregulate the cellular response to SFN treatment. Our data would suggest that in MRP-rich cell types prior induction of GST could self-limit subsequent ARE-dependent gene induction in response to repeated SFN exposure; whereas in MRP-poor cell types, the magnitude of subsequent phase II induction would remain unchanged or, perhaps, even increase.
Funding
National Institutes of Health (CA70338).
Abbreviations:
- ARE
antioxidant response element
- ATP
adenosine triphosphate
- BTT
1,3-benzodithiole-2-thione
- GSH
glutathione
- GST
glutathione S-transferase
- ITC
isothiocyanate
- Keap1
Kelch ECH-associating protein 1
- MRP
multidrug resistance or resistance-associated protein
- NQO1
NAD(P)H:quinone oxidoreductase
- Nrf2
NF-E2-related factor 2
- SFN
sulforaphane
- SFN
SG glutathione conjugate of sulforaphane
- tBHQ
tert-butylhydroquinone
Footnotes
Conflict of Interest Statement: None declared.
References
- 1.Fahey JW et al. (1997) Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl Acad. Sci. USA, 94, 10367–10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y et al. (1994) Anticarcinogenic activities of organic isothiocyanates: chemistry and mechanisms. Cancer Res, 54, 1976–1981. [PubMed] [Google Scholar]
- 3.Kwon KH et al. (2007) Cancer chemoprevention by phytochemicals: potential molecular targets, biomarkers and animal models. Acta Pharmacol. Sin, 28, 1409–1421. [DOI] [PubMed] [Google Scholar]
- 4.Talalay P et al. (2001) Phytochemicals from cruciferous plants protect against cancer by modulating marcinogen metabolism. J. Nutr, 131, 3027S–33033. [DOI] [PubMed] [Google Scholar]
- 5.Pham N-A et al. (2004) The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol. Cancer Ther, 3, 1239–1248. [PubMed] [Google Scholar]
- 6.Singh SV et al. (2005) Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J. Biol. Chem, 280, 19911–19924. [DOI] [PubMed] [Google Scholar]
- 7.Xiao D et al. (2002) Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res, 62, 3615–3619. [PubMed] [Google Scholar]
- 8.Wolf CR (2001) Chemoprevention: increased potential to bear fruit. Proc. Natl Acad. Sci. USA, 98, 2941–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yates MS et al. (2007) Keap1 eye on the target: chemoprevention of liver cancer. Acta Pharmacol. Sin, 28, 1331–1342. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y et al. (2004) A strategy for cancer prevention: stimulation of the Nrf2-ARE signaling pathway. Mol. Cancer Ther, 3, 885–893. [PubMed] [Google Scholar]
- 11.Rushmore TH et al. (2002) Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr. Drug Metab, 3, 481–490. [DOI] [PubMed] [Google Scholar]
- 12.Itoh K et al. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun, 236, 313–322. [DOI] [PubMed] [Google Scholar]
- 13.Itoh K et al. (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev, 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McWalter GK et al. (2004) Transcription factor Nrf2 is essential for induction of NAD(P)H: quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J. Nutr, 134, 3499S–33506S. [DOI] [PubMed] [Google Scholar]
- 15.McMahon M et al. (2003) Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem, 278, 21592–21600. [DOI] [PubMed] [Google Scholar]
- 16.Cullinan SB et al. (2004) The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol, 24, 8477–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinkova-Kostova AT et al. (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl Acad. Sci. USA, 99, 11908–11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong F et al. (2005) Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol, 18, 1917–1926. [DOI] [PubMed] [Google Scholar]
- 19.Wakabayashi N et al. (2004) Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl Acad. Sci. USA, 101, 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cullinan SB et al. (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol, 23, 7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang HC et al. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem, 277, 42769–42774. [DOI] [PubMed] [Google Scholar]
- 22.Iwasaki K et al. (2007) PIAS3 interacts with ATF1 and regulates the human ferritin H gene through an antioxidant-responsive element. J. Biol. Chem, 282, 22335–22343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuji Y (2005) JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene, 24, 7567–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zipper LM et al. (2003) Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol. Sci, 73, 124–134. [DOI] [PubMed] [Google Scholar]
- 25.Kolm R et al. (1995) Isothiocyanates as substrates for human glutathione transferases: structure-activity studies. Biochem. J, 311, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer D et al. (1995) Forward and reverse catalysis and product sequestration by human glutathione S-transferases in the reaction of GSH with dietary aralkyl isothiocyanates. Biochem. J, 306, 565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji Y et al. (2005) Transport of dietary phenethyl isothiocyanate is mediated by multidrug resistance protein 2 but not P-glycoprotein. Biochem. Pharmacol, 70, 640–647. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y et al. (2002) High cellular accumulation of sulphoraphane, a dietary anticarcinogen, is followed by rapid transporter-mediated export as a glutathione conjugate. Biochem. J, 364, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paumi C et al. (2004) Glutathione S-transferases (GSTs) inhibit transcriptional activation by the peroxisomal proliferator-activated receptor gamma (PPAR gamma) ligand, 15-deoxy-delta(12,14)prostaglandin J(2) (15-d-PGJ(2). Biochemistry, 43, 2345–2352. [DOI] [PubMed] [Google Scholar]
- 30.Paumi CM et al. (2003) Multidrug resistance protein (MRP) 1 and MRP3 attenuate cytotoxic and transactivating effects of the cyclopentenone prostaglandin, 15-deoxy-delta(12,14)prostaglandin J(2) in MCF7 breast cancer cells. Biochemistry, 42, 5429–5437. [DOI] [PubMed] [Google Scholar]
- 31.Habig W et al. (1974) Glutathione S-transferase: the first enzymatic step in mercapturic acid formation. J. Biol. Chem, 249, 7130–7139. [PubMed] [Google Scholar]
- 32.Morrow CS et al. (1998) Coordinated action of glutathione S-transferases (GSTs) and multidrug resistance protein 1 (MRP1) in antineoplastic drug detoxification. Mechanism of GST A1–1- and MRP1-associated resistance to chlorambucil in MCF7 breast carcinoma cells. J. Biol. Chem, 273, 20114–20120. [DOI] [PubMed] [Google Scholar]
- 33.Diah SK et al. (2001) Resistance to mitoxantrone in MRP1-expressing MCF7 breast cancer cells: characterization of mitoxantrone transport. Cancer Res, 61, 5461–5467. [PubMed] [Google Scholar]
- 34.Morrow CS et al. (1998) Multidrug resistance protein and glutathione S-transferase P1–1 act in synergy to confer protection from 4-nitroquinoline 1-oxide toxicity. Carcinogenesis, 19, 109–115. [DOI] [PubMed] [Google Scholar]
- 35.Morrow CS et al. (2000) Role of multidrug-resistance protein 2 in glutathione S-transferase P1–1-mediated resistance to 4-nitroquinoline 1-oxide toxicities in HepG2 cells. Mol. Carcinog, 29, 170–178. [DOI] [PubMed] [Google Scholar]
- 36.Skehan P et al. (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl Cancer Inst, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- 37.Paumi CM et al. (2001) Role of multidrug resistance protein 1 (MRP1) and glutathione S-transferase A1–1 in alkylating agent resistance. Kinetics of glutathione conjugate formation and efflux govern differential cellular sensitivity to chlorambucil versus melphalan toxicity. J. Biol. Chem, 276, 7952–7956. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y et al. (1996) Quantitative determination of isothiocyanates, dithiocarbamates, carbon disulfide, and related thiocarbonyl compounds by cyclocondensation with 1,2-benzenedithiol. Anal Biochem, 239, 160–167. [DOI] [PubMed] [Google Scholar]
- 39.Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 40.Tietze F (1969) Enzymatic method for quantitative determination of nanogram quantities of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem, 27, 502–522. [DOI] [PubMed] [Google Scholar]
- 41.Diah SK et al. (1999) Detoxification of 1-chloro-2,4-dinitrobenzene in MCF7 breast cancer cells expressing glutathione S-transferase P1–1 and/or multidrug resistance protein 1. Toxicol. Appl. Pharmacol, 157, 85–93. [DOI] [PubMed] [Google Scholar]
- 42.Wasserman WW et al. (1997) Functional antioxidant responsive elements. Proc. Natl Acad. Sci. USA, 94, 5361–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ausubel FM et al. (2004) Current Protocols in Molecular Biology John Wiley & Sons, New York, NY. [Google Scholar]
- 44.Nguyen T et al. (2003) Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol.Toxicol, 43, 233–260. [DOI] [PubMed] [Google Scholar]
- 45.Peklak-Scott C et al. (2005) Dynamics of glutathione conjugation and conjugate efflux in detoxification of the carcinogen, 4-nitroquinoline 1-oxide: contributions of glutathione, glutathione S-transferase, and MRP1. Biochemistry, 44, 4426–4433. [DOI] [PubMed] [Google Scholar]
- 46.Nakamura Y et al. (2003) Pivotal role of electrophilicity in glutathione S-transferase induction by tert-butylhydroquinone. Biochemistry, 42, 4300–4309. [DOI] [PubMed] [Google Scholar]
- 47.Ye LX et al. (2001) Total intracellular accumulation levels of dietary isothiocyanates determine their activity in elevation of cellular glutathione and induction of phase 2 detoxification enzymes. Carcinogenesis, 22, 1987–1992. [DOI] [PubMed] [Google Scholar]
- 48.Pugazhenthi S et al. (2007) Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by a PI3-kinase/Akt-mediated pathway in mouse beta-cells. Am. J. Physiol. Endocrinol. Metab, 293, E645–E655. [DOI] [PubMed] [Google Scholar]
- 49.Wang W et al. (2007) The p65 isoform of Nrf1 is a dominant negative inhibitor of ARE-mediated transcription. J. Biol. Chem, 282, 24670–24678. [DOI] [PubMed] [Google Scholar]
- 50.Purdom-Dickinson SE et al. (2007) Translational control of Nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol, 72, 1074–1081. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H et al. (2006) 4-Hydroxynonenal induces rat {gamma}-glutamyl transpeptidase through mitogen-activated protein kinase-mediated electrophile response element/nuclear factor erythroid 2-related factor 2 signaling. Am. J. Respir. Cell. Mol. Biol, 34, 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cullinan SB et al. (2004) PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem, 279, 20108–20117. [DOI] [PubMed] [Google Scholar]
- 53.Deeley RG et al. (2003) Multidrug resistance protein 1 (ABCC1). In Holland I,Cole S,Kuchler K and Higgins C (eds.) ABC Proteins from Bacteria to Man Academic Press, Amsterdam, pp. 393–422. [Google Scholar]
- 54.Borst P et al. (2003) The multidrug resistant proteins 3–7. In Holland I, -Cole S, Kuchler K and Higgins C (eds.) ABC Proteins from Bacteria to Man Academic Press, Amsterdam, pp. 445–458. [Google Scholar]
- 55.Konig J et al. (2003) MRP2, the apical export pump for anionic conjugates. In Holland I,Cole S,Kuchler K and Higgins C (eds.) ABC Proteins from Bacteria to Man Academic Press, Amsterdam, pp. 423–443. [Google Scholar]
- 56.Eaton DL et al. (1999) Concise review of the glutathione S-transferases and their significance to toxicology. Toxicol. Sci, 49, 156–164. [DOI] [PubMed] [Google Scholar]
- 57.Hayes JD et al. (1995) The glutathione S-transferase supergene family: regulation of GST and the contribution of the isozymes to cancer chemoprevention and drug resistance. Crit. Rev. Biochem. Mol. Biol, 30, 445–600. [DOI] [PubMed] [Google Scholar]
- 58.Svehlikova V et al. (2004) Interactions between sulforaphane and apigenin in the induction of UGT1A1 and GSTA1 in CaCo-2 cells. Carcinogenesis, 25, 1629–1637. [DOI] [PubMed] [Google Scholar]
- 59.Hayashi A et al. (2003) Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem. Biophys. Res. Commun, 310, 824–829. [DOI] [PubMed] [Google Scholar]
- 60.Maher JM et al. (2007) Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology, 46, 1597–1610. [DOI] [PubMed] [Google Scholar]