

Abstract
Background
While rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children and adolescents, past epidemiology studies of this malignancy used data that covered <30% of the US population. Therefore, we evaluated RMS incidence using data from U.S. Cancer Statistics (USCS) and survival trends using the National Program of Cancer Registries (NPCR), which covers 100% and 94% of the U.S. population, respectively.
Methods
Incidence and survival were assessed for pediatric patients diagnosed with RMS during 2003–2017 and 2001–2016, respectively. Both demographic and clinical variables were evaluated. Age‐adjusted incidence rates, average annual percent change (AAPC), and 5‐year relative survival (RS) were calculated, all with corresponding 95% confidence intervals (CIs). Cox regression models were used to evaluate the impact of demographic and clinical variables on survival.
Results
We identified 5656 primary RMS cases in USCS during 2003–2017. The age‐adjusted incidence rate was 4.58 per 1 million (95% CI: 4.46–4.70) with an AAPC of 0.3% (95% CI: −0.7 to 1.2%). In NPCR, 5‐year RS for all cases was 68.0% (95% CI: 66.6–69.3%). In multivariable analyses, non‐Hispanic (NH) Black cases had worse survival compared with NH White cases (hazard ratio [HR] = 1.16, 95% CI: 1.01–1.33).
Conclusion
The incidence and survival rates were stable in the largest and most comprehensive population‐based analysis for pediatric RMS cases in the U.S. Additionally, we observed a survival disparity among NH Black cases. Findings from this study could inform interventions to address disparities, risk stratification strategies, and clinical trial design.
Keywords: rhabdomyosarcoma, epidemiology, pediatric cancer, incidence, survival, soft tissue sarcoma
In a large population‐based analysis of pediatric rhabdomyosarcoma cases in the U.S. from 2001‐2017, the incidence and survival rates were stable compared to previous studies. Additionally, a novel survival disparity among Black cases was observed.
1. INTRODUCTION
Rhabdomyosarcoma (RMS) is a malignant tumor that originates from developing skeletal muscle cells. The most common soft tissue sarcoma in children and adolescents, RMS represents about 3–4% of all pediatric cancers, which corresponds to about 350 new cases diagnosed annually in the United States. 1 Both incidence and survival for RMS have been characterized by several different demographic and clinical factors. Historically, the two major histologic subtypes are embryonal (ERMS) and alveolar (ARMS), which make up about 70% and 20% of RMS cases, respectively. 2 Recently, due to advances in cytogenetic testing, the RMS subtype is now primarily classified according to the presence or the absence of a fusion involving the FOXO1 gene. 3 These pathological characteristics and other clinical factors—including patient age; tumor histology, size, and primary anatomic site; and the presence of metastases—have been confirmed as valid prognostic factors through both epidemiological studies and clinical trials. 4 , 5 , 6 , 7 As such, they have been incorporated into staging protocols and risk‐adapted therapy strategies in successive clinical trials from national cooperative groups. 1 , 8 , 9 Sociodemographic variables such as sex, race/ethnicity, and economic status have also been explored in several studies, with some emerging patterns but overall varying results. 10 , 11 , 12 , 13 , 14 Unfortunately, despite initial improvements in outcomes during the 1970s and 1980s, overall survival rates in the U.S. have subsequently plateaued and recent clinical trial interventions have failed to demonstrate success for patient populations across the spectrum of low‐, intermediate‐, and high‐risk disease. 3 , 15 , 16 , 17 , 18 , 19
While there have been notable registry‐based studies of pediatric RMS, including those using data from the Surveillance, Epidemiology, and End Results (SEER) Program covering <30% of the U.S. population 5 , 16 , 20 , 21 , 22 , 23 as well as international registries, 24 , 25 there are still considerable gaps in our understanding of the overall epidemiology of this clinically important malignancy. Specifically, more comprehensive and contemporary data are needed to characterize (1) incidence patterns of RMS to inform new studies evaluating the etiologies of these tumors and (2) survival trends of RMS to inform new risk stratification strategies and novel therapies. Therefore, the purpose of this study is to provide an updated and more comprehensive investigation of incidence and survival for pediatric RMS using population‐based databases from U.S. Cancer Statistics (USCS) and the National Program of Cancer Registries (NPCR), which includes data from 100% and 94% of the US population, respectively.
2. METHODS
2.1. Data source and study population
The USCS database was used to assess incidence between 2003 and 2017, whereas the NPCR survival database was used to evaluate survival for patients diagnosed between 2001 and 2016. 26 The USCS database includes all 50 states and the District of Columbia (DC), encompassing 100% of the US population. The NPCR survival database includes all states and DC except for Connecticut, Hawaii, Indiana, Iowa, Kansas, and New Mexico, covering 94% of the US population. The NPCR conducts active case follow‐up or linkage with the CDC's National Death Index.
The International Classification of Childhood Cancer, third edition (ICCC‐3) was used to define RMS cases for all individuals diagnosed at <20 years of age. 27 Within ICCC category IXa (RMS), we identified relevant International Classification of Disease for Oncology, third edition (ICD‐O‐3) morphology codes and included the following RMS subtypes in our analysis: 8900/3 (rhabdomyosarcoma NOS), 8902/3 (mixed‐type), 8910/3 (embryonal), 8912/3 (spindle cell), 8920/3 (alveolar), and 8991/3 (embryonal sarcoma). 28 Due to low numbers of cases (n < 15) and inconsistent inclusion in prior pediatric epidemiology studies, subtypes 8901/3 (pleomorphic RMS, adult type) and 8921/3 (RMS with ganglionic differentiation) were excluded. Only first‐primary, malignant tumor cases with microscopic diagnosis confirmation were included. Cases determined by autopsy or death certificate only were excluded.
2.2. Variables
Incidence and survival rates were reported overall and according to clinical and sociodemographic variables. Tumor histologies included embryonal, spindle cell, and alveolar; additionally, we created an “other” category to represent morphology codes 8900/3, 8902/3, and 8991/3, both to increase the sample size for ease of comparison, and because these morphologies are largely considered outdated. 29 Anatomic site of the primary tumor was classified into eight categories according to the definitions in the open Children‘s Oncology Group (COG) clinical trial (refer to Appendices III, V, and VI). 30 Sites considered clinically favorable include orbit, head/neck (excluding the para‐meningeal region), biliary tract/liver, and genitourinary system (excluding bladder/prostate). Unfavorable sites include the para‐meningeal region, bladder/prostate, extremity, and trunk/other (including retroperitoneum and unknown). To characterize the extent of the disease, we utilized the merged summary stage. 31 Finally, a chronological variable that divided the survival data into 5‐ or 6‐year time frames was generated to examine any potential changes in outcomes over time.
Patients were further stratified by sociodemographic characteristics that included sex, race/ethnicity, age at diagnosis, and socioeconomic status (SES) as well as the geographic census region and population density of primary residence by county. Age was categorized into known prognostic delineations for pediatric RMS based on previous reports. 4 Race/ethnicity was grouped into non‐Hispanic (NH) White, NH Black, NH Asian‐Pacific Islander (API), NH American Indian/Alaska Native (AI/AN), NH All Other Races Combined, and Hispanic. For survival analysis, the NH All Other Races Combined group was not available and 63 NH Unknown cases were excluded. SES was grouped into five categories as defined by the Appalachian Regional Commission index‐based county economic classification system and was regrouped into three categories (top 25%, 25–75%, and bottom 25%) as has been described previously. 32
2.3. Statistical analysis
Incidence rates (IRs) for each variable were expressed per 1 million persons and age‐adjusted to the 2000 U.S. standard population. Incidence rate ratios (IRRs) were reported for each variable, generally with the largest and/or the most clinically favorable group as the referent. Temporal trends in incidence were described using average annual percent change (AAPC) calculated by joinpoint regression. 33 A maximum of two joinpoints were used to determine a change in direction of the trend.
In the survival analyses, outcomes were measured using 5‐year relative survival (RS), which aims to represent cancer survival in the absence of other causes of death, or cancer‐specific survival. RS is defined as the ratio of the observed survival of patients with cancer to the expected survival of a matched cohort of cancer‐free individuals. RS is calculated using expected life tables that are stratified by age, sex, race/ethnicity, SES, geographic location, and calendar year of diagnosis; the methods of estimation have been described elsewhere. 34 All‐cause survival curves, overall and by demographic and clinical variables, were generated using the Kaplan–Meier method. Statistical testing for survival curves was performed using the log‐rank test. Univariate and multivariable Cox regression models were conducted to examine the effects of demographic and clinical variables on 5‐year all‐cause survival. Hazard ratios (HR) were generated for each variable, with a higher HR between compared groups indicating a higher risk of death. Variables were included in the multivariable model if their univariate p value was <0.20. For survival curves and regression analysis, NH API and NH AI/AN groups were combined into an “NH Other” grouping.
The SEER*Stat 8.3.8 software program was used to perform all analyses related to incidence and relative survival. 35 All‐cause survival curves and related Cox regression models were generated using SAS Version 9.4. 36 All tests were two‐sided and a p value of 0.05 was considered statistically significant. The data that support the findings of this study are available upon request by contacting uscsdata@cdc.gov. 26 The data are not publicly available due to privacy and legal restrictions.
3. RESULTS
3.1. Incidence
In the USCS database, for the period 2003–2017, 5656 cases of RMS were diagnosed in children and adolescents <20 years of age (Table 1). The overall incidence for this time period was 4.58 per million per year (95% CI: 4.46–4.70). The incidence in females was lower than in males, with a rate ratio of 0.78 (95% CI: 0.74–0.83). For individuals 10–19 years old compared with those 1–9 years of age, IRR was 0.61 (95% CI: 0.57–0.64). NH White and NH Black cases had similar incidence rates. The IRRs for both Hispanic and NH API cases were significantly lower than NH White cases, at 0.87 (95% CI: 0.81–0.93) and 0.71 (95% CI: 0.62–0.82), respectively. By SES, the incidence was also higher for those who lived in counties in the top 25% compared with the middle 25–75% of counties.
TABLE 1.
Incidence rate, incidence rate ratios, and average annual percent change of children and adolescents with rhabdomyosarcoma in the United States cancer statistics database, 2003–2017
| Variable | No. (%) | IR | 95% CI | IRR | 95% CI | p value | AAPC | 95% CI |
|---|---|---|---|---|---|---|---|---|
| Total | 5656 (100) | 4.58 | 4.46–4.70 | 0.3 | −0.7–1.2 | |||
| Sex | ||||||||
| Male | 3236 (57) | 5.12 | 4.95–5.30 | Ref | 0.2 | −0.7–1.1 | ||
| Female | 2420 (43) | 4.01 | 3.85–4.18 | 0.78 | 0.74–0.83 | <0.0001 | 0.3 | −1.3–2.0 |
| Age at diagnosis (years) | ||||||||
| <1 | 323 (6) | 5.39 | 4.82–6.02 | 0.94 | 0.84–1.05 | 0.30 | 0.9 | −1.6–3.4 |
| 1–9 | 3123 (55) | 5.74 | 5.54–5.94 | Ref | 0.3 | −0.6–1.1 | ||
| 10–19 | 2210 (39) | 3.48 | 3.34–3.63 | 0.61 | 0.57–0.64 | <0.0001 | 0.2 | −1.4–1.7 |
| Race/Ethnicity | ||||||||
| NH White | 3235 (57) | 4.71 | 4.55–4.87 | Ref | 0.2 | −0.8–1.2 | ||
| NH Black | 926 (16) | 4.91 | 4.60–5.24 | 1.04 | 0.97–1.12 | 0.26 | 0.1 | −2.0–2.2 |
| NH API | 215 (4) | 3.36 | 2.93–3.84 | 0.71 | 0.62–0.82 | <0.0001 | 2.7 | 0.0–5.5 |
| NH AI/AN | 53 (1) | 4.22 | 3.16–5.52 | 0.90 | 0.67–1.18 | 0.47 | — | — |
| NH All Other Races Combined | 73 (1) | — | — | — | — | — | — | — |
| Hispanic | 1153 (20) | 4.10 | 3.86–4.34 | 0.87 | 0.81–0.93 | <0.0001 | −0.3 | −2.0–1.4 |
| Histology (ICD‐O‐3 code) | ||||||||
| Embryonal (ERMS) | 2857 (51) | 2.32 | 2.23–2.41 | Ref | 0.7 | −0.3–1.7 | ||
| Spindle | 194 (3) | 0.16 | 0.14–0.18 | 0.07 | 0.06–0.08 | <0.0001 | 2.3 | −2.3–7.0 |
| Alveolar (ARMS) | 1667 (29) | 1.35 | 1.28–1.41 | 0.58 | 0.55–0.62 | <0.0001 | −0.9 | −2.4–0.7 |
| Others a | 938 (17) | 0.76 | 0.71–0.81 | 0.33 | 0.30–0.35 | <0.0001 | 0.5 | −1.5–2.6 |
| Primary tumor site | ||||||||
| Favorable | ||||||||
| Orbit | 375 (7) | 0.31 | 0.28–0.34 | Ref | 0.6 | −2.3–3.5 | ||
| Genitourinary system | 693 (12) | 0.56 | 0.52–0.60 | 1.81 | 1.59–2.06 | <0.0001 | −1.2 | −3.3–1.0 |
| Biliary tract/liver | 232 (4) | 0.19 | 0.17–0.22 | 0.62 | 0.52–0.73 | <0.0001 | 1.0 | −2.6–4.7 |
| Head/neck | 1083 (19) | 0.88 | 0.83–0.94 | 2.86 | 2.54–3.23 | <0.0001 | 0.9 | −1.2–3.1 |
| Unfavorable | ||||||||
| Bladder/prostate | 322 (6) | 0.26 | 0.23–0.29 | 0.84 | 0.72–0.98 | 0.03 | −2.2 | −5.4–1.1 |
| Para‐meningeal region | 477 (8) | 0.39 | 0.35–0.42 | 1.26 | 1.10–1.44 | 0.001 | −2.9 | −4.6–−1.2 |
| Extremity | 738 (13) | 0.60 | 0.56–0.64 | 1.94 | 1.71–2.21 | <0.0001 | 0.3 | −1.8–2.4 |
| Trunk/other | 1715 (30) | 1.39 | 1.32–1.45 | 4.50 | 4.02–5.05 | <0.0001 | 1.7 | 0.6–2.8 |
| Unknown | 20 (<1) | 0.02 | 0.01–0.03 | 0.05 | 0.03–0.08 | <0.0001 | — | — |
| Stage | ||||||||
| Localized | 1923 (34) | 1.56 | 1.49–1.63 | Ref | 1.3 | 0.1–2.4 | ||
| Regional | 1831 (32) | 1.48 | 1.42–1.55 | 0.95 | 0.89–1.01 | 0.12 | −0.9 | −2.4–0.8 |
| Distant | 1632 (29) | 1.32 | 1.25–1.38 | 0.84 | 0.79–0.90 | <0.0001 | 0.6 | −0.6–1.9 |
| Unknown | 270 (5) | 0.22 | 0.19–0.25 | 0.14 | 0.12–0.16 | <0.0001 | −1.4 | −6.3–3.7 |
| US Census region | ||||||||
| Northeast | 1007 (18) | 4.90 | 4.60–5.21 | Ref | 0.6 | −1.3–2.5 | ||
| Midwest | 1220 (22) | 4.55 | 4.30–4.81 | 0.93 | 0.85–1.01 | 0.09 | 0.7 | −0.9–2.4 |
| South | 2112 (37) | 4.56 | 4.37–4.76 | 0.93 | 0.86–1.01 | 0.07 | 0 | −0.9–1.0 |
| West | 1317 (23) | 4.43 | 4.19–4.67 | 0.90 | 0.83–0.98 | 0.02 | −0.1 | −1.4–1.2 |
| Socioeconomic status | ||||||||
| Top 25% | 1597 (28) | 4.82 | 4.58–5.06 | Ref | 0.5 | −0.9–2.0 | ||
| 25% ‐ 75% | 3243 (57) | 4.49 | 4.34–4.65 | 0.93 | 0.88–0.99 | 0.02 | 0.2 | −0.8–1.1 |
| Bottom 25% | 653 (12) | 4.48 | 4.15–4.84 | 0.93 | 0.85–1.02 | 0.13 | −0.1 | −1.5–1.4 |
| Unknown | 163 (3) | — | — | — | — | — | — | — |
| Population density by county | ||||||||
| Metro (population) | 4922 (87) | 4.77 | 4.64–4.90 | 0.3 | −0.7–1.3 | |||
| >1,000,000 | 3244 (57) | 4.89 | 4.72–5.06 | Ref | 0.5 | −0.9–1.8 | ||
| 250,000‐1,000,000 | 1186 (21) | 4.53 | 4.28–4.80 | 0.93 | 0.87–0.99 | 0.03 | −0.2 | −1.9–1.6 |
| <250,000 | 492 (9) | 4.61 | 4.21–5.04 | 0.94 | 0.86–1.04 | 0.24 | 0.6 | −1.3–2.5 |
| Non‐Metro | 731 (13) | 4.34 | 4.03–4.66 | 0.89 | 0.82–0.96 | 0.003 | −0.1 | −1.4–1.3 |
Note: Dash indicates unable to calculate due to sampling size/missing data.
Abbreviations: AAPC, average annual percent change; AI/AN, American Indian/Alaska Native; API, Asian‐Pacific Islander; CI, confidence interval; ICD, International Classification of Diseases; IR, incidence rate; IRR, incidence rate ratio; NH, non‐Hispanic; SEER, Surveillance, Epidemiology, and End Results.
Other histologies include RMS NOS, mixed‐type RMS, and embryonal sarcoma. Variables with missing individual cases (n): Race/Ethnicity (1), Primary tumor site group (1), Population density (3).
ERMS was the most common histologic subtype (51%), followed by ARMS (29%). The most common primary tumor site was trunk/other (30%), followed by head/neck (19%), extremity (13%), and genitourinary system (12%). The SEER stage was relatively evenly distributed among all cases. The incidence of RMS overall did not change significantly during 2003–2017 (Table 1, AAPC 0.3, 95% CI: −0.7 to 1.2), nor were their significant changes when stratified by histology, with ERMS having an AAPC 0.7 (95% CI: −0.3 – 1.7) and ARMS having an AAPC ‐0.9 (95% CI: −2.4 to 0.7). Likewise, incidence did not change by sex, age, SES, population density, or geographic region. There was an increase for patients with localized disease (AAPC 1.3, 95% CI: 0.1–2.4), however, while other SEER stages showed stable incidence.
3.2. Survival
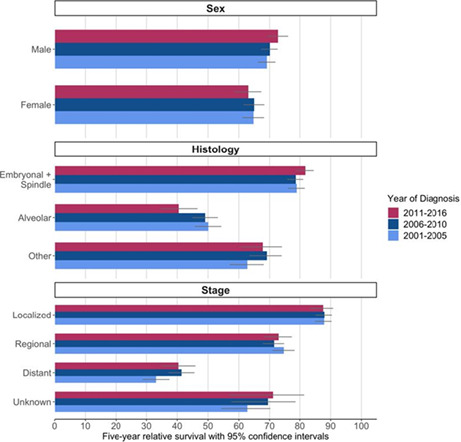
In the NPCR database, for the period 2001–2016, 5589 cases of RMS diagnosed in children and adolescents <20 years of age were identified. Overall, relative survival was 68.0% (95% CI: 66.6–69.3) (Table 2). RS in females (64.7%, 95% CI: 62.5–66.7%) was lower than in males (70.4%, 95% CI: 68.7–72.1%). Of the race/ethnicity groups with >200 patient cases, NH White cases showed the highest survival at 69.3% (95% CI: 67.6–71.0%), which was higher than NH Black cases at 64.4% (95% CI: 61.0–67.6%). Similarly, cases identified in counties with the highest SES (69.6%, 95% CI: 66.9–72.1%) had higher survival than those living in the lowest SES areas (65.5%, 95% CI: 61.6–69.1%), but confidence intervals overlapped. Relative survival was unchanged across time intervals when comparing 95% confidence intervals (Figure 1). Notably, there may be a widening trend over time in survival difference by sex, with the 2011–2016 period having the largest discrepancy (males [n = 1173]: 72.9%, 95% CI: 69.2–76.2%; females [n = 900]: 63.1%, 95% CI: 58.5–67.3%). No other evaluated variables suggest any definitive temporal changes in relative survival.
TABLE 2.
Five‐year relative survival (RS) and univariate all‐cause survival hazard ratios (HR) for children and adolescents with rhabdomyosarcoma in the national program of cancer registries, 2001–2016
| Relative survival | All‐cause survival | |||||
|---|---|---|---|---|---|---|
| Variable | No. (%) | 5‐year RS (%) | 95% CI (%) | HR | 95% CI | p value |
| Total | 5589 (100) | 68.0 | 66.6–69.3 | |||
| Sex | ||||||
| Male | 3228 (58) | 70.4 | 68.7–72.1 | Ref | ||
| Female | 2361 (42) | 64.7 | 62.5–66.7 | 1.25 | 1.13–1.38 | <0.0001 |
| Age at diagnosis (years) | ||||||
| <1 | 317 (6) | 70.6 | 64.8–75.7 | 1.45 | 1.16–1.82 | 0.001 |
| 1–9 | 3094 (55) | 77.2 | 75.5–78.7 | Ref | ||
| 10–19 | 2178 (39) | 54.4 | 52.1–56.6 | 2.30 | 2.07–2.55 | <0.0001 |
| Race/Ethnicity | ||||||
| NH White | 3269 (58) | 69.3 | 67.6–71.0 | Ref | ||
| NH Black | 941 (17) | 64.4 | 61.0–67.6 | 1.20 | 1.05–1.36 | 0.01 |
| NH API | 196 (4) | 64.8 | 56.7–71.8 | — | — | — |
| NH AI/AN | 53 (1) | 69.9 | 53.3–81.5 | — | — | — |
| Hispanic | 1130 (20) | 67.5 | 64.5–70.4 | 1.09 | 0.96–1.24 | 0.18 |
| Histology (ICD‐O‐3 code) | ||||||
| Embryonal (ERMS) | 2802 (50) | 79.2 | 77.5–80.7 | Ref | ||
| Spindle | 197 (4) | 85.4 | 79.0–90.0 | 0.66 | 0.44–0.99 | 0.04 |
| Alveolar (ARMS) | 1653 (30) | 47.9 | 45.2–50.5 | 2.96 | 2.64–3.31 | <0.0001 |
| Others a | 937 (17) | 66.9 | 63.5–70.0 | 1.76 | 1.52–2.04 | <0.0001 |
| Primary tumor site | ||||||
| Favorable | ||||||
| Orbit | 364 (7) | 93.4 | 90.0–95.7 | Ref | ||
| Genitourinary system | 711 (13) | 87.4 | 84.5–89.8 | 2.07 | 1.28–3.34 | 0.003 |
| Biliary tract/liver | 230 (4) | 82.8 | 76.8–87.4 | 3.05 | 1.77–5.23 | <0.0001 |
| Head/neck | 1066 (19) | 71.9 | 68.8–74.7 | 5.00 | 3.20–7.80 | <0.0001 |
| Unfavorable | ||||||
| Bladder/prostate | 326 (6) | 74.4 | 68.9–79.1 | 4.44 | 2.74–7.20 | <0.0001 |
| Para‐meningeal region | 493 (9) | 62.8 | 58.1–67.1 | 6.93 | 4.40–10.90 | <0.0001 |
| Extremity | 744 (13) | 54.7 | 50.7–58.6 | 8.62 | 5.54–13.43 | <0.0001 |
| Trunk/other | 1633 (29) | 55.4 | 52.7–58.0 | 8.96 | 5.80–13.84 | <0.0001 |
| Unknown | 22 (<1) | 36.4 | 17.4–55.7 | N/A | — | — |
| Stage | ||||||
| Localized | 1892 (34) | 87.9 | 86.2–89.4 | Ref | ||
| Regional | 1801 (32) | 73.0 | 70.7–75.1 | 2.45 | 2.07–2.90 | <0.0001 |
| Distant | 1575 (28) | 38.5 | 35.9–41.1 | 7.55 | 6.46–8.82 | <0.0001 |
| Unknown | 321 (6) | 66.9 | 61.0–72.0 | N/A | — | — |
| US Census region | ||||||
| Northeast | 988 (18) | 68.6 | 65.4–71.6 | Ref | ||
| Midwest | 1060 (19) | 69.9 | 66.9–72.8 | 0.95 | 0.81–1.13 | 0.57 |
| South | 2183 (39) | 67.6 | 65.4–69.7 | 1.03 | 0.90–1.19 | 0.66 |
| West | 1358 (24) | 66.6 | 63.8–69.2 | 1.08 | 0.93–1.26 | 0.32 |
| Socioeconomic status | ||||||
| Top 25% | 1480 (26) | 69.6 | 66.9–72.1 | Ref | ||
| 25%–75% | 3283 (59) | 67.6 | 65.8–69.3 | 1.12 | 0.99–1.26 | 0.07 |
| Bottom 25% | 716 (13) | 65.5 | 61.6–69.1 | 1.17 | 0.99–1.38 | 0.07 |
| Unknown | 110 (2) | 74.2 | 64.1–81.8 | 0.83 | 0.56–1.25 | 0.37 |
| Population density by county | ||||||
| Metro (population) | 4884 (87) | 67.9 | 66.4–69.3 | |||
| >1,000,000 | 3299 (59) | 68.4 | 66.7–70.1 | Ref | ||
| 250,000‐1,000,000 | 1119 (20) | 66.9 | 63.8–69.8 | 1.08 | 0.95–1.22 | 0.26 |
| <250,000 | 466 (8) | 66.1 | 61.3–70.5 | 1.13 | 0.95–1.35 | 0.17 |
| Non‐Metro | 704 (13) | 68.7 | 64.9–72.2 | 1.00 | 0.86–1.17 | 0.99 |
| Year of diagnosis | ||||||
| 2001–2005 | 1712 (31) | 67.4 | 65.1–69.5 | Ref | ||
| 2006–2010 | 1804 (32) | 67.9 | 65.7–70.0 | 0.96 | 0.86–1.08 | 0.53 |
| 2011–2016 | 2073 (37) | 68.6 | 65.7–71.2 | 0.94 | 0.83–1.06 | 0.31 |
Note: Dash indicates unable to calculate due to sampling size/missing data.
Abbreviations: AI/AN, American Indian/Alaska Native; API, Asian‐Pacific Islander; CI, confidence interval; HR, Cox hazard ratio; ICD, International Classification of Diseases; NH, non‐Hispanic; RS, relative survival; SEER, surveillance, epidemiology, and end results.
Other histologies include RMS NOS, mixed‐type RMS, and embryonal sarcoma. Patients with NH unknown race have been excluded from this analysis (n = 63). Variables with missing individual cases (n): population density (1).
FIGURE 1.
Relative survival for children and adolescents with rhabdomyosarcoma by year of diagnosis for sex, histology, and stage.
Using Kaplan–Meier Survival curves, significant differences in survival were seen for sex (Figure 2A), age (Figure 2B), race/ethnicity (Figure 2C), anatomic site, histology, and stage (Figure S1). Cases with 1–9 years of age at diagnosis, orbital or genitourinary anatomic site, spindle cell or embryonal tumors, and localized disease all had the highest survival. Significant differences in survival curves were not seen for SES, year of diagnosis, population density by county, and US Census region.
FIGURE 2.
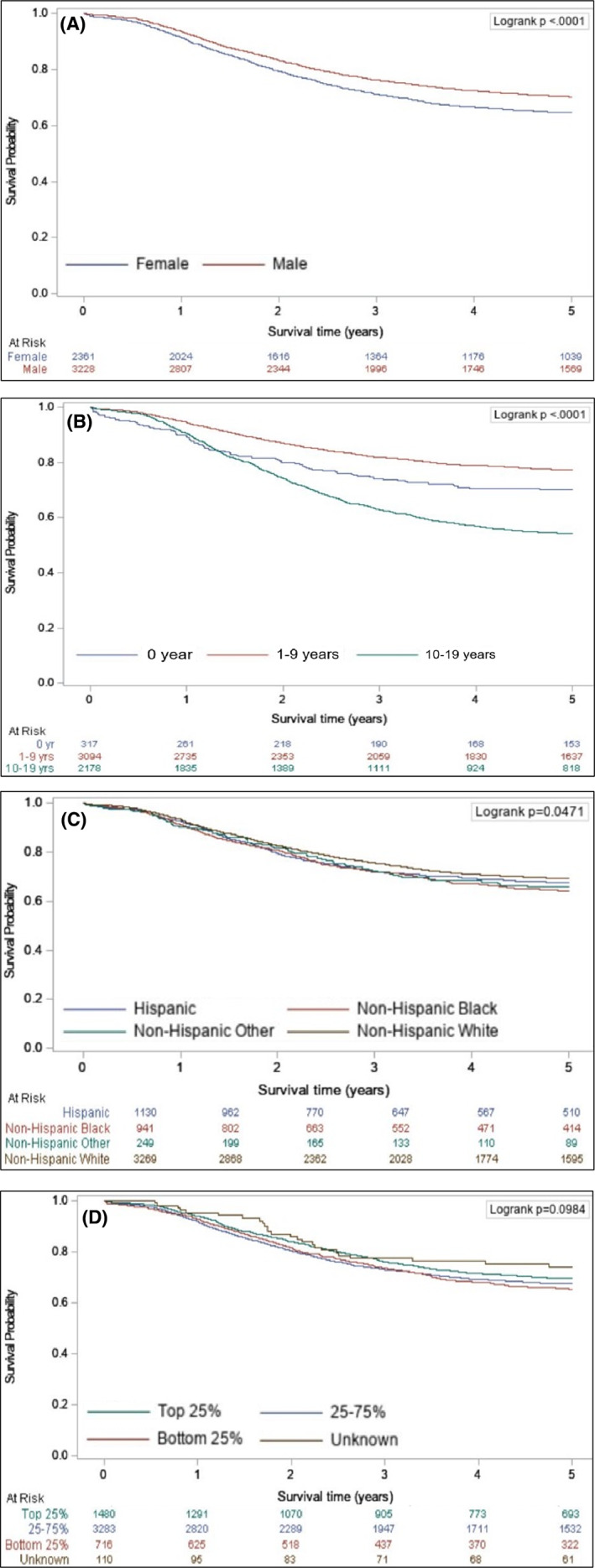
Kaplan–Meier survival estimation curve for children and adolescents with rhabdomyosarcoma by (A) sex, (B) age, (C) race/ethnicity, and (D) socioeconomic status.
In the multivariable analysis of 5‐year all‐cause survival, age <1 year or 10–19 years, ARMS histology, unfavorable anatomic site, and regional or distant stage were associated with poor survival (aHR >1.50, p < 0.001 for all) compared with the referent (Figure 3). NH Black cases had a 16% increased risk of death (95% CI: 1.01–1.33, p = 0.04) compared with NH White cases. Finally, cases identified in areas with relatively lower‐income levels had a 19% higher risk of death (95% CI: 1.00–1.42, p = 0.052) compared with those living in areas with higher‐income levels.
FIGURE 3.
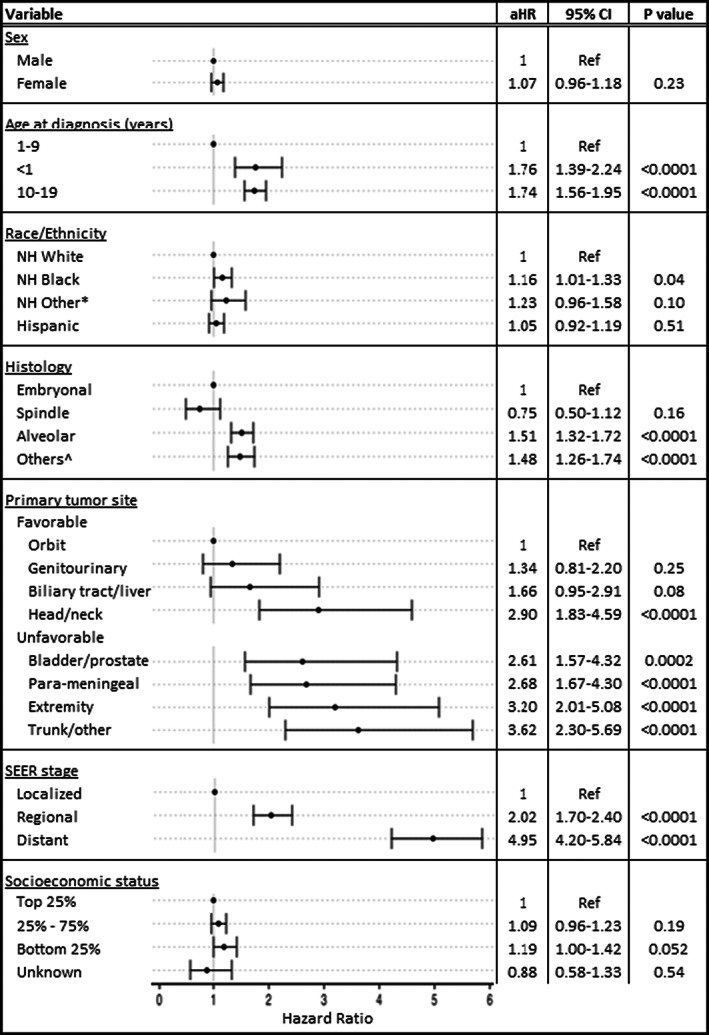
Multivariable Cox regression analysis with adjusted hazard ratios (aHR) for 5‐year all‐cause survival in children and adolescents with rhabdomyosarcoma in the National Program of Cancer Registries, 2001–2016. *Patients with NH Asian‐Pacific Islander and NH American Indian/Alaska Native race are combined into a single variable (NH Other). NH Unknown race has been excluded from this analysis (n = 63). ^Other histologies include RMS NOS, mixed‐type RMS, and embryonal sarcoma. aHR, adjusted hazard ratio; NH, non‐Hispanic; SEER, Surveillance, Epidemiology, and End Results.
4. DISCUSSION
In one of the largest population‐based studies of pediatric RMS including over 5000 cases, we found stable incidence overall and by most demographic and clinical variables for the period 2003–2017. We also did not observe an improvement in 5‐year RS for the 2001–2016 study period, which is consistent with other epidemiological reports 7 , 16 , 20 and COG clinical trials. 3 , 15 , 17 , 18 , 19 While we confirmed the role of multiple previously identified prognostic factors – such as age, stage, histology, and tumor site 4 , 7 – on survival, we also observed a novel association for race.
In our study, the incidence of RMS overall has been stable over the past two decades, which contrasts with increasing trends found in several other extracranial solid tumors such as thyroid carcinomas and renal and hepatic tumors. 37 , 38 We observed stable incidence rates for individual histologic subtypes, contrasting with two previous reports with smaller sample sizes using SEER data which showed increasing incidence for ARMS tumors diagnosed before 2006. 16 , 20 Our disparate result is likely explained by shifts in diagnostic criteria for ARMS histology in the United States in the 1990s and early 2000s, followed by the widespread adoption of more precise and comprehensive testing using immunohistochemistry and fluorescent in situ hybridization (FISH), as recently described by Rudzinski et al. 29
We observed significant differences in incidence by race/ethnicity and other sociodemographic factors. For example, we found the incidence of RMS was significantly lower in Hispanic and API cases compared with NH White cases, which is consistent with other studies. 14 , 16 Additionally, we observed a higher incidence among those living in higher SES areas. The few studies evaluating the role of SES on RMS have been equivocal, with both direct and inverse associations reported. 39 , 40 Ultimately, more targeted studies can help understand the different causal factors that may be driving these observed differences and potential strategies to reduce RMS incidence.
We observed a small but statistically significant difference in survival by race. Specifically, we found that NH Black cases with RMS had worse survival compared with NH White cases. Previous studies have produced inconsistent results in this area. Pui et al. conducted one of the earliest explorations of pediatric outcomes by race and found no differences in 5‐year survival in their regional cohort (patients diagnosed 1962–1992; RMS patients, n = 289). 10 Using Intergroup Rhabdomyosarcoma Study Group data from multiple clinical trials, Baker et al. also found no racial differences in 5‐year failure‐free survival, despite Black patients often having higher‐risk disease features (1983–1997; n = 2350). 11 Using different iterations of the SEER database, Ognjanovic et al. observed improved RS for Black patients (SEER‐9, 1996–2000; n = 166), whereas Johnson et al. reported worse outcomes by ethnicity, but not by race (SEER‐17, 1985–2005; n = 1228). 13 , 16 Importantly, none of these studies had available SES data to address confounding. More recently, Kehm et al. performed a mediation method analysis and reported an unadjusted mortality HR of 1.44 (95% CI: 1.10–1.88) for Black children and adolescents that did not hold after accounting for SES (SEER 18, 2000–2011; n = 1202). 41 Given our study's sample size and multivariable regression that controlled for SES, we provide the strongest evidence to date of this racial disparity in RMS outcomes. Such race‐/ethnicity‐based survival differences have been reported for many pediatric cancers and are largely considered multifactorial, with Pui et al. demonstrating that unequal access to care may be one major driving element regardless of cancer type. 42 , 43 , 44 Furthermore, enrollment in national cooperative trials, which is known to improve outcomes but also harbor racial disparities among children, warrants attention for potential differences. 45 , 46 , 47 However, data for RMS specifically are lacking. The most recently completed COG study for RMS, ARST0531, enrolled 62 Black patients out of 448 total (13.8%), similar to the proportion of NH Black cases in this study (16% of RMS cases in the USCS database). 18
Survival for the lowest SES group was lower than survival for the highest SES group, although the differences were not statistically significant. Lower survival has been associated with lower SES status in many cancers across all ages. 48 , 49 , 50 This is the first large‐scale population‐based study to explore two family residential variables (geographic region and population density) for children and adolescents with RMS, and we found that these do not appear to have a significant impact on outcomes. However, an additional exploration into other aspects of social and economic advantage/disadvantages, such as health insurance status, access to care, and availability of community support systems, may be needed to identify and address disparities in survival.
Our study must be considered in light of certain limitations. As with any registry‐based study, some clinical and biological data were not available, there is no central pathology review, and patient vital status can be compromised by lack of active follow‐up or incomplete reporting. Most significantly, tumor fusion status and therapy information (e.g., chemotherapy agents, local control timing and methods, trial enrollment) are known to affect survival but were unavailable in the NPCR database. Furthermore, evolving diagnostic definitions and ICD terminology can introduce disease misclassification. 28 , 32 , 51 For example, embryonal sarcoma comprised the vast majority of cases with the liver anatomic site but was kept in the analysis for consistency of comparison to previous population‐based studies. Notably, on post hoc analyses, we excluded such patients (n = 185) and found no change in our major finding of survival disparity by race on the univariate (HR 1.19, 95% CI: 1.04–1.36, p = 0.01) or multivariable (HR 1.15, 95% CI: 1.00–1.33, p = 0.044) models or the log‐rank test (p = 0.039). With conventional SEER staging, our study does not align with the combinatorial risk stratification system used by the COG. 52 , 53 Despite these limitations, the overall trends for survival in our population‐based data match well with the aforementioned results reported from COG clinical trials, suggesting a high concordance of findings despite the methodological study differences.
Strengths of this study include the broad coverage of the U.S. population afforded by the USCS and NPCR databases. To our knowledge, this is the largest and most representative exploration to date of incidence and outcomes for pediatric RMS in the U.S. The sample size provides the necessary power for detecting true differences between variables, which can aid in clarifying risk factors and trends that have inconsistent findings in previous analyses. Furthermore, the population‐based nature of the sample maximizes generalizability, since inclusion is not limited to those who are eligible for and/or have access to enrollment in a clinical trial with a formalized treatment protocol. Similar population‐based studies in Europe report the overall incidence and outcome data that generally match our results, though analyses into race/ethnicity differences in Europe are lacking. 24 , 25 Finally, with the addition of new variables such as geographic region and population density, our analysis offers a comprehensive investigation of potential factors related to incidence and survival for pediatric RMS.
Our study used updated national cancer registry data to assess incidence and outcomes over the past 20 years for the pediatric RMS population. We provide the largest, most comprehensive population‐based analysis to date for pediatric RMS. While overall incidence and survival rates were stable over the study period, potential disparities in outcome by race/ethnicity, SES, sex, and other variables merit attention and future research. With such a large and representative sample, our study results can assist clinicians and researchers in their decision‐making regarding risk stratification for individual patients, clinical trial design, and public health outreach initiatives.
AUTHOR CONTRIBUTIONS
Matthew T. McEvoy: conceptualization, methodology, project administration, formal analysis, writing—original draft, writing—review and editing. David A. Siegel: conceptualization, data curation, methodology, project administration, formal analysis, writing—review and editing. Shifan Dai: conceptualization, data curation, methodology, project administration, formal analysis, writing—review and editing. M. Fatih Okcu: conceptualization, methodology, project administration, formal analysis, writing—review and editing. Mark Zobeck: formal analysis, writing—review and editing. Rajkumar Venkatramani: formal analysis, writing—review and editing. Philip J. Lupo: conceptualization, methodology, project administration, formal analysis, writing—original draft, writing—review and editing. All authors participated in the manuscript final approval and agree to be accountable for all aspects of the work.
ETHICS APPROVAL
The current study is not considered human subjects research as only de‐identified registry data were used.
DISCLAIMER
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Supporting information
Figure S1
McEvoy MT, Siegel DA, Dai S, et al. Pediatric rhabdomyosarcoma incidence and survival in the United States: An assessment of 5656 cases, 2001–2017. Cancer Med. 2023;12:3644‐3656. doi: 10.1002/cam4.5211
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request by contacting uscsdata@cdc.gov. [26] The data are not publicly available due to privacy and legal restrictions.
REFERENCES
- 1. Skapek SX, Ferrari A, Gupta AA, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019;5(1):1. doi: 10.1038/s41572-018-0051-2 PMID: 30617281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li H, Sisoudiya SD, Martin‐Giacalone BA, et al. Germline cancer‐predisposition variants in pediatric rhabdomyosarcoma: A report from the Children's oncology group. J Natl Cancer Inst. 2020;113(7):875‐883. doi: 10.1093/jnci/djaa204. PMID: 33372952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hibbitts E, Chi YY, Hawkins DS, et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children's oncology group. Cancer Med. 2019. Oct;8(14):6437‐6448. doi: 10.1002/cam4.2504 Epub 2019 Aug 27. PMID: 31456361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joshi D, Anderson JR, Paidas C, et al. Age is an independent prognostic factor in rhabdomyosarcoma: A report from the soft tissue sarcoma Committee of the Children's oncology group. Pediatr Blood Cancer. 2004;42(1):64‐73. doi: 10.1002/pbc.10441 PMID: 14752797. [DOI] [PubMed] [Google Scholar]
- 5. Punyko JA, Mertens AC, Baker KS, Ness KK, Robison LL, Gurney JG. Long‐term survival probabilities for childhood rhabdomyosarcoma. A Population‐Based Evaluation. Cancer. 2005;103(7):1475‐1483. doi: 10.1002/cncr.20929 PMID: 15712283. [DOI] [PubMed] [Google Scholar]
- 6. Meza JL, Anderson J, Pappo AS, Meyer WH. Children's oncology group. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The Children's oncology group. J Clin Oncol. 2006;24(24):3844‐3851. doi: 10.1200/JCO.2005.05.3801 PMID: 16921036. [DOI] [PubMed] [Google Scholar]
- 7. Yang L, Takimoto T, Fujimoto J. Prognostic model for predicting overall survival in children and adolescents with rhabdomyosarcoma. BMC Cancer. 2014;5(14):654. doi: 10.1186/1471-2407-14-654 PMID: 25189734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children's oncology group (COG) soft‐tissue sarcoma committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59(1):5‐10. doi: 10.1002/pbc.24118 Epub 2012 Feb 29. PMID: 22378628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oberlin O, Rey A, Brown KL, et al. Prognostic factors for outcome in localized extremity rhabdomyosarcoma. Pooled analysis from four international cooperative groups. Pediatr Blood Cancer. 2015;62(12):2125‐2131. doi: 10.1002/pbc.25684. Epub 2015 Aug 10 PMID: 26257045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pui CH, Boyett JM, Hancock ML, Pratt CB, Meyer WH, Crist WM. Outcome of treatment for childhood cancer in black as compared with white children. The St Jude Children's research hospital experience, 1962 through 1992. Jama. 1995;273(8):633‐637. PMID: 7844873. [PubMed] [Google Scholar]
- 11. Baker KS, Anderson JR, Lobe TE, et al. Children from ethnic minorities have benefited equally as other children from contemporary therapy for rhabdomyosarcoma: a report from the intergroup rhabdomyosarcoma study group. J Clin Oncol. 2002;20(22):4428‐4433. doi: 10.1200/JCO.2002.11.131 PMID: 12431964. [DOI] [PubMed] [Google Scholar]
- 12. Linabery AM, Ross JA. Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975‐1999. Cancer. 2008;113(9):2575‐2596. doi: 10.1002/cncr.23866 PMID: 18837040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson KA, Aplenc R, Bagatell R. Survival by race among children with extracranial solid tumors in the United States between 1985 and 2005. Pediatr Blood Cancer. 2011;56(3):425‐431. doi: 10.1002/pbc.22825 PMID: 21072824. [DOI] [PubMed] [Google Scholar]
- 14. Friedrich P, Itriago E, Rodriguez‐Galindo C, Ribeiro K. Racial and ethnic disparities in the incidence of pediatric extracranial embryonal tumors. J Natl Cancer Inst. 2017;109(10):djx050. doi: 10.1093/jnci/djx050 PMID: 29117360. [DOI] [PubMed] [Google Scholar]
- 15. Arndt CAS, Bisogno G, Koscielniak E. Fifty years of rhabdomyosarcoma studies on both sides of the pond and lessons learned. Cancer Treat Rev. 2018;68:94‐101. doi: 10.1016/j.ctrv.2018.06.013 Epub 2018 Jun 19. PMID: 29940525. [DOI] [PubMed] [Google Scholar]
- 16. Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975‐2005. Cancer. 2009;115(18):4218‐4226. doi: 10.1002/cncr.24465 PMID: 19536876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walterhouse DO, Pappo AS, Meza JL, et al. Reduction of cyclophosphamide dose for patients with subset 2 low‐risk rhabdomyosarcoma is associated with an increased risk of recurrence: A report from the soft tissue sarcoma Committee of the Children's oncology group. Cancer. 2017;123(12):2368‐2375. doi: 10.1002/cncr.30613 Epub 2017 Feb 17. PMID: 28211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hawkins DS, Chi YY, Anderson JR, et al. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate‐risk rhabdomyosarcoma: A report from the Children's oncology group. J Clin Oncol. 2018;36(27):2770‐2777. doi: 10.1200/JCO.2018.77.9694 Epub 2018 Aug 9. PMID: 30091945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malempati S, Weigel BJ, Chi YY, et al. The addition of cixutumumab or temozolomide to intensive multiagent chemotherapy is feasible but does not improve outcome for patients with metastatic rhabdomyosarcoma: A report from the Children's oncology group. Cancer. 2019;125(2):290‐297. doi: 10.1002/cncr.31770 Epub 2018 Oct 23. PMID: 30351457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez EA, Kassira N, Cheung MC, Koniaris LG, Neville HL, Sola JE. Rhabdomyosarcoma in children: a SEER population based study. J Surg Res. 2011;170(2):e243‐e251. doi: 10.1016/j.jss.2011.03.001 Epub 2011 Mar 29. PMID: 21529833. [DOI] [PubMed] [Google Scholar]
- 21. Sultan I, Qaddoumi I, Yaser S, Rodriguez‐Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: An analysis of 2,600 patients. J Clin Oncol. 2009;27(20):3391‐3397. doi: 10.1200/JCO.2008.19.7483 Epub 2009 Apr 27. PMID: 19398574. [DOI] [PubMed] [Google Scholar]
- 22. Ferrari A, Sultan I, Huang TT, et al. Soft tissue sarcoma across the age spectrum: a population‐based study from the surveillance epidemiology and end results database. Pediatr Blood Cancer. 2011;57(6):943‐949. doi: 10.1002/pbc.23252 Epub 2011 Jul 25. PMID: 21793180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amer KM, Thomson JE, Congiusta D, et al. Epidemiology, incidence, and survival of rhabdomyosarcoma subtypes: SEER and ICES database analysis. J Orthop Res. 2019;37(10):2226‐2230. doi: 10.1002/jor.24387 Epub 2019 Jun 28. PMID: 31161653. [DOI] [PubMed] [Google Scholar]
- 24. Gatta G, Botta L, Rossi S, et al. Childhood cancer survival in Europe 1999–2007: Results of EUROCARE‐5—A population‐based study. Lancet Oncol. 2014;15(1):35‐47. doi: 10.1016/S1470-2045(13)70548-5 Epub 2013 Dec 5. Erratum in: Lancet Oncol. 2014 Feb;15(2):e52. PMID: 24314616. [DOI] [PubMed] [Google Scholar]
- 25. Nakata K, Ito Y, Magadi W, et al. Childhood cancer incidence and survival in Japan and England: A population‐based study (1993‐2010). Cancer Sci. 2018. Feb;109(2):422‐434. doi: 10.1111/cas.13457 Epub 2017 Dec 26. PMID: 29178401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Centers for Disease Control and Prevention . Division of Cancer Prevention and Control. National Program of Cancer Registries. 2022. Accessed March 23, 2022. https://www.cdc.gov/cancer/npcr/ [Google Scholar]
- 27. Steliarova‐Foucher E, Stiller C, Lacour B, Kaatsch P. International classification of childhood cancer, third edition. Cancer. 2005;103(7):1457‐1467. doi: 10.1002/cncr.20910 PMID: 15712273. [DOI] [PubMed] [Google Scholar]
- 28. World Health Organization , ed. International Classification of Diseases for Oncology. 3rd ed. World Health Organization; 2000. [Google Scholar]
- 29. Rudzinski ER, Kelsey A, Vokuhl C, et al. Pathology of childhood rhabdomyosarcoma: A consensus opinion document from the Children's oncology group, European Paediatric soft tissue sarcoma study group, and the cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer. 2021;68(3):e28798. doi: 10.1002/pbc.28798. Epub 2020 Dec 11 PMID: 33306276. [DOI] [PubMed] [Google Scholar]
- 30. National Library of Medicine (U.S.) . (2015), Combination chemotherapy with or without temsirolimus in treating patients with intermediate risk rhabdomyosarcoma. Identifier NCT 02567435. Accessed March 23, 2022. https://clinicaltrials.gov/ct2/show/NCT02567435
- 31. Centers for Disease Control and Prevention . Division of Cancer Prevention and Control. United States Cancer Statistics. Merged Summary Stage. Updated June 22, 2021. Accessed March 23, 2022. https://www.cdc.gov/cancer/uscs/public‐use/dictionary/data‐variable.htm?dataVarDesc=merged‐summary‐stage&dataVarAlt=merged‐stage&dbName=us
- 32. Siegel DA, Li J, Ding H, Singh SD, King JB, Pollack LA. Racial and ethnic differences in survival of pediatric patients with brain and central nervous system cancer in the United States. Pediatr Blood Cancer. 2019;66(2):e27501. doi: 10.1002/pbc.27501 Epub 2018 Oct 23. PMID: 30350913; PMCID: PMC6314020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. National Cancer Institute . Division of Cancer Control & Population Sciences. Joinpoint Trend Analysis Software. Updated March 25, 2021. Accessed March 23, 2022. https://surveillance.cancer.gov/joinpoint/
- 34. Siegel DA, Richardson LC, Henley SJ, et al. Pediatric cancer mortality and survival in the United States, 2001‐2016. Cancer. 2020;126(19):4379‐4389. doi: 10.1002/cncr.33080 Epub 2020 Jul 29. PMID: 32725630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. National Cancer Institute. Surveillance, Epidemiology, and End Results Program . SEER*Stat Software. Accessed March 23, 2022. https://seer.cancer.gov/seerstat/
- 36. SAS Institute Inc . SAS/ACCESS® 9.4 Interface to ADABAS: Reference. SAS Institute Inc; 2013. [Google Scholar]
- 37. Siegel DA, King J, Tai E, Buchanan N, Ajani UA, Li J. Cancer incidence rates and trends among children and adolescents in the United States, 2001–2009. Pediatrics. 2014. Oct;134(4):e945‐e955. doi: 10.1542/peds.2013-3926 Epub 2014 Sep 8. PMID: 25201796; PMCID: PMC4536809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Siegel DA, Li J, Henley SJ, et al. Incidence rates and trends of pediatric cancer – United States, 2001‐2014. Poster Presented at: American Society of Pediatric Hematology‐Oncology Annual Meeting. 2018; Pittsburgh, Pennsylvania, United States. [Google Scholar]
- 39. Geris JM, Spector LG. Race, ethnicity, and socioeconomic differences in incidence of pediatric embryonal tumors in the United States. Pediatr Blood Cancer. 2020;67(9):e28582. doi: 10.1002/pbc.28582 Epub 2020 Jul 16. PMID: 32672899; PMCID: PMC7674242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kehm RD, Spector LG, Poynter JN, Vock DM, Osypuk TL. Socioeconomic status and childhood cancer incidence: A population‐based multilevel analysis. Am J Epidemiol. 2018;187(5):982‐991. doi: 10.1093/aje/kwx322 PMID: 29036606; PMCID: PMC5928452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kehm RD, Spector LG, Poynter JN, Vock DM, Altekruse SF, Osypuk TL. Does socioeconomic status account for racial and ethnic disparities in childhood cancer survival? Cancer. 2018;124(20):4090‐4097. doi: 10.1002/cncr.31560 Epub 2018 Aug 20. PMID: 30125340; PMCID: PMC6234050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pui CH, Pei D, Pappo AS, et al. Treatment outcomes in black and white children with cancer: Results from the SEER database and St Jude Children's research hospital, 1992 through 2007. J Clin Oncol. 2012;30(16):2005‐2012. doi: 10.1200/JCO.2011.40.8617 Epub 2012 Apr 30. PMID: 22547602; PMCID: PMC3383176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhatia S. Disparities in cancer outcomes: Lessons learned from children with cancer. Pediatr Blood Cancer. 2011;56(6):994‐1002. doi: 10.1002/pbc.23078 Epub 2011 Feb 15. PMID: 21328525; PMCID: PMC3369622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Austin MT, Nguyen H, Eberth JM, et al. Health disparities are important determinants of outcome for children with solid tumor malignancies. J Pediatr Surg. 2015;50(1):161‐166. doi: 10.1016/j.jpedsurg.2014.10.037. PMID: 25598116; PMCID: PMC4408987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lund MJ, Eliason MT, Haight AE, Ward KC, Young JL, Pentz RD. Racial/ethnic diversity in children's oncology clinical trials: Ten years later. Cancer. 2009;115(16):3808‐3816. doi: 10.1002/cncr.24437 PMID: 19484783. [DOI] [PubMed] [Google Scholar]
- 46. Aristizabal P, Singer J, Cooper R, et al. Participation in pediatric oncology research protocols: Racial/ethnic, language and age‐based disparities. Pediatr Blood Cancer. 2015;62(8):1337‐1344. doi: 10.1002/pbc.25472 Epub 2015 Mar 8. PMID: 25755225; PMCID: PMC4482802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Faulk KE, Anderson‐Mellies A, Cockburn M, Green AL. Assessment of enrollment characteristics for Children's oncology group (COG) upfront therapeutic clinical trials 2004‐2015. PLoS One 2020;15(4):e0230824. 10.1371/journal.pone.0230824. PMID: 32324751; PMCID: PMC7179840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gupta S, Wilejto M, Pole JD, Guttmann A, Sung L. Low socioeconomic status is associated with worse survival in children with cancer: A systematic review. PLoS One. 2014;9(2):e89482. doi: 10.1371/journal.pone.0089482 PMID: 24586813; PMCID: PMC3935876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aristizabal P, Winestone LE, Umaretiya P, Bona K. Disparities in pediatric oncology: the 21st century opportunity to improve outcomes for children and adolescents with cancer. Am Soc Clin Oncol Educ Book. 2021;41:e315‐e326. doi: 10.1200/EDBK_320499 PMID: 34061564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee JM, Wang X, Ojha RP, Johnson KJ. The effect of health insurance on childhood cancer survival in the United States. Cancer. 2017;123(24):4878‐4885. doi: 10.1002/cncr.30925 Epub 2017 Sep 11. PMID: 28891067. [DOI] [PubMed] [Google Scholar]
- 51. Choi JH, Ro JY. The 2020 WHO classification of tumors of soft tissue: Selected changes and new entities. Adv Anat Pathol. 2021;28(1):44‐58. doi: 10.1097/PAP.0000000000000284 PMID: 32960834. [DOI] [PubMed] [Google Scholar]
- 52. Lawrence W, Anderson JR, Gehan EA, Maurer H. Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the intergroup rhabdomyosarcoma study group. Cancer. 1997;80:1165‐1170. [PubMed] [Google Scholar]
- 53. Raney RB, Anderson JR, Barr FG, et al. Rhabdomyosarcoma and undifferentiated sarcoma in the first two decades of life: A selective review of intergroup rhabdomyosarcoma study group experience and rationale for intergroup rhabdomyosarcoma study V. J Pediatr Hematol Oncol. 2001;23(4):215‐220. doi: 10.1097/00043426-200105000-00008 PMID: 11846299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
The data that support the findings of this study are available on request by contacting uscsdata@cdc.gov. [26] The data are not publicly available due to privacy and legal restrictions.